 Xiuchun Lin
Xiuchun Lin- College of Information and Electrical Engineering, China Agricultural University, Beijing, China
Structural variations in the genome are closely related to human health and the occurrence and development of various diseases. To understand the mechanisms of diseases, find pathogenic targets, and carry out personalized precision medicine, it is critical to detect such variations. The rapid development of high-throughput sequencing technologies has accelerated the accumulation of large amounts of genomic mutation data, including synonymous mutations. Identifying pathogenic synonymous mutations that play important roles in the occurrence and development of diseases from all the available mutation data is of great importance. In this paper, machine learning theories and methods are reviewed, efficient and accurate pathogenic synonymous mutation prediction methods are developed, and a standardized three-level variant analysis framework is constructed. In addition, multiple variation tolerance prediction models are studied and integrated, and new ideas for structural variation detection based on deep information mining are explored.
Introduction
The decreasing cost of genome sequencing has resulted in a large amount of sequence information and variation data becoming available. According to the number of mutated bases, genomic variations have been classified as: 1) single-nucleotide variations (formerly single-nucleotide polymorphisms); 2) very short insertions and deletions, usually less than 50 bp; and 3) structural variations, usually longer than 50 bp (Genome structural variati, 2011). Gene mutations are known to be closely related to the occurrence and development of diseases (Hunt et al., 2014; Alkan et al., 2011; Yin et al., 2020; Fang et al., 2019; Li et al., 2020a; He et al., 2020; Zhang et al., 2020a; Zhang et al., 2016; Hu et al., 2021; Hu et al., 1990; Hu et al., 2020). High-throughput sequencing technologies have allowed the mutations in the genomes of patients with particular diseases to be determined systematically, quickly and accurately, including common but less studied synonymous mutations in the coding regions of genomes (Meyerson et al., 2010; Li et al., 2017; Cheng et al., 2018; Zhou et al., 2019). Synonymous mutations are single-nucleotide mutations that occur in the coding regions of genes but do not change the amino acid sequence of the protein due to the degeneracy of the genetic code. Because they do not change the coded amino acids, synonymous mutations were once mistakenly thought to have no biological function (Hong et al., 2020; Tang et al., 2020; Cheng et al., 2021a). However, later systematic studies have shown that synonymous mutations are involved in a variety of biological processes and play important roles in the occurrence and development of diseases (Li et al., 2020b; Yang et al., 2021a). Whole genome sequencing using reversible terminator chemistry can generate accurate nucleotide sequences of billions of bases at low cost (Bentley et al., 2008), which greatly improves the data obtained in sequencing projects. Structural variations in genomes are closely related to the occurrence and development of many diseases that affect human health. Therefore, the detection of structural mutations is essential to understand the mechanisms of diseases, find pathogenic targets, and carry out personalized precision medicine (Guo et al., 2011; Liu et al., 2021a; Yu et al., 2021a; Yu et al., 2021b). However, detecting structural variations can be difficult, and therefore methods that can accurately and quickly predict genomic variations are urgently required.
In this paper, an efficient and accurate method for predicting disease-causing synonymous mutations based on existing research and using machine learning theories and methods is reviewed. This method not only incorporates the current understanding of the pathogenic mechanism of synonymous mutations, but also provides a theoretical basis for the diagnosis and treatment of diseases, drug development, and the development of memory precision medicine. There are three steps to this method: 1) defining a standardized variation analysis framework based entirely on genome sequencing data; 2) using computational methods to construct a variation tolerance prediction model as the classification basis, and two high-performance variation mechanisms (influence of protein solubility and metabolic stability) prediction models; and 3) developing software tools and combining multiple models to provide general variation analysis services, as well as medical research and precision medical services. For structural variation detection based on deep mining of information, the following key technologies and methods are established: 1) extract sequence features related to structural variations from different sides and establish a comprehensive representation of variation; 2) use the sequence to comparison text information and the corresponding variation feature map to generate images and amplify imbalanced images of small samples; and 3) using the powerful feature representation capabilities of deep learning, automatically extract global features, hidden features, and associated features to complete variation detection. This approach will be an effective way to improve the accuracy of genome structural variation detection, and will also help to promote the development of new structural variation detection technologies. The outline of this study is shown in Figure 1.
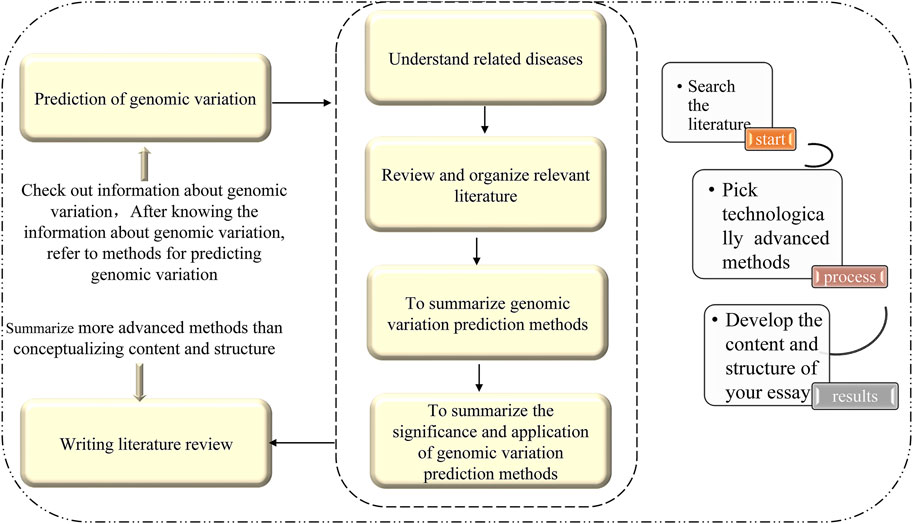
FIGURE 1. Idea map of the paper.
