 Yuhan Sun1,2S. Ram Kumar1,3
Yuhan Sun1,2S. Ram Kumar1,3 Chee Ern David Wong1
Chee Ern David Wong1 Zhiyu Tian1Haipeng Bai1,4J. Gage Crump5Ruchi Bajpai2,6
Zhiyu Tian1Haipeng Bai1,4J. Gage Crump5Ruchi Bajpai2,6 Ching Ling Lien1,3,6*
Ching Ling Lien1,3,6*- 1Saban Research Institute and Heart Institute, Children’s Hospital Los Angeles, Los Angeles, CA, United States
- 2Center for Craniofacial Molecular Biology, Ostrow School of Dentistry, University of Southern California, Los Angeles, CA, United States
- 3Department of Surgery, Keck School of Medicine, University of Southern California, Los Angeles, CA, United States
- 4State Key Laboratory of Chemical Oncogenomics, Key Laboratory of Chemical Genomics, Peking University Shenzhen Graduate School, Shenzhen, China
- 5Eli and Edythe Broad CIRM Center for Regenerative Medicine and Stem Cell Research, Keck School of Medicine, University of Southern California, Los Angeles, CA, United States
- 6Department of Biochemistry and Molecular Medicine, Keck School of Medicine, University of Southern California, Los Angeles, CA, United States
Congenital heart defects occur in almost 80% of patients with CHARGE syndrome, a sporadically occurring disease causing craniofacial and other abnormalities due to mutations in the CHD7 gene. Animal models have been generated to mimic CHARGE syndrome; however, heart defects are not extensively described in zebrafish disease models of CHARGE using morpholino injections or genetic mutants. Here, we describe the co-occurrence of craniofacial abnormalities and heart defects in zebrafish chd7 mutants. These mutant phenotypes are enhanced in the maternal zygotic mutant background. In the chd7 mutant fish, we found shortened craniofacial cartilages and extra cartilage formation. Furthermore, the length of the ventral aorta is altered in chd7 mutants. Many CHARGE patients have aortic arch anomalies. It should be noted that the aberrant branching of the first branchial arch artery is observed for the first time in chd7 fish mutants. To understand the cellular mechanism of CHARGE syndrome, neural crest cells (NCCs), that contribute to craniofacial and cardiovascular tissues, are examined using sox10:Cre lineage tracing. In contrast to its function in cranial NCCs, we found that the cardiac NCC-derived mural cells along the ventral aorta and aortic arch arteries are not affected in chd7 mutant fish. The chd7 fish mutants we generated recapitulate some of the craniofacial and cardiovascular phenotypes found in CHARGE patients and can be used to further determine the roles of CHD7.
1 Introduction
CHARGE syndrome is an autosomal dominant genetic disease that is named after the abbreviation for six standard features of this disorder: coloboma, heart disease, choanal atresia, retarded growth, genital hypoplasia, and ear anomalies and/or deafness (Hsu et al., 2014). It is the leading syndrome associated with deafness–blindness in school-aged children in the United States (Flanagan et al., 2007). Babies born with CHARGE syndrome often have life-threatening defects and require intensive treatments including many surgeries. In addition to the six standard features, craniofacial defects including cleft palate/lip and asymmetric face palsy (Pagon et al., 1981) and cardiovascular malformations including outflow tract defects, atrioventricular septal defect, persistent ductus arteriosus (Corsten-Janssen et al., 2013), and aortic arch anomalies (Corsten-Janssen and Scambler, 2017) are frequently present in CHARGE patients. CHARGE syndrome is also extremely complex in terms of symptomatology, which can vary in severity from patient to patient, making it difficult to study.
Chromodomain helicase DNA-binding protein 7 (CHD7), a member of the SNF2-protein superfamily, is an ATP-dependent chromatin remodeler (Allen et al., 2007). CHD7 has been found mutated in two-thirds of patients with CHARGE syndrome. A study involving 299 patients with pathogenic CHD7 mutations shows that 74% of these patients have heart defects (Corsten-Janssen et al., 2013). Moreover, in addition to CHARGE syndrome patients, CHD7 mutations have also been identified in non-syndromic patients with congenital heart defects (Zaidi et al., 2013; Yan et al., 2020).
To study the mechanisms underlying CHARGE syndrome in vivo, multiple Chd7 N-ethyl-N-nitrosourea (ENU) mutants and Chd7 null mouse models have been generated. While no homozygous embryos were detected beyond E10.5 (Hurd et al., 2007; Layman et al., 2010; Yan et al., 2020), the heterozygous Chd7 mice recapitulate various symptoms of CHARGE syndrome, such as coloboma, inner ear abnormalities, cleft palate, choanal defects, cardiovascular defects, genital defects, and developmental growth delays (Bosman et al., 2005; Layman et al., 2009; Layman et al., 2010; Schulz et al., 2014b; Ogier et al., 2014; Gage et al., 2015). However, the postnatal lethality of Chd7 mutant mice makes it difficult to study the overall function of CHD7 and screen the potential therapeutic compounds in mice.
Zebrafish have become an emerging model to study development and model human diseases. In situ hybridization experiments have shown that the chd7 transcripts are broadly expressed in developing zebrafish, mainly in the central nervous system, gut, tail bud, and somite borders (Jacobs-McDaniels and Albertson, 2011). To study CHARGE syndrome, a few chd7 zebrafish mutants have been generated and reported. In contrast to CHARGE patients with only one mutant allele of CHD7, chd7 heterozygous mutant fish do not display obvious phenotypes, except for the anxiety-like and aggressive behavior (Liu and Liu, 2020). Nevertheless, chd7 homozygous fish showed mild CHARGE-like phenotypes such as smaller eyes, enlarged heart with edema, failure to inflate the swim bladder (Prykhozhij et al., 2017), small head, cranial cartilage malformations, and cranial nerve defects (Jamadagni et al., 2021). In addition, decreased size of the gastrointestinal tract (Cloney et al., 2018), scattered sox10-positive cranial NCCs (Liu et al., 2019), and impaired T-cell and thymus development (Liu et al., 2018) were also reported in chd7 homozygous fish mutants. However, no detailed craniofacial cartilage and cardiovascular phenotypes in chd7 fish mutants have been reported so far.
It should be noted that the tissues affected in CHARGE syndrome, such as missing ear lobes, deafness, broad forehead, reduced dysmorphic jaw, and hypopigmentation in some patients, are mainly derived from the neural crest cell (NCC) lineage. NCC is a group of transient cells in vertebrates, which migrates out from the neural tube and then gives rise to diverse cell lineages and contributes to different tissues, including the craniofacial skeleton, cartilage, ear, cells in the great vessels, and the septum of the heart in humans (Huang and Saint-Jeannet, 2004; Achilleos and Trainor, 2012). Thus, NCCs may be one major cell type that contributes to concomitant craniofacial and congenital cardiac defects, which are present in patients with CHARGE and other genetic syndromes and can also occur in non-syndromic patients with cleft and heart defects (Munabi et al., 2022). Consistent with this notion, in situ hybridization data showed that chd7 is expressed in rhombomeres, from which some NCCs are derived, at 24 days post fertilization (dpf) and in the developing pharynx at 48 to 96 hpf (Liu et al., 2018).
NCCs migrate to seven pharyngeal arches (PAs) in zebrafish. The first one contributes to the craniofacial cartilage including the Meckel’s cartilage and part of the palatoquadrate; the second PA forms the dorsal hyosymplectics, the ventral ceratohyals, and the interhyal cartilage; and the 3–7 PAs produce pairs of ventrolateral ceratobranchial cartilages. Specifically, the seventh PA contributes to the fifth ceratobranchial cartilage that forms several ossified pharyngeal teeth (Frisdal and Trainor, 2014; Mork and Crump, 2015). In mammals, PAs 3, 4, and 6 contribute to the formation of aortic arch arteries (AAs) and then remodel into the great vessels–the aortic arch, pulmonary arteries, and carotid arteries (Vincent and Buckingham, 2010). There are only five pairs of AAs in mammals since the sixth pair is never fully formed (Bamforth et al., 2013). However, in zebrafish, all six pairs of AAs that branch out from the ventral aorta (VA) are completely formed. The initial AA formation and patterning in zebrafish are very similar to those in mammals, but the later complex remodeling into the aorta and pulmonary artery does not happen; and AAs 3–6 carry blood to the gills instead (Anderson et al., 2008). Nonetheless, zebrafish is still an excellent model to study the initial formation of AAs and the great vessels (Nagelberg et al., 2015).
Here, we report the generation and characterization of new chd7 mutants and the novel concomitant craniofacial and cardiovascular phenotypes. These defects are further enhanced in maternal zygotic chd7 mutants. We discovered that chd7 mutants have altered chondrocyte stacking, shortened craniofacial cartilages, and extra craniofacial cartilage. Furthermore, the length of the VA is decreased and the branching and patterning of AA3 (the first brachial arch artery) are impaired in chd7 mutants. Interestingly, we found that NCC-derived mural cells (pericytes and smooth muscle cells) are not affected along the VA and AAs. Our chd7 mutant provides the first model to mimic the concomitant craniofacial and cardiovascular phenotypes found in CHARGE and non-syndromic patients and can be used to further determine the underlying mechanisms of CHD7.
2 Materials and methods
2.1 Zebrafish maintenance
All procedures described have been approved by the CHLA IACUC committee. To raise zebrafish, we keep the fish at 28.5°C with a 14-h light/10 h dark cycle. Brine shrimp and commercial powder food smaller than 450 µm have been used to feed the fish. In our system, we feed the larvae with powder food smaller than 50 μm at the beginning and switch to powder food smaller than 100 µm together with brine shrimp for juvenile fish. Fish housing and maintenance are under standard care with CHLA IACUC oversight. After crossing the fish, fertilized embryos were collected and changed into the E3 media (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4, and 10–5% methylene blue). An incubator at 28.5°C is used to raise the embryos, and plus and minus 5°C may be used to adjust the speed of the development.
