 Anita V. Kumar
Anita V. Kumar Joslyn Mills
Joslyn Mills Louis R. Lapierre
Louis R. Lapierre
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol. , 14 February 2022
Sec. Molecular and Cellular Pathology
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.793328
This article is part of the Research Topic Defective Macroautophagy in Organelle Turnover: From Basic Mechanisms to Human Disease View all 9 articles
Efficient proteostasis is crucial for somatic maintenance, and its decline during aging leads to cellular dysfunction and disease. Selective autophagy is a form of autophagy mediated by receptors that target specific cargoes for degradation and is an essential process to maintain proteostasis. The protein Sequestosome 1 (p62/SQSTM1) is a classical selective autophagy receptor, but it also has roles in the ubiquitin-proteasome system, cellular metabolism, signaling, and apoptosis. p62 is best known for its role in clearing protein aggregates via aggrephagy, but it has recently emerged as a receptor for other forms of selective autophagy such as mitophagy and lipophagy. Notably, p62 has context-dependent impacts on organismal aging and turnover of p62 usually reflects active proteostasis. In this review, we highlight recent advances in understanding the role of p62 in coordinating the ubiquitin-proteasome system and autophagy. We also discuss positive and negative effects of p62 on proteostatic status and their implications on aging and neurodegeneration. Finally, we relate the link between defective p62 and diseases of aging and examine the utility of targeting this multifaceted protein to achieve proteostatic benefits.
The health and survival of an organism is reliant on efficient proteostasis, and breakdown of this process results in accumulation of toxic protein aggregates that contribute to aging and age-related diseases (Kaushik and Cuervo, 2015). The main contributors to protein quality control include chaperones for protein folding and the ubiquitin-proteasome system (UPS) and autophagy for protein degradation. The UPS degrades individual proteins with specific polyubiquitin tags including short-lived, misfolded, and damaged proteins (Rock et al., 1994), while autophagy has the capacity to degrade large proteins as well as protein aggregates and damaged organelles (reviewed by Lamb et al., 2013). Autophagy is enhanced as a compensatory mechanism for impaired proteasomes and coordination between the UPS and autophagy ensures efficient protein turnover (reviewed by Dikic, 2017). Sequestosome 1 (SQSTM1 or p62), hereafter p62, a ubiquitous and multifunctional protein, can direct ubiquitinated proteins to the proteasome (Babu et al., 2005; Myeku and Figueiredo-Pereira, 2011) or the growing autophagosome (Pankiv et al., 2007), highlighting its role as a key receptor and pivot for the two main cellular pathways of protein degradation. Here, this review discusses new knowledge in p62 biology with a focus on the role of p62 during cellular stress and aging, and in age-related diseases.
p62 has multiple conserved domains that interact with various proteins with diverse functions (Figure 1A). These domains and associated post-translational modifications have been discussed in detail in several excellent reviews (Lin et al., 2013; Emanuele et al., 2020; Berkamp et al., 2021). From N- to C-terminal, these domains include the PB1 domain for p62 homo- and hetero-dimerization and oligomerization (Moscat et al., 2006; Nakamura et al., 2010; Christian et al., 2014; Ciuffa et al., 2015; Turco et al., 2021), the ZZ domain that recognizes N′-end degrons in autophagic substrates (Cha-Molstad et al., 2017; Kwon et al., 2018), a TRAF6 binding (TB) domain (Wooten et al., 2005), the LC3- and Keap1-interacting regions (LIR and KIR, respectively) (Pankiv et al., 2007; Ichimura et al., 2013), and the ubiquitin-binding UBA domain (Isogai et al., 2011). Flanking the TB domain lie nuclear localization and nuclear export signal (NLS and NES) sequences which mediate the nucleo-cytoplasmic shuttling of p62. While p62 aggregates with cytoplasmic inclusions containing ubiquitinated proteins, nuclear p62 associates with nuclear polyubiquitinated proteins at promyelocytic leukemia (PML) bodies and accumulates when nuclear export mediated by the exportin XPO1 (CRM1) is blocked (Pankiv et al., 2010). Nuclear p62 can form condensates with ubiquitinated proteins to degrade nuclear proteins via the nuclear UPS machinery (Fu et al., 2021). A nucleolar localization sequence (NoLS) has recently been identified between the PB1 and NLS regions that causes p62 to shuttle to the nucleolus where it sequesters nuclear proteins during cellular stress (Lobb et al., 2021). Overall, p62’s domains provide a scaffold that directs substrates to autophagosomes and facilitates the autophagic process. For instance, formation of helical p62 filaments by polymeric PB1 self-assembly facilitates autophagic cargo uptake (Jakobi et al., 2020). The ZZ domain that recognizes N-degrons such as N-terminal arginine (Nt-R) mediates p62 puncta formation and autophagy (Zhang et al., 2018). The UBA domain has important phosphorylation sites S403 and S409 that, when phosphorylated, increase p62’s affinity for polyubiquitin chains (Matsumoto et al., 2011; Lim et al., 2015). In addition to autophagy, some domains cause p62 to participate in signaling events, for e.g., p62’s TB domain triggers TRAF6 polyubiquitination thereby activating the inflammatory NF-κB pathway (Wooten et al., 2005; Zotti et al., 2014). The KIR domain interacts with Keap1 which releases the transcription factor Nrf2 to translocate to the nucleus and stimulate expression of p62 and antioxidant element-responsive and proteasomal genes (Ichimura et al., 2013; Sha et al., 2018). Altogether, p62 levels and its multiple interactions have important ramifications in the onset of aging where UPS and autophagic capacities progressively decline.
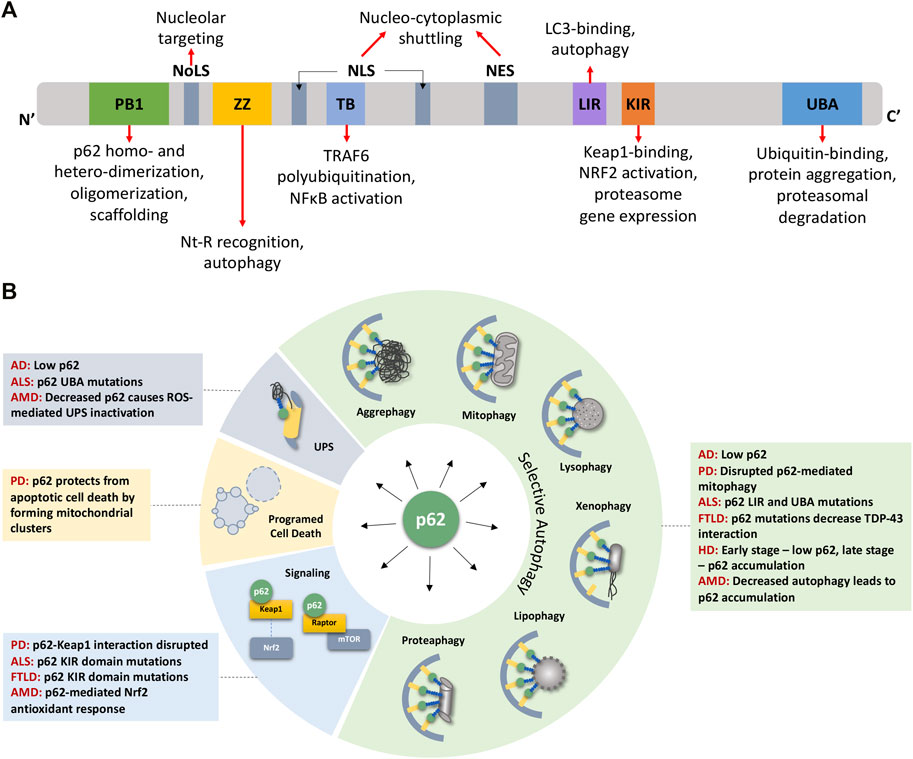
FIGURE 1. p62 domains, its multifaceted nature, and its impact on detriments associated with age-related degenerative diseases. (A) p62 protein consists of several well-characterized domains that interact with various proteins leading to p62’s involvement in diverse functions (see text for details). (B) p62 plays roles in various forms of selective autophagy, the UPS, programmed cell death, and signaling pathways. These functions are disrupted owing to mutations or aberrant expression/accumulation of p62 in several age-related degenerative diseases discussed in this review. AD Alzheimer’s Disease, ALS Amyotrophic Lateral Sclerosis, AMD Age-related Macular Degeneration, FTLD Frontotemporal Lobar Degeneration, HD Huntington’s Disease, NES Nuclear Export Sequence, NLS Nuclear Localization Sequence, NoLS Nucleolar Localization Sequence, PD Parkinson’s Disease.
