 László Juhász
László Juhász Szabolcs Péter Tallósy
Szabolcs Péter Tallósy Anna Nászai
Anna Nászai Gabriella Varga
Gabriella Varga Dániel Érces
Dániel Érces Mihály Boros
Mihály Boros
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol., 07 January 2022
Sec. Cellular Biochemistry
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.824749
This article is part of the Research TopicInsights in Intensive Care Medicine and Anesthesiology: 2021View all 41 articles
A number of studies have demonstrated explicit bioactivity for exogenous methane (CH4), even though it is conventionally considered as physiologically inert. Other reports cited in this review have demonstrated that inhaled, normoxic air-CH4 mixtures can modulate the in vivo pathways involved in oxidative and nitrosative stress responses and key events of mitochondrial respiration and apoptosis. The overview is divided into two parts, the first being devoted to a brief review of the effects of biologically important gases in the context of hypoxia, while the second part deals with CH4 bioactivity. Finally, the consequence of exogenous, normoxic CH4 administration is discussed under experimental hypoxia- or ischaemia-linked conditions and in interactions between CH4 and other biological gases, with a special emphasis on its versatile effects demonstrated in pulmonary pathologies.
In the Earth’s atmosphere, where oxygen (O2) accounts for ∼ 21% of the environmental gases, reduction-oxidation reactions provide the energy which makes complex organisms capable of sustaining life (Schmidt-Rohr, 2020). Heterotrophs, such as humans, consume organic compounds for energy production by burning O2, with carbon dioxide (CO2) and water as the ultimate end products. Through this process, the inspired O2 level in the lungs is reduced to about 14.5% by the presence of alveolar water vapour and CO2, and then the O2 levels range from 3.4 to 6.8% by the time it reaches the peripheral tissues (Carreau et al., 2011). Thus, “normoxia” corresponds to the atmospheric O2 pressure, and much lower but still physiological (“normal”) levels of O2 are found in different tissues within the organs (Carreau et al., 2011). The evolution of aerobic cells has created a range of control mechanisms for the optimal utilization of O2 for subcellular, mitochondrial respiration, where multiprotein complexes of the electron transport system (ETS) are dedicated to accepting electrons from reduced carriers and delivering them to accessible molecular O2. Three of these complexes (Complex I, III and IV) are also H+ channels, responsible for a transmembrane electrochemical gradient between the surfaces of the inner membrane and the resulting driving force for ATP synthase (Complex V), which transforms adenosine diphosphate (ADP) into adenosine triphosphate (ATP).
In this substrate-level oxidative phosphorylation (OxPhos) reaction, availability of O2 is the most critical issue. However, many other gases, oxidative or reductive metabolic by-products of this aerobic system can also influence the intra- and extramitochondrial responses. In addition, it is highly likely that the many ways in which the gases combine both in physical and biochemical ways determine the nature of organ responses in clinical conditions associated with hypoxia. The first aim of this review is to summarize the knowledge of certain possibilities through which mitochondrial activity may be modulated by exogenous biological gases, with special emphasis on pulmonary reactions. Indeed, in terms of clinical applications, pulmonary gas delivery is an attractive idea, since applying a bioactive agent either prophylactically or at the time of an operation allows for prompt, specific and local interventions at the barrier sites of the respiratory tract. In this sense, a medically important gas should be easy to apply, have the appropriate chemical and physical properties and kinetics (e.g., be dissolved in plasma), and be nontoxic and biocompatible to achieve the expected biological results. The research on bioactive gases and derivatives has been intense, leading to the listing of four essential characteristics (simplicity, availability, volatility and effectiveness) and the definition of six criteria that make a gas physiologically important or irreplaceable (Wang, 2014). To date, nitrogen monoxide (NO), hydrogen sulphide (H2S) and carbon monoxide (CO) are “officially” recognized as signalling substances and referred to as gasotransmitters (Wang, 2014). Against this background, many attempts have concentrated on the therapeutic outcomes of gas deliveries of individual gasotransmitters in various pathological conditions. Nevertheless, the consequences of a more complex interplay of intrapulmonary O2 with NO, CO or H2S have not yet been investigated systematically.
Further, it should also be taken into account that there are many other gas molecules present in the cellular environment that do not fully meet the gas mediator criteria under the current classifications. Although methane (CH4) is conventionally believed to be physiologically inert, studies cited in this review demonstrate that it can modulate the pathways involved in key events of inflammation and influence the interactions of other biological gases. Therefore, the second part deals with CH4 bioactivity, a consequence of exogenous, normoxic CH4 administration in experimental hypoxic conditions and the implications of its interactions with other gases in respiratory pathologies.
Hypoxic air induces a number of compensatory responses in the microenvironment of the lung. As the cells become less oxygenated, pulmonary mitochondria have less access to substrates (O2 and acetyl-CoA), and the uncontrolled calcium (Ca2+) influx is accompanied by reactive oxygen species (ROS) formation (Lukyanova and Kirova, 2015). More importantly, a rapid compensatory mechanism prevents or reverses acute hypoxia-induced disturbances, while a delayed mechanism is responsible for a reversible reprogramming of the regulation of mitochondrial complexes so that the mitochondrial respiratory chain switches from oxidation of NAD-related substrates (Complex I) to succinate oxidation (Complex II), thus providing proper ATP synthesis. Indeed, Complex I contributes to roughly 80% of mitochondrial respiration in normoxia, whereas, during an impeded or deficient O2 supply, this is significantly reduced in favour of Complex II [also causing mitochondrial fission through GPR91 signalling (Lu et al., 2018)], which then contributes to nearly 75–90% of the total respiration. Therefore, these hypoxic cells are able to respond in a regulated manner to reduced O2 supply; compensatory mechanisms will ensure adequate ATP synthesis until the cellular PO2 reaches a critically low ( <1%) level. It follows that a mechanism that allows cells to sense even a minimal change in O2 supply activates signalling pathways responsible for triggering adaptive responses. Cytochrome c oxidase (Complex IV) is the main enzyme that transfers electrons and binds O2 in the ETS, and thus it was proposed that cells should have O2-sensing mechanisms regardless of their bioenergetic state (Bell et al., 2005; Guzy and Schumacker, 2006; Kierans and Taylor, 2021).
