 Yuyang Guo
Yuyang Guo Hengli Zhao1,2,3
Hengli Zhao1,2,3 Qingchun Zeng
Qingchun Zeng- 1State Key Laboratory of Organ Failure Research, Department of Cardiology, Nanfang Hospital, Southern Medical University, Guangzhou, China
- 2Guangdong Provincial Key Laboratory of Shock and Microcirculation, Southern Medical University, Guangzhou, China
- 3Bioland Laboratory (Guangzhou Regenerative Medicine and Health Guangdong Laboratory), Guangzhou, China
- 4Department of Cardiopulmonary Rehabilitation, First Affiliated Hospital of Guangzhou University of Chinese Medicine, Guangzhou, China
Heme, the protoporphyrin IX iron complex is widely present in the human body and it is involved in oxygen storage, electron transfer, and enzymatic reactions. However, free heme can be toxic as it catalyzes the production of reactive oxygen species, oxidizes lipids and proteins, and causes DNA damage, thereby inducing a pro-inflammatory environment. The generation, metabolism, and degradation of heme in the human body are regulated by precise mechanisms to ensure that heme remains non-toxic. However, in several types of cardiovascular diseases, impaired metabolism and exposure to heme may occur in pathological processes, including neovascularization, internal hemorrhage, ischemia, and reperfusion. Based on years of research, in this review, we aimed to summarize the underlying mechanisms by which heme contributes to the development of cardiovascular diseases through oxidative stress, relative pathway gene expression regulation and phenotypic changes in cells. Excess heme plays a detrimental role in atherosclerosis, heart failure, myocardial ischemia-reperfusion injury, degenerative aortic valve stenosis, cardiac iron overload. Recent researches revealed that in some cases heme involved in cardiac damage though ferroptosis. Thus, heme concentrations beyond normal levels are dangerous. Further research on the role of heme in cardiovascular diseases is needed.
1 Introduction
Heme, chemically named as protoporphyrin IX iron complex, comprises an iron atom that is bound to heterocyclic tetrapyrrole porphyrin ring (Sawicki et al., 2015a). It is mainly involved in oxygen storage, transfer, and activation and electron transfer in the form of hemoglobin, myoglobin, cytochrome P450, and cytochrome c (Shimizu et al., 2019). The ability of heme to perform redox reactions is attributed to its iron atom. Iron, a d-block transition metal, easily donates and accepts electrons (Pantopoulos et al., 2012). In addition, proteins that contain heme can form different signaling molecules, enzymes and hormones (Derbyshire and Marletta, 2012).
1.1 Heme Biosynthesis
Heme is mainly generated in the mitochondria. It is formed by a series of reactions that starts with the condensation of succinyl coenzyme A and glycine, which results in the formation of the compound δ-aminolevulinic acid (ALA). This reaction is mediated by the rate-limiting enzyme, ALA synthase (ALAS) (Bonkovsky et al., 2013). ALA is transported from the mitochondria to the cytoplasm, where it is converted into coproporphyrinogen III (CP) through additional enzymatic reactions. It is then imported back to the mitochondria, in which CP forms protoporphyrin IX (PPIX). Iron is then inserted into PPIX by ferrochelatase (FECH). The newly generated heme may directly be used in the formation of respiratory chain proteins, or it may be transported into the cytosol via the feline leukemia virus subgroup C cellular receptor (FLVCR) (Chiabrando et al., 2012) for the formation of hemoglobin (Muckenthaler et al., 2017) (Figure 1).
