 Adam Green1
Adam Green1 David M. Eckmann
David M. Eckmann- 1Department of Anesthesiology, The Ohio State University, Columbus, OH, United States
- 2Center for Medical and Engineering Innovation, The Ohio State University, Columbus, OH, United States
Mitochondria are cell organelles that play pivotal roles in maintaining cell survival, cellular metabolic homeostasis, and cell death. Mitochondria are highly dynamic entities which undergo fusion and fission, and have been shown to be very motile in vivo in neurons and in vitro in multiple cell lines. Fusion and fission are essential for maintaining mitochondrial homeostasis through control of morphology, content exchange, inheritance of mitochondria, maintenance of mitochondrial DNA, and removal of damaged mitochondria by autophagy. Mitochondrial motility occurs through mechanical and molecular mechanisms which translocate mitochondria to sites of high energy demand. Motility also plays an important role in intracellular signaling. Here, we review key features that mediate mitochondrial dynamics and explore methods to advance the study of mitochondrial motility as well as mitochondrial dynamics-related diseases and mitochondrial-targeted therapeutics.
Introduction
The origins of mitochondria are widely accepted to be traced to an ancestral endosymbiotic bacterium known as alphaproteobacterium (Gray et al., 2001). Phylogenomic analyses have traced the origin of eukaryotes to a group of Archaea, coined Asgards (Spang et al., 2015), and have also confirmed the connection between the mitochondrial endosymbiont and alphaproteobacterial ancestor (Wang and Wu, 2015). The transformation into mitochondria occurred approximately 1.5 billion years ago and altered forms started to appear after the evolutionary paths began to diverge (Mentel et al., 2014). The current human mitochondrial proteome has an estimated 15%–20% relation to the original endosymbiont DNA (Gabaldon and Huynen, 2007). Theories and debates regarding the exact origin are still ongoing.
Mitochondria are commonly known as the “powerhouse” of the cell, but this overly simplistic description ignores many important aspects of these intracellular organelles. Mitochondria are indeed the intracellular site for aerobic respiration and the subsequent synthesis of ATP. Extensive research over the past half a century has revealed numerous biochemical processes involving mitochondria such as protein synthesis, cell metabolism, cell death, calcium signaling, and gene expression in the nucleus that are critically important in the survival of eukaryotes (Mposhi et al., 2017; Vakifahmetoglu-Norberg et al., 2017; Patra et al., 2021).
Mitochondria are not static structures, but in fact they are dynamic. They have the ability to change their morphology through fusion and fission events and they are also motile with the ability to relocate within the cell to meet energy demands. The field of mitochondrial dynamics was founded over a century ago through Lewis and Lewis’s observations with light microscopy showing that “any one type of mitochondria such as a granule, rod or thread may at times change into any other type or may fuse with another mitochondrium [sic], or it may divide into one or several mitochondria.” (Lewis and Lewis, 1914). Technological advancements in microscopy with dyes or targeted fluorescent proteins over the last 30–40 years have made it easier to track mitochondrial movement in live cells (Kandel et al., 2015; Higuchi-Sanabria et al., 2016; Liu et al., 2017). Mitochondrial motility has been most heavily studied in neurons, since neuronal mitochondria travel long distance in axons and dendrites along microtubule tracks via motor proteins and adaptors (Ehlers, 2013; Lin and Sheng, 2015). The strategic localization of mitochondria at particular subcellular sites is necessary for energy use and calcium signaling. Thus, neuronal activity regulates the movements of mitochondria across the axonal and dendritic arbors. Mitochondrial transport in neurons is bidirectional with anterograde and retrograde directions (Mandal and Drerup, 2019). Furthermore, movement of neuronal mitochondria is not continuous due to frequent pausing for extended periods of time in sites with high energy demand (Spillane et al., 2013) and with excess of cytosolic calcium, such as synapses (Yi et al., 2004; Zhang et al., 2010). Finally, the moving pattern of mitochondria is wiggly due to mitochondrial dynamics including constant fusion and fission.
Mitochondrial fission is the process through which mitochondria divide into two daughter organelles, whereas mitochondrial fusion is the combining of two separate mitochondria into a larger one. The significance of fission and fusion events was not well characterized or understood until the discovery of mutations in fusion and fission that caused human diseases and correlated strongly with mitophagy and apoptosis (Lin and Beal, 2006; Pieczenik and Neustadt, 2007; Chistiakov et al., 2018). The dynamic behavior of mitochondria requires a balance between fusion and fission in order to maintain equilibrium when responding to constantly changing physiological conditions. In order for this balance to be maintained, mitochondria rely on a number of key fusion and fission proteins for the creation of mitochondrial networks or fragmentation as well as movement within the cell or axon.
In this review we provide an overview of mitochondrial dynamics, including fission, fusion, and motility, while bringing focus to the molecular mechanisms driving these mitochondrial dynamic events as well as the diseases that occur when such mitochondrial dynamics are disrupted. The opportunity for therapeutic interventions based on specific molecular factors is also discussed.
Proteins involved in fusion and fission
Key proteins involved in mitochondrial fusion and fission are included in Table 1. The machinery that is directly responsible for the important dynamic activity recognized as mitochondrial fission and fusion all belongs to the same family of highly conserved large dynamin-related GTPase proteins (DRPs) that control membrane remodeling events with their ability to hydrolyze GTP and self-assemble (Praefcke and McMahon, 2004). The first-identified member of the DRP family, dynamin, is best studied for its function in clathrin-mediated endocytosis where it mediates scission of the neck of clathrin-coated vesicles from the plasma membrane (Faelber et al., 2013). Mitochondrial fission in mammals is similarly catalyzed by a cytosolic DRP known as dynamin-related protein 1 (DRP1) (Smirnova et al., 2001; Praefcke and McMahon, 2004). The yeast ortholog of DRP1, Dynamin-1 (Dnm1), is also required for mitochondrial fission (Ingerman et al., 2005). The exact mechanism through which Dnm1 drives membrane constriction and mitochondrial fission events is unclear, but it is postulated that the spiral-like structures that Dnm1 forms possess dimensions similar to that of mitochondrial constriction sites and drive mitochondrial division (Tieu and Nunnari, 2000; Ingerman et al., 2005).
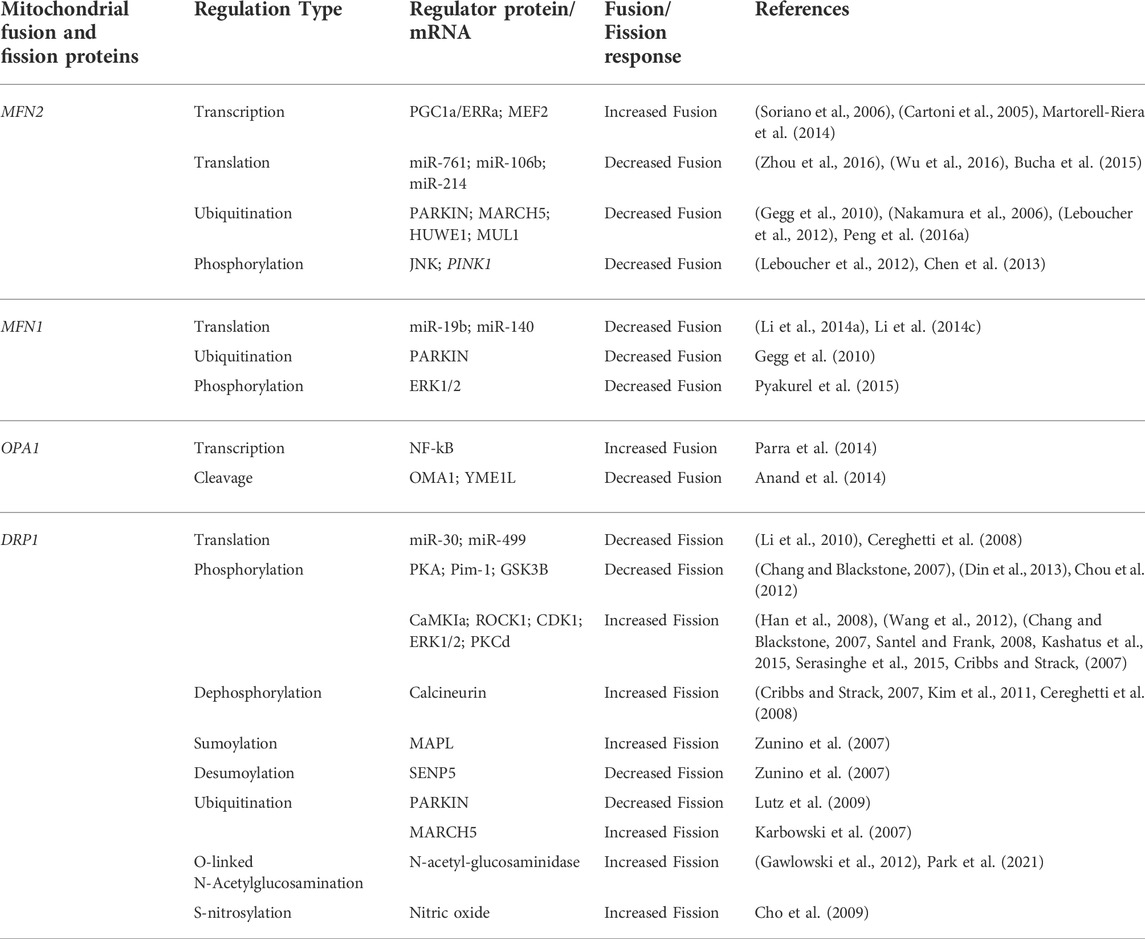
TABLE 1. Mitochondrial fusion and fission regulator proteins.
Three proteins have been discovered in yeast to have a direct effect on mitochondrial fusion. Two of these are outer-membrane proteins, fuzzy onions (Fzo1) and Ugo1 and the third is the inner-membrane protein, Mgm1. The mammalian orthologs of these proteins are mitofusins 1 (MFN1) and 2 (MFN2) in relation to Fzo1 and optic atrophy protein (OPA1) in relation to Mgm1. The loss of any of the genes for these proteins leads to a disruption in the balance of mitochondrial fission and fusion and results in severe pathophysiological consequences. Mouse models lacking MFN1, MFN2 (Chen H. et al., 2003), OPA1 (Davies et al., 2007), or DRP1 (Wakabayashi et al., 2009) are embryonic lethal.
Fzo1 and MFN1 and MFN2
A study published in 1997 showcased the Fzo1 gene, the first known regulator of mitochondrial fusion through the molecular genetic analysis of male Drosophila melanogaster (Hales and Fuller, 1997). Mutation in Fzo1 caused male sterility and defects in mitochondrial fusion leading to the finding that Fzo1 is required for mitochondrial fusion during spermatogenesis (Hermann et al., 1998; Rapaport et al., 1998). Stemming from this finding was the discovery of the core components of the mitochondrial fusion machinery - Fzo1 in yeast and its mammalian orthologs MFN1 and MFN2. As DRPs, they were originally characterized as containing a GTPase domain, a middle domain, and a C-terminal assembly or GTPase effector domain (GED) (Koshiba et al., 2004). New insights from structural studies have revised the organization of DRP1 as well as other proteins in the dynamin family (Frohlich et al., 2013). The large N-terminal GTPase domain is followed by the middle domain, a helix bundle region and two transmembrane segments. These are connected by a small and functionally important loop containing amino acids known as the variable domain (VD) in the intermembrane space at the C-terminus. Both the N and C termini of DRPs face the cytosol (Hinshaw, 2000; Chen H. et al., 2003; Eura et al., 2003). Recent information however posits that the C terminus resides in the intermembrane space instead (Mattie et al., 2018). The positions of the termini within the cell have a functional importance as described in the below sections. The positions of the protein termini within the cell have functional importance as will be described in the sections below. There remains need for additional information to be ascertained regarding this discovery in order for there to be full understanding of these structure-function relationships involved in mitochondrial fusion (Giacomello and Scorrano, 2018). Fzo1 also contains three heptad repeat regions which are likely to be important for inter- and intra-molecular interactions and mitochondrial tethering (Koshiba et al., 2004). While MFN1 and MFN2 are distinct, they are still highly structurally related. Each is able to support mitochondrial fusion independent of the other, suggesting partial redundancy in their functions (Chen H. et al., 2003). MFN1 has displayed higher GTPase activity than MFN2, whereas MFN2 has been specifically implicated in oxidative metabolism, ER-mitochondria tethering, insulin signaling, mitophagy, and apoptosis. MFN2 gene mutations can lead to autosomal dominant Charcot-Marie Tooth disease, which is discussed in greater detail in the diseases section of this review.
Mgm1 and OPA1
The proteins Mgm1 and OPA1 are DRPs which reside in the mitochondrial inner-membrane and which have exposure to the intermembrane space (Olichon et al., 2002; Wong et al., 2003; Meeusen et al., 2006). Mgm1, which is tethered to the inner-membrane by its N-terminal transmembrane domain, is necessary for both outer and inner mitochondrial membrane fusion. Any interruption of the Mgm1 gene can cause mitochondrial fragmentation and loss of mitochondrial DNA (mtDNA) (Wong et al., 2000; Liang et al., 2018). OPA1 has also been shown to be crucial for mitochondrial fusion (Alexander et al., 2000; Yarosh et al., 2008). Originally discovered in a study of gene mutation screening of autosomal dominant optic atrophy (Alexander et al., 2000), at least eight mRNA variants are generated in vivo as a result of alternative splicing of exons 4, 4b, and 5b. These alternative splices have relatively unknown functions but appear to be functionally important in preserving mtDNA content, energetics, and cristae structure, as well as having a role in mitochondrial dynamics. For more information regarding potential functions of these mRNA variants, we direct interested readers to recently published in-depth reviews (Belenguer and Pellegrini, 2013; Del Dotto et al., 2018).
OPA1 has two distinct isoforms: short and long. The long isoform is generated after OPA1 is imported into the IMM. The precursor protein contains what is known as a mitochondrial targeting sequence. Cleavage of this sequence generates membrane-anchored long OPA1 isoforms (l-OPA) (Del Dotto et al., 2018). The generation of short isoforms (s-OPA) is a result of proteolytic processing of OPA1 by OMA1 and YME1L, creating a balance between the two (Ali and McStay, 2018). It was initially believed that both l-OPA and s-OPA were required for mitochondrial fusion (Herlan et al., 2004), but the function of s-OPA is a controversial matter. It is generally considered now that the long isoform is sufficient for fusion and that the soluble short isoform may be necessary to facilitate mitochondrial fission (MacVicar and Langer, 2016; Ban et al., 2018). However, recent work still indicates that s-OPA plays a role in regulating mitochondrial fusion (Wang et al., 2021).
Mgm1 also has short and long isoforms. After outer membrane fusion has occurred, the Mgm1 isoforms come together to form a functional heterodimeric unit that mediates mitochondrial fusion (Zick et al., 2009). It is proposed that the long Mgm1 isoform is responsible for tethering the opposite inner membrane during fusion whereas the short isoform initiates lipid bilayer mixing in a GTPase-dependent manner (Meeusen et al., 2006; DeVay et al., 2009). It also contributes to local membrane bending, which is mechanically necessary for fusion to occur (Rujiviphat et al., 2015).
While the significance of the different molecular OPA1 species remains unclear, it is known that both the long and short forms are necessary for full functionality of the protein, as is also the case with Mgm1. Mgm1/OPA1 have also been shown to play a role in maintaining the structure of ATP synthase, cytochrome c storage within cristae, and cristae morphology (Amutha et al., 2004; Del Dotto et al., 2018).
Ugo1
Ugo1 is a non-DRP outer-membrane tethered protein containing three transmembrane domains and is specific to yeast belonging to the mitochondrial transporter family. It was initially detected while screening for yeast mutants that lost mtDNA in Dnm1-dependent mitochondrial fission (Sesaki and Jensen, 2001). Ugo1 is proposed to be the bridge between Fzo1 and Mgm1. The N-terminus of Ugo1 faces the cytosol and the C-terminal domain faces the intermembrane space. Interactions between Fzo1 and Mgm1 requires Ugo1 to be present (Sesaki and Jensen, 2001). Studies have shown that OPA1 requires MFN1 to be present for mitochondria to engage in fusion (Alexander et al., 2000; Cipolat et al., 2004; Yarosh et al., 2008). However, as it relates to Ugo1, there is no mammalian equivalent that has been identified as a bridge between OPA1 and MFN2. More research is needed to identify the exact mechanism of the proteins’ interactions. Future work also remains to be done to determine the specific mechanisms by which Ugo1 functions in mitochondrial fusion.
Mitochondrial fission proteins
There are four key proteins in yeast involved in mitochondrial fission: Dynamin-1 (Dnm1), Mitochondrial fission protein 1 (Fis1), and adaptor proteins Mdv1 and Caf4.
Dnm1 and DRP1
Dnm1 and its mammalian equivalent, DRP1, constitute the central components of mitochondrial fission in the majority of eukaryotic organisms. Dnm1 was originally discovered through the screening of yeast mutants with defective mitochondrial morphology (Hales and Fuller, 1997). DRP1 contains four identified functional domains. A large N-terminal GTPase domain, a middle domain, the intrinsically disordered variable domain (VD) which is also referred to as the B-insert (Lu et al., 2018), and a C-terminal GED. The middle domain and C-terminal GED form the stalk region consisting of a three-helix bundle known as the bundle signaling element (BSE) that connects to the GED to form a four-helix bundle (Wenger et al., 2013). The VD is a lipid-binding region of approximately 100 amino acids that is found between the third and fourth α-helices of the stalk (Kalia et al., 2018; Kalia and Frost, 2019; Banerjee et al., 2022). The VD a critical component for DRP1 function and regulation. Intramolecular interactions between the GTPase and GED regions are necessary for fission to be fully functional (Zhu et al., 2004). DRP1 exists mostly as a cytosolic dynamin family member and is recruited to punctate structures on the mitochondrial surface. It then forms spirals to constrict and sever both the outer and inner mitochondrial membranes. DRP1 and Dnm1 recruitment is reliant on certain accessory proteins to be successful. These accessory proteins include mitochondrial fission 1 (Fis1) and mitochondrial division protein 1 (Mdv1) (Zhang et al., 2012).
Fis1, MFF, MiD49 and MiD51
Fis1 is a protein in the outer membrane of mitochondria that recruits Dnm1 to the surface via a molecular adaptor (either Mdv1 or Caf4). Mdv1 acts as a protein bridge between Fis1 and Dnm1 by binding its N-terminus to Fis1 and its C-terminus to Dnm1 (Zhang et al., 2012). The Fis1-Mdv1-Dnm1 complex is not the only pathway for recruitment of Dnm1 to mitochondria in yeast. Caf4 is a Mdv1 paralogue with a similar structure. While Mdv1 is present, Caf4 is unnecessary for fission to occur, however, in mutants lacking Mdv1, it acts as a placeholder for fission activity (Griffin et al., 2005). The mechanism and biological importance surrounding Caf4 is still unclear and requires more research. The same can be said for the mechanisms underlying fission in mammalian models (Figure 1). Fis1 interacts with DRP1 in a similar way to its yeast ortholog Dnm1, as overexpression of Fis1 leads to the fragmentation of mitochondria and interconnected mitochondrial networks with Fis1 depletion (James et al., 2003; Yoon et al., 2003). A Mdv1 equivalent has yet to be discovered outside of yeast models, and human Fis1 knockdown experiments have demonstrated there to be little effect on DRP1 distribution in mitochondria (Lee et al., 2004). This has led to the assumption that there are alternative pathways for DRP1 recruitment in metazoans.