p62 is the first selective autophagy receptor to be characterized (Bjorkoy et al., 2005; Pankiv et al., 2007). Its transcription is modulated by the conserved autophagy and lysosomal regulator transcription factor EB (TFEB) (Sardiello et al., 2009; Settembre et al., 2011; Lapierre et al., 2013), whose nuclear localization is modulated by major nutrient sensor mTORC1 (Pena-Llopis et al., 2011; Martina et al., 2012) and nuclear export protein XPO1 (Kirli et al., 2015; Silvestrini et al., 2018). The oxidative stress transcription factor Nrf2 also induces p62 expression (Jain et al., 2010). One of the key roles of p62 is to deliver various ubiquitinated cargoes bound to its UBA domain to the autophagosome via LIR domains, ultimately leading to their degradation by the lysosome (Bjorkoy et al., 2005; Liu et al., 2016). Defective autophagy leads to p62 accumulation, and p62 levels are used as a marker for autophagic flux, along with LC3B (Mizushima et al., 2010). During recognition of aggregated polyubiquitinated cargo, p62 self-assembles and forms oligomers, resulting in clearance of misfolded proteins by a process known as aggrephagy (Wurzer et al., 2015; Galluzzi et al., 2017; Zaffagnini et al., 2018). p62 forms cytosolic inclusion bodies known as p62 bodies consisting primarily of K63-linked polyubiquitinated substrates (Bjorkoy et al., 2005; Stolz et al., 2014). Polyubiquitinated cargoes linked via lysine-63 (K63) are more apparent in p62 cluster formation than K48-linked cargoes, suggesting the K63-linked chains specifically triggered clustering, while K48-linked chains needed higher concentrations for clustering, which might occur during proteasomal inhibition (Zaffagnini et al., 2018). Recently, interaction of the chaperone UTX with a Lim-binding domain of p62 was found to increase clustering and p62 body formation (Yoon et al., 2021). Notably, p62 bodies have liquid-like properties formed by polyubiquitin chain-triggered phase separation (Sun et al., 2018; Zaffagnini et al., 2018). While originally believed to be rigid, p62-ubiquitinated protein clusters are dynamic structures in which ubiquitinated proteins can freely move within them (Sun et al., 2018). The formation of p62 bodies is mediated by autophagy receptor NBR1, which activates Nrf2 and promotes Nrf2-mediated stress response (Yang et al., 2019; Sanchez-Martin et al., 2020). The oligomerization of p62 during aggrephagy, and its consequent binding to LC3B and GABARAP, is negatively regulated by the binding of short, non-coding RNA called vault RNA1-1 to p62 (Horos et al., 2019). Thus, understanding p62 cluster dynamics and regulation could further shed light onto the range of effects of p62 in aging and diseases.
In addition to its classical role as an aggrephagy receptor, p62 is involved in several other forms of selective autophagy. Mitochondrial proteins that are damaged beyond the capacity of the unfolded protein response and mitochondria losing membrane potential (Chen et al., 2020) can be autophagically degraded via mitophagy (Geisler et al., 2010). p62 is recruited to ubiquitinated outer mitochondrial membrane proteins in Parkin-dependent mitophagy and has a role in mitochondrial ubiquitination in PARKIN-independent mitophagy (Narendra et al., 2010; Yamada et al., 2018; Yamada et al., 2019). Lipid droplets, in addition to being lipid storage organelles, are emerging as hubs of cellular proteostasis integrating cytosolic and ER-related degradation processes (Roberts and Olzmann, 2020). The selective engulfment of lipid droplets (LDs) is mediated by the autophagic machinery in a process called lipophagy (Singh et al., 2009; Roberts and Olzmann, 2020). p62-mediated autophagy targets LDs for autophagic turnover in myocytes (Lam et al., 2016), hepatic cells (Wang et al., 2017; Yan et al., 2019), and macrophage foam cells (Robichaud et al., 2021), highlighting p62 as a potential receptor for LD turnover. Xenophagy, or the targeted clearance of foreign entities such as invading pathogens by autophagy, is an important part of host immune defense (Sharma et al., 2018). Phosphorylated p62 promotes ubiquitin conjugation to xenophagy target proteins (Tsuchiya et al., 2018) and Mycobacterium tuberculosis protein (Chai et al., 2019). Targeted autophagic degradation of the proteasome itself, termed proteaphagy, occurs in mammalian cells in response to amino acid starvation (Marshall et al., 2016; Cohen-Kaplan et al., 2017). Ubiquitinated proteasomes are recognized and recruited for autophagosomal uptake by p62 via its UBA domain independent of its PB1 domain (Cohen-Kaplan et al., 2017) or could be partially sequestered into aggresomes (Choi et al., 2020). Damaged lysosomes and harmful products of lysosomal rupture are cleared by lysophagy (Yim and Mizushima, 2020), and p62 is the major receptor discovered to play a role in lysophagy. It is present on damaged lysosomes along with ubiquitin-targeted AAA + -ATPase p97, and p62 deletion impairs lysosome clearance (Papadopoulos et al., 2017). p62 is also a receptor for selective autophagy of peroxisomes, termed pexophagy, where p62 interacts with NBR1 to promote clustering of peroxisomes and enhance pexophagy (Deosaran et al., 2013; Germain and Kim, 2020). Owing to its involvement in selective autophagy of several organelles, perturbance of p62 could result in accumulation of different damaged organelles commonly observed in stress, aging, and age-related diseases.
The Ubiquitin-Proteasome System (UPS) accounts for nearly 80% of protein degradation in the cell (Lee and Goldberg, 1998). p62 colocalizes with proteasomes (Seibenhener et al., 2004; Myeku and Figueiredo-Pereira, 2011) and can shuttle polyubiquitinated substrates for degradation via the proteasome (Babu et al., 2005). While the UBA domain on p62 recognizes ubiquitinated substrates, the PB1 “oligomerization” domain interacts with Rpn10 and Rpn1, proteins of the regulatory 19S subunit of the proteasome, to facilitate delivery of ubiquitinated substrates to the proteasome by p62 (Seibenhener et al., 2004). Additionally, with the help of its two NLS domains, p62 enters the nucleus, where it has been shown to phase separate into p62 foci which recruit functional proteasomes that actively degrade nuclear proteins and unincorporated proteasome subunits (Pankiv et al., 2010; Fu et al., 2021). Formation of such condensates is responsive to various stressors (Fu et al., 2021) and could be important in recruiting components of the UPS machinery thus improving efficiency of degradation. Inhibition of proteasome activity stimulates p62 transcription along with that of proteasomal genes (Sha et al., 2018). p62 overexpression in presence of autophagy inhibition hampers proteostatic flux through the UPS without affecting proteasome catalytic activity (Korolchuk et al., 2009) indicating a block in delivery of ubiquitinated substrates to the proteasome by p62.
The UPS and autophagy are two major intracellular degradation routes and p62 is a key mediator of crosstalk between these pathways (Liu et al., 2016). Proteasome inhibition leads to proteotoxic stress that promotes p62 phosphorylation at S403 in humans. This stabilizes ubiquitinated proteins in p62 clusters and promotes their clearance by autophagy (Matsumoto et al., 2011; Lim et al., 2015). Reducing proteasomal capacity by knocking down UPS ubiquitin receptors, PSMD4 and ADRM1, stimulates the transcription of p62 via the transcription factor ATF4 and induces compensatory autophagy (Demishtein et al., 2017). Similarly, p62 can also be induced by transcription factor Nrf1 upon pharmacological inhibition of the proteasome, which promotes cell survival by sequestering ubiquitinated proteins into inclusions (Sha et al., 2018). Upregulation of the deubiquitinase TRIM44, that binds K48-linked ubiquitin chains, promotes p62 oligomerization (Lyu et al., 2021). Prolonged proteasomal inhibition and ubiquitin overexpression causes accumulation of ubiquitinated p62 that activates autophagy (Peng et al., 2017).
Like the UPS, inhibiting autophagy also causes accumulation of p62, but this accumulation delays delivery of ubiquitinated proteins to the proteasome and thus reduces flux through the UPS (Korolchuk et al., 2009). Autophagy inhibition can also impair proteasomal function by affecting proteaphagy, in which p62 recognizes ubiquitinated proteasomes, especially prevalent during starvation, and targets them for autophagic degradation (Marshall et al., 2016; Cohen-Kaplan et al., 2017). Although p62 primarily carries out aggregation-dependent clearance of damaged material, p62 can turn detrimental by exacerbating pathological aggregation and proteotoxicity during autophagy inhibition or when proteostasis is overwhelmed. Since p62 is an important receptor that delivers substrates for both proteasomal degradation and autophagy, alterations in p62 levels and function could influence the activity of UPS versus autophagy.
In addition to its well-studied role in autophagic and proteasomal degradation, p62 influences other cellular pathways, including pathogen resistance, programmed cell death, and signal transduction, through its scaffolding property brought about by its several interacting domains (Figure 1A). Along with other autophagy components such as LC3, ATG7, and ATG16L1, p62 restricts the growth of Toxoplasma gondii by encapsulating them in vesicles that do not fuse with lysosomes (Selleck et al., 2015). In addition to pathogen resistance, p62 also controls programmed cell death independent of autophagic cargo degradation. By recruiting RIPK1, a component of the necroptosis complex, p62 controls a switch from apoptosis to necroptosis in a prostate cancer model (Goodall et al., 2016). Due to its ability to provide scaffolding, p62 participates in several signal transduction cascades by bringing together pathway components (Bitto et al., 2014). Briefly, it facilitates TNF-R and IL-1βR signaling, activating the NF-κB pathway (Sanz et al., 1999; Wooten et al., 2005), enables the oxidative stress response by binding to Keap1 which allows the release of Nrf2 to induce the antioxidant response (Ichimura et al., 2013), and stimulates apoptosis by acting downstream via cullin-3 regulation of caspase-8 (Jin et al., 2009). p62 also participates in amino acid sensing by the mTORC1 pathway, which is perhaps most relevant to stress and aging. p62 associates with components of the mTORC1 complex, Raptor and Rag GTPases, which sense amino acid levels and activate mTORC1 (Duran et al., 2011). Since mTORC1 signaling regulates autophagy, p62 can influence the balance between autophagy and cell growth by its action on finetuning mTORC1 signaling (Moscat and Diaz-Meco, 2011; Komatsu et al., 2012). Through its involvement in signaling pathways governing cell growth and autophagy, p62 is an important player in tumor initiation and progression (Moscat et al., 2016; Hennig et al., 2021). Altogether, owing to its multifaceted nature and ability to modulate growth and survival mechanisms, p62 plays a pivotal role in cellular stress, aging, and various pathologies including metabolic and neurodegenerative diseases (Figure 1B).