Genomic Variation Prediction Methods
Variations in the human genome are related to human evolution and disease risk (Jiang and Liu, 2016; Jiang et al., 2017; Liu et al., 2017; Liu et al., 2018a; Liu et al., 2018b; Liu et al., 2018c; Liu et al., 2019; Wu et al., 2019; Xu et al., 2019; Deng et al., 2021). Moreover, with the systematic in-depth studies of single-nucleotide mutations, especially those that have special genetic variation patterns such as synonymous mutations, the understanding of the composition of the human genome, genetic differences between individuals, and the pathogenic mechanisms of diseases has greatly improved. The genetic variation prediction method reviewed in this paper will help to make such studies more convenient and economical by identifying variations of interest that can be targeted (Table 1).
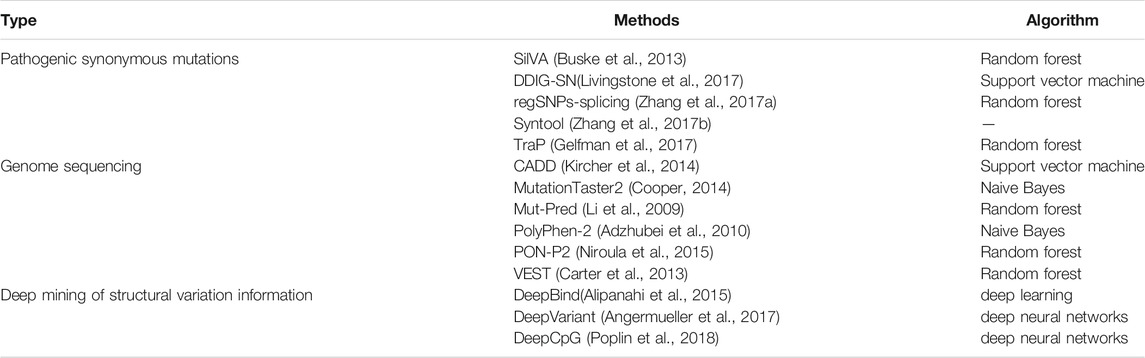
TABLE 1. Summary of genomic variation prediction methods.
Pathogenic Synonymous Mutations
In recent years, the interest and attention of researchers in the analysis and prediction of pathogenic synonymous mutations have increased. Published methods include SilVA (Buske et al., 2013), DDIG-SN (Livingstone et al., 2017), regSNPs-splicing (Zhang et al., 2017a), Syntool (Zhang et al., 2017b) and TraP (Gelfman et al., 2017). sHowever, the available prediction methods still have certain defects that need improvement. Among them, the use of machine learning methods to predict pathogenic synonymous mutations is still in the preliminary stage. The main problems that remain to be solved include: 1) positive sample data is scarce and standard negative sample data is lacking (Zhang et al., 2020b); 2) feature representation ability is weak and not easy to promote (Buske et al., 2013; Wei et al., 2018; Xiong et al., 2018; Jin et al., 2019; Shen et al., 2019; Su et al., 2019; Wei et al., 2019; Yang et al., 2020a; Zhang et al., 2020c; Peng et al., 2020; Su et al., 2020; Teng et al., 2020; Chu et al., 2021a; Cheng et al., 2021b; Chu et al., 2021b; Jin et al., 2021; Su et al., 2021); and 3) the prediction performances of existing methods need to be improved, and the results of different methods have a low degree of coincidence (Cheng et al., 2019). The methods reviewed in this article aim to solve these problems.
The aim of this project is to develop an efficient and accurate method for predicting disease-causing synonymous mutations. The main steps are as follows: 1) establish a data set using a variety of different methods and data sources; 2) analyze in detail the biological characteristic attributes related to pathogenic synonymous mutations; 3) design machine learning methods to predict mutations; and 4) develop a public service platform and corresponding software system to predict disease-related mutations. The following aspects were included in the method. 1) A pathogenic synonymous mutation database and benchmark data set are constructed. Then, pathogenic synonymous mutation data reported in the literature are collected to supplement the database, and the two types of data are integrated to improve the database. 2) A feature representation method of the pathogenic mechanism of the synonymous mutation is established. Synonymous mutations can occur in various processes of gene expression. In this paper, the pathogenic principle of synonymous mutations was fully utilized, and the method of numerical expression of pathogenic synonymous mutations was studied at the DNA, RNA, and protein levels. In addition, the same data set and the same machine learning model are combined for testing, and then the feature selection method is used to remove irrelevant features from the extracted features to select a relatively good feature representation method. 3) A prediction method for pathogenic synonymous mutations with convolutional neural network was used as the basic model. The aim was to learn the representation method of pathogenic synonymous mutation data based on deep learning, especially the efficient implicit feature representation ability. The deep feature representation method of the biological characteristics of pathogenic synonymous mutations is also used to make up for the lack of feature representation ability of shallow learning. Then, deep network structure design and deep model training optimization strategies are studied to improve the robustness and generalization performance of the model. 4) The prediction method of pathogenic synonymous mutation based on ensemble learning is evaluated. To reduce the correlation of individual classifier results, a method suitable for training and learning is selected from the existing prediction methods to obtain the individual classifier. Then, randomization is introduced in the learning and training processes to obtain diverse individual classifiers. Finally, a learning algorithm for the integrated decision-making classifier is designed to construct a suitable secondary classifier to more effectively solve the problem of pathogenic synonymous mutation prediction. How to perform ensemble pruning after the generation of cascading ensemble classifiers should also be studied to further improve the predictive performance of the cascading ensemble learning classifier and obtain better pathogenic synonymous mutation predictions. 5) Result verification and algorithm software development are performed. The predictive analysis data obtained in 3 and 4 above are analyzed and the results with high reliability and potential clinical application value are selected to carry out molecular and biochemical experiments to determine the biological functions of synonymous mutations at the cellular level and to verify the accuracy of the prediction results.