Tricaine-s/syncaine (ms 222) fish anesthetic (Syndel) was used as anesthesia (0.168 mg/ml in fish water), and euthanasia (0.4 mg/ml in fish water for more than 15 min).
2.2 Fish strains
The AB line (ZIRC and UCLA) is used as the wild type (WT) control in this experiment. The following transgenic lines were used in this study: Tg(kdrl:mTurquoise) (Harrison et al., 2019), Tg(pdgfrb:GFP) (Ando et al., 2016), Tg(Mmu.Sox10-Mmu.Fos:Cre)zf384 (Kague et al., 2012), and Tg(-3.5ubb:loxP-STOP-loxP-mCherry)el818 (Fabian et al., 2020).
2.3 chd7 mutant generation using CRISPR/Cas9 technology
The targeting sequences of CRISPR Cas9 gRNAs are shown in Table 1. Before injection, the gRNA was thawed, the mix was prepared with 1/10 EnGen Spy Cas9 Nucleases (Cas9 protein, BioLabs), 1/10 10X Buffer 3.1(NEB), 200–300 ng/μL for single gRNA injection or 125 ng/μL each for multiple gRNA injection, and RNase free ddH2O. The mix was placed at 37°C for 10 min. The gRNA was coinjected together with Cas9 protein into single-cell stage zebrafish embryos and 2 nL of injection mixture per embryo.

TABLE 1. CRISPR gRNA targeting sequencing on chd7.
gRNAs were injected together with Cas9 protein individually, and four most efficient gRNAs were then chosen to carry out co-injection to gain the highest knockout efficiency. To check the efficiency of creating mutations, the genomic DNA of the injected fish was collected at 2 days post fertilization (dpf). T7E1 assay was used to distinguish and cut at the mismatched DNA heteroduplexes (Vouillot et al., 2015). The WT band cannot be cut by T7 endonuclease I (BioLabs), while the mutant mismatched bands can be cut.
Founder fish were raised, genotyping was performed, and mutants were sequenced to confirm the mutation (Genewiz from Azenta).
2.4 qRT-PCR
Quantitative real-time reverse transcriptional PCR was carried out using 24 hpf fish and four dpf fish. The total mRNA was extracted using TRIzol (Invitrogen). An RNeasy kit (Qiagen) was used to purify the total mRNA. cDNA was generated using the SuperScriptTM III Reverse Transcriptase kit (Invitrogen), and then real-time PCR (RT-PCR) was performed using LightCycler 480 SYBR Green I master mix (Roche). Two sets of PCR primer combinations (chd7-1 and chd7-2 listed in Supplementary Table S1) were used to check chd7 expression. Data were normalized to ef1α as the reference gene.
2.5 Confocal microscopy
Embryos from 18 hpf to 5 dpf were euthanized in 0.1% Tricaine S (Syndel) and then mounted in 1% agarose with tricaine in fish water. Confocal microscopes (Leica STELLARIS 5 inverted confocal microscope and Zeiss LSM 710 inverted confocal microscope) were used to image the embryos and larvae.
2.6 Alcian blue and alizarin red staining
The larvae were euthanized with tricaine and then fixed in 2% PFA at room temperature for 1 h. Then, the larvae were washed with 100 mM Tris (pH 7.5) and 10 mM MgCl2 for 10 min (min) and incubated overnight in Alcian stain (stock: 0.1 g Alcian blue 8GX, 2.6 ml H2O, bring the volume to 50 ml by using 95% EtOH; working solution: 10 ml Alcian blue stock, 32.6 ml 95% EtOH, 5 ml Tris-HCl, pH 7.5, 0.5 ml 1M MgCl2, 0.9 ml H2O) at room temperature. The fish were destained by 80% EtOH/100 mM Tris (pH 7.5)/10 mM MgCl2, 50% EtOH/100 mM Tris (pH 7.5)/10 mM MgCl2, and 25% EtOH/100 mM Tris (pH 7.5)/10 mM MgCl2 for 5 min each. The fish were moved into a well of 24-well plates and bleached by 3% H2O2/0.5% KOH under a light source until the eyes turn to light brown. The fish were washed twice with 1 ml 25% glycerol/0.1% KOH for 10 min each and nutated in alizarin stain (stock: 0.25 g alizarin red S to 50 ml H2O, working solution: 1 ml alizarin red stock, 12.5 ml glycerol, 5 ml Tris-HCl, pH 7.5, 31.5 ml H2O) at room temperature for 1 h. The fish were destained by washing with 1 ml 50% glycerol/0.1% KOH for 10 min twice. Then gradually move them into 100 % glycerol and image the fish in glycerol using a Zeiss Axioplan Upright Microscope in a bright field.
2.7 Quantification of chondrocyte stacking arrangement
To analyze the chondrocyte arrangement, the angle between the three neighboring chondrocytes was measured. Since the cells connecting to the basibranchials are round and not as organized as the chondrocytes in the other ceratobranchials, counting one-fourth to one-third of the chondrocytes on the basibranchial side was avoided. The angles were measured from the first more organized chondrocytes if possible. If organized chondrocytes still cannot be identified, one-third of the chondrocytes were skipped on the basibranchial side. The basibranchial side was treated as the base and the other end as the tip. 1. A line vertical to the cell membrane was drawn to connect the cell to the adjacent cell (choose the one near the base side if there is more than one cell). 2. Step 1 was repeated, and the angle between these two lines was measured. 3. Within the cells that are not measured, the one which is closest to the base was chosen and steps 1 and 2 were repeated until all the cells are measured or there are less than three cells in the tip.
2.8 Survival rate quantification
For chd7+/sr5 incrossed fish, there are three offspring genotypes: WT, chd7+/sr5, and chd7sr5. Due to the difficulty in performing genotyping in young larvae, we separate the embryos into three groups randomly after the gastrula stage. Genotyping was then performed at 7 dpf, 14 dpf, and 30 dpf for each group. The entire larvae were lysed to extract genomic DNA at 7 dpf and 14 dpf, and the fin was cut and used to extract the genomic DNA at 30 dpf.
We also incrossed chd7sr5 male and female fish to get the MZchd7sr5 offspring, and WT and chd7+/sr5 fish from separate crosses were used as non-sibling controls. The fish number was counted every 3 days to draw the survival rate from 0 dpf to 30 dpf.
2.9 Aortic arch patterning quantification
To determine the asymmetry of the AAs and the location of AA3 branching points, the length of the bifurcation point of AA3 from the intersection point of the opercular artery (ORA) to the VA along the VA was measured. The value of the length on the left side is plotted as X, and the one on the right side is plotted as Y. For some chd7+/sr5 and chd7sr5 fish with abnormal AA patterning, the AA3 branched out from the ORA instead of from the VA. Thus, the length was considered negative along the VA. Scatter plots were used to show the symmetry of AA patterning. The data point with negative X or Y values marks the fish with abnormal AA3 branching. All the data points outside of the gray (15%) error interval region are considered having asymmetric AAs.
2.10 Statistical analysis
Sample sizes, statistical tests, and p-values are stated in the figure legends. Data were measured using ImageJ (Schneider et al., 2012). One-way ANOVA followed by Dunnett’s multiple comparisons test and chi-squared test were performed using GraphPad Prism version 9.0.0 for Mac OS X, GraphPad Software, San Diego, California, United States , www.graphpad.comtest. Figures were drawn using Prism. Differences were considered statistically significant at p < 0.05. For the bar chart, the error bars are generated with the mean and standard error of the measurements. For the box and whisker plots, the whiskers are generated with the minimum to maximum value and all points were shown.
3 Results
3.1 chd7 mutant fish show CHARGE like phenotypes
The CRISPR/Cas9 system was used to generate chd7 mutants by co-injecting Cas9 protein together with gRNAs into one-cell stage embryos. We modified a previously published method (Wu et al., 2018) by co-injecting two or four gRNAs targeting the same exon (Table 1) of the chd7 gene to achieve high knockout efficiency. We identified two chd7 mutant alleles; the first chd7sr5 allele contains a 55-base pair deletion in exon 5 which encodes the first CHROMO domain of Chd7. This deletion is predicted to result in a frame-shift mutation that starts at the 803rd lysine and creates a stop codon after 45 amino acids (Figures 1A,B). Mutations in the ATPase domain might have a dominant negative effect by disrupting important protein–protein interactions (Bouazoune and Kingston, 2012). Thus, we generated the second chd7sr6 allele carrying an in-frame deletion that removes two highly conserved amino acids (the 1082nd tryptophan and 1083rd threonine) in the ATPase domain of Chd7 (Figures 1A,B).
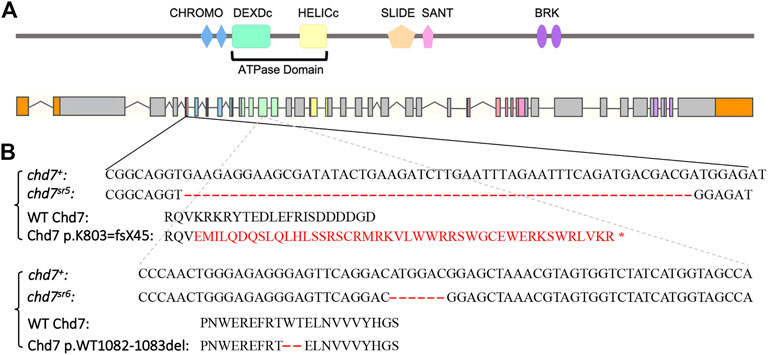
FIGURE 1. Generation of chd7 zebrafish mutants. (A) Schematics of Chd7 protein and chd7 gene structure. Chd7 protein contains double CHROMO domains, an ATPase domain including a DEXDc and HELICc domain, a SLIDE/SANT domain, and double BRK domains. (B) Sequence of the chd7sr5 and chd7sr6 mutants. Chd7sr5 contains a 55-bp deletion in the fifth exon of chd7, which generated a truncation after the first chromo domain. The deletion created a 44-amino acid frame-shift followed by a stop codon. Chd7sr6 has a mutation in the twelfth exon, which encodes the DEXDc domain, and causes a deletion of two amino acids, tryptophan and threonine.