p62 contributes to many cellular processes, so in theory, mutations, loss, or mislocalization of p62 is bound to trigger various outcomes, and the context will determine its impact on health (positive or negative). p62 expression is age- and disease-dependent, characterized by a decline of expression with age and senescence in mice (Kwon et al., 2012; Salazar et al., 2020) and flies (Aparicio et al., 2019), as well as a decrease in human and mouse brains with Alzheimer’s disease (Du et al., 2009). To this end, few p62 mutations that lead to disease have been identified, such as those in Paget’s disease of bone and Amyotrophic Lateral Sclerosis (ALS) (Kwok et al., 2014; Seton et al., 2016; Ma et al., 2019); however, the effects due to complete loss, overexpression, protein-protein interaction disruptions, or mislocalization of the protein have been explored experimentally. Much of the research investigating p62’s role has been done in p62 null or p62 overexpression backgrounds in different species, with only a few studies investigating the tissue-specific or spatio- and temporal-specific roles of the p62 protein. This section will cover what is known about the requirement of p62 for lifespan and disease prevention, how the timing, location, and protein levels are essential to the overall health of the organism, and the role of p62 in specific age-related diseases (Figure 1B).
A number of studies have shown a beneficial lifespan response to p62 overexpression, albeit with context-specific caveats. For instance, p62 overexpression has been shown to prolong lifespan in Drosophila melanogaster, but only in females that have the overexpression initiated at middle-age. This follows the endogenous expression pattern: transcript levels of p62 are increased in early adulthood, but the sharp decrease in expression after midlife can be rescued by p62 overexpression only at that stage (Aparicio et al., 2019). There is no effect on lifespan when p62 is overexpressed in early adulthood, and it is not clear why there is a sex-dependent benefit. In another example, Caenorhabditis elegans shows an extension of lifespan with the overexpression of SQST-1/p62, similar to the lifespan extension seen with hormetic heat shock. However, this lifespan extension is impaired by the loss of neuronal sqst-1/p62. Further, only the nerve-ring neurons require SQST-1/p62 for autophagosome formation (Kumsta et al., 2019). This study indicates a potential tissue-specific requirement of SQST-1/p62, which reveals differential benefits or detriments depending on the tissue target.
Other studies have demonstrated the importance of p62 by investigating the effects of p62 knockout. For example, loss of p62 in the pituitary has a detrimental effect on female mouse fertility due to impaired luteinizing hormone production through mitochondrial OXPHOS signaling (Li X. et al., 2021). Another study showed that p62 protects against glycation-derived toxicity by driving the autophagic degradation of harmful age-associated advanced glycation end products (Aragones et al., 2020). Also, loss of p62 increases the rate of aging by inducing senescence through downregulation of autophagy in vascular smooth muscle cells, suggesting a protective role of p62 in vascular disease and atherosclerosis (Salazar et al., 2020). Many other studies have found that p62 maintains health by mediating the oxidative stress response through interactions with other proteins. Notably, p62 interacts with Keap1 (Figure 1A), which prevents its inhibitory binding to Nrf2. Increased p62 leads to hyperactivation of Nrf2 target genes, which protect against oxidative damage and inflammation. The interaction between p62 and Keap1 declines with age and is lost in some neurodegenerative diseases, leading to age-associated oxidative damage and inflammation (Ma et al., 2019). Further adding to this decline is the oxidative damage to the p62 promotor, demonstrated in cells treated with H2O2, which yields lower p62 levels (Du et al., 2009). Another recent finding shows that treatment with spermidine, a lifespan-extending polyamine, upregulates p62 expression, and this induces cytoprotective autophagy of female germline stem cells (FGSCs) ex vivo. The upregulation of p62 by spermidine is indispensable to delay aging caused by oxidative stress-induced senescence (Yuan et al., 2021). Interestingly, p62 bodies can form “gel-like” droplets, which serves as a platform for the anti-oxidative stress response by sequestering Keap1 within the droplets (Kageyama et al., 2021). Overall, many studies support that the loss of p62 is generally detrimental due to its protective role in the oxidative stress response.
While it is evident that p62 plays an important role in cellular homeostasis through proteostasis and signaling pathways, it is important to highlight that elevated levels of p62 can also be detrimental. A number of studies demonstrate the harmful effects of p62 accumulation through loss of autophagy, such as the loss of oxidative stress response via FOXO1/3 (Zhao et al., 2019). Other studies show that overexpression or general induction of p62 can be damaging. For instance, tumorigenesis is associated with increased p62 (Mathew et al., 2009), or inflammation is triggered by the interaction between p62 and αPKCs to activate the NF-kB pathway which induces senescence (Ma et al., 2019). Genetic overexpression of p62 can have unfavorable effects as well. Indeed, the UPS pathway of degradation does not respond to p62 overexpression efficiently; it is not sufficient to increase proteasome activity, but it instead delays UPS substrate delivery, causing proteotoxic stress that eventually activates the autophagic pathway (Korolchuk et al., 2009). Finally, our group has found that lifelong SQST-1/p62 overexpression in C. elegans maintained under mild heat stress (25°C) causes SQST-1/p62 to accumulate, leading to a decrease in lifespan (Kumar et al., 2021). This shortened lifespan in the SQST-1/p62 overexpressing strains is due to the marked upregulation in SQST-1/p62 transcription and protein levels which exacerbates the proteotoxic stress of accumulated ubiquitinated proteins. Notably, lipid droplet accumulation restores SQST-1/p62 function and dynamics and prevents the rapid proteostatic decline. Overall, these studies highlight the need to reassess the idea that enhancing the expression of a single autophagy receptor, such as p62, is necessarily beneficial for proteostasis and lifespan, especially when the whole process of autophagy, involving more than 30 proteins (Wong et al., 2020a), is not enhanced concomitantly.
Alzheimer’s Disease (AD) is a progressive neurodegenerative disease that destroys memory and cognitive functions and is characterized by accumulation of amyloid-β (Aβ) and hyperphosphorylated tau, causing amyloid plaques and tau tangles, respectively, in the brain. Low expression of p62 has been observed in the frontal cortex of AD patients as well as in transgenic AD mouse models; however, the remaining p62 is associated with tangles, and is believed to play an important role in tau degradation (Salminen et al., 2012). In addition to decreased tau clearance, the low levels of p62 also lead to decreased Nrf2-dependent antioxidant response (Ma et al., 2019), suggesting that impaired oxidative stress resistance may significantly contribute to AD pathology.
Parkinson’s Disease (PD) is characterized by intracellular accumulation of Lewy bodies and Lewy neurites, which consist of p62-associated aggregated proteins, including α-synuclein, parkin, and ubiquitinated proteins (Shin et al., 2020). Disrupted p62-mediated mitophagy due to mutations in PINK and parkin is the most common cause of familial PD (Geisler et al., 2010). Mutations in the kinase LRRK2 disrupt the p62-LRRK2 interaction and impairs LRRK2-mediated phosphorylation of p62 and LRRK2 degradation. Phosphorylated p62 cannot interact with Keap1, which allows Keap1 to inhibit Nrf2 signaling, connecting oxidative stress to PD pathology (Park et al., 2016). Hyperactivation of Parkin/PINK1 mitophagy is also implicated in PD pathogenesis, but recent research suggests that p62 could prevent apoptotic cell death by clustering mitochondria to regulate this process (Xiao et al., 2017).
Amyotrophic Lateral Sclerosis (ALS) is characterized by ubiquitin-p62 positive intraneuronal inclusions, with increased levels of p62 in the spinal cord and motor neurons. Some cases of ALS are associated with p62 mutations in the UBA, LIR, or KIR domains. UBA domain mutations prevent interactions with ubiquitinated proteins tagged for degradation (Ma et al., 2019), and LIR domain mutations lead to reduced LC3 binding, both causing decreased p62-mediated cargo degradation (Deng et al., 2020). Finally, KIR domain mutations disrupt the interaction with Keap1, deactivating Nrf2 and preventing an effective response to oxidative stress, which may contribute to the etiology of ALS (Goode et al., 2016). There is also evidence that p62 itself might intensify ALS. For instance, autophagic induction by rapamycin treatment exacerbated the pathology in an SOD mutant mouse model of ALS, probably due to apoptosis and oxidative stress (Zhang et al., 2011). Additionally, a C. elegans model of ALS showed defective autophagy and increased levels of p62; however the removal of p62 alleviated the locomotion defect without restoring the autophagy defects (Baskoylu et al., 2022), suggesting that the autophagy defects are upstream and not dependent on p62 in C. elegans.
Frontotemporal Lobar Degeneration (FTLD) is characterized by neuronal cytoplasmic inclusions of TDP-43, which are correlated with neurodegeneration. Overexpression of p62 leads to the mislocalization of TDP-43 to the cytoplasm, causing aggregates and neuronal death (Foster et al., 2021). p62 has also been found in TDP-43-negative inclusions in a subset of FTLD patients (Al-Sarraj et al., 2011), opening the possibility that other protein aggregates contribute to FTLD pathogenesis. p62’s interaction with these aggregates suggests their degradation is mediated by p62, and mutations in p62 that decrease this interaction or any disruption in the autophagic or UPS pathways could possibly lead to the accumulation of these inclusions. Notably, the same KIR mutation in ALS is also seen in FTLD cases (Ma et al., 2019).
