
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 18 October 2022
Sec. Epigenomics and Epigenetics
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.1020958
This article is part of the Research Topic Epigenetics and Epigenomics of Gene-Environment Interactions Affecting Fitness of Organisms to the Changing Global Climate View all 4 articles
 Vijay Rani Rajpal1*
Vijay Rani Rajpal1* Priyanka Rathore2§
Priyanka Rathore2§ Sahil Mehta3‡§
Sahil Mehta3‡§ Nikita Wadhwa4§
Nikita Wadhwa4§ Priyanka Yadav2§
Priyanka Yadav2§ Eapsa Berry5§
Eapsa Berry5§ Shailendra Goel2*Vishnu Bhat2*
Shailendra Goel2*Vishnu Bhat2* Soom Nath Raina6*
Soom Nath Raina6*Recent research in plant epigenetics has increased our understanding of how epigenetic variability can contribute to adaptive phenotypic plasticity in natural populations. Studies show that environmental changes induce epigenetic switches either independently or in complementation with the genetic variation. Although most of the induced epigenetic variability gets reset between generations and is short-lived, some variation becomes transgenerational and results in heritable phenotypic traits. The short-term epigenetic responses provide the first tier of transient plasticity required for local adaptations while transgenerational epigenetic changes contribute to stress memory and help the plants respond better to recurring or long-term stresses. These transgenerational epigenetic variations translate into an additional tier of diversity which results in stable epialleles. In recent years, studies have been conducted on epigenetic variation in natural populations related to various biological processes, ecological factors, communities, and habitats. With the advent of advanced NGS-based technologies, epigenetic studies targeting plants in diverse environments have increased manifold to enhance our understanding of epigenetic responses to environmental stimuli in facilitating plant fitness. Taking all points together in a frame, the present review is a compilation of present-day knowledge and understanding of the role of epigenetics and its fitness benefits in diverse ecological systems in natural populations.
In recent years, epigenetic modifications of chromatin material have been discovered as a major player in modulating plant fitness under challenging environmental conditions. Epigenetics refers to the study of changes in gene functions that are mitotically and/or meiotically heritable and do not involve a change in DNA sequence but are based on modifications in chromatin organization (Dupont et al., 2009). Epigenetic regulation consists of covalent modifications of DNA and histone proteins affecting the accessibility of chromatin to transcriptional machinery (Iwasaki and Paszkowski, 2014). Three major types of epigenetic marks or signatures include DNA methylation, histone modifications, and small RNAs. During DNA methylation, the methyl groups get covalently bound to cytosine nucleotides (5 mC) (Bartels et al., 2018; Gomez-Cabellos et al., 2022) and along with covalent modifications like methylation, acetylation, and phosphorylation in histone proteins regulate the local chromatin landscape (Zhu et al., 2021; Tian et al., 2022). Non-coding small RNAs such as microRNAs play a significant role in post-transcriptional regulation directing DNA methylation and chromatin remodeling at their target loci (Bossdorf et al., 2008; Robertson and Wolf, 2012; Bond and Baulcombe, 2014; Ariel and Manavella, 2021). Covalent modifications on DNA and histone proteins and small RNAs impact the condensation of chromatin and its accessibility to transcriptional machinery (Meyer, 2015). Epigenetic control mainly operates at the transcriptional level and affects the activity of genes, repetitive sequences, and transposable elements, thus providing genome integrity and helping plants survive and reproduce successfully in unpredictable environments (Kumari et al., 2022). Both genetic and epigenetic variations control gene expression independently and together.
Epigenetic mechanisms help sessile plants cope with environmental changes either by acclimation or adaptation to the changing environment. Acclimation outcomes include phenotypic responses achieved by altering gene expression and physiological attributes without incurring any genotypic changes in organisms, a phenomenon called phenotypic plasticity (Pigliucci, 2005). In other words, phenotypic plasticity is the capacity for non-genetic modifications of the phenotype. On the other hand, when phenotypic alterations are hereditary and increase the reproductive fitness of a species in relation to the environment, they are deemed to be of evolutionary adaptive significance. It has been fairly reasoned that there might be a close connection between phenotypic plasticity and adaptation, stating that plants with better plastic responses might have better adaptive ability to enhance their relative fitness, a narrative that needs to be empirically tested (Wang et al., 2021).
Environmental stress response management is quite complex and involves integrating physiological, biochemical, and genetic changes in the genome and modulating vital processes including vernalization response, genomic imprinting, and mobility of transposable elements (TEs) among others. These responses of plants could be transient in the short run or may persist over the long term. For example, Jackson and colleagues (Jackson et al., 2002) found an immediate decrease in the frequency of arbuscules formation during phosphorous sufficiency in lettuce (Lactuca sativa). In the longer term, only a few plant species could survive due to higher competitive abilities under resource-limiting conditions, whereas plants that control water use by reducing sap flux sensitivity survived under long-term soil moisture changes (Grossiord et al., 2018).
The most common stress-induced changes include ion toxicity, reactive oxygen species (ROS) production, redox/osmotic imbalances, nutritional disproportionality, and lipid peroxidation that bring changes in the ultrastructure of cellular and organellar membranes, chlorophyll content, photosynthetic capacities, and turgor pressure (Anamika et al., 2019; Mehta et al., 2019; Malhi et al., 2021; Singh et al., 2022). Upon perceiving these stresses, a cascade of genetic, molecular, biochemical, anatomical, physiological, and morphological changes occur in plants. While phenotypic plasticity provides plant fitness under changing environmental conditions through the spontaneous generation of epimutations (chemical changes that result in a new epi-allelic state without changing the DNA sequence), transgenerational inheritance of these epimutations induces local adaptation after many generations. These heritable epimutations encompass DNA methylation or histone modifications of a genetic locus generating epialleles (the epigenetic equivalent of genetic alleles that are stably transmitted across generations) (Robertson and Wolf, 2012; Taudt et al., 2016). These epialleles provide another layer of heritable variation to natural genetic diversity.
During the last few decades, climate change has had an unprecedented impact, which is critically categorized as a global threat to mankind in terms of agricultural and forest productivity (Leisner, 2020; Shahzad et al., 2021). Adding to risks, the average global temperature is expected to rise by 2°C by 2100 as per Intergovernmental Panel on Climate Change (IPCC) predictions (Tollefson, 2020; Pielke et al., 2022). The global climate changes have immensely impacted both the biotic and abiotic components of the environment by bringing changes in growth, multiplication, spread, virulence, and the emergence of many plant pathogens/insect pests and escalating abiotic stresses like drought, floods, heat, and cold stresses (Tito et al., 2018; Vilela et al., 2018; Juroszek et al., 2020; Skendžić et al., 2021). These environmental stresses conjointly act as a daunting obstacle that incurs fitness costs to plants. In this context, rapid phenotypic diversity that results due to alterations in epigenetic marks related to changes in chromatin architecture plays an important role. The epigenetic effects combat both short- and long-term stresses allowing natural populations to mitigate climate-change-related stresses by enhancing plant fitness (Lämke et al., 2016a).
Although epigenetic variation can be induced in the absence of genetic variability, the presence of the latter can influence the former. The genetic makeup of an individual, for instance, can affect the epigenetic marks by altering the number of cytosines available for methylation, small RNA production, availability of transposable elements, genes controlling DNA methylation enzymatic machinery, and histone-mediated chromatin modification (Feinberg and Irizarry, 2010; Robertson and Wolf, 2012). This offers further lability and advantage to the species population and adds to the ability to show transgenerational plasticity and further fuel adaptive change (Beldade et al., 2011; Robertson and Wolf, 2012). In a nutshell, genetic and epigenetic processes may complement each other or work independently, and subtle DNA polymorphisms can affect the epigenetic processes, selection, and adaptive evolution of a species. Therefore, the present review tries to compile relevant studies dealing with epigenetic processes involved in modulating plant fitness in natural populations in changing environments. The information collated will provide a platform to plan and execute in-depth studies to unravel underlying mechanisms of epigenetic regulation of plant response to environmental changes.
The growth, reproduction, and survival of natural populations are largely determined by functional attributes like morphological traits, physiology, and phenology. Amidst global climatic changes that have resulted in accentuated biotic and abiotic stresses, plants have made fine adjustments and acquired additional stress mitigation strategies for their survival leading to the adaptive phenotypic divergence that depends upon natural selection, gene flow, and phenotypic plasticity (Hendry et al., 2002). Natural selection operates faster in populations with a high degree of gene flow and can fix genes that respond positively to changing environments (Charlesworth and Charlesworth, 2017; Charlesworth and Charlesworth, 2017). Adaptive divergence mechanisms in populations with a narrow genetic base, with no association with the habitats, on the other hand, indicate the involvement of epigenetic mechanisms independently or in complementation with the genetic mechanisms in facilitating adaptive evolution (Saze et al., 2012; Song et al., 2013; Amaral et al., 2020; Yi and Goodisman, 2021). Plants maximize their phenotypic plasticity either by increasing the natural variation through large population sizes or by modulating the gene expression patterns to produce altered gene functions and novel phenotypes (Wang et al., 2021). Ultimately, it is the availability of sufficient and meaningful plastic response that determines the capacity of plants to fuel reproductive success leading to fitness benefits in changing environments (Dar et al., 2022). Increased reproductive fitness serves as the basis for the survival of natural populations and their colonizing ability in the changing environments, through the amelioration of various kinds of stresses.
Notably, phenotypic plasticity patterns do not always follow the principles of Mendelian genetics (Foust et al., 2016; Yi and Goodisman, 2021; Guarino et al., 2022) and instead are shown to be supported largely by epigenetic mechanisms that generally originate as combinatorial effects of the interactions of genotypes with the environment (Genotype x Environment, abbreviated as GxE) (Peltier et al., 2018). The classic example showing that epigenetic mechanisms generate phenotypic variability came from studies in Linaria vulgaris (Cubas et al., 1999). Consequently, many more instances showing the contribution of epigenetic variability to phenotypic and functional diversity were reported (Manning et al., 2006; Martin et al., 2009; Miura et al., 2009; Herrera and Bazaga, 2013; Quadrana et al., 2014; Nicotra et al., 2015; Song et al., 2015; Ci et al., 2016; Lämke and Bäurle 2017; Schmid et al., 2018; Alonso et al., 2019; Chen et al., 2020; Baduel et al., 2021). The documentation of subtle phenotypic plasticity in homogeneous genetic populations of many self-pollinating and asexually reproducing plant species later strongly substantiated these observations (Preite et al., 2015; Wilschut et al., 2016). Although the molecular underpinnings of the causal relationship between the epigenetic mechanisms and phenotypic plasticity are yet to be clearly understood, epigenetic responses to environmental changes are shown to be mediated by the epigenome that works like a regulatory system and integrates the environmental and genetic variation to shape the inventory of available adaptive phenotypic variation (Marin et al., 2020). In this context, the epigenetic marks, especially changes in DNA methylation, can result in the induction of rapid epimutations much faster than genetic alterations and provide useful epi-allelic diversity that contributes to phenotypic plasticity. While the somatic epialleles are capable of conferring short-term responses, they provide a rapid and immediate means of negotiating the imposed stresses and ensuring the survival of populations; the long-term adaptations, on the other hand, are brought about by epialleles that show transgenerational inheritance (Figure 1) (Ashapkin et al., 2020; Yi and Goodisman, 2021).
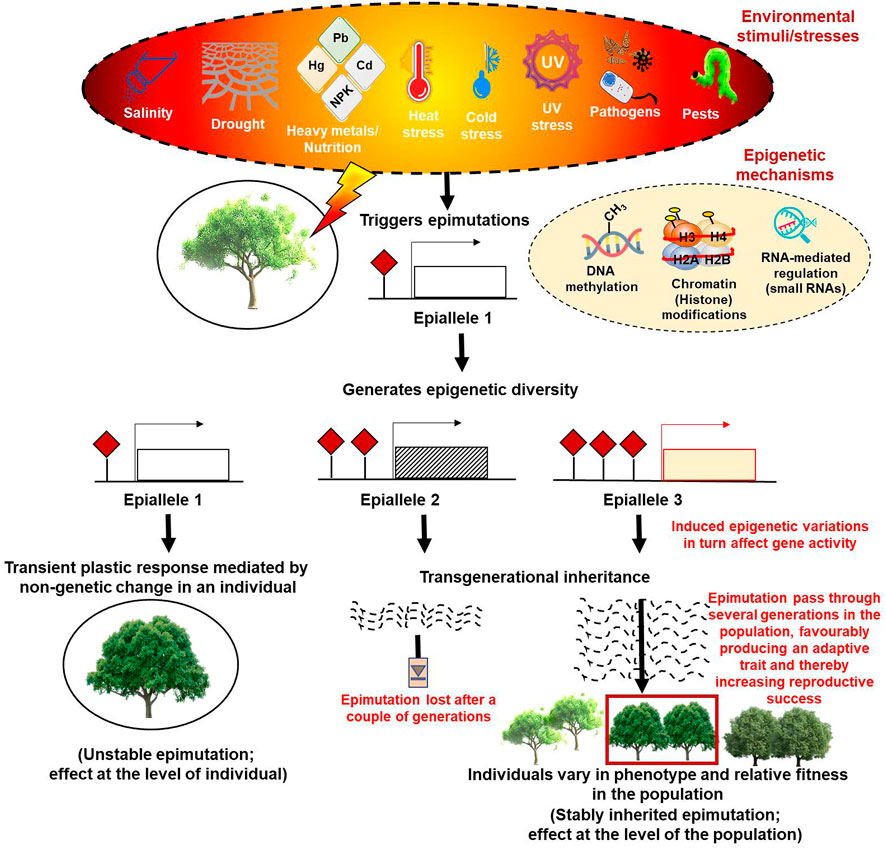
FIGURE 1. Diagrammatic representation of the epigenetic modifications based on their plastic or adaptive response to various environmental cues.
In recent decades, emerging epigenetic processes have been highlighted as promising mechanisms to facilitate the generation of the first tier of rapid phenotypic diversity required to improve the fitness and adaptive responses in the natural populations of plants (Bräutigam et al., 2013; Amaral et al., 2020; Ashe et al., 2021). A survey of published literature shows that a large number of research investigations wherein the epigenetic route has been shown to deploy rapid phenotypic plasticity for adaptation and evolution have come from studies in model plant and crop species (Ding et al., 2018; Guarino et al., 2022; Varotto et al., 2022) which is primarily due to the availability of large genomic resources accumulated in this group of plants. The advancement in sequencing technologies and their cost effectiveness, has lately facilitated the availability of genomic resources in natural populations of non-model species as well. This data has provided useful insights into the role of epigenetic mechanisms in expanding phenotypic plasticity and adaptive responses. Even for model species, efforts have begun to compare the epigenetic and genetic heritability in field conditions.
With many published reports (Table 1, Table 2, Table 3, and Table 4), it has been convincingly shown that epigenetic processes vary among populations, affect phenotypes, stress responses, adaptability, fitness, range distribution and can also become heritable across generations. Therefore, they produce an additional, first and rapid tier of ‘epi-variability’, which individually or in combination with genetic variations have been observed to play a significant role in regulating ecological and evolutionary processes in plants (Richards et al., 2017; Baduel et al., 2021; Boquete et al., 2021). The acquired epigenetic changes allow populations to initiate the mechanisms to colonize, adapt and evolve in novel environments and the adaptations achieved get stabilized slowly by long-lasting genetic variations (Yona et al., 2015; Amaral et al., 2020). The cross-talk between the epigenetic and genetic components of variability is mediated by an interplay between changes in chromatin organization and physiological responses (Ashe et al., 2021; Boquete et al., 2021) and represents one of the most important factors contributing to the adaptive success of populations under climatic stresses.
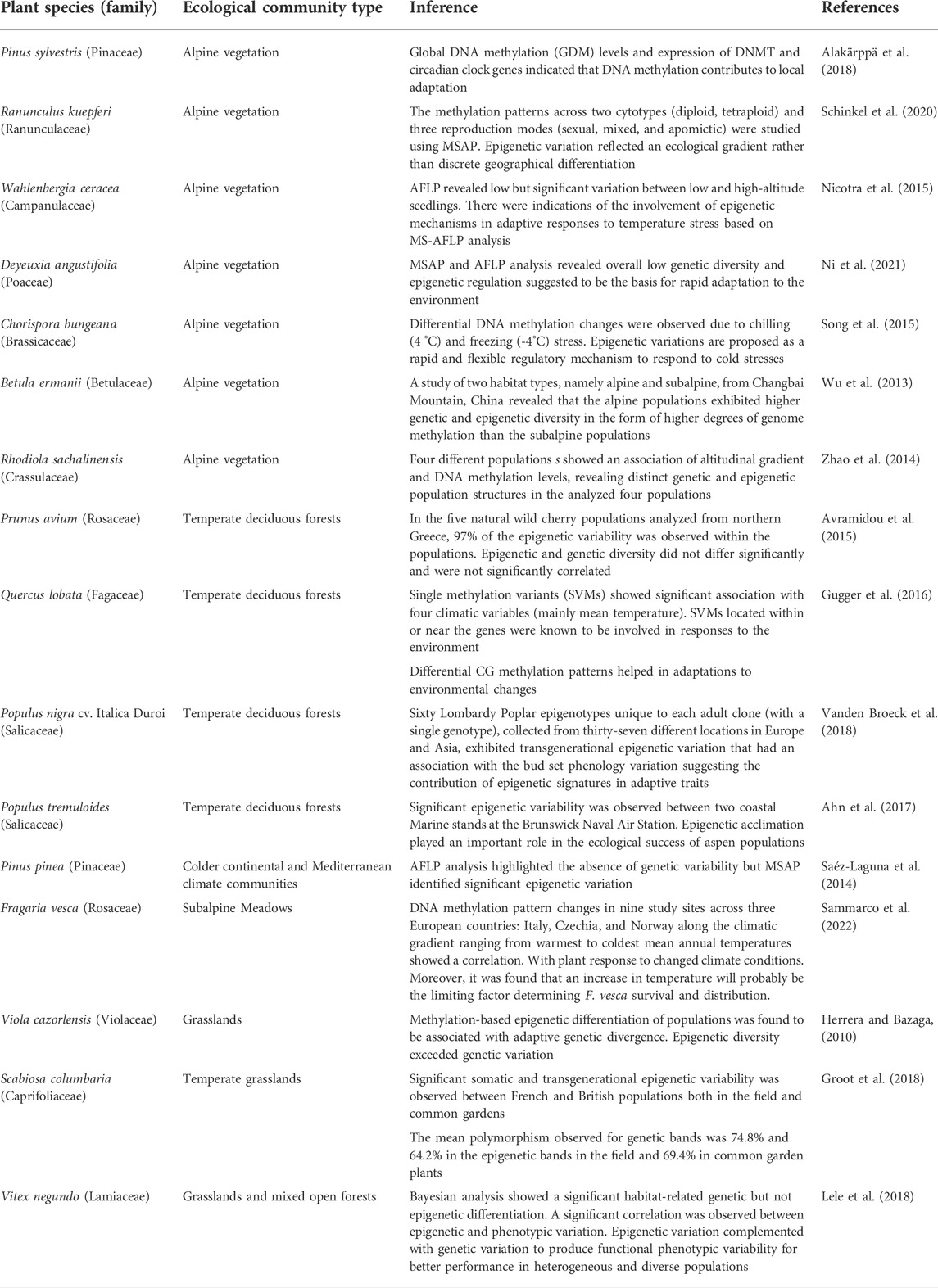
TABLE 1. Instances of epigenetic regulation in plant species from different ecological communities.
