 J. A. Carlisle
J. A. Carlisle M. A. Glenski2
M. A. Glenski2 W. J. Swanson
W. J. Swanson
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 07 April 2022
Sec. Molecular and Cellular Reproduction
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.795273
This article is part of the Research Topic Fertilization in the Spotlight: Dynamics and Mechanisms of Sperm-Egg Interaction View all 16 articles
Reproductive proteins mediating fertilization commonly exhibit rapid sequence diversification driven by positive selection. This pattern has been observed among nearly all taxonomic groups, including mammals, invertebrates, and plants, and is remarkable given the essential nature of the molecular interactions mediating fertilization. Gene duplication is another important mechanism that facilitates the generation of molecular novelty through functional divergence. Following duplication, paralogs may partition ancestral gene function (subfunctionalization) or acquire new roles (neofunctionalization). However, the contributions of duplication followed by sequence diversification to the molecular diversity of gamete recognition genes has been understudied in many models of fertilization. The marine gastropod mollusk abalone is a classic model for fertilization. Its two acrosomal proteins (lysin and sp18) are ancient gene duplicates with unique gamete recognition functions. Through detailed genomic and bioinformatic analyses we show how duplication events followed by sequence diversification has played an ongoing role in the evolution of abalone acrosomal proteins. The common ancestor of abalone had four members of its acrosomal protein family in a tandem gene array that repeatedly experienced positive selection. We find that both sp18 paralogs contain positively selected sites located in different regions of the paralogs, suggestive of functional divergence where selection acted upon distinct binding interfaces in each paralog. Further, a more recent species-specific duplication of both lysin and sp18 in the European abalone H. tuberculata is described. Despite clade-specific acrosomal protein paralogs, there are no concomitant duplications of egg coat proteins in H. tuberculata, indicating that duplication of egg proteins per se is not responsible for retention of duplicated acrosomal proteins. We hypothesize that, in a manner analogous to host/pathogen evolution, sperm proteins are selected for increased diversity through extensive sequence divergence and recurrent duplication driven by conflict mechanisms.
Despite their essential role in many organisms, genes functioning in fertilization or sexual reproduction are often rapidly diverging between closely related species including mammals, birds, fish, and invertebrates (Swanson and Vacquier, 2002; Swanson et al., 2003; Carlisle and Swanson, 2020). Some pairs of interacting sperm and egg gamete recognition proteins have been shown to be rapidly co-evolving, pointing to sexual conflict or sexual selection driving the rapid evolution of fertilization genes (Kamei and Glabe, 2003; Clark et al., 2009; Bianchi et al., 2014; Grayson, 2015). This rapid diversification of sperm and egg gamete recognition proteins at a sequence level can result in species-specific fertilization function (Zigler et al., 2005; Avella et al., 2014; Raj et al., 2017). Investigations into the evolution of reproductive genes paired with characterization of species-specific function can provide unique insights into infertility and reproductive isolation (Lehmann, 2018).
In addition to sequence evolution, gene duplication can also contribute to the molecular diversification of reproductive protein families. Drosophila seminal fluid proteins often undergo duplication and diversification, with many of these duplications being species-specific (Wagstaff and Begun, 2005; Findlay et al., 2008; Almeida and Desalle, 2009; Sirot et al., 2014; Doty et al., 2016; Wilburn et al., 2017). In mammals many well-studied reproductive proteins belong to paralogous gene families that show interesting patterns of duplication and diversification (Cai and Clapham, 2008; Aagaard et al., 2010; Grayson and Civetta, 2012; Cooper and Phadnis, 2017). The paralogous members of the mammalian CatSper gene family show recurrent patterns of positive selection (Cai and Clapham, 2008; Cooper and Phadnis, 2017). The Izumo protein family contains four paralogs associated with various reproductive functions (including the sperm fertilization gene Izumo1) and these paralogs are undergoing positive selection in differing phylogenetic groups (Grayson and Civetta, 2012). ZP2 and ZP3 are paralogous mammalian egg coat glycoproteins with differing functions in sperm-recognition and both genes have been shown to undergo positive selection in some lineages (Carlisle and Swanson, 2020). Together these examples in mammals and Drosophila point to a recurring pattern of duplication paired with clade-specific sequence divergence of reproductive proteins across animals. This process likely leads to functional diversification of reproductive protein paralogs. Post-duplication, functions may be partitioned between paralogs in a process called subfunctionalization or a paralog may acquire a new function via neofunctionalization (Rastogi and Liberles, 2005). Here, we investigate how both sequence diversification and duplication together contribute to the molecular diversification of fertilization proteins in abalone.
The marine gastropod abalone (Genus Haliotis) is a classic model system for studying the function of gamete recognition proteins and their evolution. Abalone sperm have an extremely large acrosome containing two gamete recognition proteins, lysin and sp18 (Lewis et al., 1980). Lysin is the sperm mediator of the dissolution of the egg’s vitelline envelope (VE), and sp18 is thought to mediate sperm-egg plasma membrane fusion (Swanson and Vacquier, 1995a; Wilburn et al., 2018; Carlisle and Swanson, 2020). The abalone egg VE is an elevated glycoproteinaceous layer homologous to the mammalian egg zona pellucida (ZP) (Carlisle and Swanson, 2020). The abalone VE and mammalian ZP are biochemically and structurally similar (Mozingo et al., 1995) and both contain proteins with ZP-N domains (Swanson et al., 2011; Avella et al., 2014; Raj et al., 2017; Carlisle and Swanson, 2020). Binding between lysin and the ZP-N domains of the vitelline envelope receptor for lysin (VERL) leads to the non-enzymatic dissolution of the VE (Swanson and Vacquier, 1997; Aagaard et al., 2013; Raj et al., 2017). Three-dimensional structures of lysin, VERL and their complex have been investigated using crystallography and NMR (Kresge et al., 2000, 2001; Aagaard et al., 2013; Raj et al., 2017; Wilburn et al., 2018). The highly fusogenic protein sp18 is the putative mediator of sperm-egg plasma membrane fusion in abalone (Swanson and Vacquier, 1995a; Kresge et al., 2001). The structure of sp18 has been determined via crystallography, but no binding receptor or fertilization mechanism has been identified (Kresge et al., 2001).
Investigations into the evolution of abalone reproductive proteins have provided valuable insights within the field of reproductive biology. Lysin, sp18 and VERL have each been shown to evolve under positive selection by analysis of the ratio of rates of nonsynonymous substitutions to rates of synonymous substitutions (dN/dS > 1) (Lee et al., 1995; Swanson and Vacquier, 1995a; Galindo et al., 2003). Further, population genetic analysis indicate that lysin and its binding partner VERL are coevolving with each other (Clark et al., 2009). Previous studies of abalone egg and sperm gamete recognition proteins hint at the importance of duplication events for their evolution. Despite their divergent functions in reproduction and low sequence similarity, sp18 and lysin are paralogs with similar three-dimensional protein structures (Kresge et al., 2001). While a protein with amino acid sequence similarity and functional similarity to lysin has been identified in Tegula marine snails, a homolog to sp18 has not, potentially indicating the duplication event leading to the creation of sp18 and lysin is ancestral to abalone (Hellberg and Vacquier, 1999). The common ancestor of abalone lysin and sp18 is hypothesized to have mediated both VE dissolution and sperm egg fusion, post-duplication these ancestral functions were partitioned to lysin and sp18, respectively.
Additional evidence suggests that duplication may be contributing to the evolution of abalone sperm fertilization proteins on a more recent timescale. The European abalone H. tuberculata has a species-specific duplication of lysin (Clark et al., 2007). On the egg side there is also evidence of extensive gene duplication. In addition to VERL, abalone VEs contain ∼30 homologous VEZP proteins containing ZP-N domains (Aagaard et al., 2006; Aagaard et al., 2010). Many of these VEZPs may be structural components of the VE that play no role in gamete recognition (Killingbeck and Swanson, 2018). However, one protein (VEZP-14) is a close paralog of VERL that has undergone positive selection and is capable of binding lysin (Aagaard et al., 2013). Abalone VEZPs have only been described in two species (H. rufescens, and H. fulgens) from the North American clade. It is unknown whether there is variation in gene content across more distantly related abalone species (Aagaard et al., 2006).
In this study we investigated the contributions of duplication and sequence diversification to the evolution of proteins mediating fertilization across the genus Haliotis. Using new testes and ovary transcriptomic data and published genome assemblies we discovered novel duplications of the acrosomal proteins lysin and sp18. Some of these paralogs are ancestral to abalone and others are clade-specific. Further we discover signatures of positive selection in many of the paralogs and identify differences in distributions of positively selected sites between paralogs that suggest selection for diversification in function (subfunctionalization or neofunctionalization). Our detailed evolutionary genomic analysis reveals how recurrent patterns of duplication paired with diversification led to the evolution of abalone gamete recognition proteins and their variation between species. Repeated duplications within the protein family containing lysin and sp18 parallels the duplication and diversification of other reproductive protein families, such as mammalian Izumo and CatSper families.
To identify potential transcripts present in abalone gonadal tissue, methods were adapted from the PacBio Iso-seq protocolto create cDNA libraries. Ovary and testes transcriptome libraries were prepared for PacBio sequencing. RNA was extracted from H. tuberculata ovary and testes samples by cesium chloride density gradient centrifugation (MacDonald et al., 1987). RNA samples were enriched for mRNA by using the Oligotext mRNA Mini Kit from Qiagen. Purified mRNA was used as the template for single stranded cDNA synthesis using the Clontech SMARTer cDNA Synthesis Kit. The cDNA was amplified by PCR using the AccuPrime High-Fidelity Taq system (Invitrogen, Carlsbad, CA, United States). Double stranded synthesis conditions were optimized with the following PCR program: °C for 2 min, followed by 20 cycles of 94°C for 30°s, 55°C for 30°s, and 68°C for 10 min. We used unique identifying barcoded PCR primers to amplify testes (barcode: CTGCGTGCTCTACGAC) and ovary (barcode: TCAGACGATGCGTCAT) cDNA. Because of size bias during PacBio sequencing, double stranded cDNA was fractionated using Ampure XP Beads (Beckman Coulter Life Sciences) into two fractions (Ratio of 4:1 of 0.45×: 0.6× size selection). Testis and ovary cDNA were sequenced using the PacBio RSII and was performed by the Washington State University Genomics Core.