Huntington’s Disease (HD) is characterized by neuronal degeneration associated with a CAG repeat expansion (polyQ) in the huntingtin gene (mHTT). Early work investigating mTOR inhibition by rapamycin treatment in a HD mouse model demonstrated reduce HD pathology (Ravikumar et al., 2004); however, later work shows that it is independent of autophagy and is instead due to decreased protein synthesis (King et al., 2008). In fact, autophagy is already upregulated in HD, but the defect lies in the recognition of autophagic cargo, not the process itself (Martinez-Vicente et al., 2010). Therefore, the inability for autophagosomes to recognize cargo for degradation (Martinez-Vicente et al., 2010) and p62’s sequestration of ULK into an insoluble cellular fraction (Wold et al., 2016) are likely responsible for the dysfunctional autophagy seen in HD cells. The role of p62 was investigated in a Huntington mouse model which revealed that levels of p62 were reduced in all brain regions at early stages of the disease, but then accumulated in striatal and hippocampal neurons in the late stage of the disease, in particular in the nuclei of these cells. Indeed, the increased p62 accumulated with mHTT in neuronal nuclei (Rue et al., 2013). Thus, p62 depletion reduces nuclear inclusions and ameliorates HD (Kurosawa et al., 2015), but reduction of p62 protein levels or dysfunction of p62 significantly increased cell death induced by mHTT in HD (Bjorkoy et al., 2005). Generally, p62 is upregulated in response to proteotoxic stress (Lim et al., 2015), and the subcellular localization of p62 in response to this stress may underlie the vulnerability of HD cells to cell death under proteotoxic stress (Huang et al., 2018).
Age-related macular degeneration is a progressive and degenerative eye disease, with aging and oxidative stress contributing to its pathogenesis. This disease is mainly caused by breakdown of proteostasis in Retinal Pigment Epithelium (RPE), leading to the accumulation of cellular waste such as lipofuscin. Oxidative stress induced by H2O2 leads to inhibition of proteasome activity and an increase in p62 expression in RPE cells. Silencing of p62 increases the accumulation of protein aggregates in RPE cells that showed decreased autophagy and Nrf2-mediated antioxidant response (Blasiak et al., 2019). RPE protection against oxidant-induced protein damage seems to rely on p62; however, if p62 is unable to be cleared by autophagy, the aggregates that form will likely contribute to further damage to the RPE cells, highlighting the importance of properly regulating p62 expression.
p62 is commonly upregulated in human tumors (Zatloukal et al., 2007), and the multiple roles p62 plays in cancer has been recently reviewed (Hennig et al., 2021). Suppression of tumorigenesis by autophagy is accomplished by limiting p62 accumulation and preventing activation of NF-κB. p62-induced expression of NF-κB in autophagy-defective cells is sufficient to activate the DNA damage response and enhance tumor growth (Mathew et al., 2009). Further, p62 overexpression promotes bone metastasis by stimulating migration, but not proliferation, of lung adenocarcinoma. High expression of p62 was associated with poor prognosis in patients with bone metastasis (Li D. et al., 2021). p62-induced tumorigenesis reveals an important concern when considering p62 induction as a potential therapeutic via autophagy induction.
Most degenerative diseases are characterized by protein aggregation, so naturally it would seem that targeting autophagy would be the best option to clear large aggregates. But what is the best way to induce this process when it requires coordination of multiple steps for autophagy to work? Since p62 has a multitude of roles beyond protein turnover and is also not essential for autophagy (Fan et al., 2010; Xu et al., 2015), specifically targeting p62 is not necessarily the answer. Induction of the entire autophagic pathway may instead be required. Therefore, identifying the limitations of pharmacological or genetic induction of autophagy (for example, by rapamycin treatment or TFEB activation), may offer better opportunities to therapeutically harness this process.
Loss of p62 leads to accelerated aging due to a decline in proteostasis, dysregulation of signaling pathways, and inability to sufficiently respond to oxidative stress. However, overexpression of p62 has potential detrimental effects, including accumulation of cellular p62-aggregates (Kumar et al., 2021), tumorigenesis and metastasis induction (Zatloukal et al., 2007; Mathew et al., 2009; Li D. et al., 2021), and mislocalization of the protein (Kurosawa et al., 2015). In order for p62 overexpression to be beneficial, global coordination of the induction of the 30 + autophagy genes (Wong et al., 2020b) by, for example, transcription factors FOXO and TFEB (Lapierre et al., 2015) may enhance autophagy protein levels and increase autophagic flux. In addition, the upstream ubiquitination machinery, including the E3 ligases correctly linking the poly-ubiquitin (K-63), must be functional to label appropriate cargo for degradation (Ma et al., 2019). Research remains to be done to elucidate the p62-associated changes that contribute to aging (Table 1). Additionally, it will be essential to classify what effects tissue- and spatiotemporal-specific p62 expression are accountable for. Understanding how to harness the positive and avoid the negative effects of p62 is crucial before it can be seriously considered as a therapeutic target for proteinopathies and neurodegenerative diseases.
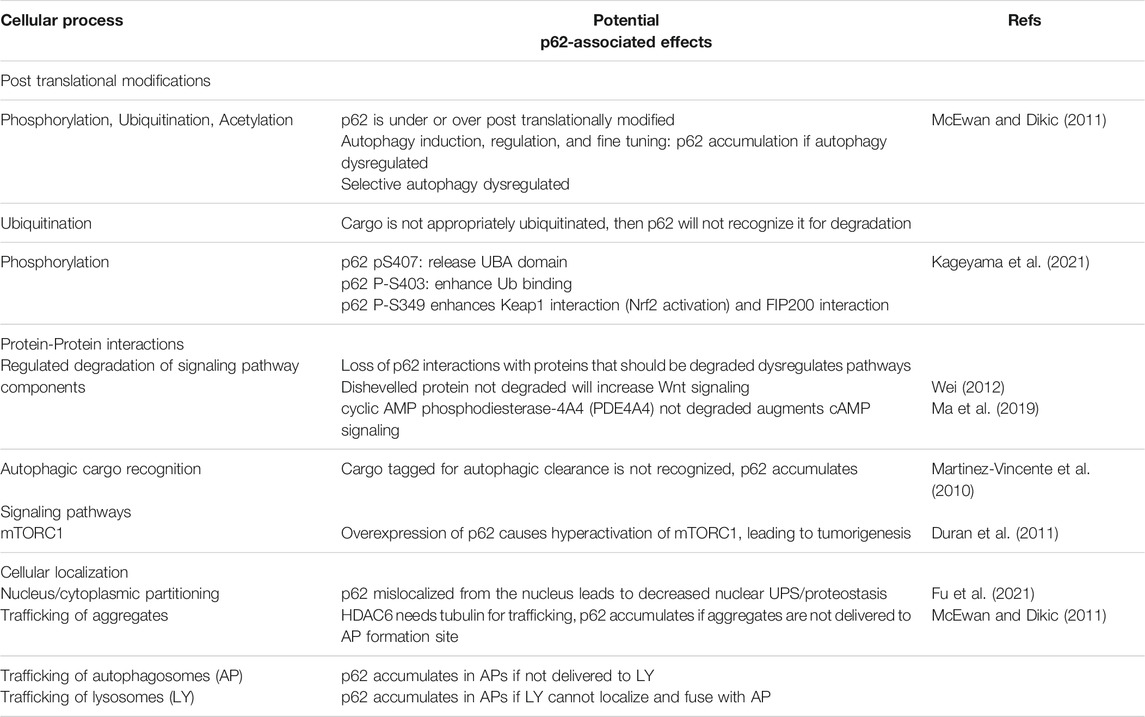
TABLE 1. Exploratory avenues of p62-associated changes.
AK and JM wrote the manuscript (equal contributions). LL provided guidance and edited the manuscript.
