
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 16 September 2022
Sec. Developmental Epigenetics
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.1021820
This article is part of the Research Topic Genetics and Epigenetics in Cellular Identity and Cell State Transitions View all 8 articles
 Xiyao Shi1,2Ying Wang3Longhui Zhang1,2Wenjie Zhao1,2
Xiyao Shi1,2Ying Wang3Longhui Zhang1,2Wenjie Zhao1,2 Xiangpeng Dai1,2*
Xiangpeng Dai1,2* Yong-Guang Yang1,2,4*
Yong-Guang Yang1,2,4* Xiaoling Zhang1,2*
Xiaoling Zhang1,2*Bromodomain and extra-terminal domain (BET) family proteins play important roles in regulating the expression of multiple proto-oncogenes by recognizing acetylation of histones and non-histone proteins including transcription factors, which subsequently promote tumor cell proliferation, survival, metastasis and immune escape. Therefore, BET family proteins are considered attractive therapeutic targets in various cancers. Currently, blocking of the BET proteins is a widely used therapeutic strategy for MYCN amplified high-risk neuroblastoma. Here, we summarized and reviewed the recent research progresses for the critical function of BET proteins, as an epigenetic reader, on tumorigenesis and the therapeutic potential of the BET/BRD4 inhibitors on MYCN amplified neuroblastoma. We also discussed the combined therapeutic strategies for BET inhibitor-resistant neuroblastoma.
Epigenetics are the hereditary changes in gene function without alterations in the DNA sequence, which eventually leads to phenotypic changes (Jeong et al., 2014; Mondal et al., 2022). Studies have shown that epigenetic regulation of oncogenes plays an important role in the expression of these genes and subsequent occurrence and development of human cancer (Hillyar et al., 2020). Furthermore, epigenetic modifications also regulate the tumor immune monitoring such as activation of T cells, generation and recognition of tumor antigens (Sun et al., 2022). Histone acetylation is one of the major epigenetic modifications. Histone acetyltransferases (HATs) is the main “writer” of histone acetylation, and histone deacetylases (HDAC) is the main “erasers” to remove acetyl groups from histone and non-histone proteins. The bromodomain and extra-terminal domain (BET) family proteins act as the “readers” of histone acetylation. Dysregulation of BET family proteins which caused a high acetylation levels promotes transcription of multiple oncogenes and participates in the occurrence and development of inflammation and cancers (Anand et al., 2013; Huang et al., 2017; Otto et al., 2019). Given the critical function of BET proteins in regulating oncogenes expression and cell proliferation, they are now considered attractive therapeutic targets in many cancer types (Chung et al., 2011). Currently, studies have shown that BET protein inhibitors exhibit significant anti-tumor effects in vitro and in vivo. A plenty of BET family protein inhibitors, such as RVX-208, I-BET 762, OTX 015, CPI0610, and TEN-010, are developed and now in the clinical trials (Herait et al., 2014; Fu et al., 2015). However, the emerged drug resistance limited the wide application of BET inhibitors and it was reported that the BET proteins abundance confer resistance to the BET inhibitor in multiple cancer types (Dai et al., 2017; Zhang et al., 2017).
Neuroblastoma (NB) is the most common extracranial solid tumor in children. Amplification of MYCN is the important driver of high-risk neuroblastoma. Therefore, MYCN is considered a remarkable target for drug development in the treatment of MYCN amplified neuroblastoma (Huang and Weiss, 2013). BRD4, the well-studied member of the BET family proteins, plays an important role in MYCN expression via binding to acetylated histones at the super enhancer sites to regulate the transcription of MYCN and c-Myc (Henssen et al., 2016; Taniguchi, 2016). JQ1, the first developed BET inhibitor, could efficiently suppress the expression of MYCN in neuroblastoma by inhibiting the binding of BRD4 with acetyl lysine of histone (Delmore et al., 2011). Importantly, the BET inhibitors, such as JQ1, I-BET726 and OTX015 exhibited efficient anti-tumor effect in NB by markedly inhibiting the growth of neuroblastoma cells and prolonging the survival time of tumor-bearing mice. Therefore, BET proteins are the potential therapeutic targets to combat MYCN amplified neuroblastoma by inhibiting the expression of MYCN and other related oncogenes.
Here, the critical function of BET proteins as epigenetic reader in cancer development and the research progress to target BET protein in neuroblastoma was complicated summarized and discussed.
Histone acetylation is a reversible biological process controlled by the acetylation “writer” histone acetyltransferases (HATs) that transfer the acetyl groups on histone tails and the “eraser” histone deacetylases (HDAC) that remove the acetyl groups from histone (Ito et al., 2011; Noguchi-Yachide, 2016). The BET family proteins are subfamily of bromodomain protein superfamily. The N-terminal of human BET family proteins contains two conserved bromodomains (BD1 and BD2) which can recognize acetylated histones and regulate gene transcription and the C-terminal contains an extra-terminal (ET) domain. Bromodomain includes four α spiral slices (αZ, αA, αB, and αC) and two rings (ZA and BC) (Vollmuth and Geyer, 2010; Tang et al., 2021). Together with α-helixes, the ring region form a hydrophobic cavity core to recognize acetyl-lysine (Shi and Vakoc, 2014; French, 2016). The BET family proteins consist of BRD2, BRD3, BRD4, and BRDT (Law et al., 2018; Carlson et al., 2019). BRD4 is the well-studied member of the BET family due to its overexpression or fusion with other genes in caner development and drug resistance. BRD4 can bind to acetyl lysine on super enhancers and promoter of histones and transcription factors, leading to BRD4 localization in chromosomes where they recruit other regulatory complexes to affect gene expression (Kulikowski et al., 2021). The regulatory complexes include the core positive transcription elongation factor b (P-TEFb) and Mediator (Shi and Vakoc, 2014; Mochizuki et al., 2021). P-TEFb, which is constituted CDK9 and cyclinT1, is an important transcription factor in the process of gene expression. The interaction with BRD4 dissociates P-TEFb from the 7SK/HEXIM complex and activates P-TEFb kinases (Jang et al., 2005; Yang et al., 2005). Subsequently, CDK9 phosphorylates C-terminal Ser2 of Pol II and forms a stable transcription elongation complex, which ultimately promotes the expression of many target oncogenes of BET family, such as MYC and Bcl2 (Zhu et al., 2016; Hogg et al., 2017; Yin et al., 2020). Moreover, enhancer RNAs (eRNA) such as PSA eRNA could affect the P-TEFb activation in different cancer types through CYCLIN T1/CDK9 complex (Schaukowitch et al., 2014; Zhao et al., 2016). BRD4 bromine domain not only binds acetylated histones, but also interacts with acetylated transcription factors (TFs) (Cheung et al., 2017). Roe et al. demonstrated that after TF was acetylated by p300, BRD4 could be recruited and located at a specific location to promote the transcription of TF (Roe et al., 2015). In addition, the ET domain of BRD4 is responsible for additional protein-protein interactions, making BETs become the function core to promote the formation of polymerized structure composed of recruited TFs and coactivators (Wang et al., 2021).
The therapeutic agents targeting chromatin “writers” and “erasers” have been successfully developed. For example, HDAC inhibitors showed positive effect on tumor cell inhibition, but their clinical application is limited due to side toxic effect. Given that many downstream targets of BET proteins are pro-oncogenes, therefore, dysregulation of BET is closely correlated with cancer development and BET proteins have become novel drug targets. In 2010, the small molecule BET inhibitor JQ1 (Filippakopoulos et al., 2010) and I-BET (GSK525762A) was developed (Nicodeme et al., 2010). They have high affinity to the bromodomain of BET family members and could competitively bind to acetylated peptides, thus block the interaction of BET protein with chromatin. The BET inhibitor I-BET726 developed by GSK has high affinity and selectivity to BRD2, BRD3 and BRD4 (Gosmini et al., 2014). In 2012, the I-BET762 was approved for phase I clinical trials. In 2013, Picaud et al. found that PFI-1 inhibitors were acetyl lysine mimics and could replace acetyl lysine to bind to BRD2 and BRD4, thereby inhibiting the recruitment of BET protein to chromatin, down-regulating the expression of MYC and Aurora B kinases (Alqahtani et al., 2019). It was reported that RVX-208 can bind to BET proteins, especially BRD4, and regulate ApoA-I expression which playing a therapeutic role in atherosclerosis (Vertessy et al., 2013). The BET inhibitor OTX015 is a selective inhibitor for BRD2, BRD3, and BRD4 by inhibiting their binding to AcH4. In mature B-cell lymphoid tumors, OTX015 showed positive inhibitory effect by regulating MYC and E2F1 genes expression and NF-κB/JAK signaling pathways (Boi et al., 2015). ABBV-075 is a novel BET inhibitor exhibited significant inhibitory effect on tumor growth in Kasumi-1 cells xenograft mice model (McDaniel et al., 2017) and prostate cancer which provides a new option for therapeutic treatment of CRPC patients (Faivre et al., 2017).