We conducted phylogenetic and molecular evolutionary analysis using a combination of pre-existing Genbank sequences, sequences identified from published genomes or transcriptomes, sequences identified from newly generated ovary and testes PacBio transcriptomes. The process for identifying sequences from pre-existing or newly generated datasets is explained below, a summary of the datasets used can be found in Supplementary Table S1. The list of sequences included in our phylogentic analysis of lysin, sp18, and their paralogs and their sources is included in Supplementary Table S2.
Sequences of sp18 and lysin from the genus Haliotis were retrieved from NCBI Genbank sequence repository (accession numbers for lysin: L26270-79, L26281, L35180-81, L36589, M34388-89, M59968-71, M98874-75, HM582239; accession numbers for sp18: L36552-54, L36589-90, MN102340-42). These sp18 and lysin sequences were used as the initial query sequences when identifying paralogs in abalone transcriptomes and genomes. Queries of the H. rufescens Illumina-based testis transcriptome (Palmer et al., 2013) and the H. tuberculata PacBio testes transcriptome were conducted with tblastn with an e-value cutoff of 1e-10. Significant matches from the testes transcriptomes were searched against the NCBI sequence repository (July 2020) using tblastn in order to confirm homology to lysin or sp18 (McGinnis and Madden, 2004). New sequences were uploaded to Genbank under accession numbers OK491874-OK491877.
Regions of publicly available abalone genomes containing novel acrosomal protein duplications of sp18 and lysin were identified by using tblastn with a e-value cutoff of 1e-10 (Nam et al., 2017; Botwright et al., 2019; Gan et al., 2019; Masonbrink et al., 2019). Samtools faidx was used to extract the region of scaffolds containing the tblastn hits and 20,000 base pairs upstream and downstream of the hit. We predicted the exonic sequences of the sp18 and lysin paralogs from these extracted regions using the Protein2 Genome command of the program Exonerate version 2.2.0 (Slater and Birney, 2005). The top scoring prediction from Exonerate was used to define the paralog’s exons. We used the same lysin and sp18 sequences from the tblastn search as query sequences. For all full-length sequences, the SignalP-5.0 prediction server was used to predict presence of functional signal peptides (Almagro Armenteros et al., 2019). Presence of signal peptides were predicted with probabilities >0.9; the signal peptide cleavage site was predicted with a probability >0.5.
The phylogenetic inference tool RAxML-NG was used to construct all phylogenetic trees with the LG substitution matrix (Le and Gascuel, 2008; Kozlov et al., 2019). RaxML-NG conducts maximum likelihood based phylogenetic inference and provides branch support using non-parametric bootstrapping (Kozlov et al., 2019). The best scoring topology of 20 starting trees (10 random and 10 parsimony-based) was chosen. RaxML-NG was used to perform non-parametric bootstrapping with 1,000 re-samplings that were used to re-infer a tree for each bootstrap replicate MSA. Finally, we mapped the bootstrap scores on the best-scoring starting tree. The Transfer Bootstrap Expectation (TBE) was used as a branch support metric (Lemoine et al., 2018).
DNA multiple sequence alignments (MSA) for phylogenies of lysin and sp18 and their respective paralogs were constructed. First the protein sequences of the genes were aligned using PROMALS3D (Pei et al., 2008a; Pei et al., 2008b). PROMALS3D may use protein three-dimensional protein structures to inform protein alignments. The representative PDB 5UTG was used for lysin and lysin paralogs and the PDB 1GAK was used for sp18 and sp18-dup sequences (Kresge et al., 2000; Wilburn et al., 2018). For alignments of VEZP sequences, no structure PDB was used. The protein MSAs were used to create DNA alignments of the same genes using the Pal2Nal sever (Suyama et al., 2006). Gaps were not removed from the alignments for our analysis. After paralog identification, new MSAs of orthologous sequences were constructed for positive selection analysis using the method described above. For phylogenetic analysis of H. rufescens and H. tuberculata VEZP sequences, the protein sequences of the C-terminal ZP modules of each of the proteins were aligned using PROMALS3D (Pei et al., 2008b).
The published genome of Haliotis rufescens is annotated with ORFs identified via transcriptomic sequencing (Masonbrink et al., 2019). We collected the sequences of 2-3 large annotated ORFs surrounding lysin, sp18, and their newly described paralogs within the H. rufescens genome. We used these sequences as BLAST queries against the H. rubra genome (Gan et al., 2019). The top hits for the H. rufescens ORFs were annotated onto the H. rubra genome and used to establish synteny between H. rubra scaffold 62 and the H. rufescens scaffolds 48 and 101. A reciprocal blast of the regions identified as orthologous ORFs in H. rubra were queried against the H. rufescens genome to verify orthology. H. rufescens and H. rubra were chosen as representative genomes from North American and Australian abalone, respectively. The genome assemblies of the North American abalone species H. sorenseni and H. fulgens are based on the H, rufescens assembly. The genome assembly of the North American abalone H. discus contains shorter scaffolds than the other published genomes, thereby preventing synteny analysis of this genomic region. The Australian abalone genomes of H. laevigata and H. rubra are similar, H. rubra was arbitrarily chosen to compare to H. rufescens, however H. laevigata shows the same syntenic relationship between sp18 and lysin paralogs.
Values of dN/dS for genes were estimated using the codeml program of PAML 4.8 (Yang, 2007). We compared models of selection using a likelihood ratio test (LRT) between neutral models and models with positive selection. Specifically, we compared M1a v. M2a, M7 v. M8, and M8a v. M8 using the codon frequency model F3X4. Likelihood ratio tests were performed where the likelihood ratio (LRT) statistic was twice the negative difference in likelihoods between nested models. For M1a v M2a or M7 v Model 8 the LRT was compared to the χ2 distribution with 2 degrees of freedom (Yang, 2007). For the M8a v. M8 comparison, twice the negative difference in likelihoods between the nested models being compared, the LRT statistic, is approximated by the 50-50 mixture distribution of 0 and χ2 with degree of freedom 1 (Swanson et al., 2003). Convergence was checked by running the analysis from multiple initial omega values. To identify specific sites in proteins evolving under positive selection, we used a Bayes Empirical Bayes threshold of [Pr (omega > 1) = 0.75].The threshold of 0.75 was chosen since it gave a sufficient number of positively selected sites in each paralog in order to perform our analysis while still reliably identifying positively selected sites. According to simulations run in Yang et al., 2005, the false positive rate of detecting positively selected sites using the BEB method is lower than 1—(Pr (omega > 1), with a threshold value of [Pr (omega > 1) = 0.7] leading to a false positive rate of 0.03 (Yang et al., 2005).
We designed three unbiased tests to determine if sites under positive selection in either sp18 or sp18-dup are differentially clustered between paralogs. First, we created a parametric test based on the Wald-Wolfowitz runs test (Magel and Wibowo, 1997) to determine whether positively selected sites in the paralogs sp18 and sp18-dup were non-randomly distributed in a protein alignment of both paralogs. The Wald-Wolfowitz runs test determines the randomness of a two-category data string by examining changes between categories by counting “runs.” We designed a parametric version of the test to allow the inclusion of three categories. The categories were sites under positive selection in sp18, sites under selection in sp18-dup, and sites under selection in both. The order in which these sites under selection in the categories appeared in an alignment of H. fulgens sp18 and H. sorenseni sp18-dup became our data string. For the data string generated from our paralog alignment we counted how many times the identity of sites in the string changed plus one. A visualization of the pipeline for preparing this data string and counting “runs” is shown in Supplementary Figure S1A.
To make a parametric version of this runs test, we generated 1,000 simulated data strings via bootstrapping based on the proportion of sites shown to be under positive selection in either paralog. Each simulated data string was created by sampling from two strings 142 times each (142 is the length of the protein alignment between paralogs). Each string simulates the chance of a site randomly being positively selected in either paralog or not based on the proportions of the observed data string. If a site is simulated as undergoing positive selection in both paralogs it is marked in the simulated data string as such. For each of these simulated data strings we also calculated the number of runs. The number of data strings with a count of “runs” less than or equal to the count of “runs” found in the data string derived from the paralog protein alignment divided by the total number of simulated data strings gives the parametric probability that by random chance categories of sites would be more or equally clustered compared to the true clustering observed. We also performed a version of the test where sites under selection in both paralogs were eliminated from the analysis. When these sites were eliminated the parametric runs test retained statistical significance (p-value <0.001).
For our second test we evaluated whether sites under positive selection in sp18 vs. sp18-dup were distributed throughout the sp18 crystal structure (1GAK) in a significantly different way. In MATLAB Online version 9.9, we identified the plane of best fit between the c-alpha carbons (the first carbon attached to the functional group of an amino acid) of the sp18 crystal structure using linear regression (Supplementary Figure S2A, Supplementary File S1). This plane divided the sp18 crystal structure into two sides that we arbitrarily designated “left” and “right” (Supplementary Figure S2B). We mapped the 62 sites in sp18 and the 30 sites in sp18-dup that are under positive selection onto the sp18 crystal structure. Given the number of amino acids sites in each side of the crystal structure, we estimated the expected number of positively selected sites from each paralog that would be expected to be located on either side as the number of amino acid sites on a side divided by the total number of amino acid sites in the molecule and multiplied by the number of positively selected sites in a sp18 paralog. We used a chi-square test to examine whether the real distribution of sites between categories rejected the null expectation. This test determined whether the distribution of sites under selection in either paralog was not distributed similarly between the sides of the protein. We also created a plane perpendicular to the plane of best fit to divide the sp18 crystal structure into the categories “top” and “bottom” (Supplementary Figure S2C). We repeated an analysis for this new pair of categories that is identical to what was described previously. This analysis gave us a sense if sites under selection in either paralog were clustered nonrandomly throughout the crystal structure in different ways.