Genome Sequencing
Methods to rapidly and comprehensively interpret the various new variations identified in genome sequencing are lacking. Therefore, it is not yet possible to associate variations with possible diseases or health issues, which greatly reduces the value of genome sequencing. A primary task of sequencing research is how to analyze the sequence data, especially the variation information that it contains. Prediction methods can effectively solve this problem (Castrense et al., 2019; Gang et al., 2019; Liu et al., 2020a; Jin et al., 2021; Yin et al., 2021). The published methods include CADD (Kircher et al., 2014), MutationTaster2 (Cooper, 2014),Mut-Pred (Li et al., 2009),PolyPhen-2 (Adzhubei et al., 2010),PON-P2 (Niroula et al., 2015) and VEST (Carter et al., 2013). The aim of the project was to design a standard variation analysis framework for genomic variation prediction. Specifically, a variation tolerance prediction model is constructed based on the genome sequencing data and calculation method, and a relatively high-performance variation mechanism is also constructed based on the influence of protein solubility and metabolic stability. The following aspects are included. 1) A general prediction model of variation tolerance classification is constructed based on the existing variation data for all kinds of variations (e.g., replacement, insertion, and deletion) using only the sequence information as the basic and important step of the analysis services. A highly accurate predictive model ProtSol is constructed. New data will be collected and sorted out, the training data set will be integrated, and input features will be selected. Then, the classification algorithm will be optimized and a protein solubility impact prediction model with higher accuracy and greater generalization will be established for variation analysis. 3) A model ProtMS to predict the effect of variation on protein metabolism stability is constructed. Classification and regression models based on sequence information will be established to predict the impact of variations on metabolic stability. These models will serve as important parts of the mechanism analysis. In addition, browser/server architecture variant analysis software tools will be developed and released to provide online services for researchers and clinical medical staff.
Deep Mining of Structural Variation Information
Structural variations in the human genome can cause diseases (Feuk, et al., 2006). Structural variations include translocation, inversion, deletion, and duplication of genes, and accurate detection of genetic variations or genetic testing can contribute to the exploration and analysis of diseases and life processes. The obtained structural variant information can be applied, for example, to target drugs to tumors and to provide a reliable reference for clinical applications (Xu et al., 2018; Xue et al., 2018; Tang et al., 2019; Liu et al., 2020b; Zhang et al., 2020d; An and Yu, 2021; Liu et al., 2021b; Wang et al., 2021; Wu and Yu, 2021). Therefore, we need to mine structural variation information accurately. Published methods include DeepBind (Alipanahi et al., 2015), DeepVariant (Angermueller et al., 2017), and DeepCpG (Poplin et al., 2018). Deep mining of the human genomic structural variation information includes the following aspects. 1) Comprehensive characterization of features using three basic detection methods: sequence features related to structural variation from different aspects, definition of representation modes of different types of variation, and construction of classification and combined expression of feature descriptions. Then, the comprehensive characteristics are obtained by combining three types of detection methods (i.e., double-terminal mapping-based, split-comparison-based, and mapping depth-based methods) to form a complete comprehensive characterization of variation features. 2) Feature image generation whereby images are generated to discover comprehensive variation information from two main aspects, pixel composition and color expression. Pixel composition includes base stacking and fragment tiled, and color expression includes stacked base color expression and tiled fragment color expression. 3) Data amplification to ensure that the generated images can be used for deep learning training and recognition. The main purpose of data amplification is to avoid the problem of network overfitting or a decline in the recognition of minority samples. This step includes mainly studying the method of amplification of mutated images different from natural images; studying sufficiently clear and high-resolution amplified images, and studying the speed of image generation to ensure the network is better able to learn the mapping of complex functions. A schematic diagram of this method of deep mining of structural information is given in Figure 2.
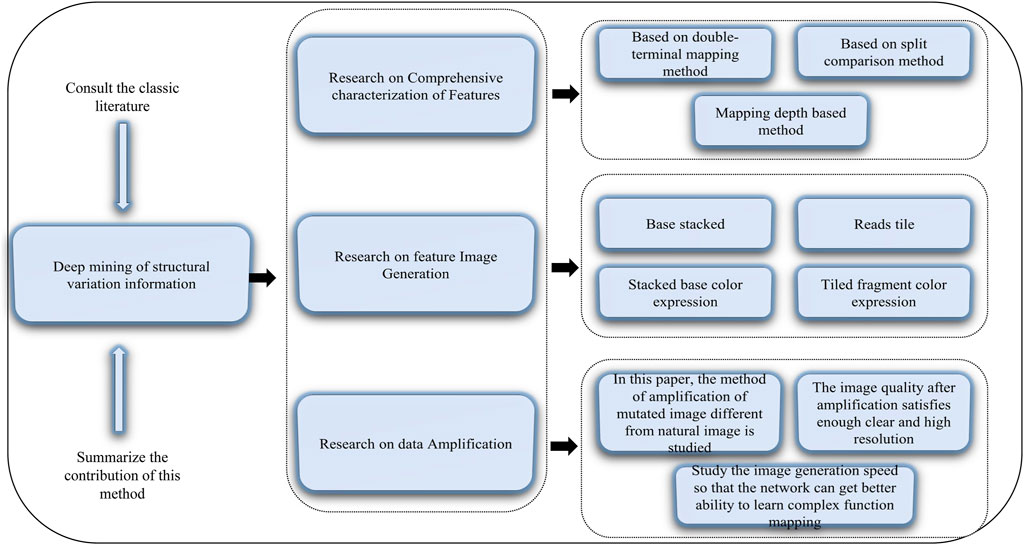
FIGURE 2. Diagram of deep mining of structural variation information.
Literature Contribution
Early studies of the human genome focused mainly on collecting data and understanding structural variation at the genome level. In this paper, the literature closely related to diseases and current medical fields was reviewed, including genome prediction methods and many aspects of genome variation. The genomic variants associated with the diseases are summarized in Table 2. Bioinformatics analysis studies of pathogenic synonymous mutations aim to integrate different data sources and numerical types and detect reliable characteristics for feature representation methods, and also to accurately characterize feature coding methods that are intrinsically linked to pathogenic synonymous mutations. For ensemble learning methods, it is necessary to propose a pruning mechanism for the primary classifier learning algorithm with adaptive learning ability. Genome sequencing studies aim to establish a set of standardized solutions and integrate existing databases and multiple prediction models. The goal is to solve the versatility and generalization of variation analysis models, to solve the data imbalance problem that is common in variation data, and to find sequence-based biological characteristics in different variation prediction models. The contribution of the deep mining of structural variation information is to develop a new way of generating variation feature images from sequence comparison text information, a method of data amplification for small samples of variation images to achieve a balance, and an accurate detection technology framework by digging deep into the genomic structural variation information. The proposed points can take genomic variation prediction research one step further, and provide medication recommendations for the treatment of specific diseases, thereby reducing the adverse effects on patients due to improper medication strategies. This is one of the reasons why genomic variation is an important area of study.