Quantitative reverse transcription PCR (qRT-PCR) of chd7 in these two mutants at 4 dpf showed dramatically decreased chd7 mRNA levels in chd7sr5 homozygous mutants (chd7sr5) but not in chd7sr5 heterozygous (chd7+/sr5) or chd7sr6 homozygous (chd7sr6) larvae (Figure 2A; Supplementary Tables S1, S2). This result suggests that chd7 mRNA is degraded in the chd7sr5 mutant. Upon further analyses, we observed significantly impaired survival of chd7sr5. We uncovered that 6% fish are surviving chd7sr5 mutants compared to 25%, the predicted Mendelian ratio by 30 dpf (p-value <0.001) (Figure 2B). Despite the impaired survival, the surviving chdsr5 fish are able to reproduce normally and display very mild gross phenotypes (Figure 2C). Since chd7 transcripts can be detected in zygotes and 2-cell stage embryos (Rauch et al., 2003) and maternal chd7 mRNAs could play important roles in early development processes, we examined whether chd7sr5 might show maternal zygotic mutant phenotypes. We found that maternal zygotic chd7sr5 (MZchd7sr5) mutants show impaired survival similar to the zygotic chd7sr5 mutant larvae (Figure 2B). The number of MZchd7 sr5 larvae dramatically decreased after 10 dpf (∼90% died), while only ∼15% of chd7+/sr5 and WT siblings died by 30 dpf (Supplementary Figure S1A). The chd7 transcripts are also significantly decreased in MZchd7sr5 mutants (Supplementary Figure S1B).
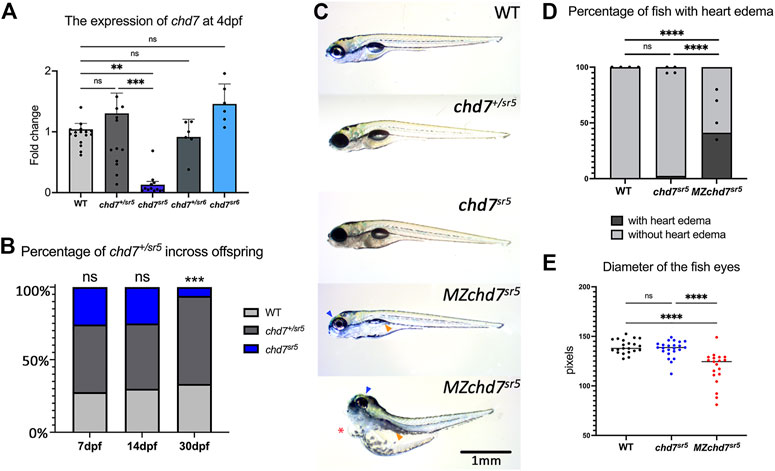
FIGURE 2. Overall phenotypes of chd7 mutants. (A) Expression of chd7 in chd7sr5 and chd7sr6 larvae at 4 dpf. The fold change of chd7 expression compared to WT was shown. (B) Percentage of the surviving offspring from chd7sr5incross (n > 40 for each time point). (C) Bright field images of chd7sr5 and MZchd7 sr5 mutants at 7 dpf. MZchd7 sr5 fish have a smaller swim bladder (yellow arrowhead), pericardial edema (red asterisk), and small eyes (blue arrowhead). (D) Quantification of heart edema. Nearly 40% of MZchd7 sr5 fish have heart edema; however, chd7 sr5 fish do not show heart edema phenotypes. WT: n = 879, chd7sr5: n = 747, MZchd7sr5: n = 751. (E) Quantification of the small eye phenotype. The longest diameter of the dissected eye was measured. The eye size of the MZchd7 sr5 mutants is significantly reduced (n = 22 for each group). Statistical analyses were performed using the ordinary one-way ANOVA test for (A,E) and chi-square test for (B,D). ns: p ≥ 0.05, *: p < 0.05, **: p < 0.01, ***: p < 0.001, and ****: p < 0.0001.
As we expected, the MZchd7sr5 mutants showed more severe CHARGE-like phenotypes such as small head, small eyes, and cardiac edema (Figure 2C). This is consistent with other chd7 fish mutants reported (Prykhozhij et al., 2017; Liu et al., 2018; Jamadagni et al., 2021). When comparing the pericardial edema in WT (n = 879), chd7 sr5 (n = 747), and MZchd7 sr5 (n = 751), the MZchd7 sr5 larvae have significantly more heart edema (∼40%) (Figure 2D). These data suggested that maternal chd7 mRNA might function during the early development in chd7sr5 larvae. This might also account for the mild phenotypes in previously published fish lines (Prykhozhij et al., 2017; Liu and Liu, 2020; Jamadagni et al., 2021). MZchd7 sr5 fish have smaller eyes compared to those of the WT (n = 22 for each group), but chd7 sr5 fish do not show this phenotype (n = 22) (Figure 2E). This small-eye phenotype was also observed in other chd7 mutants (Prykhozhij et al., 2017). In summary, chd7sr5 mutants showed mild gross CHARGE phenotypes, which can be enhanced in the MZchd7sr5 mutants.
3.2 The chd7sr5 mutants show CHARGE-like phenotypes in craniofacial structure
Many patients with the CHARGE syndrome have a typical CHARGE face, which is asymmetric with a broad prominent forehead, a prominent nasal bridge with square root, a prominent nasal columella, flat midface, small mouth, and occasional small chin. In zebrafish, the neurocranium and viscerocranium together form the craniofacial cartilage. In chd7sr5 and MZchd7sr5 fish, the structure of neurocranium cartilage does not have dramatic defects compared to that of the WT. However, they have mild defects in the viscerocranium cartilage structure, especially in the basihyal, the ceratobranchials, and the teeth (Figures 3A–C) (Mork and Crump, 2015).
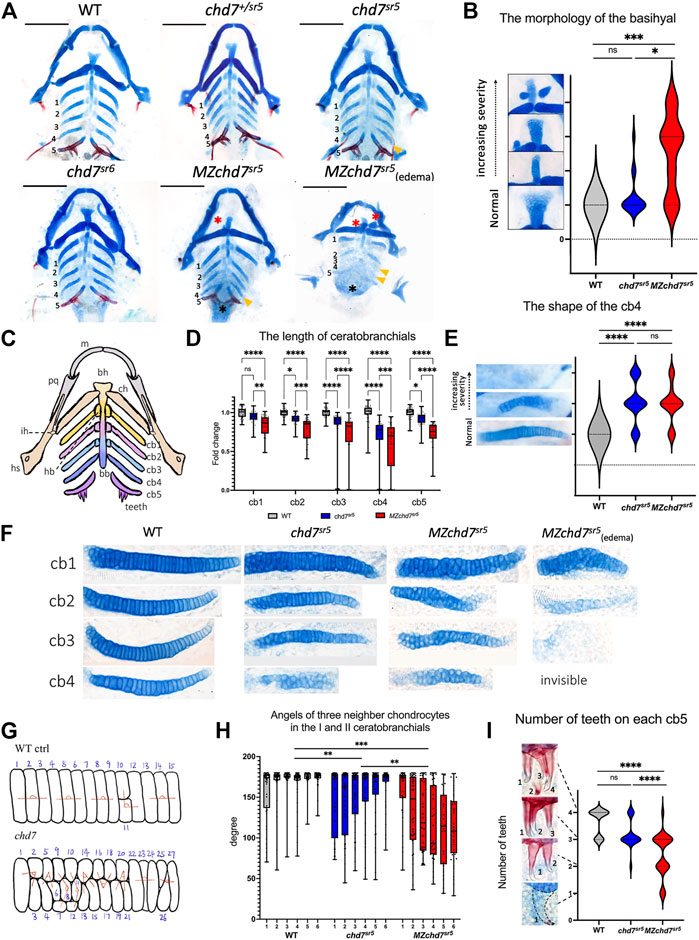
FIGURE 3. Craniofacial phenotype characterization in chd7sr5 mutants. (A) Viscerocranium morphology. The viscerocranium was dissected out from the Alcian blue and Alizarin red costained WT, chd7sr5, and MZchd7 sr5 mutants with or without heart edema at 7 dpf. Yellow arrowheads indicate the reduction in the IV ceratobranchial; black asterisks mark the defects in the teeth; red asterisks mark the abnormalities in the basihyal. (B) Quantification of the different types of the morphology of basihyal in WT, chd7sr5, and MZchd7 sr5. (WT: n = 16; chd7sr5: n = 16; MZchd7sr5: n = 15). The statistical analysis was performed using the chi-square test. (C) Schematic of the viscerocranium cartilage at 7 dpf. bb, basibranchial; bh, basihyal; cb, ceratobranchial; ch, ceratohyal; hb, hypobranchial; hs, hyosymplectic; ih, interhyal; m, Meckel’s; and pq, palatoquadrate. (D) Quantification of the length of I to V ceratobranchials in WT, chd7sr5, and MZchd7 sr5. (WT: n = 24; chd7sr5: n = 20; MZchd7sr5: n = 23). The statistical significance was performed using the ordinary one-way ANOVA test. (E) Quantification of different types of the morphology of the IV ceratobranchial. (n = 22 for each group). The statistical analysis was performed using the chi-square test. (F) High-magnification image shows that chondrocyte stacking is disrupted in chd7sr5 mutants, and severely disorganized stacking has been found in MZchd7sr5 mutants. (G) Schematic of chondrocytes’ stacking measurement. (H) Angles of three neighbor chondrocytes in I and II ceratobranchials. Each number indicates a different fish. (n = 6 for each group). The significance was calculated between different groups. chd7 sr5 mutants have mild disorganization in the chondrocyte arrangement, but MZchd7 sr5 mutants have more severe defects in the chondrocytes. The statistical analysis was performed using the ordinary one-way ANOVA test. (I) Alizarin red-stained teeth are reduced in chd7sr5 mutants at 7 dpf. Nearly 50% of MZchd7sr5 fish have defects in teeth number. (WT: n = 23; chd7sr5: n = 20; MZchd7sr5: n = 38). The statistical significance was performed using the chi-square test. ns: p ≥ 0.05, *: p < 0.05, **: p < 0.01, ***: p < 0.001, and ****: p < 0.0001.