For our last clustering test we determined whether sites under positive selection in a paralog were more likely to be close in proximity in three-dimensional space to another site under positive selection in the same paralog rather than a site under positive selection in the other paralog. For each site that was under selection in a paralog, we identified the closest positively selected site in three-dimensional space that belonged to either paralog. We then calculated the expected number of times by chance the closest adjacent site for each site under positive selection would belong to the same gene rather than the other paralog. We used a chi-square test to determine whether observed sites under selection in one paralog were statistically more likely to be close to positively selected sites belonging to the same paralog than what would be expected by chance. This test has four categories of sites (for each paralog the nearest site could belong to the same paralog or not), and therefore three degrees of freedom were used to determine the p-value. Sites that were under selection in both paralogs were counted twice in this analysis since these sites were undergoing positive selection independently in both paralogs. When the closest adjacent site to a positively selected site in one paralog was undergoing positive selection in both paralogs, the adjacent site was treated as belonging to the same paralog.
We also examine the sequence divergence of species-specific duplications of lysin and sp18 in H. tuberculata (Figure 3). In Figure 3A, sites that diverge between sp18 and lysin H. tuberculata paralogs are mapped onto the sp18 crystal structure (1GAK) and the lowest energy NMR ensemble of lysin (5UTG) (Kresge et al., 2001; Wilburn et al., 2018). Since lysin has been shown not to crystallize in its native formation (Wilburn et al., 2018), the NMR structure was chosen to better examine the clustering of sites that are diverging between H. tuberculata lysin paralogs at lysin’s VERL binding interface.
H. tuberculata testis tissue was homogenized in 1% sodium dodecyl sulfate with BME at 70°C for 30 min. Testis samples were separated by SDS-PAGE using a Tris-Tricine buffering system with discontinuous 4% resolving/15% separating acrylamide gels. Samples were electrophoresed at 50 V for 15 min followed by 100 V for 90 min. The gel was run with the BioRad Broad Range Ladder and stained with Coomassie Blue R-250 for 15 min. Using the ladder as reference, the lysin and sp18-containing region (∼14–22 kDa) of the polyacrylamide gel was excised using a clean scalpel, with multiple rounds of perfusion with an ammonium bicarbonate solution followed by acetonitrile to extract detergents and salts. Trypsin proteolysis of immobilized proteins was by perfusion of a Trypsin solution (40 μg/ml stock Trypsin 1:10 in 50 mM ammonium bicarbonate) and incubation at 37°C overnight. The supernatant from the digest was collected along with the supernatant from two rounds of hydration with ammonium bicarbonate and extraction with 50% acetonitrile. The collected supernatant containing the liberated peptides was concentrated to a dry pellet using a vacuum centrifuge then reconstituted in 0.1% FA for liquid chromatography tandem mass spectrometry (LC/MS-MS). Unique peptides for sp18 copy #1 were identified in the sample using the Crux toolkit comet command (Park et al., 2008). The protein sequence database was composed of a six-frame translation of the H. tuberculata testis transcriptome.
An exhaustive BLAST search of the H. tuberculata ovary transcriptome identified all cDNA sequences with homology to H. rufescens VEZPs. A previous study used a similar approach to originally identify known VEZPs in H. rufescens indicating that this approach should be sufficient to identify novel VEZPs (Aagaard et al., 2010). All cDNA sequences that matched VEZPs were filtered for duplicates using CD-HIT-EST with a threshold of 0.9 sequence identity (Huang et al., 2010). The longest sequence from each cluster created by CD-HIT-EST was chosen as the cluster’s representative sequence. All H. tuberculata sequences from this filtering process were translated and the C-terminal ZP modules were identified by identifying conserved cysteine residues. The ZP module protein sequences from both H. tuberculata and H. rufescens were aligned using PROMALS3D (Pei et al., 2008b). The MSA of these ZP modules from were used to construct a VEZP homolog protein phylogeny using the same RAXML-NG protocol described above for lysin and sp18 paralog phylogenies. New sequences were uploaded to Genbank under accession numbers OK491878- OK491909.
By pairing phylogenetic and genomic analysis of abalone species belonging to the North American clade (H. rufescens, H. sorenseni, H. discus) and the Australian clade (H. rubra, H. laevigata), we identifed ancestral duplications of both sp18 and lysin (Figure 1; sp18-dup and lysin-dup, respectively). We calculated maximum likelihood DNA phylogenies independently for lysin and sp18 with their paralogs and rooted the phylogenies by orthology (Figures 1A,B). Predicted intron/exon boundaries of the novel acrosomal protein paralogs were shared with lysin and sp18 (Metz et al., 1998). No mutations causing pseudogenization were detected within the predicted CDS of either paralog. For the abalone species with published genomes, only one (H. rufescens) has a published testes transcriptome (Palmer et al., 2013). Full-length sequences of lysin, sp18, and sp18-dup are expressed in the testes transcriptome of H. rufescens; however, lysin-dup was not detected.
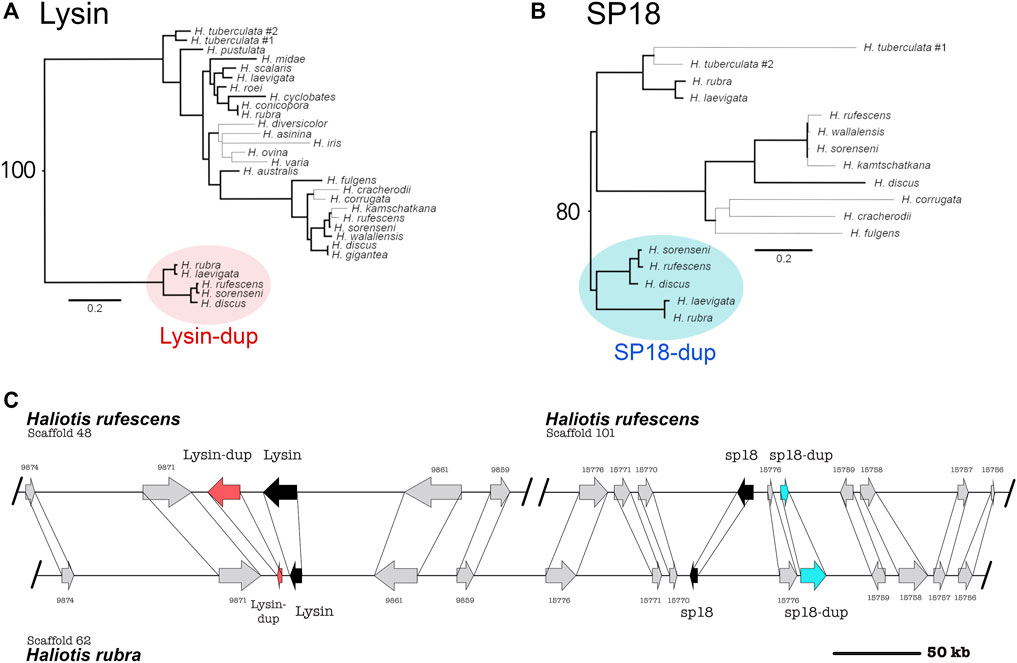
FIGURE 1. Syntenic and phylogenetic analysis indicate that four tandem acrosomal proteins are ancestral to all abalone. (A) Ancestral duplication led to the paralogs lysin and lysin-dup (Red). Bold lines indicate greater than or equal to 80% bootstrap branch support. Bootstrap support for the node connecting lysin and lysin-dup sequences is 100. (B) Ancestral duplication led to the paralogs sp18 and sp18-dup (blue). A clade specific duplication of sp18 is present in the H. tuberculata testes transcriptome. Bold lines indicate greater than or equal to 80% bootstrap branch support. Bootstrap support for the node connecting sp18 and sp18-dup sequences is 80. (C) Lysin (black), sp18 (black) and their paralogs lysin-dup (red) and sp18-dup (blue) are found in the genomes of North American and Australian abalone. Syntenic analysis indicates that paralogs are located near each other in the abalone genome, although all four paralogs are only genetically linked in Australian abalone genome assemblies. In the North American abalone H. rufescens genome assembly, lysin and sp18 are found on separate scaffolds linked with their paralogs. The scaffolds extend beyond the breakpoint shown in the (C).
Sequence analysis is consistent with the sp18-dup gene encoding a functional reproductive protein ancestral to Haliotis. Sp18-dup is predicted to have a signal peptide sequence and maintains a pair of cysteine residues involved in forming a structurally important disulfide-bond in sp18 (Kresge et al., 2000, 2001). Sp18-dup has not been identified in previous analysis due to the high divergence between it and sp18 (27.5% sequence identity between H. rufescens sp18 paralogs) obscuring homology.