FIGURE 1. Mitochondrial Fission. (A) Fis1 and potential DRP1 recruitment candidates MiD 49, MiD 51, and MFF reside on the OMM with most of the protein facing the cytosol. DRP1 exists in both the cytosol and in punctate spots on mitochondria (not shown). (B) An initial constriction of mitochondrial tubules occurs at sites of endoplasmic reticulum contact independent of DRP1. (C) Mitochondrial fission proceeds as DRP1 is recruited to the OMM and further constricts the mitochondrial tubule.
Other studies have shown promise for possible candidates for recruiting DRP1, including Mitochondrial Fission Factor (MFF) (Gandre-Babbe and Van der bliek, 2008; Otera et al., 2010), and Mitochondrial Dynamics proteins 49 (MiD49) and 51 (MiD51) (Palmer et al., 2011). The complex interplay between these various species makes the physiological role of Fis1 in mitochondrial fission, mitophagy, and apoptosis a subject of controversy, given that the dominance and specific roles of Fis1, MFF, MiD49/51 continue to be clarified. Fis1 has historically been regarded as a key component of mitochondrial fission due to its role in trafficking DRP1 to the mitochondria. However, this is being challenged since Fis1 deletion in some, but not all cell types, is associated with mitochondrial elongation (Ihenacho et al., 2021). This imbalance between fusion and fission has increased interest in the roles of MFF, MiD49 and MiD51. MFF is currently regarded as the most important regulator of fission as a result of knockdown studies presenting more extreme elongation of mitochondria (Loson et al., 2013). Studies within the last decade of MFF’s role in mitochondrial fission indicate its ability to recruit DRP1 independent of Fis1 (Loson et al., 2013). In addition, MiD49 and MiD51 can act independently of both Fis1 and MFF in trafficking (recruitment of) DRP1 (Loson et al., 2013). Studies suggest that MiD49 and MiD51 interact with DRP1 and MFF, serving as an adaptor to link them together (Palmer et al., 2013; Yu et al., 2017). The data reveal that they work in tandem and that the level of MiD49/51 creates a balance between mitochondrial fusion and fission. While the particular implications for their mitochondrial dynamics influences remain to be demonstrated, greater detail regarding the roles of MFF, MiD49 and MiD51 in interactions with fission machinery can be accessed in several excellent publications (Loson et al., 2013; Kraus and Ryan, 2017; Samangouei et al., 2018; Ihenacho et al., 2021).
Organelle and cytoskeletal interactions
Endoplasmic reticulum
The endoplasmic reticulum (ER) interacts with mitochondrial membranes at ER contact sites (see Figure 1). These contact sites have been shown to be critical in calcium signaling, phospholipid synthesis, and mitochondrial constriction (i.e., membrane indentation), marking a mitochondrion for fission (de Brito and Scorrano, 2010; Friedman et al., 2011). Initiation of mitochondrial constriction induced by contact with the ER is an essential step toward mitochondrial fission, as DRP1 oligomers alone are unable to induce fission due to mitochondrial size (Friedman et al., 2011). MFN2 has also been found to localize at mitochondrial-ER contact sites where it promotes the fusion of mitochondrial membranes (Koshiba et al., 2004; Abrisch et al., 2020). The emergence of information regarding MFN2 as a crucial regulator of mitochondrial-ER tethering as well as mitophagy has promoted interest in conducting studies regarding mitochondrial diseases and therapeutics, particularly in regard to aging and age-related illness (Moltedo et al., 2019).
Microtubule interactions
The highly motile nature of mitochondria in mammalian cells is chiefly motivated by molecular motors that actively translocate mitochondria along the microtubule cytoskeleton. In some cell types, approximately 80% of the cell volume is explored by mitochondria within 15 min (Valm et al., 2017). Microtubules are radially organized with their plus ends spreading outward toward the cell periphery, and their less dynamic minus ends clustered at or close to the microtubule-organizing center. Observation of mitochondrial movement in neurons identified proteins, kinesin and dynein, that regulate movement toward these microtubule ends. Dynein and kinesin are in a balancing act with each other and the net directionality of mitochondrial movement resulting from this “tug-of-war” is ultimately determined by the relative number of attached and active dynein and kinesin molecules.
Dyneins
Cytoplasmic dynein and its activator dynactin, an elaborate multiprotein complex, drive motility inward toward the minus-end of microtubules and act as the primary retrograde motor determined through the same functional analysis techniques as with kinesin-1 (Schnapp and Reese, 1989; Vale, 2003; Pilling et al., 2006), described below. Dyneins are structurally more complex than kinesins and contain a large number of light chains. Dynactin associates with microtubule plus and minus ends and acts as a link to dynein resulting in a dynein-dynactin complex (Schroer, 2004). Additionally, coiled-coil proteins known as activating adaptors are required for the activation of dynein-dynactin motility (Reck-Peterson et al., 2018). Mutations in dynein have been linked to neurological diseases consistent with mitochondrial defects. There is a study however that claims that, in Drosophila, dynein heavy chain 64C is the primary motor for both anterograde and retrograde transport of mitochondria (Melkov et al., 2016). This requires further study before any definitive conclusion can be reached.
Kinesins
Kinesins, which face the synaptic terminal of a nerve cell ending, are the proteins that direct mitochondrial motion toward the plus-ends of axons (Hirokawa et al., 2009). Kinesin-1 was identified as the primary anterograde motor for long-distance intracellular trafficking according to functional inhibition and biochemical studies (Hurd and Saxton, 1996; Pilling et al., 2006). These studies, however, also showed a significant reduction in retrograde flux, controlled by cytoplasmic dynein, in the mutant Drosophila. This suggests a possible functional interdependence between mitochondria-associated dynein and kinesin-1. Since the kinesin-1 mutant showed no signs of mitochondrial fission in the axons, dynein-dynein retrograde transport does not occur because kinesin-1 is necessary for mitochondria to be carried into the axon (Pilling et al., 2006). This theory is backed up by showing a 50.5% decrease in the number of mitochondria in the mutant nerves. It should be noted as well that in tissue culture specifically, there are studies that have shown that kinesin-3 motors Klp6 and Kif1b also transport mitochondria, leading researchers to conclude that it also does so in axons (Nangaku et al., 1994; Tanaka et al., 2011).
Milton and Miro
The best-studied and understood adaptors for associating kinesin-1 with mitochondria are Milton and Miro. Kinesin-1 lies within the kinesin heavy chain (KHC) and interacts with Milton, a kinesin-binding protein crucial for the mediation of mitochondrial motility discovered through genetic screening for Drosophila mutants with impaired neuronal function (Stowers et al., 2002). Milton binds directly to the C-terminal cargo-binding domain of the KHC. Studies also indicate that a mutated Milton isoform inhibits retrograde mitochondrial transport from nurse cells into future oocytes during oogenesis in Drosophila (Cox and Spradling, 2006). This shows that Milton may exert an effect on dynein specific to that isoform.
The mammalian homologs, trafficking kinesin proteins 1 (TRAK1) and 2 (TRAK2), similarly engage with kinesin-1 and dynein/dynactin to induce mitochondrial motility. We have demonstrated that TRAK1 plays an important role in mitochondrial trafficking in a study that identified deleterious variants in TRAK1 from encephalopathic patients (Barel et al., 2017). TRAK1-deficient fibroblasts from this study displayed unorthodox mitochondrial distribution, diminished membrane potential, modified mitochondrial motility, and reduced mitochondrial respiration. TRAK2 has recently been demonstrated to form a complex containing both kinesin and dynein-dynactin through co-immunoprecipitation and colocalization experiments. Thus it is thought that TRAK2 acts as an interdependent motor complex that provides integrated control over the two opposing motors (Fenton et al., 2021).
Miro is a mitochondrial Rho-like GTPase containing calcium binding motifs. It is an integral mitochondrial outer membrane protein that interacts with the scaffolding proteins Milton/TRAKs 1 and 2 (Fransson et al., 2003; Guo et al., 2005). Unique to other Rho GTPases, Miro-1 and Miro-2 have been shown to have roles in mitochondrial homeostasis, most likely affecting the trafficking apparatus (Vale, 2003). Miro may be preferentially needed for retrograde mitochondrial transport (Melkov et al., 2016). Mutation of either Milton or Miro genes halts any anterograde mitochondrial axon transport, depleting mitochondria in the axon and synapse leading to an aggregation of mitochondria and eventual apoptosis. Miro has also been found to be required for mitochondrial transport from axons to synapses in Drosophila (Guo et al., 2005). Mutant flies lacking Miro display mislocalized mitochondria in both muscles and neurons as well as a lack of presynaptic mitochondria in neuromuscular junctions. These findings suggest that Miro plays an important role in transporting mitochondria to nerve ends.
Other adaptor complexes that have been associated with a linkage between kinesin and mitochondria include kinectin, an integral endoplasmic reticulum (ER) membrane protein that targets kinesin to the ER membrane (Santama et al., 2004), and syntabulin, which contains a syntaxin-independent mitochondrial binding site. When syntabulin is inhibited, anterograde mitochondrial movement is disrupted (Cai et al., 2005).
Syntaphilin
Syntaphilin (Snph) anchors mitochondria to microtubules, rendering them stationary. Snph behaves as an engine off-switch by detecting Miro-Ca2+ and it acts as a brake by tethering mitochondria to the microtubule track (Ohno et al., 2014). Snph facilitates the survival of demyelinated axons by increasing the volume of static mitochondria. The deletion of axonal Snph significantly increases the deterioration of demyelinated central nervous system axons indicating a need for mitochondrial stationary sites to meet the increased energy demands of an unmyelinated axon (Ohno et al., 2014).
Intermediate filaments
Links between intermediate filaments (IFs) and mitochondria appear in the colocalization of mitochondria with neuronal IFs (Wagner et al., 2003). There is also evidence that IFs are involved in mitochondrial tethering. IFs interact with mitochondria by surrounding the organelle by confining or binding the mitochondrial membrane via a protein known as plectin (Schwarz and Leube, 2016). Mutations in IF proteins result in the disruption of IF networks. The deletion of desmin, an IF protein, alters the morphology, distribution, and respiratory function of mitochondria in striated muscle cells (Milner et al., 2000). In addition, desmin deletion inhibits kinesin-1 recruitment to mitochondria in heart muscle (Linden et al., 2001). Vimentin IFs (VimIFs) affect the transportation and tethering of mitochondria. Knock-Out of VimIFs in fibroblasts showed altered mitochondrial distribution, and increased motility (Nekrasova et al., 2011). The modulation of mitochondrial motility due to VimIFs in cultured cells is directly related to its interactions between mitochondria and cytoskeletal structures. More research needs to be done regarding the regulation between IFs and mitochondria. As of now there is no conclusive evidence for IF-associated motor molecules and IFs might have more influence over microtubule motors than is in the literature.
Actin interactions
A 1995 study was the first to show that mitochondria directly interact with the actin cytoskeleton, and that there is an implication in motility (Simon et al., 1995). The actin cytoskeleton guides mitochondrial translocation in simple eukaryotes such as budding yeast and plays pivotal and diverse roles mediating the mitochondrial network and function. While long-range mitochondrial motility is directed by microtubules, the actin cytoskeleton specializes in coordinated short range mitochondrial motion (Quintero et al., 2009) and anchoring (Pathak et al., 2010). This is carried out by myosins, which are a family of actin-dependent motor molecules.
Myosin
Myosin is a motor protein known mostly for its role in muscle contraction and is ATP-dependent and responsible for actin-based motility. Myosin directs movement toward actin filament plus-ends, such as myosin V (Altmann et al., 2008), or minus-ends like myosin VI (Lister et al., 2004). There are also some indications that Myo2 may be involved in mitochondrial motility more than indirectly (Altmann et al., 2008). While there is a large number of actin-mitochondria interactions, there lacks critical information as to how mitochondria are linked to the filamentous actin (F-actin) cytoskeleton. The best evidence for actin-based mitochondrial motility comes from the myosin motor Myosin XIX (Myo19). Similar to myosin V, it is a plus-end directed motor but it is known to localize specifically to the OMM to drive mitochondrial motility (Lu et al., 2014). While it is an example of an actin binding protein interacting directly with the OMM, its occurrence is limited to the starvation induced filopodia in select cell lines, and does not provide any further information as to how mitochondria are tied to actin in other situations. Actin-based mitochondrial motility is still under much scrutiny as the mechanism by which it affects motility is ambiguous. Studies provide information on opposing motor dynamics without information as to how it occurs. Myosin V is said to promote motility in one study (Hollenbeck, 1996) while F-actin and Myosin V are said to be resistors of mitochondrial motility (Pathak et al., 2010; Gutnick et al., 2019).
Evidence for mitochondrial dynamics
Advances in imaging techniques and technology have enabled mitochondrial fusion and fission events to be quantitatively ascertained and visually observed. The best indirect methods for the assessment of mitochondrial fusion and fission in mammalian cells include cell-cell fusion, photoactivatable mitochondrially target Green Fluorescence Protein (mitoGFP), and fluorescence recovery after photobleaching (FRAP). Cell-cell fusion is a technique used to observe the mixing of mitochondrial matrices in mammalian cells. It was experimentally induced between two cell populations labeled with two differing mitochondrial markers, either mitoGFP or mitochondrially targeted Red Fluorescence Protein (Legros et al., 2002; Malka et al., 2005).
FRAP was developed in order to observe mitochondria under in vivo-like conditions by avoiding potential artifacts from membrane-altering agents. FRAP is performed by photobleaching fluorescent molecules within a subcellular area and monitoring its recovery of fluorescence in the bleached zone by observing the movement of organellar structures, in this case mitochondria, from an unbleached area. Information acquired from such experiments includes the mobility of the fluorescent molecule, mitochondrial continuity, and mitochondrial dynamics (e.g., motility, fusion, fission). Mitochondrial connectivity is confirmed with rapid recovery of fluorescence while a failure of recovery is an indication of mitochondrial fragmentation or discontinuity (Collins et al., 2002). An example of this approach is its use in determining that CED-9, a C. elegans Bcl-2 homolog, bolsters mitochondrial fusion in HeLa cells (Delivani et al., 2006).
Use of photoactivatable fluorophores such as mitoGFP is widely used to assess mitochondrial fusion and fission events. Photoactivatable mitoGFP is designed to display a 100-fold increase in green fluorescence after laser activation (Patterson and Lippincott-Schwartz, 2002) and can be fused to a mitochondrial targeting sequence. This approach was used in a study that showed Bcl-2 family proteins BAX and BCL2 antagonist/killer (BAK) to have direct roles in mitochondrial fusion (Karbowski et al., 2006). An observation of a loss of photoactivated fluorescence in an area can be indicative of mitochondrial fusion events if one assumes that this loss is a representation of fluorescent diffusion into a neighboring mitochondria, while the opposite can be said for mitochondrial fission. The best markers are considered to be mitoGFP, mitoRFP, and mitochondrially targeted Yellow Fluorescence Protein (mitoYFP), as they have excitation peaks at a longer wavelength to avoid possible mitochondrial damage that a more energetic and shorter wavelength would provide during recording of a time-lapse microscopy-based assessment.
A more direct method uses photoconvertible fluorophores, which differ from photoactivatable fluorophores by emitting fluorescence in their non-converted state. Dendra2 was the first monomeric red-to-green photoconvertible protein to be commercially developed, and it was used to create the mito-Dendra2 mouse model. The mito-Dendra2 mouse model has been extensively used to track mitochondrial fusion and fission in vivo and ex vivo in skeletal muscle fiber (Mishra et al., 2015), cerebral vasculature (Rutkai et al., 2020), and hematopoietic stem cells (Takihara et al., 2019), among many other cells and tissues. The mito-Dendra2 mouse has also been used to display mitochondrial transfer activities from early stage erythroblasts to macrophages in vitro (Yang et al., 2021), and axonal mitochondrial dynamics in regenerative response to injured neurons (Han et al., 2016).
Characteristics of mitochondrial motility
The ability to quantify mitochondrial motility has been largely driven by advances in live cell imaging techniques and image processing. A number of fluorophores have been developed which are specific for mitochondria or which accumulate in mitochondria based on membrane potential. These dyes can be used to localize mitochondria within cells. When coupled with particle tracking-based image analysis of time-sequentially generated microscopy frames, motion of individual mitochondria can be determined. Kandel et al. (Kandel et al., 2015) utilized this approach and were able to demonstrate that a population of hundreds of mitochondria tracked simultaneously followed a log-normal distribution in their net displacement over time. These investigators found that motility was dependent on cytoskeletal structure, with motility increasing in the presence of depolymerized actin and motility decreasing with inhibition of microtubule production. Further work has demonstrated alterations in mitochondrial motility in cells undergoing various forms of environmental or chemical stress such as rapid decompression from hyperbaric conditions (Jang et al., 2018) and carbon monoxide poisoning (Owiredu et al., 2020) as well as critical illness (Jang et al., 2019). Both molecular and environmental therapies have been shown to have capabilities to restore disturbed motility to baseline values (Ranganathan et al., 2020; Green et al., 2021). These findings demonstrate that motility characterization is potentially a useful clinical index of mitochondrial dynamics dysfunction and this raises the possibility of using motility assessment for surveillance of disease progression or effectiveness of therapeutic intervention.
Methods to assess mitochondrial motility
Assessment of mitochondrial motility relies primarily on imaging-based methods to track individual mitochondrial movements. Mitochondrial tracking is technically challenging owing to the small size and complexity of their movements. Tracking individual mitochondrial motion manually is time-consuming and extremely prone to errors. Most studies have traditionally focused on the tracking of neuronal mitochondria because they are more sparse and hence can be individually well-resolved, making them most suitable to be tracked. Some methods for ascertaining mitochondrial motility in generalized cell types are described in a paper from De Vos and Sheetz (De Vos and Sheetz, 2007). The first method relies on use of two consecutive images of a cell from which the non-overlapping region stained with a fluorescent dye is measured as a whole-cell index of mitochondrial motility. Another method utilizes a kymograph, which evaluates space versus time images for individual mitochondria.
Confocal microscopy (Koopman et al., 2006; Bros et al., 2015) and time-lapse fluorescence microscopy imaging (Kandel et al., 2015; Chalmers et al., 2016) are the most commonly used methods to image cells that are stained with a fluorescent dye specific to mitochondria. Successive images are acquired over varying amounts of time depending on the specific methodology being used. In a majority of the techniques, the sequence of images is made into a stack that is processed by a third-party program or algorithm for image analysis. Resultant visualizations of individual mitochondria can be observed and specific measurements of mitochondrial displacements can be determined.
For purposes of analyzing mitochondrial motility through image processing, there are a few noteworthy methods. The Mitochondrial Network Analysis (MiNA) (Valente et al., 2017), which can be accessed at https://github.com/StuartLab/MiNA, utilizes the freeware program ImageJ to quantitatively describe the morphology of mitochondria present in fluorescence micrographs. This software is optimized for 2D images, but it has some limited functionality for 3D, especially if it is used for simple visualization purposes. MiNA does not enable any temporal analysis to be performed since it only outputs cell-wide aggregate mitochondrial data.