Interestingly, it has been reported that BD1 and BD2 domains can also regulate different gene sets by specifically recognizing acetylated lysine residues (Westermann et al., 2008; Gilan et al., 2020; Fu et al., 2021). For example, BRD4-BD2 could recruit transcription factor TWIST and BRD4-BD1 is responsible for the binding to chromatin (Shi et al., 2014). BRDT-BD1, but not BRDT-BD2, is necessary for spermatogenesis (Shang et al., 2007). BRD4-BD1 covalent inhibitors, such as Olinone, ZL0580, MS436, inhibit the transcription of BET target genes to retard the proliferation of tumor cells (Tang et al., 2021). However, the ABBV-744, an efficient BRD4-BD2 domain inhibitor, inhibits AR-dependent transcription of genes in prostate cancer xenograft model (Faivre et al., 2020).
Aberrant c-Myc expression is frequently found in inflammation and cancer (Leal et al., 2017; White et al., 2019) and MYC is thus considered a promising therapeutic target but it is also an “undruggable” target. Mechanismly, BET inhibitors exert their anti-tumor effect mainly through the inhibition of MYC, a downstream gene of BRD4, in many cancer types. In myeloma cell line MM1.S, JQ1 treatment results in downregulation of MYC expression and inhibition of cell proliferation (French, 2016). JQ1 and OTX015 treatment reduced c-MYC expression and led to cell growth inhibition, cell cycle arrest and apoptosis in acute leukemia cells and patient-derived leukemic cells (Coude et al., 2015). In addition, BET inhibitors are also effective in inhibiting medulloblastoma (Henssen et al., 2013) and hepatocellular carcinoma (Li et al., 2016) by downregulating MYC expression.
However, BET inhibitor could inhibit tumorigenesis in an MYC independent manner. NUT midline carcinoma (NMC) is caused by translocation-derived fusion proteins BRD4-NUT or BRD3-NUT. JQ1 treatment can detach BRD4 and BRD4-NUT from chromatin (Filippakopoulos et al., 2010; French, 2016). In addition, I-BET151, OTX015 and JQ1 inhibit the interaction between BRD4 and the acetylated NF-κB subunit RelA at lysine 310 site, which subsequently reduced the transcriptional activation of NF-κB (Wu et al., 2013; Alqahtani et al., 2019). After androgen ablation, androgen receptor (AR) signal is the main driver for the development of castration-resistant prostate cancer (CRPC), JQ1 treatment inhibited the interaction between BRD4 and AR and subsequently hindered AR-mediated gene transcription (Asangani et al., 2014; Sahai et al., 2016). Furthermore, JQ1 showed markedly inhibitory effect on lung adenocarcinoma through downregulating FOSL1 and its targets (Lockwood et al., 2012). Moreover, JQ1 also demonstrated anti-tumor effects via inhibiting the expression of Forkhead box protein M1 (FoxM1) in ovarian cancer (Zhang et al., 2016) and aurora A kinase in triple-negative breast cancer (Sahni et al., 2016), respectively. Importantly, Donati et al. complicated summarized and discussed the key BRD4 target genes in normal and tumor cells such as embryonic cells, somatic cells, neuron cells, cardiac cells and cancer cells. The genes are related to osteogenesis, abiogenesis, myogenesis, NF-kB signaling pathway, estrogen and androgen receptor target genes including TNFα, IL8, GREB1, TFF1, PSA, HOXB13, CAMKK2, CCND1, MAPK8/10, and FOSL1 (Donati et al., 2018). Therefore, exploring more BRD4 downstream targets will help to understand the underlying mechanisms of the oncogenic function of BRD4 and other BET family members.
Altogether, BET inhibitors play important roles in cancer treatment by downregulating the expression of various BET target oncogenes (Table 1).
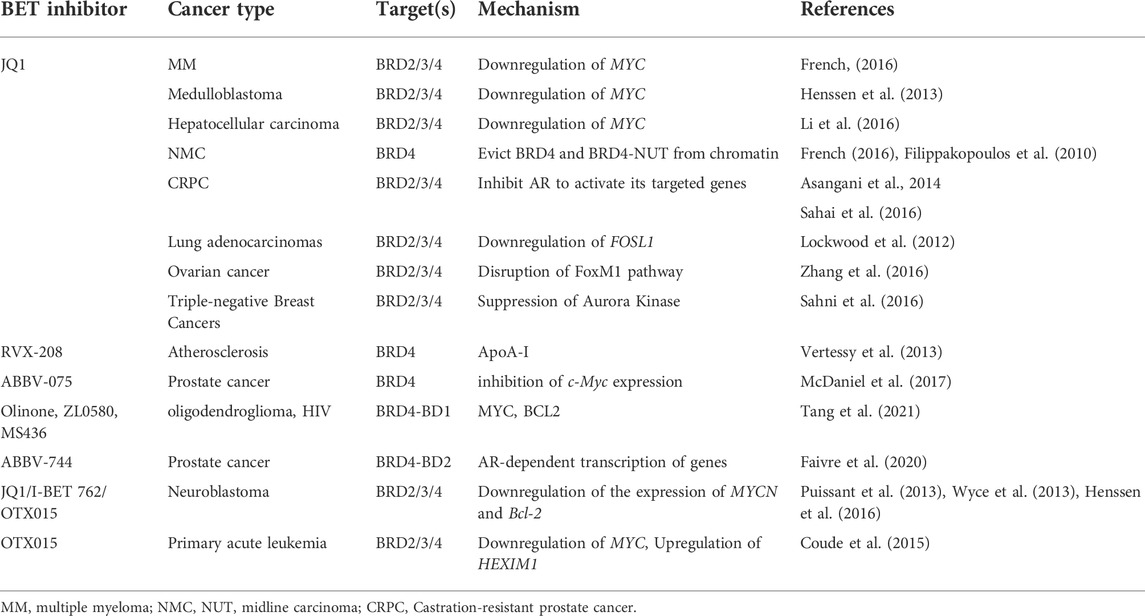
TABLE 1. BET inhibitors are used in multiple cancer types.
Neuroblastoma (NB), the most common extracranial solid tumor in children and newborns, originates from neural crest progenitors especially the adrenal glands (Brodeur, 2003; Zafar et al., 2021). The 5-years survival rate of patients with high-risk neuroblastoma are less than 50% (London et al., 2005). MYC-family transcription factors, including c-Myc, N-Myc and L-Myc, regulate cell proliferation and survival in multiple cancer types (Albihn et al., 2010). Among them, MYCN amplification accounts for 20% of primary neuroblastoma and it is closely associated with advanced NB and resistance to treatment (Hansford et al., 2004; Westermann et al., 2008). Moreover, MYCN amplification occurs in 40%–50% of high-risk NB (Bell et al., 2010). However, the amplification of other MYC family members such as MYCL and MYC is infrequently observed in NB patients. Furthermore, the expression of MYC is inversely correlated with MYCN expression (Breit and Schwab, 1989). Importantly, in mice embryos, Myc is ubiquitously expressed through the developmental stages but MYCN strictly expressed in the hematopoietic stem cells and cells of developing nervous system (Zimmerman et al., 1986; Trumpp et al., 2001). The restricted expression profile of MYCN might be mirrored the human NB which arise from the undifferentiated neural crest cells. Transgenic mouse model indicated that dysregulation of MYCN expression in neural crest was sufficient to induce tumorigenesis (Weiss et al., 1997). Therefore, MYCN has been considered a strong predictor of poor prognosis and mortality and an attractive target for therapeutic intervention in high-risk neuroblastoma (Westermark et al., 2011).