Using Alcian blue staining to visualize the cartilages, we found that the structure of the basihyal cartilage is impaired in chd7sr5 and the defect is more severe in MZchd7sr5 mutants. Based on their various severities, we categorized the morphology of the basihyal into four types (Figure 3B). Normal basihyal is triangle shaped; while in the fish with mild defects, the basihyal cartilage is narrowed. For larvae with moderate defects, an extra cartilage could also be found near the narrowed basihyal cartilage. For the larvae with the most severe defects, two extra cartilages were found on both sides of the narrowed basihyal. A few chd7sr5 mutants have defects in the basihyal (3 in 16). However, this number is increased to around 2/3 in MZchd7sr5 mutants (11 in 15). Moreover, more fish have moderate and severe phenotypes in MZchd7sr5 (9 in 15) compared to chd7sr5 mutants (1 in 16) (Figure 3B).
In addition to the basihyal phenotype, the quantification of the length of ceratobranchials I to V showed that both chd7 sr5 and MZchd7sr5 mutants have shorter ceratobranchial cartilage (Figure 3D). Interestingly, the III and IV ceratobranchials are more severely decreased but not the V ceratobranchial, indicating that Chd7 may have more specific roles in the 3–6th PAs but not in the 7th PA. In addition to the shorter length, some of the IV ceratobranchials also have an abnormal shovel shape or are completely missing in both chd7sr5 and MZchd7sr5 mutants (Figure 3E). However, we cannot rule out the possibility that significantly shortened ceratobranchials in MZchd7sr5 mutants is secondary to the heart edema phenotypes (Figure 3F).
We further investigated the cartilage defects in chd7sr5 mutants by examining the chondrocyte arrangement. During maturation, the prechondrocytes go through a process called convergence-extension. Rather than via cell proliferation, an elongation of cartilages is driven from bulky precartilaginous condensations into stacks of disk-shaped chondrocytes (Mork and Crump, 2015). When checking the chondrocytes at higher magnification, we found that their stacking is disorganized in chd7sr5 and MZchd7sr5 mutants (Figure 3F). To quantify this disorganization, we measured the angles between three neighboring chondrocytes (Figure 3G) in ceratobranchials I and II using a method as described (Shull et al., 2022). The results indicated that chd7sr5 mutants have abnormal chondrocyte stacking (Figure 3H) compared to the WT. Moreover, this phenotype is significantly enhanced in MZchd7 sr5 mutants.
CHARGE patients can have one or more congenital dental abnormalities (Inchingolo et al., 2014). Therefore, we checked if chd7 sr5 mutants have craniofacial phenotype in teeth. Like other vertebrates, the teeth of zebrafish arise through epithelial–mesenchymal interactions (Verstraeten et al., 2010). We analyzed the tooth number of the fifth ceratobranchial. The chd7 sr5 mutants do not show a significantly reduced number of teeth. However, MZchd7 sr5 mutants have a large variation in the number of teeth (Figure 3I). It should be noted that many of the MZchd7sr5 fish with heart edema lost the alizarin red-stained bone tissue, and only the tip of the teeth are stained red (Figure 3I), and this is caused by developmental delay. Overall, chd7sr5 mutants have abnormalities in chondrocytes and osteocytes. Moreover, some of these defects are more severe in MZchd7 sr5 mutants. These results indicated that the early development process affected by the maternal chd7 mRNA may also contribute to the later craniofacial cartilage, eye, and tooth formation.
3.3 The chd7sr5 mutant fish have defects in ventral aorta and aortic arch artery
Some of the patients with CHD7 mutations have defects in the outflow tract (OFT) and aortic arch artery (AA) (Corsten-Janssen et al., 2013). For instance, the aberrant subclavian artery has been observed in around 8% of CHARGE patients with congenital heart defects (Corsten-Janssen et al., 2013). Similarly, Chd7 knockout mice have been shown to display abnormal AA development and AA4 is hypoplastic (Randall et al., 2009). To observe AA morphology, we utilized a kdrl:mTurquoise reporter line (Harrison et al., 2019) to label the endothelial cells of the vessel (Figure 4A). In zebrafish, the OFT is mainly composed of the bulbus arteriosus that connects the heart ventricle and the ventral aorta (VA) (Figure 4B). At 102 hpf, several AAs can be observed with the kdrl:mTurquoise reporter, which are the mandibular aortic arch (AA1), opercular artery (ORA), and branchial aortic arch arteries (AA3 and AA4) (Figures 4A,B).
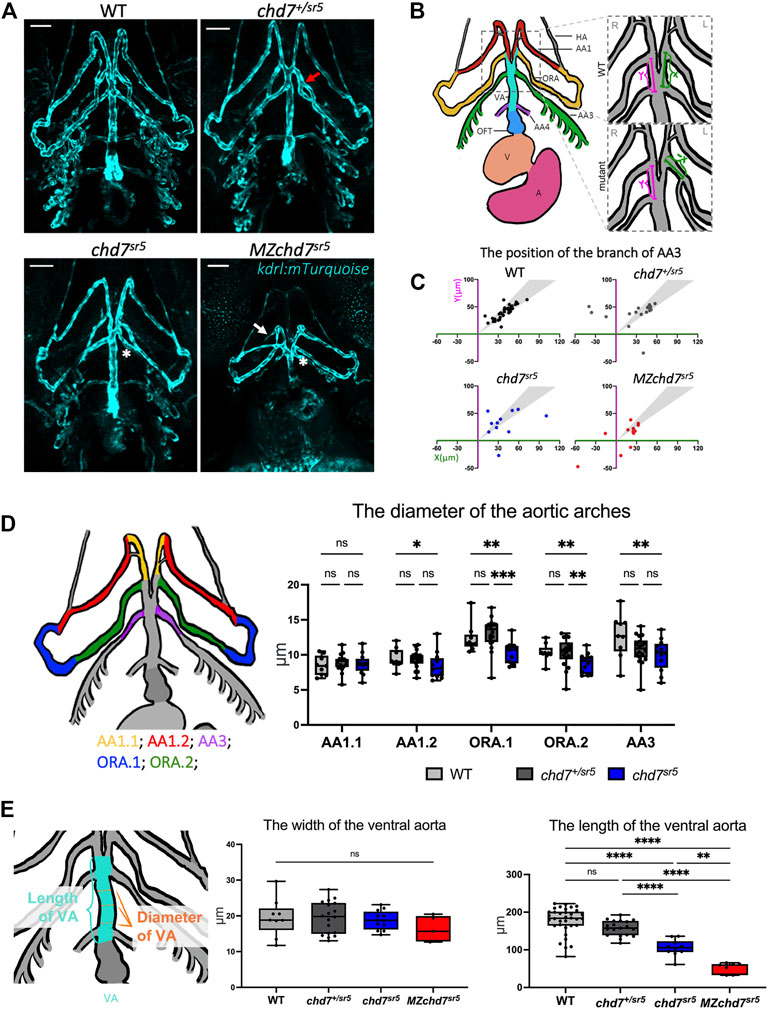
FIGURE 4. chd7sr5 mutants show defects in arch artery patterning and ventral aorta length. (A) Ventral view of the kdrl:mTurquois fish at 102 hpf. 20x stacked confocal image, number of stacks: WT n = 25 (interval = 5 µm), chd7sr5/+ n = 17 (interval = 7.5 µm), chd7sr5 n = 20 (interval = 7.5 µm), and MZchd7sr5 n = 21 (interval = 7.5 µm). Scale bar = 50 µm. (B) Schematic of ventral view of the zebrafish vasculature system at 102 hpf. The enlarged/zoomed in schematic illustrated the abnormal AA3 branching observed in some of the chd7 mutants. The distance between the branching points of the ORA and AA3 along the VA was measured. X = the length on the left side of the fish, Y = the length on the right side of the fish. If the AA3 branches off from the ORA instead of the VA, a negative value is used to quantify the length. AA, aortic arch; AA1, mandibular arch; ORA, opercular artery; AA3, first branchial arch; AA4, second branchial arch; HA, hypobranchial artery; VA, ventral aorta; OFT, outflow tract; V, ventricle; and A, atrium. (C) Quantification of the branching positions of the AA3 on the left and right side of WT, chd7sr5, and MZchd7 sr5 as X and Y coordinates. Each spot in the scatter diagram indicates one single fish. The gray area represents the range of symmetric distributions, in which the differences of the distance between X and Y is less than 15%. (WT: n = 34, chd7+/sr5: n = 19, chd7 sr5: n = 11, and MZchd7 sr5: n = 14). (D) Schematic and data quantification of the diameter of the AA1, ORA, and AA3. Due to the variation in the diameter at different segments of the vessels, AA1 and ORA were separated into AA1.1 and AA1.2 and ORA.1 and ORA.2, respectively. (WT: n = 9, chd7+/sr5: n = 18, and chd7 sr5: n = 12). The statistical analysis was performed using the two-way ANOVA test (mixed-effects analysis). (E) Schematic and data quantification of the width and length of the OFT. The mean diameter of the VA was used for quantification. The statistical analysis was performed using the ordinary one-way ANOVA test. (WT: n = 34, chd7+/sr5: n = 19, chd7 sr5: n = 11, and MZchd7 sr5: n = 14). ns: p ≥ 0.05, *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001.