LL is funded by grants from the National Institute on Aging (Nos. R01 AG051810 and R21 AG068922).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Al-Sarraj, S., King, A., Troakes, C., Smith, B., Maekawa, S., Bodi, I., et al. (2011). p62 Positive, TDP-43 Negative, Neuronal Cytoplasmic and Intranuclear Inclusions in the Cerebellum and hippocampus Define the Pathology of C9orf72-Linked FTLD and MND/ALS. Acta Neuropathol. 122, 691–702. doi:10.1007/s00401-011-0911-2
Aparicio, R., Rana, A., and Walker, D. W. (2019). Upregulation of the Autophagy Adaptor p62/SQSTM1 Prolongs Health and Lifespan in Middle-Aged Drosophila. Cel Rep. 28, 1029–1040. doi:10.1016/j.celrep.2019.06.070
Aragonès, G., Dasuri, K., Olukorede, O., Francisco, S. G., Renneburg, C., Kumsta, C., et al. (2020). Autophagic Receptor P62 Protects against Glycation‐derived Toxicity and Enhances Viability. Aging Cell 19, e13257. doi:10.1111/acel.13257
Babu, J. R., Geetha, T., and Wooten, M. W. (2005). Sequestosome 1/p62 Shuttles Polyubiquitinated Tau for Proteasomal Degradation. J. Neurochem. 94, 192–203. doi:10.1111/j.1471-4159.2005.03181.x
Baskoylu, S. N., Chapkis, N., Unsal, B., Lins, J., Schuch, K., Simon, J., et al. (2022). Disrupted Autophagy and Neuronal Dysfunction in C. elegans Knock-In Models of FUS Amyotrophic Lateral Sclerosis. Cell Rep. 38, 110195. doi:10.1016/j.celrep.2021.110195
Berkamp, S., Mostafavi, S., and Sachse, C. (2021). Structure and Function of p62/SQSTM1 in the Emerging Framework of Phase Separation. FEBS J. 288, 6927–6941. doi:10.1111/febs.15672
Bitto, A., Lerner, C. A., Nacarelli, T., Crowe, E., Torres, C., and Sell, C. (2014). P62/SQSTM1 at the Interface of Aging, Autophagy, and Disease. Age 36, 9626. doi:10.1007/s11357-014-9626-3
Bjørkøy, G., Lamark, T., Brech, A., Outzen, H., Perander, M., Øvervatn, A., et al. (2005). p62/SQSTM1 Forms Protein Aggregates Degraded by Autophagy and Has a Protective Effect on Huntingtin-Induced Cell Death. J. Cel Biol 171, 603–614. doi:10.1083/jcb.200507002
Blasiak, J., Pawlowska, E., Szczepanska, J., and Kaarniranta, K. (2019). Interplay between Autophagy and the Ubiquitin-Proteasome System and its Role in the Pathogenesis of Age-Related Macular Degeneration. Ijms 20, 210. doi:10.3390/ijms20010210
Cha-Molstad, H., Yu, J. E., Feng, Z., Lee, S. H., Kim, J. G., Yang, P., et al. (2017). p62/SQSTM1/Sequestosome-1 Is an N-Recognin of the N-End Rule Pathway Which Modulates Autophagosome Biogenesis. Nat. Commun. 8, 102. doi:10.1038/s41467-017-00085-7
Chai, Q., Wang, X., Qiang, L., Zhang, Y., Ge, P., Lu, Z., et al. (2019). A Mycobacterium tuberculosis Surface Protein Recruits Ubiquitin to Trigger Host Xenophagy. Nat. Commun. 10, 1973. doi:10.1038/s41467-019-09955-8
Chen, G., Kroemer, G., and Kepp, O. (2020). Mitophagy: An Emerging Role in Aging and Age-Associated Diseases. Front. Cel Dev. Biol. 8, 200. doi:10.3389/fcell.2020.00200
Choi, W. H., Yun, Y., Park, S., Jeon, J. H., Lee, J., Lee, J. H., et al. (2020). Aggresomal Sequestration and STUB1-Mediated Ubiquitylation during Mammalian Proteaphagy of Inhibited Proteasomes. Proc. Natl. Acad. Sci. USA 117, 19190–19200. doi:10.1073/pnas.1920327117
Christian, F., Krause, E., Houslay, M. D., and Baillie, G. S. (2014). PKA Phosphorylation of p62/SQSTM1 Regulates PB1 Domain Interaction Partner Binding. Biochim. Biophys. Acta (Bba) - Mol. Cel Res. 1843, 2765–2774. doi:10.1016/j.bbamcr.2014.07.021
Ciuffa, R., Lamark, T., Tarafder, A. K., Guesdon, A., Rybina, S., Hagen, W. J. H., et al. (2015). The Selective Autophagy Receptor P62 Forms a Flexible Filamentous Helical Scaffold. Cel Rep. 11, 748–758. doi:10.1016/j.celrep.2015.03.062
Cohen-Kaplan, V., Ciechanover, A., and Livneh, I. (2017). Stress-induced Polyubiquitination of Proteasomal Ubiquitin Receptors Targets the Proteolytic Complex for Autophagic Degradation. Autophagy 13, 759–760. doi:10.1080/15548627.2016.1278327
Demishtein, A., Fraiberg, M., Berko, D., Tirosh, B., Elazar, Z., and Navon, A. (2017). SQSTM1/p62-mediated Autophagy Compensates for Loss of Proteasome Polyubiquitin Recruiting Capacity. Autophagy 13, 1697–1708. doi:10.1080/15548627.2017.1356549
Deng, Z., Lim, J., Wang, Q., Purtell, K., Wu, S., Palomo, G. M., et al. (2020). ALS-FTLD-linked Mutations of SQSTM1/p62 Disrupt Selective Autophagy and NFE2L2/NRF2 Anti-oxidative Stress Pathway. Autophagy 16, 917–931. doi:10.1080/15548627.2019.1644076
Deosaran, E., Larsen, K. B., Hua, R., Sargent, G., Wang, Y., Kim, S., et al. (2013). NBR1 Acts as an Autophagy Receptor for Peroxisomes. J. Cel Sci 126, 939–952. doi:10.1242/jcs.114819
Dikic, I. (2017). Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 86, 193–224. doi:10.1146/annurev-biochem-061516-044908
Du, Y., Wooten, M. C., Gearing, M., and Wooten, M. W. (2009). Age-associated Oxidative Damage to the P62 Promoter: Implications for Alzheimer Disease. Free Radic. Biol. Med. 46, 492–501. doi:10.1016/j.freeradbiomed.2008.11.003
Duran, A., Amanchy, R., Linares, J. F., Joshi, J., Abu-Baker, S., Porollo, A., et al. (2011). p62 Is a Key Regulator of Nutrient Sensing in the mTORC1 Pathway. Mol. Cel 44, 134–146. doi:10.1016/j.molcel.2011.06.038
Emanuele, S., Lauricella, M., D’Anneo, A., Carlisi, D., De Blasio, A., Di Liberto, D., et al. (2020). p62: Friend or Foe? Evidences for OncoJanus and NeuroJanus Roles. Ijms 21, 5029. doi:10.3390/ijms21145029
Fan, W., Tang, Z., Chen, D., Moughon, D., Ding, X., Chen, S., et al. (2010). Keap1 Facilitates P62-Mediated Ubiquitin Aggregate Clearance via Autophagy. Autophagy 6, 614–621. doi:10.4161/auto.6.5.12189
Foster, A. D., Flynn, L. L., Cluning, C., Cheng, F., Davidson, J. M., Lee, A., et al. (2021). p62 Overexpression Induces TDP-43 Cytoplasmic Mislocalisation, Aggregation and Cleavage and Neuronal Death. Sci. Rep. 11, 11474. doi:10.1038/s41598-021-90822-2
Fu, A., Cohen-Kaplan, V., Avni, N., Livneh, I., and Ciechanover, A. (2021). p62-containing, Proteolytically Active Nuclear Condensates, Increase the Efficiency of the Ubiquitin-Proteasome System. Proc. Natl. Acad. Sci. USA 118, e2107321118. doi:10.1073/pnas.2107321118
Galluzzi, L., Baehrecke, E. H., Ballabio, A., Boya, P., Bravo‐San Pedro, J. M., Cecconi, F., et al. (2017). Molecular Definitions of Autophagy and Related Processes. EMBO J. 36, 1811–1836. doi:10.15252/embj.201796697
Geisler, S., Holmström, K. M., Skujat, D., Fiesel, F. C., Rothfuss, O. C., Kahle, P. J., et al. (2010). PINK1/Parkin-mediated Mitophagy Is Dependent on VDAC1 and p62/SQSTM1. Nat. Cel Biol 12, 119–131. doi:10.1038/ncb2012
Germain, K., and Kim, P. K. (2020). Pexophagy: A Model for Selective Autophagy. Ijms 21, 578. doi:10.3390/ijms21020578
Goodall, M. L., Fitzwalter, B. E., Zahedi, S., Wu, M., Rodriguez, D., Mulcahy-Levy, J. M., et al. (2016). The Autophagy Machinery Controls Cell Death Switching between Apoptosis and Necroptosis. Dev. Cel 37, 337–349. doi:10.1016/j.devcel.2016.04.018
Goode, A., Rea, S., Sultana, M., Shaw, B., Searle, M. S., and Layfield, R. (2016). ALS-FTLD Associated Mutations of SQSTM1 Impact on Keap1-Nrf2 Signalling. Mol. Cell Neurosci. 76, 52–58. doi:10.1016/j.mcn.2016.08.004
Hennig, P., Fenini, G., Di Filippo, M., Karakaya, T., and Beer, H.-D. (2021). The Pathways Underlying the Multiple Roles of P62 in Inflammation and Cancer. Biomedicines 9, 707. doi:10.3390/biomedicines9070707