Mitograph (Viana et al., 2015), accessible at https://github.com/vianamp/MitoGraph, is a program that is fully automated for analyzing the 3D morphology, volume and topology of mitochondria in living cells. This software has been optimized and validated for tubular mitochondrial networks in budding yeast (Viana et al., 2020), which may limit its use for other applications. With additional validation it may be applied to the study of other cell types. Only a single temporal frame can be viewed with this package, thus restricting the use of this imaging method for making any measurements of motility. The program runs exclusively through command lines and is a free software written in C++; it does not have a graphical user interface (GUI).
Mitoe (Lihavainen et al., 2012), a freely available resource, is online, located at https://sites.google.com/view/andreribeirolab/home/software,. It can be used to analyze mitochondrial dynamics and structure from 2D fluorescence microscopy images. It is not clear how a temporal sequence of images is handled by the optical flow algorithm, which analyzes two adjacent frames at a time but apparently does not record displacement tracks established between frames. Also, multiple image stacks cannot be analyzed simultaneously with this software, as only a single frame from each temporal stack of images gets processed. Individual analysis of stacks must be performed first and then they can subsequently be manually aggregated. The program is written in MATLAB, however a standalone Windows version that does not require MATLAB is available. One user challenge with this software is that the MATLAB GUI is not fully operational.
Our own Mitochondrial Single Particle Tracking (MitoSPT) (Kandel et al., 2015), is available for download at https://github.com/kandelj/MitoSPT. This program is written in MATLAB and has a user-friendly GUI. The algorithm, which is clearly annotated, utilizes analysis of 2D imaging to provide a depiction of mitochondrial motility, rates of fusion and fission, morphometric parameters (mitochondrial number, size) and statistical analysis for comparison between image stacks obtained from individual experimental conditions. In addition, the software has provision for subdivision of the measured parameters to be analyzed in two distinct intracellular regions, a thin perinuclear region and the remaining cell periphery, as depicted in Figure 2, rather than on a whole-cell basis (Jang et al., 2019; Green et al., 2021). This software was instrumental in the initial determination that mitochondrial motility follows a lognormal distribution as seen in Figure 2 (Kandel et al., 2015) and in demonstrating that motility in the cell perinuclear and peripheral regions is not identical, as is shown in Figure 2 (Jang et al., 2019; Green et al., 2021).
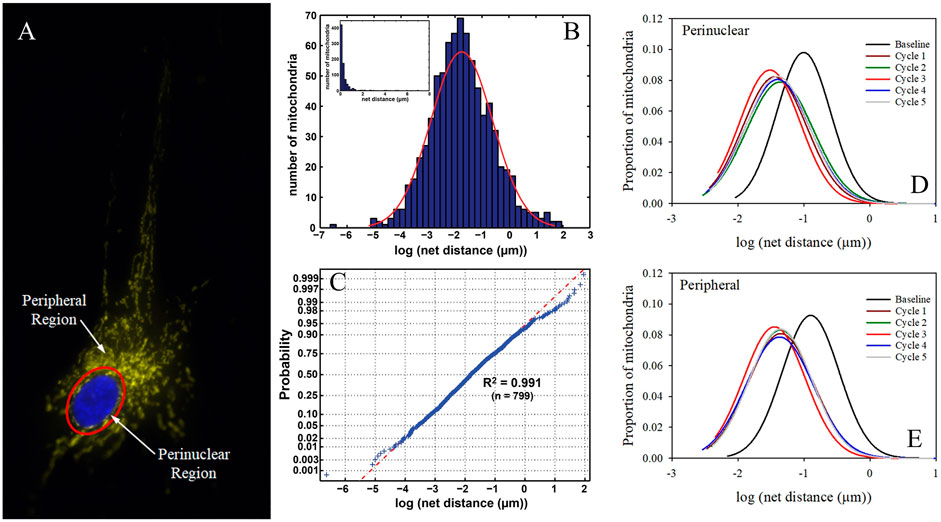
FIGURE 2. Intracellular partitioning of mitochondria and lognormal distribution of mitochondrial motility. (A) The image of a fibroblast shows a nucleus stained with DAPI (blue) and TMRM-stained mitochondria (yellow). The mitochondria are separated into peripheral and perinuclear regions by the image-analysis derived partition (red). (B) Example of distribution of net distances traversed by hundreds of mitochondria on a log scale, with inset showing linear scaling of the same data (Kandel et al., 2015). (C) Normal probability distribution of the log values of net mitochondrial distances traversed indicating that motility follows a lognormal distribution (Kandel et al., 2015). (D) Lognormal distribution of mitochondrial motility in cellular perinuclear region at baseline and following multiple hyperbaric oxygen exposures as detailed in (Green et al., 2021). (E) Lognormal distribution of mitochondrial motility in cellular peripheral region at baseline and following multiple hyperbaric oxygen exposures as detailed in (Green et al., 2021).
QuoVadoPro (QVP) (Basu and Schwarz, 2020) is also available online and can be found at https://github.com/ThomasSchwarzLab/QuoVadoPro, utilizes ImageJ macros to quantify movement of a fluorescently tagged organelle. The extent of intracellular movement is inferred by quantifying the variance of pixel illumination over a series of time-lapse images. QVP gives only a motility score as a motility metric on an image-wide basis and not by frame-by-frame quantification. Mitochondrial morphometrics are not analyzed nor are fission and fusion events. QVP is implemented as an ImageJ plugin.
Mitochondrial Segmentation Network (MitoSegNet) (Fischer et al., 2020), found at https://github.com/MitoSegNet, uses a pre trained deep learning model that segments mitochondria and analyzes mitochondrial morphometrics. There is no need for user-input parameters with this software, which handles batch input and thus eliminates bias. MitoSegNet analyzes 2D images from fluorescence microscopy but does not perform analysis of temporal variation. It can be used only to visualize and quantify individual mitochondrial morphometrics. This program is based in Python and runs on a standalone software platform.
MTrack2, found at https://github.com/fiji/MTrack2, utilizes ImageJ’s particle analyzer plugin to link particles between frames of 2D images. This is a fairly simple 2D tracking software package that gives centroid positions of objects in a binary image and tracks them over time. No morphology or fission and fusion events can be analyzed through this software. MTrack2 is an ImageJ plugin written in Java, and its GUI is relatively limited because it relies on ImageJ’s templates and results page to provide results output.
Regulation of mitochondrial morphology
Mitochondria form tubular networks that undergo changes in morphology to correspond with cellular needs through fusion and fission events. The shape of mitochondria can have an effect on their distribution to specific subcellular locations. This has been found to be especially important in neurons, in which small mitochondria are more efficiently transported to nerve terminals. It is however worth noting that smaller mitochondria do not always engage in efficient transport and that fragmented mitochondria resulting from a fusion impairment show signs of critical transport defects (Chen H. C. et al., 2003). Mitochondrial size and shape may also be directly related to bioenergetic function as elongated mitochondria have been shown to be correlated with more effective ATP production. The distinct connection between the shape of mitochondria and bioenergetic function still remains unclear.
Mitochondria are known to be responsive to cues that change their morphology. Under conditions of starvation and stress, stress-induced mitochondrial hyperfusion (SIHM) is triggered. During SIHM episodes, mitochondria will hyperfuse into large complexes to offer cytoprotection and enhanced resistance to apoptosis, increasing opportunity for the cell to return to homeostasis (Tondera et al., 2009). Apoptosis activation, oxidative stress, and cell senescence have also been shown to induce mitochondrial elongation. The elongation of mitochondria during the G1-S phase might play a role as a mediator in stress-induced premature senescence (Yoon et al., 2006). Nutrient excess on the other hand leads to mitochondrial network fragmentation, an apparent response to the luxury of available bioenergetics substrate (Liesa and Shirihai, 2013).
Content exchange in mitochondria
A primary function of mitochondrial fission and fusion is the promotion of the membrane and contents mixing between mitochondria. Kiss-and-run fusion events, or transient fusion, progress content exchange without having an effect on mitochondrial morphology (Liu et al., 2009). Labeled mitochondria have been shown to exchange matrix contents during these transient fusion events, which constitute nearly half of all fusion events resulting in the exchange of matrix proteins between mitochondria. This suggests that fusion pores between two mitochondria can open and close rapidly and furthers the suggestion that aside from the regulation of morphology, fusion plays a large role in content exchange between mitochondria.
The benefits of content exchange include the promotion of homogenization of the mitochondrial population as well as amelioration of injurious effects of heteroplasmic mtDNA mutations. Since the mtDNA only encodes for 13 polypeptides, most of the mitochondria’s proteasome is imported from the cytosol. The inhibition of mitochondrial fusion causes individual mitochondria to deviate in their properties, indicating that fusion assists in the reduction of variability between organelles. As most mtDNA mutations are recessive in nature, the mutational load must attain high levels of around 60–90% heteroplasmy before any appreciable dysfunction in the respiratory chain is encountered. The capacity to withstand these excessive levels of mtDNA mutations is extremely reliant on mitochondrial fusion (Chen et al., 2010).
Mitochondrial inheritance
Mitochondria cannot be created de novo; thus, they must be inherited by daughter cells through mitosis. Elongation of mitochondria occurs during the G1-S phase in tissue culture cells but then fragmentation begins at the G2 and M phases (Mitra et al., 2009). Mitotic phosphorylation of DRP1 promotes this fragmentation that enhances the rate of fission and results in the production of many more mitochondria during mitosis (Taguchi et al., 2007). This is important because mitochondria have affiliations with elements in the cytoskeleton that are dispersed throughout the cell (see above), and this fragmentation might assist with the even dispersal of mitochondria between the two daughter cells.
Mitochondrial morphology changes that are cell-cycle dependent are not as noticeable in yeast because their mitochondria are inherited in an organized manner by cytoskeleton-dependent bud-directed transport. A lack of Dnm1 in yeast mutants does not show a notable growth defect despite having dramatic morphological changes, even though, during cell division, their interconnected mitochondrial network must undergo division (Sesaki et al., 2014). As division still occurs, the mitochondria sustain no loss in functionality. Additionally, the knockout of DRP1 in mouse embryonic fibroblasts does not affect cytokinesis progression, however this results in embryonic lethality in mice (Ishihara et al., 2009). It is unclear whether the division of mitochondria in mutated cells is regulated by mechanical forces that act on the mitochondria during cytokinesis or by an unknown division machinery. Since mice lacking DRP1 die before they are birthed, there is an indication that mitochondrial dynamics are critical for development and survival (Song et al., 2017).
Disruption of fusion in yeast is associated with a rapid loss of mitochondrial genome and resultant defects in respiration. This is most likely due to fragmentation of mitochondria producing multiple mitochondrial fragments with many of them lacking mtDNA. The progeny produced from the partitioning of these mitochondria to daughter cells results in mitochondrial genomes being absent from the population after a few generations. Mitochondrial fusion makes certain that the loss of gene products and the mitochondrial genome lost due to fission are replaced before full functionality is impaired.
Mitochondrial DNA maintenance
Mitochondrial fission and fusion are both essential in the maintenance of mtDNA. With the loss of mitochondrial fusion through the silencing or removal of necessary proteins, cells display an extreme reduction in the amount of mtDNA they contain. Many of the mitochondria in mutant mammalian cells lack evidence of any mtDNA, which renders them unable to maintain oxidative phosphorylation (OXPHOS) activity (Chen et al., 2007). The loss of mitochondrial fusion in many cell types reduces OXPHOS activity, however it is unclear whether this defect is a product of the reduced mtDNA levels or if an additional mechanism akin to fusion is involved.
When mitochondrial fission is absent, the mtDNA nucleoids begin to aggregate and form large structures that distort mitochondrial tubules (Ban-Ishihara et al., 2013). This aggregation causes an unbalanced dispersion of mtDNA in the mitochondria, with studies using cardiomyocytes having shown that this results in mosaic OXPHOS deficits that promote cardiac arrhythmia during aging (Ishihara et al., 2015).
Mitophagy and apoptosis
Mitochondrial fission acts as a regulator, identifying poorly functioning organelles that display reduced mitochondrial membrane potential (Twig et al., 2008). Mitochondria may recover as discussed above through fusion and fission. Alternatively, mitochondria may be targeted for mitophagy, which is the degeneration of mitochondria by autophagy (Nguyen et al., 2016; Dikic and Elazar, 2018). Mitophagy is an adaptive survival mechanism that prevents the cell from undergoing apoptosis and allows the cell to maintain a healthy pool of mitochondria for sustained energy production. Autophagic machinery within the cell recognizes dysfunctional mitochondria, which become engulfed by autophagosomes and are subsequently delivered to lysosomes for degradation. There has been important recent work clarifying the relationship between mitochondrial fission and mitophagy. While DRP1 and mitochondrial fission have previously been thought to be necessary for mitophagy to occur in mammalian cells, some recent research findings demonstrate that certain forms of mitophagy do in fact occur independent of DRP1 (Murakawa et al., 2015; Yamashita et al., 2016). Another study has revealed that the loss of DRP1 actually enhances mitophagy in vitro (Burman et al., 2017). This form of mitochondrial quality control actively functions in both pathological and physiological conditions as a protector from damage and stress inflicted beyond the protein level. Dysfunctional mitochondria which are beyond a level amenable to mitochondrial dynamics repair must be culled from the mitochondrial population. Mitophagy is likely to have a functional association with mitochondrial dynamics as interactions between mitochondrial dynamics factors and LC3 adapters, specifically optineurin, become more apparent (Moore and Holzbaur, 2016). The PINK1/Parkin pathway, while better known for being genetic factors of Parkinson’s disease, is now known to also play a role in mitophagy (Nguyen et al., 2016; Yoo and Jung, 2018). The biomolecular specifics of the association between mitophagy and mitochondrial dynamics remain to be determined.
Mitochondrial fragmentation through fission has also been well observed in cells undergoing apoptosis, a form of programmed cell death that is critical for development and for adult tissue homeostasis in all multicellular organisms. A key event during the apoptotic process is mitochondrial outer membrane permeabilization (MOMP). As a result of MOMP, there is a release of pro-apoptotic factors including cytochrome c from the intermembrane space to enter into the cytosol, and this triggers downstream cell death pathways. BAX is responsible for executing this event (Brady and Gil-Gomez, 1998). BAX is recruited to the mitochondrial outer membrane after activation in the cytosol, which leads to oligomer formation and MOMP. This brings the cell to the ‘point of no return’ in the apoptotic pathway. BAK has been observed interacting with DRP1 and mitofusins. This indicates that there does exist crosstalk between the separate machineries of mitochondrial dynamics and apoptosis. While experimental results have shown that the BAX oligomerization is promoted by the membrane tethering activity of DRP1, DRP1 itself is not necessary for the initial recruitment of BAX to the outer mitochondrial membrane (OMM) (Jenner et al., 2022). This points toward an initiation of MOMP by BAX that is independent of the function of DRP1, however, DRP1 promotes the further aggregation of BAX in later phases of recruitment. Additional studies are needed to determine exactly how mitochondrial fusion and fission components actively participate in apoptosis.
Regulation of mitochondrial fusion and fission
Fusion
The fusion and fission proteins discussed in the previous section are targets for a number of post-translational modifications (PTMs). PTMs facilitate quick responses to alterations in physiological demands and are common mechanisms to modify protein activity. The most studied modifications include acetylation, phosphorylation, ubiquitination, O-GlcNAcylation, and SUMOylation. Mitochondrial fusion is the result of adjacent mitochondria tethering to one another followed by the fusion of the outer mitochondrial membrane (OMM) and the inner mitochondrial membrane (IMM), in that order (Mattie et al., 2019). The regulation of mitochondrial fusion events is carried out through transcription and post-transcriptional and post-translational modifications.
The fusion of adjacent mitochondria is under the regulation of Mitofusins 1 and 2 as discussed previously in this article. A study done in 2006 in Barcelona showed upregulation of MFN2 expression in mice due to an increased demand for energy by the protein peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1a), such as skeletal muscle due to increased mechanical load/exercise or brown adipose tissue reacting to cold environments (Soriano et al., 2006). It has also been demonstrated in vitro that PGC-1a stimulates the activity of MFN2 through an interaction with estrogen related receptor-alpha (ERR alpha) (Cartoni et al., 2005). Another disruptor of mitochondrial fusion is seen during the excitotoxicity of neuronal mitochondria. The transcription factor MEF2 has been shown to regulate basal expression of MFN2 in neurons and during excitotoxicity, MEF2 is degraded resulting in neuronal mitochondria fragmentation and a downregulation of MFN2 is seen (Martorell-Riera et al., 2014).
Some notable miRNAs have been shown to reduce the expression of MFN2, such as mi2R-761 in hepatoma cells (Zhou et al., 2016), miR-106B in breast cancer cells (Wu et al., 2016), and miR-214 in neuroblastoma cells (Bucha et al., 2015). Post-translational modifications that influence mitochondrial fusion include the ubiquitination of MFN2 by E3-ubiquiting ligases, marking MFN2 for proteasomal degradation and an overall inhibition of mitochondrial fusion. These include PARKIN (Gegg et al., 2010), MARCH5 (Nakamura et al., 2006), HUWE1 (Leboucher et al., 2012), and MUL1 (Peng et al., 2016a). In the cellular response to stress signals, MFN2 can also be phosphorylated by PINK1 and JNK (Leboucher et al., 2012; Chen et al., 2013). They are subsequently marked for ubiquitination by PARKIN and HUWE1 respectively.
MiRNAs are also at the forefront of regulating MFN1 expression. MiR-140 is known to increase in response to genotoxic or oxidative stress, negatively regulating MFN1 translation in cardiomyocytes (Li J. et al., 2014). This of course induces mitochondrial fragmentation. MFN1 has also been found to be downregulated in osteosarcoma cells by miR-19b (Li X. et al., 2014). PARKIN is also involved in the ubiquitination of MFN1 (Gegg et al., 2010). Post-translational phosphorylation by extracellular-signal-regulated kinase (ERK) also leads to a decrease in mitochondrial fusion (Pyakurel et al., 2015).
The upregulation of mitochondrial fusion is a common response to cellular stress (Tondera et al., 2009; Rambold et al., 2011), with oxidative processes serving as a common pathway for cellular stress to occur. Oxidized glutathione is known to be a primary cellular stress indicator. Elevation of the intracellular level of oxidized glutathione has been shown to induce mitochondrial fusion by promoting the activity of mitofusins (Shutt et al., 2012; Mattie et al., 2018). Results of in vivo experiments have demonstrated that exposure to sublethal or low doses of hydrogen peroxide can lead to mitochondrial hyperfusion occurring (Yoon et al., 2006; Shutt et al., 2012). Additionally, there is evidence that some minimum level of ROS or oxidation may be required for the activation of fusion to occur (Shutt et al., 2012).
Other biomolecules play important roles in the regulation of mitofusion activity. The soluble form of BAX, the proapoptotic protein discussed previously, localizes MFN2 to fusion sites and is a positive regulator of mitochondrial fusion (Hoppins et al., 2011). In vitro studies indicate that Bcl2 proteins may play important mitochondrial dynamics housekeeping roles (Karbowski et al., 2006). Mitochondrial carrier homologue 2 (MTCH2) is known as a regulator of mitochondrial metabolism and apoptosis as it is a repressor of mitochondrial OXPHOS (Yuan et al., 2021). However, in vitro studies involving embryonic stem cells (ESCs) show that MTCH2 may serve as a novel regulator of mitochondrial fusion, because knockout of MTCH2 from ESCs results in failure of mitochondria to elongate (Bahat et al., 2018). A link between mitochondrial fusion and lipogenesis by way of MTCH2 has also been reported (Labbe et al., 2021), however to date, just how MTCH2 regulates mitochondrial fusion and morphology has not been resolved.