In chd7sr5 fish, we found that the patterning of ORA and AA3 is abnormal. In WT (n = 34), all AA3 branched out from the VA; however, in some chd7+/sr5 larvae, AA3 branched out from the ORA instead, and this may affect the blood circulation (Figure 4A). Moreover, the AA3 branching patterns are asymmetric in chd7+/sr5 and chd7sr5 mutants (Figure 4A). To quantify the asymmetric patterning and branching defects, the length of the branching point of the AA3 from the intersection point of ORA and VA along the VA was measured for both left and right sides of the fish. Compared to the WT, chd7+/sr5, chd7sr5, and MZchd7sr5 fish have more asymmetric branching points, and some also have abnormal AA3 branching (Figure 4C). In chd7+/sr5, four out of 19 fish have abnormal AA3 branching. Furthermore, in MZchd7sr5, nearly one-third have AA3 bifurcated from the ORA (four out of 14) (Figure 4C). These data indicated that the AA patterning is affected even in the chd7+/sr5 fish and blood circulation to the craniofacial region and gill could be altered due to this abnormality.
To check whether other AAs are also affected in our chd7 mutants, the diameter of the AAs was measured along different segments of the vessels (Figure 4D). Except for the first segment of the AA1 (AA1.1), we found that all other segments of the AA1 (AA1.2), ORA, and AA3 are thinner in chd7sr5 fish but not in chd7+/sr5 fish. Since the VA connects the OFT and AAs, we also measured the width and length of the VA (Figure 4E). The width of the VA does not show a significant difference between the WT and chdsr5 mutants. In contrast, the length of the VA is significantly reduced in chd7 sr5 mutants and is even more decreased in MZchd7 sr5 fish (Figure 4E). In summary, AA patterning is impaired and VA length is decreased in chd7 sr5 mutants, and all these abnormalities together could influence blood circulation in chd7 sr5 mutants.
3.4 The cardiac neural crest cells and mural cells are not affected in chd7sr5 ventral aorta
CHD7 has been shown to have cell-autonomous functions in human embryonic stem cell-derived NCCs in vitro (Bajpai et al., 2010). However, its roles in cardiac NCCs are controversial. A previous report showed that two waves of cardiac NCC migrations are observed in zebrafish, and the second wave of cardiac NCCs contributes to the smooth muscle cells of the VA (Cavanaugh et al., 2015). To determine whether cardiac NCC-derived cells are affected in chd7sr5 mutants, we used Tg [(Mmu.Sox10-Mmu.Fos:Cre) zf384, -3.5ubb:loxP-STOP-loxP-mCherry) el818] (sox10:switch for short) to label and follow these cells. Furthermore, a pdgfrb:GFP reporter line was also used to label mural cells that include vascular smooth muscle cells and pericytes covering the endothelial cells and regulate vascular stability and homeostasis (Ando et al., 2016; Kapuria et al., 2022).
As expected, sox10:switch labeled the cells around the VA, partially overlapping with the pdgfrb:GFP positive mural cells but not with the kdrl:mTurquoise positive endothelial cells (Figure 5A). Interestingly, the distribution of these sox10:switch positive cells is neither reduced nor increased in chd7sr5 (Figure 5A) and MZchd7sr5 (Supplementary Figure S2) mutants. On the other hand, although the size of the OFT remains the same in both WT and chd7sr5 mutants (Figure 5B), the shape of the OFT changes significantly in chd7+/sr5 and chd7sr5 fish. The OFT is narrowed and elongated in the mutants (Figures 5C,D). Similar to the previous report (Cavanaugh et al., 2015), very few sox10:switch cells contribute to the smooth muscle cells in the OFT in zebrafish (Figure 5D). Therefore, the abnormally elongated shape of the OFT in chd7+/sr5 and chd7sr5 fish is not a result of changes in NCC-derived mural cells.
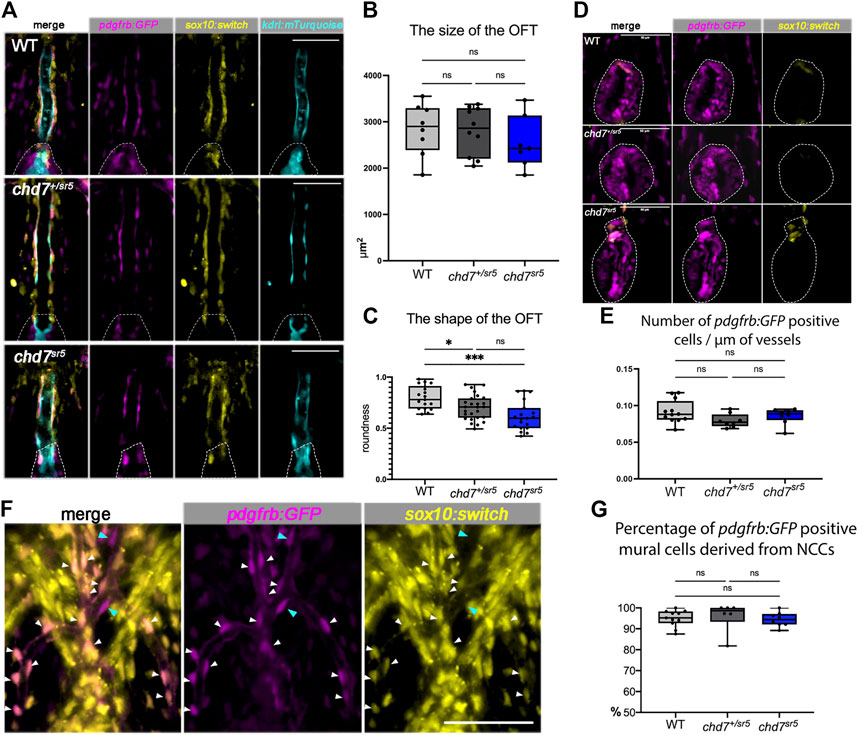
FIGURE 5. Cardiac NCCs and mural cells are not affected in the VA and OFT of chd7sr5 mutants at 7 dpf. (A) sox10:switch (pseudo-colored yellow) positive NCCs contribute to pdgfrb:GFP (pseudo-colored magenta) positive mural cells around the VA in WT, chd7+/sr5, and chd7sr5 fish. 40X single-slice confocal images at the thickest z position of the VA. Dotted lines circled the OFT. (B) Quantification of the size of the OFT in WT, chd7+/sr5, and chd7sr5 fish. (WT: n = 8, chd7+/sr5: n = 10, and chd7sr5: n = 7). (C) Roundness analysis of the shape of OFT in WT, chd7+/sr5, and chd7sr5 fish. (WT: n = 17, chd7+/sr5: n = 26, chd7sr5: n = 19). (D) Very few sox10:switch-labeled NCCs contribute to the OFT (circled by the dotted lines). 40X single-slide confocal images at the widest z position of the OFT. (E) Quantification of the distribution of the pdgfrb:GFP-positive cells along the vessel. The distribution is determined using the total length of the vessel divided by the total pdgfrb:GFP-positive cells. (WT: n = 12, chd7+/sr5: n = 8; chd7sr5: n = 8). (F) Representative images of the pdgfrb:GFP and sox10:switch-positive cells along the aortic arches. White arrowheads indicate NCC-derived (sox10:switch) pdgfrb:GFP-labeled mural cells; blue arrowheads indicate the pdgfrb:GFP single positive mural cells. 40X confocal stacked images, stack number n = 2, interval = 6 µm. (G) Quantification of the percentage of pdgfrb: GFP positive mural cells derived from NCCs (sox10: switch positive) along the AAs. Scale bar = 50 µm for all images. The statistical analysis was performed using the ordinary one-way ANOVA test for all data quantifications. (WT: n = 11, chd7+/sr5: n = 6; chd7sr5: n = 8). ns: p ≥ 0.05, *: p < 0.05, **: p < 0.01, ***: p < 0.001, and ****: p < 0.0001.
Although AA patterning is affected in chd7sr5 fish, the distribution of the pdgfrb: GFP-positive cells on AAs is not affected (Figure 5E). Consistent with previously published data (Ando et al., 2016), we also observed that the NCC lineage traced (sox10:switch positive) cells contributing to the pdgfrb: GFP-labeled mural cells on AAs. Around 95% of pdgfrb:GFP positive cells are NCC-derived (sox10:switch positive) (Figure 5F), and the percentage is not affected in chd7sr5 mutants (Figure 5G). These data together suggested that the cardiac NCCs and the NCC-derived mural cells are not affected in chd7sr5 mutants. Thus, the abnormal AA branching may not be caused by the defects in NCC-derived mural cells.
3.5 chd7sr5 mutants have extra craniofacial cartilage
Previously, pdgfrb has been shown to be expressed in cranial NCCs within the PAs in zebrafish and the perichondrial layer surrounding the hyoid bone and nasal cartilage in E13.5 and E14.5 mice (McCarthy et al., 2016). Using the pdgfrb:GFP reporter fish, we found that the pdgfrb expression level is higher in the posterior part of the ceratohyal cartilage and the peripheral basihyal, but not in the anterior part of the ceratohyal and the central part of the basihyal (Figure 6A).