Horos, R., Büscher, M., Kleinendorst, R., Alleaume, A.-M., Tarafder, A. K., Schwarzl, T., et al. (2019). The Small Non-coding Vault RNA1-1 Acts as a Riboregulator of Autophagy. Cell 176, 1054–1067. doi:10.1016/j.cell.2019.01.030
Huang, N., Erie, C., Lu, M. L., and Wei, J. (2018). Aberrant Subcellular Localization of SQSTM1/p62 Contributes to Increased Vulnerability to Proteotoxic Stress Recovery in Huntington's Disease. Mol. Cell Neurosci. 88, 43–52. doi:10.1016/j.mcn.2017.12.005
Ichimura, Y., Waguri, S., Sou, Y.-s., Kageyama, S., Hasegawa, J., Ishimura, R., et al. (2013). Phosphorylation of P62 Activates the Keap1-Nrf2 Pathway during Selective Autophagy. Mol. Cel 51, 618–631. doi:10.1016/j.molcel.2013.08.003
Isogai, S., Morimoto, D., Arita, K., Unzai, S., Tenno, T., Hasegawa, J., et al. (2011). Crystal Structure of the Ubiquitin-Associated (UBA) Domain of P62 and its Interaction with Ubiquitin. J. Biol. Chem. 286, 31864–31874. doi:10.1074/jbc.m111.259630
Jain, A., Lamark, T., Sjøttem, E., Bowitz Larsen, K., Atesoh Awuh, J., Øvervatn, A., et al. (2010). p62/SQSTM1 Is a Target Gene for Transcription Factor NRF2 and Creates a Positive Feedback Loop by Inducing Antioxidant Response Element-Driven Gene Transcription. J. Biol. Chem. 285, 22576–22591. doi:10.1074/jbc.m110.118976
Jakobi, A. J., Huber, S. T., Mortensen, S. A., Schultz, S. W., Palara, A., Kuhm, T., et al. (2020). Structural Basis of p62/SQSTM1 Helical Filaments and Their Role in Cellular Cargo Uptake. Nat. Commun. 11, 440. doi:10.1038/s41467-020-14343-8
Jin, Z., Li, Y., Pitti, R., Lawrence, D., Pham, V. C., Lill, J. R., et al. (2009). Cullin3-based Polyubiquitination and P62-dependent Aggregation of Caspase-8 Mediate Extrinsic Apoptosis Signaling. Cell 137, 721–735. doi:10.1016/j.cell.2009.03.015
Kageyama, S., Gudmundsson, S. R., Sou, Y.-S., Ichimura, Y., Tamura, N., Kazuno, S., et al. (2021). p62/SQSTM1-droplet Serves as a Platform for Autophagosome Formation and Anti-oxidative Stress Response. Nat. Commun. 12, 16. doi:10.1038/s41467-020-20185-1
Kaushik, S., and Cuervo, A. M. (2015). Proteostasis and Aging. Nat. Med. 21, 1406–1415. doi:10.1038/nm.4001
King, M. A., Hands, S., Hafiz, F., Mizushima, N., Tolkovsky, A. M., and Wyttenbach, A. (2008). Rapamycin Inhibits Polyglutamine Aggregation Independently of Autophagy by Reducing Protein Synthesis. Mol. Pharmacol. 73, 1052–1063. doi:10.1124/mol.107.043398
Kırlı, K., Karaca, S., Dehne, H. J., Samwer, M., Pan, K. T., Lenz, C., et al. (2015). A Deep Proteomics Perspective on CRM1-Mediated Nuclear export and Nucleocytoplasmic Partitioning. Elife 4, e11466. doi:10.7554/eLife.11466
Komatsu, M., Kageyama, S., and Ichimura, Y. (2012). p62/SQSTM1/A170: Physiology and Pathology. Pharmacol. Res. 66, 457–462. doi:10.1016/j.phrs.2012.07.004
Korolchuk, V. I., Mansilla, A., Menzies, F. M., and Rubinsztein, D. C. (2009). Autophagy Inhibition Compromises Degradation of Ubiquitin-Proteasome Pathway Substrates. Mol. Cel 33, 517–527. doi:10.1016/j.molcel.2009.01.021
Kumar, A. V., Mills, J., Parker, W. M., Leitão, J. A., Ng, C., Patel, R., et al. (2021). Lipid Droplets Modulate Proteostasis, SQST-1/SQSTM1 Dynamics, and Lifespan in C. elegans. bioRxiv. doi:10.1101/2021.04.22.440991
Kumsta, C., Chang, J. T., Lee, R., Tan, E. P., Yang, Y., Loureiro, R., et al. (2019). The Autophagy Receptor p62/SQST-1 Promotes Proteostasis and Longevity in C. elegans by Inducing Autophagy. Nat. Commun. 10, 5648. doi:10.1038/s41467-019-13540-4
Kurosawa, M., Matsumoto, G., Kino, Y., Okuno, M., Kurosawa-Yamada, M., Washizu, C., et al. (2015). Depletion of P62 Reduces Nuclear Inclusions and Paradoxically Ameliorates Disease Phenotypes in Huntington's Model Mice. Hum. Mol. Genet. 24, 1092–1105. doi:10.1093/hmg/ddu522
Kwok, C. T., Morris, A., and De Belleroche, J. S. (2014). Sequestosome-1 (SQSTM1) Sequence Variants in ALS Cases in the UK: Prevalence and Coexistence of SQSTM1 Mutations in ALS kindred with PDB. Eur. J. Hum. Genet. 22, 492–496. doi:10.1038/ejhg.2013.184
Kwon, D. H., Park, O. H., Kim, L., Jung, Y. O., Park, Y., Jeong, H., et al. (2018). Insights into Degradation Mechanism of N-End Rule Substrates by p62/SQSTM1 Autophagy Adapter. Nat. Commun. 9, 3291. doi:10.1038/s41467-018-05825-x
Kwon, J., Han, E., Bui, C. B., Shin, W., Lee, J., Lee, S., et al. (2012). Assurance of Mitochondrial Integrity and Mammalian Longevity by the P62-Keap1-Nrf2-Nqo1 cascade. EMBO Rep. 13, 150–156. doi:10.1038/embor.2011.246
Lam, T., Harmancey, R., Vasquez, H., Gilbert, B., Patel, N., Hariharan, V., et al. (2016). Reversal of Intramyocellular Lipid Accumulation by Lipophagy and a P62-Mediated Pathway. Cel Death Discov. 2, 16061. doi:10.1038/cddiscovery.2016.61
Lamb, C. A., Yoshimori, T., and Tooze, S. A. (2013). The Autophagosome: Origins Unknown, Biogenesis Complex. Nat. Rev. Mol. Cel Biol 14, 759–774. doi:10.1038/nrm3696
Lapierre, L. R., De Magalhaes Filho, C. D., Mcquary, P. R., Chu, C.-C., Visvikis, O., Chang, J. T., et al. (2013). The TFEB Orthologue HLH-30 Regulates Autophagy and Modulates Longevity in Caenorhabditis elegans. Nat. Commun. 4, 2267. doi:10.1038/ncomms3267
Lapierre, L. R., Kumsta, C., Sandri, M., Ballabio, A., and Hansen, M. (2015). Transcriptional and Epigenetic Regulation of Autophagy in Aging. Autophagy 11, 867–880. doi:10.1080/15548627.2015.1034410
Lee, D. H., and Goldberg, A. L. (1998). Proteasome Inhibitors: Valuable New Tools for Cell Biologists. Trends Cel Biol. 8, 397–403. doi:10.1016/s0962-8924(98)01346-4
Li, D., He, C., Ye, F., Ye, E., He, H., Chen, G., et al. (2021). p62 Overexpression Promotes Bone Metastasis of Lung Adenocarcinoma Out of LC3-dependent Autophagy. Front. Oncol. 11, 609548. doi:10.3389/fonc.2021.609548
Li, X., Zhou, L., Peng, G., Liao, M., Zhang, L., Hu, H., et al. (2021). Pituitary P62 Deficiency Leads to Female Infertility by Impairing Luteinizing Hormone Production. Exp. Mol. Med. 53, 1238–1249. doi:10.1038/s12276-021-00661-4
Lim, J., Lachenmayer, M. L., Wu, S., Liu, W., Kundu, M., Wang, R., et al. (2015). Proteotoxic Stress Induces Phosphorylation of p62/SQSTM1 by ULK1 to Regulate Selective Autophagic Clearance of Protein Aggregates. Plos Genet. 11, e1004987. doi:10.1371/journal.pgen.1004987
Lin, X., Li, S., Zhao, Y., Ma, X., Zhang, K., He, X., et al. (2013). Interaction Domains of P62: a Bridge between P62 and Selective Autophagy. DNA Cel Biol. 32, 220–227. doi:10.1089/dna.2012.1915
Liu, W. J., Ye, L., Huang, W. F., Guo, L. J., Xu, Z. G., Wu, H. L., et al. (2016). p62 Links the Autophagy Pathway and the Ubiqutin-Proteasome System upon Ubiquitinated Protein Degradation. Cell Mol Biol Lett 21, 29. doi:10.1186/s11658-016-0031-z
Lobb, I. T., Morin, P., Martin, K., Thoms, H. C., Wills, J. C., Lleshi, X., et al. (2021). A Role for the Autophagic Receptor, SQSTM1/p62, in Trafficking NF-κB/RelA to Nucleolar Aggresomes. Mol. Cancer Res. 19, 274–287. doi:10.1158/1541-7786.mcr-20-0336
Lyu, L., Chen, Z., and Mccarty, N. (2021). TRIM44 Links the UPS to SQSTM1/p62-dependent Aggrephagy and Removing Misfolded Proteins. Autophagy, 1–16. doi:10.1080/15548627.2021.1956105
Ma, S., Attarwala, I. Y., and Xie, X.-Q. (2019). SQSTM1/p62: A Potential Target for Neurodegenerative Disease. ACS Chem. Neurosci. 10, 2094–2114. doi:10.1021/acschemneuro.8b00516
Marshall, R. S., Mcloughlin, F., and Vierstra, R. D. (2016). Autophagic Turnover of Inactive 26S Proteasomes in Yeast Is Directed by the Ubiquitin Receptor Cue5 and the Hsp42 Chaperone. Cel Rep. 16, 1717–1732. doi:10.1016/j.celrep.2016.07.015