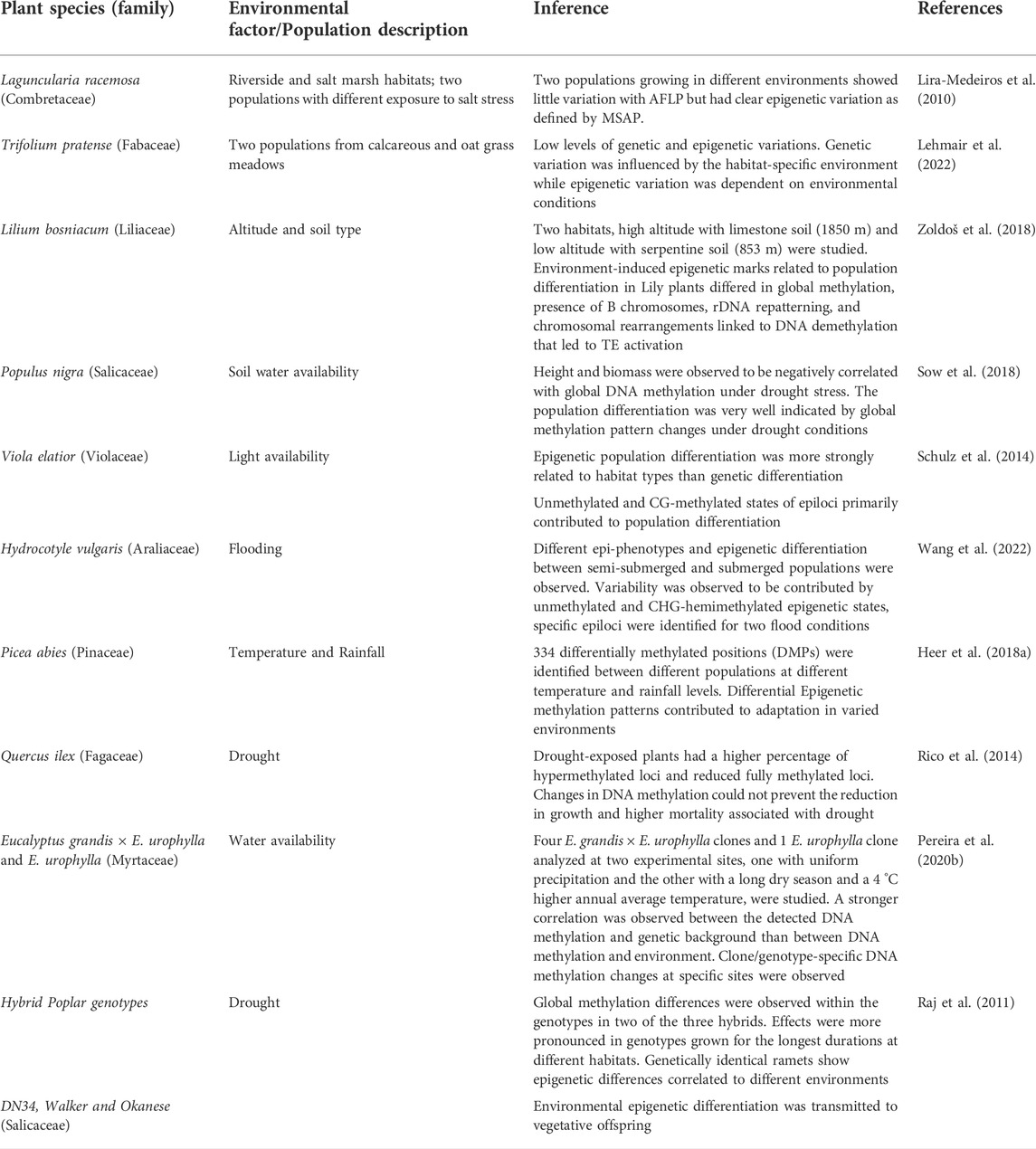
TABLE 2. The instances of epigenetic diversity and its relationship with environmental factors.
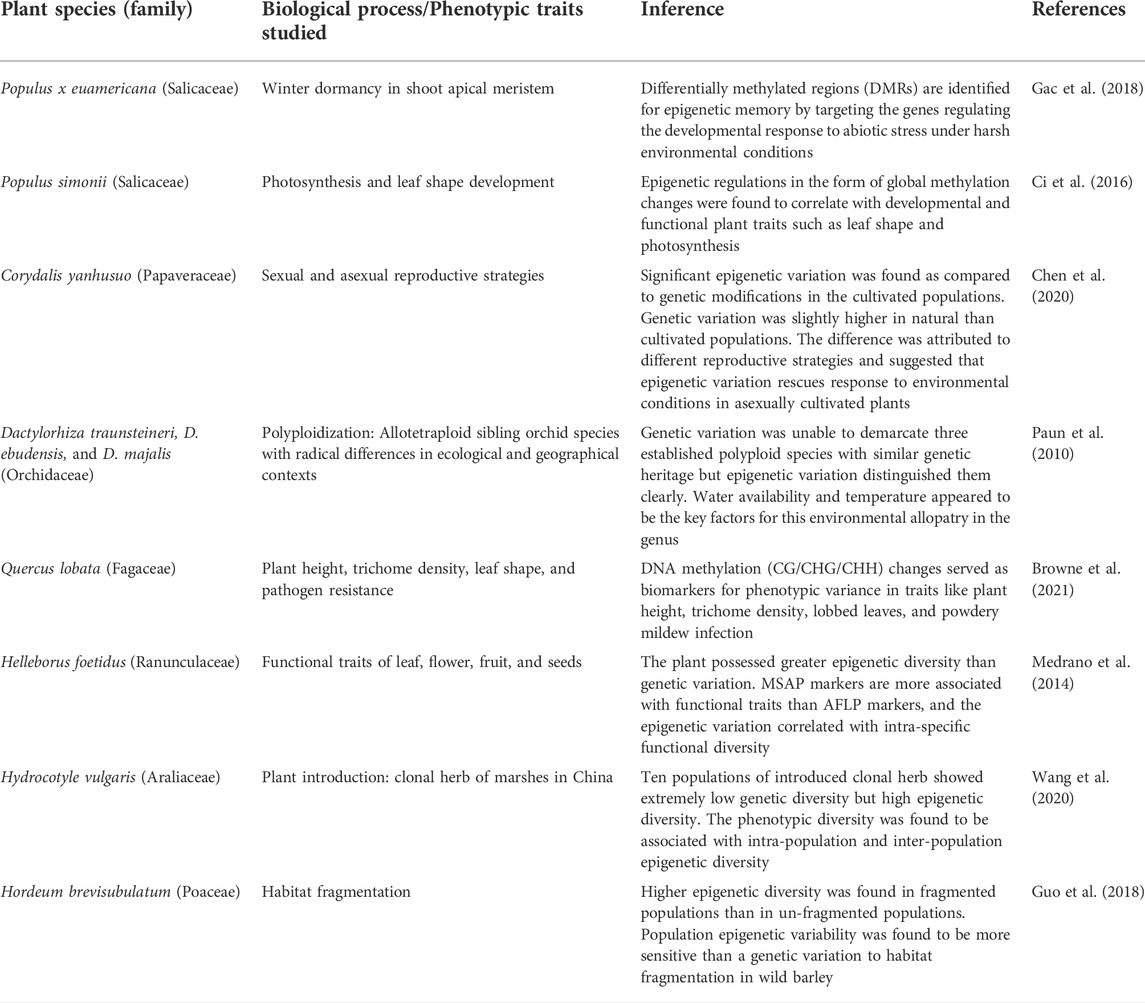
TABLE 3. Epigenetic regulation during various biological processes and functional traits development in natural plant populations.

TABLE 4. Studies on epigenetic regulation in invasive plant species.
Plants being stationary get exposed to various periodic (such as seasonal variations) or sudden (such as biotic stresses or climate factors) environmental changes. Epigenetic mechanisms help plants in generating plastic responses to stressful environments (Lämke and Bäurle 2017; Ashapkin et al., 2020; Yi and Goodisman, 2021; Guarino et al., 2022). The plants may remember the past stress(es) and use their past responses’ memory to mitigate the recurring stress(es) by generating a more pronounced and/or faster response through a phenomenon known as ‘defense priming’ or ‘stress priming’ (Lämke and Bäurle, 2017; Liu et al., 2022). It is primarily achieved by activation of stress-related genetic pathways induced by modification of the epigenetic marks cued by the environment (Bruce et al., 2007; Lämke and Bäurle, 2017; Ashapkin et al., 2020). There are two categories of genes involved in stress memory genetic pathways, namely stress-responsive genes (non-memory genes) and stress-memory genes. While the stress-responsive genes express at the same level during each stress exposure, the expression of stress-memory genes increases significantly upon repeated exposure to the same stress (Ding et al., 2012; Ashapkin et al., 2020). Table 5 collates various genes that have been observed to express in plants under stress conditions. The whole process of recognition of previous stress cues, retaining the information, and processing it to a further modified response is termed ‘stress memory’. When the stress memory is short-term and ranges from days to weeks it is referred to as ‘somatic stress memory’. However, stress memory may also become heritable and get transmitted to the offspring as ‘transgenerational stress memory’.
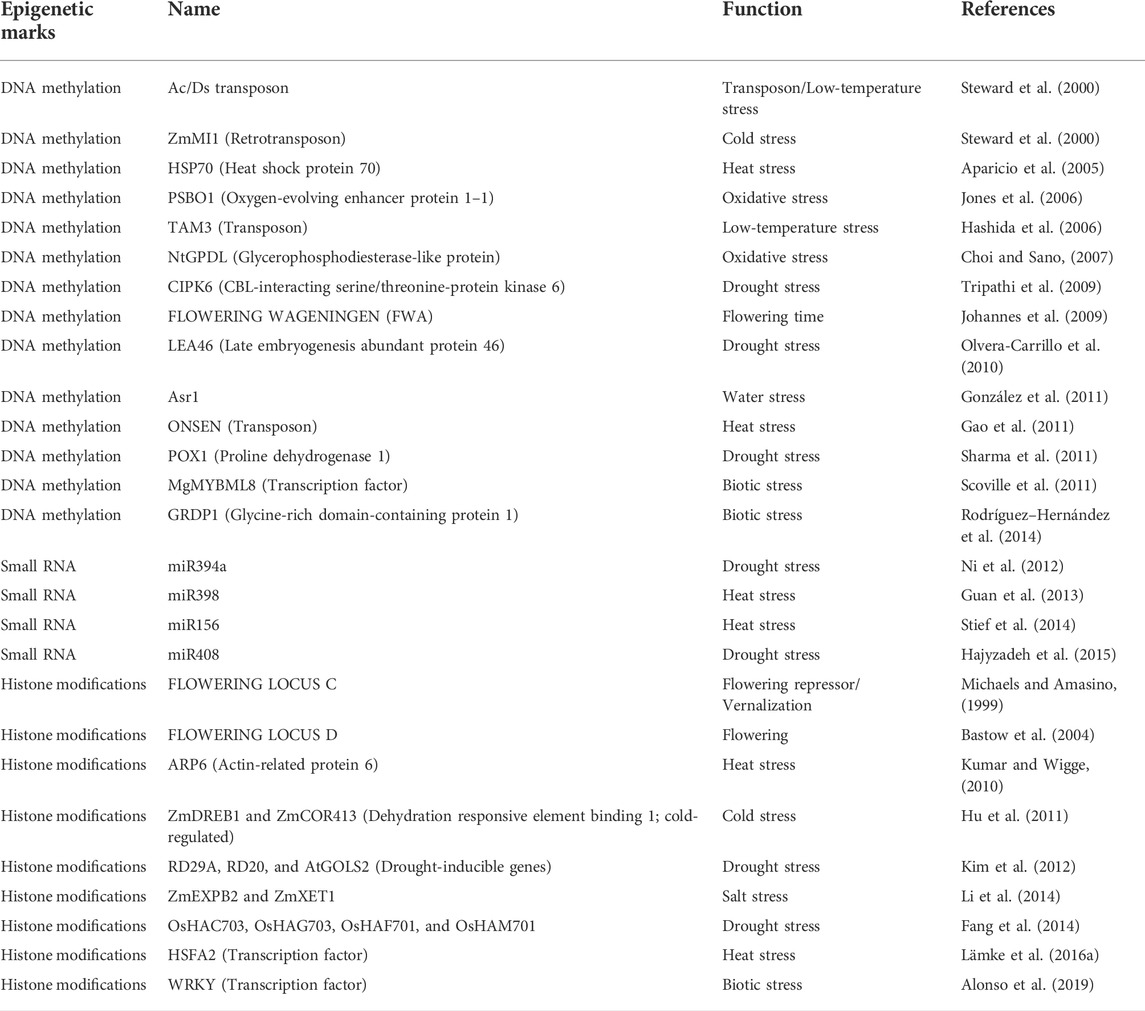
TABLE 5. List of candidate genes regulated by epigenetics in GxE interactions.
Somatic stress memory involves short-term sustained or pronounced responses that are often accompanied by chromatin modification, histone H3K4 methylation, and reduced nucleosome occupancy (Bäurle and Trindade, 2020). These responses result in phenotypic plasticity which affects only the somatic tissues, and the phenotypic changes revert to their previous normal states when the stress conditions get over. This type of resetting is advantageous when the stress condition does not prevail in the long term because to maintain the primed response, plants need to reallocate resources that cost energy (Lämke and Bäurle, 2017). Somatic stress memory provides a specific fitness advantage under short-term stress conditions.
Most of the reports pertaining to epigenetic regulation of somatic stress memory have been derived from research investigations undertaken in the model plant Arabidopsis under different temperature and dehydration stress conditions. The somatic stress memory in these cases has been shown to vary from a few days to months and is observed to be associated with epigenetic changes in DNA methylation, histone proteins, and small miRNAs (Ding et al., 2012; Kim et al., 2012; Stief et al., 2014; Ashapkin et al., 2020). For instance, the trimethylation of lysine four of histone H3 (H3K4me3) has been observed in several abiotic stress-responsive genes in Arabidopsis. It has been observed that the accumulation of memory genes in dehydration stress regulated by H3K4me3 in the first priming stimulus is maintained throughout the memory phase (Ding et al., 2012). Similarly, accumulation of histone acetylation and activation of the COLD RESPONSIVE (COR) genes pervaded during cold stress conditions (Pavangadkar et al., 2010; Park et al., 2018). Kim et al. (2012) reported the accumulation of RD29A, RD20, and AtGOLS2 transcripts during drought stress conditions and the reduction of these transcripts to the basal level during rehydration (Table 5). This is due to the stress memory associated with acetylation of lysine nine of histone H3 (H3K9ac) and the recruitment of RNA polymerase II to these genic regions suggesting that chromatin marks play a role in transcription memory during stress conditions. Furthermore, H3K4me3 methyltransferase ARABIDOPSIS TRITHORAX1 (ATX1) which controls several genes in response to drought stress, showed abscisic acid (ABA) production that promoted drought tolerance in plants by the production of more ABA and closing of the stomata (Bäurle and Trindade, 2020).
Transcription factors are identified as key components for maintaining transcriptional memory and recruiting chromatin-regulatory proteins to their target loci. HEAT SHOCK FACTOR A2 (HSFA2) has been found to be responsible for the methylation of H3K4me in response to heat stress (Lämke et al., 2016b). Vernalization is another example of environmentally induced somatic epigenetic memory. It refers to the induction of flowering in vegetative plants upon cold exposition, a response that the plants remember as vernalization memory. This memory allows plants to remember a previous exposure to cold temperatures. For example, the FLOWERING LOCUS C gene FLC has been shown to accumulate H3K27 trimethylation in response to cold temperatures in Arabidopsis. This histone modification at the FLC locus is brought about by a Polycomb group complex recruited by VERNALIZATION INSENSITIVE 3 (VIN3) and VERNALIZATION 5 (VRN5) genes, and long non-coding RNAs (COLDAIR and COOLAIR) activated by low temperatures (Heo and Sung, 2011; Bäurle and Trindade, 2020). From the reports cited above, it has been shown that overall variation in environmental conditions can lead to local adaptation of plant populations in short term, while long-term adaptive fitness is largely derived from transgenerational effects (Lämke et al., 2016a; Lämke and Bäurle, 2017; González et al., 2018).
Transgenerational stress memory is transmitted from stressed parental generations to unstressed offspring (Bruce et al., 2007; Robertson and Wolf 2012) to help prepare future generations to mitigate environments with the same stress (es). The first instance of phenotypic variation linked to the epigenetic down-regulation of a gene (MgMYBML8) was related to the transmission of simulated herbivory (leaf damage) induced trichome production on the underside of leaves in Mimulus guttatus. The epimutation was transgenerational and the unstressed offspring also exhibited increased trichome production, as a response showing preparedness for future herbivory (Scoville et al., 2011). Following this, there have been many ecologically relevant intergenerational and transgenerational stress responses that are associated with epigenetic processes induced by the modification of epigenetic marks cued by the environment (Huang et al., 2010; Pecinka et al., 2010; Ding et al., 2012; Lämke et al., 2016b; González et al., 2018; Ashe et al., 2021). For instance, chromatin modification induced transcriptional activation of a repetitive element in heat-stressed Arabidopsis thaliana (Pecinka et al., 2010), switching from C3 to crassulacean acid metabolism (CAM) photosynthetic pathway assisted by the down-regulation of associated loci in Mesembryanthemum crystallinum induced by acute salt-stress (Huang et al., 2010), an increase in drought tolerance in Oryza sativa induced by drought-induced methylation changes (Wang et al., 2011), retention of more water by Arabidopsis plants subjected to multiple cycles of dehydration than the plants exposed to dehydration stress for the first time (Ding et al., 2012), sustained induction and persistence of several ‘stress memory genes’ in response to heat stress correlated with different histones’ methylation (H3K4me3 and H3K4me2) (Lämke et al., 2016a) represents some stress amelioration instances mediated by epigenetic processes. The epigenetic transgenerational effects may occur across generations of both asexually (Verhoeven et al., 2010) and sexually reproducing species (González et al., 2018) and modify phenotypes of offspring generations. These changes can set a rapid pace of adaptation and microevolution in a population (Ashe et al., 2021).
In long-term stress conditions, stress memory has been observed to be inherited by successive generations mainly through the maternal parent (Wibowo et al., 2016). Expression of DNA glycosylase DEMETER (DME) in the male gametes inhibits paternal inheritance of methylation marks as they are removed by DEMETER in the absence of the hyperosmotic stress in Arabidopsis while in dme mutants the transmission of the stress memory through the paternal is restored in next generations (Wibowo et al., 2016; Lämke and Bäurle, 2017). In the natural populations of a mangrove species Rhizophora mangle, Mounger et al. (2021) observed a low genetic and high epigenetic diversity. About 25% of the epigenetic differences were observed among the offspring of the maternal trees and the inheritance of epigenetic variations among them was regulated by the stressed maternal environments.