Lysin-dup was identified in all abalone genomes investigated but was not detected in the testes illumina transcriptome of H. rufescens (Palmer et al., 2013). The absence of lysin-dup in the testes transcriptome could indicate insufficient read depth, differences in tissue-expression, or potentially pseudogenization. Since the full-length sequence of lysin-dup was not identified within the H. rufescens testis transcriptome, lysin sequences were used instead to identify lysin-dup exons within abalone genomes. However, divergence between lysin and lysin-dup likely prevented the identification of full-length coding sequence from abalone genomes. Only exons 2-4 could be identified (79% of query sequence) within H. rufescens and H.rubra. The missing exons 1 and 5 contain the signal peptide and the N- and C-termini of the molecule. In lysin, the N- and C-terminus are under strong positive selection promoting extensive divergence that reduces the ability to identify these exons using homology-based approaches (Lee et al., 1995; Lyon and Vacquier, 1999).
In the Australian abalone genomes lysin, lysin-dup, sp18, and sp18-dup are all found within a single contig with 233 kb separating the paralog pair of lysin and lysin-dup from the paralog pair of sp18 and sp18-dup. But in the genome of the North American abalone species H. rufescens, the paralog pair of lysin and lysin-dup are on a separate scaffold from the paralog pair of sp18 and sp18-dup. We compared the Australian contig containing the four acrosomal protein paralogs with the two H. rufescens contigs containing the lysin and sp18 paralog pairs respectively (Figure 1C). We found several ORFs surrounding each paralog pair in H. rufescens that were found in the same order between in H. rubra, indicating synteny between scaffolds. All four acrosomal proteins being located near each other in the same scaffold in the H. rubra genome suggests that tandem duplication led to recurrent duplications of this protein family (Reams and Roth, 2015). The sp18 ORF codes in a different direction than the other paralogs, suggesting that transposition and inversion may have also contributed to duplications within this protein family (Reams and Roth, 2015). In H, rufescens, the sp18 paralog pair and lysin paralog pair are found in separate scaffolds. H. rufescens scaffolds 48 and 101 extend beyond the breaking point shown in Figure 1. Therefore, the paralogs being found in separate scaffolds cannot be attributed to a fragmented genome assembly. Rather, we hypothesize that recombination led to the separation of the paralogs within H. rufescens.
Lysin, sp18, and sp18-dup all contained sites detected to be subjected to positive selection. (Table 1). Lysin-dup did not show signatures of positive selection, though this could be due to having insufficient sequences to provide the statistical power to conduct the test (Table 1) (Anisimova et al., 2001). Clustering and distribution of amino acid sites undergoing positive selection can identify regions important to the function of rapidly evolving genes (Anisimova et al., 2001). For example, many of the sites in lysin that are undergoing positive selection (11/23) are in a region of the molecule that binds its egg receptor VERL (Wilburn et al., 2018). We investigated the distribution of sites undergoing positive selection in sp18 and sp18-dup. Similar regions of the molecule undergoing positive selection in both paralogs would suggest a shared biochemical mechanism while differences in distributions of positively selected sites would indicate divergence in biochemical mechanism.
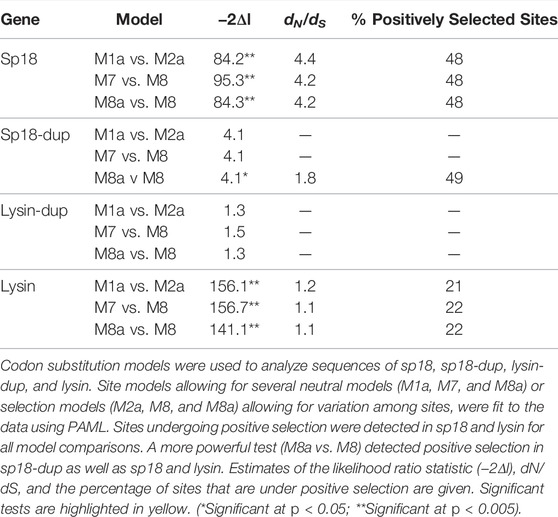
TABLE 1. Acrosomal protein paralogs are evolving under positive selection.
By mapping sites under positive selection onto a protein alignment of sp18 and sp18-dup, we determined that sites under positive selection in either paralog are non-randomly distributed across the protein alignment and differentially clustered. We analyzed the selected sites in the primary sequence alignment with a parametric adaptation of the runs test (Wald-Wolfowitz test) (Figure 2A) (Magel and Wibowo, 1997). This analysis showed that there were significant runs of sites undergoing positive selection in either paralog (p-value = 0.0331), consistent with different regions evolving under positive selection among paralogs. Positive selection acting on different regions of the protein alignment is consistent with functional divergence of paralogs.
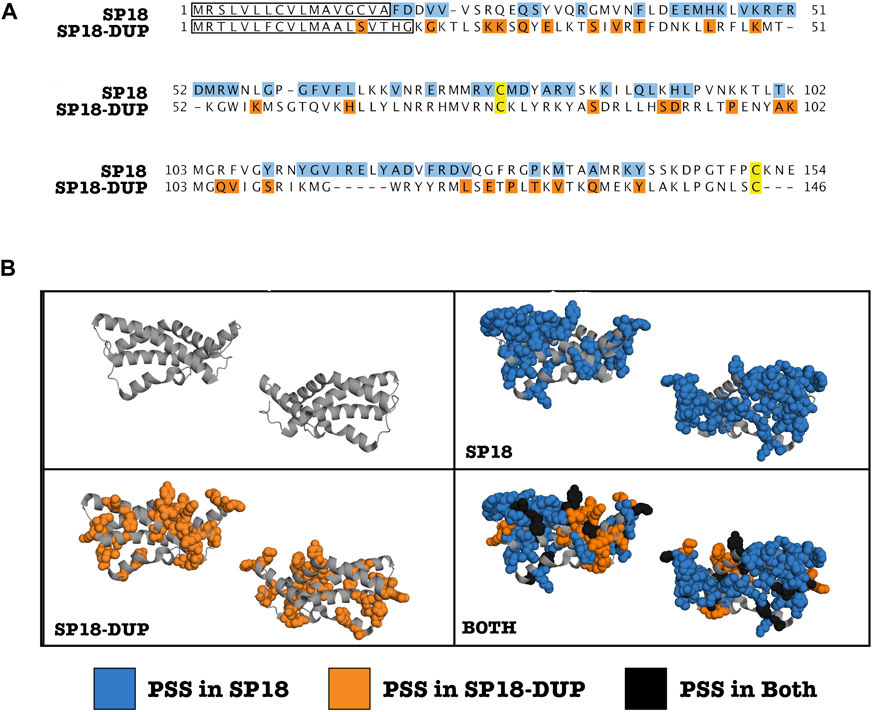
FIGURE 2. Clustering of positively selected sites differs between SP18 and SP18-dup. (A) Alignment of H. fulgens sp18 and H. sorenseni sp18-dup mature protein sequences. Sites under positive selection are highlighted. Blue (Sp18), Orange (Sp18-dup), and Black (both). A modified parametric runs test was used to determine that there were statistically significant runs in linear sequence space of sites under positive selection in sp18 or sp18-dup (p-value—0.0331). Yellow sites indicate conserved cysteines shown to be involved in forming a disulfide bond in SP18. (B) Sites under positive selection in sp18 or sp18-dup were mapped onto the crystal structure of sp18 from H. fulgens and appear clustered in 3-D space.
We investigated clustering of positively selected sites in three-dimensional space. By mapping sp18 and sp18-dup positively selected sites onto the sp18 structure, it is visually apparent that there are distinct clusters of sites under selection between paralogs (Figure 2B). Using the plane of best fit through the crystal structure we divided the molecule into “left” and “right” sides agnostic to the location of positively selected sites. To define the “top” and “bottom” of the molecule we used a plane perpendicular to the plane of best fit. Sites under positive selection in sp18-dup were statistically more likely to be on the “right” side than on the “left” (p-value <0.05), however, sp18-dup positively selected sites were not statistically significantly enriched at either the “top” or “bottom” of the molecule (Supplementary Figure S2). Sp18 sites, using the same tests, showed no statistically significant difference from the null distribution. These tests show that sites under selection in sp18 and sp18-dup are distributed differently across their three-dimensional structures.
We also developed a test to examine whether sites under selection in sp18 and sp18-dup were statistically more likely to be adjacent to a site under selection from the same paralog. Such a pattern of clustering would indicate a spatial relationship between positively selected sites belonging to a particular paralog. For each site under positive selection in sp18 or sp18-dup, we identified whether the closest positively selected site in three-dimensional space was significantly more likely to belong to the same paralog. We found that sites under selection in both paralogs were more likely to have the closest positively selected site belong to the same paralog rather than the other paralog according to a chi-squared test (p-value <0.01). Together, the runs test analysis and three-dimensional analyses point to diversifying selection post-duplication of these proteins to promote functional diversification. However, it should be noted that there is some uncertainty in the prediction of positively selected sites which is unaccounted for in our clustering analyses.
Because Lysin-dup was not detected to be under positive selection, we did not test for differences in sites under selection between lysin paralogs. However, we did evaluate how the lysin-dup sequence diverged from lysin. Many of the sites shown to be undergoing positive selection in lysin differ in sequence from lysin-dup (13/14) when comparing the H. rufescens sequences (Figure 3B). Although this comparison is not significant (p = 0.088), this suggests similar sites driving the diversification in sequence of lysin between species and between lysin and its paralog lysin-dup.