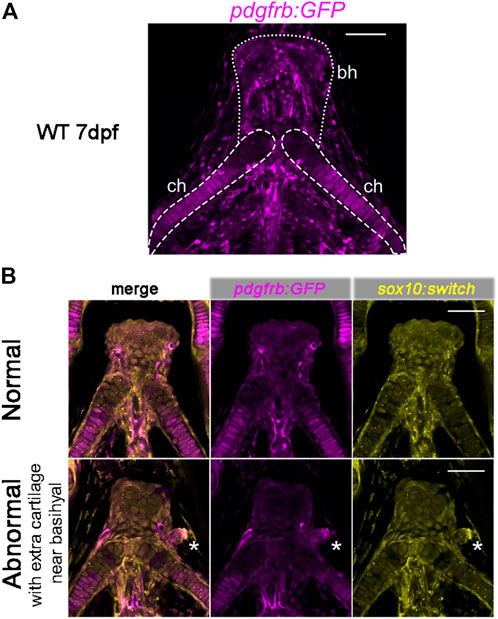
FIGURE 6. Expression of pdgfrb:GFP in craniofacial cartilages. (A) pdgfrb:GFP (pseudo-colored magenta) expression level is higher in the posterior part of the ceratohyal. 20X confocal stacked images, number of stacks n = 7, interval = 12 µm. (B) Representative images for the normal and abnormal basihyal in chd7sr5 mutant fish. The abnormal one has extra cartilage near the basihyal cartilage, which expresses more pdgfrb:GFP compared to the basihyal cartilage and the nearby ceratohyal. sox10:switch (pseudo-colored yellow) labels NCCs. The white asterisks mark the extra cartilage. 20X single-slice confocal image. ch: ceratohyal; bh: basihyal (the number of fish with extra cartilage: WT: 0 in 6, chd7+/sr5: 1 in 13, and chd7sr5: 4 in 14). Scale bar = 50 µm for all images.
Intriguingly, in some of the chd7sr5and MZchd7sr5mutant fish, one or two extra isolated cartilages have been found next to the basihyal cartilage. These extra cartilages express more pdgfrb:GFP than the basihyal and the nearby anterior ceratohyal (Figure 6B). This enhanced pdgfrb expression may reflect a homeotic transformation of these cartilages from the posterior side into a more anterior position.
4 Discussion
CHARGE syndrome is a complex genetic disease. It is not only the leading cause of deafness–blindness disease in school-age children in the US but is also a disorder of the craniofacial structure and congenital heart problem that could be life-threatening and needs treatments immediately after birth. CHD7 is the gene mutated in two-thirds of CHARGE patients. We have generated new chd7 mutants and characterized novel concomitant craniofacial and cardiovascular phenotypes. We found that these defects are further enhanced in maternal zygotic chd7 mutants. Although 75%–80% of the patients with CHARGE syndrome have congenital heart diseases, the heart defects are not extensively studied in previous zebrafish CHARGE models. We observed aberrant branching of the first branchial arch in both chd7 heterozygous and homozygous fish mutants for the first time. Moreover, our zebrafish CHARGE model can be used to investigate the pathogenesis and identify potential therapeutic targets in the future.
4.1 Genetic compensation may cause weaker CHARGE-like phenotype
Morpholino (MO) was previously used to block chd7 expression in zebrafish. chd7-MO injected at a higher dose (5–10 ng) induced lethality within 24 hpf, and embryos injected at a lower dose showed phenotypes such as eye abnormalities, otolith abnormalities, craniofacial defects (Patten et al., 2012; Asad et al., 2016), heart defects, and pectoral fin hypoplasia (Balasubramanian et al., 2014). None of these phenotypes can be rescued by coinjecting chd7 and p53 morpholinos (Patten et al., 2012; Balow et al., 2013), indicating that these phenotypes are not caused by random cell apoptosis. Due to concerns of off-target effects causing more severe phenotypes in the morphants, several chd7 mutants were generated by different groups. However, none of the mutants has shown similar severity of CHARGE-like symptoms comparable to that of the morphants.
Genetic compensation triggered by nonsense-mediated decay of mutant mRNA has been reported to occur in zebrafish CRISPR mutants and might result in no or weak phenotypes (El-Brolosy et al., 2019). The previously published three chd7 mutants and the chd7sr5 described here all cause frameshift mutations. RT-PCR data showed that the chd7 transcripts in the chd7sr5 allele are degraded. Thus, genetic compensation might be triggered in chd7sr5 mutants, and this might result in the weak CHARGE-like phenotypes we observed. The chd7 transcripts in the chd7sr6 allele are not degraded and thus should not trigger the genetic compensation. Therefore, chd7sr6 mutants could, in theory, display more severe phenotypes. Yet, the craniofacial cartilage phenotypes in chd7sr6 mutants are also mild. It is unknown whether the in-frame deletion in chd7sr6 mutants does not abolish the Chd7 function completely. Nonetheless, CHARGE patients display large variations in symptoms, which may be due to different CHD7 mutations. Analyzing patient-specific mutants mimicking patients’ alleles in the future will provide more insight into the pathogenesis.
4.2 Potential candidate genes that can compensate for loss of chd7
CHARGE syndrome has been shown to share some phenotypes overlapping with other congenital diseases such as Kabuki, DiGeorge, Alagille, Pallister–Hall, Feingold syndromes (Lettieri et al., 2021), and Mowat–Wilson syndromes. KMT2D, the gene mutated in Kabuki syndrome, has been suggested to function in the same chromatin modification machinery (Schulz et al., 2014a). Furthermore, it was reported that CHD7LOF and KMT2DLOF DNA methylation signatures share common CpG targets, specifically within HOXA5 and SLITRK5 (Butcher et al., 2017), providing more evidence that CHD7 and KMT2D could have overlapping functions and may compensate each other. ZEB2, the gene mutated in Mowat–Wilson syndromes (Birkhoff et al., 2021), may compensate for CHD7. The zeb2 is one of the downstream target genes of chd7 (He et al., 2016) in zebrafish. Moreover, ZEB2 regulates NCC formation and can bind to SMAD proteins (Birkhoff et al., 2021) as CHD7 (Lettieri et al., 2021). These data suggest that zeb2 could be another candidate gene to compensate for the loss of chd7. SOX2, which is mutated in Alagille, Pallister–Hall, and Feingold syndromes, regulates similar genes that are misexpressed in neural stem cells after shRNA-mediated knockdown with Chd7 (Lettieri et al., 2021). Although CHD7 displays broader genomic binding sites than SOX2, SOX2 could be a possible candidate gene to partially compensate for the defects caused by CHD7 depletion.
Other candidates are the other members in the CHD family. In Drosophila, kismet is the only gene related to the subgroup III CHD members, and its function is overtaken in mammals by CHD6–CHD9 (Batsukh et al., 2010). The previous publication showed that CHD7 interacts with CHD8 to build a core component of a complex of similar function to kismet (Batsukh et al., 2010). Similar combinations of two members from the same CHD subgroup to form a complex have also been observed between CHD3 and CHD4 (Hall and Georgel, 2007). Therefore, we could not rule out the possibility that other members from the subgroup III CHD family (CHD6, CHD8, and CHD9) could bind to each other and compensate for CHD7’s function.
4.3 Chd7 does not show cell-autonomous roles in cardiac NCC
Chd7 was previously shown as required in the pharyngeal ectoderm for normal AA development (Randall et al., 2009). Remarkably, the abnormal AA development in Chd7 knockout mice cannot be rescued by NCC-specific expression of Chd7 (Randall et al., 2009). Moreover, in a conditional Chd7 knockout mouse model using an improved Wnt1:Cre (Lewis et al., 2013), the conotruncal defects are found to be caused by a dramatically reduced NCC migration into the proximal OFT cushions at E11.5 but not by impaired migration of NCCs into AAs (Yan et al., 2020). These results raised the question about the cell-autonomous roles of Chd7 in cardiac NCCs in different structures.
In mammals, a subpopulation of NCC continues to migrate to the OFT of the heart (Vincent and Buckingham, 2010). In zebrafish, two waves of cardiac NCC migrations were observed, and the second wave of cardiac NCCs contribute to the VA (Cavanaugh et al., 2015). Unlike in mice and humans, we found that very few cardiac NCCs contribute to the OFT in zebrafish. Although the data using Chd7 conditional knockout mice suggest a cell-autonomous function of Chd7 in cardiac NCC (Yan et al., 2020), the proximal region of OFT, the conus arteriosus, is thought to be lost or form a segment distinct from the bulbus during the evolution in fish (Icardo, 2006), and the septation of the heart also does not occur (Grant et al., 2017). Interestingly, cardiac NCCs have been found to contribute to cardiomyocytes in the ventricle (Cavanaugh et al., 2015; Tang et al., 2019). This anatomical difference might explain the cardiac NCC contribution in fish and mammals. Furthermore, we found that the NCC-derived mural cells in both the VA and AAs are not affected in chd7sr5 zebrafish. On the other hand, craniofacial NCCs have been shown as defective in both chd7 morphant fish (Asad et al., 2016) and human cells in vitro (Bajpai et al., 2010). Thus, CHD7 could have different cell-autonomous or non-cell-autonomous roles in cranial and cardiac NCCs.
5 Conclusion and significance
In summary, we generated two chd7 mutant fish lines as the CHARGE syndrome disease model. Here, we describe the co-occurrence of craniofacial abnormalities and heart defects in new zebrafish chd7 mutants. We described the possible maternal zygotic function of chd7 for the first time. Furthermore, we investigated the cardiovascular phenotypes in detail and observed aberrant branching of the first branchial arch for the first time in chd7 fish mutants. Many CHARGE patients have aortic arch artery anomalies. Intriguingly, we found extra cartilage formation in chd7 mutants, which may be a homeotic transformation and worth further investigation for craniofacial phenotypes. Our zebrafish CHARGE model can be used to investigate the pathogenesis and identify potential therapeutic targets in the future.
Data availability statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee of Children’s Hospital, Los Angeles.
Author contributions
YS, SRK, RB, and C-LL conceived the project and designed experiments. RB and GC provided advice on experiment design. YS, CW, and ZT performed experiments. YS performed the measurements and data analysis. C-LL procured funding and supervised the study. YS and C-LL wrote and edited the manuscript. HB helped optimize the CRISPR procedures. GC shared reporter lines. YS, SRK, and C-LL revised the manuscript. All authors provided comments on the manuscript and gave final approval.
Funding
Team Science Award from Saban Research Institute C-LL (PI) and R01HL130172 (C-LL).