Mechanismly, as a transcription factor, MYCN can regulate the expression of many target genes, thereby regulating the basic processes of cell proliferation, protein generation, apoptosis and differentiation (Eilers and Eisenman, 2008). For example, in MYCN-amplified neuroblastoma, MYCN can bind to the promoter of telomerase catalytic subunit TERT, up-regulate and activate the expression of TERT which is a key function of amplified MYCN (Nikiforov et al., 2002; Pugh et al., 2013). Importantly, MYCN can also inhibit the expression of many cell adhesion related genes and cell cycle negative regulators (Westermark et al., 2011). Studies have shown that downregulation of MYCN by RNA interference led to cell cycle arrest and induction of apoptosis (Westermark et al., 2011). In addition, blocking the upstream signaling pathway that can regulate MYCN expression and protein stability and targeting the key downstream targets of MYCN may be another method to attenuate the effect of MYCN (Bell et al., 2010; Gustafson and Weiss, 2010). For example, inhibiting PI3K or mTOR can reduce the protein level of MYCN due to that PI3K/Akt/mTOR can maintain the stability of MYCN (Segerstrom et al., 2011).
Therefore, MYCN plays an important role in promoting the development of NB by activating or upregulating the multiple downstream genes.
Pan-cancer genome studies have shown that the mutation rate of genes in childhood cancer is significantly lower than that in adult cancer. Few recurrent mutations are detected in pediatric neuroblastoma which suggests that epigenetic disorders might play an important role in the development of childhood cancer (Iniguez et al., 2018). The result of chromatin immunoprecipitation (ChIP) indicated that BRD4 is enriched in MYCN promoter and enhancer regions to facilitate the expression of MYCN. Therefore, blocking the BET proteins by molecule inhibitors could be an efficiently therapeutic strategy to inhibit MYCN function. Currently, three BET small molecule inhibitors, JQ1, I-BET726, and OTX015, have been examined and showed significant inhibitory effect on the growth of neuroblastoma, which provides important support for the clinical application of BET inhibitors in the treatment of neuroblastoma patients. JQ1 treatment blocked the enrichment of BRD4 on the MYCN promoter region, then downregulated the transcription activity of MYCN and the expression of MYCN target genes. Furthermore, the transcription activity of MDM2 was also reduced upon MYCN inhibition which subsequently increased p53 expression that eventually led to apoptosis (Chen et al., 2010; Mazar et al., 2020). Moreover, JQ1 could induce cell cycle arrest in MYCN-amplified neuroblastoma in vitro and block tumor growth in vivo by inhibiting MYCN expression (Puissant et al., 2013). Lee et al. demonstrated that JQ1 treatment significantly increased the expression of key differentiation markers in the NB cell lines, indicating that the therapeutic effect of JQ1 could be achieved by promoting the differentiation of neuroblastoma (Lee et al., 2015). I-BET726, a novel BET small molecule inhibitor, binds to the hydrophobic cavity of BET family proteins to block the interaction of BRD4 and acetylated histone which downregulates MYCN and BCL2 expression to retard NB cell growth, promote differentiation and apoptosis (Galderisi et al., 1999; Jiang et al., 2011; Wyce et al., 2013). Henssen et al. found that MYCN-amplified neuroblastoma cell lines were more sensitive to OTX015 treatment than those MYCN non-amplification NB cells in their MYCN-driven neuroblastoma xenograft model (Henssen et al., 2016).
Altogether, targeting BET proteins such as BRD4 by small molecule inhibitors of BET proteins is potent therapeutic strategy to efficiently inhibit the high-risk NB by downregulating MYCN expression (Liu et al., 2016) (Figure 1).
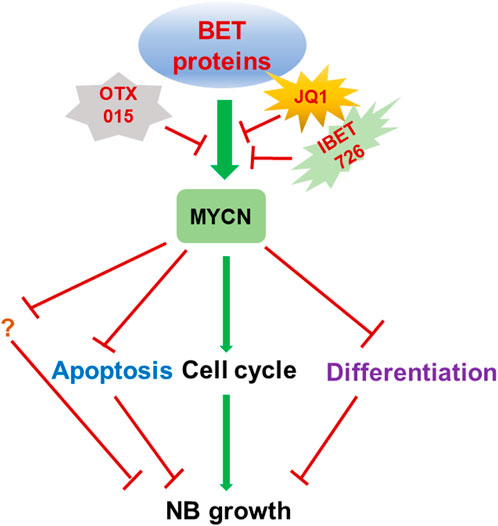
FIGURE 1. Targeting BET proteins inhibits NB growth through MYCN. Targeting BET proteins by different BET protein inhibitors such as JQ1, i-BET726 and OTX015 is therapeutic strategies which are widely tested in multiple cancer types including MYCN amplified NB. BET inhibitor treatment could induce cell cycle inhibition, apoptosis and differentiation by regulating the MYCN target genes.
It was well known that, resistance to chemotherapy are frequently found in multiple cancer types and chemotherapy strategies. Therefore, the drug resistance of NB tumor cells to BET inhibitors is also occurred which hampers the clinical application of the BET inhibitors on NB patients (Felgenhauer et al., 2018; Iniguez et al., 2018). For example, the abnormal activation of ERK1/2 signal in JQ1 resistant neuroblastoma cells attenuated the antitumor role of BET inhibitor by stabilizing MYCN protein (Liu et al., 2021). Furthermore, the PI3K pathway activation is another resistance mechanism to BET inhibitors in NB cells (Iniguez et al., 2018). Anastasia Wyce et al. found the expression level of BCL2 might affect the sensitivity of NB cells to BET inhibitor GSK1324726A (Wyce et al., 2013). Moreover, the activation of NOTCH1 and the expression of GNAS, MDM2, and NF2 might be the predictors of resistance to BET inhibitors (Puissant et al., 2013). Although the gene expression signatures related with sensitivity to BET inhibitors in NB cells have been examined, but further investigation for their effect in clinical trials are still needed (Stathis and Bertoni, 2018).
The emerging resistance to BET inhibitors affects the therapeutic effects on cancer patients and the activation of key oncogenes or inactivation of tumor suppressors are important mechanisms of BET inhibitor resistance. Therefore, it is important to explore the new therapeutic strategies by combining BET inhibitors with other drugs or methods. However, although the combination for BET and MEK inhibition showed markedly synergistic effect on tumor cells growth and apoptosis in multiple NB cell lines, but their synergistic effect on tumor growth in vivo is limited (Healy et al., 2020). In 2016, Shahbazi et al. found that JQ1 could play a synergistic role with HDAC inhibitor panobinostat to significantly decrease the expression of MYCN by reducing the transcription of LIN28B in their in vivo and in vitro models (Shahbazi et al., 2016). Moreover, the combined application of BRD4 inhibitor I-BET151 and AURKA inhibitor alisertib significantly inhibited the neuroblastoma cells growth and dramatically prolonged the survival time of neuroblastoma xenograft mice (Felgenhauer et al., 2018). Furthermore, PI3K inhibitors could overcome the resistance of NGP cell to JQ1 which indicated that PI3K inhibitors and BET inhibitors have a strong synergistic effect (Iniguez et al., 2018). Interestingly, the proteasome inhibitor carfilzomib showed a synergistic anti-tumor effect with BET inhibitor OTX015. Since OTX015 is currently in phase II clinical trials and carfilzomib is an approved anti-tumor drug, the combination of OTX015 and carfilzomib is likely to be the first targeted therapy in the clinical trials for patients with TERT-rearrangement neuroblastoma (Chen et al., 2021). Moreover, JQ1 and CDK inhibitor dinaciclib showed synergistic effect on the induction of cytotoxicity in MYCN amplified NB cells but the combination of AZD5153 and dinaciclib reduced the tumor size in mice models in vivo through increasing the tumor necrosis and lymphocyte infiltration (Wood et al., 2021). Specifically, JQ1 synergized with CDK2 inhibitor Milciclib which has been used in clinical trials to induce apoptosis and inhibit MYCN amplified NB cell growth by downregulating the MYC target genes (Bolin et al., 2018). Although the retinoic acids (RAs) were applied in the high-risk NB differentiation therapy but the effectiveness is limited, low dose of JQ1 and RA could synergistically inhibit NB cells proliferation and induce differentiation which indicated BET inhibitor and RA might be a combination therapy in combating NB (Alleboina et al., 2021). Importantly, p53 inactivation is frequently observed in the NB tumors after relapse, therefore, the combination of MDM2 inhibitor CGM097 and BET inhibitor OTX015 exhibited a synergistic inhibition of NB cell growth by activating p53 and decreasing expression of MYC family proteins (Maser et al., 2020) (Figure 2).