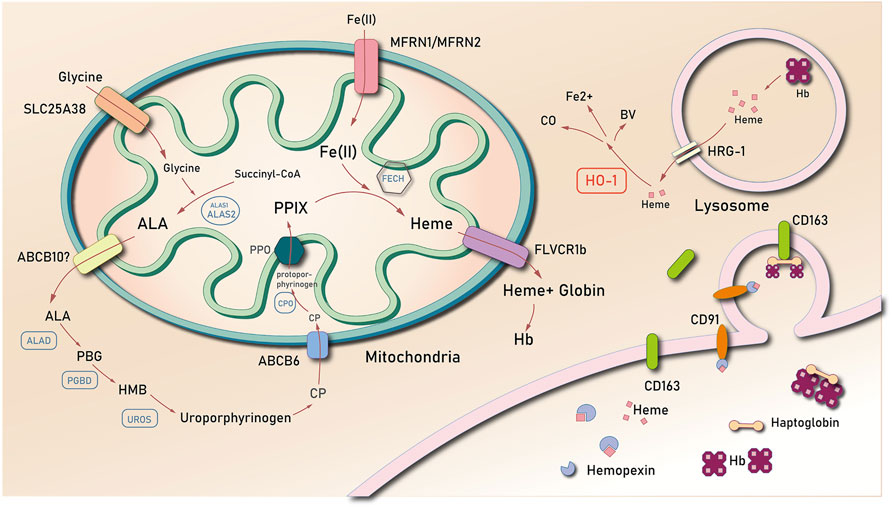
FIGURE 1. The biosynthesis and degradation of heme. Abbreviations: δ-aminolevulinic acid (ALA); coproporphyrinogen III (CP); protoporphyrin IX (PPIX); ferrochelatase (FECH); feline leukemia virus subgroup C cellular receptor (FLVCR); heme oxygenase 1 (HO-1); hemopexin (Hx); low-density lipoprotein receptor-related protein/CD91 (LDR/CD91); heme-responsive gene 1 (HRG-1); ATP binding cassette subfamily B member 6 (ABCB6); ATP binding cassette subfamily B member 10 (ABCB10); Mitoferrin-1 (Mfrn1); ALA dehydrase (ALAD); uroporphyrinogen III synthase (UROS); hydroxy-methyl bilane (HMB); porphobilinogen (PBG); PBG deaminase (PGBD); hemoglobin (Hb).
1.2 Heme Degradation
Heme is predominantly concentrated in the red blood cells (RBCs) of mammals. This implies that there is a higher proportion of heme degradation, which results from the liberation of hemoglobin from senescent erythrocytes. Heme is catabolized by the macrophages of the reticuloendothelial system (de Back et al., 2014). In most cases, senescent RBCs are phagocytosed by specialized macrophages in the spleen, liver, and bone marrow (de Back et al., 2014). Phagosomes carrying RBCs combine with lysosomal vesicles to form erythrocyte-containing lysosomes, wherein RBCs are degraded to release heme. Heme is then exported into the cytosol via the lysosome heme transporter, HRG1 (Rajagopal et al., 2008; Yuan et al., 2012). Notably, free heme is released into the cytoplasm, which when present in high concentrations, causes downregulation of Bach1, a mammalian heme-responsive transcription factor that suppresses the activation of the heme oxygenase 1 (HO-1) gene (Sassa, 2004). There are at least two functionally active heme oxygenase isozymes: HO-1 and HO-2. The former is inducible and is the first and rate-limiting enzyme of the heme degradation pathway, whereas the latter is relatively constitutive.
Heme is degraded into ferrous iron and equimolar amounts of biliverdin-IX-α and carbon monoxide. This reaction is catalyzed by heme oxygenase (Ponka et al., 2017). HO-1 catalyzes the cleavage of the alpha-methylene linked carbon on the heme molecule to release the products. The body therefore maintains free heme concentrations at an extremely low level that is undetectable in the plasma of healthy individuals. Furthermore, the concentration of labile hemin is maintained to as low as 20–40 nM in the cytosol of Saccharomyces cerevisiae (Hanna et al., 2018) and to even lower concentrations in the mitochondria and nucleus (<2.5 nM) (Hanna et al., 2016).
During intravascular hemolysis, macrophages phagocytose free heme. This process is mediated by hemopexin (Hx), a circulating latent plasma carrier protein mainly synthesized in the liver with an extremely high affinity to heme (Nielsen et al., 2010). As a heme scavenger, Hx binds with heme, which results in the increased solubility of heme. The soluble heme diminishes its oxidative activity (Eskew et al., 1999). The Hx-heme complex combines with low-density lipoprotein receptor-related protein/CD91 (LDR/CD91), a transmembrane protein expressed in many types of cells, including macrophages, fibroblasts, hepatocytes and adipocytes, adsorbing the Hx-heme complex from the circulation (Moestrup et al., 1992; Hvidberg et al., 2005) and mediating endocytosis of the complex. When the Hx-Heme complex is degraded in the lysosome, LRP/CD91 dissociates from the complex and is then recycled to the cell membrane. LRP/CD91 might be the key protein to import the Hx-heme complex since the heme-induced increasement of HO-1 mRNA transcription was LRP/CD91-dependent (Hvidberg et al., 2005).