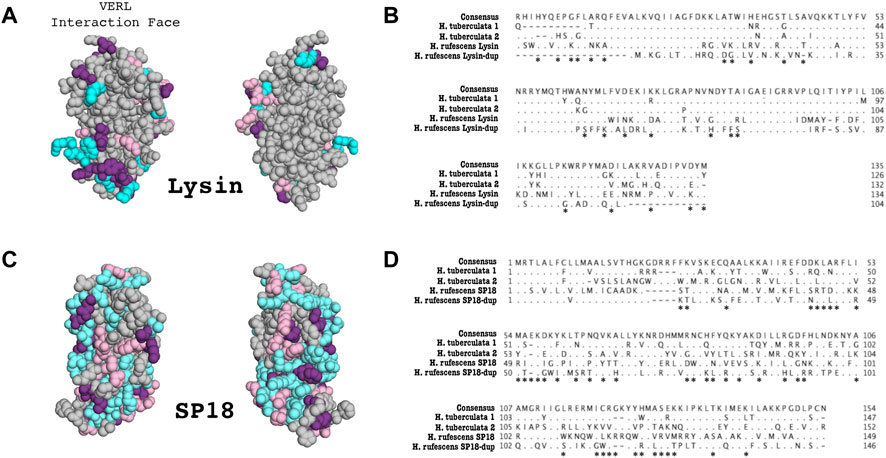
FIGURE 3. Duplication and divergence of lysin and sp18 paralogs in H. tuberculata. (A) Sites that differentiate H. tuberculata lysin paralogs from H. rufescens lysin are mapped onto the H. rufescens NMR structure (PDB 5UTG). Blue–Sites that differ between H. rufescens lysin and H. tuberculata Copy #1, Pink–Sites that differ between H. rufescens lysin and H. tuberculata Copy #2, Purple–Sites that differ between H. rufescens lysin and both H. tuberculata lysin paralogs. Notably the face of the lysin molecule that interacts with VERL harbors most substitutions between H. tuberculata paralogs, suggestive of a shared interaction face for both H. tuberculata lysin paralogs. (B) Alignment of Lysin paralogs in H. rufescens and H. tuberculata. Both sets of paralogs arose from independent duplication events. Asterisks indicate sites that are shown to be under positive selection in H. rufescens lysin. (C) Sites that differ between H. tuberculata sp18 paralogs are mapped onto the crystal structure of sp18 from H. fulgens (PDB 1GAK). Blue–Sites that differ between H. rufescens sp18 and H. tuberculata Copy #1, Pink–Sites that differ between H. rufescens sp18 and H. tuberculata Copy #2, Purple–Sites that differ between H. rufescens sp18 and both H. tuberculata sp18 paralogs. (D) Alignment of sp18 paralogs in H. rufescens and H. tuberculata. Both sets of paralogs arose from independent duplication events. Asterisks indicate sites that are shown to be under positive selection in H. rufescens sp18.
Previous work described a lysin duplication unique to H. tuberculata (Clark et al., 2007). The lysin paralogs were shown to be evolving under positive selection and to be maintained in the testis proteome (Clark et al., 2007). Sites that vary between European lysin paralogs are largely located on the face of the molecules interacting with lysin receptor VERL (Figure 3A). To investigate the presence of additional sp18 and lysin paralogs in H. tuberculata, we constructed a long-read PacBio testis transcriptome. Performing tBLASTN searches of the H. tuberculata transcriptome for lysin and sp18 revealed the previously described species-specific duplication of lysin (H. tuberculata lysin copy #1 and copy #2) and a novel duplication of sp18 (H. tuberculata sp18 copy #1 and copy #2).
Phylogenetic analysis indicates the H. tuberculata sp18 paralogs are the result of a recent duplication and not ancestral to Haliotis. Sequence information from other abalone species is needed to determine whether this duplication is species-specific to H. tuberculata; it appears to be specific to the abalone clade containing the European species. The signal sequences of the H. tuberculata sp18 paralogs are more similar to each other than to signal sequences from other species’ sp18 paralogs. Signal sequences are not part of the mature protein and not subjected to the same evolutionary pressures driving rapid divergence, therefore these sequences show more conservation between closely related paralogs. This similarity in signal sequence between H. tuberculata sp18 paralogs (15/19 sites are identical) further supports that these paralogs are the result of a non-ancestral duplication. Despite being a more recent duplication of sp18, the paralogs have a low sequence identity (39%), lower than that of the H. tuberculata lysin paralogs (83%). This is consistent with sp18 having a higher dN/dS and evolving more rapidly than lysin (Table 1). H. tuberculata sp18 paralogs maintain a pair of structurally important cysteine residues involved in forming a disulfide bond. Rapid sequence divergence, no premature stop codons, and both genes being expressed in the testis transcriptome are all indicators that both sp18 paralogs (referred to here as H. tuberculata copy #1 and copy #2) are likely to be functional.
The H. tuberculata sp18 copy #1 is the more divergent to the ancestral sequence than copy #2, as indicated by its long branch in the sp18 phylogeny (Figure 1B). When comparing the sequence identity of H. tuberculata sp18 paralogs to H. rubra sp18 (an outgroup sequence), copy #1 shows a lower sequence identity (41%) than copy #2 (69%). This rapid sequence divergence of copy #1 without accruing mutations causing pseudogenization suggests strong positive selection. However pairwise dN/dS between H. tuberculata sp18 paralogs could not be reliably estimated due to extensive divergence resulting in saturation (multiple substitutions per site) (Swanson and Vacquier, 1995a). Maintenance of both paralogs in the testis proteome despite the observed sequence divergence would indicate that both paralogs are being selected for functions, presumably related to fertilization. We used data dependent acquisition mass spectrometry to identify peptides belonging to either paralog in the H. tuberculata testes proteome. Diagnostic peptides were detected for copy #1 but not copy #2. Despite being the more divergent sp18 sequence, copy #1 is maintained in the proteome. This result indicates that copy #1 is likely important for fulfilling sp18’s membrane fusion function in H. tuberculata.
We generated an ovary PacBio transcriptome for H. tuberculata to identify VEZP proteins. Using the 33 VEZP and ZP-domain sequences from the H. rufescens ovary transcriptome as the initial query sequences, exhaustive tBlastn searches of the ovary transcriptome were used to identify all cDNA sequences with sequence similarity to any H. rufescens VEZP. ZP module protein sequences were extracted from our H. tuberculata cDNA hits and the 33 H. rufescens VEZPs and then were aligned to construct a phylogeny (Figure 4). Clustering of H. tuberculata and H. rufescens ZP module sequences indicate that these distantly related abalone species have the same complement of ZP-proteins in their transcriptomes. In H. tuberculata’s ovary transcriptome, orthologs of 32 of the 33 H. rufescens ovary ZP-domain proteins were identified. No VEZPs, including VERL and its most closely related paralogs VEZP14 and VEZP9 were duplicated. The only missing sequence belonged to ZPC, a gene whose cDNA sequence contains a premature stop codon in H. rufescens and for which no peptides were detected in the H. rufescens VE proteome (Aagaard et al., 2010). Therefore, ZPC is likely pseudogenized in H. rufescens and its expression its expression is no longer maintained in European abalone. Remarkably, no new ZP-module-containing proteins were identified in H. tuberculata despite the species having multiple clade-specific duplications of acrosomal proteins. These results suggest that the clade-specific maintenance of duplicated sperm acrosomal proteins found in the European abalone H. tuberculata are unlikely to be the result of duplicated egg proteins.
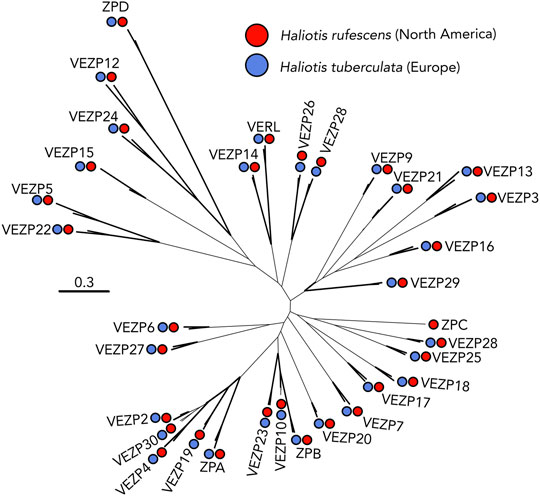
FIGURE 4. VEZP proteins are conserved across abalone species. The distantly related abalone species H. rufescens (Red) and H. tuberculata (Blue) share the same vitelline envelope zona pellucida (VEZP) proteins within their ovary transcriptomes. Although there are species-specific duplications of lysin and sp18 in H. tuberculata, there is no evidence of species-specific duplications of VERL or other VEZP proteins. Bold lines indicate greater than or equal to 80% bootstrap branch support.
Despite decades of research examining the evolution of abalone fertilization genes, only recently have genomic resources been available that enable a broad investigation into the evolution of the protein families to which lysin and VERL belong. Here, we explore the contributions of duplication and sequence divergence to the evolution of abalone fertilization genes across the genus Haliotis. For our investigation we generated ovary and testes transcriptomes from the European abalone H. tuberculata and utilized recently published North American and Australian abalone genomes and a North American abalone testes transcriptome (Palmer et al., 2013; Nam et al., 2017; Botwright et al., 2019; Gan et al., 2019; Masonbrink et al., 2019). We discovered novel duplications of both lysin and sp18 ancestral to abalone, indicating that abalone lysin and sp18 are members of an ancestral abalone protein family with four members. The newly discovered sp18 paralog (sp18-dup) was shown to be undergoing positive selection, like lysin and sp18, and expressed in the testes of North American abalone. Further, differences in clustering of positively selected sites in sp18-dup compared to sp18 is potentially consistent with a model of subfunctionalization where a distinct binding interface is undergoing positive selection in sp18-dup but not sp18. However, it is also possible that this pattern of sequence divergence could be explained by neofunctionalization and sp18-dup is acquiring a different reproductive function. We investigated whether there are clade-specific duplications of abalone VEZPs or acrosomal proteins. In addition to a species-specific lysin duplication described in a previous publication (Clark et al., 2007), the H. tuberculata testes transcriptome contains a clade-specific duplication of sp18 not found in Australian or North American abalone species. However, no duplications of VERL or other VEZPs were observed between North American or European abalone, indicating that VEZP gene content is conserved across the genus Haliotis. Together, this data demonstrates that recurrent duplication and diversification driven by positive selection drives the evolution of an acrosomal protein family involved in fertilization in Haliotis.