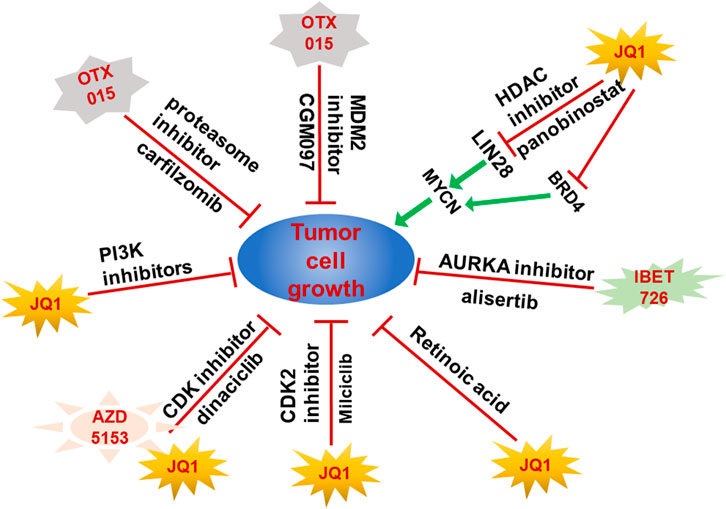
FIGURE 2. Combination of BET proteins inhibitors and other drugs achieved better therapeutic effect on NB growth. Resistance to BET proteins inhibitors hampered their clinical application. Combination of BET protein inhibitors with other small molecular inhibitors exhibited synergistic inhibition effect on NB tumors by targeting both MYCN and other genes upregulated or activated in resistance NB tumors.
Except the BET inhibitors which recognize and bind to BET proteins to block the interaction of BRD4 with acetylated histone, the selective BET inhibitors which specifically bind to BD domain of BRD4 are also developed (Shi et al., 2014). In 2020, Slavish et al. demonstrated that SJ432, a BD2 Selective inhibitors, could reduce the MYC protein level in neuroblastoma cell line. Furthermore, the NB mouse models treated with SJ432 showed a smaller tumor volume and longer survival time than those mice treated with JQ1 (Slavish et al., 2020).
Therefore, the exploration of combined therapeutic strategy and novel selective inhibitors for BET proteins will help to efficiently combat the neuroblastoma in a more precise manner.
In this review, the basic function of BET protein in regulating gene expression as epigenetic “reader” and the therapeutic effect of BET inhibitors on cancer development is complicated discussed. More importantly, we also emphasized the critical role of BET inhibitors in combating neuroblastoma through regulating MYCN.
It is well known that patients with high-risk NB have a less than 50% 5-years survival rate (Kang et al., 2006). The amplification of various oncogenes favorable NB development are commonly observed in high-risk NB patients. Notably, the amplification of MYCN is found in 20% of high-risk NB patients and amplification of MYCN is considered the best genetic marker and therapeutic target for high-risk NB (Weiss et al., 1997; Valentijn et al., 2012). However, MYCN is thought to be a “undruggable” target which make it impossible to directly target MYCN proteins. Therefore, inhibiting MYCN expression by blocking MYCN transcription regulators such as the epigenetic reader proteins is an important therapeutic strategy. BET proteins, the epigenetic readers, contain two bromodomains which can recognize acetylated histones and regulate gene transcription (French, 2016). BRD4 plays an important role in MYCN expression. BRD4 can bind to acetylated histones at the super enhancer sites and regulate the transcription of MYCN and c-Myc (Henssen et al., 2016). The three BET inhibitors, JQ1, I-BET726 and OTX015 exhibited efficiently antitumor effect in NB in the in vitro cells and in vivo xenograft models. However, the results of phase I clinical trials of BET inhibitors in human cancer patients did not show ideal therapeutic benefits. Moreover, the expression level of targeted genes might be an important factor to induce drug resistance. It was reported that the triple-negative breast cancer (TNBC) cell lines with higher expression level of MYCN are more sensitive to BET inhibitor (Schafer et al., 2020). Furthermore, Alexandre et al. conducted a high-throughput, cell-based screening of different NB cancer cell lines, and they found the MYCN amplification was a strong predictor of sensitivity to JQ1 treatment (Puissant et al., 2013) which indicated a marked correlation between the MYCN amplification status and the sensitivity to BET inhibitor.
Given that the side effects and drug resistance might be an important obstacle for the clinical utilization of BET inhibitors (Bechter and Schoffski, 2020). The combination of BET inhibitors with other targeted and epigenetic therapies might be an alternative therapeutic strategy to conquer the side effect of single administration of BET inhibitor or drug resistance. The combination of BET inhibition with PI3K/AKT/PTEN pathway inhibition can overcome the drug resistance caused by single usage of the two drugs (Alqahtani et al., 2019). In addition, BRD4 can be inhibited together with other epigenetic regulators, such as HDAC, to achieve the best anti-cancer effect (Mohammad et al., 2019). It should be noted that the overlapping toxicity should be carefully considered during the application of combination therapy. Recent study showed that combined BET and MEK inhibition showed synergistic effect in inhibiting multiple NB cells growth and survival in vitro but the antitumor activity in vivo was limited which is possibly caused by the expression level of MYCN and other oncogenes such as NF1 (Healy et al., 2020). More importantly, the Proteolysis Targeting Chimeric (PROTAC) technology was developed to promote BRD4 for proteasome mediated degradation which show different mechanisms from the BET protein inhibitors. Winter et al. created a bifunctional JQ1 molecule fused with thalidomide, called dBET which can specifically target BET protein for E3 ligase mediated degradation, resulting in a higher level of apoptosis in primary AML cells than those treated with JQ1 alone (Winter et al., 2015). Importantly, Li et al. found the PROTAC ARV-825 exhibited profound anti-tumor activity in NB cell lines and NB xenograft mice models by inhibiting the expression of MYCN or c-Myc (Li et al., 2020). Furthermore, BET degraders caused more marked cytotoxic effect on multiple cancer types by degrading the BET proteins than the BET protein inhibitors (Lu et al., 2015; Raina et al., 2016; Winter et al., 2017). Whether the BET degraders could more efficiently inhibit the NB development than BET protein inhibitors or are valuable in combating drug resistance in NB need further in deep investigation.
In summary, BET proteins are the important upstream regulators of MYCN and inhibiting BET proteins by the BET inhibitors alone or combined with other therapeutic strategies might be efficient methods to inhibit NB development by attenuating the expression of MYCN. In addition, exploring the new inhibitors of BET family proteins and the novel combined therapeutic strategies have made valuable contributions to the treatment of NB patients.
XS wrote the manuscript and drew the pictures with partial help from YW, LZ, and WZ. Y-GY, XZ, and XD edited and revised the manuscript. All authors approved the final manuscript.
This work was partly supported by the National Natural Science Foundation of China (No: 81972558), the “Startup funding of First Hospital, JLU,” the Natural Science Foundation of Jilin Province (Nos: 20200201473JC, 20200201367JC, 20210204165YY).