The activation of many genetic components is involved in the induction of transgenerational epigenetic effects that combat long-term stress conditions allowing species to mitigate climate-change-related problems by enhancing plant fitness (Lämke et al., 2016b). Many genes have been reported in Arabidopsis that are influenced by GxE effects and regulated by epigenetics (Table 5). For example, Kim et al. (2012) reported the accumulation of RD29A, RD20, and AtGOLS2 transcripts during drought stress and the reduction of their amounts to the basal level during rehydration (Table 5). In another set of studies, novel phenotypes were induced in Arabidopsis thaliana and were linked to epigenomic patterns inherited (Schmid et al., 2018). Xu et al. (2021) showed retention of transgenerational epigenetic memory in Arabidopsis thaliana. A great deal of epigenetic regulation has also been observed through the activation of transposable elements (TEs) in Arabidopsis. The retrotransposon ONSEN which remains suppressed by RdDM (RNA-dependent DNA methylation) mechanism gets activated under heat stress in Arabidopsis. RdDM plays an important role in heat stress defense as mutations in RdDM machinery can limit the heat stress tolerance of plants by activating heat stress-responsive genes (Popova et al., 2013) that have been observed to be carried to future generations (Erdmann and Picard, 2020). Baduel and Colot (2021), also observed TEs-associated epi-variation that persisted for at least eight generations in Arabidopsis under drought, salt, UV, heat, and cold stress (Lang–Mladek et al., 2010; Ganguly et al., 2017).
In wild-type Arabidopsis, siRNAs target the methylation of ∼2.6 kb upstream region of the HKT1 (HIGH-AFFINITY K+ CHANNEL 1) gene. In the mutants of small RNA biogenesis pathway genes such as the rdr2 mutant, the methylation is reduced at CHG and CHH contexts. In a mutant of another gene involved in DNA methylation MET1 (Methyltransferase 1), methylation is reduced in all three contexts (Baek et al., 2011). During salt stress, met1 plants were observed to be hypersensitive while the rdr2 mutant showed normal salt sensitivity. Therefore, non-CG methylation regulates the expression of HKT1 in leaves, which may be essential in providing long-term adaptation of plants to salinity stress (Ashapkin et al., 2020).
To combat heavy metal stress, treatment of rice seedlings with heavy metals inhibited the shoot and root development in response to changing DNA methylation patterns of TEs and different protein-encoding genes (Cong et al., 2019; Ashapkin et al., 2020). In Hg2+-treated plants (F0 generation), changed methylation (CHG hypomethylation) was observed in most studied DNA sequences. In the progeny, stable inheritance of the modified methylation patterns was observed. These studies showed that the altered epigenetic state induced by heavy metal stress is heritable between plant generations for providing long-term adaptations. Further, Salicylic acid has been observed to induce epigenetic changes in the F2 progeny of dandelions (Preite et al., 2018).
The above studies indicate that epigenetic variation induced in response to climate changes can increase plant fitness and show transgenerational inheritance influencing long-term adaptation in populations. Thus, epigenetic mechanisms seem to play an important role in the survival, adaptation, and microevolution of plant species under challenging climatic conditions. However, future research efforts are required to establish the extent of stress response, phenotypic plasticity, their transgenerational capacity, and the mechanistic and ecological impact of epigenetics on plant adaptations in natural populations. Nonetheless, even if the induced epigenetic changes are not heritable, these rapid stress responses play a crucial role in generating transient fitness ensuring survival without switching on the expensive constitutive expression of stress-tolerant genes.
During the last decade, a myriad of studies has signified the role of epigenetics in plant adaptations and its comparison with the innate genetic diversity in diverse taxa. These studies have compared genetic and epigenetic variation in natural populations of plants in contrasting environments (discussed in the next section). An alternate approach involves generating epigenetic variability in systems with little genetic diversity and has been adopted in model plants where the availability of detailed biological information, genetic resources, and immense genomic data allows answering complex questions. Many such studies have been conducted on model plants like Arabidopsis thaliana, Oryza sativa, and Zea mays (Richards et al., 2017).
Moreover, A. thaliana is ideally suited for studies of natural phenotypic variation and is a good experimental system to explore the mechanistic link between genetic and epigenetic variation, especially with regard to cytosine methylation (Schmitz and Ecker 2012). Epigenetic studies in A. thaliana have largely involved analysis of genome-wide methylation and epiRILs. One such study used demethylating agent 5-azacytidine and revealed differential responses in genotypes in terms of plant trait and plasticity suggesting that epigenetic variation contributes significantly to plant fitness in addition to genetic variability (Bossdorf et al., 2010). Furthermore, the investigators created epigenetic recombinant inbred lines (epiRILs) by crossing a ddm1 mutant of A. thaliana (col-ddm1) with its wild type resulting in lines that were homozygous for ddm1. These epiRILs were genetically nearly identical but differed in the pattern of DNA methylation (Latzel et al., 2013) and exhibited functional diversity having an effect similar to genetic diversity and leading to phenotypic variations. These two studies suggest that epigenetic diversity is an important component of functional biodiversity and can contribute to evolutionary adaptation by responding to selection at the community level (Richards et al., 2017). In a fitness-based study, the A. thaliana population subjected to selection in rapidly changing environments revealed a reduced epigenetic diversity suggesting that epigenetic variation is subjected to selection (Schmid et al., 2018). A study on Swedish accessions of A. thaliana revealed that CHH methylation is temperature sensitive while CpG gene body methylation was not affected by growth temperature. Moreover, the study concluded that local adaptation involves changes in the epigenetic landscape (Dubin et al., 2015). All these studies indicate that epigenetic diversity is providing an additional layer of variation that helps in local adaptation just like genetic variation when subjected to selection.
The studies in rice and maize are not as detailed as in A. thaliana, but some interesting comparative studies can be found on natural populations. These studies draw similar conclusions as from the A. thaliana-based studies. Targeted DNA methylation profiling has been carried out on 263 maize inbred lines (Xu et al., 2019). The study identified 16,000 differentially methylated regions (DMRs) that were not tagged by SNPs, suggesting that these DMRs possess unique information. The epigenetic variation has also been linked to inbreeding in maize (Han et al., 2021), showing hypermethylation of CHH genomic regions. DMRs have also been identified based on genome-wide DNA methylation profiles of 20 maize inbred lines. The study found that DMRs are heritable and are associated with genetic variation identified near transposable elements that may contribute to the variation in DNA methylation (Eichten et al., 2013). Similarly, MSAP marker genotyping was carried out on 50 accessions of japonica rice varieties to show more epigenetic and phenotypic similarities between the accessions grown at the same latitude (Zhang et al., 2022).
Besides these model/crop plants, several studies conducted on non-model systems exposed to diverse environments have revealed significant plasticity and can answer questions related to plant adaptations in varied environments (Thiebaut et al., 2019). These plants can serve as model plants for such studies. Examples of such systems can be identified in Table 1, Table 2, Table 3, and Table 4.
Traditionally, studies on ecological adaptations in natural populations have been comprehended through biological processes, namely genetic drift, mutation, and natural selection that work upon populations and the pre-existing genetic diversity therein. These processes are slow and take a long time to engender evolutionary divergence. To cope with the fast-changing climate and novel stresses, populations need rapid responses like phenotypic plasticity to provide the required reproductive success. It has been shown that stresses can modify epigenetic marks and generate a rapid epigenetic response(s) in the form of a wide range of novel and stable ecological phenotypes in just a few generations through gene expression regulation (Grativol et al., 2012; Heer et al., 2018a; Ashapkin et al., 2020; Ashe et al., 2021; Baduel et al., 2021; Boquete et al., 2021; García–García et al., 2022). Among all the known epigenetic mechanisms, DNA methylation is relatively stable with transgenerational heritability and contributes importantly to phenotypic variability under stress. In this context, it is extremely important to understand the mechanisms that generate phenotypic plasticity and convert local phenotypic adjustments into adaptive variability of ecological and evolutionary significance.
Rapid strides have been made in recent years to understand the role of various biological processes and other ecological factors in orchestrating strong epigenetic support to species in natural populations in generating phenotypic plasticity and acquiring evolutionary adaptations (Heer et al., 2018b; Paun et al., 2019; Amaral et al., 2020; Ashapkin et al., 2020; Campoy et al., 2022; Sun et al., 2022). The conclusive testament of their involvement in modulating epigenetic adaptive divergence in plants has come from studies conducted in plant resources representing genetically uniform backgrounds such as recombinant inbred lines (RILs and epi-RILs), asexually reproducing plant species (apomictic, and vegetative propagation) growing in varied habitats, and rapid adaptations shown by a single genotype of invasive plant species in different alien invaded habitats. Additionally, various other factors like pollination type and mating systems, polyploidy and hybridization, sympatry and allopatry as well as contrasting habitats of communities have been observed to influence the epigenetic dynamics of the genome in natural populations. The effect of these biological processes on the epigenetic landscape is reflected in the form of modifications in 1) reproductive mechanisms, 2) nuclear-cellular ratio, 3) copy number of genes altering the genome organization, 4) alterations in the expression of genes, and 5) alterations in phenotypes producing ecotypes (see examples in Tables 1, 2, 3, and 4). Studies have been undertaken to understand the spectrum of epigenetic variability in natural populations thriving under the influence of the above factors and to better comprehend the regulatory dynamics of adaptations. The findings have been tabulated for clarity, better readability, and comparisons (Table 1, Table 2, Table 3, and Table 4).
The magnitudes of heritable phenotypic variation in epigenetic recombinant inbred lines (epi-RILs) of Arabidopsis thaliana differing in DNA methylation but with little divergence in DNA sequence was compared with standard RILs that had considerable DNA sequence variation (Zhang et al., 2018). The lines were grown in the same environment and assessed for heritable variation in growth, phenology, and fitness. Results indicated that in some traits the phenotypic variation among epi-RILs was comparable to that among RILs and natural ecotypes, indicating that phenotypic variability can have an epigenetic basis.
Since the asexually reproducing plants lack meiotic recombination, their narrow genetic base demonstrates poor prospects for genetic adaptations. For this reason, asexually reproducing plants have even been considered as evolutionary dead ends (Lynch et al., 1993). Notwithstanding this consideration, however, there are many clonally reproducing species adapted to varied environments. Some of the most successful invasive plants of the world, for example, are clonally reproducing and many a times, a single genotype is observed to expand and thrive in diverse habitats successfully. Among the 468 invasive plants listed in the IUCN Global Invasive Species Database (http://www.issg.org/database), as many as 70% reproduce clonally. Some of the most successful invasive plants are found to be genetically uniform in their introduced ranges. Invasive clonal plant species like knotweeds in the United States, Europe alligator weed (Alternanthera philoxeroides) in China, Spartina anglica in Europe, and water hyacinth (Eichhornia crassipes) outside of South America show almost no detectable genetic diversity even after their establishment into a new environment (Salmon et al., 2005; Gao et al., 2010), which is primarily attributed to genetic bottlenecks and founder effects (Dlugosch and Parker, 2008). The expansion and adaptive success of invasive species with clonal uniformity and narrow genetic base in novel habitats is perplexing and challenges our understanding of the process of adaptation and indicates the involvement of additional mechanisms (Richards et al., 2012). For this reason, the study of ecology and evolutionary epigenetics has lately attracted a lot of attention in invasive plants (Salmon et al., 2005; Richards et al., 2008, 2012; Gao et al., 2010; Preite et al., 2015; Foust et al., 2016; Wilschut et al., 2016; Robertson et al., 2017, 2020; Liu et al., 2018; Alvarez et al., 2020; Qiu et al., 2021; Shen et al., 2021; Su et al., 2021; Campoy et al., 2022).
To elaborate on a few instances, Shen and coworkers (2021) reported the incidence of higher epigenetic diversity compared to genetic diversity in Mikania micrantha which contributed to its invasive success. They could also predict that under global warming, the populations are expanding northward, emphasizing the usefulness of such a study in predicting future invasions. The clonal plants collected from distinct habitat types of invasive Japanese knotweed (Fallopia japonica) in the northeastern United States showed habitat-specific phenotypes when grown in common environments (Richards et al., 2008). Further, the epigenetic differentiation with MSAP to habitat type was also found in these clonal genotypes that showed no AFLP variation (Richards et al., 2012) implying thereby that epigenetic variation allows a single clonal genotype to adjust its phenotype to different environments. Induction of environment-specific epigenetic modifications is suggested by observations in Alligator weed (Alternanthera philoxeroides), an invasive weed in China that reproduces clonally through stolons. MS-AFLP analysis of natural field-grown plants showed very little genetic but high epigenetic variation. Even after 10 asexual generations, some of the epigenetic differences appeared to be induced by environmental differences (Gao et al., 2010).
The aquatic invasive plant, Ludwigia grandiflora subsp. hexapetala, is classified as being harmful to European rivers. In French wet meadows, this species has shown a rapid transition from aquatic to terrestrial environments with the emergence of two distinct morphotypes over a period of 5 years. Both genetic and epigenetic mechanisms have been involved in adjustment to the new environment, and are supposed to be possible contributors to this fast terrestrial transition. Artificial hypomethylation was induced on both morphotypes to assess the role of epigenetics. The hypomethylation of the aquatic morphotype mimicked the characteristics of the terrestrial morphotype. This suggests that DNA methylation in a new adaptive genetic capacity is playing a key role in L. grandiflora subsp. hexapetala plasticity during its rapid aquatic to terrestrial transition (Genitoni et al., 2020).
The studies conducted on epigenetic regulation of adaptations in invasive taxa have been collated in Table 4 and show that plants adapt to different environments in many ways, and genomic and epigenomic variation provides the basis for plant adaptation and invasiveness. Epigenetic variation such as induction of DNA methylation is projected to be involved in response to diverse local environments, thus representing one crucial mechanism to promote invasion success.
Epigenetic inheritance has also been observed to be influenced by reproductive strategies, for instance, pollination type, and the kind and time of events leading to germline development. In plants, late germline segregation results in a longer temporal window during which epigenetic alterations caused by the environment can be integrated. A study by Preite et al. (2018) showed that alteration in DNA methylation and ncRNA expression induced by drought was inherited for two to three generations in unexposed offspring in apomictic dandelions (Taraxacum spp.). The study underlines the potential of long-term epigenetic inheritance in species reproducing without fertilization. Similarly, self-pollinating plants show long-term persistence of epigenetic inheritance compared to cross-pollinated plants (Wang H et al., 2009). This persistence of epigenetic inheritance might be a rescue mechanism to tackle the deprivation of genetic diversity in self-pollinating plants. Self-pollination has been thought to be derived from cross-pollination and generally leads to a loss in genetic diversity in self-pollinating plants (Stebbins, 1957). The evolutionary dynamics in self-pollinated and vegetatively reproducing plants that lack a germline have remained mystifying scientific queries for a long time. Based on the recent evidence, it is reasonable to argue that there seems to be an evolutionary correlation between the presence of self-pollination and epigenetic marks across angiosperms (Figure 2). It seems that a dearth of genetic variability is compensated by substantial epigenetic structure in a plant species (Meller et al., 2018; Kuźnicki et al., 2019; Perrin et al., 2020).
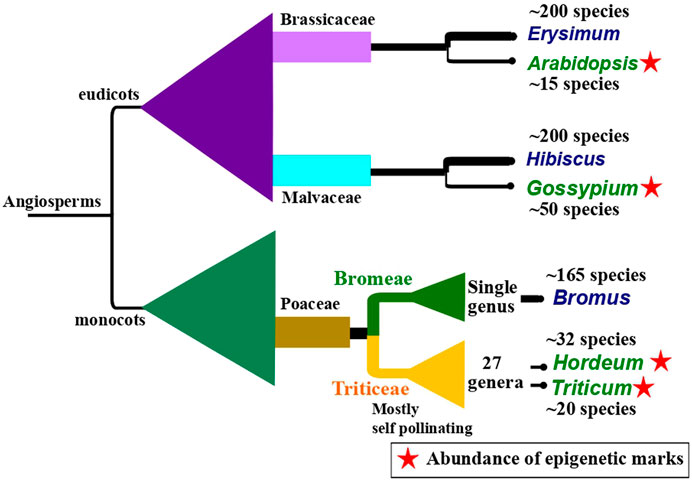
FIGURE 2. An illustration of a hypothesis of evolutionary correlation between the occurrence of self-pollinating species and the presence of abundant epigenetic marks citing three examples found across angiosperm. The illustration shows closely related pairs of sister taxa in which one is predominantly self-pollinating (taxa name in green) and the other is cross-pollinating (taxa name in blue). The predominantly self-pollinating species, namely, Arabidopsis (Klosinska et al., 2016), Gossypium (Rehman et al., 2021), Hordeum (Guo et al., 2018), and Triticum (Qiu et al., 2017) show high epigenetic marks. The phylogenetic background is drawn to show the relative systematic positions. It is generally assumed that self-pollination has evolved from cross-pollination and self-pollinating taxa have low genetic variability. Higher values in species numbers in predominantly cross-pollinating taxa might be associated with higher genetic variability in these species. The approximate species number for each taxon reported so far, has been taken from World Flora Online (World Flora Online, 2022), except for Bromus (Soreng et al., 2017).
Further, the time of germline development can also affect epigenetic patterns. The mature nuclei of pollen grains that are associated with chromatin organization showed different epigenetic marks reflecting the plants’ fertilization timing strategies. For instance, DNA cytosine hypermethylation comparison of the MROS1 gene between the vegetative and generative nuclei in pollen grains of Silene latifolia (Caryophyllaceae) indicated hypermethylation of DNA sequences in the vegetative nucleus during pollen grain maturation. Hypermethylation was essential for the long-term survival of the pollen until it reaches the stigma (Janousek et al., 2002). In contrast, a low level of DNA methylation was observed in the vegetative nuclei of Quercus suber (Fagaceae), a wind-pollinated tree, leading to active chromatin and hence a higher potential for transcription, compensating for the chromatin silencing of the generative nuclei. Further, the chromatin compaction level and transcription level of generative nuclei of Q. suber were greater than that of Lilium sps. which can be linked to differences in their pollination or fertilization processes (Ribeiro et al., 2009).
Polyploid species show increased methylated DNA, and activation of several transposons which may be essential for phenotypic divergence, adaptation, and biotic interactions (Paun et al., 2007; Fulnecek et al., 2009; Li et al., 2011; Mirouze and Paskowski, 2011; Alonso et al., 2016; Song et al., 2017; Picazo–Aragonés et al., 2020). Besides cell size, biomass, seed weight, and seed size, polyploidization is also known to affect flowering phenology between diploid and polyploid cytotypes. For example, partitioning due to ploidy differences influenced the persistence of a novel cytotype of Anacamptis pyramidalis that might otherwise have been out-competed by its diploid progenitor (Pegoraro et al., 2016). Niche differentiation of allopolyploid species relative to their progenitors could help the survival of invasive allopolyploid hybrids.