The transcriptional activator hypoxia-inducible factor (HIF-1) is responsible for regulating oxygenation and is required for the increased expression of more than 60 genes under hypoxia. In aerobic conditions, cells express the COX4-1 regulatory subunit of Complex IV under HIF-1 regulation but switch to the COX4-2 subunit in hypoxic conditions (Semenza, 2011). The stability, subcellular localization and transcriptional activity of HIF-1α are also strongly affected by changes in O2 levels. In normoxia, the transcriptional activity of HIF-1α is inhibited by ubiquitous proteases (Figure 1). In this process, HIF-1α can bind to the von Hippel–Lindau tumour suppressor protein (pVHL) after hydroxylation of prolyl (PHD 1,3) (with 2 oxoglurate and Fe2+ as cofactors), which promotes ubiquitin-mediated degradation (Bell et al., 2005). In O2-deficient states, hydroxylation does not occur, so pVHL cannot bind to HIF-1α, leading to a decrease in degradation processes. In the normoxic state, the binding of the transcription cofactors p300 and CBP to HIF-1α is inhibited so that, in contrast to the hypoxic state, further transcription processes are also prevented (Bell et al., 2005).
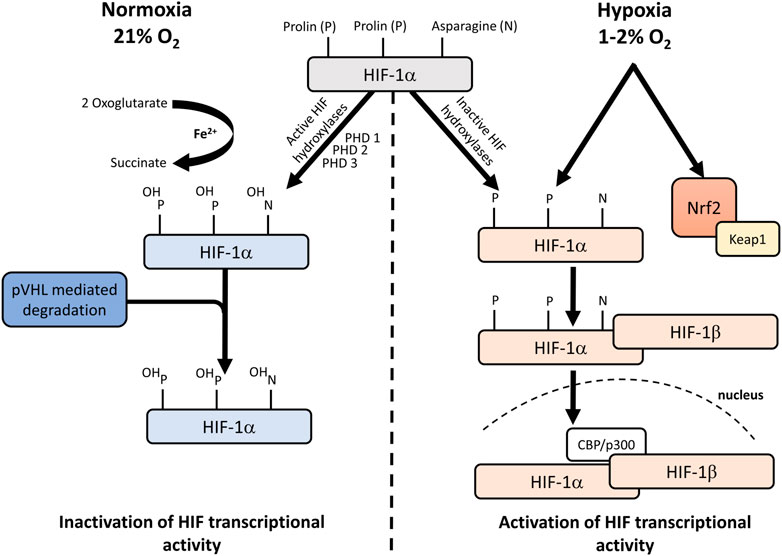
FIGURE 1. Hypoxic regulation of HIF-1. Under normoxic conditions, NO impairs the activity of HIF-1α prolyl hydroxylases and inhibits HIF-1α ubiquitination and interaction with pVHL. Under hypoxic conditions, NO induces Keap1 signalling and suppresses HIF-1α hypoxic stabilization. CH4 increases the expression of Nrf2. HIF: hypoxia-inducible factor; pVHL: von Hippel–Lindau protein; Ub: ubiquitin; NO: nitrogen monoxide; PHD: prolin hydroxylases; Keap1: kelch-like ECH-associated protein-1; Nrf2: nuclear factor-erythroid factor 2-related factor 2.
HIF-1α is regulated by mitochondria in two different ways. Firstly, if the respiration is inhibited, an intracellular O2 level of around 1% is still sufficient for the hydroxylation of HIF-1α. Hydroxylation is only reduced when O2 tensions are below 1% so that OxPhos or the ability to respire is not related to the regulation of hypoxic stabilization of HIF-1α. The second is that ROS production during hypoxia is required for HIF-1α protein stabilization (Brunelle et al., 2005). It would logically follow that in hypoxic conditions, ROS formation is reduced in the absence of O2, but the levels paradoxically increase during hypoxia (Guzy and Schumacker, 2006). It has been shown that ROS generated at Complex III stabilize HIF-1α during hypoxia (Solaini et al., 2010) and that HIF-1α expression is reduced when Complex V is inhibited with oligomycin (Gong and Agani, 2005). Taken together, the available data do not allow us to clearly establish the exact role of mitochondrial ROS in the regulation of HIF-1α, but the pathway that stabilizes HIF-1α can undoubtedly be considered mitochondria-dependent. According to some authors, mitochondrial ROS can also stabilize HIF-1α under hypoxic conditions (via a transcriptional regulatory cascade) via the nuclear factor E2-related factor 2 (Nrf2) pathway (Lacher et al., 2018; Potteti et al., 2021).
Together with Complex III and IV, the Complex I (NADH: ubiquinone oxidoreductase) is involved in proton pumping from the matrix to the intermembrane space (extruding four hydrogen ions per NADH). Through this action, Complex I collects the Krebs cycle-derived reducing equivalents and participates in redox energy conversion, and the proton gradient across the membrane is then used for energy production by the ATP synthase during OxPhos (Ramsay, 2019). More importantly, considerable amounts of ROS can be generated by Complex I in the mitochondrial matrix when electrons flow both in the forward (forward electron transport; FET) or reverse (reverse electron transport; RET) direction. To date, flavin mononucleotide (FMN), Q-binding site and the iron–sulphur cluster N2 have been identified in mitochondrial superoxide generation. Although RET was long considered as an in vitro phenomenon, the in vivo role in ROS generation has recently been demonstrated (Scialò et al., 2017).
Complex I is one of the largest membrane-bound enzymes (1 MDa MW), with a FMN-containing protein and a number of (eight) iron–sulphur centres. The L-shaped structure consists of two major parts, with the components embedded in the inner membrane and the peripheral arm located in the cytoplasm or mitochondrial matrix (Martin and Matyushov, 2017). When a hydride ion is transferred from NADH to FMN at the peripheral arm, two electrons pass through the iron–sulphur clusters (chain of electron transfer cofactors; Fe2S2 and seven Fe4S2; terminal cofactor N2) to ubiquinone (membrane domain), where the proton extrusion is carried out through the membrane. A unique characteristic of Complex I has recently gained much attention as it has been demonstrated that the limited in vivo O2 availability deactivates Complex I, which is required for the catalytic activity of ETS enzymes (Maklashina et al., 2002; Hernansanz-Agustín et al., 2017). Given a lack of available substrate, Complex I spontaneously forms a deactive (D) form that can be re-activated by exogenous NADH and ubiquinone administration (Galkin and Moncada, 2017; Blaza et al., 2018). The active (A) state catalyses the rapid NADH oxidation at a linear rate, while a lag phase is present during the D→A transition. The lag phase is prolonged at alkaline pH or in the presence of divalent cations, such as Ca2+ or Mg2+. Most notably, the transition from catalytically active to dormant D form also occurs during acute hypoxia or ischaemia. The biological consequences of the conformational change are not fully mapped, but is has been shown that it fine-tunes ETS, may reduce oxidative/nitrosative stress and switches the NADH:ubiquinone oxidoreductase activity to a sodium-proton (Na+/H+) antiporter through its hydrophobic membrane-bound domains (ND2, ND4 and ND5 subunits) (Roberts and Hirst, 2012; Babot et al., 2014; Hernansanz-Agustín et al., 2017). In addition, the D-form is more sensitive to ischaemia/reperfusion (IR)-mediated oxidative injury than the A-form. Therefore, modulation of the dormant form may also be a protective strategy during ischaemia/hypoxia (Chouchani et al., 2013; Gorenkova et al., 2013).