As discussed previously, OPA1 is in charge of fusion of the IMM. In cardiomyocytes, the transcription factor NF-kB regulates OPA1 in response to insulin by the way of the Akt-mTOR signaling pathway (Parra et al., 2014). IMM fusion and morphology relies on maintaining the balance between the long and short OPA1 isoforms. Post-transcriptional and post-translational regulation plays a role in the cleavage of OPA1 to produce these isoforms. Mitochondrial proteases m-AAA (OMA1) and YME-like protein 1 (YME1L) have been shown to cleave OPA1 (Anand et al., 2014). Proteolytic processing of OPA1 by YME1L in vitro has been found to be regulated by the level of OXPHOS (Mishra et al., 2014). Under high levels of OXPHOS, OPA1 is more efficiently cleaved.
Fission
Translation, signaling molecules and post-translational modifications promote fragmentation of mitochondrial networks and accelerate mitochondrial fission. The main protein involved in mitochondrial fission regulation is DRP1 as discussed previously. The miRNA miR-30 helps to activate DRP1 expression during apoptosis as decreasing levels of miR-30 cause an upregulation of p53, a well-known tumor suppressor protein that transcriptionally activates DRP1 (Li et al., 2010). Post-translational modifications that play a role in DRP1 activity and translocation to mitochondria include phosphorylation, ubiquitination, SUMOylation, nitrosylation, and O-GlcNAcylation. Serine 616 (SER616) in mice and Serine 637 (SER637) in human DRP1 are the most well studied sites of regulation. SER616 is phosphorylated by cdk1, PKC, ERK1-2, and CaMKII resulting in DRP1-induced mitochondrial fission. On the other hand, this process is inhibited by PKA phosphorylation of SER637 (Chang and Blackstone, 2007; Cribbs and Strack, 2007; Santel and Frank, 2008; Kashatus et al., 2015; Serasinghe et al., 2015). SER637 can also be dephosphorylated by calcineurin and produces an opposite effect as the blockage of calcineurin results in a prevention of translocation (Cribbs and Strack, 2007; Cereghetti et al., 2008; Kim et al., 2011). It has also been shown that miR-499 is involved in repressing calcineurin and inhibiting pro-fission activity (Cereghetti et al., 2008). Downregulation of calcineurin by miR-499 has been shown to provide protection from mitochondrial fragmentation in cardiomyocytes in response to ischemia-reperfusion (Wang et al., 2011). Other kinases involved in the phosphorylation of DRP1 that result in a decrease of DRP1 levels and translocation to mitochondria include Pim-1 at SER637 (Din et al., 2013) and GSK3b at SER693 (Chou et al., 2012). Kinases that resulted in an increase in DRP1 and the promotion of mitochondrial fission include ROCK1 (Wang et al., 2012) and CaMKIa (Han et al., 2008), both at SER637, as components of the response to hyperglycemia or increased levels of calcium.
SUMOylation plays an important role in regulating DRP1 protein stability. MAPL, a mitochondria-anchored SUMO E3-ligase, SUMOylates DRP1 resulting in mitochondrial fission (Braschi et al., 2009). SENp5, a SUMO protease, alternatively deSUMOylates DRP1 by removing SUMO-1 from DRP1 inhibiting mitochondrial fission (Zunino et al., 2007). PARKIN and MARCH5 are also involved in fission processes by mediating the ubiquitination of DRP1. In neurons, PARKIN has been shown to degrade DRP1 and suppress mitochondrial fission (Lutz et al., 2009). The opposite occurs with MARCH5 as it regulates subcellular trafficking of DRP1 leading to an increase in mitochondrial fission (Karbowski et al., 2007). The most likely explanation is that it affects the disassembly of fission complexes or the precise assembly at scission sites. O-GlcNAcylation of DRP1 in cardiomyocytes has also been shown to have an effect on mitochondrial fission. The first study to display this showed that during high glucose treatment of N-acetyl-glucosaminidase inhibition, Threonine 585 (THR585) and Threonine 586 (THR586) are O-GlcNAcylated leading to a reduction in the phosphorylation of DRP1 at SER637. SER637 as discussed above, inhibits DRP1 activity, so a reduction in phosphorylation at SER637 is followed by an increase in mitochondrial fragmentation and a reduction in mitochondrial membrane potential (Gawlowski et al., 2012). Another more recent study on O-GlcNAcylation of DRP1 discussed the possibility that amyloid-beta regulates mitochondrial fission in neuronal cells (Park et al., 2021). Nitric oxide is also known to be a regulator of mitochondrial fission and is an important signaling molecule which can mediate neuronal injury when in excess. It is produced as a byproduct of the amyloid-beta protein via S-nitrosylation of DRP1 (SNO-DRP1) (Cho et al., 2009). This study revealed that SNO-DRP1 increased in the brains of humans with Alzheimer’s disease, leading to synaptic loss, mitochondrial fission, and neuronal damage.
Regulation of mitochondrial motility
The mechanics behind mitochondrial motility have been best studied in the neuron and the molecular mechanisms that engage mitochondria in their motility are well established. As neurons are postmitotic cells that survive as long as the organism is alive, mitochondria need to be cycled when they are subject to dysfunction or age. Conditions of stress and impaired integrity also lead to shifts in mitochondrial motility. Regulation of mitochondrial motility is therefore incredibly important to meet the necessary metabolic requirements to adapt to stressors, as well as to remove dysfunctional and aged mitochondria while replenishing the cell’s supply of healthy ones. The implications of a defect in mitochondrial transport are seen in the pathogenesis of a multitude of major neurological disorders. The coordination between these mechanisms to distribute mitochondria in neurons and in other cells, however, is not very well understood.
The bidirectional transport of mitochondria along microtubules is carried out by kinesin and dynein motors as discussed previously, and is frequently described as a “tug of war.” Our mechanistic understanding of how motor proteins, adapters, and corresponding regulatory proteins interact at the molecular level is lacking. With the amount of research that has been done, we cannot accurately predict how disturbances at a cellular level play a role in the determination of net retrograde or anterograde movement, but there are hypotheses as to how this occurs. Much of the research done regarding how kinesins and dyneins regulate movement of a cargo being transported along a microtubule is based on mechanics and the velocity or static behavior of mitochondria are dependent on the balance of forces. Retrograde and anterograde velocity is thought to be regulated by the amount of kinesin and dynein molecules acting on a mitochondria as well as the overall load force the motors need to act against, such as the dynein stall force (Barnhart, 2016; Ohashi et al., 2019). Other factors may include specific protein-protein interactions between Snph and mitochondria (Kang et al., 2008), myosin activity opposing the axonal transport of mitochondria (Pathak et al., 2010), and even viscous drag due to high microtubule density (Wortman et al., 2014). Furthermore, the post-translational modifications of microtubules may play a role in controlling motor protein velocity and processivity (Sirajuddin et al., 2014) with a study showing that there is a possible kinesin motility bias toward the axon due to increased acetylation of axonal microtubules (Hammond et al., 2010). It still remains unclear though the extent at which post-translational modifications actually has on mitochondrial motility.
Mitochondrial distribution is heavily correlated with the demand for energy. Mitochondria are typically clustered at sites of high energy demand because of a higher need for ATP generation, such as near synapses or surrounding the cell nucleus (Jang et al., 2019; Green et al., 2021), which lacks its own mitochondria. Elevated intracellular Ca2+ levels draw mitochondria to synapses by activation of the voltage-gated calcium channels at presynaptic terminals or NMDA receptors at postsynaptic sites (Rintoul et al., 2003; Chang et al., 2006; Szabadkai et al., 2006). Studies over the past decade focused heavily on how mitochondria are recruited and arrested at synapses and new insights revealed that synaptic Ca2+ levels play a role in the regulation of mitochondrial trafficking and anchoring when looking into KIF5-TRAK-Miro complexes (Sheng, 2014). Miro specifically was found to serve as a regulator of mitochondrial motility by sensing Ca2+ levels. Miro calcium-binding via EF hands either inactivates or disassembles the KIF5-TRAK-Miro transport machinery arresting mitochondria at active synapses (Saotome et al., 2008; Macaskill et al., 2009b; Wang and Schwarz, 2009). These studies purport that there are two differing Mito-Ca2+ sensing models as of now to explain how this occurs (Figure 3). One model shows that the direct interaction between Miro and KIF5 is inhibited by the binding of calcium, which results in mitochondria uncoupling from the transport machinery and the potential halting of the molecular motors (Macaskill et al., 2009b). A second model discusses that under normal calcium levels, Miro binds to KIF5 via Milton while during high levels of calcium, the kinesin motor domain unbinds from the microtubule and turns upward to directly bind to Miro on the mitochondria, effectively uncoupling the mitochondria from the microtubule transport pathway (Wang and Schwarz, 2009). It has also been purported that the binding of calcium to Miro prompts a conformational change that either reduces microtubule engagement of Milton-bound kinesin or decreases the volume of kinesin molecules mitochondrially-bound (Macaskill et al., 2009b). One hypothesis is that Miro calcium sensing facilitates the localization of mitochondria to active synapses by decreasing the amount of kinesin motors actively driving motility. Miro expression elevation has also been shown to increase mitochondrial motility through the recruitment of TRAK and motors to mitochondria (MacAskill et al., 2009a).
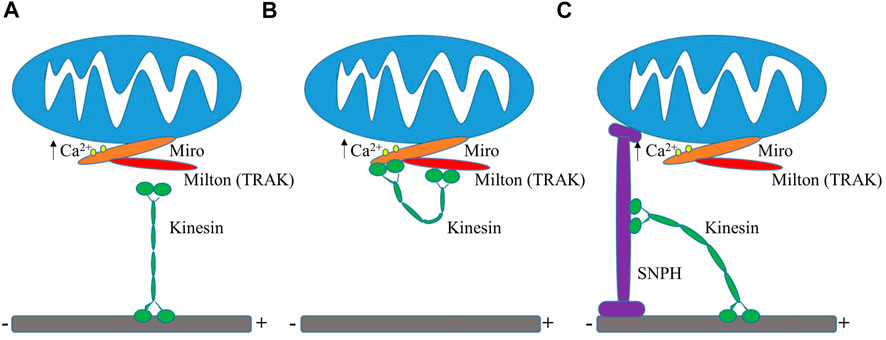
FIGURE 3. Mito-Ca2+ sensing models. (A) Ca2+ binding to Miro results in the uncoupling of mitochondria from kinesin and the possible deactivation of motors (Macaskill et al., 2009b). (B) High levels of Ca2+ uncouple kinesin machinery from the microtubule, leading to its direct binding to Miro (Wang and Schwarz, 2009). (C) Display of the “Engine-Switch and Brake” model in which kinesin detaches from Miro and subsequently interacts with the mitochondrial docking protein Snph (Chen and Sheng, 2013).
The mitochondrial docking protein Snph as discussed before immobilizes axonal mitochondria. Mitochondrial movement is arrested by binding kinesin and inhibiting ATPase activity through the reduction of active kinesin motors attached to a singular mitochondrion as well as creating a greater force that any remaining motors must overcome. Snph also becomes important for calcium-induced mitochondrial arrest in axons. The mechanisms that regulate Snph-mediated mitochondrial docking are unknown for the most part but there have been some recently proposed models such as the “Engine-Switch and Brake” model where Snph serves as an engine-off switch when it senses Miro GTPase-Ca2+ and as a brake when it anchors mitochondria to the microtubule track (Chen and Sheng, 2013). The arrest of mitochondrial motility can also be affected by nutrient availability as seen by the increase of extracellular glucose concentrations mediated by post-translational modifications of the protein Milton by the enzyme O-GlcNAcylation transferase (Pekkurnaz et al., 2014). The GTPase Rac1 regulates VimIFs and their binding to mitochondria (Matveeva et al., 2015).
Mitochondrial diseases and mitochondrial dynamics
As discussed earlier in this review, fission allows for the segregation of damaged mitochondria while fusion exchanges materials between damaged and functional mitochondria. The balance between fission and fusion is what allows mitochondria to remain healthy and supply enough energy to remain fully functional. Alterations to mitochondrial dynamics therefore play a critical role in the manifestation of mitochondrial disorders.
Charcot-Marie-Tooth disease type 2A
Charcot-Marie-Tooth disease (CMT) is a rare heterogenous inherited neuropathy. Those affected experience peripheral neuropathy, hypotonia and severe progressive muscle weakness in the distal limbs resulting in gait defects. The most common type, CMT Type 2A (CMT2A), is known to be caused by defects to the inner mitochondrial membrane fusion protein, MFN2, and remains an incurable condition (Kijima et al., 2005; Cho et al., 2007). Current models are looking into mitochondrial transport defects as a cause for CMT2A because MFN2 has been reported to interact with the Milton/Miro/kinesin complex in neurons (Misko et al., 2010). MFN2 mutations associated with CMT2A have been shown to create mitochondrial clusters when overexpressed in cell cultures (Detmer and Chan, 2007). This clustering suggests that the MFN2 mutations do indeed have an effect on mitochondrial locomotion. Transgenic mouse models overexpressing T105M or R94W mutations confirm this model as the clumping of mitochondria is associated with these phenotypes and there is a decrease in mitochondrial motility in neurons with a sparsity of action in the axons (Detmer and Chan, 2007).
The authors of one interesting study regarding the role of MFN2 in cells concluded that the defect in locomotion was not a result of the depletion of MFN2, but instead due to the incorrect mitochondrial shaping. They came to this conclusion after seeing that mouse embryonic fibroblasts that were isolated from MFN2 knockout embryos exhibited normal mitochondrial locomotion. Both short tubular and round mitochondria were present and even though the round mitochondria seemed to lose their directed movement, they retained the ability to fuse with the mitochondrial network to create mitochondria having undirected movement (Chen H. C. et al., 2003). There is a possibility that MFN2 plays a role in quality control to ensure that only healthy mitochondria with the ability to fuse are transported (Cartoni and Martinou, 2009).
Another function of MFN2 is its association with ER-mitochondrial tethering. Many key cellular functions occur due to this connection including lipid and calcium homeostasis and fission of mitochondria. This model is speculative as it is disputed over whether this tethering is promoted or inhibited by MFN2 (Filadi et al., 2015; Naon et al., 2016). More studies regarding this behavior need to be conducted to establish the definitive role of MFN2.
Dominant optic atrophy
DOA is considered to be the most common inherited optic neuropathy. Most cases of DOA are tied to heterozygous mutations in OPA1, which, as discussed previously, is necessary for IMM fusion and maintenance of OXPHOS supercomplexes and cristae membrane ultrastructure (Alexander et al., 2000; Yarosh et al., 2008). The overall lack of OPA1 functionality leads towards increased mitochondrial fission and mitochondrial network fragmentation. This in turn increases reactive oxygen species, alters calcium homeostasis, and impairs oxidative phosphorylation. The progressive loss of retinal ganglion cells located in the inner retina and subsequent atrophy of the optic nerves cause bilateral loss of vision resulting in pallor of the optic disk (Kline and Glaser, 1979; Delettre et al., 2002).
Mitochondrial dynamics and neurodegenerative diseases
Mitochondrial dysfunction is a prominent early marker and pathological feature for a number of neurodegenerative diseases such as Huntington’s disease (HD), Parkinson’s disease (PD), Alzheimer’s disease (AD), and amyotrophic lateral sclerosis (ALS). Abnormal mitochondrial dynamics lead to the progressive loss of structure and function of neurons within the central and peripheral nervous systems. With the discovery of fusion and fission proteins, namely DRP1, OPA1, and MFN2, mitochondrial dynamics regulators and their functions have been increasingly studied to try and find novel therapeutic strategies for these well-known and incurable diseases. There is a lack of research regarding mitochondrial motility and how this plays a role in the development of disorders related to mitochondrial dynamics. While difficult to observe and assess, this could be an important avenue of research that can shed more light on these diseases.
HD is an autosomal dominant neurodegenerative disease caused by the expansion of the CAG triplet nucleotide responsible for encoding a polyglutamine tract in exon 1 of the HTT (huntington) gene (Warby et al., 2009). The mutant HTT variant activates DRP1 through the enhancement of its GTP-hydrolyzing activity (Song et al., 2011), thus causing polyglutamine accumulation and mitochondrial fragmentation. These observations are further backed up through ameliorative results in models of Huntington’s disease in which DRP1 is inhibited or MFN2 is augmented in a way to restore fusion preventing cell death (Wang et al., 2009). Mutant HTT has also been shown to interact with motor proteins by inactivating them or by disrupting the connection between microtubules and motor proteins resulting in mitochondrial trafficking impairment (Trushina et al., 2004).
PD has been extensively studied as it is the second most prominent neurodegenerative disease behind AD. Symptoms include bradykinesia, resting tremors, and rigidity. Mitophagy related proteins PINK1 and Parkin have been linked to PD and mutations in either result in impaired mitophagy and early accumulation of DRP1 in neurons. The excessive amount of fission leads to excessive fragmentation accompanied by oxidative stress and reduced ATP production (Dagda et al., 2009; Lutz et al., 2009). Some missense mutations in OPA1 (G488R, A495V) have also been seen in patients with syndromic parkinsonism as well as dementia (Carelli et al., 2015).
AD experimental models display altered expressions of mitochondrial fusion and fission regulators like DRP1, MFN 1 and 2, OPA1, and Fis1; however, detailed mechanisms physically connecting mitochondrial dynamics regulators and AD have yet to be determined. There is mounting evidence regarding autophagy and its important role in the pathogenesis of AD (Zare-Shahabadi et al., 2015). This shows that abnormal mitochondrial dynamics and morphological changes may be important pathways with contributions to mitochondrial dysfunction, and by association, neuronal dysfunction in the brains of those with AD.
ALS is a rare and heritable form of disease that progressively degenerates motor neurons in the brainstem and spinal cord. ALS causes issues with muscle strength, atrophy, swallowing, difficulty in speaking, and breathing. Abnormal mitochondrial morphology as well as mitochondrial fragmentation have been documented in both ALS cell and animal models (Rodriguez et al., 2012; Peng et al., 2016b). Mutations in superoxide dismutase 1 (SOD1) have shown signs of mitochondrial fragmentation due to the changed expression of a few notable mitochondrial fission and fusion proteins such as DRP1, MFN1, OPA1, and Fis1 (Vande Velde et al., 2011; Liu et al., 2013; Song et al., 2013; Vinsant et al., 2013). Miro downregulation in the spinal cords of ALS patients and mutant SOD1 and TDP-43 transgenic mice also seems to contribute to mitochondrial motility impairments prominent in ALS (Zhang et al., 2015). Despite all of this, mitochondrial fragmentation in these experimental models and patients suffering from ALS is likely to be a result, and not a cause, of the onset of the disease, given that there are no reported physical associations between ALS associated proteins and mitochondrial dynamics proteins.