Hybridization and introgression also occur extensively in natural populations and contribute to diversification and speciation. Along with genomic changes, hybridization can result in epigenomic changes like activation of transposable elements (Kashkush and Khasdan, 2007; Rathore et al., 2020), small RNA changes, as well as changes in DNA methylation and histone modifications (Ha et al., 2009; Marfil et al., 2009; Kraitshtein et al., 2010; Zhao et al., 2011) which can lead to the establishment of new species via these stabilizing mechanisms (Feldman and Levy, 2005). In genus Spartina, the hybridization of an European native and an American hexaploid resulted in two genetically uniform hybrids that showed massive methylation changes in their genomes compared to their ancestors (Salmon et al., 2005). In general, allopolyploids show high epigenetic variation due to which many novel phenotypes are observed with increased species fitness (Jackson, 2017). It has also been reported that synthetic hybridization may induce chromosomal instability and hence changes in epigenetic patterns (Comai 2000; Li et al., 2010; Cara et al., 2013). Accumulating evidence also shows the significant contribution of epigenetics to heterosis.
Epigenetic regulation studies have been carried out in various ecological communities like grasslands, temperate deciduous forests, and alpine biomes (Table 1) to further understand the effect of contrasting habitats on ecological adaptations. In this context, alpine biomes represent extreme environments with harsh abiotic conditions such as extremely low temperatures, dryness, low oxygen concentration, strong ultraviolet (UV) radiation, and violent winds. Plant life in such habitats is challenging, as environmental influences can alter the conditions for development and reproduction. All these factors make alpine habitats a fragile ecosystem, ideal for studying the effect of the environment on epigenomes. Many studies in alpine plants have indicated that epigenetic modifications play an important role in the adaptive response to climatic changes and local adaptations (Wu et al., 2013; Zhao et al., 2014; Nicotra et al., 2015; Song et al., 2015; Alakärppä et al., 2018; Schinkel et al., 2020; Ni et al., 2021) (Table 1). Further, studies have also revealed interesting observations of epigenetic regulation of fitness and adaptability of populations in ecological communities like grasslands (Herrera and Bazaga, 2010), temperate deciduous forests (Avramidou et al., 2015; Gugger et al., 2016; Ahn et al., 2017; Vanden Broeck et al., 2018) and communities from colder continental and Mediterranean climates (Saéz-Laguna et al., 2014) (Table 1).
To study the effect of various environmental factors on the regulation of epigenetic dynamics, many studies have combined investigations involving field screenings of natural populations in contrasting habitats. Epigenetic variations have been enlisted in populations of selected species under ecological habitats of salt-marsh conditions against the riverside habitats or contrasting altitudes, soil, water, temperature, or rainfall conditions (Table 2). Efforts have also been done to study epigenetic regulation during various biological processes and functional traits’ development in natural plant populations (Table 3).
All the above settings have provided valuable insights into epigenetic differentiation and the role of epigenetic mechanisms in regulating trait variations. An exhaustive collation of epigenetic research conducted in natural populations in relation to various biological processes, ecological factors, communities, mating types, reproductive behaviors, and habitats will help provide a better understanding of how epigenetics can get affected by environmental stimuli and regulate phenotypic plasticity and adaptive responses during changing climates. The findings strongly propound the involvement of epigenetic regulation of phenotypic plasticity induced by environmental stimuli and substantiate the viewpoints that epigenetics plays a major role in the adaptive evolution of natural populations alone or in conjunction with genetic variability.
As evident, natural populations have been observed to show distinct genetic and epigenetic population structures. Even when there is low genetic variation in the population, higher epigenetic variation could be observed and vice versa. In some instances, epigenetic variation aligned more strongly with habitat than genetic variation indicating the link between DNA methylation patterns and the environment. We hypothesize that the interaction between the reproductive strategy of the population and its inhabited environment is crucial in determining whether the genetic and epigenetic variability exists as a balancing determiner through trade-offs or reinforce each other through the contributions of both mutations and epimutations. The plant strategizes to invest predominantly in asexual reproduction, self-pollination, or cross-pollination based on the inhabited environment that could be stable (homogenous or heterogenous) or unstable. Based on these factors, genetic and epigenetic variability finds a balance for providing a competitive edge to the plant, and finally these two pools of variability either show a trade-off or reinforcement with each other (Figure 3).
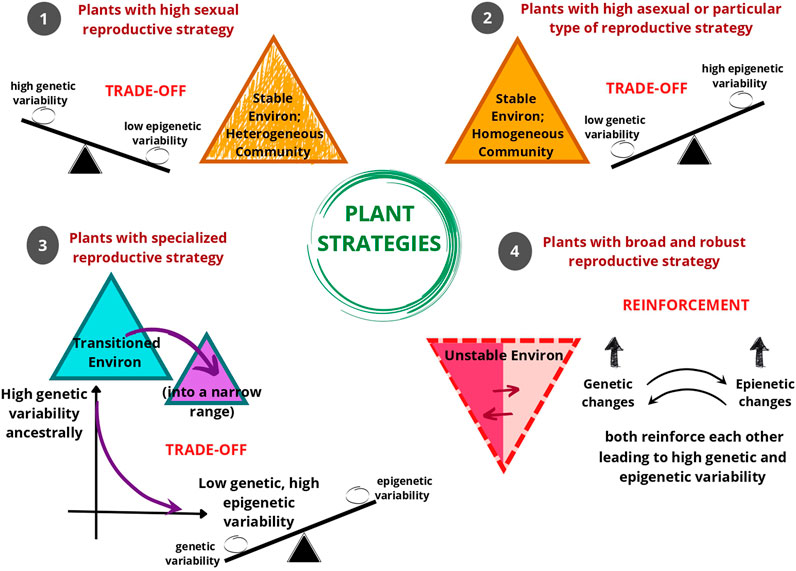
FIGURE 3. An illustration of the hypothesis that as a response to environmental conditions, different reproductive strategies of plants are associated with the coordination of genetic and epigenetic variability within the gene pool. Mostly, plant reproductive strategies show trade-offs, i.e. balancing of genetic and epigenetic variability. Environmental conditions (or Environ) 1, 2, and 3 show a trade-off between genetic and epigenetic variability in the gene pool. In Environment (1), the environment is stable with a heterogenous plant community, where plants generally tend to reproduce sexually. In this type of environment, the plant tends to have high genetic variability and low epigenetic variability. In Environment (2), the environment is stable with a homogeneous plant community, where plants generally tend to reproduce asexually or only through self-pollination. In this type of environment, the plant tends to have high epigenetic variability and low genetic variability. In Environment (3), when the plant transitions into an environment that is peculiar in some characteristic then the plant tends to perform specialized reproductive strategy suited for that narrow-ranged environment. In this type of environment too, there is a trend of a decrease in genetic variability and an increase in epigenetic variability. In Environment (4) opportunistic or invasive plants with diverse (i.e. broad) and robust reproductive responses can survive in unstable or fluctuating environments, here the genetic variability and epigenetic variability reinforce each other.
Epigenetics plays an important role in developing strategies for crop breeding and domestication through the selection of elite crop varieties by generating more phenotypes. The induction of novel epi-alleles as plants respond to changing climates provides an additional tier of epigenetic diversity to select desirable crop traits and improve resilience. This is important as the genetic base of most of today’s crop cultivars has become narrow due to recurrent selections (Gallusci et al., 2017). The availability of sufficient variability in the gene pools is pivotal for executing meaningful and successful crop improvement programs to ameliorate various biotic and abiotic stresses arising due to the changing stressful global climates (Samantara et al., 2021; Krishnaswami et al., 2022). Stress exposure cascades a response-signaling cycle in plants that along with genetic modifications leads to many epigenetic changes routed through mechanisms such as chromatin remodeling, DNA methylation, histone modification, and non-coding RNAs (Turcotte et al., 2022). These epigenetic mechanisms have resulted in novel epialleles associated with morphological, metabolic, and developmental traits in plants (Tirnaz and Batley 2019) that can be exploited for developing new climate-resilient crop varieties to mitigate the challenges of changing environments (Springer and Schmitz, 2017; Kawakatsu and Ecker, 2019; Varotto et al., 2020; Kakoulidou et al., 2021; Guarino et al., 2022) and help in crop domestication by targeting epigenetic diversity to engineer crop plants for optimal crop production (Ding and Chen, 2018).
The epigenetic variability has shown tremendous potential for the selection of agronomic traits and QTLs in Arabidopsis and other important crops like rice, rapeseed, soybean, cotton, maize, and tomato. The bilateral symmetry of flower development in Linaria cycloidea, FWA locus involved in regulating flowering time in Arabidopsis, colorless non-ripening locus in tomato, vitamin E accumulation in tomato, improved anthocyanin production in apple, dwarf plant stature in rice, sex determination in melon, slower fruit ripening in mature tomato, somaclonal variation in oil palm, increased embryogenesis in pineapple, high heterosis in pigeon pea, altered oil content in oilseed rape, enhanced yield in soybean and adaptation of upland domesticated rice in response to osmotic stress among others exemplify some of the epigenetic variants showing useful traits in crops (Kakutani, 1997; Cubas et al., 1999; Manning et al., 2006; Martin et al., 2009; Miura et al., 2009; Telias et al., 2011; Zhong et al., 2013; Nonogaki, 2014; Quadrana et al., 2014; van Zanten et al., 2014; Ong-Abdullah et al., 2015; Xia et al., 2016; Ding and Chen, 2018; Luan et al., 2020; Sinha et al., 2020).
In rice, changes in DNA methylation and/or histone methylation and acetylation patterns have resulted in many epialleles linked to desired crop traits like increased panicle branching and yield (OsSPL14) (Miura et al., 2010), delayed flowering (SDG724 induced loss of function mutant Ivp1), increase in grain size, weight, height and yield (down- and upregulation of the genes OsLAC and OsglHAT1, respectively) (Song et al., 2015) and grain yield (OsPCF7) (Li et al., 2020). Likewise, while an increase in DNA methylation mitigated cold stress in tomato, heat and salinity stress in rapeseed, drought stress in faba bean and controlled photosynthetic activity in rice (Wei et al., 2017; Samantara et al., 2021), the hypomethylation of ESP gene regulated the improved panicle architecture in rice (Luan et al., 2019). The methylation of histone H3 lysine 36 (H3K36), acetylation of H3K9, and histone H3 lysine 27 trimethylation (H3K27me) have played key roles in bringing about many epigenetic changes in rice (Gupta and Salgotra 2022). Similarly, alterations in H3K4me2 in maize helped in mitigating abiotic and biotic stresses (Samantara et al., 2021) and histone acetylation was shown to regulate drought stress in tomato and Arabidopsis (González et al., 2013; Zheng et al., 2016). It has been demonstrated that histone deacetylase HDA9 plays a key role in regulating drought stress in plants (Turcotte et al., 2022). Further, miRNAs regulated the expression of the OsSPL14 gene affecting the panicle branching and increasing the rice yield (Miura et al., 2010).
Further, a comprehensive epigenomic and functional analysis of domesticated allotetraploid cotton and their tetraploid and diploid relatives revealed differentially methylated cytosines regions that contribute to domestication traits such as flowering time and seed dormancy in cotton (Song et al., 2017). In another study, domestication was exhibited through DMRs using 45 soybean accessions. Interestingly, no association was observed with the genetic variation enriched in carbohydrate metabolic pathways (Shen et al., 2018). In rice, flowering time is crucial for seed set which is regulated by histone methyltransferase (HMTase) genes such as SET DOMAIN GENE 724 (SDG724) that promotes flowering by methylating the histone H3 lysine 36 (H3K36) (Sun et al., 2012; Gupta and Salgotra, 2022). The useful epigenetic diversity in many other crops has been collated well in recent review articles (Varotto et al., 2020; Kakoulidou et al., 2021; Alfalahi et al., 2022; Gill et al., 2022; Guarino et al., 2022; Gupta and Salgotra 2022; Sharma et al., 2022; Villagómez-Aranda et al., 2022). All these studies suggest that the identification of agronomic traits regulated through epigenetics will provide new opportunities for crop improvement and domestication.
Since it has been established that the epigenetic mechanisms aid plants in stressed conditions (Chachar et al., 2022), efforts are underway to understand the mechanism of epigenetic enzymatic systems that help in recognizing, installing, and erasing the epigenetic marks. The availability of cutting-edge sequencing technologies coupled with computing methods like machine learning and deep learning is geared towards decoding the role of epigenetic regulation in crop species (Ali et al., 2022). Although many instances of spontaneous epi-allelic diversity have been documented, efforts are also underway to induce epigenetic diversity in crops. In this context, ‘Eustressors’ that involve low-dose exposures to stress factors to trigger positive responses in plants through effects on their physiology, biochemistry, genetics, and epigenetics have emerged as potent breeding tools (Villagómez-Aranda et al., 2022). This is an emerging area of research and future efforts aimed at identifying eustressors capable of generating stable epigenetic marks, understanding mechanisms leading to trans-generational memory to stimulate a priming state and their adaptability potential are essential before they can be used to induce epigenetic variability for the advancement of important agronomic traits in crop plants. In the nutshell, emerging epigenetics-based technologies highlight the prospects of novel epi-alleles to broaden the genetic base of crop plants and open vistas to use epi-variability to support future crop epi-breeding and domestication programs. Integrating epigenetic methods into traditional and modern crop breeding and domestication exercises can serve as an expanded toolbox to address the agricultural productivity challenges.
Traditionally, the study of epigenetic changes has been based on DNA methylation profiling, methylation-sensitive amplification polymorphism (MSAP) (Waalwijk and Flavell, 1978), and bisulfite sequencing (Cokus et al., 2008; Kurdyukov and Bullock, 2016), though the advent of techniques like chromatin immunoprecipitation (ChIP), next generation sequencing (NGS), and bisulfite reduction sequencing (bsRAD) (Trucchi et al., 2016) in the recent years has changed the scenario of data generation in studying epigenetic aspects. Significant efforts have been undertaken for technological simplification to study DNA methylation changes in natural populations or species with large genomes (Chwialkowska et al., 2017). The methods used in epigenetic studies are listed in Table 6.

TABLE 6. Commonly used NGS-based techniques for epigenetic analysis.
Of all the methods discovered so far, MSAP remains the most widely used due to the associated ease of performance and the simplicity of the technique. It is similar to amplified fragment length polymorphism (AFLP) in procedure but uses two isoschizomers HpaII and Msp1, that differ in their sensitivity to the methylation state of cytosine (Waalwijk and Flavell, 1978). The advent of NGS has allowed MSAP to become a high-throughput method known as methylation-sensitive amplification polymorphism sequencing (MSAP-Seq) (Chwialkowska et al., 2017). However, poor efficiency of the restriction enzymes to cut at CHH and CHG contexts than CG context is a limitation associated with this technique as this can portray an incorrect methylation status of the genome.
The bisulfite treatment is the next most widely used technology after MSAP. It is used for the detection of cytosine methylation both at specific loci and genome-wide levels. Bisulfite treatment of genomic DNA results in the deamination of unmethylated cytosines to uracil but methylated cytosines are not prone to this treatment and still read as C and can be distinguished from unmethylated cytosines (Jones, 2012). The downstream analysis after bisulfite treatment can be done by using several methods such as direct sequencing of bisulfite PCR product, sub-cloning of PCR product followed by sequencing, combined bisulfite restriction analysis (COBRA), methylation-specific PCR (MSP), and pyrosequencing (Li and Tollefsbol, 2011).
The advent of NGS has made it possible to look at DNA methylation changes at the whole genome level using whole genome bisulfite sequencing (WGBS) (Meissner et al., 2008; Lister et al., 2009). However, the required depth becomes a bottleneck for large, complex genomes, resulting in increased costs. To address this problem, a method like reduced representation bisulfite sequencing (RRBS) was developed, where an enzyme is used to target a specific portion of the genome, which often is an enzyme like MspI, which targets 5′CCGG3′ sites often found in promoters (Meissner et al., 2005). RRBS allows working with large genome sizes as it constitutes genome fragmentation that reduces the complexity of genomes (Davey et al., 2011).
The methods have also been developed to target particular sequences for DNA methylation analysis such as bisulfite padlock probes (BSPP) (Diep et al., 2012). Although the bisulfite-based NGS methods have provided a single base resolution of epigenetic changes, it fails to provide information on specific cells when it comes to complex tissues. In this context, the recently developed, single-cell bisulfite sequencing (Smallwood et al., 2014) technique provides whole epigenetic insights into individual cells.
The utility of bisulfite sequencing in natural populations became possible after adapting the RADseq and GBS protocols to procure the methylation profile of reduced genome libraries for species where a reference genome is not available. Three RRBS protocols namely epiRAseq (Schield et al., 2016), bsRADseq (Trucchi et al., 2016), and epiGBS (van Gurp et al., 2016) find great utility in this context. Methylation-sensitive, MS-DArT (Pereira et al., 2020a) has emerged as another promising technique to study DNA methylation profiling. It is a genotyping technology that involves double digestion preceded by adaptor ligation and NGS. The technology is attractive and amenable to use for any species as the availability of a reference genome is not imperative, though it is useful to establish the genomic context of methylated bases.
Another technique for accurately studying DNA methylation has been through the deployment of antibodies developed against 5-methylcytidine (Mohn et al., 2009). This method is called Methylated DNA immunoprecipitation (MeDIP) which can be combined with simple PCR, microarrays, or NGS to create a powerful tool for the unbiased detection of methylated DNA. MeDIP can be combined with MRE-seq (methylation-sensitive restriction enzyme sequencing) for more accuracy in methylation studies (Maunakea et al., 2010). MRE-seq involves fragmentation of DNA using methylation-sensitive restriction enzymes followed by sequencing. Methyl-binding domain isolated genome sequencing (MBDiGs) is another technique that involves recombinant Methyl Binding Domain (MBD) and MBD2 proteins for the enrichment of methyl-rich DNA fragments (Serre et al., 2010).
Antibodies have also been used to develop various other methods for epigenetic analysis to look at DNA-protein interaction and protein modifications including those present in histone proteins. Chromatin immunoprecipitation (ChIP) can be used to identify protein modifications and DNA-protein interactions. It involves in vivo cross-linking of proteins to DNA with formaldehyde and then isolation of crosslinked chromatin from nuclei. The isolated chromatin is further sheared into smaller fragments followed by immunoprecipitation with the help of specific antibodies. The isolated DNA with this procedure can be investigated with specific primers or combined with high throughput NGS creating powerful tools like ChIP-Seq (Diaz et al., 2017). Various useful modifications like sequential ChIP where QPCR and ChIP are used in combination to assess the modifications present on histone proteins H3 and H4 on the target location (Tollefsbol, 2011) and ChlP-chip/ChlP-microarray have been added. Coupled with whole genome microarrays, ChlPs allow the determination of all DNA-binding sites of any protein.
The latest innovations in technology development are anticipated to change the dynamics of DNA methylation research investigations in the future. Efforts have been undertaken to develop methods to study epigenomes where high-quality reference genome sequences are not available. One such transformational technology launched recently (April 2022) is Sequel II/IIe Platform which includes methylation calling in native DNA (MCND) at the same time sequencing is done (https://www.pacb.com). Moreover, the expression of small RNAs can be studied through RNA-seq, RT-qPCRs northern blot, and in situ hybridization (Wang Z et al., 2009). All these techniques enable the study of expression and identification of small RNAs and their target predictions. Databases of ncRNAs sequences and their functional annotation from plants like CANTATAdb 2.01 (Szcześniak et al., 2016) with over 200,000 lncRNAs from 39 plant species, MiRbase and TAIR (Rhee et al., 2003; Kozomara et al., 2019) have been very important resources.