Although inhaled NO has been successfully tested in neonates and adult patients with acute respiratory distress syndrome (Feng et al., 2021; Lotz et al., 2021; Safaee Fakhr et al., 2021), the clinical benefit of intrapulmonary administration is still subject to much debate (Sokol et al., 2016; Vieira et al., 2021). In this line, the oxygenation of the tissues is a main factor when the rather controversial results of gasotransmitter reactions are discussed. For example, the most important physiological mechanism linked to NO metabolism requires proper O2 concentrations; under normal or higher O2 tension, the half-life of NO is shorter, while in hypoxic environments NO will be eliminated after a significantly longer time with a number of prolonged effects (Kuschman et al., 2021). Likewise, a combination of low O2 tension with mitochondria-derived ROS and higher NO flows leads to peroxynitrite formation (Thomas et al., 2008) with nitroxidative stress and post-translational protein modification (Campolo et al., 2020). Further, like other inhibitors of mitochondrial respiration, NO prevents the stabilization of HIF-1α. A recent key finding has revealed a novel role for Complex I in this process, as prior A/D conversion is necessary for S-nitrosothiols and peroxynitrite to interfere with the respiratory activity of mitochondria (Babot et al., 2014; Galkin and Moncada, 2017). Therefore, the hypoxia-linked mitochondrial duality may explain, at least partly, the controversial clinical results and the narrow range of effectiveness of NO inhalation.
H2S is the next gas mediator, with Janus-faced characteristics being clearly present at the mitochondrial level. It is toxic when inhaled in high concentrations, while it is anti-inflammatory and cytoprotective at low partial pressure (Elrod et al., 2007; Cui et al., 2016; Scheid et al., 2021). Inhalation of 80–150 ppm H2S induces a suspended animation state with reduced metabolic rate, which leads to an increased resistance to severe hypoxia (5% FiO2) (Blackstone and Roth, 2007). In an oxygenated environment and in low (less than 1 µM) H2S concentrations, the regular substrates of the respiratory chain are used for biological oxidation. However, as soon as H2S content is increased (to less than 10 µM) an active sulphide quinone reductase (as the immediate electron acceptor) is available, H2S acts as an alternative electron source for the respiratory chain. A H2S concentration of over 10 µM impairs the mitochondrial function with the inhibition of Complex IV (Bouillaud and Blachier, 2011). Here it should be added that cancer cells may utilize this phenomenon by up-regulating H2S, thus producing enzymes to stimulate mitochondrial ATP synthesis and maintain mitochondrial function (Szabo, 2021).
Carbon monoxide (CO) is the third gas in the sequence of gasotransmitters, again with dual properties: low levels exert cyto- and tissue protective effects, but in higher concentrations systemic toxicity comes to the fore. Due to its affinity to bind to the haem iron centre of haemoglobin, carboxyhaemoglobin (CO-Hb) formation ensues with cellular hypoxia. A number of signal transduction pathways have been recognized as potential targets of low concentrations of inhaled CO via its anti-inflammatory (Otterbein et al., 2000), anti-apoptotic (Ryter et al., 2018), anti-oxidative (Parfenova et al., 2012) and anti-proliferative effects. CO binds primarily to haem iron and may activate soluble guanylate cyclase, although with lower efficacy than NO (Sharma and Magde, 1999). Through the modulation of the mitogen-activated protein kinase pathway, CO inhibits the expression of several pro-inflammatory cytokines, such as tumour necrosis factor-alpha (TNF-⍺) and interleukin-1beta (IL-1β), and increases the expression of the anti-inflammatory cytokine interleukin-10 (IL-10) (Otterbein et al., 2000). The mitochondrium is also one of the recognized cellular targets for CO, with physiological concentrations of CO increasing mitochondrial ROS generation, which activates cellular endogenous mechanisms of defence involved in preconditioning and cytoprotection (Bilban et al., 2008). Furthermore, CO prevents apoptotic cell death by limiting mitochondrial membrane permeabilization, which inhibits the release of pro-apoptotic factors into the cytosol; both events are ROS-dependent (R. Oliveira et al., 2016). The protective effects of low concentrations (of up to 500 ppm) of inhaled CO have been observed in a number of lung injury models (Otterbein et al., 1999; Morse et al., 2003; Kohmoto et al., 2006). However, most of the clinical studies in various disease trials have been terminated because the expected primary outcomes had not been met.
The bioactivity of all recognized gas mediators is related to their tendency to react chemically with biologically important target molecules. Therefore, and precisely due to this characteristic, these compounds are also categorized as toxic asphyxiants in environmental chemistry. For example, and according to current knowledge on NO, CO and H2S biochemistry, these gaseous substances all readily inhibit mitochondrial O2 consumption by Complex IV (Figure 2). It is therefore important to consider that physiologically important gases that trigger vital functional changes will have profound adverse effects in any cellular system given sufficient exposure, and many of the unfavourable consequences are directly linked to inhibition of mitochondrial function.