Mitochondrial dynamics and cardiovascular disease
The heart relies on mitochondria to produce enough ATP to sustain its contractile functions. Approximately 30% of the total cell volume is occupied by mitochondria alone (Hall et al., 2014). Mitochondrial dynamics are being studied more recently in association with varying fields of cardiovascular biology including cardiomyopathies (Fang et al., 2007), heart development (Martin-Fernandez and Gredilla, 2016), myocardial infarction (Davidson et al., 2014) and ischemia (Ashrafian et al., 2010).
Mitochondrial dynamic impairment represented by a decrease in mitochondrial fission as mediated by DRP1 expression can be associated with a decrease in ATP production. In addition, a reduction in mitochondrial fission negatively impacts mitochondrial quality control, and as discussed previously, leads to an accumulation of damaged mitochondria. Quality control of mitochondria has been shown to be extremely critical when it comes to cardiovascular pathologies (Marzetti et al., 2013).
Mitochondrial therapeutics
An emerging field of therapeutics is developing new treatment strategies to remedy mutations, dysfunctions, and diseases affecting mitochondrial biogenesis, dynamics, and healthy aging. Therapeutics targeted at mitochondrial dysfunction have the potential to make a very broad clinical impact. In particular, treatments that focus on the amelioration of derangements in mitochondrial dynamics observed in a multitude of mitochondrial diseases, have the potential to significantly impact cellular function and survival, thus markedly improving patient outcomes. As has been previously discussed, maintenance of balanced mitochondrial fusion and fission events and normal mitochondrial motility are essential to enjoying good health.
Dysfunctional mitochondria can be distinguished based on their morphology and can be degraded through selective autophagy (Dufour et al., 2011). Pro-autophagic agents have been used as a therapy in recent years. Rapamycin, an inhibitor of the mammalian target of rapamycin (mTOR) pathway, and its derivatives are among the most studied therapeutic agents for the treatment of neurodegenerative diseases, including PD and HD (Sarkar et al., 2008; Johnson et al., 2013; Villa-Cuesta et al., 2014). Some recent studies showcase positive results of rapamycin intervention in LS mouse and Drosophila models (Li Q. et al., 2014; Wang et al., 2016). The pharmacological inhibition of TOR rescues the shortened lifespan, neurodegeneration, and neurological symptoms in the mouse models of LS. Using the Drosophila model, investigators found that the increased lifespan occurred in an autophagy-independent manner. The authors of these papers concluded that the improvement could potentially have been the result of an immunosuppressive effect of the rapamycin as opposed to a direct effect on mitochondrial function. More research is needed to discern any direct effects that rapamycin and related autophagic agents have on mitochondrial dysfunction.
Another popular strategy for mitochondrial disease therapy is the use of pharmacological agents to inhibit mitochondrial fission. The inhibition of mitochondrial fission proteins DRP1/Fis1 has been shown to be reduce excessive mitochondrial fission and heart damage in cultured murine cardiac myocytes from whole rat heart models of ischemia and reperfusion (IR) injury (Sharp et al., 2014). Use of fission inhibitors Mdivi-1, calcineurin inhibitor, DRP1 siRNA, and therapeutic hypothermia were shown to reverse mitochondrial swelling and fragmentation in these IR models. The application of these small molecules that inhibit mitochondrial fragmentation are important therapeutic factors to be further studied because uncontrolled mitochondrial fusion is a significant contributor to a number of neurodegenerative diseases (Chen and Chan, 2009). Mdivi-1, a DRP1 inhibitor, and the selective peptide inhibitor P110 have also been reported to rescue mitochondrial morphological and functional defects induced by mutations in PINK1 (Qi et al., 2013). The continual study of small molecule mitochondrial fission inhibitors may be useful in the treatment and prevention of mitochondrial disorders.
Some lifestyle interventions that are recognized to have promising effects are regular exercise and caloric restriction. Many studies have shown that exercise has an effect on mitochondrial fission and fusion as well as mitochondrial biogenesis. Mitochondrial biogenesis and turnover rates decrease as organisms age causing a myriad of health problems. Exercise has become recognized as an intervention to increase mitochondrial biogenesis as well as carbohydrate levels and fatty acid oxidation in skeletal muscle (Holloszy and Booth, 1976). Exercise leads to an increase in stress signals which appear to be responsible for activation of mitochondrial biogenesis post-exercise. These signals include increased levels of cytosolic Ca2+ (Baar et al., 2003), AMP/AMPK (McConell et al., 2010), and ROS (Irrcher et al., 2009). Exercise also stimulates PGC-1a, a transcription coactivator. Deficiency shows blunted expression of genes for oxidative phosphorylation and decreased mitochondrial function (Zechner et al., 2010).
Caloric restriction (CR) is an effective nutritional intervention that helps in the prevention of age-related metabolic disorders (Omodei and Fontana, 2011). The molecular mechanisms of CR-induced benefits are still under further investigation. Many studies have demonstrated that CR reduces the overproduction of ROS and oxidative damage (Barja and Herrero, 2000; Barja, 2002) which leads to the enhancement of mitochondrial function in humans. CR also increases mitochondrial biogenesis through the activation of PGC-1a, a key regulator of energy metabolism (Lopez-Lluch et al., 2008).
Other pharmacological interventions for mitochondrial disorders include mitochondria-targeted antioxidants and a CR mimetic, resveratrol, which has been demonstrated to affect mitochondrial biogenesis in liver, muscle, and brain. Resveratrol has also been shown to promote the expression of proteins involved in mitochondrial fission and fusion resulting in a protective effect in mitochondria (Peng et al., 2016b). Oxidative damage from mitochondria is associated with numerous metabolic disorders and antioxidant therapies pose a potential route for treatment. In the past few years, antioxidant compounds containing ubiquinone (MitoQ) or vitamin E specifically targeted to mitochondria have been used in the treatment of mitochondrial dysfunctions (Kelso et al., 2001). Some recent studies have shown it to be effective by inhibiting oxidative stress and apoptosis (Chen et al., 2020) and the restoration of mitochondrial respiration in heart failure induced by pressure overload (Ribeiro Junior et al., 2018). These pharmacological interventions are not directly tied to mitochondrial dynamics and are more so related to mitochondrial membrane potential and relieving oxidative stress making them important for mitochondrial health.
Discussion
Mitochondria are complex and dynamic intracellular organelles that undergo highly regulated motile interactions involving numerous complexes (e.g., kinesins, cytoskeletal elements). They are also subject to fusion and fission events under the control of regulating proteins. Mitochondrial fission and fusion maintain the functionality of mitochondria and plays an incredibly important role in cell fate. Fission is responsible for proper distribution and number of mitochondria while fusion ensures the exchange of content for optimal mitochondrial activity. Disruption of mitochondrial motility or an imbalance in mitochondrial fusion/fission is associated with organ dysfunction and diseases known to involve the neurological, vascular, and skeletal systems. With an evolving understanding of mitochondrial dynamics, some therapeutic avenues are being researched to combat dysfunctional dynamics with many trying to reverse excess fission (Mdivi-1, calcineurin inhibitor, etc.). There is a lack in knowledge of the molecular events involved in mitochondrial fusion and fission, requiring more mechanistic understanding. Further studies regarding how fusion and fission is related to mitophagy is promising as it relates to mitochondrial disorders and cancer. The field of mitochondrial dynamics is expanding and more research regarding mitochondrial motility and its role in the pathogenesis of disease states and dysfunction is necessary. An expanding pool of experimental techniques and imaging tools offer potential for novel discoveries in mitochondrial dynamics understanding as well as novel treatment approaches for mitochondrial diseases involving errors of dynamic functions.
Author contributions
AG—Discussing the manuscript structure, writing a draft, preparing figures, TH—Discussing the manuscript structure, editing a draft, DE—discussing the manuscript structure, writing a draft, editing and finalizing the manuscript.
Funding
This work was funded by grant N000142212170 from the Office of Naval Research (DE).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abrisch, R. G., Gumbin, S. C., Wisniewski, B. T., Lackner, L. L., and Voeltz, G. K. (2020). Fission and fusion machineries converge at ER contact sites to regulate mitochondrial morphology. J. Cell Biol. 219. 1.
Alexander, C., Votruba, M., Pesch, U. E., Thiselton, D. L., Mayer, S., Moore, A., et al. (2000). OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 26, 211–215.
Ali, S., and Mcstay, G. P. (2018). Regulation of mitochondrial dynamics by proteolytic processing and protein turnover. Antioxidants (Basel) 7, 15. doi:10.3390/antiox7010015
Altmann, K., Frank, M., Neumann, D., Jakobs, S., and Westermann, B. (2008). The class V myosin motor protein, Myo2, plays a major role in mitochondrial motility in Saccharomyces cerevisiae. J. Cell Biol. 181, 119–130. doi:10.1083/jcb.200709099
Amutha, B., Gordon, D. M., Gu, Y., and Pain, D. (2004). A novel role of Mgm1p, a dynamin-related GTPase, in ATP synthase assembly and cristae formation/maintenance. Biochem. J. 381, 19–23. doi:10.1042/BJ20040566
Anand, R., Wai, T., Baker, M. J., Kladt, N., Schauss, A. C., Rugarli, E., et al. (2014). The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 204, 919–929. doi:10.1083/jcb.201308006
Ashrafian, H., Docherty, L., Leo, V., Towlson, C., Neilan, M., Steeples, V., et al. (2010). A mutation in the mitochondrial fission gene Dnm1l leads to cardiomyopathy. PLoS Genet. 6, e1001000. doi:10.1371/journal.pgen.1001000
Baar, K., Song, Z., Semenkovich, C. F., Jones, T. E., Han, D. H., Nolte, L. A., et al. (2003). Skeletal muscle overexpression of nuclear respiratory factor 1 increases glucose transport capacity. FASEB J. 17, 1666–1673. doi:10.1096/fj.03-0049com
Bahat, A., Goldman, A., Zaltsman, Y., Khan, D. H., Halperin, C., Amzallag, E., et al. (2018). MTCH2-mediated mitochondrial fusion drives exit from naive pluripotency in embryonic stem cells. Nat. Commun. 9, 5132. doi:10.1038/s41467-018-07519-w
Ban, T., Kohno, H., Ishihara, T., and Ishihara, N. (2018). Relationship between OPA1 and cardiolipin in mitochondrial inner-membrane fusion. Biochim. Biophys. Acta. Bioenerg. 1859, 951–957. doi:10.1016/j.bbabio.2018.05.016
Ban-Ishihara, R., Ishihara, T., Sasaki, N., Mihara, K., and Ishihara, N. (2013). Dynamics of nucleoid structure regulated by mitochondrial fission contributes to cristae reformation and release of cytochrome c. Proc. Natl. Acad. Sci. U. S. A. 110, 11863–11868. doi:10.1073/pnas.1301951110
Banerjee, R., Mukherjee, A., and Nagotu, S. (2022). Mitochondrial dynamics and its impact on human health and diseases: Inside the DRP1 blackbox. J. Mol. Med. 100, 1–21. doi:10.1007/s00109-021-02150-7
Barel, O., Malicdan, M. C. V., Ben-Zeev, B., Kandel, J., Pri-Chen, H., Stephen, J., et al. (2017). Deleterious variants in TRAK1 disrupt mitochondrial movement and cause fatal encephalopathy. Brain 140, 568–581. doi:10.1093/brain/awx002
Barja, G. (2002). Endogenous oxidative stress: Relationship to aging, longevity and caloric restriction. Ageing Res. Rev. 1, 397–411. doi:10.1016/s1568-1637(02)00008-9
Barja, G., and Herrero, A. (2000). Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. FASEB J. 14, 312–318. doi:10.1096/fasebj.14.2.312
Barnhart, E. L. (2016). Mechanics of mitochondrial motility in neurons. Curr. Opin. Cell Biol. 38, 90–99. doi:10.1016/j.ceb.2016.02.022
Basu, H., and Schwarz, T. L. (2020). QuoVadoPro, an autonomous tool for measuring intracellular dynamics using temporal variance. Curr. Protoc. Cell Biol. 87, e108. doi:10.1002/cpcb.108
Belenguer, P., and Pellegrini, L. (2013). The dynamin GTPase OPA1: More than mitochondria? Biochim. Biophys. Acta 1833, 176–183. doi:10.1016/j.bbamcr.2012.08.004
Brady, H. J., and Gil-Gomez, G. (1998). Bax. The pro-apoptotic Bcl-2 family member, Bax. Int. J. Biochem. Cell Biol. 30, 647–650. doi:10.1016/s1357-2725(98)00006-5
Braschi, E., Zunino, R., and Mcbride, H. M. (2009). MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep. 10, 748–754. doi:10.1038/embor.2009.86
Bros, H., Hauser, A., Paul, F., Niesner, R., and Infante-Duarte, C. (2015). Assessing mitochondrial movement within neurons: Manual versus automated tracking methods. Traffic 16, 906–917. doi:10.1111/tra.12291
Bucha, S., Mukhopadhyay, D., and Bhattacharyya, N. P. (2015). Regulation of mitochondrial morphology and cell cycle by microRNA-214 targeting Mitofusin2. Biochem. Biophys. Res. Commun. 465, 797–802. doi:10.1016/j.bbrc.2015.08.090
Burman, J. L., Pickles, S., Wang, C., Sekine, S., Vargas, J. N. S., Zhang, Z., et al. (2017). Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol. 216, 3231–3247. doi:10.1083/jcb.201612106
Cai, Q., Gerwin, C., and Sheng, Z. H. (2005). Syntabulin-mediated anterograde transport of mitochondria along neuronal processes. J. Cell Biol. 170, 959–969. doi:10.1083/jcb.200506042
Carelli, V., Musumeci, O., Caporali, L., Zanna, C., LA Morgia, C., Del Dotto, V., et al. (2015). Syndromic parkinsonism and dementia associated with OPA1 missense mutations. Ann. Neurol. 78, 21–38. doi:10.1002/ana.24410
Cartoni, R., Leger, B., Hock, M. B., Praz, M., Crettenand, A., Pich, S., et al. (2005). Mitofusins 1/2 and ERRalpha expression are increased in human skeletal muscle after physical exercise. J. Physiol. 567, 349–358. doi:10.1113/jphysiol.2005.092031
Cartoni, R., and Martinou, J. C. (2009). Role of mitofusin 2 mutations in the physiopathology of Charcot-Marie-Tooth disease type 2A. Exp. Neurol. 218, 268–273. doi:10.1016/j.expneurol.2009.05.003
Cereghetti, G. M., Stangherlin, A., Martins DE Brito, O., Chang, C. R., Blackstone, C., Bernardi, P., et al. (2008). Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. U. S. A. 105, 15803–15808. doi:10.1073/pnas.0808249105
Chalmers, S., Saunter, C. D., Girkin, J. M., and Mccarron, J. G. (2016). Age decreases mitochondrial motility and increases mitochondrial size in vascular smooth muscle. J. Physiol. 594, 4283–4295. doi:10.1113/JP271942
Chang, C. R., and Blackstone, C. (2007). Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 282, 21583–21587. doi:10.1074/jbc.C700083200
Chang, D. T., Honick, A. S., and Reynolds, I. J. (2006). Mitochondrial trafficking to synapses in cultured primary cortical neurons. J. Neurosci. 26, 7035–7045. doi:10.1523/JNEUROSCI.1012-06.2006
Chen, H. C., Detmer, S. A., Ewald, A. J., Griffin, E. E., Fraser, S. E., and Chan, D. C. (2003b). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 160, 189–200. doi:10.1083/jcb.200211046
Chen, H., and Chan, D. C. (2009). Mitochondrial dynamics-fusion, fission, movement, and mitophagy-in neurodegenerative diseases. Hum. Mol. Genet. 18, R169–R176. doi:10.1093/hmg/ddp326
Chen, H. C., Mccaffery, J. M., and Chan, D. C. (2007). Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 130, 548–562. doi:10.1016/j.cell.2007.06.026
Chen, H. C., Vermulst, M., Wang, Y. E., Chomyn, A., Prolla, T. A., Mccaffery, J. M., et al. (2010). Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141, 280–289. doi:10.1016/j.cell.2010.02.026
Chen, H., Detmer, S. A., Ewald, A. J., Griffin, E. E., Fraser, S. E., and Chan, D. C. (2003a). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 160, 189–200. doi:10.1083/jcb.200211046
Chen, X. J., Wang, L., and Song, X. Y. (2020). Mitoquinone alleviates vincristine-induced neuropathic pain through inhibiting oxidative stress and apoptosis via the improvement of mitochondrial dysfunction. Biomed. Pharmacother. 125, 110003. doi:10.1016/j.biopha.2020.110003
Chen, Y., and Dorn, G. W. (2013). PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340, 471–475. doi:10.1126/science.1231031
Chen, Y., and Sheng, Z. H. (2013). Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. J. Cell Biol. 202, 351–364. doi:10.1083/jcb.201302040
Chistiakov, D. A., Shkurat, T. P., Melnichenko, A. A., Grechko, A. V., and Orekhov, A. N. (2018). The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 50, 121–127. doi:10.1080/07853890.2017.1417631