TABLE 2. Summary of genomic variations associated with disease.
Conclusion
The study of structural variations in genomes can promote research on genome evolution, significant biological phenotypic changes (Yang et al., 2020b; Yang et al., 2020c; Yin et al., 2020), the treatment of many diseases (Li et al., 2018; Yang et al., 2021b; Long et al., 2021), and recommendations for therapeutic drugs (Wei et al., 2014; Ding et al., 2020a; Ding et al., 2020b; Wang et al., 2020; Wei et al., 2020). The accurate prediction of genomic variation is of great importance to studies of many diseases, which indicates the significance of this literature review through which existing variation data were integrated and collected, and a tolerance classification model of various variations was constructed based on sequence information. Furthermore, all the literature is experimenting around key scientific issues. It is essential to accurately predict individual genomic variation events that are conducive to systematically inferring the process of variation formation, so that the results can be confidently used for the clinical application of precision medicine. Finally, accurate predictions also help in analyzing the functions of synonymous mutations and can guide relevant experiments. Therefore, genomic variation prediction is of great significance to drug design and precision medicine.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Margaret Biswas, PhD, from Liwen Bianji (Edanz) (www.liwenbianji.cn/) for editing the English text of a draft of this manuscript.
References
Abdel-Rahman, M. H., Pilarski, R., Cebulla, C. M., Massengill, J. B., Christopher, B. N., Boru, G., et al. (2011). Germline BAP1 Mutation Predisposes to Uveal Melanoma, Lung Adenocarcinoma, Meningioma, and Other Cancers. J. Med. Genet. 48 (12), 856–859. doi:10.1136/jmedgenet-2011-100156
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A Method and Server for Predicting Damaging Missense Mutations. Nat Methods. 7(4), 248–9. doi:10.1038/nmeth0410-248
Alipanahi, B., Delong, A., Weirauch, M. T., and Frey, B. J. (2015). Predicting the Sequence Specificities of DNA- and RNA-Binding Proteins by Deep Learning. Nat. Biotechnol. doi:10.1038/nbt.3300
An, Q., and Yu, L. (2021). A Heterogeneous Network Embedding Framework for Predicting Similarity-Based Drug-Target Interactions. Brief. Bioinformatics. 22, bbab275. doi:10.1093/bib/bbab275
Angermueller, C., Lee, H. J., Reik, W., and Stegle, O. (2017). Erratum to: DeepCpG: Accurate Prediction of Single-Cell DNA Methylation States Using Deep Learning. Genome Biol. 18 (1), 90. doi:10.1186/s13059-017-1233-z
Bentley, D. R., Balasubramanian, S., Swerdlow, H. P., Smith, G. P., Milton, J., Brown, C. G., et al. (2008). Accurate Whole Human Genome Sequencing Using Reversible Terminator Chemistry. Nature 456 (7218), 53–59. doi:10.1038/nature07517
Bonberg, N., Taeger, D., Gawrych, K., Johnen, G., Banek, S., Schwentner, C., et al. (2013). Chromosomal Instability and Bladder Cancer: the UroVysionTMtest in the UroScreen Study. BJU Int. 112 (4), E372–E382. doi:10.1111/j.1464-410x.2012.11666.x
Buske, O. J., Manickaraj, A., Mital, S., Ray, P. N., and Brudno, M. (2013). Identification of Deleterious Synonymous Variants in Human Genomes. Bioinformatics 29, 1843–1850. doi:10.1093/bioinformatics/btt308
Carter, H., Douville, C., Stenson, P. D., Cooper, D. N., and Karchin, R. (2013). Identifying Mendelian Disease Genes with the Variant Effect Scoring Tool. BMC Genomics 14 (3), S3–S16. doi:10.1186/1471-2164-14-S3-S3
Castrense, S., Giulia, B., Samuele, B., Emidio, C., Pier, L. M., and Rita, C. (2019). Are Machine Learning Based Methods Suited to Address Complex Biological Problems? Lessons from CAGI‐5 Challenges. Hum. Mutat. 40, 1455–1462. doi:10.1002/humu.23784
Cheng, L., Qi, C., Yang, H., Lu, M., Cai, Y., Fu, T., et al. (2021). gutMGene: a Comprehensive Database for Target Genes of Gut Microbes and Microbial Metabolites. Nucleic Acids Res. 9, gkab786. doi:10.1093/nar/gkab786
Cheng, L., Han, X., Zhu, Z., Qi, C., Wang, P., and Zhang, X. (2021). Functional Alterations Caused by Mutations Reflect Evolutionary Trends of SARS-CoV-2. Brief. Bioinformatics 22 (2), 1442–1450. doi:10.1093/bib/bbab042
Cheng, L., Hu, Y., Sun, J., Zhou, M., and Jiang, Q. (2018). DincRNA: a Comprehensive Web-Based Bioinformatics Toolkit for Exploring Disease Associations and ncRNA Function. Bioinformatics 34 (11), 1953–1956. doi:10.1093/bioinformatics/bty002
Cheng, N., Li, M., Zhao, L., Zhang, B., Yang, Y., Zheng, C. H., et al. (2019). Comparison and Integration of Computational Methods for Deleterious Synonymous Mutation Prediction. Brief. Bioinformatics. 21, 970–981. doi:10.1093/bib/bbz047
Chu, Y., Wang, X., Dai, Q., Wang, Y., Wang, Q., Peng, S., et al. (2021). MDA-GCNFTG: Identifying miRNA-Disease Associations Based on Graph Convolutional Networks via Graph Sampling through the Feature and Topology Graph. Brief Bioinform.