1.3 Heme Transporters
There have been several studies on heme transmembrane transport, which have resulted in the discovery of a number of potential heme transporters. In 2005, Shayeghi et al. (2005) identified a transmembrane protein called SLC46A1, a heme carrier protein 1 (HCP1). HCP1 is highly localized in the apical membrane of duodenal enterocytes and is found to promote heme absorption in the intestines (Shayeghi et al., 2005; Chung et al., 2012). The intracellular location of HCP1 makes it sensitive to changes in iron stores. Most HCP1 proteins are localized in the membranes of the brush borders of duodenal enterocytes under iron-deficient conditions. Contrastingly, HCP1 proteins retreat into the cytoplasm in iron-loaded circumstances (Shayeghi et al., 2005). However, Qiu et al. (2006) demonstrated that HCP1, which is previously thought to carry heme, is a proton-coupled folate transporter (PCFT) (Qiu et al., 2006; Ponka et al., 2017). According to the results of the Km test performed by Qiu et al. (2006), the affinity of methyl tetrahydrofolate to SLC46A1 is higher than that of heme (Qiu et al., 2006; Andrews, 2007). Studies have shown that in families with hereditary folate malabsorption whose SLC46A1 gene carries a deficient mutation, the affected children are required to take high doses of supplemental folate to thrive. However, these children have no apparent defect in iron metabolism. These findings implicate SLC46A1 as a folate transporter rather than a heme carrier protein (Qiu et al., 2006; Andrews, 2007).
The heme-responsive gene 1 (HRG-1) is a conserved, membrane-bound permease that binds with and translocates heme in Caenorhabditis elegans and humans (Yuan et al., 2012). For instance, Rajagopal et al. (2008) found that the reactive high expression of HRG-1 in a low heme environment promotes the importation of heme in C. elegans. The mammalian homolog of HRG-1, which is expressed in various tissues, including the brain, heart, and kidney, is also a heme transporter from the extracellular space (Sawicki et al., 2015a). Delaby et al. (2012) found that HRG-1 proteins transport heme to the cytoplasm from lysosome. HRG-1 proteins are recruited to the erythrophagosome during erythrophagocytosis, while heme is derived from the digestion of ingested hemoproteins in the lysosome (Rajagopal et al., 2008; Yuan et al., 2012).
Feline leukemia virus subgroup C receptor (FLCRV1/2), a member of the SLC49 family, is a cell surface transporter that is crucial for erythrocyte generation and intracellular iron homeostasis (Duffy et al., 2010; Khan and Quigley, 2013). FLCRV1 acts as a heme exporter. The dysfunction of FLVCR1 arises from a mutation that impedes the development of erythroid progenitors due to heme toxicity, thereby resulting in Diamond-Blackfan anemia (Khan and Quigley, 2013). Notably, there are two different isoforms of FLVCR1. FLVCR1a is expressed on the plasma membrane, whereas FLVCR1b is expressed only in the mitochondria. Heme is exported by extracellular heme-binding proteins, such as albumin or hemopexin (Hpx), via FLVCR1a. Furthermore, the heme-export efficiency of FLVCR1a is directly proportional to the affinity of the extracellular protein for heme (Yang et al., 2010). The subcellular localization and function of FLVCR1b remain elusive. Contrastingly, FLVCR2 is more likely to mediate heme uptake (Duffy et al., 2010). Mammalian cells and Xenopus laevis oocytes that express FLVCR2 show an increase in heme uptake and a more sensitive response to heme toxicity. Heme toxicity is decreased after silencing FLVCR2 via siRNA or by binding the FLVCR2 receptor with the specific inhibitor called the FY981 FeLV envelope protein (Duffy et al., 2010). These observations suggest that FLVCR2 might induced toxicity through the import of excess heme.
1.4 Toxicity of Heme
Heme is poorly soluble in aqueous solution (Smith, 1975). The most widely used method to solubilize heme is to dissolve it in 0.001–0.01 M sodium hydroxide (Smith, 1975) or in dimethyl sulfoxide (DMSO) (Bohle et al., 2012). The oxidation state of iron in heme complexes is diverse, and the two most common are ferric protoporphyrin IX (Fe (III) complex), which is commonly known as hemin and hematin, and ferrous protoporphyrin IX (Fe(II) complex), which is commonly known as heme. In this review, heme is used as a general term to refer to either of the iron oxidation states. Free heme can be toxic at concentrations that exceed 1 µM because it promotes the formation of reactive oxygen species (ROS) (Sassa, 2004), oxidizes lipids and proteins, damages DNA (Tappel, 1953; Aft and Mueller, 1983; Aft and Mueller, 1984; Vincent, 1989; Chiabrando et al., 2014; Roumenina et al., 2016), and induces pro-inflammatory reactions in various cells. It is also suggested that heme catalyzes the production of hydroxyl radicals through the Fenton reaction in the same way as that of free Fe2+ (Sadrzadeh et al., 1984). However, based on the ESR spin trapping technique, Schmitt et al. (1993) found that heme likely reacts with lipids and alkanes rather than with H2O2 and hydroxyl radicals. However, the hypothesis of the heme-induced Fenton reaction remains contradictory. In aqueous buffers under physiological conditions, heme forms a stable and inert oxygen-bridged dimer through an Fe-O-Fe linkage (Brown et al., 1969; Silver and Lukas, 1983). Further research supports that heme causes cellular oxidative damage by embedding in the phospholipid biological membrane and by inducing the catalytic oxidation of organic compounds (Liu et al., 1985; Schmitt et al., 1993). This review further discusses the mechanism by which heme affects cardiovascular diseases.