In the H. rubra abalone genome, the paralogs lysin, lysin-dup, sp18, and sp18-dup are found on a single scaffold. This clustering within the genome indicates that ancestral tandem duplication events occurred leading to the creation of this acrosomal protein family (Reams and Roth, 2015). Further, three of the four ancestral paralogs were shown to be maintained in the testis transcriptome and to be evolving under positive selection, a common characteristic of reproductive proteins.
This evolutionary pattern of duplication paired with sequence diversification found in the abalone acrosomal protein family can be compared to protein families in other taxa which contain sperm proteins mediating fertilization. Notably, the mammalian Izumo gene family contains four ancestral paralogs whose members all show testes-specific tissue expression in humans (Grayson and Civetta, 2012). Izumo1 is an essential gene for sperm-egg plasma membrane fusion in mammals that functions by binding the egg plasma membrane protein JUNO (Bianchi et al., 2014). There is evidence that the other three Izumo paralogs may also possess important, although potentially varied, functions in fertility (Ellerman et al., 2009). All four paralogs have been shown to be undergoing rapid sequence evolution in at least one mammalian lineage, for Izumo1, 2, and 3 this is driven by positive selection and for Izumo4 this appears to be driven by relaxed selection (Grayson and Civetta, 2012; Grayson, 2015). Given that both the abalone acrosomal protein family and the mammalian Izumo family both contain multiple paralogs showing testis-specific function, subfunctionalization may be a common driver of the evolution of fertilization and reproductive genes across taxa. Understanding how fertilization proteins emerge and evolve can be important for identifying and understanding mechanisms of fertilization across diverse taxa.
Differences in optimal mating rates for sperm and eggs can drive antagonistic coevolution of reproductive proteins. Under this sexual conflict scenario, evolution of egg coat proteins interacting with sperm acrosomal proteins could lead to constrained evolution on the sperm side (Gavrilets and Waxman, 2002). Duplication followed by diversification of sperm fertilization proteins can be an important means of sperm escaping evolutionary constraints imposed by egg protein evolution. For two duplication events within the abalone acrosomal protein family there is evidence of functional divergence from either functional experiments [lysin vs. sp18, (Swanson and Vacquier, 1995a, b, 1997; Kresge et al., 2001; Aagaard et al., 2010)] or site-clustering analysis (sp18 vs. sp18-dup, current manuscript).
Plasma membrane fusion in fertilization or other contexts is traditionally thought to consist of two steps, binding and fusion (Bianchi and Wright, 2020). In sea urchins, both steps are mediated by different regions of the same protein, su-bindin (Vacquier and Moy, 1977; Ulrich et al., 1998; Vacquier and Swanson, 2011). However, in abalone these steps may have been partitioned between sp18 and sp18-dup via subfunctionalization. Abalone eggs have a thin layer directly overlaying the surface of the plasma membrane which morphologically resembles a duplication of the elevated VE (Mozingo et al., 1995). Just as lysin binds the VE protein VERL, sp18 may bind a VEZP protein found within the thin layer overlaying the abalone egg plasma membrane. Indeed, in addition, to having a strong fusagenic function, sp18 has been demonstrated to bind to VEZP proteins, an unsurprising trait for a lysin paralog (Swanson and Vacquier, 1995a; Aagaard et al., 2010). One possibility is that the subfunctionalization of sp18 and sp18-dup may have been driven by the separation of the steps of plasma membrane binding and fusion between paralogs. Additional functional characterization of each paralog’s fusagenic function and ability to bind VEZPs is necessary to examine this hypothesis.
While the paralogs lysin-dup and lysin do show high sequence divergence, we were only able to detect evidence of positive selection in lysin. Therefore, the observed sequence divergence is likely driven by lysin’s evolution post-duplication. Unlike the other acrosomal protein family paralogs discussed in this paper, lysin-dup is not detected in the testis transcriptome. However, there is an appealing hypothesis as to its potential function. In abalone egg coats there are two VEZP proteins capable of binding lysin, VERL the major binding partner of lysin and VEZP-14 the most recent paralog of VERL (Aagaard et al., 2013). It is possible that lysin-dup may be the binding partner of VEZP-14 and if true this could explain why lysin shows correlated evolution with VERL but not VEZP-14 (Aagaard et al., 2013). Currently there is insufficient data to test for correlated rates of evolution between lysin-dup and VEZP-14. However, further molecular and biochemical characterization through binding kinetic analysis could test the hypothesis that lysin-dup and VEZP-14 interact.
Previous work described a lysin duplication unique to H. tuberculata and maintained in the testis proteome (Clark et al., 2007). In this study a clade-specific duplication of sp18 was discovered within the H. tuberculata transcriptome. Despite having two acrosomal protein duplications, European abalone’s ovary transcriptome did not reveal any novel VEZP protein sequences indicative of a duplication event. While gene duplications are an ongoing contributor to the evolution of sperm fertilization genes in abalone, this may not be true for egg fertilization genes. Our data suggests that it is not duplications on the egg side driving the duplication of abalone acrosomal proteins in H. tuberculata. This could be explained by different selective pressures on the sperm and the egg, such as sperm competition and polyspermy risk (Carlisle and Swanson, 2020). Further, it does not seem that a process of gene birth and loss explains the evolution of abalone’s acrosomal protein family since all paralogs are maintained in the transcriptome and have accrued no pseudogenizing mutations. A hypothesis for the duplication and diversification of acrosomal protein paralogs in H. tuberculata is that paralogs are specialized for different binding sites of their egg receptor or different allelic variants of their receptor. For example, H. rufescens VERL has 22 tandem ZP-N domains with three unique amino acid sequences, H. tuberculata VERL may show similar differences in ZP-N sequences and H. tuberculata lysin paralogs may be optimized for binding different ZP-N sequences (Galindo et al., 2002). In addition, the abalone H. tuberculata VERL may be polymorphic, as seen for H. corrugata VERL, and lysin paralogs are optimized for VERL allelic variants (Clark et al., 2009). This study observed that sites that vary between H. tuberculata lysin paralogs are largely located on the face of the molecules interacting with lysin receptor VERL (Figure 3A). Unlike the distribution of positively selected sites between sp18 and sp18-dup where sites are differentially clustered on the protein structure. This pattern of diversification may be suggestive of specialization of function, such as interacting with different VERL allelic variants or VERL ZP-N domains. Further characterization of VERL in H. tuberculata and population-level variation is necessary to explore these hypotheses.
This study characterizes duplication events of a sperm acrosomal protein family with functions directly associated with fertilization. Although lysin was one of the first fertilization proteins discovered and the first for which an egg binding partner was defined, its evolutionary origins are unknown. By placing duplication events of lysin and sp18 within their genomic context and identifying clade-specific duplication events, this study has revealed the importance of duplication for the evolution of this protein family that has previously been unknown. We describe six acrosomal protein paralogs arising from both ancestral and clade-specific duplication events (Figure 5). Recurrent duplication events of sperm acrosomal proteins have occurred throughout the evolutionary history of abalone. For the two abalone species with transcriptomic data both have paralogs maintained in the testis transcriptome. Remarkably none of these genes have been pseudogenized and many are undergoing strong positive selection consistent with maintenance of their function in abalone reproduction. Further inquiry is required to investigate why these proteins are undergoing duplication, the functional consequences of these duplication events, and whether other fertilization proteins in other species (as also seen for the mammalian Izumo family) are undergoing recurrent duplication events.
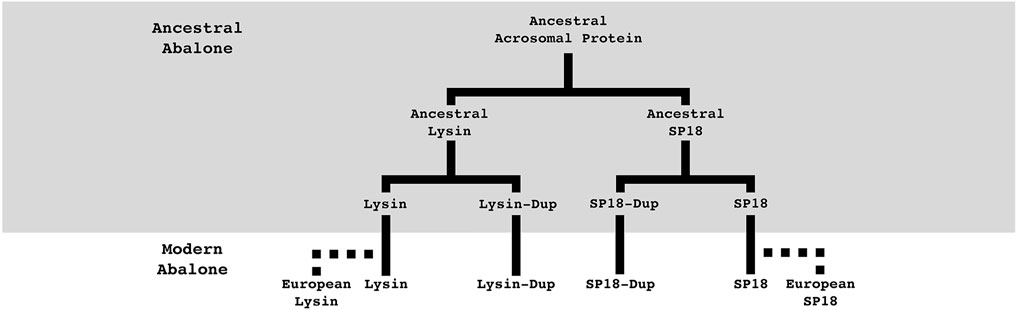
FIGURE 5. Summary of abalone acrosomal duplications. Abalone acrosomal proteins have duplicated multiple times over the course of abalone evolution. Five duplication events can describe the currently identified protein family. Most duplications are ancestral to all abalone, but species-specific duplications in European abalone point to duplication paired with positive selection still playing an important role in the evolution of abalone fertilization pathways. Solid lines indicate orthologs present in all abalone, dotted lines indicate species-specific paralogs.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, OK491874-OK491909.
JC and WS designed the research. JC and MG performed the research. JC and WS analyzed the data. JC wrote the paper.