Chu, Y., Kaushik, A. C., Wang, X., Wang, W., Zhang, Y., Shan, X., et al. (2021). DTI-CDF: a cascade Deep forest Model towards the Prediction of Drug-Target Interactions Based on Hybrid Features. Brief Bioinform 22 (1), 451–462. doi:10.1093/bib/bbz152
Deng, L., Li, W., and Zhang, J. (2021). LDAH2V: Exploring Meta-Paths across Multiple Networks for lncRNA-Disease Association Prediction. Ieee/acm Trans. Comput. Biol. Bioinf. 18 (4), 1572–1581. doi:10.1109/tcbb.2019.2946257
Ding, Y., Tang, J., and Guo, F. (2020). Identification of Drug-Target Interactions via Dual Laplacian Regularized Least Squares with Multiple Kernel Fusion. Knowledge-Based Syst. 204, 106254. doi:10.1016/j.knosys.2020.106254
Ding, Y., Tang, J., and Guo, F. (2020). Identification of Drug-Target Interactions via Fuzzy Bipartite Local Model. Neural Comput. Applic 32, 10303–10319. doi:10.1007/s00521-019-04569-z
Fang, S., Pan, J., Zhou, C., Tian, H., He, J., Shen, W., et al. (2019). Circular RNAs Serve as Novel Biomarkers and Therapeutic Targets in Cancers. Cgt 19 (2), 125–133. doi:10.2174/1566523218666181109142756
Feuk, L., Carson, A. R., and Scherer, S. W. (2006). Structural Variation in the Human Genome. Nat. Rev. Genet. 7, 85–97. doi:10.1038/nrg1767
Freemantle, N., Blonde, L., Duhot, D., Hompesch, M., Eggertsen, R., Hobbs, F. D. R., et al. (2005). Availability of Inhaled Insulin Promotes Greater Perceived Acceptance of Insulin Therapy in Patients with Type 2 Diabetes. Diabetes care 28, 427–428. doi:10.2337/diacare.28.2.427
Gang, H., Liu, G., Zhang, M., Zhao, Y., Jiang, J., and Chen, S. (2019). Comprehensive Characterization of T-DNA Integration Induced Chromosomal Rearrangement in a Birch T-DNA Mutant. BMC Genomics 20 (1), 311. doi:10.1186/s12864-019-5636-y
Gelfman, S., Wang, Q., Mcsweeney, K. M., Ren, Z., La Carpia, F., Halvorsen, M., et al. (2017). Annotating Pathogenic Non-coding Variants in Genic Regions. Nat. Commun. 8 (1), 236. doi:10.1038/s41467-017-00141-2
Alkan, C., Coe, B., and Eichler, E. E. (2011). Genome Structural Variation Discovery and Genotyping. Nat Rev Genet. 12, 363–76. doi:10.1038/nrg2958
Guo, F., and Wang, L. (2011). “Computing the Protein Binding Sites,”. Bioinformatics Research and Applications. Editors J. Chen, and J. X. Wang (Changsha, China: Zelikovsky A), 6674, 25–36. doi:10.1007/978-3-642-21260-4_7
He, B., Lang, J., Wang, B., Liu, X., Lu, Q., He, J., et al. (2020). TOOme: A Novel Computational Framework to Infer Cancer Tissue-Of-Origin by Integrating Both Gene Mutation and Expression. Front. Bioeng. Biotechnol. 8, 394. doi:10.3389/fbioe.2020.00394
Hong, J., Luo, Y., Zhang, Y., Ying, J., Xue, W., Xie, T., et al. (2020). Protein Functional Annotation of Simultaneously Improved Stability, Accuracy and False Discovery Rate Achieved by a Sequence-Based Deep Learning. Brief Bioinform 21 (4), 1437–1447. doi:10.1093/bib/bbz081
Hu, Y., Qiu, S., and Cheng, L. (2021). Integration of Multiple-Omics Data to Analyze the Population-specific Differences for Coronary Artery Disease. Comput. Math. Methods Med. 2021, 7036592. doi:10.1155/2021/7036592
Hu, Y., Sun, J. Y., Zhang, Y., Zhang, H., Gao, S., Wang, T., et al. (1990). Variant Associates with Alzheimer's Disease and Regulates TMEM106B Expression in Human Brain Tissues. BMC Med. 19 (1), 11.
Hu, Y., Zhang, H., Liu, B., Gao, S., Wang, T., Han, Z., et al. (2020). rs34331204 Regulates TSPAN13 Expression and Contributes to Alzheimer's Disease with Sex Differences. Brain 143 (11), e95. doi:10.1093/brain/awaa302
Hunt, R. C., Simhadri, V. L., Iandoli, M., Sauna, Z. E., and Kimchi-Sarfaty, C. (2014). Exposing Synonymous Mutations. Trends Genet. 30, 308-21. doi:10.1016/j.tig.2014.04.006
Jiang, Q., and Liu, G. (2016). Lack of Association between MC1R Variants and Parkinson's Disease in European Descent. Ann. Neurol. 79, 866–868. doi:10.1002/ana.24627
Jiang, Q., Jin, S., Jiang, Y., Liao, M., Feng, R., Zhang, L., et al. (2017). Alzheimer's Disease Variants with the Genome-wide Significance Are Significantly Enriched in Immune Pathways and Active in Immune Cells. Mol. Neurobiol. 54 (1), 594–600. doi:10.1007/s12035-015-9670-8
Jin, Q., Cui, H., Sun, C., Meng, Z., and Su, R. (2021). Free-form Tumor Synthesis in Computed Tomography Images via Richer Generative Adversarial Network. Knowledge-Based Syst. 218, 106753. doi:10.1016/j.knosys.2021.106753
Jin, Q., Meng, Z., Pham, T. D., Chen, Q., Wei, L., Su, R., et al. (2019). DUNet: A Deformable Network for Retinal Vessel Segmentation. Knowledge-Based Syst. 178, 149–162. doi:10.1016/j.knosys.2019.04.025
Kircher, M., Witten, D. M., Jain, P., O'Roak, B. J., Cooper, G. M., and Shendure, J. (2014). A General Framework for Estimating the Relative Pathogenicity of Human Genetic Variants. Nat. Genet. 46 (3), 310–315. doi:10.1038/ng.2892