While heme causes cytotoxicity in various ways, it is a strong inducer of heme oxygenase-1 (HO-1), a protein generally considered cytoprotective against oxidative injury and other cellular stresses in various disease settings (Ortiz de Montellano, 2000). Carbon monoxide (CO) and biliverdin (BV) are the by-products of heme degradation via heme oxygenase. They control cellular processes such as inflammation, apoptosis, and antioxidant defense (Aft and Mueller, 1983; Stocker et al., 1987; Jansen et al., 2010; Jansen and Daiber, 2012; Almeida et al., 2015). For instance, HO-1 and its by-products play a protective role against the progression of atherosclerosis through the inhibition of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB activation), which results in the attenuation of tumor necrosis factor (TNF)-α-induced upregulation of vascular cell adhesion molecule 1 (VCAM-1) and E-selectin in endothelial cells (ECs) and in the decreased expression of TNF-α, interleukin (IL)-1β, and macrophage inflammatory protein-1β in macrophages (Wu et al., 2011; Barbagallo et al., 2013; Cheng et al., 2014; Fredenburgh et al., 2015). Simultaneously, Fe2+ generated from heme degradation increases the intracellular labile iron pool (LIP), thus leading to iron-catalyzed ROS (Kakhlon and Cabantchik, 2002; Yuan et al., 2004; Lane et al., 2015). In response to increasing LIP, cells rapidly express ferritin, an intracellular iron storage protein with ferroxidase activity and iron sequestration capability (Clegg et al., 1980; Lane et al., 2015; Hagen et al., 2017). Therefore, in various diseases and in different research models, determining whether heme is harmful or protective is complicated. Its harmful and protective effects may be closely related to its concentration, time, and the environmental conditions in which it is exposed to. The protective effects of HO-1 and heme metabolites are not discussed in detail in this review.
2 HEME AND ATHEROSCLEROSIS
2.1 Heme Accumulation in Atherosclerosis
The atherosclerotic plaques, especially the necrotic core of unstable plaques, contain apoptotic macrophages, erythrocytes and its metabolites (heme and hemoglobin), and cytotoxic substances (cholesterol crystals, cholesterol esters, oxidized lipids, fibrin, inorganic mineral-like hydroxyapatite, iron, and calcium) (Li et al., 2006). In the early 80 s, studies have demonstrated that there is a complicated microvascular network that has extended from the adventitia into the thickened intima. These neovessels have incomplete endothelial lining and have no adhesion and support of smooth muscle cells. They are too weak to maintain their integrity, thereby leading to leaks and recurrent hemorrhages (Virmani et al., 1998). Kolodgie et al. (2003) found that plaques with cores or thin caps in the late stage of necrosis have a marked increase in glycophorin A, an erythrocyte surface antigen, and iron deposits in regions of cholesterol clefts compared with those in the early stage of necrosis. It has also been suggested that RBCs and their related metabolites enter the plaques through fissures on the surface of atherosclerotic lesions (Nagy et al., 2010). According to the histological examination of atherosclerotic carotid lesions collected from patients undergoing carotid endarterectomy, Hellings et al. (2010) demonstrated that an increase in neovascularization and intraplaque hemorrhage is correlated with adverse clinical cardiovascular outcomes. However, this increase is independent of other clinical risk factors and medications, such as previous vascular intervention, bilateral carotid stenosis, hypertension, statin and dipyridamole use, and C-reactive protein and high-density lipoprotein levels (Hellings et al., 2010). Intraplaque neovascularization and intraplaque hemorrhages are crucial processes in the conversion of plaques from stable to unstable. Numerous studies have shown that RBCs, RBC membranes, hemoglobin, heme, iron, and other RBC products that enter the plaque contribute to plaque inflammation and toxicity (Nagy et al., 2010). In this review, we elaborate the relevant mechanisms induced by heme.