We thank other members of the Yang laboratory for critical reading of the manuscript and useful discussions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor TL declared a past co-authorship with the author XD.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Albihn, A., Johnsen, J. I., and Arsenian Henriksson, M. (2010). MYC in oncogenesis and as a target for cancer therapies. Adv. Cancer Res. 107, 163–224. doi:10.1016/S0065-230X(10)07006-5 | |
Alleboina, S., Aljouda, N., Miller, M., and Freeman, K. W. (2021). Therapeutically targeting oncogenic CRCs facilitates induced differentiation of NB by RA and the BET bromodomain inhibitor. Mol. Ther. Oncol. 23, 181–191. doi:10.1016/j.omto.2021.09.004 |
Alqahtani, A., Choucair, K., Ashraf, M., Hammouda, D. M., Alloghbi, A., Khan, T., et al. (2019). Bromodomain and extra-terminal motif inhibitors: a review of preclinical and clinical advances in cancer therapy. Future Sci. OA 5, FSO372. doi:10.4155/fsoa-2018-0115 | |
Anand, P., Brown, J. D., Lin, C. Y., Qi, J., Zhang, R., Artero, P. C., et al. (2013). BET bromodomains mediate transcriptional pause release in heart failure. Cell 154, 569–582. doi:10.1016/j.cell.2013.07.013 | |
Asangani, I. A., Dommeti, V. L., Wang, X., Malik, R., Cieslik, M., Yang, R., et al. (2014). Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 510, 278–282. doi:10.1038/nature13229 | |
Bechter, O., and Schoffski, P. (2020). Make your best BET: The emerging role of BET inhibitor treatment in malignant tumors. Pharmacol. Ther. 208, 107479. doi:10.1016/j.pharmthera.2020.107479 | |
Bell, E., Chen, L. D., Liu, T., Marshall, G. M., Lunec, J., and Tweddle, D. A. (2010). MYCN oncoprotein targets and their therapeutic potential. Cancer Lett. 293, 144–157. doi:10.1016/j.canlet.2010.01.015 | |
Boi, M., Gaudio, E., Bonetti, P., Kwee, I., Bernasconi, E., Tarantelli, C., et al. (2015). The BET bromodomain inhibitor OTX015 affects pathogenetic pathways in preclinical B-cell tumor models and synergizes with targeted drugs. Clin. Cancer Res. 21, 1628–1638. doi:10.1158/1078-0432.CCR-14-1561 | |
Bolin, S., Borgenvik, A., Persson, C. U., Sundstrom, A., Qi, J., Bradner, J. E., et al. (2018). Combined BET bromodomain and CDK2 inhibition in MYC-driven medulloblastoma. Oncogene 37, 2850–2862. doi:10.1038/s41388-018-0135-1 | |
Breit, S., and Schwab, M. (1989). Suppression of MYC by high expression of NMYC in human neuroblastoma cells. J. Neurosci. Res. 24, 21–28. doi:10.1002/jnr.490240105 | |
Brodeur, G. M. (2003). Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 3, 203–216. doi:10.1038/nrc1014 | |
Carlson, A. S., Cui, H., Divakaran, A., Johnson, J. A., Brunner, R. M., Pomerantz, W. C. K., et al. (2019). Systematically mitigating the p38α activity of triazole-based BET inhibitors. ACS Med. Chem. Lett. 10, 1296–1301. doi:10.1021/acsmedchemlett.9b00227 | |
Chen, L. D., Iraci, N., Gherardi, S., Gamble, L. D., Wood, K. M., Perini, G., et al. (2010). p53 is a direct transcriptional target of MYCN in neuroblastoma. Cancer Res. 70, 1377–1388. doi:10.1158/0008-5472.CAN-09-2598 | |
Chen, J., Nelson, C., Wong, M., Tee, A. E., Liu, P. Y., La, T., et al. (2021). Targeted therapy of TERT-rearranged neuroblastoma with BET bromodomain inhibitor and proteasome inhibitor combination therapy. Clin. Cancer Res. 27, 1438–1451. doi:10.1158/1078-0432.CCR-20-3044 | |
Cheung, K., Lu, G., Sharma, R., Vincek, A., Zhang, R., Plotnikov, A. N., et al. (2017). BET N-terminal bromodomain inhibition selectively blocks Th17 cell differentiation and ameliorates colitis in mice. Proc. Natl. Acad. Sci. U. S. A. 114, 2952–2957. doi:10.1073/pnas.1615601114 | |
Chung, C. W., Coste, H., White, J. H., Mirguet, O., Wilde, J., Gosmini, R. L., et al. (2011). Discovery and characterization of small molecule inhibitors of the BET family bromodomains. J. Med. Chem. 54, 3827–3838. doi:10.1021/jm200108t | |
Coude, M.-M., Braun, T., Berrou, J., Dupont, M., Bertrand, S., Masse, A., et al. (2015). BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget 6, 17698–17712. doi:10.18632/oncotarget.4131 | |
Dai, X., Gan, W., Li, X., Wang, S., Zhang, W., Huang, L., et al. (2017). Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat. Med. 23, 1063–1071. doi:10.1038/nm.4378 | |
Delmore, J. E., Issa, G. C., Lemieux, M. E., Rahl, P. B., Shi, J., Jacobs, H. M., et al. (2011). BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917. doi:10.1016/j.cell.2011.08.017 | |
Donati, B., Lorenzini, E., and Ciarrocchi, A. (2018). BRD4 and cancer: going beyond transcriptional regulation. Mol. Cancer 17, 164. doi:10.1186/s12943-018-0915-9 | |
Eilers, M., and Eisenman, R. N. (2008). Myc's broad reach. Genes Dev. 22, 2755–2766. doi:10.1101/gad.1712408 | |
Faivre, E. J., Wilcox, D., Lin, X., Hessler, P., Torrent, M., He, W., et al. (2017). Exploitation of castration-resistant prostate cancer transcription factor dependencies by the novel BET inhibitor ABBV-075. Mol. Cancer Res. 15, 35–44. doi:10.1158/1541-7786.MCR-16-0221 | |
Faivre, E. J., McDaniel, K. F., Albert, D. H., Mantena, S. R., Plotnik, J. P., Wilcox, D., et al. (2020). Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 578, 306–310. doi:10.1038/s41586-020-1930-8 | |
Felgenhauer, J., Tomino, L., Selich-Anderson, J., Bopp, E., and Shah, N. (2018). Dual BRD4 and AURKA inhibition is synergistic against MYCN-amplified and nonamplified neuroblastoma. Neoplasia 20, 965–974. doi:10.1016/j.neo.2018.08.002 | |
Filippakopoulos, P., Qi, J., Picaud, S., Shen, Y., Smith, W. B., Fedorov, O., et al. (2010). Selective inhibition of BET bromodomains. NATURE 468, 1067–1073. doi:10.1038/nature09504 | |
French, C. A. (2016). Small-molecule targeting of BET proteins in cancer. Adv. Cancer Res. 131, 21–58. doi:10.1016/bs.acr.2016.04.001 | |
Fu, L. L., Tian, M., Li, X., Li, J. J., Huang, J., Ouyang, L., et al. (2015). Inhibition of BET bromodomains as a therapeutic strategy for cancer drug discovery. Oncotarget 6, 5501–5516. doi:10.18632/oncotarget.3551 | |
Fu, Y., Zhang, Y., and Sun, H. (2021). Progress in the development of domain selective inhibitors of the bromo and extra terminal domain family (BET) proteins. Eur. J. Med. Chem. 226, 113853. doi:10.1016/j.ejmech.2021.113853 | |
Galderisi, U., Di Bernardo, G., Cipollaro, M., Peluso, G., Cascino, A., Cotrufo, R., et al. (1999). Differentiation and apoptosis of neuroblastoma cells: Role of N-myc gene product. J. Cell. Biochem. 73, 97–105. doi:10.1002/(SICI)1097-4644(19990401)73:1<97:AID-JCB11>3.0.CO;2-M | |