Li, B., Krishnan, V. G., Mort, M. E., Xin, F., Kamati, K. K., Cooper, D. N., et al. (2009). Automated Inference of Molecular Mechanisms of Disease from Amino Acid Substitutions. Bioinformatics 25, 2744–2750. doi:10.1093/bioinformatics/btp528
Li, B., Tang, J., Yang, Q., Li, S., Cui, X., Li, Y., et al. (2017). NOREVA: Normalization and Evaluation of MS-based Metabolomics Data. Nucleic Acids Res. 45 (W1), W162–W170. doi:10.1093/nar/gkx449
Li, F., Zhou, Y., Zhang, X., Tang, J., Yang, Q., Zhang, Y., et al. (2020). SSizer: Determining the Sample Sufficiency for Comparative Biological Study. J. Mol. Biol. 432 (11), 3411–3421. doi:10.1016/j.jmb.2020.01.027
Li, Y. H., Li, X. X., Hong, J. J., Wang, Y. X., Fu, J. B., Yang, H., et al. (2020). Clinical Trials, Progression-Speed Differentiating Features and Swiftness Rule of the Innovative Targets of First-In-Class Drugs. Brief. Bioinformatics 21 (2), 649–662. doi:10.1093/bib/bby130
Li, Y. H., Yu, C. Y., Li, X. X., Zhang, P., Tang, J., Yang, Q., et al. (2018). Therapeutic Target Database Update 2018: Enriched Resource for Facilitating Bench-To-Clinic Research of Targeted Therapeutics. Nucleic Acids Res. 46 (D1), D1121–D1127. doi:10.1093/nar/gkx1076
Liu, G., Hu, Y., Han, Z., Jin, S., and Jiang, Q. (2019). Genetic Variant Rs17185536 Regulates SIM1 Gene Expression in Human Brain Hypothalamus. Proc. Natl. Acad. Sci. USA 116 (9), 3347–3348. doi:10.1073/pnas.1821550116
Liu, G., Hu, Y., Jin, S., and Jiang, Q. (2017). Genetic Variant Rs763361 Regulates Multiple Sclerosis CD226 Gene Expression. Proc. Natl. Acad. Sci. USA 114 (6), E906–E907. doi:10.1073/pnas.1618520114
Liu, G., Jin, S., Hu, Y., and Jiang, Q. (2018). Disease Status Affects the Association between Rs4813620 and the Expression of Alzheimer's Disease Susceptibility geneTRIB3. Proc. Natl. Acad. Sci. USA 115 (45), E10519–E10520. doi:10.1073/pnas.1812975115
Liu, G., Wang, T., Tian, R., Hu, Y., Han, Z., Wang, P., et al. (2018). Alzheimer's Disease Risk Variant Rs2373115 Regulates GAB2 and NARS2 Expression in Human Brain Tissues. J. Mol. Neurosci. 66 (1), 37–43. doi:10.1007/s12031-018-1144-9
Liu, G., Zhang, Y., Wang, L., Xu, J., Chen, X., Bao, Y., et al. (2018). Alzheimer's Disease Rs11767557 Variant Regulates EPHA1 Gene Expression Specifically in Human Whole Blood. Jad 61 (3), 1077–1088. doi:10.3233/jad-170468
Liu, H., Zhang, W., Zou, B., Wang, J., Deng, Y., and Deng, L. (2020). DrugCombDB: a Comprehensive Database of Drug Combinations toward the Discovery of Combinatorial Therapy. Nucleic Acids Res. 48 (D1), D871–D881. doi:10.1093/nar/gkz1007
Liu, J., Liu, S., Liu, C., Zhang, Y., Pan, Y., Wang, Z., et al. (2021). Nabe: an Energetic Database of Amino Acid Mutations in Protein-Nucleic Acid Binding Interfaces. Database (Oxford) 2021, 2021. doi:10.1093/database/baab050
Liu, J., Su, R., Zhang, J., and Wei, L. (2021). Classification and Gene Selection of Triple-Negative Breast Cancer Subtype Embedding Gene Connectivity Matrix in Deep Neural Network. LID - Bbaa395 [pii] LID -. Briefings in Bioinformatics, 2021, 1477–4054. doi:10.1093/bib/bbaa395
Liu, Y., Huang, Y., Wang, G., and Wang, Y. (2020). A Deep Learning Approach for Filtering Structural Variants in Short Read Sequencing Data. Brief Bioinform.
Livingstone, M., Folkman, L., Yang, Y., Zhang, P., Mort, M., Cooper, D. N., et al. (2017). Investigating DNA-, RNA-, and Protein-Based Features as a Means to Discriminate Pathogenic Synonymous Variants. Hum. Mutat. 38 (10), 1336–1347. doi:10.1002/humu.23283
Long, J., Yang, H., Yang, Z., Jia, Q., Liu, L., Kong, L., et al. (2021). Integrated Biomarker Profiling of the Metabolome Associated with Impaired Fasting Glucose and Type 2 Diabetes Mellitus in Large-Scale Chinese Patients. Clin. Transl Med. 11 (6), e432. doi:10.1002/ctm2.432
Meyerson, M., Gabriel, S., Getz, G., Meyerson, M., Gabriel, S., and GAdvances, Getz. (2010). Advances in Understanding Cancer Genomes through Second-Generation Sequencing. Nat. Rev. Genet. 11 (10), 685–696. doi:10.1038/nrg2841
Niroula, A., Urolagin, S., and Vihinen, M. (2015). PON-P2: Prediction Method for Fast and Reliable Identification of Harmful Variants. Plos One 10 (2), e0117380. doi:10.1371/journal.pone.0117380
Peng, L., Zhou, D., Liu, W., Zhou, L., Wang, L., Zhao, B., et al. (2020). Prioritizing Human Microbe-Disease Associations Utilizing a Node-Information-Based Link Propagation Method. IEEE Access 8, 31341–31349. doi:10.1109/access.2020.2972283
Poplin, R., Chang, P-C., Alexander, D., Schwartz, S., Colthurst, T., Ku, A., et al. (2018). Creating a Universal SNP and Small Indel Variant Caller with Deep Neural Networks. bioRxiv, 092890.