Cho, D. H., Nakamura, T., Fang, J., Cieplak, P., Godzik, A., Gu, Z., et al. (2009). S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 324, 102–105. doi:10.1126/science.1171091
Cho, H. J., Sung, D. H., Kim, B. J., and Ki, C. S. (2007). Mitochondrial GTPase mitofusin 2 mutations in Korean patients with Charcot-Marie-Tooth neuropathy type 2. Clin. Genet. 71, 267–272. doi:10.1111/j.1399-0004.2007.00763.x
Chou, C. H., Lin, C. C., Yang, M. C., Wei, C. C., Liao, H. D., Lin, R. C., et al. (2012). GSK3beta-mediated Drp1 phosphorylation induced elongated mitochondrial morphology against oxidative stress. PLoS One 7, e49112. doi:10.1371/journal.pone.0049112
Cipolat, S., Martins DE Brito, O., Dal Zilio, B., and Scorrano, L. (2004). OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. U. S. A. 101, 15927–15932. doi:10.1073/pnas.0407043101
Collins, T. J., Berridge, M. J., Lipp, P., and Bootman, M. D. (2002). Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 21, 1616–1627. doi:10.1093/emboj/21.7.1616
Cox, R. T., and Spradling, A. C. (2006). Milton controls the early acquisition of mitochondria by Drosophila oocytes. Development 133, 3371–3377. doi:10.1242/dev.02514
Cribbs, J. T., and Strack, S. (2007). Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 8, 939–944. doi:10.1038/sj.embor.7401062
Dagda, R. K., Cherra, S. J., 3R. D., Kulich, S. M., Tandon, A., Park, D., and Chu, C. T. (2009). Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 284, 13843–13855. doi:10.1074/jbc.M808515200
Davidson, S. M., Lopaschuk, G. D., Spedding, M., and Beart, P. M. (2014). Mitochondrial pharmacology: Energy, injury and beyond. Br. J. Pharmacol. 171, 1795–1797. doi:10.1111/bph.12679
Davies, V. J., Hollins, A. J., Piechota, M. J., Yip, W., Davies, J. R., White, K. E., et al. (2007). Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum. Mol. Genet. 16, 1307–1318. doi:10.1093/hmg/ddm079
De Brito, O. M., and Scorrano, L. (2010). An intimate liaison: Spatial organization of the endoplasmic reticulum-mitochondria relationship. EMBO J. 29, 2715–2723. doi:10.1038/emboj.2010.177
De Vos, K. J., and Sheetz, M. P. (2007). Visualization and quantification of mitochondrial dynamics in living animal cells. Methods Cell Biol. 80, 627–682. doi:10.1016/S0091-679X(06)80030-0
Del Dotto, V., Fogazza, M., Carelli, V., Rugolo, M., and Zanna, C. (2018). Eight human OPA1 isoforms, long and short: What are they for? Biochim. Biophys. Acta. Bioenerg. 1859, 263–269. doi:10.1016/j.bbabio.2018.01.005
Delettre, C., Lenaers, G., Pelloquin, L., Belenguer, P., and Hamel, C. P. (2002). OPA1 (kjer type) dominant optic atrophy: A novel mitochondrial disease. Mol. Genet. Metab. 75, 97–107. doi:10.1006/mgme.2001.3278
Delivani, P., Adrain, C., Taylor, R. C., Duriez, P. J., and Martin, S. J. (2006). Role for CED-9 and Egl-1 as regulators of mitochondrial fission and fusion dynamics. Mol. Cell 21, 761–773. doi:10.1016/j.molcel.2006.01.034
Detmer, S. A., and Chan, D. C. (2007). Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J. Cell Biol. 176, 405–414. doi:10.1083/jcb.200611080
Devay, R. M., Dominguez-Ramirez, L., Lackner, L. L., Hoppins, S., Stahlberg, H., and Nunnari, J. (2009). Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion. J. Cell Biol. 186, 793–803. doi:10.1083/jcb.200906098
Dikic, I., and Elazar, Z. (2018). Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 19, 349–364. doi:10.1038/s41580-018-0003-4
Din, S., Mason, M., Volkers, M., Johnson, B., Cottage, C. T., Wang, Z., et al. (2013). Pim-1 preserves mitochondrial morphology by inhibiting dynamin-related protein 1 translocation. Proc. Natl. Acad. Sci. U. S. A. 110, 5969–5974. doi:10.1073/pnas.1213294110
Dufour, M., Dormond-Meuwly, A., Demartines, N., and Dormond, O. (2011). Targeting the mammalian target of rapamycin (mTOR) in cancer therapy: Lessons from past and future perspectives. Cancers (Basel) 3, 2478–2500. doi:10.3390/cancers3022478
Ehlers, M. D. (2013). Dendritic trafficking for neuronal growth and plasticity. Biochem. Soc. Trans. 41, 1365–1382. doi:10.1042/BST20130081
Eura, Y., Ishihara, N., Yokota, S., and Mihara, K. (2003). Two mitofusin proteins, mammalian homologues of FZO, with distinct functions are both required for mitochondrial fusion. J. Biochem. 134, 333–344. doi:10.1093/jb/mvg150
Faelber, K., Gao, S., Held, M., Posor, Y., Haucke, V., Noe, F., et al. (2013). Oligomerization of dynamin superfamily proteins in health and disease. Prog. Mol. Biol. Transl. Sci. 117, 411–443. doi:10.1016/B978-0-12-386931-9.00015-5
Fang, L., Moore, X. L., Gao, X. M., Dart, A. M., Lim, Y. L., and DU, X. J. (2007). Down-regulation of mitofusin-2 expression in cardiac hypertrophy in vitro and in vivo. Life Sci. 80, 2154–2160. doi:10.1016/j.lfs.2007.04.003
Fenton, A. R., Jongens, T. A., and Holzbaur, E. L. F. (2021). Mitochondrial adaptor TRAK2 activates and functionally links opposing kinesin and dynein motors. Nat. Commun. 12, 4578. doi:10.1038/s41467-021-24862-7
Filadi, R., Greotti, E., Turacchio, G., Luini, A., Pozzan, T., and Pizzo, P. (2015). Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc. Natl. Acad. Sci. U. S. A. 112, E2174–E2181. doi:10.1073/pnas.1504880112
Fischer, C. A., Besora-Casals, L., Rolland, S. G., Haeussler, S., Singh, K., Duchen, M., et al. (2020). MitoSegNet: Easy-to-use deep learning segmentation for analyzing mitochondrial morphology. iScience 23, 101601. doi:10.1016/j.isci.2020.101601
Fransson, A., Ruusala, A., and Aspenstrom, P. (2003). Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis. J. Biol. Chem. 278, 6495–6502. doi:10.1074/jbc.M208609200
Friedman, J. R., Lackner, L. L., West, M., Dibenedetto, J. R., Nunnari, J., and Voeltz, G. K. (2011). ER tubules mark sites of mitochondrial division. Science 334, 358–362. doi:10.1126/science.1207385
Frohlich, C., Grabiger, S., Schwefel, D., Faelber, K., Rosenbaum, E., Mears, J., et al. (2013). Structural insights into oligomerization and mitochondrial remodelling of dynamin 1-like protein. EMBO J. 32, 1280–1292. doi:10.1038/emboj.2013.74
Gabaldon, T., and Huynen, M. A. (2007). From endosymbiont to host-controlled organelle: The hijacking of mitochondrial protein synthesis and metabolism. PLoS Comput. Biol. 3, e219. doi:10.1371/journal.pcbi.0030219
Gandre-Babbe, S., and Van der bliek, A. M. (2008). The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol. Biol. Cell 19, 2402–2412. doi:10.1091/mbc.e07-12-1287
Gawlowski, T., Suarez, J., Scott, B., Torres-Gonzalez, M., Wang, H., Schwappacher, R., et al. (2012). Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-beta-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. J. Biol. Chem. 287, 30024–30034. doi:10.1074/jbc.M112.390682
Gegg, M. E., Cooper, J. M., Chau, K. Y., Rojo, M., Schapira, A. H., and Taanman, J. W. (2010). Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 19, 4861–4870. doi:10.1093/hmg/ddq419
Giacomello, M., and Scorrano, L. (2018). The INs and OUTs of mitofusins. J. Cell Biol. 217, 439–440. doi:10.1083/jcb.201801042
Gray, M. W., Burger, G., and Lang, B. F. (2001). The origin and early evolution of mitochondria. Genome Biol. 2, REVIEWS1018. doi:10.1186/gb-2001-2-6-reviews1018
Green, A., Hossain, T., and Eckmann, D. M. (2021). Hyperbaric oxygen alters intracellular bioenergetics distribution in human dermal fibroblasts. Life Sci. 278, 119616. doi:10.1016/j.lfs.2021.119616
Griffin, E. E., Graumann, J., and Chan, D. C. (2005). The WD40 protein Caf4p is a component of the mitochondrial fission machinery and recruits Dnm1p to mitochondria. J. Cell Biol. 170, 237–248. doi:10.1083/jcb.200503148
Guo, X. F., Macleod, G. T., Wellington, A., Hu, F., Panchumarthi, S., Schoenfield, M., et al. (2005). The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron 47, 379–393. doi:10.1016/j.neuron.2005.06.027
Gutnick, A., Banghart, M. R., West, E. R., and Schwarz, T. L. (2019). The light-sensitive dimerizer zapalog reveals distinct modes of immobilization for axonal mitochondria. Nat. Cell Biol. 21, 768–777. doi:10.1038/s41556-019-0317-2
Hales, K. G., and Fuller, M. T. (1997). Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell 90, 121–129. doi:10.1016/s0092-8674(00)80319-0
Hall, A. R., Burke, N., Dongworth, R. K., and Hausenloy, D. J. (2014). Mitochondrial fusion and fission proteins: Novel therapeutic targets for combating cardiovascular disease. Br. J. Pharmacol. 171, 1890–1906. doi:10.1111/bph.12516
Hammond, J. W., Huang, C. F., Kaech, S., Jacobson, C., Banker, G., and Verhey, K. J. (2010). Posttranslational modifications of tubulin and the polarized transport of kinesin-1 in neurons. Mol. Biol. Cell 21, 572–583. doi:10.1091/mbc.e09-01-0044
Han, S. M., Baig, H. S., and Hammarlund, M. (2016). Mitochondria localize to injured axons to support regeneration. Neuron 92, 1308–1323. doi:10.1016/j.neuron.2016.11.025
Han, X. J., Lu, Y. F., Li, S. A., Kaitsuka, T., Sato, Y., Tomizawa, K., et al. (2008). CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J. Cell Biol. 182, 573–585. doi:10.1083/jcb.200802164
Herlan, M., Bornhovd, C., Hell, K., Neupert, W., and Reichert, A. S. (2004). Alternative topogenesis of Mgm1 and mitochondrial morphology depend on ATP and a functional import motor. J. Cell Biol. 165, 167–173. doi:10.1083/jcb.200403022
Hermann, G. J., Thatcher, J. W., Mills, J. P., Hales, K. G., Fuller, M. T., Nunnari, J., et al. (1998). Mitochondrial fusion in yeast requires the transmembrane GTPase Fzo1p. J. Cell Biol. 143, 359–373. doi:10.1083/jcb.143.2.359
Higuchi-Sanabria, R., Swayne, T. C., Boldogh, I. R., and Pon, L. A. (2016). Live-cell imaging of mitochondria and the actin cytoskeleton in budding yeast. Methods Mol. Biol. 1365, 25–62. doi:10.1007/978-1-4939-3124-8_2
Hinshaw, J. E. (2000). Dynamin and its role in membrane fission. Annu. Rev. Cell Dev. Biol. 16, 483–519. doi:10.1146/annurev.cellbio.16.1.483
Hirokawa, N., Noda, Y., Tanaka, Y., and Niwa, S. (2009). Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell Biol. 10, 682–696. doi:10.1038/nrm2774
Hollenbeck, P. J. (1996). The pattern and mechanism of mitochondrial transport in axons. Front. Biosci. 1, d91–d102. doi:10.2741/a118
Holloszy, J. O., and Booth, F. W. (1976). Biochemical adaptations to endurance exercise in muscle. Annu. Rev. Physiol. 38, 273–291. doi:10.1146/annurev.ph.38.030176.001421
Hoppins, S., Edlich, F., Cleland, M. M., Banerjee, S., Mccaffery, J. M., Youle, R. J., et al. (2011). The soluble form of Bax regulates mitochondrial fusion via MFN2 homotypic complexes. Mol. Cell 41, 150–160. doi:10.1016/j.molcel.2010.11.030
Hurd, D. D., and Saxton, W. M. (1996). Kinesin mutations cause motor neuron disease phenotypes by disrupting fast axonal transport in Drosophila. Genetics 144, 1075–1085. doi:10.1093/genetics/144.3.1075
Ihenacho, U. K., Meacham, K. A., Harwig, M. C., Widlansky, M. E., and Hill, R. B. (2021). Mitochondrial fission protein 1: Emerging roles in organellar form and function in health and disease. Front. Endocrinol. 12, 660095. doi:10.3389/fendo.2021.660095
Ingerman, E., Perkins, E. M., Marino, M., Mears, J. A., Mccaffery, J. M., Hinshaw, J. E., et al. (2005). Dnm1 forms spirals that are structurally tailored to fit mitochondria. J. Cell Biol. 170, 1021–1027. doi:10.1083/jcb.200506078
Irrcher, I., Ljubicic, V., and Hood, D. A. (2009). Interactions between ROS and AMP kinase activity in the regulation of PGC-1alpha transcription in skeletal muscle cells. Am. J. Physiol. Cell Physiol. 296, C116–C123. doi:10.1152/ajpcell.00267.2007
Ishihara, N., Nomura, M., Jofuku, A., Kato, H., Suzuki, S. O., Masuda, K., et al. (2009). Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 11, 958–966. doi:10.1038/ncb1907
Ishihara, T., Ban-Ishihara, R., Maeda, M., Matsunaga, Y., Ichimura, A., Kyogoku, S., et al. (2015). Dynamics of mitochondrial DNA nucleoids regulated by mitochondrial fission is essential for maintenance of homogeneously active mitochondria during neonatal heart development. Mol. Cell. Biol. 35, 211–223. doi:10.1128/MCB.01054-14
James, D. I., Parone, P. A., Mattenberger, Y., and Martinou, J. C. (2003). hFis1, a novel component of the mammalian mitochondrial fission machinery. J. Biol. Chem. 278, 36373–36379. doi:10.1074/jbc.M303758200
Jang, D. H., Greenwood, J. C., Owiredu, S., Ranganathan, A., and Eckmann, D. M. (2019). Mitochondrial networking in human blood cells with application in acute care illnesses. Mitochondrion 44, 27–34. doi:10.1016/j.mito.2017.12.009
Jang, D. H., Owiredu, S., Ranganathan, A., and Eckmann, D. M. (2018). Acute decompression following simulated dive conditions alters mitochondrial respiration and motility. Am. J. Physiol. Cell Physiol. 315, C699–C705. doi:10.1152/ajpcell.00243.2018
Jenner, A., Pena-Blanco, A., Salvador-Gallego, R., Ugarte-Uribe, B., Zollo, C., Ganief, T., et al. (2022). DRP1 interacts directly with BAX to induce its activation and apoptosis. EMBO J. 41, e108587. doi:10.15252/embj.2021108587
Johnson, S. C., Yanos, M. E., Kayser, E. B., Quintana, A., Sangesland, M., Castanza, A., et al. (2013). mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 342, 1524–1528. doi:10.1126/science.1244360
Kalia, R., and Frost, A. (2019). Open and cut: Allosteric motion and membrane fission by dynamin superfamily proteins. Mol. Biol. Cell 30, 2097–2104. doi:10.1091/mbc.E16-10-0709
Kalia, R., Wang, R. Y., Yusuf, A., Thomas, P. V., Agard, D. A., Shaw, J. M., et al. (2018). Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 558, 401–405. doi:10.1038/s41586-018-0211-2
Kandel, J., Chou, P., and Eckmann, D. M. (2015). Automated detection of whole-cell mitochondrial motility and its dependence on cytoarchitectural integrity. Biotechnol. Bioeng. 112, 1395–1405. doi:10.1002/bit.25563
Kang, J. S., Tian, J. H., Pan, P. Y., Zald, P., Li, C., Deng, C., et al. (2008). Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 132, 137–148. doi:10.1016/j.cell.2007.11.024
Karbowski, M., Neutzner, A., and Youle, R. J. (2007). The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J. Cell Biol. 178, 71–84. doi:10.1083/jcb.200611064
Karbowski, M., Norris, K. L., Cleland, M. M., Jeong, S. Y., and Youle, R. J. (2006). Role of bax and bak in mitochondrial morphogenesis. Nature 443, 658–662. doi:10.1038/nature05111
Kashatus, J. A., Nascimento, A., Myers, L. J., Sher, A., Byrne, F. L., Hoehn, K. L., et al. (2015). Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol. Cell 57, 537–551. doi:10.1016/j.molcel.2015.01.002
Kelso, G. F., Porteous, C. M., Coulter, C. V., Hughes, G., Porteous, W. K., Ledgerwood, E. C., et al. (2001). Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 276, 4588–4596. doi:10.1074/jbc.M009093200
Kijima, K., Numakura, C., Izumino, H., Umetsu, K., Nezu, A., Shiiki, T., et al. (2005). Mitochondrial GTPase mitofusin 2 mutation in Charcot-Marie-Tooth neuropathy type 2A. Hum. Genet. 116, 23–27. doi:10.1007/s00439-004-1199-2
Kim, H., Scimia, M. C., Wilkinson, D., Trelles, R. D., Wood, M. R., Bowtell, D., et al. (2011). Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol. Cell 44, 532–544. doi:10.1016/j.molcel.2011.08.045
Kline, L. B., and Glaser, J. S. (1979). Dominant optic atrophy. The clinical profile. Arch. Ophthalmol. 97, 1680–1686. doi:10.1001/archopht.1979.01020020248013
Koopman, W. J., Visch, H. J., Smeitink, J. A., and Willems, P. H. (2006). Simultaneous quantitative measurement and automated analysis of mitochondrial morphology, mass, potential, and motility in living human skin fibroblasts. Cytom. A 69, 1–12. doi:10.1002/cyto.a.20198
Koshiba, T., Detmer, S. A., Kaiser, J. T., Chen, H., Mccaffery, J. M., and Chan, D. C. (2004). Structural basis of mitochondrial tethering by mitofusin complexes. Science 305, 858–862. doi:10.1126/science.1099793
Kraus, F., and Ryan, M. T. (2017). The constriction and scission machineries involved in mitochondrial fission. J. Cell Sci. 130, 2953–2960. doi:10.1242/jcs.199562
Labbe, K., Mookerjee, S., LE Vasseur, M., Gibbs, E., Lerner, C., and Nunnari, J. (2021). The modified mitochondrial outer membrane carrier MTCH2 links mitochondrial fusion to lipogenesis. J. Cell Biol. 220, e202103122. doi:10.1083/jcb.202103122
Leboucher, G. P., Tsai, Y. C., Yang, M., Shaw, K. C., Zhou, M., Veenstra, T. D., et al. (2012). Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol. Cell 47, 547–557. doi:10.1016/j.molcel.2012.05.041
Lee, Y. J., Jeong, S. Y., Karbowski, M., Smith, C. L., and Youle, R. J. (2004). Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Biol. Cell 15, 5001–5011. doi:10.1091/mbc.e04-04-0294
Legros, F., Lombes, A., Frachon, P., and Rojo, M. (2002). Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol. Biol. Cell 13, 4343–4354. doi:10.1091/mbc.e02-06-0330
Lewis, M. R., and Lewis, W. H. (1914). Mitochondria in tissue culture. Science 39, 330–333. doi:10.1126/science.39.1000.330
Li, J., Donath, S., Li, Y., Qin, D., Prabhakar, B. S., and Li, P. (2010). miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. 6, e1000795. doi:10.1371/journal.pgen.1000795