Acknowledgments
We thank Jau-nian Chen for the Tg(sox10:GAL4-UAS-Cre) fish line, Naoki Mochizuki and Koji Ando for the pdgfrb:GFP line, Stefan Schulte–Merker for the kdrl:mTurquoise line, and Subir Kapuria for technical support and suggestions. We also thank other laboratory members, Xidi Feng, Siqi Tao, Stanislao Travisano, Raquel Rodriguez, and Hui-Yu Chen, in the Lien laboratory for helpful discussions; Esteban Fernandez and the Cellular Imaging Core of Saban Research Institute of Children’s Hospital Los Angeles for technical support; and Peter Fabian, Pengfei Xu, D’Juan Farmer, and Olivia Hung-Jhen Chen in Crump laboratory for help and suggestions. We also thank the Animal Care Facility of Children’s Hospital, Los Angeles for fish maintenance and TSRI Team Science Research Award (C-LL) and R01HL130172 (C-LL.) for supporting this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2022.1030587/full#supplementary-material
References
Achilleos, A., and Trainor, P. A. (2012). Neural crest stem cells: discovery, properties and potential for therapy. Cell Res. 22 (2), 288–304. doi:10.1038/cr.2012.11
Allen, M. D., Religa, T. L., Freund, S. M., and Bycroft, M. (2007). Solution structure of the BRK domains from CHD7. J. Mol. Biol. 371 (5), 1135–1140. doi:10.1016/j.jmb.2007.06.007
Anderson, M. J., Pham, V. N., Vogel, A. M., Weinstein, B. M., and Roman, B. L. (2008). Loss of unc45a precipitates arteriovenous shunting in the aortic arches. Dev. Biol. 318 (2), 258–267. doi:10.1016/j.ydbio.2008.03.022
Ando, K., Fukuhara, S., Izumi, N., Nakajima, H., Fukui, H., Kelsh, R. N., et al. (2016). Clarification of mural cell coverage of vascular endothelial cells by live imaging of zebrafish. Development 143 (8), 1328–1339. doi:10.1242/dev.132654
Asad, Z., Pandey, A., Babu, A., Sun, Y., Shevade, K., Kapoor, S., et al. (2016). Rescue of neural crest-derived phenotypes in a zebrafish CHARGE model by Sox10 downregulation. Hum. Mol. Genet. 25 (16), 3539–3554. doi:10.1093/hmg/ddw198
Bajpai, R., Chen, D. A., Rada-Iglesias, A., Zhang, J., Xiong, Y., Helms, J., et al. (2010). CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 463 (7283), 958–962. doi:10.1038/nature08733
Balasubramanian, R., Choi, J. H., Francescatto, L., Willer, J., Horton, E. R., Asimacopoulos, E. P., et al. (2014). Functionally compromised CHD7 alleles in patients with isolated GnRH deficiency. Proc. Natl. Acad. Sci. U. S. A. 111 (50), 17953–17958. doi:10.1073/pnas.1417438111
Balow, S. A., Pierce, L. X., Zentner, G. E., Conrad, P. A., Davis, S., Sabaawy, H. E., et al. (2013). Knockdown of fbxl10/kdm2bb rescues chd7 morphant phenotype in a zebrafish model of CHARGE syndrome. Dev. Biol. 382 (1), 57–69. doi:10.1016/j.ydbio.2013.07.026
Bamforth, S. D., Chaudhry, B., Bennett, M., Wilson, R., Mohun, T. J., Van Mierop, L. H., et al. (2013). Clarification of the identity of the mammalian fifth pharyngeal arch artery. Clin. Anat. 26 (2), 173–182. doi:10.1002/ca.22101
Batsukh, T., Pieper, L., Koszucka, A. M., von Velsen, N., Hoyer-Fender, S., Elbracht, M., et al. (2010). CHD8 interacts with CHD7, a protein which is mutated in CHARGE syndrome. Hum. Mol. Genet. 19 (14), 2858–2866. doi:10.1093/hmg/ddq189
Birkhoff, J. C., Huylebroeck, D., and Conidi, A. (2021). ZEB2, the mowat-wilson syndrome transcription factor: Confirmations, novel functions, and continuing surprises. Genes (Basel) 12 (7), 1037. doi:10.3390/genes12071037
Bosman, E. A., Penn, A. C., Ambrose, J. C., Kettleborough, R., Stemple, D. L., and Steel, K. P. (2005). Multiple mutations in mouse Chd7 provide models for CHARGE syndrome. Hum. Mol. Genet. 14 (22), 3463–3476. doi:10.1093/hmg/ddi375
Bouazoune, K., and Kingston, R. E. (2012). Chromatin remodeling by the CHD7 protein is impaired by mutations that cause human developmental disorders. Proc. Natl. Acad. Sci. U. S. A. 109 (47), 19238–19243. doi:10.1073/pnas.1213825109
Butcher, D. T., Cytrynbaum, C., Turinsky, A. L., Siu, M. T., Inbar-Feigenberg, M., Mendoza-Londono, R., et al. (2017). CHARGE and Kabuki syndromes: Gene-specific DNA methylation signatures identify epigenetic mechanisms linking these clinically overlapping conditions. Am. J. Hum. Genet. 100 (5), 773–788. doi:10.1016/j.ajhg.2017.04.004
Cavanaugh, A. M., Huang, J., and Chen, J. N. (2015). Two developmentally distinct populations of neural crest cells contribute to the zebrafish heart. Dev. Biol. 404 (2), 103–112. doi:10.1016/j.ydbio.2015.06.002
Cloney, K., Steele, S. L., Stoyek, M. R., Croll, R. P., Smith, F. M., Prykhozhij, S. V., et al. (2018). Etiology and functional validation of gastrointestinal motility dysfunction in a zebrafish model of CHARGE syndrome. FEBS J. 285 (11), 2125–2140. doi:10.1111/febs.14473
Corsten-Janssen, N., Kerstjens-Frederikse, W. S., du Marchie Sarvaas, G. J., Baardman, M. E., Bakker, M. K., Bergman, J. E., et al. (2013). The cardiac phenotype in patients with a CHD7 mutation. Circ. Cardiovasc. Genet. 6 (3), 248–254. doi:10.1161/CIRCGENETICS.113.000054
Corsten-Janssen, N., and Scambler, P. J. (2017). Clinical and molecular effects of CHD7 in the heart. Am. J. Med. Genet. C Semin. Med. Genet. 175 (4), 487–495. doi:10.1002/ajmg.c.31590
El-Brolosy, M. A., Kontarakis, Z., Rossi, A., Kuenne, C., Günther, S., Fukuda, N., et al. (2019). Genetic compensation triggered by mutant mRNA degradation. Nature 568 (7751), 193–197. doi:10.1038/s41586-019-1064-z
Fabian, P., Tseng, K. C., Smeeton, J., Lancman, J. J., Dong, P. D. S., Cerny, R., et al. (2020). Lineage analysis reveals an endodermal contribution to the vertebrate pituitary. Science 370 (6515), 463–467. doi:10.1126/science.aba4767
Flanagan, J. F., Blus, B. J., Kim, D., Clines, K. L., Rastinejad, F., and Khorasanizadeh, S. (2007). Molecular implications of evolutionary differences in CHD double chromodomains. J. Mol. Biol. 369 (2), 334–342. doi:10.1016/j.jmb.2007.03.024
Frisdal, A., and Trainor, P. A. (2014). Development and evolution of the pharyngeal apparatus. Wiley Interdiscip. Rev. Dev. Biol. 3 (6), 403–418. doi:10.1002/wdev.147
Gage, P. J., Hurd, E. A., and Martin, D. M. (2015). Mouse models for the dissection of CHD7 functions in eye development and the molecular basis for ocular defects in CHARGE syndrome. Invest. Ophthalmol. Vis. Sci. 56 (13), 7923–7930. doi:10.1167/iovs.15-18069
Grant, M. G., Patterson, V. L., Grimes, D. T., and Burdine, R. D. (2017). Modeling syndromic congenital heart defects in zebrafish. Curr. Top. Dev. Biol. 124, 1–40. doi:10.1016/bs.ctdb.2016.11.010
Hall, J. A., and Georgel, P. T. (2007). CHD proteins: a diverse family with strong ties. Biochem. Cell Biol. 85 (4), 463–476. doi:10.1139/O07-063
Harrison, M. R., Feng, X., Mo, G., Aguayo, A., Villafuerte, J., Yoshida, T., et al. (2019). Late developing cardiac lymphatic vasculature supports adult zebrafish heart function and regeneration. Elife 8, e42762. doi:10.7554/eLife.42762
He, D., Marie, C., Zhao, C., Kim, B., Wang, J., Deng, Y., et al. (2016). Chd7 cooperates with Sox10 and regulates the onset of CNS myelination and remyelination. Nat. Neurosci. 19 (5), 678–689. doi:10.1038/nn.4258
Hsu, P., Ma, A., Wilson, M., Williams, G., Curotta, J., Munns, C. F., et al. (2014). CHARGE syndrome: a review. J. Paediatr. Child. Health 50 (7), 504–511. doi:10.1111/jpc.12497
Huang, X., and Saint-Jeannet, J. P. (2004). Induction of the neural crest and the opportunities of life on the edge. Dev. Biol. 275 (1), 1–11. doi:10.1016/j.ydbio.2004.07.033
Hurd, E. A., Capers, P. L., Blauwkamp, M. N., Adams, M. E., Raphael, Y., Poucher, H. K., et al. (2007). Loss of Chd7 function in gene-trapped reporter mice is embryonic lethal and associated with severe defects in multiple developing tissues. Mamm. Genome 18 (2), 94–104. doi:10.1007/s00335-006-0107-6
Icardo, J. M. (2006). Conus arteriosus of the teleost heart: dismissed, but not missed. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 288 (8), 900–908. doi:10.1002/ar.a.20361
Inchingolo, F., Pacifici, A., Gargari, M., Acitores Garcia, J. I., Amantea, M., Marrelli, M., et al. (2014). CHARGE syndrome: an overview on dental and maxillofacial features. Eur. Rev. Med. Pharmacol. Sci. 18 (15), 2089–2093.