Shen, Y., Tang, J., and Guo, F. (2019). Identification of Protein Subcellular Localization via Integrating Evolutionary and Physicochemical Information into Chou's General PseAAC. J. Theor. Biol. 462, 230–239. doi:10.1016/j.jtbi.2018.11.012
Su, R., Hu, J., Zou, Q., Manavalan, B., and Wei, L. (2020). Empirical Comparison and Analysis of Web-Based Cell-Penetrating Peptide Prediction Tools. Brief. Bioinformatics 21 (2), 408–420. doi:10.1093/bib/bby124
Su, R., Liu, X., Jin, Q., Liu, X., and Wei, L. (2021). Identification of Glioblastoma Molecular Subtype and Prognosis Based on Deep MRI Features. Knowledge-Based Syst. 232, 107490. doi:10.1016/j.knosys.2021.107490
Su, R., Liu, X., Wei, L., and Zou, Q. (2019). Deep-Resp-Forest: A Deep forest Model to Predict Anti-cancer Drug Response. Methods 166, 91–102. doi:10.1016/j.ymeth.2019.02.009
Tang, J., Fu, J., Wang, Y., Li, B., Li, Y., Yang, Q., et al. (2020). ANPELA: Analysis and Performance Assessment of the Label-free Quantification Workflow for Metaproteomic Studies. Brief. Bioinformatics 21 (2), 621–636. doi:10.1093/bib/bby127
Tang, J., Fu, J., Wang, Y., Luo, Y., Yang, Q., Li, B., et al. (2019). Simultaneous Improvement in the Precision, Accuracy, and Robustness of Label-free Proteome Quantification by Optimizing Data Manipulation Chains*. Mol. Cell Proteomics 18 (8), 1683–1699. doi:10.1074/mcp.ra118.001169
Teng, H., Wei, W., Li, Q., Xue, M., Shi, X., Li, X., et al. (2020). Prevalence and Architecture of Posttranscriptionally Impaired Synonymous Mutations in 8,320 Genomes across 22 Cancer Types. Nucleic Acids Res. 48 (3), 1192–1205. doi:10.1093/nar/gkaa019
Thuresson, A. C., Van Buggenhout, G., Sheth, F., Kamate, M., Andrieux, J., Clayton Smith, J., et al. (2016). Whole Gene Duplication of SCN2A and SCN3A Is Associated with Neonatal Seizures and a normal Intellectual Development. Clin. Genet. 91 (1), 106–110. doi:10.1111/cge.12797
Wang, J., Liu, X., Shen, S., Deng, L., and Liu, H. (2021). DeepDDS: Deep Graph Neural Network with Attention Mechanism to Predict Synergistic Drug Combinations. Brief. Bioinformatics. doi:10.1093/bib/bbab390
Wang, Y., Zhang, S., Li, F., Zhou, Y., Zhang, Y., Wang, Z., et al. (2020). Therapeutic Target Database 2020: Enriched Resource for Facilitating Research and Early Development of Targeted Therapeutics. Nucleic Acids Res. 48 (D1), D1031–D1041. doi:10.1093/nar/gkz981
Wei, L., Hu, J., Li, F., Song, J., Su, R., and Zou, Q. (2020). Comparative Analysis and Prediction of Quorum-sensing Peptides Using Feature Representation Learning and Machine Learning Algorithms. Brief. Bioinformatics 21 (1), 106–119.
Wei, L., Zhou, C., Chen, H., Song, J., and Su, R. (2018). ACPred-FL: a Sequence-Based Predictor Using Effective Feature Representation to Improve the Prediction of Anti-cancer Peptides. Bioinformatics 34 (23), 4007–4016. doi:10.1093/bioinformatics/bty451
Wei, L., Liao, M., Gao, Y., Ji, R., He, Z., and Zou, Q. (2014). Improved and Promising Identification of Human MicroRNAs by Incorporating a High-Quality Negative Set. Ieee/acm Trans. Comput. Biol. Bioinf. 11 (1), 192–201. doi:10.1109/tcbb.2013.146
Wei, L., Xing, P., Shi, G., Ji, Z., and Zou, Q. (2019). Fast Prediction of Protein Methylation Sites Using a Sequence-Based Feature Selection Technique. Ieee/acm Trans. Comput. Biol. Bioinf. 16 (4), 1264–1273. doi:10.1109/tcbb.2017.2670558
Wu, X., and Yu, L. (2021). EPSOL: Sequence-Based Protein Solubility Prediction Using Multidimensional Embedding. Oxford, England: Bioinformatics.