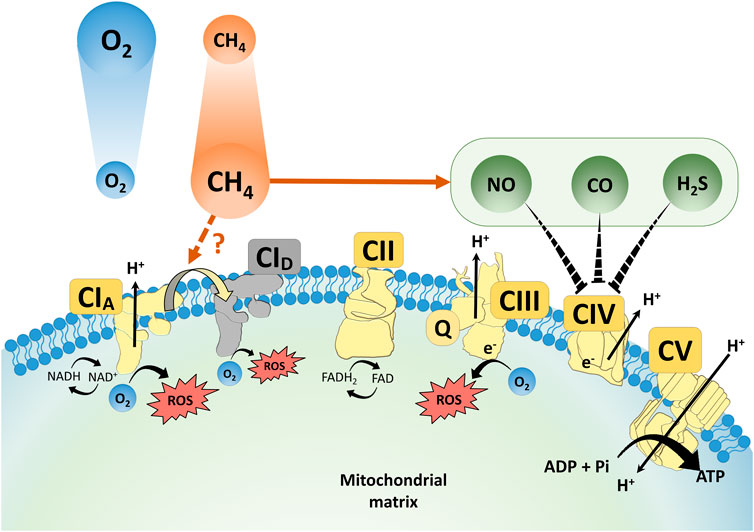
FIGURE 2. A scheme for the proposed interaction of CH4, NO, CO and H2S at mitochondrial respiratory complexes. ROS: reactive oxygen species; NO: nitrogen monoxide; CO: carbon monoxide; NAD/NADH: nicotinamide adenine dinucleotide/dihydronicotinamide adenine dinucleotide; CI–V: Complex I–V; TCA cycle: tricarboxylic acid cycle; FAD/FADH2: flavin adenine dinucleotide/dihydroflavine-adenine dinucleotide; Q: ubiquinone.
On the other hand, simple asphyxiants, such as CH4, act by physically limiting the utilization of O2, without producing cytotoxic effects (Boros et al., 2018). Tissue hypoxia may indeed occur when CH4 displaces the air and hence O2 in a restricted space. However, in such cases, respiratory distress is not due to the chemical specificity of the gas, but to the decreased O2 content (CH4 in the inspired air should be present at about 14% or 140,000 parts per million by volume (ppmv) to reduce O2 to 18%) (Boros and Keppler, 2019). Information on the respiratory effects of CH4 is sparse but the inhalation of normoxic artificial air containing 21% O2 and 2.5% CH4 had no side-effects on the blood gas chemistry and mean arterial blood pressure in normotensive unstressed animals (Boros et al., 2012; Zaorska et al., 2021). Likewise, the administration of CH4-enriched saline did not affect cytochrome c release in rats (Wang et al., 2017).
Under standard conditions for temperature and pressure, the solubility of CH4 in blood is rather low (a blood:air partition coefficient of 0.066) but significantly higher in membrane bilayers (a partition coefficient of 0.20) [as reviewed by Boros and Keppler (2018)]. Therefore, the concentration of CH4 in the tissues rapidly reaches equilibrium with that in the inspired air, and this equilibrium remains stable even with prolonged exposure time (Watanabe and Morita, 1998). It follows that inhaled CH4 will move readily from the alveoli into the circulation, throughout which it is distributed rapidly and may accumulate transiently at cell membrane interfaces, thereby changing the relationship between gases and the in situ functionality within this environment. Without a new exogenous supply, CH4 will enter the circulation again and then be excreted through the lungs if its partial pressure is higher than that in the atmosphere.
The outcome of exogenous CH4 respiration in the human body under stress conditions has not yet been evaluated. Nevertheless, a wealth of data is available in plants and animals in such situations and also on the links between CH4 and gas messengers. The effects of CH4 supplementation to CO, H2S and NO biology were repeatedly shown during the adaptation to abiotic stress and germination inhibition in plant species, which confirmed that CO, NO and H2S signalling mechanisms are involved in the molecular basis of CH4-induced stress tolerance (Cui et al., 2015; Qi et al., 2017; Kou et al., 2018). Apart from plant pathophysiology, several series of in vivo analyses have demonstrated that CH4-containing normoxic artificial air has anti-inflammatory effects by decreasing the biochemical, functional and structural consequences of nitroxidative stress [(Boros et al., 2012), (Mészáros et al., 2017a), (Poles et al., 2018)]. Data show that NO can directly inhibit mitochondrial functions via several pathways and that NO-influenced or mediated inhibition can be reversed with 2.2–2.5 %v/v CH4-containing gas mixtures. Notably, it has been demonstrated that normoxic CH4 ventilation decreases tyrosine nitrosylation after IR injury, a process which involved NO and peroxynitrite formation. In addition, exogenous CH4 administration reduced the xanthine oxidoreductase (XOR)-linked nitrate reductase activity, the generation of nitrogen-centred radicals and the damage to nitrergic neurons during a standardized IR challenge (Poles et al., 2018). Along these lines, it has been shown that higher concentrations of exogenous CH4 can lead to direct anti-cytokine effects via master switches, such as Nrf2/Keap1 and NF-κB (Mészáros et al., 2017b). More recently, the addition of 2.5% v/v CH4-normoxic air mixture to the oxygenator sweep gas reduced the systemic inflammatory response to extracorporeal circulation in a clinically relevant large animal model. In this study, the inotropic demand was significantly lower, the renal arterial flow was significantly higher, and the hour diuresis remained in the low normal range as compared to the oliguria in the non-treated animals (Bari et al., 2019) (Supplementary Table S1).
In this line, many studies have also explored the relationship among CH4 actions in the context of mitochondrial biology. Inhaled CH4 reduced cytochrome c release and preserved the mitochondrial respiratory capacity in vivo and in transient anoxia-treated cell cultures as well (Strifler et al., 2016; Jász et al., 2021). Recently, we carried out a sequential study with exogenous normoxic CH4 in simulated IR environments using a high-resolution respirometry system to quantify the ETS responses (Jász et al., 2021). In this protocol, CH4 treatment restricted the forward electron transfer within Complex I in control mitochondria while effectively restricting RET in post-anoxic mitochondria, thus it could be concluded that interaction with Complex I occupies a key position in the protective mechanism of CH4 against a hypoxia/reoxygenation injury (Jász et al., 2021). Parallel in vivo studies have also shown that the CH4 content of an organ preservation solution effectively influenced several components of the endoplasmic reticulum (ER) stress-mitochondria-related pro-apoptotic signalling pathways (Benke et al., 2021). The myocardial OxPhos capacity was more preserved and cytochrome c release was decreased as a result of CH4-enriched storage, with the relative mRNA expression for hypoxia- and ER stress-associated genes (including HIF-1α) also being significantly reduced (Benke et al., 2021). Indeed, several previous studies demonstrated that exogenous CH4 modulates the intrinsic, mitochondrial pathway of pro-apoptotic activation in model experiments (Ye et al., 2015; Chen et al., 2016; Liu et al., 2016; Jia et al., 2018) and CH4 administration exhibited anti-apoptotic effects and protected the pulmonary epithelial cells in a murine model of ovalbumin-induced allergic asthma as well (Zhang et al., 2019). More importantly, the anti-apoptotic properties of CH4 inhalation were associated with improved pulmonary compliance and surfactant production in a rodent model of lung IR injury (Zhang et al., 2021). In summary, a possible indirect way in which CH4 supplementation modulates apoptosis is by reducing cytochrome c release from the inner mitochondrial membrane, which has already been demonstrated in several tissues (Chen et al., 2016; Strifler et al., 2016; Wang et al., 2017). It seems that further knowledge of inhaled CH4 and other gaseous molecular species with their mitochondrial targets, most importantly of Complex I, has the potential to increase the understanding of the mechanism of pathological processes at work in the pulmonary alveoli and capillaries (Figure 2).