Martina, J. A., Chen, Y., Gucek, M., and Puertollano, R. (2012). MTORC1 Functions as a Transcriptional Regulator of Autophagy by Preventing Nuclear Transport of TFEB. Autophagy 8, 903–914. doi:10.4161/auto.19653
Martinez-Vicente, M., Talloczy, Z., Wong, E., Tang, G., Koga, H., Kaushik, S., et al. (2010). Cargo Recognition Failure Is Responsible for Inefficient Autophagy in Huntington's Disease. Nat. Neurosci. 13, 567–576. doi:10.1038/nn.2528
Mathew, R., Karp, C. M., Beaudoin, B., Vuong, N., Chen, G., Chen, H.-Y., et al. (2009). Autophagy Suppresses Tumorigenesis through Elimination of P62. Cell 137, 1062–1075. doi:10.1016/j.cell.2009.03.048
Matsumoto, G., Wada, K., Okuno, M., Kurosawa, M., and Nukina, N. (2011). Serine 403 Phosphorylation of p62/SQSTM1 Regulates Selective Autophagic Clearance of Ubiquitinated Proteins. Mol. Cel 44, 279–289. doi:10.1016/j.molcel.2011.07.039
McEwan, D. G., and Dikic, I. (2011). The Three Musketeers of Autophagy: phosphorylation, ubiquitylation and acetylation. Trends Cell Biol. 21, 195–201. doi:10.1016/j.tcb.2010.12.006
Mizushima, N., Yoshimori, T., and Levine, B. (2010). Methods in Mammalian Autophagy Research. Cell 140, 313–326. doi:10.1016/j.cell.2010.01.028
Moscat, J., Diaz-Meco, M. T., Albert, A., and Campuzano, S. (2006). Cell Signaling and Function Organized by PB1 Domain Interactions. Mol. Cel 23, 631–640. doi:10.1016/j.molcel.2006.08.002
Moscat, J., and Diaz-Meco, M. T. (2011). Feedback on Fat: P62-mTORC1-Autophagy Connections. Cell 147, 724–727. doi:10.1016/j.cell.2011.10.021
Moscat, J., Karin, M., and Diaz-Meco, M. T. (2016). p62 in Cancer: Signaling Adaptor beyond Autophagy. Cell 167, 606–609. doi:10.1016/j.cell.2016.09.030
Myeku, N., and Figueiredo-Pereira, M. E. (2011). Dynamics of the Degradation of Ubiquitinated Proteins by Proteasomes and Autophagy. J. Biol. Chem. 286, 22426–22440. doi:10.1074/jbc.m110.149252
Nakamura, K., Kimple, A. J., Siderovski, D. P., and Johnson, G. L. (2010). PB1 Domain Interaction of p62/Sequestosome 1 and MEKK3 Regulates NF-Κb Activation. J. Biol. Chem. 285, 2077–2089. doi:10.1074/jbc.m109.065102
Narendra, D., Kane, L. A., Hauser, D. N., Fearnley, I. M., and Youle, R. J. (2010). p62/SQSTM1 Is Required for Parkin-Induced Mitochondrial Clustering but Not Mitophagy; VDAC1 Is Dispensable for Both. Autophagy 6, 1090–1106. doi:10.4161/auto.6.8.13426
Pankiv, S., Clausen, T. H., Lamark, T., Brech, A., Bruun, J.-A., Outzen, H., et al. (2007). p62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. J. Biol. Chem. 282, 24131–24145. doi:10.1074/jbc.m702824200
Pankiv, S., Lamark, T., Bruun, J.-A., Øvervatn, A., Bjørkøy, G., and Johansen, T. (2010). Nucleocytoplasmic Shuttling of p62/SQSTM1 and its Role in Recruitment of Nuclear Polyubiquitinated Proteins to Promyelocytic Leukemia Bodies. J. Biol. Chem. 285, 5941–5953. doi:10.1074/jbc.m109.039925
Papadopoulos, C., Kirchner, P., Bug, M., Grum, D., Koerver, L., Schulze, N., et al. (2017). VCP/p97 Cooperates with YOD 1, UBXD 1 and PLAA to Drive Clearance of Ruptured Lysosomes by Autophagy. EMBO J. 36, 135–150. doi:10.15252/embj.201695148
Park, S., Han, S., Choi, I., Kim, B., Park, S. P., Joe, E.-H., et al. (2016). Interplay between Leucine-Rich Repeat Kinase 2 (LRRK2) and p62/SQSTM-1 in Selective Autophagy. PLoS ONE 11, e0163029. doi:10.1371/journal.pone.0163029
Peña-Llopis, S., Vega-Rubin-De-Celis, S., Schwartz, J. C., Wolff, N. C., Tran, T. A. T., Zou, L., et al. (2011). Regulation of TFEB and V-ATPases by mTORC1. EMBO J. 30, 3242–3258. doi:10.1038/emboj.2011.257
Peng, H., Yang, J., Li, G., You, Q., Han, W., Li, T., et al. (2017). Ubiquitylation of P62/sequestosome1 Activates its Autophagy Receptor Function and Controls Selective Autophagy upon Ubiquitin Stress. Cell Res 27, 657–674. doi:10.1038/cr.2017.40
Ravikumar, B., Vacher, C., Berger, Z., Davies, J. E., Luo, S., Oroz, L. G., et al. (2004). Inhibition of mTOR Induces Autophagy and Reduces Toxicity of Polyglutamine Expansions in Fly and Mouse Models of Huntington Disease. Nat. Genet. 36, 585–595. doi:10.1038/ng1362
Roberts, M. A., and Olzmann, J. A. (2020). Protein Quality Control and Lipid Droplet Metabolism. Annu. Rev. Cel Dev. Biol. 36, 115–139. doi:10.1146/annurev-cellbio-031320-101827
Robichaud, S., Fairman, G., Vijithakumar, V., Mak, E., Cook, D. P., Pelletier, A. R., et al. (2021). Identification of Novel Lipid Droplet Factors that Regulate Lipophagy and Cholesterol Efflux in Macrophage Foam Cells. Autophagy 17, 3671–3689. doi:10.1080/15548627.2021.1886839
Rock, K. L., Gramm, C., Rothstein, L., Clark, K., Stein, R., Dick, L., et al. (1994). Inhibitors of the Proteasome Block the Degradation of Most Cell Proteins and the Generation of Peptides Presented on MHC Class I Molecules. Cell 78, 761–771. doi:10.1016/s0092-8674(94)90462-6
Rué, L., López-Soop, G., Gelpi, E., Martínez-Vicente, M., Alberch, J., and Pérez-Navarro, E. (2013). Brain Region- and Age-dependent Dysregulation of P62 and NBR1 in a Mouse Model of Huntington's Disease. Neurobiol. Dis. 52, 219–228. doi:10.1016/j.nbd.2012.12.008
Salazar, G., Cullen, A., Huang, J., Zhao, Y., Serino, A., Hilenski, L., et al. (2020). SQSTM1/p62 and PPARGC1A/PGC-1alpha at the Interface of Autophagy and Vascular Senescence. Autophagy 16, 1092–1110. doi:10.1080/15548627.2019.1659612
Salminen, A., Kaarniranta, K., Haapasalo, A., Hiltunen, M., Soininen, H., and Alafuzoff, I. (2012). Emerging Role of P62/sequestosome-1 in the Pathogenesis of Alzheimer's Disease. Prog. Neurobiol. 96, 87–95. doi:10.1016/j.pneurobio.2011.11.005
Sánchez‐Martín, P., Sou, Y. s., Kageyama, S., Koike, M., Waguri, S., and Komatsu, M. (2020). NBR 1‐mediated P62‐liquid Droplets Enhance the Keap1‐Nrf2 System. EMBO Rep. 21, e48902. doi:10.15252/embr.201948902
Sanz, L., Sanchez, P., Lallena, M. J., Diaz-Meco, M. T., and Moscat, J. (1999). The Interaction of P62 with RIP Links the Atypical PKCs to NF-Kappa B Activation. EMBO J. 18, 3044–3053. doi:10.1093/emboj/18.11.3044
Sardiello, M., Palmieri, M., Di Ronza, A., Medina, D. L., Valenza, M., Gennarino, V. A., et al. (2009). A Gene Network Regulating Lysosomal Biogenesis and Function. Science 325, 473–477. doi:10.1126/science.1174447
Seibenhener, M. L., Babu, J. R., Geetha, T., Wong, H. C., Krishna, N. R., and Wooten, M. W. (2004). Sequestosome 1/p62 Is a Polyubiquitin Chain Binding Protein Involved in Ubiquitin Proteasome Degradation. Mol. Cel Biol 24, 8055–8068. doi:10.1128/mcb.24.18.8055-8068.2004
Selleck, E. M., Orchard, R. C., Lassen, K. G., Beatty, W. L., Xavier, R. J., Levine, B., et al. (2015). A Noncanonical Autophagy Pathway Restricts Toxoplasma Gondii Growth in a Strain-specific Manner in IFN-γ-Activated Human Cells. mBio 6, e01157–15. doi:10.1128/mBio.01157-15
Seton, M., Hansen, M., and Solomon, D. H. (2016). The Implications of the Sequestosome 1 Mutation P392L in Patients with Paget's Disease in a United States Cohort. Calcif Tissue Int. 98, 489–496. doi:10.1007/s00223-015-0103-5
Settembre, C., Di Malta, C., Polito, V. A., Arencibia, M. G., Vetrini, F., Erdin, S., et al. (2011). TFEB Links Autophagy to Lysosomal Biogenesis. Science 332, 1429–1433. doi:10.1126/science.1204592