Diverse studies on natural populations of plants including investigations in 1) species from various ecological communities, 2) model and non-model species, 3) species of contrasting habits, 4) species from extreme habitats, 5) species from varied climatic gradients, 6) species with asexual reproduction, and 7) invasive taxa have empirically pointed out the involvement of epigenetic phenomenon in facilitating adaptative ability in plants to cope with the changing environments. Epigenetic mechanisms, either individually or in accompaniment with genetic variation, represent long-term stress responses as compared to transient metabolic and physiological modifications. While short-term responses ensure survival, long-term genetic and epigenetic changes contribute toward plastic and adaptive responses of evolutionary significance. The incidence of phenotypic plasticity in genetically homogeneous populations strongly supports its epigenetic ontogeny, although the causal relationship between the stresses and plasticity still remains to be deciphered. The ability to modulate gene expression to produce stable and heritable alternate phenotypes that affect reproductive success strongly suggests the role of epigenetics in developing adaptability in natural populations in changing and stressful climates. Furthermore, epiallelic transgenerational diversity resulting in novel phenotypes seems to play a major role in helping plants to adapt to new environments by aiding them in expanding their habitat range.
To sum up, evidence indicates that 1) epigenetic marks lead to phenotypic changes in plants and serve as important drivers to rapidly widen the total phenotypic plasticity and phenotypic variance of natural populations, 2) plant epigenomes show high diversity and rapidly generate responses to external stimuli, 3) epigenetic mechanisms help to provide the required variability for selection and expand the plants’ repertoire of adaptive responses 4) epi-variability creates epialleles, that serve as an additional tier of diversity for adaptive evolution 5) epigenetic variability is amenable to selection and may eventually become heritable, 6) epigenetic and genetic fields may complement each other for better execution of phenotypic plasticity and adaptive responses and 7) various biological processes, ecological factors and habitats strongly influence the epigenetic regulatory processes in natural populations (Robertson and Wolf 2012; Trucchi et al., 2016; Amaral et al., 2020; Ashapkin et al., 2020; Ashe et al., 2021; Baduel et al., 2021; Boquete et al., 2021; Qiu et al., 2021; García–García et al., 2022). It has been shown that both genetic and epigenetic variations contribute to environmental stress-induced heritable phenotypic divergence. Many studies have shown higher epigenetic variability than genetic variation (Medrano et al., 2014; Foust et al., 2016; Guo et al., 2018; Wang et al., 2020). Although the population-level studies of epigenetic variability in species with high genetic diversity distributed across different habitats are limited (Robertson et al., 2017; Qiu et al., 2021), future studies in such species would further enhance our knowledge of the interactions between genetic and epigenetic domains of the genome that coordinate adaptive responses.
Conceptualization and methodology: VRR and SNR. Data collection: PR, SM, NW, and PY under the guidance of VRR. Data analysis: PR, SM, NW, and PY under the guidance of VRR, SG, VB, and SNR. Original draft preparation: PR, SM, NW, PY, and EB. Review and editing: VRR, SG, VB, EB, and SNR. Draft finalization: VRR, Project administration: VRR and SNR. All authors listed have made a substantial and intellectual contribution to the manuscript writing, reviewing, and editing. All authors have read and approved the publication of the manuscript in its current form.
VRR acknowledges a research grant support number BT/PR34491/NDB/39/678/2020 provided by the Department of Biotechnology (DBT), Government of India. SG acknowledges the lab support through the grant provided by the Department of Biotechnology, Government of India under the sanction orders “BT/PR29259/FCB/125/2/2018 and grant provided by the University of Delhi under the scheme, “Institute of Eminence, sanction order IoE-DU/MRP/2022/056. EB is supported by a Maharishi Kanad Postdoctoral Fellowship, Institution of Eminence, University of Delhi.
The authors would like to thank the editors as well as reviewers for their critical comments that helped to improve the MS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ahn, J., Franklin, S. B., and Douhovnikoff, V. (2017). Epigenetic variation in clonal stands of aspen. Folia Geobot. 52, 443–449. doi:10.1007/s12224–017–9308–x
Alakärppä, E., Salo, H. M., Valledor, L., Cañal, M. J., Häggman, H., and Vuosku, J. (2018). Natural variation of DNA methylation and gene expression may determine local adaptations of Scots pine populations. J. Exp. Bot. 69, 5293–5305. doi:10.1093/jxb/ery292
Alfalahi, A. O., Hussein, Z. T., Khalofah, A., Sadder, M. T., Qasem, J. R., Al-Khayri, J. M., et al. (2022). Epigenetic variation as a new plant breeding tool: A review. J. King Saud Univ. - Sci. 34, 102302. doi:10.1016/j.jksus.2022.102302
Ali, S., Khan, N., and Tang, Y. (2022). Epigenetic marks for mitigating abiotic stresses in plants. J. Plant Physiol. 275, 153740. doi:10.1016/j.jplph.2022.153740
Alonso, C., Balao, F., Bazaga, P., and Pérez, R. (2016). Epigenetic contribution to successful polyploidizations: Variation in global cytosine methylation along an extensive ploidy series in dianthus broteri (Caryophyllaceae). New Phytol. 212, 571–576. doi:10.1111/nph.14138
Alonso, C., Ramos-Cruz, D., and Becker, C. (2019). The role of plant epigenetics in biotic interactions. New Phytol. 221, 731–737. doi:10.1111/nph.15408
Alvarez, M., Robertson, M., van Gurp, T., Wagemaker, N., Giraud, D., Ainouche, M. L., et al. (2020). Reduced representation characterization of genetic and epigenetic differentiation to oil pollution in the foundation plant Spartina alterniflora. bioRxiv 426569. doi:10.1101/426569
Amaral, M. N., Auler, P. A., Rossatto, T., Barros, P. M., Oliveira, M. M., and Braga, E. J. B. (2020). Long–term somatic memory of salinity unveiled from physiological, biochemical and epigenetic responses in two contrasting rice genotypes. Physiol. Plant. 170, 248–268. doi:10.1111/ppl.13149
Amaral, J., Ribeyre, Z., Vigneaud, J., Sow, M. D., Fichot, R., Messier, C., et al. (2020). Advances and promises of epigenetics for forest trees. Forests 11, 976. doi:10.3390/f11090976
Anamika, , , Mehta, S., Singh, B., Patra, A., and Islam, M. (2019). in Databases: A weapon from the arsenal of bioinformatics for plant abiotic stress research” in recent approaches in omics for plant resilience to climate change. Editor S. Wani (Cham: Springer), 135–169. doi:10.1007/978–3–030–21687–0_7
Aparicio, F., Thomas, C. L., Lederer, C., Niu, Y., Wang, D., and Maule, A. J. (2005). Virus induction of heat shock protein 70 reflects a general response to protein accumulation in the plant cytosol. Plant Physiol. 138, 529–536. doi:10.1104/pp.104.058958
Ariel, F. D., and Manavella, P. A. (2021). When junk DNA turns functional: Transposon–derived non–coding RNAs in plants. J. Exp. Bot. 72, 4132–4143. doi:10.1093/jxb/erab073
Ashapkin, V. V., Kutueva, L. I., Aleksandrushkina, N. I., and Vanyushin, B. F. (2020). Epigenetic mechanisms of plant adaptation to biotic and abiotic stresses. Int. J. Mol. Sci. 21, 7457. doi:10.3390/ijms21207457
Ashe, A., Colot, V., and Oldroyd, B. P. (2021). How does epigenetics influence the course of evolution? Philos. Trans. R. Soc. Lond. B Biol. Sci. 376, 20200111. doi:10.1098/rstb.2020.0111
Avramidou, E. V., Ganopoulos, I. V., Doulis, A. G., Tsaftaris, A. S., and Aravanopoulos, F. A. (2015). Beyond population genetics: Natural epigenetic variation in wild cherry (Prunus avium). Tree Genet. Genomes 11, 95–99. doi:10.1007/s11295–015–0921–7
Baduel, P., and Colot, V. (2021). The epiallelic potential of transposable elements and its evolutionary significance in plants. Philos. Trans. R. Soc. Lond. B Biol. Sci. 376, 20200123. doi:10.1098/rstb.2020.0123
Baduel, P., Leduque, B., Gil, J., Loudet, O., Vincent, C., Quadrana, L., et al. (2021). Genetic and environmental modulation of transposition shapes the evolutionary potential of Arabidopsis thaliana. Genome Biol. 22, 138. doi:10.1186/s13059–021–02348–5
Baek, D., Jiang, J., Chung, J. S., Wang, B., Chen, J., Xin, Z., et al. (2011). Regulated AtHKT1 gene expression by a distal enhancer element and DNA methylation in the promoter plays an important role in salt tolerance. Plant Cell Physiol. 52, 149–161. doi:10.1093/pcp/pcq182
Bartels, A., Han, Q., Nair, P., Stacey, L., Gaynier, H., Mosley, M., et al. (2018). Dynamic DNA methylation in plant growth and development. Int. J. Mol. Sci. 19, 2144. doi:10.3390/ijms19072144
Bastow, R., Mylne, J. S., Lister, C., Lippman, Z., Martienssen, R. A., and Dean, C. (2004). Vernalization requires epigenetic silencing of FLC by histone methylation. Nature 427, 164–167. doi:10.1038/nature02269
Bäurle, I., and Trindade, I. (2020). Chromatin regulation of somatic abiotic stress memory. J. Exp. Bot. 71, 5269–5279. doi:10.1093/jxb/eraa098
Beldade, P., Mateus, A. R. A., and Keller, R. A. (2011). Evolution and molecular mechanisms of adaptive developmental plasticity. Mol. Ecol. 20, 1347–1363. doi:10.1111/j.1365–294X.2011.05016.x
Bond, D. M., and Baulcombe, D. C. (2014). Small RNAs and heritable epigenetic variation in plants. Trends Cell Biol. 24, 100–107. doi:10.1016/j.tcb.2013.08.001
Boquete, M. T., Muyle, A., and Alonso, C. (2021). Plant epigenetics: Phenotypic and functional diversity beyond the DNA sequence. Am. J. Bot. 108, 553–558. doi:10.1002/ajb2.1645
Bossdorf, O., Arcuri, D., Richards, C. L., and Pigliucci, M. (2010). Experimental alteration of DNA methylation affects the phenotypic plasticity of ecologically relevant traits in Arabidopsis thaliana. Evol. Ecol. 24, 541–553. doi:10.1007/s10682-010-9372-7
Bossdorf, O., Richards, C. L., and Pigliucci, M. (2008). Epigenetics for ecologists. Ecol. Lett. 11, 106–115. doi:10.1111/j.1461–0248.2007.01130.x
Bräutigam, K., Vining, K. J., Lafon-Placette, C., Fossdal, C. G., Mirouze, M., Marcos, J. G., et al. (2013). Epigenetic regulation of adaptive responses of forest tree species to the environment. Ecol. Evol. 3, 399–415. doi:10.1002/ece3.461
Browne, L., MacDonald, B., Fitz–Gibbon, S., Wright, J. W., and Sork, V. L. (2021). Genome–wide variation in DNA methylation predicts variation in leaf traits in an ecosystem–foundational oak species. Forests 12, 569. doi:10.3390/f12050569
Bruce, T. J., Matthes, M. C., Napier, J. A., and Pickett, J. A. (2007). Stressful “memories” of plants: Evidence and possible mechanisms. Plant Sci. 173, 603–608. doi:10.1016/j.plantsci.2007.09.002
Campoy, J. G., Sobral, M., Carro, B., Lema, M., Barreiro, R., and Retuerto, R. (2022). Epigenetic and phenotypic responses to experimental climate change of native and invasive Carpobrotus edulis. Front. Plant Sci. 13, 888391. doi:10.3389/fpls.2022.888391
Cara, N., Marfil, C. F., and Masuelli, R. W. (2013). Epigenetic patterns newly established after interspecific hybridization in natural populations of Solanum. Ecol. Evol. 3, 3764–3779. doi:10.1002/ece3.758
Chachar, S., Chachar, M., Riaz, A., Shaikh, A. A., Li, X., Li, X., et al. (2022). Epigenetic modification for horticultural plant improvement comes of age. Sci. Hortic. 292, 110633. doi:10.1016/j.scienta.2021.110633
Charlesworth, B., and Charlesworth, D. (2018). Neutral variation in the context of selection. Mol. Biol. Evol. 35, 1359–1361. doi:10.1093/molbev/msy062
Charlesworth, B., and Charlesworth, D. (2017). Population genetics from 1966 to 2016. Heredity 118, 2–9. doi:10.1038/hdy.2016.55
Chen, C., Zheng, Z., Bao, Y., Zhang, H., Richards, C. L., Li, J., et al. (2020). Comparisons of natural and cultivated populations of Corydalis yanhusuo indicate divergent patterns of genetic and epigenetic variation. Front. Plant Sci. 11, 985. doi:10.3389/fpls.2020.00985
Choi, C. S., and Sano, H. (2007). Abiotic–stress induces demethylation and transcriptional activation of a gene encoding a glycerophosphodiesterase–like protein in tobacco plants. Mol. Genet. Genomics 277, 589–600. doi:10.1007/s00438–007–0209–1
Chwialkowska, K., Korotko, U., Kosinska, J., Szarejko, I., and Kwasniewski, M. (2017). Methylation sensitive amplification polymorphism sequencing (MSAP–Seq)—a method for high–throughput analysis of differentially methylated CCGG sites in plants with large genomes. Front. Plant Sci. 8, 2056. doi:10.3389/fpls.2017.02056
Ci, D., Song, Y., Du, Q., Tian, M., Han, S., and Zhang, D. (2016). Variation in genomic methylation in natural populations of Populus simonii is associated with leaf shape and photosynthetic traits. J. Exp. Bot. 67, 723–737. doi:10.1093/jxb/erv485
Cokus, S. J., Feng, S., Zhang, X., Chen, Z., Merriman, B., Haudenschild, C. D., et al. (2008). Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452, 215–219. doi:10.1038/nature06745
Comai, L. (2000). Genetic and epigenetic interactions in allopolyploid plants. Plant Mol. Biol. 43, 387–399. doi:10.1023/a:1006480722854
Cong, W., Miao, Y., Xu, L., Zhang, Y., Yuan, C., Wang, J., et al. (2019). Transgenerational memory of gene expression changes induced by heavy metal stress in rice (Oryza sativa L.). BMC Plant Biol. 19, 282. doi:10.1186/s12870–019–1887–7
Cubas, P., Vincent, C., and Coen, E. (1999). An epigenetic mutation responsible for natural variation in floral symmetry. Nature 401, 157–161. doi:10.1038/43657
Dar, F. A., Mushtaq, N. U., Saleem, S., Rehman, R. U., Dar, T. U. H., and Hakeem, K. R. (2022). Role of epigenetics in modulating phenotypic plasticity against abiotic stresses in plants. Int. J. Genomics 2022, 1092894. doi:10.1155/2022/1092894
Davey, J. W., Hohenlohe, P. A., Etter, P. D., Boone, J. Q., Catchen, J. M., and Blaxter, M. L. (2011). Genome–wide genetic marker discovery and genotyping using next–generation sequencing. Nat. Rev. Genet. 12, 499–510. doi:10.1038/nrg3012
David, S. A., Mersch, M., Foissac, S., Collin, A., Pitel, F., and Coustham, V. (2017). Genome–wide epigenetic studies in chicken: A review. Epigenomes 1, 20. doi:10.3390/epigenomes1030020
Diaz, R. E., Sanchez, A., Anton Le Berre, V., and Bouet, J. Y. (2017). High–resolution chromatin immunoprecipitation: ChIP–sequencing” in the bacterial nucleoid: Methods in molecular Biology. Editor O. Espéli (New York: Humana Press), 61–73. Vol. 1624. doi:10.1007/978–1–4939–7098–8_6
Diep, D., Plongthongkum, N., Gore, A., Fung, H. L., Shoemaker, R., and Zhang, K. (2012). Library–free methylation sequencing with bisulfite padlock probes. Nat. Methods 9, 270–272. doi:10.1038/nmeth.1871
Ding, L., Wang, S., Song, Z. T., Jiang, Y., Han, J. J., Lu, S. J., et al. (2018). Two B-box domain proteins, BBX18 and BBX23, interact with ELF3 and regulate thermomorphogenesis in Arabidopsis. Cell Rep. 25, 1718–1728. doi:10.1016/j.celrep.2018.10.060
Ding, M., and Chen, Z. J. (2018). Epigenetic perspectives on the evolution and domestication of polyploid plant and crops. Curr. Opin. Plant Biol. 42, 37–48. doi:10.1016/j.pbi.2018.02.003
Ding, Y., Fromm, M., and Avramova, Z. (2012). Multiple exposures to drought ‘train' transcriptional responses in Arabidopsis. Nat. Commun. 3, 740. doi:10.1038/ncomms1732
Dlugosch, K. M., and Parker, I. M. (2008). Invading populations of an ornamental shrub show rapid life history evolution despite genetic bottlenecks. Ecol. Lett. 11, 701–709. doi:10.1111/j.1461–0248.2008.01181.x
Dubin, M. J., Zhang, P., Meng, D., Remigereau, M. S., Osborne, E. J., Casale, F. P., et al. (2015). DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. elife 4, e05255. doi:10.7554/eLife.05255
Dupont, C., Armant, D. R., and Brenner, C. A. (2009). Epigenetics: Definition, mechanisms and clinical perspective. Semin. Reprod. Med. 27, 351–357. doi:10.1055/s–0029–1237423
Eichten, S. R., Briskine, R., Song, J., Li, Q., Swanson-Wagner, R., Hermanson, P. J., et al. (2013). Epigenetic and genetic influences on DNA methylation variation in maize populations. Plant Cell 25, 2783–2797. doi:10.1105/tpc.113.114793
Erdmann, R. M., and Picard, C. L. (2020). RNA–directed DNA methylation. PLoS Genet. 16, e1009034. doi:10.1371/journal.pgen.1009034