This study was supported by NIH Grant HD076862 to WS and a National Science Foundation Graduate Research Fellowship to JC (2140004). MG was supported by University of Washington School of Medicine-Gonzaga University Regional Health Partnership.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Thank you to France Haliotis for help acquiring H. tuberculata samples, to Evan Cox, Dr. Daniel Promislow, Dr. Josh Schraiber, and Dr. Damien Wilburn for help with data analysis, and to Alberto Rivera, Dr. Jan Aagaard, and Dr. Bryce Taylor for useful discussions and comments. All research was performed on the traditional lands of the Duwamish Tribe. To learn more about the Duwamish Tribe and their continuing legacy, please visit https://www.duwamishtribe.org/.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2022.795273/full#supplementary-material
Aagaard, J. E., Springer, S. A., Soelberg, S. D., and Swanson, W. J. (2013). Duplicate Abalone Egg Coat Proteins Bind Sperm Lysin Similarly, but Evolve Oppositely, Consistent with Molecular Mimicry at Fertilization. Plos Genet. 9, e1003287. doi:10.1371/journal.pgen.1003287
Aagaard, J. E., Vacquier, V. D., MacCoss, M. J., and Swanson, W. J. (2010). ZP Domain Proteins in the Abalone Egg Coat Include a Paralog of VERL under Positive Selection that Binds Lysin and 18-kDa Sperm Proteins. Mol. Biol. Evol. 27, 193–203. doi:10.1093/molbev/msp221
Aagaard, J. E., Yi, X., MacCoss, M. J., and Swanson, W. J. (2006). Rapidly Evolving Zona Pellucida Domain Proteins Are a Major Component of the Vitelline Envelope of Abalone Eggs. Proc. Natl. Acad. Sci. 103, 17302–17307. doi:10.1073/pnas.0603125103
Almagro Armenteros, J. J., Tsirigos, K. D., Sønderby, C. K., Petersen, T. N., Winther, O., Brunak, S., et al. (2019). SignalP 5.0 Improves Signal Peptide Predictions Using Deep Neural Networks. Nat. Biotechnol. 37, 420–423. doi:10.1038/s41587-019-0036-z
Almeida, F. C., and Desalle, R. (2009). Orthology, Function and Evolution of Accessory Gland Proteins in the Drosophila repleta Group. Genetics 181, 235–245. doi:10.1534/genetics.108.096263
Anisimova, M., Bielawski, J. P., and Yang, Z. (2001). Accuracy and Power of the Likelihood Ratio Test in Detecting Adaptive Molecular Evolution. Mol. Biol. Evol. 18, 1585–1592. doi:10.1093/oxfordjournals.molbev.a003945
Avella, M. A., Baibakov, B., and Dean, J. (2014). A Single Domain of the ZP2 Zona Pellucida Protein Mediates Gamete Recognition in Mice and Humans. J. Cell Biol 205, 801–809. doi:10.1083/jcb.201404025
Bianchi, E., Doe, B., Goulding, D., and Wright, G. J. (2014). Juno Is the Egg Izumo Receptor and Is Essential for Mammalian Fertilization. Nature 508, 483–487. doi:10.1038/nature13203
Bianchi, E., and Wright, G. J. (2020). Find and Fuse: Unsolved Mysteries in Sperm-Egg Recognition. Plos Biol. 18, e3000953. doi:10.1371/journal.pbio.3000953
Botwright, N. A., Zhao, M., Wang, T., McWilliam, S., Colgrave, M. L., Hlinka, O., et al. (2019). Greenlip Abalone (Haliotis Laevigata) Genome and Protein Analysis Provides Insights into Maturation and Spawning. G3 (Bethesda) 9, 3067–3078. doi:10.1534/g3.119.400388
Cai, X., and Clapham, D. E. (2008). Evolutionary Genomics Reveals Lineage-specific Gene Loss and Rapid Evolution of a Sperm-specific Ion Channel Complex: CatSpers and CatSperβ. PLoS One 3, e3569. doi:10.1371/journal.pone.0003569
Carlisle, J. A., and Swanson, W. J. (2020). Molecular Mechanisms and Evolution of Fertilization Proteins. J. Exp. Zool B Mol. Dev. Evol. 336 (8), 652–665. doi:10.1002/jez.b.23004
Clark, N. L., Findlay, G. D., Yi, X., MacCoss, M. J., and Swanson, W. J. (2007). Duplication and Selection on Abalone Sperm Lysin in an Allopatric Population. Mol. Biol. Evol. 24, 2081–2090. doi:10.1093/molbev/msm137
Clark, N. L., Gasper, J., Sekino, M., Springer, S. A., Aquadro, C. F., and Swanson, W. J. (2009). Coevolution of Interacting Fertilization Proteins. Plos Genet. 5, e1000570. doi:10.1371/journal.pgen.1000570
Cooper, J. C., and Phadnis, N. (2017). Parallel Evolution of Sperm Hyper-Activation Ca2+ Channels. Genome Biol. Evol. 9, 1938–1949. doi:10.1093/gbe/evx131
Doty, K. A., Wilburn, D. B., Bowen, K. E., Feldhoff, P. W., and Feldhoff, R. C. (2016). Co-option and Evolution of Non-olfactory Proteinaceous Pheromones in a Terrestrial Lungless Salamander. J. Proteomics 135, 101–111. doi:10.1016/j.jprot.2015.09.019
Ellerman, D. A., Pei, J., Gupta, S., Snell, W. J., Myles, D., and Primakoff, P. (2009). Izumo Is Part of a Multiprotein Family Whose Members Form Large Complexes on Mammalian Sperm. Mol. Reprod. Dev. 76, 1188–1199. doi:10.1002/mrd.21092
Findlay, G. D., Yi, X., Maccoss, M. J., and Swanson, W. J. (2008). Proteomics Reveals Novel Drosophila Seminal Fluid Proteins Transferred at Mating. Plos Biol. 6, e178. doi:10.1371/journal.pbio.0060178
Galindo, B. E., Moy, G. W., Swanson, W. J., and Vacquier, V. D. (2002). Full-length Sequence of VERL, the Egg Vitelline Envelope Receptor for Abalone Sperm Lysin. Gene 288, 111–117. doi:10.1016/s0378-1119(02)00459-6
Galindo, B. E., Vacquier, V. D., and Swanson, W. J. (2003). Positive Selection in the Egg Receptor for Abalone Sperm Lysin. Proc. Natl. Acad. Sci. 100, 4639–4643. doi:10.1073/pnas.0830022100
Gan, H. M., Tan, M. H., Austin, C. M., Sherman, C. D. H., Wong, Y. T., Strugnell, J., et al. (2019). Best Foot Forward: Nanopore Long Reads, Hybrid Meta-Assembly, and Haplotig Purging Optimizes the First Genome Assembly for the Southern Hemisphere Blacklip Abalone (Haliotis Rubra). Front. Genet. 10, 889. doi:10.3389/fgene.2019.00889
Gavrilets, S., and Waxman, D. (2002). Sympatric Speciation by Sexual Conflict. Proc. Natl. Acad. Sci. 99, 10533–10538. doi:10.1073/pnas.152011499
Grayson, P., and Civetta, A. (2012). Positive Selection and the Evolution of Izumo Genes in Mammals. Int. J. Evol. Biol. 2012, 958164. doi:10.1155/2012/958164
Grayson, P. (2015). Izumo1 and Juno: the Evolutionary Origins and Coevolution of Essential Sperm-Egg Binding Partners. R. Soc. Open Sci. 2, 150296. doi:10.1098/rsos.150296
Hellberg, M. E., and Vacquier, V. D. (1999). Rapid Evolution of Fertilization Selectivity and Lysin cDNA Sequences in Teguline Gastropods. Mol. Biol. Evol. 16, 839–848. doi:10.1093/oxfordjournals.molbev.a026168
Huang, Y., Niu, B., Gao, Y., Fu, L., and Li, W. (2010). CD-HIT Suite: a Web Server for Clustering and Comparing Biological Sequences. Bioinformatics 26, 680–682. doi:10.1093/bioinformatics/btq003
Kamei, N., and Glabe, C. G. (2003). The Species-specific Egg Receptor for Sea Urchin Sperm Adhesion Is EBR1,a Novel ADAMTS Protein. Genes Dev. 17, 2502–2507. doi:10.1101/gad.1133003
Killingbeck, E. E., and Swanson, W. J. (2018). Egg Coat Proteins across Metazoan Evolution. Curr. Top. Dev. Biol. 130, 443–488. doi:10.1016/bs.ctdb.2018.03.005
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B., and Stamatakis, A. (2019). RAxML-NG: a Fast, Scalable and User-Friendly Tool for Maximum Likelihood Phylogenetic Inference. Bioinformatics 35, 4453–4455. doi:10.1093/bioinformatics/btz305
Kresge, N., Vacquier, V. D., and Stout, C. D. (2001). The Crystal Structure of a Fusagenic Sperm Protein Reveals Extreme Surface Properties,. Biochemistry 40, 5407–5413. doi:10.1021/bi002779v
Kresge, N., Vacquier, V. D., and Stout, C. D. (2000). The High Resolution crystal Structure of green Abalone Sperm Lysin: Implications for Species-specific Binding of the Egg Receptor 1 1Edited by R. Huber. J. Mol. Biol. 296, 1225–1234. doi:10.1006/jmbi.2000.3533
Le, S. Q., and Gascuel, O. (2008). An Improved General Amino Acid Replacement Matrix. Mol. Biol. Evol. 25, 1307–1320. doi:10.1093/molbev/msn067
Lee, Y. H., Ota, T., and Vacquier, V. D. (1995). Positive Selection Is a General Phenomenon in the Evolution of Abalone Sperm Lysin. Mol. Biol. Evol. 12, 231–238. doi:10.1093/oxfordjournals.molbev.a040200
Lehmann, R. (2018). Matchmaking Molecule for Egg and Sperm. Science 361, 974–975. doi:10.1126/science.aau8356