Gilan, O., Rioja, I., Knezevic, K., Bell, M. J., Yeung, M. M., Harker, N. R., et al. (2020). Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science 368, 387–394. doi:10.1126/science.aaz8455 | |
Gosmini, R., Van Loc, N., Toum, J., Simon, C., Brusq, J.-M. G., Krysa, G., et al. (2014). The discovery of I-BET726 (GSK1324726A), a potent tetrahydroquinoline ApoA1 up-regulator and selective BET bromodomain inhibitor. J. Med. Chem. 57, 8111–8131. doi:10.1021/jm5010539 | |
Gustafson, W. C., and Weiss, W. A. (2010). Myc proteins as therapeutic targets. Oncogene 29, 1249–1259. doi:10.1038/onc.2009.512 | |
Hansford, L. M., Thomas, W. D., Keating, J. M., Burkhart, C. A., Peaston, A. E., Norris, M. D., et al. (2004). Mechanisms of embryonal tumor initiation: distinct roles for MycN expression and MYCN amplification. Proc. Natl. Acad. Sci. U. S. A. 101, 12664–12669. doi:10.1073/pnas.0401083101 | |
Healy, J. R., Hart, L. S., Shazad, A. L., Gagliardi, M. E., Tsang, M., Elias, J., et al. (2020). Limited antitumor activity of combined BET and MEK inhibition in neuroblastoma. Pediatr. Blood Cancer 67. e28267. doi:10.1002/pbc.28267 | |
Henssen, A., Thor, T., Odersky, A., Heukamp, L., El-Hindy, N., Beckers, A., et al. (2013). BET bromodomain protein inhibition is a therapeutic option for medulloblastoma. Oncotarget 4, 2080–2095. doi:10.18632/oncotarget.1534 | |
Henssen, A., Althoff, K., Odersky, A., Beckers, A., Koche, R., Speleman, F., et al. (2016). Targeting MYCN-driven transcription by BET-bromodomain inhibition. Clin. Cancer Res. 22, 2470–2481. doi:10.1158/1078-0432.CCR-15-1449 | |
Herait, P. E., Berthon, C., Thieblemont, C., Raffoux, E., Magarotto, V., Stathis, A., et al. (2014). Abstract CT231: BET-bromodomain inhibitor OTX015 shows clinically meaningful activity at nontoxic doses: Interim results of an ongoing phase I trial in hematologic malignancies. Cancer Res. 74, CT231. doi:10.1158/1538-7445.AM2014-CT231 |
Hillyar, C., Rallis, K. S., and Varghese, J. (2020). Advances in epigenetic cancer therapeutics. Cureus 12, e11725. doi:10.7759/cureus.11725 | |
Hogg, S. J., Vervoort, S. J., Deswal, S., Ott, C. J., Li, J., Cluse, L. A., et al. (2017). BET-bromodomain inhibitors engage the host immune system and regulate expression of the immune checkpoint ligand PD-L1. Cell Rep. 18, 2162–2174. doi:10.1016/j.celrep.2017.02.011 | |
Huang, M., and Weiss, W. A. (2013). Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 3, a014415. doi:10.1101/cshperspect.a014415 | |
Huang, M., Zeng, S., Zou, Y., Shi, M., Qiu, Q., Xiao, Y., et al. (2017). The suppression of bromodomain and extra-terminal domain inhibits vascular inflammation by blocking NF-κB and MAPK activation. Br. J. Pharmacol. 174, 101–115. doi:10.1111/bph.13657 | |
Iniguez, A. B., Alexe, G., Wang, E. J., Roti, G., Patel, S., Chen, L., et al. (2018). Resistance to epigenetic-targeted therapy engenders tumor cell vulnerabilities associated with enhancer remodeling. Cancer Cell 34, 922–938.e7. doi:10.1016/j.ccell.2018.11.005 | |
Ito, T., Umehara, T., Sasaki, K., Nakamura, Y., Nishino, N., Terada, T., et al. (2011). Real-time imaging of histone H4K12-specific acetylation determines the modes of action of histone deacetylase and bromodomain inhibitors. Chem. Biol. 18, 495–507. doi:10.1016/j.chembiol.2011.02.009 | |
Jang, M. K., Mochizuki, K., Zhou, M. S., Jeong, H. S., Brady, J. N., and Ozato, K. (2005). The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 19, 523–534. doi:10.1016/j.molcel.2005.06.027 | |
Jeong, H. M., Kwon, M. J., and Shin, Y. K. (2014). Overexpression of cancer-associated genes via epigenetic derepression mechanisms in gynecologic cancer. Front. Oncol. 4, 12. doi:10.3389/fonc.2014.00012 | |
Jiang, R. C., Xue, S. F., and Jin, Z. L. (2011). Stable knockdown of MYCN by lentivirus-based RNAi inhibits human neuroblastoma cells growth in vitro and in vivo. Biochem. Biophys. Res. Commun. 410, 364–370. doi:10.1016/j.bbrc.2011.06.020 | |
Kang, J. H., Rychahou, P. G., Ishola, T. A., Qiao, J., Evers, B. M., and Chung, D. H. (2006). MYCN silencing induces differentiation and apoptosis in human neuroblastoma cells. Biochem. Biophys. Res. Commun. 351, 192–197. doi:10.1016/j.bbrc.2006.10.020 | |
Kulikowski, E., Rakai, B. D., and Wong, N. C. W. (2021). Inhibitors of bromodomain and extra-terminal proteins for treating multiple human diseases. Med. Res. Rev. 41, 223–245. doi:10.1002/med.21730 | |
Law, R. P., Atkinson, S. J., Bamborough, P., Chung, C. W., Demont, E. H., Gordon, L. J., et al. (2018). Discovery of tetrahydroquinoxalines as bromodomain and extra-terminal domain (BET) inhibitors with selectivity for the second bromodomain. J. Med. Chem. 61, 4317–4334. doi:10.1021/acs.jmedchem.7b01666 | |
Leal, A. S., Williams, C. R., Royce, D. B., Pioli, P. A., Sporn, M. B., and Liby, K. T. (2017). Bromodomain inhibitors, JQ1 and I-BET 762, as potential therapies for pancreatic cancer. Cancer Lett. 394, 76–87. doi:10.1016/j.canlet.2017.02.021 | |
Lee, S., Rellinger, E. J., Kim, K. W., Craig, B. T., Romain, C. V., Qiao, J., et al. (2015). Bromodomain and extraterminal inhibition blocks tumor progression and promotes differentiation in neuroblastoma. Surgery 158, 819–826. doi:10.1016/j.surg.2015.04.017 | |
Li, G.-Q., Guo, W.-Z., Zhang, Y., Seng, J.-J., Zhang, H.-P., Ma, X.-X., et al. (2016). Suppression of BRD4 inhibits human hepatocellular carcinoma by repressing MYC and enhancing BIM expression. Oncotarget 7, 2462–2474. doi:10.18632/oncotarget.6275 | |
Li, Z., Lim, S. L., Tao, Y., Li, X., Xie, Y., Yang, C., et al. (2020). PROTAC bromodomain inhibitor ARV-825 displays anti-tumor activity in neuroblastoma by repressing expression of MYCN or c-myc. Front. Oncol. 10, 574525. doi:10.3389/fonc.2020.574525 | |
Liu, P. Y., Atmadibrata, B., Mondal, S., Tee, A. E., and Liu, T. (2016). NCYM is upregulated by lncUSMycN and modulates N-Myc expression. Int. J. Oncol. 49, 2464–2470. doi:10.3892/ijo.2016.3730 | |
Liu, C., Gen, Y., Tanimoto, K., Muramatsu, T., Inoue, J., and Inazawa, J. (2021). Concurrent targeting of MAP3K3 and BRD4 by miR-3140-3p overcomes acquired resistance to BET inhibitors in neuroblastoma cells. Mol. Ther. Nucleic Acids 25, 83–92. doi:10.1016/j.omtn.2021.05.001 | |
Lockwood, W. W., Zejnullahu, K., Bradner, J. E., and Varmus, H. (2012). Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc. Natl. Acad. Sci. U. S. A. 109, 19408–19413. doi:10.1073/pnas.1216363109 | |