Fang, H., Liu, X., Thorn, G., Duan, J., and Tian, L. (2014). Expression analysis of histone acetyltransferases in rice under drought stress. Biochem. Biophys. Res. Commun. 443, 400–405. doi:10.1016/j.bbrc.2013.11.102
Feinberg, A. P., and Irizarry, R. A. (2010). Evolution in health and medicine Sackler colloquium: Stochastic epigenetic variation as a driving force of development, evolutionary adaptation, and disease. Proc. Natl. Acad. Sci. U.S.A. 107, 1757–1764. doi:10.1073/pnas.0906183107
Feldman, M., and Levy, A. A. (2005). Allopolyploidy – A shaping force in the evolution of wheat genomes. Cytogenet. Genome Res. 109, 250–258. doi:10.1159/000082407
Foust, C. M., Preite, V., Schrey, A. W., Alvarez, M., Robertson, M. H., Verhoeven, K. J. F., et al. (2016). Genetic and epigenetic differences associated with environmental gradients in replicate populations of two salt marsh perennials. Mol. Ecol. 25, 1639–1652. doi:10.1111/mec.13522
Fulnecek, J., Matyásek, R., and Kovarík, A. (2009). Faithful inheritance of cytosine methylation patterns in repeated sequences of the allotetraploid tobacco correlates with the expression of DNA methyltransferase gene families from both parental genomes. Mol. Genet. Genomics 281, 407–420. doi:10.1007/s00438–008–0420–8
Gac, A. L., Lafon–Placette, C., Chauveau, D., Segura, V., Delaunay, A., Fichot, R., et al. (2018). Winter–dormant shoot apical meristem in poplar trees shows environmental epigenetic memory. J. Exp. Bot. 69, 4821–4837. doi:10.1093/jxb/ery271
Gallusci, P., Dai, Z., Génard, M., Gauffretau, A., Leblanc-Fournier, N., Richard-Molard, C., et al. (2017). Epigenetics for plant improvement: Current knowledge and modeling avenues. Trends Plant Sci. 22, 610–623. doi:10.1016/j.tplants.2017.04.009
Ganguly, D. R., Crisp, P. A., Eichten, S. R., and Pogson, B. J. (2017). The Arabidopsis DNA methylome is stable under transgenerational drought stress. Plant Physiol. 175, 1893–1912. doi:10.1104/pp.17.00744
Gao, G., Ying, F., Chen, B., Li, H., Yan, G., Xu, K., et al. (2011). DNA methylation of seed in response to heat stress in Brassica rapa L. A. A. S. 37, 1597–1604. doi:10.3724/SP.J.1006.2011.01597
Gao, L., Geng, Y., Li, B. O., Chen, J., and Yang, J. I. (2010). Genome wide DNA methylation alterations of Alternanthera philoxeroides in natural and manipulated habitats: Implications for epigenetic regulation of rapid responses to environmental fluctuation and phenotypic variation. Plant Cell Environ. 33, 1820–1827. doi:10.1111/j.1365–3040.2010.02186.x
García–García, I., Méndez–Cea, B., Martín–Gálvez, D., Seco, J. I., Gallego, F. J., and Linares, J. C. (2022). Challenges and perspectives in the epigenetics of climate change–induced forests decline. Front. Plant Sci. 12, 797958. doi:10.3389/fpls.2021.797958
Genitoni, J., Vassaux, D., Delaunay, A., Citerne, S., Portillo Lemus, L., Etienne, M. P., et al. (2020). Hypomethylation of the aquatic invasive plant, Ludwigia grandiflora subsp. hexapetala mimics the adaptive transition into the terrestrial morphotype. Physiol. Plant. 170, 280–298. doi:10.1111/ppl.13162
Gill, S. S., Macovei, A., Jimenez-Bremont, J. F., Khan, N. A., and Tuteja, N. (2022). Biotechnology and crop improvement under changing environment: Current interventions. Plant gene. 32, 100376. doi:10.1016/j.plgene.2022.100376
Gomez–Cabellos, S., Toorop, P. E., Cañal, M. J., Iannetta, P. P., Fernández–Pascual, E., Pritchard, H. W., et al. (2022). Global DNA methylation and cellular 5–methylcytosine and H4 acetylated patterns in primary and secondary dormant seeds of Capsella bursa–pastoris (L.) Medik. (shepherd’s purse). Protoplasma 259, 595–614. doi:10.1007/s00709–021–01678–2
González, A. P. R., Preite, V., Verhoeven, K. J., and Latzel, V. (2018). Transgenerational effects and epigenetic memory in the clonal plant Trifolium repens. Front. Plant Sci. 9, 1677. doi:10.3389/fpls.2018.01677
González, R. M., Ricardi, M. M., and Iusem, N. D. (2011). Atypical epigenetic mark in an atypical location: Cytosine methylation at asymmetric (CNN) sites within the body of a non–repetitive tomato gene. BMC Plant Biol. 11, 94. doi:10.1186/1471–2229–11–94
González, R. M., Ricardi, M. M., and Iusem, N. D. (2013). Epigenetic marks in an adaptive water stress-responsive gene in tomato roots under normal and drought conditions. Epigenetics 8, 864–872. doi:10.4161/epi.25524
Grativol, C., Hemerly, A. S., and Ferreira, P. C. (2012). Genetic and epigenetic regulation of stress responses in natural plant populations. Biochim. Biophys. Acta 1819, 176–185. doi:10.1016/j.bbagrm.2011.08.010
Groot, M. P., Wagemaker, N., Ouborg, N. J., Verhoeven, K. J., and Vergeer, P. (2018). Epigenetic population differentiation in field- and common garden-grown Scabiosa columbaria plants. Ecol. Evol. 8, 3505–3517. doi:10.1002/ece3.3931
Grossiord, C., Sevanto, S., Limousin, J. M., Meir, P., Mencuccini, M., Pangle, R. E., et al. (2018). Manipulative experiments demonstrate how long–term soil moisture changes alter controls of plant water use. Environ. Exp. Bot. 152, 19–27. doi:10.1016/j.envexpbot.2017.12.010
Guan, Q., Lu, X., Zeng, H., Zhang, Y., and Zhu, J. (2013). Heat stress induction of miR398 triggers a regulatory loop that is critical for thermotolerance in Arabidopsis. Plant J. 74, 840–851. doi:10.1111/tpj.12169
Guarino, F., Cicatelli, A., Castiglione, S., Agius, D. R., Orhun, G. E., Fragkostefanakis, S., et al. (2022). An epigenetic alphabet of crop adaptation to climate change. Front. Genet. 13, 818727. doi:10.3389/fgene.2022.818727
Gugger, P. F., Fitz-Gibbon, S., PellEgrini, M., and Sork, V. L. (2016). Species-wide patterns of DNA methylation variation in Quercus lobata and their association with climate gradients. Mol. Ecol. 25, 1665–1680. doi:10.1111/mec.13563
Guo, W., Hussain, N., Wu, R., and Liu, B. (2018). High hypomethylation and epigenetic variation in fragmented populations of wild barley (Hordeum brevisubulatum). Pak. J. Bot. 50. 1379–1386.
Gupta, C., and Salgotra, R. K. (2022). Epigenetics and its role in effecting agronomical traits. Front. Plant Sci. 13, 925688. doi:10.3389/fpls.2022.925688
Ha, M., Lu, J., Tian, L., Ramachandran, V., Kasschau, K. D., Chapmane, E. J., et al. (2009). Small RNAs serve as a genetic buffer against genomic shock in Arabidopsis interspecific hybrids and allopolyploids. Proc. Natl. Acad. Sci. U.S.A. 106, 17835–17840. doi:10.1073/pnas.0907003106
Hajyzadeh, M., Turktas, M., Khawar, K. M., and Unver, T. (2015). miR408 overexpression causes increased drought tolerance in chickpea. Gene 555, 186–193. doi:10.1016/j.gene.2014.11.002
Han, T., Wang, F., Song, Q., Ye, W., Liu, T., Wang, L., et al. (2021). An epigenetic basis of inbreeding depression in maize. Sci. Adv. 7, eabg5442. doi:10.1126/sciadv.abg5442
Hashida, S. N., Uchiyama, T., Martin, C., Kishima, Y., Sano, Y., and Mikami, T. (2006). The temperature–dependent change in methylation of the Antirrhinum transposon Tam3 is controlled by the activity of its transposase. Plant Cell 18, 104–118. doi:10.1105/tpc.105.037655
Heer, K., Mounger, J., Boquete, M. T., Richards, C. L., and Opgenoorth, L. (2018a). The diversifying field of plant epigenetics. New Phytol. 217, 988–992. doi:10.1111/nph.14985
Heer, K., Ullrich, K. K., Hiss, M., Liepelt, S., Schulze Brüning, R., Zhou, J., et al. (2018b). Detection of somatic epigenetic variation in Norway spruce via targeted bisulfite sequencing. Ecol. Evol. 8, 9672–9682. doi:10.1002/ece3.4374
Hendry, A. P., Taylor, E. B., and McPhail, J. D. (2002). Adaptive divergence and the balance between selection and gene flow: Lake and stream stickleback in the misty system. Evolution 56, 1199–1216. doi:10.1111/j.0014–3820.2002.tb01432.x
Heo, J. B., and Sung, S. (2011). Vernalization–mediated epigenetic silencing by a long intronic noncoding RNA. Science 331, 76–79. doi:10.1126/science.1197349
Herrera, C. M., and Bazaga, P. (2013). Epigenetic correlates of plant phenotypic plasticity: DNA methylation differs between prickly and nonprickly leaves in heterophyllous Ilex aquifolium (Aquifoliaceae) trees. Bot. J. Linn. Soc. 171, 441–452. doi:10.1111/boj.12007
Herrera, C. M., and Bazaga, P. (2010). Epigenetic differentiation and relationship to adaptive genetic divergence in discrete populations of the violet Viola cazorlensis. New Phytol. 187, 867–876. doi:10.1111/j.1469–8137.2010.03298.x
Hu, Y., Zhang, L., Zhao, L., Li, J., He, S., Zhou, K., et al. (2011). Trichostatin A selectively suppresses the cold–induced transcription of the ZmDREB1 gene in maize. PLoS One 6, e22132. doi:10.1371/journal.pone.0022132
Huang, N. C., Li, C. H., Lee, J. Y., and Yen, H. E. (2010). Cytosine methylation changes in the ice plant Ppc1 promoter during transition from C3 to Crassulacean acid metabolism. Plant Sci. 178, 41–46. doi:10.1016/j.plantsci.2009.10.005
Iwasaki, M., and Paszkowski, J. (2014). Epigenetic memory in plants. EMBO J. 33, 1987–1998. doi:10.15252/embj.201488883
Jackson, L. E., Miller, D., and Smith, S. E. (2002). Arbuscular mycorrhizal colonization and growth of wild and cultivated lettuce in response to nitrogen and phosphorus. Sci. Hortic. 94, 205–218. doi:10.1016/S0304–4238(01)00341–7
Jackson, S. A. (2017). Epigenomics: Dissecting hybridization and polyploidization. Genome Biol. 18, 117. doi:10.1186/s13059–017–1254–7
Janousek, B., Matsunaga, S., Kejnovsky, E., Ziuvova, J., and Vyskot, B. (2002). DNA methylation analysis of a male reproductive organ specific gene (MROS1) during pollen development. Genome 45, 930–938. doi:10.1139/g02–052
Johannes, F., Porcher, E., Teixeira, F. K., Saliba–Colombani, V., Simon, M., Agier, N., et al. (2009). Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet. 5, e1000530. doi:10.1371/journal.pgen.1000530
Jones, A. M., Thomas, V., Bennett, M. H., Mansfield, J., and Grant, M. (2006). Modifications to the Arabidopsis defense proteome occur prior to significant transcriptional change in response to inoculation with Pseudomonas syringae. Plant Physiol. 142 (4), 1603–1620. doi:10.1104/pp.106.086231
Jones, P. A. (2012). Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 13, 484–492. doi:10.1038/nrg3230
Juroszek, P., Racca, P., Link, S., Farhumand, J., and Kleinhenz, B. (2020). Overview on the review articles published during the past 30 years relating to the potential climate change effects on plant pathogens and crop disease risks. Plant Pathol. 69, 179–193. doi:10.1111/ppa.13119
Kakoulidou, I., Avramidou, E. V., Baránek, M., Brunel-Muguet, S., Farrona, S., Johannes, F., et al. (2021). Epigenetics for crop improvement in times of global change. Biology 10, 766. doi:10.3390/biology10080766
Kakutani, T. (1997). Genetic characterization of late-flowering traits induced by DNA hypomethylation mutation in Arabidopsis thaliana. Plant J. 12, 1447–1451. doi:10.1046/j.1365-313x.1997.12061447.x
Kashkush, K., and Khasdan, V. (2007). Large–scale survey of cytosine methylation of retrotransposons and the impact of readout transcription from long terminal repeats on expression of adjacent rice genes. Genetics 177, 1975–1985. doi:10.1534/genetics.107.080234
Kawakatsu, T., and Ecker, J. R. (2019). Diversity and dynamics of DNA methylation: Epigenomic resources and tools for crop breeding. Breed. Sci. 69, 191–204. doi:10.1270/jsbbs.19005
Kim, J. M., To, T. K., Ishida, J., Matsui, A., Kimura, H., and Seki, M. (2012). Transition of chromatin status during the process of recovery from drought stress in Arabidopsis thaliana. Plant Cell Physiol. 53, 847–856. doi:10.1093/pcp/pcs053
Klosinska, M., Picard, C. L., and Gehring, M. (2016). Conserved imprinting associated with unique epigenetic signatures in the Arabidopsis genus. Nat. Plants 2, 16145. doi:10.1038/nplants.2016.145
Kozomara, A., Birgaoanu, M., and Griffiths–Jones, S. (2019). miRBase: from microRNA sequences to function. Nucleic Acids Res. 47, D155–D162. doi:10.1093/nar/gky1141
Kraitshtein, Z., Yaakov, B., Khasdan, V., and Kashkush, K. (2010). Genetic and epigenetic dynamics of a retrotransposon after allopolyploidization of wheat. Genetics 186, 801–812. doi:10.1534/genetics.110.120790
Krishnaswami, V., Kumar, M., and Vijayaraghavalu, S. (2022). in Advances in epigenetics for crop improvement and sustainable agriculture” in plant genomics for sustainable agriculture. Editors R. L. Singh, S. Mondal, A. Parihar, and P. K. Singh (Singapore: Springer), 351–370. doi:10.1007/978-981-16-6974-3_14
Kumar, S. V., and Wigge, P. A. (2010). H2A. Z–containing nucleosomes mediate the thermosensory response in Arabidopsis. Cell 140, 136–147. doi:10.1016/j.cell.2009.11.006
Kumari, P., Khan, S., Wani, I. A., Gupta, R., Verma, S., Alam, P., et al. (2022). Unravelling the role of epigenetic modifications in development and reproduction of angiosperms: A critical appraisal. Front. Genet. 13, 819941. doi:10.3389/fgene.2022.819941
Kurdyukov, S., and Bullock, M. (2016). DNA methylation analysis: Choosing the right method. Biology 5, 3. doi:10.3390/biology5010003
Kuźnicki, D., Meller, B., Arasimowicz–Jelonek, M., Braszewska–Zalewska, A., Drozda, A., and Floryszak–Wieczorek, J. (2019). BABA–Induced DNA methylome adjustment to intergenerational defense priming in potato to Phytophthora infestans. Front. Plant Sci. 10, 650. doi:10.3389/fpls.2019.00650
Lämke, J., and Bäurle, I. (2017). Epigenetic and chromatin–based mechanisms in environmental stress adaptation and stress memory in plants. Genome Biol. 18, 124. doi:10.1186/s13059–017–1263–6
Lämke, J., Brzezinka, K., Altmann, S., and Bäurle, I. (2016a). A hit-and-run heat shock factor governs sustained histone methylation and transcriptional stress memory. EMBO J. 35, 162–175. doi:10.15252/embj.201592593
Lämke, J., Brzezinka, K., and Bäurle, I. (2016b). HSFA2 orchestrates transcriptional dynamics after heat stress in Arabidopsis thaliana. Transcription 7, 111–114. doi:10.1080/21541264.2016.1187550
Lang–Mladek, C., Popova, O., Kiok, K., Berlinger, M., Rakic, B., Aufsatz, W., et al. (2010). Transgenerational inheritance and resetting of stress–induced loss of epigenetic gene silencing in Arabidopsis. Mol. Plant 3, 594–602. doi:10.1093/mp/ssq014
Latzel, V., Allan, E., Bortolini Silveira, A., Colot, V., Fischer, M., and Bossdorf, O. (2013). Epigenetic diversity increases the productivity and stability of plant populations. Nat. Commun. 4, 2875. doi:10.1038/ncomms3875
Lehmair, T. A., Poschlod, P., and Reisch, C. (2022). The impact of environment on genetic and epigenetic variation in Trifolium pratense populations from two contrasting semi–natural grasslands. R. Soc. Open Sci. 9, 211406. doi:10.1098/rsos.211406
Leisner, C. P. (2020). Review: Climate change impacts on food security-focus on perennial cropping systems and nutritional value. Plant Sci. 293, 110412. doi:10.1016/j.plantsci.2020.110412
Lele, L., Ning, D., Cuiping, P., Xiao, G., and Weihua, G. (2018). Genetic and epigenetic variations associated with adaptation to heterogeneous habitat conditions in a deciduous shrub. Ecol. Evol. 8, 2594–2606. doi:10.1002/ece3.3868
Li, A., Hu, B. Q., Xue, Z. Y., Chen, L., Wang, W. X., Song, W. Q., et al. (2011). DNA methylation in genomes of several annual herbaceous and woody perennial plants of varying ploidy as detected by MSAP. Plant Mol. Biol. Rep. 29, 784–793. doi:10.1007/s11105–010–0280–3
Li, H., Yan, S., Zhao, L., Tan, J., Zhang, Q., Gao, F., et al. (2014). Histone acetylation associated up–regulation of the cell wall related genes is involved in salt stress induced maize root swelling. BMC Plant Biol. 14, 105–114. doi:10.1186/1471–2229–14–105
Li, W., Chen, G., Xiao, G., Zhu, S., Zhou, N., Zhu, P., et al. (2020). Overexpression of TCP transcription factor OsPCF7 improves agronomic trait in rice. Mol Breeding 40, 48. doi:10.1007/s11032-020-01129-5
Li, X., Guo, W., Wang, B., Li, X., Chen, H., Wei, L., et al. (2010). Instability of chromosome number and DNA methylation variation induced by hybridization and amphidiploid formation between Raphanus sativus L. and Brassica alboglabra Bailey. BMC Plant Biol. 10, 207. doi:10.1186/1471–2229–10–207
Li, Y., and Tollefsbol, T. O. (2011). in DNA methylation detection: Bisulfite genomic sequencing analysis” in epigenetics protocols. Editors T. Tollefsbol, and Humana (Totowa, NJ, 11–21. doi:10.1007/978–1–61779–316–5_2
Lira–Medeiros, C. F., Parisod, C., Fernandes, R. A., Mata, C. S., Cardoso, M. A., and Ferreira, P. C. G. (2010). Epigenetic variation in mangrove plants occurring in contrasting natural environment. PLoS One 5, e10326. doi:10.1371/journal.pone.0010326