The prosthetic group of heme is tightly bound to hemoglobin. This bond is weakened in oxidized forms of hemoglobin (Hrkal et al., 1974). Both methemoglobin (metHb, HbFe3+αβ) and ferryl hemoglobin (HbFe4+αβ) release heme, which is captured by albumin, α1-microglobulin, and lipoproteins such as low-density lipoprotein (LDL) and high-density lipoprotein (HDL) in the absence of the plasma heme scavenger Hx (Chistiakov et al., 2015). When exposed to oxidized atherosclerotic plaque materials, erythrocytes are lysed; the liberated hemoglobin is oxidized; and heme dissociates from the oxidized hemoglobin (Nagy et al., 2010).
2.2 Impact of Heme in Atherosclerosis
Heme is a well-known pro-oxidant (Sadrzadeh et al., 1984). The lipophilic properties of heme enable its combination with low reactive organics (ROOH), facilitate the production of large amounts of highly reactive alkoxyl (RO•) and peroxyl (ROO•) radicals (Van der Zee et al., 1996), and ultimately stimulate the autocatalytic lipid peroxidation cascades, which have a key role in the formation of atherosclerotic lesions. Aside from increasing the production of oxidized LDLs (ox-LDLs), heme also stimulates nicotinamide adenine dinucleotide phosphate oxidase (NADPHox)-dependent ROS generation in a protein kinase C (PKC)-dependent manner in a variety of cell types (Arruda et al., 2006; Moraes et al., 2012). In neutrophils, NADPHox-derived ROS upregulate NF-κB activity and B-cell lymphoma-extra large [Bcl-X (L)] accumulation and inhibit Bax insertion into the mitochondria, thus resulting in pro-inflammatory and anti-apoptotic responses to neutrophils (Moraes et al., 2012).
In vascular smooth muscle cells (VSMCs), NADPHox-derived ROS activate redox-sensitive signaling pathways, such as ERK-2, leading to free-heme-induced concentration-dependent migration and proliferation of VSMCs (Moraes et al., 2012). However, several contrasting results suggest that heme-derived HO-1 overexpression and heme decomposition products, including carbon monoxide (CO) and biliverdin, can suppress the proliferation of VSMCs, especially when heme concentration is maintained in vitro at a low level (5 μM) for 7–21 days (Chang et al., 2008). Gáll et al. (2018) found that heme triggers ER stress in a time- and dose-dependent manner in human aortic smooth muscle cells (HAoSMCs) via all three endoplasmic reticulum (ER)-related classical pathways, which include activating transcription factor (ATF)4 expression, X Box Binding Protein 1 (XBP1) mRNA splicing, and ATF6 cleavage.
The effects of heme on endothelial cells have also been explored. In the 1990s, Balla et al. carried out many essential studies on the toxicity of heme to vascular endothelial cells. They found that heme exposure synergistically amplified the cytotoxic effects of oxidants in endothelial cells, exacerbating their damage by polymorphonuclear leukocytes, a kind of cells that tended to marginate along endothelial surfaces in the presence of inflammatory mediators (Balla et al., 1991a). Free heme released from ferri (FeIII)hemoglobin (also named methemoglobin) might involve in atherogenesis by spontaneous inserting into LDL particles and promoting oxidation of low-density lipoprotein (LDL) into cytotoxic oxidized products to endothelial cells (Balla et al., 1991b; Balla et al., 1995; Jeney et al., 2002). Substances that strengthened heme-globin liganding, like haptoglobin or cyanide could prevent heme release from ferrihemoglobin were proved to reduce the generation of oxidized LDL (Jeney et al., 2002). Interestingly, Balla et al. proved that ferrihemoglobin, but not ferro (FeII)hemoglobin, could increase the susceptibility of LDL to oxidative modification and amplify the cytotoxic effects of oxidants in endothelial cells (Balla et al., 2000), which was probably because ferrihemoglobin released its heme moieties much more readily than ferrohemoglobin according to Bunn and Jandl’s research in 1968 (Bunn and Jandl, 1968). Instead, LDL-associated lipid hydroperoxides was able to convert ferro hemoglobin to ferrihemoglobin in a dose-dependent manner (Nagy et al., 2005). Ferryl (FeIII/FeIV = O)hemoglobin, a kind of short-lived hemoglobin forming under exposure of extracellular reactive oxygen species or lipid hydroperoxide (Giulivi and Davies, 1994), was found to accumulate in the atherosclerotic lesion (Nagy et al., 2010) and activate the inflammatory response in the resident cells of the arterial wall (Silva et al., 2009), leading to increased endothelial cell permeability and enhanced monocyte adhesion (Silva et al., 2009; Potor et al., 2013). Latest research reported that even hemoglobin-derived peptide fragments of α and β subunits had effects similar to those of ferrylhemoglobin in atherosclerotic lesions, provoking endothelial dysfunction (Posta et al., 2020). Noticeably, FerrylHb specifically induced α1-microglobulin (A1M) secretion in aortic ECs, SMCs and macrophages, which was a radical-scavenging and heme-binding protein predominantly synthesized in the liver (Berggård et al., 1997). Studies showed A1M markedly inhibited Hb oxidation and heme-driven oxidation of LDL and plaque lipids derived from atheromas (Pethő et al., 2021).