Beyond O2 and CO2, many gases are biologically active. Signalling roles were demonstrated for NO, CO and H2S and it has become clear that these simple, volatile molecules can influence the cellular biology in various ways. Likewise, the human diagnostic relevance of detection of exhaled gases, as signatures of oxido-reductive stress responses, is emerging as well (Paardekooper et al., 2017). Several aspects of mitochondrial respiration, such as energy production, Ca2+ homeostasis and intrinsic apoptosis, may also be targets of intertwined gaseous pathways but it is less clear how mitochondrial functions are altered if the membership of this molecular club changes and how the signals of a mixed gaseous input are translated to downstream manifestations of cellular reactions.
As the examples illustrate, bioactivity is not limited to those gases that have inherited the textbook characteristics of gasotransmitters. There is ample evidence that other, less prominent components of the endogenous gaseous network, such as molecular hydrogen (H2) or CH4, are also able to modulate mitochondrial respiration [(Mészáros et al., 2017b), (Hirano et al., 2021)]. As an analogy, other gaseous compounds, such as NO, H2S and CO, were previously thought to be toxic pollutants without any physiologic effects in eukaryotes. CH4 has a long evolutionary history on Earth (Hancock, 2017). It is permanent part of the gaseous environment, a nontoxic asphyxiant, which can change the symbiosis with other gas molecules within the internal milieu of aerobic cells. In this scheme, the recognized bioactivity suggests a role for exogenous CH4 to modulate the hypoxia-linked pro-inflammatory signals towards resting conditions.
The manuscript concept was designed by MB, LJ, ST, AN, GV, DÉ, and MB wrote the article. All the authors discussed and commented on the manuscript at all stages and approved the submitted version.
This study was funded by National Research Development and Innovation Office grant NKFI K12023.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, orclaim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.824749/full#supplementary-material
Babot, M., Birch, A., Labarbuta, P., and Galkin, A. (2014). Characterisation of the Active/de-Active Transition of Mitochondrial Complex I. Biochim. Biophys. Acta (Bba) - Bioenerg. 1837, 1083–1092. doi:10.1016/j.bbabio.2014.02.018
Bari, G., Érces, D., Varga, G., Szűcs, S., Varga, Z., Bogáts, G., et al. (2019). Methane Inhalation Reduces the Systemic Inflammatory Response in a Large Animal Model of Extracorporeal Circulation. Eur. J. Cardiothorac. Surg. 56, 135–142. doi:10.1093/ejcts/ezy453
Bell, E. L., Emerling, B. M., and Chandel, N. S. (2005). Mitochondrial Regulation of Oxygen Sensing. Mitochondrion 5, 322–332. doi:10.1016/j.mito.2005.06.005
Benke, K., Jász, D. K., Szilágyi, Á. L., Baráth, B., Tuboly, E., Márton, A. R., et al. (2021). Methane Supplementation Improves Graft Function in Experimental Heart Transplantation. J. Heart Lung Transplant. 40, 183–192. doi:10.1016/j.healun.2020.11.003
Bilban, M., Haschemi, A., Wegiel, B., Chin, B. Y., Wagner, O., and Otterbein, L. E. (2008). Heme Oxygenase and Carbon Monoxide Initiate Homeostatic Signaling. J. Mol. Med. 86, 267–279. doi:10.1007/s00109-007-0276-0
Blackstone, E., and Roth, M. B. (2007). Suspended Animation-like State Protects Mice from Lethal Hypoxia. Shock 27, 370–372. doi:10.1097/SHK.0b013e31802e27a0
Blaza, J. N., Vinothkumar, K. R., and Hirst, J. (2018). Structure of the Deactive State of Mammalian Respiratory Complex I. Structure 26, 312–319. doi:10.1016/j.str.2017.12.014
Boros, M., Ghyczy, M., Érces, D., Varga, G., Tőkés, T., Kupai, K., et al. (2012). The Anti-inflammatory Effects of Methane*. Crit. Care Med. 40, 1269–1278. doi:10.1097/CCM.0b013e31823dae05
Boros, M., and Keppler, F. (2018). “Chapter 8. Production and Signaling of Methane,” in Gasotransmitters. Editor R. Wang (Croydon, United Kingdom: Royal Society of Chemistry), 192–234. doi:10.1039/978178801300010.1039/9781788013000-00192
Boros, M., and Keppler, F. (2019). Methane Production and Bioactivity-A Link to Oxido-Reductive Stress. Front. Physiol. 10, 1244. doi:10.3389/fphys.2019.01244
Bouillaud, F., and Blachier, F. (2011). Mitochondria and Sulfide: a Very Old story of Poisoning, Feeding, and Signaling? Antioxid. Redox Signaling 15, 379–391. doi:10.1089/ars.2010.3678
Brunelle, J. K., Bell, E. L., Quesada, N. M., Vercauteren, K., Tiranti, V., Zeviani, M., et al. (2005). Oxygen Sensing Requires Mitochondrial ROS but Not Oxidative Phosphorylation. Cel Metab. 1, 409–414. doi:10.1016/j.cmet.2005.05.002