Lister, R., and Ecker, J. R. (2009). Finding the fifth base: Genome–wide sequencing of cytosine methylation. Genome Res. 19, 959–966. doi:10.1101/gr.083451.108
Lister, R., Pelizzola, M., Dowen, R. H., Hawkins, R. D., Hon, G., Tonti–Filippini, J., et al. (2009). Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322. doi:10.1038/nature08514
Liu, L., Pei, C., Liu, S., Guo, X., Du, N., and Guo, W. (2018). Genetic and epigenetic changes during the invasion of a cosmopolitan species (Phragmites australis). Ecol. Evol. 8, 6615–6624. doi:10.1002/ece3.4144
Liu, X., Quan, W., and Bartels, D. (2022). Stress memory responses and seed priming correlate with drought tolerance in plants: An overview. Planta 255, 45–14. doi:10.1007/s00425–022–03828–z
Luan, A., Chen, C., Xie, T., He, J., and He, Y. (2020). Methylation analysis of CpG islands in pineapple SERK1 promoter. Genes 11, 425. doi:10.3390/genes11040425
Luan, X., Liu, S., Ke, S., Dai, H., Xie, X. M., Hsieh, T. F., et al. (2019). Epigenetic modification of ESP, encoding a putative long noncoding RNA, affects panicle architecture in rice. Rice 12, 20. doi:10.1186/s12284-019-0282-1
Lynch, M., Bürger, R., Butcher, D., and Gabriel, W. (1993). The mutational meltdown in asexual populations. J. Hered. 84, 339–344. doi:10.1093/oxfordjournals.jhered.a111354
Malhi, G. S., Kaur, M., and Kaushik, P. (2021). Impact of climate change on agriculture and its mitigation strategies: A review. Sustainability 13, 1318. doi:10.3390/su13031318
Manning, K., Tör, M., Poole, M., Hong, Y., Thompson, A. J., King, G. J., et al. (2006). A naturally occurring epigenetic mutation in a gene encoding an SBP–box transcription factor inhibits tomato fruit ripening. Nat. Genet. 38, 948–952. doi:10.1038/ng1841
Marfil, C. F., Camadro, E. L., and Masuelli, R. W. (2009). Phenotypic instability and epigenetic variability in a diploid potato of hybrid origin, Solanum ruiz–lealii. BMC Plant Biol. 9, 21. doi:10.1186/1471–2229–9–21
Marin, P., Genitoni, J., Barloy, D., Maury, S., Gibert, P., Ghalambor, C. K., et al. (2020). Biological invasion: The influence of the hidden side of the (epi)genome. Funct. Ecol. 34, 385–400. doi:10.1111/1365–2435.13317
Martin, A., Troadec, C., Boualem, A., Rajab, M., Fernandez, R., Morin, H., et al. (2009). A transposon–induced epigenetic change leads to sex determination in melon. Nature 461, 1135–1138. doi:10.1038/nature08498
Maunakea, A. K., Nagarajan, R. P., Bilenky, M., Ballinger, T. J., D'Souza, C., Fouse, S. D., et al. (2010). Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466, 253–257. doi:10.1038/nature09165
Medrano, M., Herrera, C. M., and Bazaga, P. (2014). Epigenetic variation predicts regional and local intraspecific functional diversity in a perennial herb. Mol. Ecol. 23, 4926–4938. doi:10.1111/mec.12911
Mehta, S., James, D., and Reddy, M. K. (2019). in Omics technologies for abiotic stress tolerance in plants: Current status and prospects” in recent approaches in omics for plant resilience to climate change. Editor S. Wani (Cham: Springer), 1–34. doi:10.1007/978–3–030–21687–0_1
Meissner, A., Gnirke, A., Bell, G. W., Ramsahoye, B., Lander, E. S., and Jaenisch, R. (2005). Reduced representation bisulfite sequencing for comparative high–resolution DNA methylation analysis. Nucleic Acids Res. 33, 5868–5877. doi:10.1093/nar/gki901
Meissner, A., Mikkelsen, T. S., Gu, H., Wernig, M., Hanna, J., Sivachenko, A., et al. (2008). Genome–scale DNA methylation maps of pluripotent and differentiated cells. Nature 454, 766–770. doi:10.1038/nature07107
Meller, B., Kuźnicki, D., Arasimowicz–Jelonek, M., Deckert, J., and Floryszak–Wieczorek, J. (2018). BABA–Primed histone modifications in potato for intergenerational resistance to Phytophthora infestans. Front. Plant Sci. 9, 1228. doi:10.3389/fpls.2018.01228
Meyer, P. (2015). Epigenetic variation and environmental change. J. Exp. Bot. 66, 3541–3548. doi:10.1093/jxb/eru502
Michaels, S. D., and Amasino, R. M. (1999). Flowering Locus C encodes a novel MADS domain protein that acts as a repressor of flowering. Plant Cell 11, 949–956. doi:10.1105/tpc.11.5.949
Mirouze, M., and Paszkowski, J. (2011). Epigenetic contribution to stress adaptation in plants. Curr. Opin. Plant Biol. 14, 267–274. doi:10.1016/j.pbi.2011.03.004
Miura, K., Agetsuma, M., Kitano, H., Yoshimura, A., Matsuoka, M., Jacobsen, S. E., et al. (2009). A metastable DWARF1 epigenetic mutant affecting plant stature in rice. Proc. Natl. Acad. Sci. U.S.A. 106, 11218–11223. doi:10.1073/pnas.0901942106
Miura, K., Ikeda, M., Matsubara, A., Song, X. J., Ito, M., Asano, K., et al. (2010). OsSPL14 promotes panicle branching and higher grain productivity in rice. Nat. Genet. 42, 545–549. doi:10.1038/ng.592
Mohn, F., Weber, M., Schübeler, D., and Roloff, T. C. (2009). Methylated DNA immunoprecipitation (MeDIP)” in methods in molecular Biology. Editor J. Tost (Switzerland: Springer Nature), 55–64. Vol. 507. doi:10.1007/978–1–59745–522–0_5
Mounger, J., Boquete, M. T., Schmid, M. W., Granado, R., Robertson, M. H., Voors, S. A., et al. (2021). Inheritance of DNA methylation differences in the mangrove Rhizophora mangle. Evol. Dev. 23, 351–374. doi:10.1111/ede.12388
Ni, B., You, J., Li, J., Du, Y., Zhao, W., and Chen, X. (2021). Genetic and epigenetic changes during the upward expansion of Deyeuxia angustifolia kom. In the alpine tundra of the changbai mountains, China. Plants 10, 291. doi:10.3390/plants10020291
Ni, Z., Hu, Z., Jiang, Q., and Zhang, H. (2012). Overexpression of gma–MIR394a confers tolerance to drought in transgenic Arabidopsis thaliana. Biochem. Biophys. Res. Commun. 427, 330–335. doi:10.1016/j.bbrc.2012.09.055
Nicotra, A. B., Segal, D. L., Hoyle, G. L., Schrey, A. W., Verhoeven, K. J., and Richards, C. L. (2015). Adaptive plasticity and epigenetic variation in response to warming in an Alpine plant. Ecol. Evol. 5, 634–647. doi:10.1002/ece3.1329
Nonogaki, H. (2014). Seed dormancy and germination—Emerging mechanisms and new hypotheses. Front. Plant Sci. 5, 233. doi:10.3389/fpls.2014.00233
Olvera–Carrillo, Y., Campos, F., Reyes, J. L., Garciarrubio, A., and Covarrubias, A. A. (2010). Functional analysis of the group 4 late embryogenesis abundant proteins reveals their relevance in the adaptive response during water deficit in Arabidopsis. Plant Physiol. 154, 373–390. doi:10.1104/pp.110.158964
Ong-Abdullah, M., Ordway, J. M., Jiang, N., Ooi, S. E., Kok, S. Y., Sarpan, N., et al. (2015). Loss of Karma transposon methylation underlies the mantled somaclonal variant of oil palm. Nature 525, 533–537. doi:10.1038/nature15365
Park, J., Lim, C. J., Shen, M., Park, H. J., Cha, J. Y., Iniesto, E., et al. (2018). Epigenetic switch from repressive to permissive chromatin in response to cold stress. Proc. Natl. Acad. Sci. U.S.A. 115, E5400–E5409. doi:10.1073/pnas.1721241115
Paun, O., Bateman, R. M., Fay, M. F., Hedrén, M., Civeyrel, L., and Chase, M. W. (2010). Stable epigenetic effects impact adaptation in allopolyploid orchids (Dactylorhiza: Orchidaceae). Mol. Biol. Evol. 27, 2465–2473. doi:10.1093/molbev/msq150
Paun, O., Fay, M. F., Soltis, D. E., and Chase, M. W. (2007). Genetic and epigenetic alterations after hybridization and genome doubling. Taxon 56, 649–656. doi:10.2307/25065849
Paun, O., Verhoeven, K., and Richards, C. L. (2019). Opportunities and limitations of reduced representation bisulfite sequencing in plant ecological epigenomics. New Phytol. 221, 738–742. doi:10.1111/nph.15388
Pavangadkar, K., Thomashow, M. F., and Triezenberg, S. J. (2010). Histone dynamics and roles of histone acetyltransferases during cold–induced gene regulation in Arabidopsis. Plant Mol. Biol. 74, 183–200. doi:10.1007/s11103–010–9665–9
Pecinka, A., Dinh, H. Q., Baubec, T., Rosa, M., Lettner, N., and Scheid, O. M. (2010). Epigenetic regulation of repetitive elements is attenuated by prolonged heat stress in Arabidopsis. Plant Cell 22, 3118–3129. doi:10.1105/tpc.110.078493
Pegoraro, L., Cafasso, D., Rinaldi, R., Cozzolino, S., and Scopece, G. (2016). Habitat preference and flowering-time variation contribute to reproductive isolation between diploid and autotetraploid Anacamptis pyramidalis. J. Evol. Biol. 29, 2070–2082. doi:10.1111/jeb.12930
Peltier, E., Sharma, V., Martí Raga, M., Roncoroni, M., Bernard, M., Jiranek, V., et al. (2018). Dissection of the molecular bases of genotype x environment interactions: A study of phenotypic plasticity of Saccharomyces cerevisiae in grape juices. BMC Genomics 19, 1–20. doi:10.1186/s12864–018–5145–4
Pereira, W. J., Pappas, M. D. C. R., Campoe, O. C., Stape, J. L., Grattapaglia, D., and Pappas, G. J. (2020a). Patterns of DNA methylation changes in elite Eucalyptus clones across contrasting environments. For. Ecol. Manage. 474, 118319. doi:10.1016/j.foreco.2020.118319
Pereira, W. J., Pappas, M. D. C. R., Grattapaglia, D., and Pappas, G. J. (2020b). A cost–effective approach to DNA methylation detection by Methyl Sensitive DArT sequencing. PLoS One 15, e0233800. doi:10.1371/journal.pone.0233800
Perrin, A., Daccord, N., Roquis, D., Celton, J. M., Vergne, E., and Bucher, E. (2020). Divergent DNA methylation signatures of juvenile seedlings, grafts and adult apple trees. Epigenomes 4, 4. doi:10.3390/epigenomes4010004
Picazo–Aragonés, J., Terrab, A., and Balao, F. (2020). Plant volatile organic compounds evolution: Transcriptional regulation, epigenetics and polyploidy. Int. J. Mol. Sci. 21, 1–18. doi:10.3390/ijms21238956
Pielke, R., Burgess, M. G., and Ritchie, J. (2022). Plausible 2005–2050 emissions scenarios project between 2° C and 3° C of warming by 2100. Environ. Res. Lett. 17, 024027. doi:10.1088/1748–9326/ac4ebf
Pigliucci, M. (2005). Evolution of phenotypic plasticity: Where are we going now? Trends Ecol. Evol. 20, 481–486. doi:10.1016/j.tree.2005.06.001
Popova, O. V., Dinh, H. Q., Aufsatz, W., and Jonak, C. (2013). The RdDM pathway is required for basal heat tolerance in Arabidopsis. Mol. Plant 6, 396–410. doi:10.1093/mp/sst023
Preite, V., Oplaat, C., Biere, A., Kirschner, J., van der Putten, W. H., and Verhoeven, K. J. (2018). Increased transgenerational epigenetic variation, but not predictable epigenetic variants, after environmental exposure in two apomictic dandelion lineages. Ecol. Evol. 8, 3047–3059. doi:10.1002/ece3.3871
Preite, V., Snoek, L. B., Oplaat, C., Biere, A., van der Putten, W. H., and Verhoeven, K. J. (2015). The epigenetic footprint of poleward range-expanding plants in apomictic dandelions. Mol. Ecol. 24, 4406–4418. doi:10.1111/mec.13329
Qiu, T., Liu, Z., Yang, Y., and Liu, B. (2021). Epigenetic variation associated with responses to different habitats in the context of genetic divergence in Phragmites australis. Ecol. Evol. 11, 11874–11889. doi:10.1002/ece3.7954
Quadrana, L., Almeida, J., Asís, R., Duffy, T., Dominguez, P. G., Bermúdez, L., et al. (2014). Natural occurring epialleles determine vitamin E accumulation in tomato fruits. Nat. Commun. 5, 3027. doi:10.1038/ncomms5027
Raj, S., Bräutigam, K., Hamanishi, E. T., Wilkins, O., Thomas, B. R., Schroeder, W., et al. (2011). Clone history shapes Populus drought responses. Proc. Natl. Acad. Sci. U.S.A. 108, 12521–12526. doi:10.1073/pnas.1103341108
Rathore, P., Raina, S. N., Kumar, S., and Bhat, V. (2020). Retro-element gypsy-163 is differentially methylated in reproductive tissues of apomictic and sexual plants of Cenchrus ciliaris. Front. Genet. 11, 795. doi:10.3389/fgene.2020.00795
Rehman, A. U., Dang, T., Qamar, S., Ilyas, A., Fatema, R., Kafle, M., et al. (2021). Revisiting plant heterosis—from field scale to molecules. Genes 12, 1688. doi:10.3390/genes12111688
Rhee, S. Y., Beavis, W., Berardini, T. Z., Chen, G., Dixon, D., Doyle, A., et al. (2003). The Arabidopsis Information Resource (TAIR): A model organism database providing a centralized, curated gateway to Arabidopsis biology, research materials and community. Nucleic Acids Res. 31 (1), 224–228. doi:10.1093/nar/gkg076
Ribeiro, T., Viegas, W., and Morais–Cecílio, L. (2009). Epigenetic marks in the mature pollen of Quercus suber L. (Fagaceae). Sex. Plant Reprod. 22, 1–7. doi:10.1007/s00497–008–0083–y
Richards, C. L., Alonso, C., Becker, C., Bossdorf, O., Bucher, E., Colomé-Tatché, M., et al. (2017). Ecological plant epigenetics: Evidence from model and non-model species, and the way forward. Ecol. Lett. 20, 1576–1590. doi:10.1111/ele.12858
Richards, C. L., Schrey, A. W., and Pigliucci, M. (2012). Invasion of diverse habitats by few Japanese knotweed genotypes is correlated with epigenetic differentiation. Ecol. Lett. 15, 1016–1025. doi:10.1111/j.1461–0248.2012.01824.x
Richards, C. L., Walls, R. L., Bailey, J. P., Parameswaran, R., George, T., and Pigliucci, M. (2008). Plasticity in salt tolerance traits allows for invasion of novel habitat by Japanese knotweed s. l. (Fallopia japonica and F. x bohemica, Polygonaceae). Am. J. Bot. 95, 931–942. doi:10.3732/ajb.2007364
Rico, L., Ogaya, R., Barbeta, A., and Peñuelas, J. (2014). Changes in DNA methylation fingerprint of Quercus ilex trees in response to experimental field drought simulating projected climate change. Plant Biol. 16, 419–427. doi:10.1111/plb.12049
Robertson, A. L., and Wolf, D. E. (2012). The role of epigenetics in plant adaptation. Trends Evol. Biol. 4, 4. doi:10.4081/eb.2012.e4
Robertson, M., Alvarez, M., van Gurp, T., Wagemaker, C. A., Yu, F., Amador, D. M., et al. (2020). Combining epiGBS markers with long read transcriptome sequencing to assess epigenetic differentiation associated with habitat in Reynoutria (aka Fallopia). bioRxiv, 317966. doi:10.1101/2020.09.30.317966
Robertson, M., Schrey, A., Shayter, A., Moss, C. J., and Richards, C. (2017). Genetic and epigenetic variation in Spartina alterniflora following the Deepwater Horizon oil spill. Evol. Appl. 10, 792–801. doi:10.1111/eva.12482
Rodríguez–Hernández, A. A., Ortega–Amaro, M. A., Delgado–Sánchez, P., Salinas, J., and Jiménez–Bremont, J. F. (2014). AtGRDP1 gene encoding a glycine–rich domain protein is involved in germination and responds to ABA signalling. Plant Mol. Biol. Rep. 32, 1187–1202. doi:10.1007/s11105–014–0714–4
Saéz–Laguna, E., Guevara, M. Á., Diáz, L. M., Sańchez–Gómez, D., Collada, C., Aranda, I., et al. (2014). Epigenetic variability in the genetically uniform forest tree species Pinus pinea L. PLoS One 9, e103145. doi:10.1371/journal.pone.0103145
Salmon, A., Ainouche, M. L., and Wendel, J. F. (2005). Genetic and epigenetic consequences of recent hybridization and polyploidy in Spartina (Poaceae). Mol. Ecol. 14, 1163–1175. doi:10.1111/j.1365–294X.2005.02488.x
Samantara, K., Shiv, A., de Sousa, L. L., Sandhu, K. S., Priyadarshini, P., and Mohapatra, S. R. (2021). A comprehensive review on epigenetic mechanisms and application of epigenetic modifications for crop improvement. Environ. Exp. Bot. 188, 104479. doi:10.1016/j.envexpbot.2021.104479
Sammarco, I., Münzbergová, Z., and Latzel, V. (2022). DNA methylation can mediate local adaptation and response to climate change in the clonal plant Fragaria vesca: Evidence from a European–scale reciprocal transplant experiment. Front. Plant Sci. 13, 827166. doi:10.3389/fpls.2022.827166
Saze, H., Tsugane, K., Kanno, T., and Nishimura, T. (2012). DNA methylation in plants: Relationship to small RNAs and histone modifications, and functions in transposon inactivation. Plant Cell Physiol. 53, 766–784. doi:10.1093/pcp/pcs008
Schield, D. R., Walsh, M. R., Card, D. C., Andrew, A. L., Adams, R. H., Castoe, T. A., et al. (2015). EpiRADseq: Scalable analysis of genomewide patterns of methylation using next-generation sequencing. Methods Ecol. Evol. 7, 60–69. doi:10.1111/2041-210x.12435
Schinkel, C. C., Syngelaki, E., Kirchheimer, B., Dullinger, S., Klatt, S., and Hörandl, E. (2020). Epigenetic patterns and geographical parthenogenesis in the alpine plant species Rranunculus kuepferi (Ranunculaceae). Int. J. Mol. Sci. 21, 3318. doi:10.3390/ijms21093318
Schmid, M. W., Heichinger, C., Coman Schmid, D., Guthörl, D., Gagliardini, V., Bruggmann, R., et al. (2018). Contribution of epigenetic variation to adaptation in Arabidopsis. Nat. Commun. 9, 4446. doi:10.1038/s41467–018–06932–5
Schmitz, R. J., and Ecker, J. R. (2012). Epigenetic and epigenomic variation in Arabidopsis thaliana. Trends Plant Sci. 17, 149–154. doi:10.1016/j.tplants.2012.01.001
Schulz, B., Eckstein, R. L., and Durka, W. (2014). Epigenetic variation reflects dynamic habitat conditions in a rare floodplain herb. Mol. Ecol. 23, 3523–3537. doi:10.1111/mec.12835