Wu, Y., Lu, X., Shen, B., and Zeng, Y. (2019). The Therapeutic Potential and Role of miRNA, lncRNA, and circRNA in Osteoarthritis. Cgt 19 (4), 255–263. doi:10.2174/1566523219666190716092203
Xiong, Y., Wang, Q., Yang, J., Zhu, X., and Wei, D.-Q. (2018). PredT4SE-Stack: Prediction of Bacterial Type IV Secreted Effectors from Protein Sequences Using a Stacked Ensemble Method. Front. Microbiol. 9, 2571. doi:10.3389/fmicb.2018.02571
Xu, L., Liang, G., Liao, C., Chen, G. D., Chang, C. C., and k-Skip-n-Gram-Rf, (2019). K-Skip-N-Gram-RF: A Random Forest Based Method for Alzheimer's Disease Protein Identification. Front. Genet. 10 (33), 33. doi:10.3389/fgene.2019.00033
Xu, L., Liang, G., Wang, L., and Liao, C. (2018). A Novel Hybrid Sequence-Based Model for Identifying Anticancer Peptides. Genes 9 (3), 158. doi:10.3390/genes9030158
Xue, W., Yang, F., Wang, P., Zheng, G., Chen, Y., Yao, X., et al. (2018). What Contributes to Serotonin-Norepinephrine Reuptake Inhibitors' Dual-Targeting Mechanism? the Key Role of Transmembrane Domain 6 in Human Serotonin and Norepinephrine Transporters Revealed by Molecular Dynamics Simulation. ACS Chem. Neurosci. 9 (5), 1128–1140. doi:10.1021/acschemneuro.7b00490
Yang, H., Ding, Y., Tang, J., and Guo, F. (2021). Drug-disease Associations Prediction via Multiple Kernel-Based Dual Graph Regularized Least Squares. Appl. Soft Comput. 112, 107811. doi:10.1016/j.asoc.2021.107811
Yang, H., Ding, Y., Tang, J., and Guo, F. (2021). Identifying Potential Association on Gene-Disease Network via Dual Hypergraph Regularized Least Squares. BMC Genomics 22 (1), 605. doi:10.1186/s12864-021-07864-z
Yang, L., LiuLiu, B., Huang, B., Deng, J., Li, H., Yu, B., et al. (2013). A Functional Copy Number Variation in the WWOX Gene Is Associated with Lung Cancer Risk in Chinese. Hum. Mol. Genet. 22 (9), 1886–1894. doi:10.1093/hmg/ddt019
Yang, Q., Hong, J., Li, Y., Xue, W., Li, S., Yang, H., et al. (2020). A Novel Bioinformatics Approach to Identify the Consistently Well-Performing Normalization Strategy for Current Metabolomic Studies. Brief. Bioinformatics 21 (6), 2142–2152. doi:10.1093/bib/bbz137
Yang, Q., Li, B., Tang, J., Cui, X., Wang, Y., Li, X., et al. (2020). Consistent Gene Signature of Schizophrenia Identified by a Novel Feature Selection Strategy from Comprehensive Sets of Transcriptomic Data. Brief. Bioinformatics 21 (3), 1058–1068. doi:10.1093/bib/bbz049
Yang, Q., Wang, Y., Zhang, Y., Li, F., Xia, W., Zhou, Y., et al. (2020). NOREVA: Enhanced Normalization and Evaluation of Time-Course and Multi-Class Metabolomic Data. Nucleic Acids Res. 48 (W1), W436–W448. doi:10.1093/nar/gkaa258
Yin, J., Li, F., Zhou, Y., Mou, M., Lu, Y., Chen, K., et al. (2021). INTEDE: Interactome of Drug-Metabolizing Enzymes. Nucleic Acids Res. 49 (D1), D1233–D1243. doi:10.1093/nar/gkaa755
Yin, J., Sun, W., Li, F., Hong, J., Li, X., Zhou, Y., et al. (2020). VARIDT 1.0: Variability of Drug Transporter Database. Nucleic Acids Res. 48 (D1), D1042–D1050. doi:10.1093/nar/gkz779
Yu, L., Xia, M., and An, Q. (2021). A Network Embedding Framework Based on Integrating Multiplex Network for Drug Combination Prediction. Brief. Bioinformatics. doi:10.1093/bib/bbab364
Yu, L., Wang, M., Yang, Y., Xu, F., Zhang, X., Xie, F., et al. (2021). Predicting Therapeutic Drugs for Hepatocellular Carcinoma Based on Tissue-specific Pathways. Plos Comput. Biol. 17 (2), e1008696. doi:10.1371/journal.pcbi.1008696
Zhang, J., Zhang, Z., Pu, L., Tang, J., and Guo, F. (2020). AIEpred: an Ensemble Predictive Model of Classifier Chain to Identify Anti-inflammatory Peptides. Ieee/acm Trans. Comput. Biol. Bioinform, 1. doi:10.1109/TCBB.2020.2968419
Zhang, S., Su, M., Sun, Z., Lu, H., and Zhang, Y. (2020). The Signature of Pharmaceutical Sensitivity Based on ctDNA Mutation in Eleven Cancers. Exp. Biol. Med. (Maywood) 245 (8), 720–732. doi:10.1177/1535370220906518
Zhang, T., Hu, Y., Wu, X., Ma, R., Jiang, Q., and Wang, Y. (2016). Identifying Liver Cancer-Related Enhancer SNPs by Integrating GWAS and Histone Modification ChIP-Seq Data. Biomed. Res. Int. 2016, 2395341. doi:10.1155/2016/2395341
Zhang, T., Wu, Y., Lan, Z., Shi, Q., Yang, Y., Guo, J., et al. (2017). Syntool: A Novel Region-Based Intolerance Score to Single Nucleotide Substitution for Synonymous Mutations Predictions Based on 123,136 Individuals. Biomed. Res. Int. 2017, 5096208. doi:10.1155/2017/5096208
Zhang, X., Li, M., Lin, H., Rao, X., Feng, W., Yang, Y., et al. (2017). regSNPs-Splicing: a Tool for Prioritizing Synonymous Single-Nucleotide Substitution. Hum. Genet. 136 (Suppl. 9), 1279–1289. doi:10.1007/s00439-017-1783-x
Zhang, Z.-M., Tan, J.-X., Wang, F., Dao, F.-Y., Zhang, Z.-Y., and Lin, H. (2020). Early Diagnosis of Hepatocellular Carcinoma Using Machine Learning Method. Front. Bioeng. Biotechnol. 8, 254. doi:10.3389/fbioe.2020.00254
Zhang, Z.-M., Wang, J.-S., Zulfiqar, H., Lv, H., Dao, F.-Y., and Lin, H. (2020). Early Diagnosis of Pancreatic Ductal Adenocarcinoma by Combining Relative Expression Orderings with Machine-Learning Method. Front. Cel Dev. Biol. 8, 582864. doi:10.3389/fcell.2020.582864
Keywords: genome, variation, machine learning, genomic mutation, prediction
Citation: Lin X (2021) Genomic Variation Prediction: A Summary From Different Views. Front. Cell Dev. Biol. 9:795883. doi: 10.3389/fcell.2021.795883
Received: 15 October 2021; Accepted: 11 November 2021;
Published: 25 November 2021.
Edited by:
Liang Cheng, Harbin Medical University, ChinaReviewed by:
Fei Guo, Tianjin University, ChinaHao Lin, University of Electronic Science and Technology of China, China
Copyright © 2021 Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiuchun Lin, bGlueGMxMjRAMTYzLmNvbQ==, MTI0MTU2Mjg1M0BxcS5jb20=