Campolo, N., Issoglio, F. M., Estrin, D. A., Bartesaghi, S., and Radi, R. (2020). 3-Nitrotyrosine and Related Derivatives in Proteins: Precursors, Radical Intermediates and Impact in Function. Essays Biochem. 64, 111–133. doi:10.1042/EBC20190052
Carreau, A., Hafny-Rahbi, B. E., Matejuk, A., Grillon, C., and Kieda, C. (2011). Why Is the Partial Oxygen Pressure of Human Tissues a Crucial Parameter? Small Molecules and Hypoxia. J. Cel Mol Med 15, 1239–1253. doi:10.1111/j.1582-4934.2011.01258.x
Chen, O., Ye, Z., Cao, Z., Manaenko, A., Ning, K., Zhai, X., et al. (2016). Methane Attenuates Myocardial Ischemia Injury in Rats through Anti-oxidative, Anti-apoptotic and Anti-inflammatory Actions. Free Radic. Biol. Med. 90, 1–11. doi:10.1016/j.freeradbiomed.2015.11.017
Chouchani, E. T., Methner, C., Nadtochiy, S. M., Logan, A., Pell, V. R., Ding, S., et al. (2013). Cardioprotection by S-Nitrosation of a Cysteine Switch on Mitochondrial Complex I. Nat. Med. 19, 753–759. doi:10.1038/nm.3212
Cui, W., Qi, F., Zhang, Y., Cao, H., Zhang, J., Wang, R., et al. (2015). Methane-rich Water Induces Cucumber Adventitious Rooting through Heme Oxygenase1/carbon Monoxide and Ca2+ Pathways. Plant Cel Rep 34, 435–445. doi:10.1007/s00299-014-1723-3
Cui, W., Zhang, Y., Yang, C., Sun, Y., Zhang, M., and Wang, S. (2016). Hydrogen Sulfide Prevents Abeta-Induced Neuronal Apoptosis by Attenuating Mitochondrial Translocation of PTEN. Neuroscience 325, 165–174. doi:10.1016/j.neuroscience.2016.03.053
Elrod, J. W., Calvert, J. W., Morrison, J., Doeller, J. E., Kraus, D. W., Tao, L., et al. (2007). Hydrogen Sulfide Attenuates Myocardial Ischemia-Reperfusion Injury by Preservation of Mitochondrial Function. Proc. Natl. Acad. Sci. 104, 15560–15565. doi:10.1073/pnas.0705891104
Feng, W. X., Yang, Y., Wen, J., Liu, Y. X., Liu, L., and Feng, C. (2021). Implication of Inhaled Nitric Oxide for the Treatment of Critically Ill COVID-19 Patients with Pulmonary Hypertension. ESC Heart Fail. 8, 714–718. doi:10.1002/ehf2.13023
Galkin, A., and Moncada, S. (2017). Modulation of the Conformational State of Mitochondrial Complex I as a Target for Therapeutic Intervention. Interf. Focus. 7, 20160104. doi:10.1098/rsfs.2016.0104
Gong, Y., and Agani, F. H. (2005). Oligomycin Inhibits HIF-1α Expression in Hypoxic Tumor Cells. Am. J. Physiology-Cell Physiol. 288, C1023–C1029. doi:10.1152/ajpcell.00443.2004
Gorenkova, N., Robinson, E., Grieve, D. J., and Galkin, A. (2013). Conformational Change of Mitochondrial Complex I Increases ROS Sensitivity during Ischemia. Antioxid. Redox Signaling 19, 1459–1468. doi:10.1089/ars.2012.4698
Guzy, R. D., and Schumacker, P. T. (2006). Oxygen Sensing by Mitochondria at Complex III: the Paradox of Increased Reactive Oxygen Species during Hypoxia. Exp. Physiol. 91, 807–819. doi:10.1113/expphysiol.2006.033506
Hancock, J. T. (2017). Harnessing Evolutionary Toxins for Signaling: Reactive Oxygen Species, Nitric Oxide and Hydrogen Sulfide in Plant Cell Regulation. Front. Plant Sci. 8, 189. doi:10.3389/fpls.2017.00189
Hernansanz-Agustín, P., Ramos, E., Navarro, E., Parada, E., Sánchez-López, N., Peláez-Aguado, L., et al. (2017). Mitochondrial Complex I Deactivation Is Related to Superoxide Production in Acute Hypoxia. Redox Biol. 12, 1040–1051. doi:10.1016/j.redox.2017.04.025
Hirano, S.-i., Ichikawa, Y., Sato, B., Yamamoto, H., Takefuji, Y., and Satoh, F. (2021). Potential Therapeutic Applications of Hydrogen in Chronic Inflammatory Diseases: Possible Inhibiting Role on Mitochondrial Stress. Ijms 22 (5), 2549. doi:10.3390/ijms22052549
Jász, D. K., Szilágyi, Á. L., Tuboly, E., Baráth, B., Márton, A. R., Varga, P., et al. (2021). Reduction in Hypoxia-Reoxygenation-Induced Myocardial Mitochondrial Damage with Exogenous Methane. J. Cel Mol Med 25, 5113–5123. doi:10.1111/jcmm.16498
Jia, Y., Li, Z., Liu, C., and Zhang, J. (2018). Methane Medicine: A Rising Star Gas with Powerful Anti-inflammation, Antioxidant, and Antiapoptosis Properties. Oxid. Med. Cell Longev., 2018, 1912746. doi:10.1155/2018/1912746
Kierans, S. J., and Taylor, C. T. (2021). Regulation of Glycolysis by the Hypoxia-Inducible Factor (HIF): Implications for Cellular Physiology. J. Physiol. 599, 23–37. doi:10.1113/JP280572
Kohmoto, J., Nakao, A., Kaizu, T., Tsung, A., Ikeda, A., Tomiyama, K., et al. (2006). Low-dose Carbon Monoxide Inhalation Prevents Ischemia/reperfusion Injury of Transplanted Rat Lung Grafts. Surgery 140, 179–185. doi:10.1016/j.surg.2006.03.004
Kou, N., Xiang, Z., Cui, W., Li, L., and Shen, W. (2018). Hydrogen Sulfide Acts Downstream of Methane to Induce Cucumber Adventitious Root Development. J. Plant Physiol. 228, 113–120. doi:10.1016/j.jplph.2018.05.010
Kuschman, H. P., Palczewski, M. B., and Thomas, D. D. (2021). Nitric Oxide and Hydrogen Sulfide: Sibling Rivalry in the Family of Epigenetic Regulators. Free Radic. Biol. Med. 170, 34–43. doi:10.1016/j.freeradbiomed.2021.01.010