Sha, Z., Schnell, H. M., Ruoff, K., and Goldberg, A. (2018). Rapid Induction of P62 and GABARAPL1 upon Proteasome Inhibition Promotes Survival before Autophagy Activation. J. Cel Biol 217, 1757–1776. doi:10.1083/jcb.201708168
Sharma, V., Verma, S., Seranova, E., Sarkar, S., and Kumar, D. (2018). Selective Autophagy and Xenophagy in Infection and Disease. Front. Cel Dev. Biol. 6, 147. doi:10.3389/fcell.2018.00147
Shin, W. H., Park, J. H., and Chung, K. C. (2020). The central Regulator P62 between Ubiquitin Proteasome System and Autophagy and its Role in the Mitophagy and Parkinson's Disease. BMB Rep. 53, 56–63. doi:10.5483/bmbrep.2020.53.1.283
Silvestrini, M. J., Johnson, J. R., Kumar, A. V., Thakurta, T. G., Blais, K., Neill, Z. A., et al. (2018). Nuclear Export Inhibition Enhances HLH-30/TFEB Activity, Autophagy, and Lifespan. Cel Rep. 23, 1915–1921. doi:10.1016/j.celrep.2018.04.063
Singh, R., Kaushik, S., Wang, Y., Xiang, Y., Novak, I., Komatsu, M., et al. (2009). Autophagy Regulates Lipid Metabolism. Nature 458, 1131–1135. doi:10.1038/nature07976
Stolz, A., Ernst, A., and Dikic, I. (2014). Cargo Recognition and Trafficking in Selective Autophagy. Nat. Cel Biol 16, 495–501. doi:10.1038/ncb2979
Sun, D., Wu, R., Zheng, J., Li, P., and Yu, L. (2018). Polyubiquitin Chain-Induced P62 Phase Separation Drives Autophagic Cargo Segregation. Cel Res 28, 405–415. doi:10.1038/s41422-018-0017-7
Tsuchiya, M., Ogawa, H., Koujin, T., Mori, C., Osakada, H., Kobayashi, S., et al. (2018). p62/SQSTM1 Promotes Rapid Ubiquitin Conjugation to Target Proteins after Endosome Rupture during Xenophagy. FEBS Open Bio 8, 470–480. doi:10.1002/2211-5463.12385
Turco, E., Savova, A., Gere, F., Ferrari, L., Romanov, J., Schuschnig, M., et al. (2021). Reconstitution Defines the Roles of P62, NBR1 and TAX1BP1 in Ubiquitin Condensate Formation and Autophagy Initiation. Nat. Commun. 12, 5212. doi:10.1038/s41467-021-25572-w
Wang, L., Zhou, J., Yan, S., Lei, G., Lee, C.-H., and Yin, X.-M. (2017). Ethanol-triggered Lipophagy Requires SQSTM1 in AML12 Hepatic Cells. Sci. Rep. 7, 12307. doi:10.1038/s41598-017-12485-2
Wei, W., Li, M., Wang, J., Nie, F., and Li, L. (2012). The E3 ubiquitin ligase ITCH negatively regulates canonical Wnt signaling by targeting dishevelled protein. Mol. Cell Biol. 32, 3903–3912. doi:10.1128/MCB.00251-12
Wold, M. S., Lim, J., Lachance, V., Deng, Z., and Yue, Z. (2016). ULK1-mediated Phosphorylation of ATG14 Promotes Autophagy and Is Impaired in Huntington's Disease Models. Mol. Neurodegeneration 11, 76. doi:10.1186/s13024-016-0141-0
Wong, S. Q., Kumar, A. V., Mills, J., and Lapierre, L. R. (2020a). Autophagy in Aging and Longevity. Hum. Genet. 139, 277–290. doi:10.1007/s00439-019-02031-7
Wong, S. Q., Kumar, A. V., Mills, J., and Lapierre, L. R. (2020b). “C. elegans to Model Autophagy-Related Human Disorders,” in Progress In Molecular Biology And Translational Science. Editors A.B. Martinez, and L. Galluzzi (Cambridge: Academic Press), 325–373. doi:10.1016/bs.pmbts.2020.01.007In
Wooten, M. W., Geetha, T., Seibenhener, M. L., Babu, J. R., Diaz-Meco, M. T., and Moscat, J. (2005). The P62 Scaffold Regulates Nerve Growth Factor-Induced NF-Κb Activation by Influencing TRAF6 Polyubiquitination. J. Biol. Chem. 280, 35625–35629. doi:10.1074/jbc.c500237200
Wurzer, B., Zaffagnini, G., Fracchiolla, D., Turco, E., Abert, C., Romanov, J., et al. (2015). Oligomerization of P62 Allows for Selection of Ubiquitinated Cargo and Isolation Membrane during Selective Autophagy. Elife 4, e08941. doi:10.7554/elife.08941
Xiao, B., Deng, X., Lim, G. G. Y., Zhou, W., Saw, W.-T., Zhou, Z. D., et al. (2017). p62-Mediated Mitochondrial Clustering Attenuates Apoptosis Induced by Mitochondrial Depolarization. Biochim. Biophys. Acta (Bba) - Mol. Cel Res. 1864, 1308–1317. doi:10.1016/j.bbamcr.2017.04.009
Xu, T., Nicolson, S., Denton, D., and Kumar, S. (2015). Distinct Requirements of Autophagy-Related Genes in Programmed Cell Death. Cell Death Differ 22, 1792–1802. doi:10.1038/cdd.2015.28
Yamada, T., Dawson, T. M., Yanagawa, T., Iijima, M., and Sesaki, H. (2019). SQSTM1/p62 Promotes Mitochondrial Ubiquitination Independently of PINK1 and PRKN/parkin in Mitophagy. Autophagy 15, 2012–2018. doi:10.1080/15548627.2019.1643185
Yamada, T., Murata, D., Adachi, Y., Itoh, K., Kameoka, S., Igarashi, A., et al. (2018). Mitochondrial Stasis Reveals P62-Mediated Ubiquitination in Parkin-independent Mitophagy and Mitigates Nonalcoholic Fatty Liver Disease. Cel Metab. 28, 588–604. doi:10.1016/j.cmet.2018.06.014
Yan, Y., Wang, H., Wei, C., Xiang, Y., Liang, X., Phang, C.-W., et al. (2019). HDAC6 Regulates Lipid Droplet Turnover in Response to Nutrient Deprivation via P62-Mediated Selective Autophagy. J. Genet. Genomics 46, 221–229. doi:10.1016/j.jgg.2019.03.008
Yang, Y., Willis, T. L., Button, R. W., Strang, C. J., Fu, Y., Wen, X., et al. (2019). Cytoplasmic DAXX Drives SQSTM1/p62 Phase Condensation to Activate Nrf2-Mediated Stress Response. Nat. Commun. 10, 3759. doi:10.1038/s41467-019-11671-2
Yim, W. W.-Y., and Mizushima, N. (2020). Lysosome Biology in Autophagy. Cell Discov 6 (6). doi:10.1038/s41421-020-0141-7
Yoon, M. J., Choi, B., Kim, E. J., Ohk, J., Yang, C., Choi, Y.-G., et al. (2021). UXT Chaperone Prevents Proteotoxicity by Acting as an Autophagy Adaptor for P62-dependent Aggrephagy. Nat. Commun. 12, 1955. doi:10.1038/s41467-021-22252-7
Yuan, X., Tian, G. G., Pei, X., Hu, X., and Wu, J. (2021). Spermidine Induces Cytoprotective Autophagy of Female Germline Stem Cells In Vitro and Ameliorates Aging Caused by Oxidative Stress through Upregulated Sequestosome-1/p62 Expression. Cell Biosci 11, 107. doi:10.1186/s13578-021-00614-4
Zaffagnini, G., Savova, A., Danieli, A., Romanov, J., Tremel, S., Ebner, M., et al. (2018). p62 Filaments Capture and Present Ubiquitinated Cargos for Autophagy. EMBO J. 37, e98308. doi:10.15252/embj.201798308
Zatloukal, K., French, S. W., Stumptner, C., Strnad, P., Harada, M., Toivola, D. M., et al. (2007). From Mallory to Mallory-Denk Bodies: what, How and Why? Exp. Cel Res. 313, 2033–2049. doi:10.1016/j.yexcr.2007.04.024
Zhang, X., Li, L., Chen, S., Yang, D., Wang, Y., Zhang, X., et al. (2011). Rapamycin Treatment Augments Motor Neuron Degeneration in SOD1G93Amouse Model of Amyotrophic Lateral Sclerosis. Autophagy 7, 412–425. doi:10.4161/auto.7.4.14541
Zhang, Y., Mun, S. R., Linares, J. F., Ahn, J., Towers, C. G., Ji, C. H., et al. (2018). ZZ-dependent Regulation of p62/SQSTM1 in Autophagy. Nat. Commun. 9, 4373. doi:10.1038/s41467-018-06878-8
Zhao, L., Li, H., Wang, Y., Zheng, A., Cao, L., and Liu, J. (2019). Autophagy Deficiency Leads to Impaired Antioxidant Defense via P62-Foxo1/3 Axis. Oxidative Med. Cell Longevity 2019, 2526314. doi:10.1155/2019/2526314
Keywords: p62 (sequestosome 1(SQSTM1)), autophagy, proteasome, aging, neurodegenerative diseases
Citation: Kumar AV, Mills J and Lapierre LR (2022) Selective Autophagy Receptor p62/SQSTM1, a Pivotal Player in Stress and Aging. Front. Cell Dev. Biol. 10:793328. doi: 10.3389/fcell.2022.793328
Received: 12 October 2021; Accepted: 19 January 2022;
Published: 14 February 2022.
Edited by:
Javier Calvo Garrido, Karolinska Institutet (KI), SwedenReviewed by:
Paula Daza-Navarro, Sevilla University, SpainCopyright © 2022 Kumar, Mills and Lapierre. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Louis R. Lapierre, bG91aXNfbGFwaWVycmVAYnJvd24uZWR1
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.