Jacobs-McDaniels, N. L., and Albertson, R. C. (2011). Chd7 plays a critical role in controlling left-right symmetry during zebrafish somitogenesis. Dev. Dyn. 240 (10), 2272–2280. doi:10.1002/dvdy.22722
Jamadagni, P., Breuer, M., Schmeisser, K., Cardinal, T., Kassa, B., Parker, J. A., et al. (2021). Chromatin remodeller CHD7 is required for GABAergic neuron development by promoting PAQR3 expression. EMBO Rep. 22, e50958. doi:10.15252/embr.202050958
Kague, E., Gallagher, M., Burke, S., Parsons, M., Franz-Odendaal, T., and Fisher, S. (2012). Skeletogenic fate of zebrafish cranial and trunk neural crest. PLoS One 7 (11), e47394. doi:10.1371/journal.pone.0047394
Kapuria, S., Bai, H., Fierros, J., Huang, Y., Ma, F., Yoshida, T., et al. (2022). Heterogeneous pdgfrb+ cells regulate coronary vessel development and revascularization during heart regeneration. Development 149 (4), dev199752. doi:10.1242/dev.199752
Layman, W. S., Hurd, E. A., and Martin, D. M. (2010). Chromodomain proteins in development: lessons from CHARGE syndrome. Clin. Genet. 78 (1), 11–20. doi:10.1111/j.1399-0004.2010.01446.x
Layman, W. S., McEwen, D. P., Beyer, L. A., Lalani, S. R., Fernbach, S. D., Oh, E., et al. (2009). Defects in neural stem cell proliferation and olfaction in Chd7 deficient mice indicate a mechanism for hyposmia in human CHARGE syndrome. Hum. Mol. Genet. 18 (11), 1909–1923. doi:10.1093/hmg/ddp112
Lettieri, A., Oleari, R., Paganoni, A. J. J., Gervasini, C., Massa, V., Fantin, A., et al. (2021). Semaphorin regulation by the chromatin remodeler CHD7: An emerging genetic interaction shaping neural cells and neural crest in development and cancer. Front. Cell Dev. Biol. 9, 638674. doi:10.3389/fcell.2021.638674
Lewis, A. E., Vasudevan, H. N., O'Neill, A. K., Soriano, P., and Bush, J. O. (2013). The widely used Wnt1-Cre transgene causes developmental phenotypes by ectopic activation of Wnt signaling. Dev. Biol. 379 (2), 229–234. doi:10.1016/j.ydbio.2013.04.026
Liu, H., and Liu, Z. Z. (2020). Aggressive-like behavior and increased glycine transporters in a zebrafish model of CHARGE syndrome. Behav. Brain Res. 378, 112293. doi:10.1016/j.bbr.2019.112293
Liu, Z. Z., Guo, J., Lu, Y., Liu, W., Fu, X., Yao, T., et al. (2019). Sema3E is required for migration of cranial neural crest cells in zebrafish: Implications for the pathogenesis of CHARGE syndrome. Int. J. Exp. Pathol. 100 (4), 234–243. doi:10.1111/iep.12331
Liu, Z. Z., Wang, Z. L., Choi, T. I., Huang, W. T., Wang, H. T., Han, Y. Y., et al. (2018). Chd7 is critical for early T-cell development and thymus organogenesis in zebrafish. Am. J. Pathol. 188 (4), 1043–1058. doi:10.1016/j.ajpath.2017.12.005
McCarthy, N., Liu, J. S., Richarte, A. M., Eskiocak, B., Lovely, C. B., Tallquist, M. D., et al. (2016). Pdgfra and Pdgfrb genetically interact during craniofacial development. Dev. Dyn. 245 (6), 641–652. doi:10.1002/dvdy.24403
Mork, L., and Crump, G. (2015). Zebrafish craniofacial development: A window into early patterning. Curr. Top. Dev. Biol. 115, 235–269. doi:10.1016/bs.ctdb.2015.07.001
Munabi, N. C. O., Mikhail, S., Toubat, O., Webb, M., Auslander, A., Sanchez-Lara, P. A., et al. (2022). High prevalence of deleterious mutations in concomitant nonsyndromic cleft and outflow tract heart defects. Am. J. Med. Genet. A 188 (7), 2082–2095. doi:10.1002/ajmg.a.62748
Nagelberg, D., Wang, J., Su, R., Torres-Vázquez, J., Targoff, K. L., Poss, K. D., et al. (2015). Origin, specification, and plasticity of the great vessels of the heart. Curr. Biol. 25 (16), 2099–2110. doi:10.1016/j.cub.2015.06.076
Ogier, J. M., Carpinelli, M. R., Arhatari, B. D., Symons, R. C., Kile, B. T., and Burt, R. A. (2014). CHD7 deficiency in "Looper", a new mouse model of CHARGE syndrome, results in ossicle malformation, otosclerosis and hearing impairment. PLoS One 9 (5), e97559. doi:10.1371/journal.pone.0097559
Pagon, R. A., Graham, J. M., Zonana, J., and Yong, S. L. (1981). Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J. Pediatr. 99 (2), 223–227. doi:10.1016/s0022-3476(81)80454-4
Patten, S. A., Jacobs-McDaniels, N. L., Zaouter, C., Drapeau, P., Albertson, R. C., and Moldovan, F. (2012). Role of Chd7 in zebrafish: a model for CHARGE syndrome. PLoS One 7 (2), e31650. doi:10.1371/journal.pone.0031650
Prykhozhij, S. V., Steele, S. L., Razaghi, B., and Berman, J. N. (2017). A rapid and effective method for screening, sequencing and reporter verification of engineered frameshift mutations in zebrafish. Dis. Model. Mech. 10 (6), 811–822. doi:10.1242/dmm.026765
Randall, V., McCue, K., Roberts, C., Kyriakopoulou, V., Beddow, S., Barrett, A. N., et al. (2009). Great vessel development requires biallelic expression of Chd7 and Tbx1 in pharyngeal ectoderm in mice. J. Clin. Invest. 119 (11), 3301–3310. doi:10.1172/JCI37561
Rauch, G. J., Lyons, D. A., Middendorf, I., Friedlander, B., Arana, N., Reyes, T., et al. (2003) Submission and curation of gene expression data, [Lecture].unpublished.
Schneider, C. A., Rasband, W. S., and Eliceiri, K. W. (2012). NIH image to ImageJ: 25 years of image analysis. Nat. Methods 9 (7), 671–675. doi:10.1038/nmeth.2089
Schulz, Y., Freese, L., Mänz, J., Zoll, B., Völter, C., Brockmann, K., et al. (2014a). CHARGE and Kabuki syndromes: a phenotypic and molecular link. Hum. Mol. Genet. 23 (16), 4396–4405. doi:10.1093/hmg/ddu156
Schulz, Y., Wehner, P., Opitz, L., Salinas-Riester, G., Bongers, E. M., van Ravenswaaij-Arts, C. M., et al. (2014b). CHD7, the gene mutated in CHARGE syndrome, regulates genes involved in neural crest cell guidance. Hum. Genet. 133 (8), 997–1009. doi:10.1007/s00439-014-1444-2
Shull, L. C., Lencer, E. S., Kim, H. M., Goyama, S., Kurokawa, M., Costello, J. C., et al. (2022). PRDM paralogs antagonistically balance Wnt/β-catenin activity during craniofacial chondrocyte differentiation. Development 149 (4), dev200082. doi:10.1242/dev.200082
Tang, W., Martik, M. L., Li, Y., and Bronner, M. E. (2019). Cardiac neural crest contributes to cardiomyocytes in amniotes and heart regeneration in zebrafish. Elife 8, e47929. doi:10.7554/eLife.47929
Verstraeten, B., Sanders, E., van Hengel, J., and Huysseune, A. (2010). Zebrafish teeth as a model for repetitive epithelial morphogenesis: dynamics of E-cadherin expression. BMC Dev. Biol. 10, 58. doi:10.1186/1471-213X-10-58
Vincent, S. D., and Buckingham, M. E. (2010). How to make a heart: the origin and regulation of cardiac progenitor cells. Curr. Top. Dev. Biol. 90, 1–41. doi:10.1016/S0070-2153(10)90001-X
Vouillot, L., Thélie, A., and Pollet, N. (2015). Comparison of T7E1 and surveyor mismatch cleavage assays to detect mutations triggered by engineered nucleases. G3 (Bethesda) 5 (3), 407–415. doi:10.1534/g3.114.015834
Wu, R. S., Lam, I. I., Clay, H., Duong, D. N., Deo, R. C., and Coughlin, S. R. (2018). A rapid method for directed gene knockout for screening in G0 zebrafish. Dev. Cell 46 (1), 112–125. doi:10.1016/j.devcel.2018.06.003
Yan, S., Thienthanasit, R., Chen, D., Engelen, E., Brühl, J., Crossman, D. K., et al. (2020). CHD7 regulates cardiovascular development through ATP-dependent and -independent activities. Proc. Natl. Acad. Sci. U. S. A. 117 (46), 28847–28858. doi:10.1073/pnas.2005222117
Keywords: CHD7, CHARGE syndrome, craniofacial development, cardiovascular, aortic arch arteries, zebrafish
Citation: Sun Y, Kumar SR, Wong CED, Tian Z, Bai H, Crump JG, Bajpai R and Lien CL (2022) Craniofacial and cardiac defects in chd7 zebrafish mutants mimic CHARGE syndrome. Front. Cell Dev. Biol. 10:1030587. doi: 10.3389/fcell.2022.1030587
Received: 29 August 2022; Accepted: 03 November 2022;
Published: 07 December 2022.
Edited by:
Cheol-Hee Kim, Chungnam National University, South KoreaReviewed by:
Hyun-Taek Kim, Soonchunhyang University, South KoreaBen Lovely, University of Louisville, United States
Copyright © 2022 Sun, Kumar, Wong, Tian, Bai, Crump, Bajpai and Lien. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ching Ling Lien, Y2xpZW5AY2hsYS51c2MuZWR1