Lacher, S. E., Levings, D. C., Freeman, S., and Slattery, M. (2018). Identification of a Functional Antioxidant Response Element at the HIF1A Locus. Redox Biol. 19, 401–411. doi:10.1016/j.redox.2018.08.014
Liu, L., Sun, Q., Wang, R., Chen, Z., Wu, J., Xia, F., et al. (2016). Methane Attenuates Retinal Ischemia/reperfusion Injury via Anti-oxidative and Anti-apoptotic Pathways. Brain Res. 1646, 327–333. doi:10.1016/j.brainres.2016.05.037
Lotz, C., Muellenbach, R. M., Meybohm, P., Mutlak, H., Lepper, P. M., Rolfes, C. B., et al. (2021). Effects of Inhaled Nitric Oxide in COVID-19-Induced ARDS - Is it Worthwhile? Acta Anaesthesiol. Scand. 65, 629–632. doi:10.1111/aas.13757
Lu, Y.-T., Li, L.-Z., Yang, Y.-L., Yin, X., Liu, Q., Zhang, L., et al. (2018). Succinate Induces Aberrant Mitochondrial Fission in Cardiomyocytes through GPR91 Signaling. Cell Death Dis 9, 672. doi:10.1038/s41419-018-0708-5
Lukyanova, L. D., and Kirova, Y. I. (2015). Mitochondria-controlled Signaling Mechanisms of Brain protection in Hypoxia. Front. Neurosci. 9, 320. doi:10.3389/fnins.2015.00320
Maklashina, E., Sher, Y., Zhou, H.-Z., Gray, M. O., Karliner, J. S., and Cecchini, G. (2002). Effect of Anoxia/reperfusion on the Reversible Active/de-Active Transition of NADH-Ubiquinone Oxidoreductase (Complex I) in Rat Heart. Biochim. Biophys. Acta (Bba) - Bioenerg. 1556, 6–12. doi:10.1016/s0005-2728(02)00280-3
Martin, D. R., and Matyushov, D. V. (2017). Electron-transfer Chain in Respiratory Complex I. Sci. Rep. 7, 5495. doi:10.1038/s41598-017-05779-y
Mészáros, A. T., Büki, T., Fazekas, B., Tuboly, E., Horváth, K., Poles, M. Z., et al. (2017). Inhalation of Methane Preserves the Epithelial Barrier during Ischemia and Reperfusion in the Rat Small Intestine. Surgery 161, 1696–1709. doi:10.1016/j.surg.2016.12.040
Mészáros, A. T., Szilágyi, Á. L., Juhász, L., Tuboly, E., Érces, D., Varga, G., et al. (2017). Mitochondria as Sources and Targets of Methane. Front. Med. 4, 195. doi:10.3389/fmed.2017.00195
Morse, D., Pischke, S. E., Zhou, Z., Davis, R. J., Flavell, R. A., Loop, T., et al. (2003). Suppression of Inflammatory Cytokine Production by Carbon Monoxide Involves the JNK Pathway and AP-1. J. Biol. Chem. 278, 36993–36998. doi:10.1074/jbc.M302942200
Otterbein, L. E., Bach, F. H., Alam, J., Soares, M., Tao Lu, H., Wysk, M., et al. (2000). Carbon Monoxide Has Anti-inflammatory Effects Involving the Mitogen-Activated Protein Kinase Pathway. Nat. Med. 6, 422–428. doi:10.1038/74680
Otterbein, L. E., Mantell, L. L., and Choi, A. M. K. (1999). Carbon Monoxide Provides protection against Hyperoxic Lung Injury. Am. J. Physiology-Lung Cell Mol. Physiol. 276, L688–L694. doi:10.1152/ajplung.1999.276.4.L688
Paardekooper, L. M., van den Bogaart, G., Kox, M., Dingjan, I., Neerincx, A. H., Bendix, M. B., et al. (2017). Ethylene, an Early Marker of Systemic Inflammation in Humans. Sci. Rep. 7, 6889. doi:10.1038/s41598-017-05930-9
Parfenova, H., Leffler, C. W., Basuroy, S., Liu, J., and Fedinec, A. L. (2012). Antioxidant Roles of Heme Oxygenase, Carbon Monoxide, and Bilirubin in Cerebral Circulation during Seizures. J. Cereb. Blood Flow Metab. 32, 1024–1034. doi:10.1038/jcbfm.2012.13
Poles, M. Z., Bódi, N., Bagyánszki, M., Fekete, É., Mészáros, A. T., Varga, G., et al. (2018). Reduction of Nitrosative Stress by Methane: Neuroprotection through Xanthine Oxidoreductase Inhibition in a Rat Model of Mesenteric Ischemia-Reperfusion. Free Radic. Biol. Med. 120, 160–169. doi:10.1016/j.freeradbiomed.2018.03.024
Potteti, H. R., Noone, P. M., Tamatam, C. R., Ankireddy, A., Noel, S., Rabb, H., et al. (2021). Nrf2 Mediates Hypoxia-Inducible HIF1α Activation in Kidney Tubular Epithelial Cells. Am. J. Physiology-Renal Physiol. 320, F464–F474. doi:10.1152/ajprenal.00501.2020
Qi, F., Xiang, Z., Kou, N., Cui, W., Xu, D., Wang, R., et al. (2017). Nitric Oxide Is Involved in Methane-Induced Adventitious Root Formation in Cucumber. Physiol. Plantarum 159, 366–377. doi:10.1111/ppl.12531
Ramsay, R. R. (2019). Electron Carriers and Energy Conservation in Mitochondrial Respiration. ChemTexts 5, 9. doi:10.1007/s40828-019-0085-4
Roberts, P. G., and Hirst, J. (2012). The Deactive Form of Respiratory Complex I from Mammalian Mitochondria Is a Na+/H+ Antiporter. J. Biol. Chem. 287, 34743–34751. doi:10.1074/jbc.M112.384560
R. Oliveira, S. S., Queiroga, C. S. F., and Vieira, H. L. A. (2016). Mitochondria and Carbon Monoxide: Cytoprotection and Control of Cell Metabolism - a Role for Ca2+? J. Physiol. 594, 4131–4138. doi:10.1113/JP270955
Ryter, S. W., Ma, K. C., and Choi, A. M. K. (2018). Carbon Monoxide in Lung Cell Physiology and Disease. Am. J. Physiology-Cell Physiol. 314, C211–C227. doi:10.1152/ajpcell.00022.2017