Li, J., Li, Y., Jiao, J., Wang, J., Li, Y., Qin, D., et al. (2014a). Mitofusin 1 is negatively regulated by microRNA 140 in cardiomyocyte apoptosis. Mol. Cell. Biol. 34, 1788–1799. doi:10.1128/MCB.00774-13
Li, Q., Zhang, T., Wang, J., Zhang, Z., Zhai, Y., Yang, G. Y., et al. (2014b). Rapamycin attenuates mitochondrial dysfunction via activation of mitophagy in experimental ischemic stroke. Biochem. Biophys. Res. Commun. 444, 182–188. doi:10.1016/j.bbrc.2014.01.032
Li, X., Wang, F. S., Wu, Z. Y., Lin, J. L., Lan, W. B., and Lin, J. H. (2014c). MicroRNA-19b targets Mfn1 to inhibit Mfn1-induced apoptosis in osteosarcoma cells. Neoplasma 61, 265–273. doi:10.4149/neo_2014_034
Liang, C., Zhang, B., Cui, L., Li, J., Yu, Q., and Li, M. (2018). Mgm1 is required for maintenance of mitochondrial function and virulence in Candida albicans. Fungal Genet. Biol. 120, 42–52. doi:10.1016/j.fgb.2018.09.006
Liesa, M., and Shirihai, O. S. (2013). Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 17, 491–506. doi:10.1016/j.cmet.2013.03.002
Lihavainen, E., Makela, J., Spelbrink, J. N., and Ribeiro, A. S. (2012). Mytoe: Automatic analysis of mitochondrial dynamics. Bioinformatics 28, 1050–1051. doi:10.1093/bioinformatics/bts073
Lin, M. T., and Beal, M. F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. doi:10.1038/nature05292
Lin, M. Y., and Sheng, Z. H. (2015). Regulation of mitochondrial transport in neurons. Exp. Cell Res. 334, 35–44. doi:10.1016/j.yexcr.2015.01.004
Linden, M., Li, Z. L., Paulin, D., Gotow, T., and Leterrier, J. F. (2001). Effects of desmin gene knockout on mice heart mitochondria. J. Bioenerg. Biomembr. 33, 333–341. doi:10.1023/a:1010611408007
Lister, I., Roberts, R., Schmitz, S., Walker, M., Trinick, J., Veigel, C., et al. (2004). Myosin VI: A multifunctional motor. Biochem. Soc. Trans. 32, 685–688. doi:10.1042/BST0320685
Liu, W., Yamashita, T., Tian, F., Morimoto, N., Ikeda, Y., Deguchi, K., et al. (2013). Mitochondrial fusion and fission proteins expression dynamically change in a murine model of amyotrophic lateral sclerosis. Curr. Neurovasc. Res. 10, 222–230. doi:10.2174/15672026113109990060
Liu, X. G., Weaver, D., Shirihai, O., and Hajnoczky, G. (2009). Mitochondrial 'kiss-and-run': Interplay between mitochondrial motility and fusion-fission dynamics. Embo J. 28, 3074–3089. doi:10.1038/emboj.2009.255
Liu, X., Yang, L., Long, Q., Weaver, D., and Hajnoczky, G. (2017). Choosing proper fluorescent dyes, proteins, and imaging techniques to study mitochondrial dynamics in mammalian cells. Biophys. Rep. 3, 64–72. doi:10.1007/s41048-017-0037-8
Lopez-Lluch, G., Irusta, P. M., Navas, P., and DE Cabo, R. (2008). Mitochondrial biogenesis and healthy aging. Exp. Gerontol. 43, 813–819. doi:10.1016/j.exger.2008.06.014
Loson, O. C., Song, Z., Chen, H., and Chan, D. C. (2013). Fis1, mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 24, 659–667. doi:10.1091/mbc.E12-10-0721
Lu, B., Kennedy, B., Clinton, R. W., Wang, E. J., Mchugh, D., Stepanyants, N., et al. (2018). Steric interference from intrinsically disordered regions controls dynamin-related protein 1 self-assembly during mitochondrial fission. Sci. Rep. 8, 10879. doi:10.1038/s41598-018-29001-9
Lu, Z., Ma, X. N., Zhang, H. M., Ji, H. H., Ding, H., Zhang, J., et al. (2014). Mouse myosin-19 is a plus-end-directed, high-duty ratio molecular motor. J. Biol. Chem. 289, 18535–18548. doi:10.1074/jbc.M114.569087
Lutz, A. K., Exner, N., Fett, M. E., Schlehe, J. S., Kloos, K., Lammermann, K., et al. (2009). Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J. Biol. Chem. 284, 22938–22951. doi:10.1074/jbc.M109.035774
Macaskill, A. F., Brickley, K., Stephenson, F. A., and Kittler, J. T. (2009a). GTPase dependent recruitment of Grif-1 by Miro1 regulates mitochondrial trafficking in hippocampal neurons. Mol. Cell. Neurosci. 40, 301–312. doi:10.1016/j.mcn.2008.10.016
Macaskill, A. F., Rinholm, J. E., Twelvetrees, A. E., Arancibia-Carcamo, I. L., Muir, J., Fransson, A., et al. (2009b). Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 61, 541–555. doi:10.1016/j.neuron.2009.01.030
Macvicar, T., and Langer, T. (2016). OPA1 processing in cell death and disease - the long and short of it. J. Cell Sci. 129, 2297–2306. doi:10.1242/jcs.159186
Malka, F., Guillery, O., Cifuentes-Diaz, C., Guillou, E., Belenguer, P., Lombes, A., et al. (2005). Separate fusion of outer and inner mitochondrial membranes. EMBO Rep. 6, 853–859. doi:10.1038/sj.embor.7400488
Mandal, A., and Drerup, C. M. (2019). Axonal transport and mitochondrial function in neurons. Front. Cell. Neurosci. 13, 373. doi:10.3389/fncel.2019.00373
Martin-Fernandez, B., and Gredilla, R. (2016). Mitochondria and oxidative stress in heart aging. Age (Dordr) 38, 225–238. doi:10.1007/s11357-016-9933-y
Martorell-Riera, A., Segarra-Mondejar, M., Munoz, J. P., Ginet, V., Olloquequi, J., Perez-Clausell, J., et al. (2014). Mfn2 downregulation in excitotoxicity causes mitochondrial dysfunction and delayed neuronal death. EMBO J. 33, 2388–2407. doi:10.15252/embj.201488327
Marzetti, E., Csiszar, A., Dutta, D., Balagopal, G., Calvani, R., and Leeuwenburgh, C. (2013). Role of mitochondrial dysfunction and altered autophagy in cardiovascular aging and disease: From mechanisms to therapeutics. Am. J. Physiol. Heart Circ. Physiol. 305, H459–H476. doi:10.1152/ajpheart.00936.2012
Mattie, S., Krols, M., and Mcbride, H. M. (2019). The enigma of an interconnected mitochondrial reticulum: New insights into mitochondrial fusion. Curr. Opin. Cell Biol. 59, 159–166. doi:10.1016/j.ceb.2019.05.004
Mattie, S., Riemer, J., Wideman, J. G., and Mcbride, H. M. (2018). A new mitofusin topology places the redox-regulated C terminus in the mitochondrial intermembrane space. J. Cell Biol. 217, 507–515. doi:10.1083/jcb.201611194
Matveeva, E. A., Venkova, L. S., Chernoivanenko, I. S., and Minin, A. A. (2015). Vimentin is involved in regulation of mitochondrial motility and membrane potential by Rac1. Biol. Open 4, 1290–1297. doi:10.1242/bio.011874
Mcconell, G. K., Ng, G. P., Phillips, M., Ruan, Z., Macaulay, S. L., and Wadley, G. D. (2010). Central role of nitric oxide synthase in AICAR and caffeine-induced mitochondrial biogenesis in L6 myocytes. J. Appl. Physiol. 108, 589–595. doi:10.1152/japplphysiol.00377.2009
Meeusen, S., Devay, R., Block, J., Cassidy-Stone, A., Wayson, S., Mccaffery, J. M., et al. (2006). Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell 127, 383–395. doi:10.1016/j.cell.2006.09.021
Melkov, A., Baskar, R., Alcalay, Y., and Abdu, U. (2016). A new mode of mitochondrial transport and polarized sorting regulated by Dynein, Milton and Miro. Development 143, 4203–4213. doi:10.1242/dev.138289
Mentel, M., Rottger, M., Leys, S., Tielens, A. G., and Martin, W. F. (2014). Of early animals, anaerobic mitochondria, and a modern sponge. Bioessays 36, 924–932. doi:10.1002/bies.201400060
Milner, D. J., Mavroidis, M., Weisleder, N., and Capetanaki, Y. (2000). Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J. Cell Biol. 150, 1283–1298. doi:10.1083/jcb.150.6.1283
Mishra, P., Carelli, V., Manfredi, G., and Chan, D. C. (2014). Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 19, 630–641. doi:10.1016/j.cmet.2014.03.011
Mishra, P., Varuzhanyan, G., Pham, A. H., and Chan, D. C. (2015). Mitochondrial dynamics is a distinguishing feature of skeletal muscle fiber types and regulates organellar compartmentalization. Cell Metab. 22, 1033–1044. doi:10.1016/j.cmet.2015.09.027
Misko, A., Jiang, S., Wegorzewska, I., Milbrandt, J., and Baloh, R. H. (2010). Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci. 30, 4232–4240. doi:10.1523/JNEUROSCI.6248-09.2010
Mitra, K., Wunder, C., Roysam, B., Lin, G., and Lippincott-Schwartz, J. (2009). A hyperfused mitochondrial state achieved at G(1)-S regulates cyclin E buildup and entry into S phase. Proc. Natl. Acad. Sci. U. S. A. 106, 11960–11965. doi:10.1073/pnas.0904875106
Moltedo, O., Remondelli, P., and Amodio, G. (2019). The mitochondria-endoplasmic reticulum contacts and their critical role in aging and age-associated diseases. Front. Cell Dev. Biol. 7, 172. doi:10.3389/fcell.2019.00172
Moore, A. S., and Holzbaur, E. L. (2016). Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc. Natl. Acad. Sci. U. S. A. 113, E3349–E3358. doi:10.1073/pnas.1523810113
Mposhi, A., Van der wijst, M. G., Faber, K. N., and Rots, M. G. (2017). Regulation of mitochondrial gene expression, the epigenetic enigma. Front. Biosci. 22, 1099–1113. doi:10.2741/4535
Murakawa, T., Yamaguchi, O., Hashimoto, A., Hikoso, S., Takeda, T., Oka, T., et al. (2015). Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat. Commun. 6, 7527. doi:10.1038/ncomms8527
Nakamura, N., Kimura, Y., Tokuda, M., Honda, S., and Hirose, S. (2006). MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep. 7, 1019–1022. doi:10.1038/sj.embor.7400790
Nangaku, M., Satoyoshitake, R., Okada, Y., Noda, Y., Takemura, R., Yamazaki, H., et al. (1994). Kif1b, a novel microtubule plus end-directed monomeric motor protein for transport of mitochondria. Cell 79, 1209–1220. doi:10.1016/0092-8674(94)90012-4
Naon, D., Zaninello, M., Giacomello, M., Varanita, T., Grespi, F., Lakshminaranayan, S., et al. (2016). Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc. Natl. Acad. Sci. U. S. A. 113, 11249–11254. doi:10.1073/pnas.1606786113
Nekrasova, O. E., Mendez, M. G., Chernoivanenko, I. S., Tyurin-Kuzmin, P. A., Kuczmarski, E. R., Gelfand, V. I., et al. (2011). Vimentin intermediate filaments modulate the motility of mitochondria. Mol. Biol. Cell 22, 2282–2289. doi:10.1091/mbc.E10-09-0766
Nguyen, T. N., Padman, B. S., and Lazarou, M. (2016). Deciphering the molecular signals of PINK1/parkin mitophagy. Trends Cell Biol. 26, 733–744. doi:10.1016/j.tcb.2016.05.008
Ohashi, K. G., Han, L., Mentley, B., Wang, J., Fricks, J., and Hancock, W. O. (2019). Load-dependent detachment kinetics plays a key role in bidirectional cargo transport by kinesin and dynein. Traffic 20, 284–294. doi:10.1111/tra.12639
Ohno, N., Chiang, H., Mahad, D. J., Kidd, G. J., Liu, L. P., Ransohoff, R. M., et al. (2014). Mitochondrial immobilization mediated by syntaphilin facilitates survival of demyelinated axons. Proc. Natl. Acad. Sci. U. S. A. 111, 9953–9958. doi:10.1073/pnas.1401155111
Olichon, A., Emorine, L. J., Descoins, E., Pelloquin, L., Brichese, L., Gas, N., et al. (2002). The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett. 523, 171–176. doi:10.1016/s0014-5793(02)02985-x
Omodei, D., and Fontana, L. (2011). Calorie restriction and prevention of age-associated chronic disease. FEBS Lett. 585, 1537–1542. doi:10.1016/j.febslet.2011.03.015
Otera, H., Wang, C., Cleland, M. M., Setoguchi, K., Yokota, S., Youle, R. J., et al. (2010). Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 191, 1141–1158. doi:10.1083/jcb.201007152
Owiredu, S., Ranganathan, A., Eckmann, D. M., Shofer, F. S., Hardy, K., Lambert, D. S., et al. (2020). Ex vivo use of cell-permeable succinate prodrug attenuates mitochondrial dysfunction in blood cells obtained from carbon monoxide-poisoned individuals. Am. J. Physiol. Cell Physiol. 319, C129–C135. doi:10.1152/ajpcell.00539.2019
Palmer, C. S., Elgass, K. D., Parton, R. G., Osellame, L. D., Stojanovski, D., and Ryan, M. T. (2013). Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J. Biol. Chem. 288, 27584–27593. doi:10.1074/jbc.M113.479873
Palmer, C. S., Osellame, L. D., Laine, D., Koutsopoulos, O. S., Frazier, A. E., and Ryan, M. T. (2011). MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 12, 565–573. doi:10.1038/embor.2011.54
Park, S. J., Bae, J. E., Jo, D. S., Kim, J. B., Park, N. Y., Fang, J., et al. (2021). Increased O-GlcNAcylation of Drp1 by amyloid-beta promotes mitochondrial fission and dysfunction in neuronal cells. Mol. Brain 14, 6. doi:10.1186/s13041-020-00727-w
Parra, V., Verdejo, H. E., Iglewski, M., Del Campo, A., Troncoso, R., Jones, D., et al. (2014). Insulin stimulates mitochondrial fusion and function in cardiomyocytes via the Akt-mTOR-NFκB-Opa-1 signaling pathway. Diabetes 63, 75–88. doi:10.2337/db13-0340
Pathak, D., Sepp, K. J., and Hollenbeck, P. J. (2010). Evidence that myosin activity opposes microtubule-based axonal transport of mitochondria. J. Neurosci. 30, 8984–8992. doi:10.1523/JNEUROSCI.1621-10.2010
Patra, S., Mahapatra, K. K., Praharaj, P. P., Panigrahi, D. P., Bhol, C. S., Mishra, S. R., et al. (2021). Intricate role of mitochondrial calcium signalling in mitochondrial quality control for regulation of cancer cell fate. Mitochondrion 57, 230–240. doi:10.1016/j.mito.2021.01.002
Patterson, G. H., and Lippincott-Schwartz, J. (2002). A photoactivatable GFP for selective photolabeling of proteins and cells. Science 297, 1873–1877. doi:10.1126/science.1074952
Pekkurnaz, G., Trinidad, J. C., Wang, X., Kong, D., and Schwarz, T. L. (2014). Glucose regulates mitochondrial motility via Milton modification by O-GlcNAc transferase. Cell 158, 54–68. doi:10.1016/j.cell.2014.06.007
Peng, J., Ren, K. D., Yang, J., and Luo, X. J. (2016a). Mitochondrial E3 ubiquitin ligase 1: A key enzyme in regulation of mitochondrial dynamics and functions. Mitochondrion 28, 49–53. doi:10.1016/j.mito.2016.03.007
Peng, K., Tao, Y., Zhang, J., Wang, J., Ye, F., Dan, G., et al. (2016b2016). Resveratrol regulates mitochondrial biogenesis and fission/fusion to attenuate rotenone-induced neurotoxicity. Oxid. Med. Cell. Longev. 2016, 6705621. doi:10.1155/2016/6705621
Pieczenik, S. R., and Neustadt, J. (2007). Mitochondrial dysfunction and molecular pathways of disease. Exp. Mol. Pathol. 83, 84–92. doi:10.1016/j.yexmp.2006.09.008
Pilling, A. D., Horiuchi, D., Lively, C. M., and Saxton, W. M. (2006). Kinesin-1 and dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol. Biol. Cell 17, 2057–2068. doi:10.1091/mbc.e05-06-0526
Praefcke, G. J., and Mcmahon, H. T. (2004). The dynamin superfamily: Universal membrane tubulation and fission molecules? Nat. Rev. Mol. Cell Biol. 5, 133–147. doi:10.1038/nrm1313
Pyakurel, A., Savoia, C., Hess, D., and Scorrano, L. (2015). Extracellular regulated kinase phosphorylates mitofusin 1 to control mitochondrial morphology and apoptosis. Mol. Cell 58, 244–254. doi:10.1016/j.molcel.2015.02.021
Qi, X., Qvit, N., Su, Y. C., and Mochly-Rosen, D. (2013). A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J. Cell Sci. 126, 789–802. doi:10.1242/jcs.114439
Quintero, O. A., Divito, M. M., Adikes, R. C., Kortan, M. B., Case, L. B., Lier, A. J., et al. (2009). Human Myo19 is a novel myosin that associates with mitochondria. Curr. Biol. 19, 2008–2013. doi:10.1016/j.cub.2009.10.026
Rambold, A. S., Kostelecky, B., Elia, N., and Lippincott-Schwartz, J. (2011). Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. U. S. A. 108, 10190–10195. doi:10.1073/pnas.1107402108
Ranganathan, A., Owiredu, S., Jang, D. H., and Eckmann, D. M. (2020). Prophylaxis of mitochondrial dysfunction caused by cellular decompression from hyperbaric exposure. Mitochondrion 52, 8–19. doi:10.1016/j.mito.2020.02.002
Rapaport, D., Brunner, M., Neupert, W., and Westermann, B. (1998). Fzo1p is a mitochondrial outer membrane protein essential for the biogenesis of functional mitochondria in Saccharomyces cerevisiae. J. Biol. Chem. 273, 20150–20155. doi:10.1074/jbc.273.32.20150
Reck-Peterson, S. L., Redwine, W. B., Vale, R. D., and Carter, A. P. (2018). Publisher Correction: The cytoplasmic dynein transport machinery and its many cargoes. Nat. Rev. Mol. Cell Biol. 19, 479. doi:10.1038/s41580-018-0021-2
Ribeiro Junior, R. F., Dabkowski, E. R., Shekar, K. C., Ka, O. C., Hecker, P. A., and Murphy, M. P. (2018). MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radic. Biol. Med. 117, 18–29. doi:10.1016/j.freeradbiomed.2018.01.012
Rintoul, G. L., Filiano, A. J., Brocard, J. B., Kress, G. J., and Reynolds, I. J. (2003). Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J. Neurosci. 23, 7881–7888. doi:10.1523/jneurosci.23-21-07881.2003
Rodriguez, G. E., Gonzalez, D. M., Monachelli, G. M., Costa, J. J., Nicola, A. F., and Sica, R. E. (2012). Morphological abnormalities in mitochondria of the skin of patients with sporadic amyotrophic lateral sclerosis. Arq. Neuropsiquiatr. 70, 40–44. doi:10.1590/s0004-282x2012000100010
Rujiviphat, J., Wong, M. K., Won, A., Shih, Y. L., Yip, C. M., and Mcquibban, G. A. (2015). Mitochondrial genome maintenance 1 (Mgm1) protein alters membrane topology and promotes local membrane bending. J. Mol. Biol. 427, 2599–2609. doi:10.1016/j.jmb.2015.03.006
Rutkai, I., Evans, W. R., Bess, N., Salter-Cid, T., Cikic, S., Chandra, P. K., et al. (2020). Chronic imaging of mitochondria in the murine cerebral vasculature using in vivo two-photon microscopy. Am. J. Physiol. Heart Circ. Physiol. 318, H1379–H1386. doi:10.1152/ajpheart.00751.2019
Samangouei, P., Crespo-Avilan, G. E., Cabrera-Fuentes, H., Hernandez-Resendiz, S., Ismail, N. I., Katwadi, K. B., et al. (2018). MiD49 and MiD51: New mediators of mitochondrial fission and novel targets for cardioprotection. Cond. Med. 1, 239–246.