Lemoine, F., Domelevo Entfellner, J.-B., Wilkinson, E., Correia, D., Dávila Felipe, M., De Oliveira, T., et al. (2018). Renewing Felsenstein's Phylogenetic Bootstrap in the Era of Big Data. Nature 556, 452–456. doi:10.1038/s41586-018-0043-0
Lewis, C. A., Leighton, D. L., and Vacquier, V. D. (1980). Morphology of Abalone Spermatozoa before and after the Acrosome Reaction. J. Ultrastruct. Res. 72, 39–46. doi:10.1016/s0022-5320(80)90133-1
Lyon, J. D., and Vacquier, V. D. (1999). Interspecies Chimeric Sperm Lysins Identify Regions Mediating Species-specific Recognition of the Abalone Egg Vitelline Envelope. Dev. Biol. 214, 151–159. doi:10.1006/dbio.1999.9411
MacDonald, R. J., Swift, G. H., Przybyla, A. E., and Chirgwin, J. M. (1987). [20] Isolation of RNA Using Guanidinium Salts. Methods Enzymol. 152, 219–227. doi:10.1016/0076-6879(87)52023-7
Magel, R. C., and Wibowo, S. H. (1997). Comparing the Powers of the Wald-Wolfowitz and Kolmogorov-Smirnov Tests. Biom. J. 39, 665–675. doi:10.1002/bimj.4710390605
Masonbrink, R. E., Purcell, C. M., Boles, S. E., Whitehead, A., Hyde, J. R., Seetharam, A. S., et al. (2019). An Annotated Genome for Haliotis rufescens (Red Abalone) and Resequenced Green, Pink, Pinto, Black, and White Abalone Species. Genome Biol. Evol. 11, 431–438. doi:10.1093/gbe/evz006
McGinnis, S., and Madden, T. L. (2004). BLAST: at the Core of a Powerful and Diverse Set of Sequence Analysis Tools. Nucleic Acids Res. 32, W20–W25. doi:10.1093/nar/gkh435
Metz, E. C., Robles-Sikisaka, R., and Vacquier, V. D. (1998). Nonsynonymous Substitution in Abalone Sperm Fertilization Genes Exceeds Substitution in Introns and Mitochondrial DNA. Proc. Natl. Acad. Sci. 95, 10676–10681. doi:10.1073/pnas.95.18.10676
Mozingo, N. M., Vacquier, V. D., and Chandler, D. E. (1995). Structural Features of the Abalone Egg Extracellular Matrix and its Role in Gamete Interaction during Fertilization. Mol. Reprod. Dev. 41, 493–502. doi:10.1002/mrd.1080410412
Nam, B. H., Kwak, W., Kim, Y. O., Kim, D. G., Kong, H. J., Kim, W. J., et al. (2017). Genome Sequence of pacific Abalone (Haliotis Discus Hannai): the First Draft Genome in Family Haliotidae. Gigascience 6, 1–8. doi:10.1093/gigascience/gix014
Palmer, M. R., McDowall, M. H., Stewart, L., Ouaddi, A., MacCoss, M. J., and Swanson, W. J. (2013). Mass Spectrometry and Next-Generation Sequencing Reveal an Abundant and Rapidly Evolving Abalone Sperm Protein. Mol. Reprod. Dev. 80, 460–465. doi:10.1002/mrd.22182
Park, C. Y., Klammer, A. A., Käll, L., MacCoss, M. J., and Noble, W. S. (2008). Rapid and Accurate Peptide Identification from Tandem Mass Spectra. J. Proteome Res. 7, 3022–3027. doi:10.1021/pr800127y
Pei, J., Kim, B.-H., and Grishin, N. V. (2008a). PROMALS3D: a Tool for Multiple Protein Sequence and Structure Alignments. Nucleic Acids Res. 36, 2295–2300. doi:10.1093/nar/gkn072
Pei, J., Tang, M., and Grishin, N. V. (2008b). PROMALS3D Web Server for Accurate Multiple Protein Sequence and Structure Alignments. Nucleic Acids Res. 36, W30–W34. doi:10.1093/nar/gkn322
Raj, I., Sadat Al Hosseini, H., Dioguardi, E., Nishimura, K., Han, L., Villa, A., et al. (2017). Structural Basis of Egg Coat-Sperm Recognition at Fertilization. Cell 169, 1315–1326. doi:10.1016/j.cell.2017.05.033
Rastogi, S., and Liberles, D. A. (2005). Subfunctionalization of Duplicated Genes as a Transition State to Neofunctionalization. BMC Evol. Biol. 5, 28. doi:10.1186/1471-2148-5-28
Reams, A. B., and Roth, J. R. (2015). Mechanisms of Gene Duplication and Amplification. Cold Spring Harb Perspect. Biol. 7, a016592. doi:10.1101/cshperspect.a016592
Sirot, L. K., Findlay, G. D., Sitnik, J. L., Frasheri, D., Avila, F. W., and Wolfner, M. F. (2014). Molecular Characterization and Evolution of a Gene Family Encoding Both Female- and Male-specific Reproductive Proteins in Drosophila. Mol. Biol. Evol. 31, 1554–1567. doi:10.1093/molbev/msu114
Slater, G., and Birney, E. (2005). Automated Generation of Heuristics for Biological Sequence Comparison. BMC Bioinformatics 6, 31. doi:10.1186/1471-2105-6-31
Suyama, M., Torrents, D., and Bork, P. (2006). PAL2NAL: Robust Conversion of Protein Sequence Alignments into the Corresponding Codon Alignments. Nucleic Acids Res. 34, W609–W612. doi:10.1093/nar/gkl315
Swanson, W. J., Aagaard, J. E., Vacquier, V. D., Monné, M., Sadat Al Hosseini, H., and Jovine, L. (2011). The Molecular Basis of Sex: Linking Yeast to Human. Mol. Biol. Evol. 28, 1963–1966. doi:10.1093/molbev/msr026
Swanson, W. J., Nielsen, R., and Yang, Q. (2003). Pervasive Adaptive Evolution in Mammalian Fertilization Proteins. Mol. Biol. Evol. 20, 18–20. doi:10.1093/oxfordjournals.molbev.a004233
Swanson, W. J., and Vacquier, V. D. (1995a). Extraordinary Divergence and Positive Darwinian Selection in a Fusagenic Protein Coating the Acrosomal Process of Abalone Spermatozoa. Proc. Natl. Acad. Sci. 92, 4957–4961. doi:10.1073/pnas.92.11.4957
Swanson, W. J., and Vacquier, V. D. (1995b). Liposome Fusion Induced by a Mr 18 000 Protein Localized to the Acrosomal Region of Acrosome-Reacted Abalone Spermatozoa. Biochemistry 34, 14202–14208. doi:10.1021/bi00043a026
Swanson, W. J., and Vacquier, V. D. (1997). The Abalone Egg Vitelline Envelope Receptor for Sperm Lysin Is a Giant Multivalent Molecule. Proc. Natl. Acad. Sci. 94, 6724–6729. doi:10.1073/pnas.94.13.6724
Swanson, W. J., and Vacquier, V. D. (2002). The Rapid Evolution of Reproductive Proteins. Nat. Rev. Genet. 3, 137–144. doi:10.1038/nrg733
Ulrich, A. S., Otter, M., Glabe, C. G., and Hoekstra, D. (1998). Membrane Fusion Is Induced by a Distinct Peptide Sequence of the Sea Urchin Fertilization Protein Bindin. J. Biol. Chem. 273, 16748–16755. doi:10.1074/jbc.273.27.16748
Vacquier, V. D., and Moy, G. W. (1977). Isolation of Bindin: the Protein Responsible for Adhesion of Sperm to Sea Urchin Eggs. Proc. Natl. Acad. Sci. 74, 2456–2460. doi:10.1073/pnas.74.6.2456
Vacquier, V. D., and Swanson, W. J. (2011). Selection in the Rapid Evolution of Gamete Recognition Proteins in marine Invertebrates. Cold Spring Harbor Perspect. Biol. 3, a002931. doi:10.1101/cshperspect.a002931
Wagstaff, B. J., and Begun, D. J. (2005). Comparative Genomics of Accessory Gland Protein Genes in Drosophila melanogaster and D. pseudoobscura. Mol. Biol. Evol. 22, 818–832. doi:10.1093/molbev/msi067
Wilburn, D. B., Arnold, S. J., Houck, L. D., Feldhoff, P. W., and Feldhoff, R. C. (2017). Gene Duplication, Co-option, Structural Evolution, and Phenotypic Tango in the Courtship Pheromones of Plethodontid Salamanders. Herpetologica 73, 206–219. doi:10.1655/herpetologica-d-16-00082.1
Wilburn, D. B., Tuttle, L. M., Klevit, R. E., and Swanson, W. J. (2018). Solution Structure of Sperm Lysin Yields Novel Insights into Molecular Dynamics of Rapid Protein Evolution. Proc. Natl. Acad. Sci. USA 115, 1310–1315. doi:10.1073/pnas.1709061115
Yang, Z. (2007). PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 24, 1586–1591. doi:10.1093/molbev/msm088
Yang, Z., Wong, W. S., and Nielsen, R. (2005). Bayes Empirical Bayes Inference of Amino Acid Sites under Positive Selection. Mol. Biol. Evol. 22, 1107–1118. doi:10.1093/molbev/msi097
Keywords: fertilization, duplication, paralogs, molecular evolution, genome evolution, testes and epididymis, reproduction, sperm
Citation: Carlisle JA, Glenski MA and Swanson WJ (2022) Recurrent Duplication and Diversification of Acrosomal Fertilization Proteins in Abalone. Front. Cell Dev. Biol. 10:795273. doi: 10.3389/fcell.2022.795273
Received: 14 October 2021; Accepted: 21 February 2022;
Published: 07 April 2022.
Edited by:
Enrica Bianchi, University of York, United KingdomReviewed by:
Cameron Weadick, University of Exeter, United KingdomCopyright © 2022 Carlisle, Glenski and Swanson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: J. A. Carlisle, amNhcmxpc2xAdXcuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.