Safaee Fakhr, B., Di Fenza, R., Gianni, S., Wiegand, S. B., Miyazaki, Y., Araujo Morais, C. C., et al. (2021). Inhaled High Dose Nitric Oxide Is a Safe and Effective Respiratory Treatment in Spontaneous Breathing Hospitalized Patients with COVID-19 Pneumonia. Nitric Oxide 116, 7–13. doi:10.1016/j.niox.2021.08.003
Scheid, S., Goeller, M., Baar, W., Wollborn, J., Buerkle, H., Schlunck, G., et al. (2021). Hydrogen Sulfide Reduces Ischemia and Reperfusion Injury in Neuronal Cells in a Dose- and Time-dependent Manner. Ijms 22, 10099. doi:10.3390/ijms221810099
Schmidt-Rohr, K. (2020). Oxygen Is the High-Energy Molecule Powering Complex Multicellular Life: Fundamental Corrections to Traditional Bioenergetics. ACS Omega 5, 2221–2233. doi:10.1021/acsomega.9b03352
Scialò, F., Fernández-Ayala, D. J., and Sanz, A. (2017). Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 8, 428. doi:10.3389/fphys.2017.00428
Semenza, G. L. (2011). Hypoxia-inducible Factor 1: Regulator of Mitochondrial Metabolism and Mediator of Ischemic Preconditioning. Biochim. Biophys. Acta (Bba) - Mol. Cel Res. 1813, 1263–1268. doi:10.1016/j.bbamcr.2010.08.006
Sharma, V. S., and Magde, D. (1999). Activation of Soluble Guanylate Cyclase by Carbon Monoxide and Nitric Oxide: a Mechanistic Model. Methods 19, 494–505. doi:10.1006/meth.1999.0892
Sokol, G. M., Konduri, G. G., and Van Meurs, K. P. (2016). Inhaled Nitric Oxide Therapy for Pulmonary Disorders of the Term and Preterm Infant. Semin. Perinatology 40, 356–369. doi:10.1053/j.semperi.2016.05.007
Solaini, G., Baracca, A., Lenaz, G., and Sgarbi, G. (2010). Hypoxia and Mitochondrial Oxidative Metabolism. Biochim. Biophys. Acta (Bba) - Bioenerg. 1797, 1171–1177. doi:10.1016/j.bbabio.2010.02.011
Strifler, G., Tuboly, E., Szél, E., Kaszonyi, E., Cao, C., Kaszaki, J., et al. (2016). Inhaled Methane Limits the Mitochondrial Electron Transport Chain Dysfunction during Experimental Liver Ischemia-Reperfusion Injury. PLoS One 11, e0146363. doi:10.1371/journal.pone.0146363
Szabo, C. (2021). Hydrogen Sulfide, an Endogenous Stimulator of Mitochondrial Function in Cancer Cells. Cells 10, 220. doi:10.3390/cells10020220
Thomas, D. D., Ridnour, L. A., Isenberg, J. S., Flores-Santana, W., Switzer, C. H., Donzelli, S., et al. (2008). The Chemical Biology of Nitric Oxide: Implications in Cellular Signaling. Free Radic. Biol. Med. 45, 18–31. doi:10.1016/j.freeradbiomed.2008.03.020
Vieira, F., Makoni, M., Szyld, E., and Sekar, K. (2021). The Controversy Persists: Is There a Qualification Criterion to Utilize Inhaled Nitric Oxide in Pre-term Newborns? Front. Pediatr. 9, 631765. doi:10.3389/fped.2021.631765
Wang, L., Yao, Y., He, R., Meng, Y., Li, N., Zhang, D., et al. (2017). Methane Ameliorates Spinal Cord Ischemia-Reperfusion Injury in Rats: Antioxidant, Anti-inflammatory and Anti-apoptotic Activity Mediated by Nrf2 Activation. Free Radic. Biol. Med. 103, 69–86. doi:10.1016/j.freeradbiomed.2016.12.014
Wang, R. (2014). Gasotransmitters: Growing Pains and Joys. Trends Biochem. Sci. 39, 227–232. doi:10.1016/j.tibs.2014.03.003
Watanabe, T., and Morita, M. (1998). Asphyxia Due to Oxygen Deficiency by Gaseous Substances. Forensic Sci. Int. 96, 47–59. doi:10.1016/s0379-0738(98)00112-1
Ye, Z., Chen, O., Zhang, R., Nakao, A., Fan, D., Zhang, T., et al. (2015). Methane Attenuates Hepatic Ischemia/Reperfusion Injury in Rats through Antiapoptotic, Anti-inflammatory, and Antioxidative Actions. Shock 44, 181–187. doi:10.1097/SHK.0000000000000385
Zaorska, E., Gawryś-Kopczyńska, M., Ostaszewski, R., Koszelewski, D., and Ufnal, M. (2021). Methane, a Gut Bacteria-Produced Gas, Does Not Affect Arterial Blood Pressure in Normotensive Anaesthetized Rats. BioRxiv. doi:10.1101/2021.03.31.437828
Zhang, B., Tian, X., Li, G., Zhao, H., Wang, X., Yin, Y., et al. (2021). Methane Inhalation Protects against Lung Ischemia-Reperfusion Injury in Rats by Regulating Pulmonary Surfactant via the Nrf2 Pathway. Front. Physiol. 12, 615974. doi:10.3389/fphys.2021.615974
Keywords: normoxia, hypoxia, bioactive gases, methane, mitochondria
Citation: Juhász L, Tallósy SP, Nászai A, Varga G, Érces D and Boros M (2022) Bioactivity of Inhaled Methane and Interactions With Other Biological Gases. Front. Cell Dev. Biol. 9:824749. doi: 10.3389/fcell.2021.824749
Received: 30 November 2021; Accepted: 14 December 2021;
Published: 07 January 2022.
Edited by:
Fernando Antunes, University of Lisbon, PortugalReviewed by:
John Hancock, University of the West of England, United KingdomCopyright © 2022 Juhász, Tallósy, Nászai, Varga, Érces and Boros. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mihály Boros, Ym9yb3MubWloYWx5QG1lZC51LXN6ZWdlZC5odQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.