Santama, N., Er, C. P. N., Ong, L. L., and Yu, H. (2004). Distribution and functions of kinectin isoforms. J. Cell Sci. 117, 4537–4549. doi:10.1242/jcs.01326
Santel, A., and Frank, S. (2008). Shaping mitochondria: The complex posttranslational regulation of the mitochondrial fission protein DRP1. IUBMB Life 60, 448–455. doi:10.1002/iub.71
Saotome, M., Safiulina, D., Szabadkai, G., Das, S., Fransson, A., Aspenstrom, P., et al. (2008). Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc. Natl. Acad. Sci. U. S. A. 105, 20728–20733. doi:10.1073/pnas.0808953105
Sarkar, S., Krishna, G., Imarisio, S., Saiki, S., O'Kane, C. J., and Rubinsztein, D. C. (2008). A rational mechanism for combination treatment of Huntington's disease using lithium and rapamycin. Hum. Mol. Genet. 17, 170–178. doi:10.1093/hmg/ddm294
Schnapp, B. J., and Reese, T. S. (1989). Dynein is the motor for retrograde Axonal-transport of organelles. Proc. Natl. Acad. Sci. U. S. A. 86, 1548–1552. doi:10.1073/pnas.86.5.1548
Schroer, T. A. (2004). Dynactin. Annu. Rev. Cell Dev. Biol. 20, 759–779. doi:10.1146/annurev.cellbio.20.012103.094623
Schwarz, N., and Leube, R. E. (2016). Intermediate filaments as organizers of cellular space: How they affect mitochondrial structure and function. Cells 5, E30. doi:10.3390/cells5030030
Serasinghe, M. N., Wieder, S. Y., Renault, T. T., Elkholi, R., Asciolla, J. J., Yao, J. L., et al. (2015). Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol. Cell 57, 521–536. doi:10.1016/j.molcel.2015.01.003
Sesaki, H., Adachi, Y., Kageyama, Y., Itoh, K., and Iijima, M. (2014). In vivo functions of Drp1: Lessons learned from yeast genetics and mouse knockouts. Biochim. Biophys. Acta 1842, 1179–1185. doi:10.1016/j.bbadis.2013.11.024
Sesaki, H., and Jensen, R. E. (2001). UGO1 encodes an outer membrane protein required for mitochondrial fusion. J. Cell Biol. 152, 1123–1134. doi:10.1083/jcb.152.6.1123
Sharp, W. W., Fang, Y. H., Han, M., Zhang, H. J., Hong, Z., Banathy, A., et al. (2014). Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: Therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. 28, 316–326. doi:10.1096/fj.12-226225
Sheng, Z. H. (2014). Mitochondrial trafficking and anchoring in neurons: New insight and implications. J. Cell Biol. 204, 1087–1098. doi:10.1083/jcb.201312123
Shutt, T., Geoffrion, M., Milne, R., and Mcbride, H. M. (2012). The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep. 13, 909–915. doi:10.1038/embor.2012.128
Simon, V. R., Swayne, T. C., and Pon, L. A. (1995). Actin-dependent mitochondrial motility in mitotic yeast and cell-free systems: Identification of a motor activity on the mitochondrial surface. J. Cell Biol. 130, 345–354. doi:10.1083/jcb.130.2.345
Sirajuddin, M., Rice, L. M., and Vale, R. D. (2014). Regulation of microtubule motors by tubulin isotypes and post-translational modifications. Nat. Cell Biol. 16, 335–344. doi:10.1038/ncb2920
Smirnova, E., Griparic, L., Shurland, D. L., and VAN DER Bliek, A. M. (2001). Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 12, 2245–2256. doi:10.1091/mbc.12.8.2245
Song, M., Franco, A., Fleischer, J. A., Zhang, L., and Dorn, G. W.2ND (2017). Abrogating mitochondrial dynamics in mouse hearts accelerates mitochondrial senescence. Cell Metab. 26, 872–883. doi:10.1016/j.cmet.2017.09.023
Song, W., Chen, J., Petrilli, A., Liot, G., Klinglmayr, E., Zhou, Y., et al. (2011). Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat. Med. 17, 377–382. doi:10.1038/nm.2313
Song, W., Song, Y., Kincaid, B., Bossy, B., and Bossy-Wetzel, E. (2013). Mutant SOD1G93A triggers mitochondrial fragmentation in spinal cord motor neurons: Neuroprotection by SIRT3 and PGC-1α. Neurobiol. Dis. 51, 72–81. doi:10.1016/j.nbd.2012.07.004
Soriano, F. X., Liesa, M., Bach, D., Chan, D. C., Palacin, M., and Zorzano, A. (2006). Evidence for a mitochondrial regulatory pathway defined by peroxisome proliferator-activated receptor-gamma coactivator-1 alpha, estrogen-related receptor-alpha, and mitofusin 2. Diabetes 55, 1783–1791. doi:10.2337/db05-0509
Spang, A., Saw, J. H., Jorgensen, S. L., Zaremba-Niedzwiedzka, K., Martijn, J., Lind, A. E., et al. (2015). Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 521, 173–179. doi:10.1038/nature14447
Spillane, M., Ketschek, A., Merianda, T. T., Twiss, J. L., and Gallo, G. (2013). Mitochondria coordinate sites of axon branching through localized intra-axonal protein synthesis. Cell Rep. 5, 1564–1575. doi:10.1016/j.celrep.2013.11.022
Stowers, R. S., Megeath, L. J., Gorska-Andrzejak, J., Meinertzhagen, I. A., and Schwarz, T. L. (2002). Axonal transport of mitochondria to synapses depends on Milton, a novel Drosophila protein. Neuron 36, 1063–1077. doi:10.1016/s0896-6273(02)01094-2
Szabadkai, G., Simoni, A. M., Bianchi, K., DE Stefani, D., Leo, S., Wieckowski, M. R., et al. (2006). Mitochondrial dynamics and Ca2+ signaling. Biochim. Biophys. Acta 1763, 442–449. doi:10.1016/j.bbamcr.2006.04.002
Taguchi, N., Ishihara, N., Jofuku, A., Oka, T., and Mihara, K. (2007). Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 282, 11521–11529. doi:10.1074/jbc.M607279200
Takihara, Y., Nakamura-Ishizu, A., Tan, D. Q., Fukuda, M., Matsumura, T., Endoh, M., et al. (2019). High mitochondrial mass is associated with reconstitution capacity and quiescence of hematopoietic stem cells. Blood Adv. 3, 2323–2327. doi:10.1182/bloodadvances.2019032169
Tanaka, K., Sugiura, Y., Ichishita, R., Mihara, K., and Oka, T. (2011). KLP6: A newly identified kinesin that regulates the morphology and transport of mitochondria in neuronal cells. J. Cell Sci. 124, 2457–2465. doi:10.1242/jcs.086470
Tieu, Q., and Nunnari, J. (2000). Mdv1p is a WD repeat protein that interacts with the dynamin-related GTPase, Dnm1p, to trigger mitochondrial division. J. Cell Biol. 151, 353–366. doi:10.1083/jcb.151.2.353
Tondera, D., Grandemange, S., Jourdain, A., Karbowski, M., Mattenberger, Y., Herzig, S., et al. (2009). SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 28, 1589–1600. doi:10.1038/emboj.2009.89
Trushina, E., Dyer, R. B., Badger, J. D., Ure, D., Eide, L., Tran, D. D., et al. (2004). Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol. Cell. Biol. 24, 8195–8209. doi:10.1128/MCB.24.18.8195-8209.2004
Twig, G., Elorza, A., Molina, A. J. A., Mohamed, H., Wikstrom, J. D., Walzer, G., et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. Embo J. 27, 433–446. doi:10.1038/sj.emboj.7601963
Vakifahmetoglu-Norberg, H., Ouchida, A. T., and Norberg, E. (2017). The role of mitochondria in metabolism and cell death. Biochem. Biophys. Res. Commun. 482, 426–431. doi:10.1016/j.bbrc.2016.11.088
Vale, R. D. (2003). The molecular motor toolbox for intracellular transport. Cell 112, 467–480. doi:10.1016/s0092-8674(03)00111-9
Valente, A. J., Maddalena, L. A., Robb, E. L., Moradi, F., and Stuart, J. A. (2017). A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem. 119, 315–326. doi:10.1016/j.acthis.2017.03.001
Valm, A. M., Cohen, S., Legant, W. R., Melunis, J., Hershberg, U., Wait, E., et al. 2017. Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature, 546, 162-167. doi:10.1038/nature22369
Vande Velde, C., Mcdonald, K. K., Boukhedimi, Y., Mcalonis-Downes, M., Lobsiger, C. S., Bel Hadj, S., et al. (2011). Misfolded SOD1 associated with motor neuron mitochondria alters mitochondrial shape and distribution prior to clinical onset. PLoS One 6, e22031. doi:10.1371/journal.pone.0022031
Viana, M. P., Brown, A. I., Mueller, I. A., Goul, C., Koslover, E. F., and Rafelski, S. M. (2020). Mitochondrial fission and fusion dynamics generate efficient, robust, and evenly distributed network topologies in budding yeast cells. Cell Syst. 10, 287–297. doi:10.1016/j.cels.2020.02.002
Viana, M. P., Lim, S., and Rafelski, S. M. (2015). Quantifying mitochondrial content in living cells. Methods Cell Biol. 125, 77–93. doi:10.1016/bs.mcb.2014.10.003
Villa-Cuesta, E., Holmbeck, M. A., and Rand, D. M. (2014). Rapamycin increases mitochondrial efficiency by mtDNA-dependent reprogramming of mitochondrial metabolism in Drosophila. J. Cell Sci. 127, 2282–2290. doi:10.1242/jcs.142026
Vinsant, S., Mansfield, C., Jimenez-Moreno, R., Del Gaizo Moore, V., Yoshikawa, M., Hampton, T. G., et al. (2013). Characterization of early pathogenesis in the SOD1(g93a) mouse model of ALS: Part I, background and methods. Brain Behav. 3, 335–350. doi:10.1002/brb3.143
Wagner, O. I., Lifshitz, J., Janmey, P. A., Linden, M., Mcintosh, T. K., and Leterrier, J. F. (2003). Mechanisms of mitochondria-neurofilament interactions. J. Neurosci. 23, 9046–9058. doi:10.1523/jneurosci.23-27-09046.2003
Wakabayashi, J., Zhang, Z., Wakabayashi, N., Tamura, Y., Fukaya, M., Kensler, T. W., et al. (2009). The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J. Cell Biol. 186, 805–816. doi:10.1083/jcb.200903065
Wang, A., Mouser, J., Pitt, J., Promislow, D., and Kaeberlein, M. (2016). Rapamycin enhances survival in a Drosophila model of mitochondrial disease. Oncotarget 7, 80131–80139. doi:10.18632/oncotarget.12560
Wang, H., Lim, P. J., Karbowski, M., and Monteiro, M. J. (2009). Effects of overexpression of huntingtin proteins on mitochondrial integrity. Hum. Mol. Genet. 18, 737–752. doi:10.1093/hmg/ddn404
Wang, J. X., Jiao, J. Q., Li, Q., Long, B., Wang, K., Liu, J. P., et al. (2011). miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat. Med. 17, 71–78. doi:10.1038/nm.2282
Wang, R., Mishra, P., Garbis, S. D., Moradian, A., Sweredoski, M. J., and Chan, D. C. (2021). Identification of new OPA1 cleavage site reveals that short isoforms regulate mitochondrial fusion. Mol. Biol. Cell 32, 157–168. doi:10.1091/mbc.E20-09-0605
Wang, W., Wang, Y., Long, J., Wang, J., Haudek, S. B., Overbeek, P., et al. (2012). Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 15, 186–200. doi:10.1016/j.cmet.2012.01.009
Wang, X., and Schwarz, T. L. (2009). The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell 136, 163–174. doi:10.1016/j.cell.2008.11.046
Wang, Z., and Wu, M. (2015). An integrated phylogenomic approach toward pinpointing the origin of mitochondria. Sci. Rep. 5, 7949. doi:10.1038/srep07949
Warby, S. C., Montpetit, A., Hayden, A. R., Carroll, J. B., Butland, S. L., Visscher, H., et al. (2009). CAG expansion in the Huntington disease gene is associated with a specific and targetable predisposing haplogroup. Am. J. Hum. Genet. 84, 351–366. doi:10.1016/j.ajhg.2009.02.003
Wenger, J., Klinglmayr, E., Frohlich, C., Eibl, C., Gimeno, A., Hessenberger, M., et al. (2013). Functional mapping of human dynamin-1-like GTPase domain based on x-ray structure analyses. PLoS One 8, e71835. doi:10.1371/journal.pone.0071835
Wong, E. D., Wagner, J. A., Gorsich, S. W., Mccaffery, J. M., Shaw, J. M., and Nunnari, J. (2000). The dynamin-related GTPase, Mgm1p, is an intermembrane space protein required for maintenance of fusion competent mitochondria. J. Cell Biol. 151, 341–352. doi:10.1083/jcb.151.2.341
Wong, E. D., Wagner, J. A., Scott, S. V., Okreglak, V., Holewinske, T. J., Cassidy-Stone, A., et al. (2003). The intramitochondrial dynamin-related GTPase, Mgm1p, is a component of a protein complex that mediates mitochondrial fusion. J. Cell Biol. 160, 303–311. doi:10.1083/jcb.200209015
Wortman, J. C., Shrestha, U. M., Barry, D. M., Garcia, M. L., Gross, S. P., and Yu, C. C. (2014). Axonal transport: How high microtubule density can compensate for boundary effects in small-caliber axons. Biophys. J. 106, 813–823. doi:10.1016/j.bpj.2013.12.047
Wu, H., Li, Z., Wang, Y., Yang, P., Li, Z., Li, H., et al. (2016). MiR-106b-mediated Mfn2 suppression is critical for PKM2 induced mitochondrial fusion. Am. J. Cancer Res. 6, 2221–2234.
Yamashita, S. I., Jin, X., Furukawa, K., Hamasaki, M., Nezu, A., Otera, H., et al. (2016). Mitochondrial division occurs concurrently with autophagosome formation but independently of Drp1 during mitophagy. J. Cell Biol. 215, 649–665. doi:10.1083/jcb.201605093
Yang, C., Endoh, M., Tan, D. Q., Nakamura-Ishizu, A., Takihara, Y., Matsumura, T., et al. (2021). Mitochondria transfer from early stages of erythroblasts to their macrophage niche via tunnelling nanotubes. Br. J. Haematol. 193, 1260–1274. doi:10.1111/bjh.17531
Yarosh, W., Monserrate, J., Tong, J. J., Tse, S., LE, P. K., Nguyen, K., et al. (2008). The molecular mechanisms of OPA1-mediated optic atrophy in Drosophila model and prospects for antioxidant treatment. PLoS Genet. 4, e6. doi:10.1371/journal.pgen.0040006
Yi, M., Weaver, D., and Hajnoczky, G. (2004). Control of mitochondrial motility and distribution by the calcium signal: A homeostatic circuit. J. Cell Biol. 167, 661–672. doi:10.1083/jcb.200406038
Yoo, S. M., and Jung, Y. K. (2018). A molecular approach to mitophagy and mitochondrial dynamics. Mol. Cells 41, 18–26. doi:10.14348/molcells.2018.2277
Yoon, Y., Krueger, E. W., Oswald, B. J., and Mcniven, M. A. (2003). The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol. Cell. Biol. 23, 5409–5420. doi:10.1128/mcb.23.15.5409-5420.2003
Yoon, Y. S., Yoon, D. S., Lim, I. K., Yoon, S. H., Chung, H. Y., Rojo, M., et al. (2006). Formation of elongated giant mitochondria in DFO-induced cellular senescence: Involvement of enhanced fusion process through modulation of Fis1. J. Cell. Physiol. 209, 468–480. doi:10.1002/jcp.20753
Yu, R., Liu, T., Jin, S. B., Ning, C., Lendahl, U., Nister, M., et al. (2017). MIEF1/2 function as adaptors to recruit Drp1 to mitochondria and regulate the association of Drp1 with Mff. Sci. Rep. 7, 880. doi:10.1038/s41598-017-00853-x
Yuan, Q., Yang, W., Zhang, S., Li, T., Zuo, M., Zhou, X., et al. (2021). Inhibition of mitochondrial carrier homolog 2 (MTCH2) suppresses tumor invasion and enhances sensitivity to temozolomide in malignant glioma. Mol. Med. 27, 7. doi:10.1186/s10020-020-00261-4
Zare-Shahabadi, A., Masliah, E., Johnson, G. V., and Rezaei, N. (2015). Autophagy in Alzheimer's disease. Rev. Neurosci. 26, 385–395. doi:10.1515/revneuro-2014-0076
Zechner, C., Lai, L., Zechner, J. F., Geng, T., Yan, Z., Rumsey, J. W., et al. (2010). Total skeletal muscle PGC-1 deficiency uncouples mitochondrial derangements from fiber type determination and insulin sensitivity. Cell Metab. 12, 633–642. doi:10.1016/j.cmet.2010.11.008
Zhang, C. L., Ho, P. L., Kintner, D. B., Sun, D., and Chiu, S. Y. (2010). Activity-dependent regulation of mitochondrial motility by calcium and Na/K-ATPase at nodes of Ranvier of myelinated nerves. J. Neurosci. 30, 3555–3566. doi:10.1523/JNEUROSCI.4551-09.2010
Zhang, F., Wang, W., Siedlak, S. L., Liu, Y., Liu, J., Jiang, K., et al. (2015). Miro1 deficiency in amyotrophic lateral sclerosis. Front. Aging Neurosci. 7, 100. doi:10.3389/fnagi.2015.00100
Zhang, Y., Chan, N. C., Ngo, H. B., Gristick, H., and Chan, D. C. (2012). Crystal structure of mitochondrial fission complex reveals scaffolding function for mitochondrial division 1 (Mdv1) coiled coil. J. Biol. Chem. 287, 9855–9861. doi:10.1074/jbc.M111.329359
Zhou, X., Zhang, L., Zheng, B., Yan, Y., Zhang, Y., Xie, H., et al. (2016). MicroRNA-761 is upregulated in hepatocellular carcinoma and regulates tumorigenesis by targeting Mitofusin-2. Cancer Sci. 107, 424–432. doi:10.1111/cas.12904
Zhu, P. P., Patterson, A., Stadler, J., Seeburg, D. P., Sheng, M., and Blackstone, C. (2004). Intra- and intermolecular domain interactions of the C-terminal GTPase effector domain of the multimeric dynamin-like GTPase Drp1. J. Biol. Chem. 279, 35967–35974. doi:10.1074/jbc.M404105200
Zick, M., Duvezin-Caubet, S., Schafer, A., Vogel, F., Neupert, W., and Reichert, A. S. (2009). Distinct roles of the two isoforms of the dynamin-like GTPase Mgm1 in mitochondrial fusion. FEBS Lett. 583, 2237–2243. doi:10.1016/j.febslet.2009.05.053
Keywords: mitochondria, motility, fusion, fission, mitochondrial DNA, live-cell imaging, disease, therapeutics
Citation: Green A, Hossain T and Eckmann DM (2022) Mitochondrial dynamics involves molecular and mechanical events in motility, fusion and fission. Front. Cell Dev. Biol. 10:1010232. doi: 10.3389/fcell.2022.1010232
Received: 02 August 2022; Accepted: 06 October 2022;
Published: 19 October 2022.
Edited by:
Anne Chiaramello, George Washington University, United StatesReviewed by:
Prashant Mishra, University of Texas Southwestern Medical Center, United StatesRajesh Ramachandran, Case Western Reserve University, United States
Copyright © 2022 Green, Hossain and Eckmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David M. Eckmann, ZGF2aWQuZWNrbWFubkBvc3VtYy5lZHU=