Scoville, A. G., Barnett, L. L., Bodbyl-Roels, S., Kelly, J. K., and Hileman, L. C. (2011). Differential regulation of a MYB transcription factor is correlated with transgenerational epigenetic inheritance of trichome density in Mimulus guttatus. New Phytol. 191, 251–263. doi:10.1111/j.1469–8137.2011.03656.x
Serre, D., Lee, B. H., and Ting, A. H. (2010). MBD–isolated Genome Sequencing provides a high–throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Res. 38, 391–399. doi:10.1093/nar/gkp992
Shahzad, A., Ullah, S., Dar, A. A., Sardar, M. F., Mehmood, T., Tufail, M. A., et al. (2021). Nexus on climate change: Agriculture and possible solution to cope future climate change stresses. Environ. Sci. Pollut. Res. Int. 28, 14211–14232. doi:10.1007/s11356–021–12649–8
Sharma, N., Russell, S. D., Bhalla, P. L., and Singh, M. B. (2011). Putative cis–regulatory elements in genes highly expressed in rice sperm cells. BMC Res. Notes 4, 319. doi:10.1186/1756–0500–4–319
Sharma, S., Chauhan, A., Dobbal, S., and Kumar, R. (2022). Biology of plants coping stresses: Epigenetic modifications and genetic engineering. S. Afr. J. Bot. 144, 270–283. doi:10.1016/j.sajb.2021.08.031
Shen, J., Wang, Z., Su, Y., and Wang, T. (2021). Associations between population epigenetic differentiation and environmental factors in the exotic weed mile–a–minute (Mikania micrantha). Weed Sci. 69, 307–332. doi:10.1017/wsc.2021.13
Shen, Y., Zhang, J., Liu, Y., Liu, S., Liu, Z., Duan, Z., et al. (2018). DNA methylation footprints during soybean domestication and improvement. Genome Biol. 19, 128. doi:10.1186/s13059-018-1516-z
Singh, A., Mehta, S., Yadav, S., Nagar, G., Ghosh, R., Roy, A., et al. (2022). How to cope with the challenges of environmental stresses in the era of global climate change: An update on ROS stave off in plants. Int. J. Mol. Sci. 23, 1995. doi:10.3390/ijms23041995
Sinha, P., Singh, V. K., Saxena, R. K., Kale, S. M., Li, Y., Garg, V., et al. (2020). Genome-wide analysis of epigenetic and transcriptional changes associated with heterosis in pigeonpea. Plant Biotechnol. J. 18, 1697–1710. doi:10.1111/pbi.13333
Skendžić, S., Zovko, M., Živković, I. P., Lešić, V., and Lemić, D. (2021). The impact of climate change on agricultural insect pests. Insects 12, 440. doi:10.3390/insects12050440
Smallwood, S. A., Lee, H. J., Angermueller, C., Krueger, F., Saadeh, H., Peat, J., et al. (2014). Single–cell genome–wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods 11, 817–820. doi:10.1038/nmeth.3035
Song, C. X., Szulwach, K. E., Dai, Q., Fu, Y., Mao, S. Q., Lin, L., et al. (2013). Genome–wide profiling of 5–formylcytosine reveals its roles in epigenetic priming. Cell 153, 678–691. doi:10.1016/j.cell.2013.04.001
Song, Q., Zhang, T., Stelly, D. M., and Chen, Z. J. (2017). Epigenomic and functional analyses reveal roles of epialleles in the loss of photoperiod sensitivity during domestication of allotetraploid cottons. Genome Biol. 18, 99. doi:10.1186/s13059–017–1229–8
Song, Y., Liu, L., Feng, Y., Wei, Y., Yue, X., He, W., et al. (2015). Chilling– and freezing–induced alterations in cytosine methylation and its association with the cold tolerance of an alpine subnival plant, Chorispora bungeana. PLoS One 10, e0135485. doi:10.1371/journal.pone.0135485
Soreng, R. J., Peterson, P. M., Romaschenko, K., Davidse, G., Teisher, J. K., Clark, L. G., et al. (2017). A worldwide phylogenetic classification of the Poaceae (Gramineae) II: An update and a comparison of two 2015 classifications. J. Syst. Evol. 55, 259–290. doi:10.1111/jse.12262
Sow, M. D., Segura, V., Chamaillard, S., Jorge, V., Delaunay, A., Lafon–Placette, C., et al. (2018). Narrow–sense heritability and PST estimates of DNA methylation in three Populus nigra L. populations under contrasting water availability. Tree Genet. Genomes 14, 78–12. doi:10.1007/s11295–018–1293–6
Springer, N. M., and Schmitz, R. (2017). Exploiting induced and natural epigenetic variation for crop improvement. Nat. Rev. Genet. 18, 563–575. doi:10.1038/nrg.2017.45
Stebbins, G. L. (1957). Self fertilization and population variability in the higher plants. Am. Nat. 91, 337–354. doi:10.1086/281999
Steward, N., Kusano, T., and Sano, H. (2000). Expression of ZmMET1, a gene encoding a DNA methyltransferase from maize, is associated not only with DNA replication in actively proliferating cells, but also with altered DNA methylation status in cold–stressed quiescent cells. Nucleic Acids Res. 28, 3250–3259. doi:10.1093/nar/28.17.3250
Stief, A., Altmann, S., Hoffmann, K., Pant, B. D., Scheible, W. R., and Bäurle, I. (2014). Arabidopsis miR156 regulates tolerance to recurring environmental stress through SPL transcription factors. Plant Cell 26, 1792–1807. doi:10.1105/tpc.114.123851
Su, Y., Huang, Q., Wang, Z., and Wang, T. (2021). High genetic and epigenetic variation of transposable elements: Potential drivers to rapid adaptive evolution for the noxious invasive weed Mikania micrantha. Ecol. Evol. 11, 13501–13517. doi:10.1002/ece3.8075
Sun, C., Fang, J., Zhao, T., Xu, B., Zhang, F., Liu, L., et al. (2012). The histone methyltransferase SDG724 mediates H3K36me2/3 deposition at MADS50 and RFT1 and promotes flowering in rice. Plant Cell 24, 3235–3247. doi:10.1105/tpc.112.101436
Sun, M., Yang, Z., Liu, L., and Duan, L. (2022). DNA methylation in plant responses and adaption to abiotic stresses. Int. J. Mol. Sci. 23 (13), 6910. doi:10.3390/ijms23136910
Szcześniak, M. W., Rosikiewicz, W., and Makałowska, I. (2016). CANTATAdb: A collection of plant long non–coding RNAs. Plant Cell Physiol. 57, e8. doi:10.1093/pcp/pcv201
Taudt, A., Colomé–Tatché, M., and Johannes, F. (2016). Genetic sources of population epigenomic variation. Nat. Rev. Genet. 17, 319–332. doi:10.1038/nrg.2016.45
Telias, A., Lin-Wang, K., Stevenson, D. E., Cooney, J. M., Hellens, R. P., Allan, A. C., et al. (2011). Apple skin patterning is associated with differential expression of MYB10. BMC Plant Biol. 11, 93. doi:10.1186/1471-2229-11-93
Thiebaut, F., Hemerly, A. S., and Ferreira, P. C. G. (2019). A role for epigenetic regulation in the adaptation and stress responses of non-model plants. Front. Plant Sci. 10, 246. doi:10.3389/fpls.2019.00246
Tian, Y., Hou, Y., and Song, Y. (2022). LncRNAs elevate plant adaptation under low temperature by maintaining local chromatin landscape. Plant Signal. Behav. 17, 2014677. doi:10.1080/15592324.2021.2014677
Tirnaz, S., and Batley, J. (2019). Epigenetics: Potentials and challenges in crop breeding. Mol. Plant 12, 1309–1311. doi:10.1016/j.molp.2019.09.006
Tito, R., Vasconcelos, H. L., and Feeley, K. J. (2018). Global climate change increases risk of crop yield losses and food insecurity in the tropical Andes. Glob. Chang. Biol. 24, e592e592–e602. doi:10.1111/gcb.13959
Tollefsbol, T. O. (2011). Advances in epigenetic technology” in epigenetics protocols: Methods in molecular Biology. Editor T. Tollefsbol (Switzerland: Springer Nature), 1–10. Vol. 791. doi:10.1007/978–1–61779–316–5_1
Tollefson, J. (2020). How hot will Earth get by 2100? Nature 580, 443–445. doi:10.1038/d41586–020–01125–x
Tripathi, V., Parasuraman, B., Laxmi, A., and Chattopadhyay, D. (2009). CIPK6, a CBL-interacting protein kinase is required for development and salt tolerance in plants. Plant J. 58, 778–790. doi:10.1111/j.1365–313X.2009.03812.x
Trucchi, E., Mazzarella, A. B., Gilfillan, G. D., Lorenzo, M. T., Schönswetter, P., and Paun, O. (2016). Bs RAD seq: Screening DNA methylation in natural populations of non-model species. Mol. Ecol. 25, 1697–1713. doi:10.1111/mec.13550
Turcotte, H., Hooker, J., Samanfar, B., and Parent, J. S. (2022). Can epigenetics guide the production of better adapted cultivars? Agronomy 12, 838. doi:10.3390/agronomy12040838
van Gurp, T. P., Wagemaker, N. C., Wouters, B., Vergeer, P., Ouborg, J. N., and Verhoeven, K. J. (2016). epiGBS: reference–free reduced representation bisulfite sequencing. Nat. Methods 13, 322–324. doi:10.1038/nmeth.3763
van Zanten, M., Zöll, C., Wang, Z., Philipp, C., Carles, A., Li, Y., et al. (2014). Histone Deacetylase 9 represses seedling traits in Arabidopsis thaliana dry seeds. Plant J. 80, 475–488. doi:10.1111/tpj.12646
Vanden Broeck, A., Cox, K., Brys, R., Castiglione, S., Cicatelli, A., Guarino, F., et al. (2018). Variability in DNA methylation and generational plasticity in the Lombardy poplar, a single genotype worldwide distributed since the eighteenth century. Front. Plant Sci. 9, 1635. doi:10.3389/fpls.2018.01635
Varotto, S., Krugman, T., Aiese Cigliano, R., Kashkush, K., Kondić-Špika, A., Aravanopoulos, F. A., et al. (2022). Exploitation of epigenetic variation of crop wild relatives for crop improvement and agrobiodiversity preservation. Theor. Appl. Genet. 1, 1–17. doi:10.1007/s00122-022-04122-y
Varotto, S., Tani, E., Abraham, E., Krugman, T., Kapazoglou, A., Melzer, R., et al. (2020). Epigenetics: Possible applications in climate-smart crop breeding. J. Exp. Bot. 71, 5223–5236. doi:10.1093/jxb/eraa188
Verhoeven, K. J., Jansen, J. J., Van Dijk, P. J., and Biere, A. (2010). Stress-induced DNA methylation changes and their heritability in asexual dandelions. New Phytol. 185, 1108–1118. doi:10.1111/j.1469–8137.2009.03121.x
Vilela, A. A., Del Claro, V. T. S., Torezan–Silingardi, H. M., and Del–Claro, K. (2018). Climate changes affecting biotic interactions, phenology, and reproductive success in a savanna community over a 10–year period. Arthropod. Plant. Interact. 12, 215–227. doi:10.1007/s11829–017–9572–y
Villagómez-Aranda, A. L., Feregrino-Pérez, A. A., García-Ortega, L. F., González-Chavira, M. M., Torres-Pacheco, I., and Guevara-González, R. G. (2022). Activating stress memory: Eustressors as potential tools for plant breeding. Plant Cell Rep. 41, 1481–1498. doi:10.1007/s00299-022-02858-x
Waalwijk, C., and Flavell, R. A. (1978). MspI, an isoschizomer of hpaII which cleaves both unmethylated and methylated hpaII sites. Nucleic Acids Res. 5, 3231–3236. doi:10.1093/nar/5.9.3231
Wang, H., Chai, Y., Chu, X., Zhao, Y., Wu, Y., Zhao, J., et al. (2009). Molecular characterization of a rice mutator–phenotype derived from an incompatible cross–pollination reveals transgenerational mobilization of multiple transposable elements and extensive epigenetic instability. BMC Plant Biol. 9, 63. doi:10.1186/1471–2229–9–63
Wang, M. Z., Li, H. L., Li, J. M., and Yu, F. H. (2020). Correlations between genetic, epigenetic and phenotypic variation of an introduced clonal herb. Heredity 124, 146–155. doi:10.1038/s41437–019–0261–8
Wang, M. Z., Li, H. L., Tang, M., and Yu, F. H. (2022). DNA methylation correlates with responses of experimental Hydrocotyle vulgaris populations to different flood regimes. Front. Plant Sci. 13, 831175. doi:10.3389/fpls.2022.831175
Wang, W. S., Pan, Y. J., Zhao, X. Q., Dwivedi, D., Zhu, L. H., Ali, J., et al. (2011). Drought–induced site–specific DNA methylation and its association with drought tolerance in rice (Oryza sativa L.). J. Exp. Bot. 62, 1951–1960. doi:10.1093/jxb/erq391
Wang, Y., Zhang, P., Guo, W., Liu, H., Li, X., Zhang, Q., et al. (2021). A deep learning approach to automate whole-genome prediction of diverse epigenomic modifications in plants. New Phytol. 232, 880–897. doi:10.1111/nph.17630
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA–Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi:10.1038/nrg2484
Wei, X., Song, X., Wei, L., Tang, S., Sun, J., Hu, P., et al. (2017). An epiallele of rice AK1 affects photosynthetic capacity. J. Integr. Plant Biol. 59, 158–163. doi:10.1111/jipb.12518
Wibowo, A., Becker, C., Marconi, G., Durr, J., Price, J., Hagmann, J., et al. (2016). Hyperosmotic stress memory in Arabidopsis is mediated by distinct epigenetically labile sites in the genome and is restricted in the male germline by DNA glycosylase activity. Elife 5, e13546. doi:10.7554/eLife.13546
Wilschut, R. A., Oplaat, C., Snoek, L. B., Kirschner, J., and Verhoeven, K. J. (2016). Natural epigenetic variation contributes to heritable flowering divergence in a widespread asexual dandelion lineage. Mol. Ecol. 25, 1759–1768. doi:10.1111/mec.13502
World Flora Online, (2022). World Flora online. Available at: http://www.worldfloraonline.com (Accessed Aug 14, 2022).
Wu, W. Q., Yi, M. R., Wang, X. F., Ma, L. L., Jiang, L., Li, X. W., et al. (2013). Genetic and epigenetic differentiation between natural Betula ermanii (Betulaceae) populations inhabiting contrasting habitats. Tree Genet. Genomes 9, 1321–1328. doi:10.1007/s11295–013–0641–9
Xia, H., Huang, W., Xiong, J., Tao, T., Zheng, X., Wei, H., et al. (2016). Adaptive epigenetic differentiation between upland and lowland rice ecotypes revealed by methylation-sensitive amplified polymorphism. PLoS One 11, e0157810. doi:10.1371/journal.pone.0157810
Xu, J., Chen, G., Hermanson, P. J., Xu, Q., Sun, C., Chen, W., et al. (2019). Population-level analysis reveals the widespread occurrence and phenotypic consequence of DNA methylation variation not tagged by genetic variation in maize. Genome Biol. 20, 243. doi:10.1186/s13059-019-1859-0
Xu, P., Chen, H., Hu, J., and Cai, W. (2021). Potential evidence for transgenerational epigenetic memory in Arabidopsis thaliana following spaceflight. Commun. Biol. 4, 835. doi:10.1038/s42003–021–02342–4
Yi, S. V., and Goodisman, M. A. (2021). The impact of epigenetic information on genome evolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 376, 20200114. doi:10.1098/rstb.2020.0114
Yona, A. H., Frumkin, I., and Pilpel, Y. (2015). A relay race on the evolutionary adaptation spectrum. Cell 163, 549–559. doi:10.1016/j.cell.2015.10.005
Zhang, G., Li, N., Zhang, D., Li, Z., Zhang, A., and Guo, X. (2022). Exploring japonica rice epigenetic diversity in the main production regions of Heilongjiang Province. Sci. Rep. 12, 4592. doi:10.1038/s41598-022-08683-2
Zhang, Y. Y., Latzel, V., Fischer, M., and Bossdorf, O. (2018). Understanding the evolutionary potential of epigenetic variation: A comparison of heritable phenotypic variation in epiRILs, RILs, and natural ecotypes of Arabidopsis thaliana. Heredity 121, 257–265. doi:10.1038/s41437–018–0095–9
Zhao, N., Zhu, B., Li, M., Wang, L., Xu, L., Zhang, H., et al. (2011). Extensive and heritable epigenetic remodeling and genetic stability accompany allohexaploidization of wheat. Genetics 188, 499–510. doi:10.1534/genetics.111.127688
Zhao, W., Shi, X., Li, J., Guo, W., Liu, C., and Chen, X. (2014). Genetic, epigenetic, and HPLC fingerprint differentiation between natural and ex situ populations of Rhodiola sachalinensis from Changbai Mountain, China. PLoS One 9, e112869. doi:10.1371/journal.pone.0112869
Zheng, Y., Ding, Y., Sun, X., Xie, S., Wang, D., Liu, X., et al. (2016). Histone deacetylase HDA9 negatively regulates salt and drought stress responsiveness in Arabidopsis. J. Exp. Bot. 67, 1703–1713. doi:10.1093/jxb/erv562
Zhong, S., Fei, Z., Chen, Y.-R., Zheng, Y., Huang, M., Vrebalov, J., et al. (2013). Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat. Biotechnol. 31, 154–159. doi:10.1038/nbt.2462
Zhu, Z., Han, Z., Halabelian, L., Yang, X., Ding, J., Zhang, N., et al. (2021). Identification of lysine isobutyrylation as a new histone modification mark. Nucleic Acids Res. 49, 177–189. doi:10.1093/nar/gkaa1176
Keywords: epigenetic variability, DNA methylation, phenotypic plasticity, fitness, epigenetic memory, adaptive evolution, natural populations, climate change
Citation: Rajpal VR, Rathore P, Mehta S, Wadhwa N, Yadav P, Berry E, Goel S, Bhat V and Raina SN (2022) Epigenetic variation: A major player in facilitating plant fitness under changing environmental conditions. Front. Cell Dev. Biol. 10:1020958. doi: 10.3389/fcell.2022.1020958
Received: 16 August 2022; Accepted: 21 September 2022;
Published: 18 October 2022.
Edited by:
Suresh Kumar, Indian Agricultural Research Institute (ICAR), IndiaReviewed by:
Hui Xia, Shanghai Agrobiological Gene Center, ChinaCopyright © 2022 Rajpal, Rathore, Mehta, Wadhwa, Yadav, Berry, Goel, Bhat and Raina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vijay Rani Rajpal, dmlqYXlyYW5pMkBnbWFpbC5jb20=, dnJyYWpwYWxAaHJjLmR1LmFjLmlu; Shailendra Goel, c2hhaWxlbmRyYWdvZWxAZ21haWwuY29t; Vishnu Bhat, dmJoYXRAYm90YW55LmR1LmFjLmlu; Soom Nath Raina, c29vbXJAeWFob28uY29t
†ORCID: Vijay Rani Rajpal, orcid.org/0000-0001-6240-5028; Soom Nath Raina, orcid.org/0000-0002-4916-3359
‡Present address: Sahil Mehta, Department of Botany, Hansraj College, University of Delhi, Delhi, India
§These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.