London, W. B., Castleberry, R. P., Matthay, K. K., Look, A. T., Seeger, R. C., Shimada, H., et al. (2005). Evidence for an age cutoff greater than 365 days for neuroblastoma risk group stratification in the Children's Oncology Group. J. Clin. Oncol. 23, 6459–6465. doi:10.1200/JCO.2005.05.571 | |
Lu, J., Qian, Y., Altieri, M., Dong, H., Wang, J., Raina, K., et al. (2015). Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 22, 755–763. doi:10.1016/j.chembiol.2015.05.009 | |
Maser, T., Zagorski, J., Kelly, S., Ostrander, A., Goodyke, A., Nagulapally, A., et al. (2020). The MDM2 inhibitor CGM097 combined with the BET inhibitor OTX015 induces cell death and inhibits tumor growth in models of neuroblastoma. Cancer Med. 9, 8144–8158. doi:10.1002/cam4.3407 | |
Mazar, J., Gordon, C., Naga, V., and Westmoreland, T. J. (2020). The killing of human neuroblastoma cells by the small molecule JQ1 occurs in a p53-dependent manner. Anticancer. Agents Med. Chem. 20, 1613–1625. doi:10.2174/1871520620666200424123834 | |
McDaniel, K. F., Wang, L., Soltwedel, T., Fidanze, S. D., Hasvold, L. A., Liu, D., et al. (2017). Discovery of N-(4-(2, 4-Difluorophenoxy)-3-(6-methyl-7-oxo-6, 7-dihydro-1H-pyrrolo[2, 3-c]pyridin-4-yl)phenyl)ethanesulfonamide (ABBV-075/Mivebresib), a potent and orally available bromodomain and extraterminal domain (BET) family bromodomain inhibitor. J. Med. Chem. 60, 8369–8384. doi:10.1021/acs.jmedchem.7b00746 | |
Mochizuki, K., Ishiyama, S., Hariya, N., and Goda, T. (2021). Regulation of carbohydrate-responsive metabolic genes by histone acetylation and the acetylated histone reader BRD4 in the gene body region. Front. Mol. Biosci. 8, 682696. doi:10.3389/fmolb.2021.682696 | |
Mohammad, H. P., Barbash, O., and Creasy, C. L. (2019). Targeting epigenetic modifications in cancer therapy: erasing the roadmap to cancer. Nat. Med. 25, 403–418. doi:10.1038/s41591-019-0376-8 | |
Mondal, P., Natesh, J., Penta, D., and Meeran, S. M. (2022). Progress and promises of epigenetic drugs and epigenetic diets in cancer prevention and therapy: A clinical update. Semin. Cancer Biol. 83, 503–522. doi:10.1016/j.semcancer.2020.12.006 | |
Nicodeme, E., Jeffrey, K. L., Schaefer, U., Beinke, S., Dewell, S., Chung, C.-w., et al. (2010). Suppression of inflammation by a synthetic histone mimic. Nature 468, 1119–1123. doi:10.1038/nature09589 | |
Nikiforov, M. A., Chandriani, S., Park, J., Kotenko, I., Matheos, D., Johnsson, A., et al. (2002). TRRAP-dependent and TRRAP-independent transcriptional activation by Myc family oncoproteins. Mol. Cell. Biol. 22, 5054–5063. doi:10.1128/MCB.22.14.5054-5063.2002 | |
Noguchi-Yachide, T. (2016). BET bromodomain as a target of epigenetic therapy. Chem. Pharm. Bull. 64, 540–547. doi:10.1248/cpb.c16-00225 | |
Otto, C., Schmidt, S., Kastner, C., Denk, S., Kettler, J., Muller, N., et al. (2019). Targeting bromodomain-containing protein 4 (BRD4) inhibits MYC expression in colorectal cancer cells. Neoplasia 21, 1110–1120. doi:10.1016/j.neo.2019.10.003 | |
Pugh, T. J., Morozova, O., Attiyeh, E. F., Asgharzadeh, S., Wei, J. S., Auclair, D., et al. (2013). The genetic landscape of high-risk neuroblastoma. Nat. Genet. 45, 279–284. doi:10.1038/ng.2529 | |
Puissant, A., Frumm, S. M., Alexe, G., Bassil, C. F., Qi, J., Chanthery, Y. H., et al. (2013). Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 3, 308–323. doi:10.1158/2159-8290.Cd-12-0418 | |
Raina, K., Lu, J., Qian, Y., Altieri, M., Gordon, D., Rossi, A. M., et al. (2016). PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. U. S. A. 113, 7124–7129. doi:10.1073/pnas.1521738113 | |
Roe, J. S., Mercan, F., Rivera, K., Pappin, D. J., and Vakoc, C. R. (2015). BET bromodomain inhibition suppresses the function of hematopoietic transcription factors in acute myeloid leukemia. Mol. Cell 58, 1028–1039. doi:10.1016/j.molcel.2015.04.011 | |
Sahai, V., Redig, A. J., Collier, K. A., Eckerdt, F. D., and Munshi, H. G. (2016). Targeting bet bromodomain proteins in solid tumors. Oncotarget 7, 53997–54009. doi:10.18632/oncotarget.9804 | |
Sahni, J. M., Gayle, S. S., Bonk, K. L., Vite, L. C., Yori, J. L., Webb, B., et al. (2016). Bromodomain and extraterminal protein inhibition blocks growth of triple-negative breast cancers through the suppression of aurora kinases. J. Biol. Chem. 291, 23756–23768. doi:10.1074/jbc.M116.738666 | |
Schafer, J. M., Lehmann, B. D., Gonzalez-Ericsson, P. I., Marshall, C. B., Beeler, J. S., Redman, L. N., et al. (2020). Targeting MYCN-expressing triple-negative breast cancer with BET and MEK inhibitors. Sci. Transl. Med. 12, eaaw8275. doi:10.1126/scitranslmed.aaw8275 | |
Schaukowitch, K., Joo, J. Y., Liu, X., Watts, J. K., Martinez, C., and Kim, T. K. (2014). Enhancer RNA facilitates NELF release from immediate early genes. Mol. Cell 56, 29–42. doi:10.1016/j.molcel.2014.08.023 | |
Segerstrom, L., Baryawno, N., Sveinbjornsson, B., Wickstrom, M., Elfman, L., Kogner, P., et al. (2011). Effects of small molecule inhibitors of PI3K/Akt/mTOR signaling on neuroblastoma growth in vitro and in vivo. Int. J. Cancer 129, 2958–2965. doi:10.1002/ijc.26268 | |
Shahbazi, J., Liu, P. Y., Atmadibrata, B., Bradner, J. E., Marshall, G. M., Lock, R. B., et al. (2016). The bromodomain inhibitor JQ1 and the histone deacetylase inhibitor panobinostat synergistically reduce N-myc expression and induce anticancer effects. Clin. Cancer Res. 22, 2534–2544. doi:10.1158/1078-0432.CCR-15-1666 | |
Shang, E., Nickerson, H. D., Wen, D., Wang, X., and Wolgemuth, D. J. (2007). The first bromodomain of Brdt, a testis-specific member of the BET sub-family of double-bromodomain-containing proteins, is essential for male germ cell differentiation. Development 134, 3507–3515. doi:10.1242/dev.004481 | |
Shi, J., and Vakoc, C. R. (2014). The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell 54, 728–736. doi:10.1016/j.molcel.2014.05.016 | |
Shi, J., Wang, Y. F., Zeng, L., Wu, Y. D., Deng, J., Zhang, Q., et al. (2014). Disrupting the interaction of BRD4 with diacetylated twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell 25, 210–225. doi:10.1016/j.ccr.2014.01.028 | |
Slavish, P. J., Chi, L., Yun, M. K., Tsurkan, L., Martinez, N. E., Jonchere, B., et al. (2020). Bromodomain-selective BET inhibitors are potent antitumor agents against MYC-driven pediatric cancer. Cancer Res. 80, 3507–3518. doi:10.1158/0008-5472.CAN-19-3934 | |
Stathis, A., and Bertoni, F. (2018). BET proteins as targets for anticancer treatment. Cancer Discov. 8, 24–36. doi:10.1158/2159-8290.Cd-17-0605 | |
Sun, H., Huang, B., Cao, J., Yan, Q., and Yin, M. (2022). Editorial: Epigenetic regulation and tumor immunotherapy. Front. Oncol. 12, 893157. doi:10.3389/fonc.2022.893157 | |
Tang, P., Zhang, J., Liu, J., Chiang, C. M., and Ouyang, L. (2021). Targeting bromodomain and extraterminal proteins for drug discovery: From current progress to technological development. J. Med. Chem. 64, 2419–2435. doi:10.1021/acs.jmedchem.0c01487 | |
Taniguchi, Y. (2016). The bromodomain and extra-terminal domain (BET) family: Functional anatomy of BET paralogous proteins. Int. J. Mol. Sci. 17, E1849. doi:10.3390/ijms17111849 | |
Trumpp, A., Refaeli, Y., Oskarsson, T., Gasser, S., Murphy, M., Martin, G. R., et al. (2001). c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 414, 768–773. doi:10.1038/414768a | |
Valentijn, L. J., Koster, J., Haneveld, F., Aissa, R. A., van Sluis, P., Broekmans, M. E. C., et al. (2012). Functional MYCN signature predicts outcome of neuroblastoma irrespective of MYCN amplification. Proc. Natl. Acad. Sci. U. S. A. 109, 19190–19195. doi:10.1073/pnas.1208215109 | |
Vertessy, B. G., McLure, K. G., Gesner, E. M., Tsujikawa, L., Kharenko, O. A., Attwell, S., et al. (2013). RVX-208, an inducer of ApoA-I in humans, is a BET bromodomain antagonist. PLoS ONE 8, e83190. doi:10.1371/journal.pone.0083190 | |
Vollmuth, F., and Geyer, M. (2010). Interaction of propionylated and butyrylated histone H3 lysine marks with Brd4 bromodomains. Angew. Chem. Int. Ed. Engl. 49, 6768–6772. doi:10.1002/anie.201002724 | |
Wang, N., Wu, R., Tang, D., and Kang, R. (2021). The BET family in immunity and disease. Signal Transduct. Target. Ther. 6, 23. doi:10.1038/s41392-020-00384-4 | |
Weiss, W. A., Aldape, K., Mohapatra, G., Feuerstein, B. G., and Bishop, J. M. (1997). Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 16, 2985–2995. doi:10.1093/emboj/16.11.2985 | |
Westermann, F., Muth, D., Benner, A., Bauer, T., Henrich, K. O., Oberthuer, A., et al. (2008). Distinct transcriptional MYCN/c-MYC activities are associated with spontaneous regression or malignant progression in neuroblastomas. Genome Biol. 9, R150. doi:10.1186/gb-2008-9-10-r150 | |
Westermark, U. K., Wilhelm, M., Frenzel, A., and Henriksson, M. A. (2011). The MYCN oncogene and differentiation in neuroblastoma. Semin. Cancer Biol. 21, 256–266. doi:10.1016/j.semcancer.2011.08.001 | |
White, M. E., Fenger, J. M., and Carson, W. E. (2019). Emerging roles of and therapeutic strategies targeting BRD4 in cancer. Cell. Immunol. 337, 48–53. doi:10.1016/j.cellimm.2019.02.001 | |
Winter, G. E., Buckley, D. L., Paulk, J., Roberts, J. M., Souza, A., Dhe-Paganon, S., et al. (2015). DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381. doi:10.1126/science.aab1433 | |
Winter, G. E., Mayer, A., Buckley, D. L., Erb, M. A., Roderick, J. E., Vittori, S., et al. (2017). BET bromodomain proteins function as master transcription elongation factors independent of CDK9 recruitment. Mol. Cell 67, 5–18.e19. doi:10.1016/j.molcel.2017.06.004 | |
Wood, L., Huang, M., Zeki, J., Gong, M., Taylor, J., Shimada, H., et al. (2021). Combining inhibitors of Brd4 and cyclin-dependent kinase can decrease tumor growth in neuroblastoma with MYCN amplification. J. Pediatr. Surg. 56, 1199–1202. doi:10.1016/j.jpedsurg.2021.03.037 | |
Wu, X. W., Qi, J., Bradner, J. E., Xiao, G. T., and Chen, L. F. (2013). Bromodomain and extraterminal (BET) protein inhibition suppresses human T cell leukemia virus 1 (HTLV-1) tax protein-mediated tumorigenesis by inhibiting nuclear factor kappa B (NF-kappa B) signaling. J. Biol. Chem. 288, 36094–36105. doi:10.1074/jbc.M113.485029 | |
Wyce, A., Ganji, G., Smitheman, K. N., Chung, C. W., Korenchuk, S., Bai, Y., et al. (2013). BET inhibition silences expression of MYCN and BCL2 and induces cytotoxicity in neuroblastoma tumor models. PLoS One 8, e72967. doi:10.1371/journal.pone.0072967 | |
Yang, Z. Y., Yik, J. H. N., Chen, R. C., He, N. H., Jang, M. K., Ozato, K., et al. (2005). Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein brd4. Mol. Cell 19, 535–545. doi:10.1016/j.molcel.2005.06.029 | |
Yin, M., Guo, Y., Hu, R., Cai, W. L., Li, Y., Pei, S., et al. (2020). Potent BRD4 inhibitor suppresses cancer cell-macrophage interaction. Nat. Commun. 11, 1833. doi:10.1038/s41467-020-15290-0 | |
Zafar, A., Wang, W., Liu, G., Wang, X., Xian, W., McKeon, F., et al. (2021). Molecular targeting therapies for neuroblastoma: Progress and challenges. Med. Res. Rev. 41, 961–1021. doi:10.1002/med.21750 | |
Zhang, Z., Ma, P., Jing, Y., Yan, Y., Cai, M. C., Zhang, M., et al. (2016). BET bromodomain inhibition as a therapeutic strategy in ovarian cancer by downregulating FoxM1. Theranostics 6, 219–230. doi:10.7150/thno.13178 | |
Zhang, P., Wang, D., Zhao, Y., Ren, S., Gao, K., Ye, Z., et al. (2017). Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT-mTORC1 activation. Nat. Med. 23, 1055–1062. doi:10.1038/nm.4379 | |
Zhao, Y., Wang, L., Ren, S., Wang, L., Blackburn, P. R., McNulty, M. S., et al. (2016). Activation of P-TEFb by androgen receptor-regulated enhancer RNAs in castration-resistant prostate cancer. Cell Rep. 15, 599–610. doi:10.1016/j.celrep.2016.03.038 | |
Zhu, H., Bengsch, F., Svoronos, N., Rutkowski, M. R., Bitler, B. G., Allegrezza, M. J., et al. (2016). BET bromodomain inhibition promotes anti-tumor immunity by suppressing PD-L1 expression. Cell Rep. 16, 2829–2837. doi:10.1016/j.celrep.2016.08.032 | |
Keywords: BET family proteins, BRD4 inhibitors, neuroblastoma, epigenetic regulation, MYCN
Citation: Shi X, Wang Y, Zhang L, Zhao W, Dai X, Yang Y-G and Zhang X (2022) Targeting bromodomain and extra-terminal proteins to inhibit neuroblastoma tumorigenesis through regulating MYCN. Front. Cell Dev. Biol. 10:1021820. doi: 10.3389/fcell.2022.1021820
Received: 17 August 2022; Accepted: 01 September 2022;
Published: 16 September 2022.
Edited by:
Tianda Li, Chinese Academy of Sciences (CAS), ChinaCopyright © 2022 Shi, Wang, Zhang, Zhao, Dai, Yang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiangpeng Dai, ZGFpeGlhbmdwZW5nQGpsdS5lZHUuY24=; Yong-Guang Yang, eW9uZ2dAamx1LmVkdS5jbg==; Xiaoling Zhang, eGlhb2xpbmd6aGFuZ0BqbHUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.