The effects of heme exposure on endothelial cells was proved time-dependent dichotomous. Brief exposure to heme about 1 h produced an endothelium susceptibility to oxidant damage, a prolonged exposure to heme for about 16 increased heme oxygenase-1 and ferritin expression for approximately 50-fold and 10-fold respectively, which stimulated high resistance of endothelial cells to oxidant-mediated injury (Balla et al., 1992; Balla et al., 1993). Increased intracellular ferritin without any increment in heme oxygenase activity were also proved to raise marked protection against oxidant challenge in endothelial cells in a dose-responsive way, which was evidently attributable to the ferroxidase activity of the heavy (H) subunit, that catalyzes the oxidation of ferrous iron to ferric iron to allow intracellular iron storage in biological systems (Lawson et al., 1989; Harrison and Arosio, 1996). Studies revealed that abundant ferritin and up-regulation of heme oxygenase-1 occurred specifically in coronary atherosclerotic, which might reflect cellular response to heme or heme-iron-generated lipid peroxidation products in the atherosclerotic lesions (Juckett et al., 1995; Wang et al., 1998). In 2001, Ishikawa et al. revealed that heme-induced (intraperitoneal injections of hemin) HO-1 expression significantly decreased atherosclerotic lesions in LDL-receptor knockout mice fed high-fat diets (Ishikawa et al., 2001). It seems heme help alleviate coronary atherosclerotic in the long run. However more researches suggested that it was oxidized LDL instead of heme itself ascribed as the cause for the induction of heme oxygenase-1 and ferritin, since when catalyzing the oxidation of LDL, heme itself underwent degradation (Nath et al., 1995; Agarwal et al., 1996; Hill-Kapturczak et al., 2003). Wagener et al. (1997) found that the expression levels of ICAM-1, VCAM-1, and E-selectin proteins in vascular endothelial cells are consistently upregulated by heme. Erdei et al. (2018) demonstrated that heme, instead of protoporphyrin IX, induces ROS-mediated Nod-like receptor family pyrin domain containing 3 (NLRP3) inflammasome activation in human endothelial cells, which stimulates the secretion of active IL-1β. Weibel-Palade bodies (WPBs) are specific secretory organelles of endothelial cells. They contain the von Willebrand factor (VWF) and a variety of other proteins that are implicated in inflammation, neovascularization, and tissue recovery (Valentijn et al., 2011). Research (Belcher et al., 2014) involving transgenic sickle cell disease mice has shown that heme-mediated nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidase, protein kinase C (PKC), and oxidants activate endothelial toll-like receptor 4 (TLR4)/NF-κB signaling and trigger vaso-occlusion through WPB degranulation and adhesion molecule expression. Hemin significantly upregulates microvascular endothelium IL-8 secretion. This upregulation is independent from the oxidant-sensitive transcription factors, NF-κB or AP-1 (Natarajan et al., 2007).
Classically, macrophages are divided into two types: M1 and M2, which are generally considered pro-inflammatory and anti-inflammatory, respectively. Except for the two classical types of M1 and M2 macrophages, multiple phenotypes of macrophages have been reported in atherosclerotic plaques, such as Mhem. Boyle et al. (2009) defined a certain macrophage phenotype that is associated with hemorrhagic intraplaque hemorrhage (IPH), a hemorrhage-associated macrophage population (HA-mac), which is characterized by elevated expression of CD163 and HO-1, active secretion of IL-10, and low levels of human leukocyte antigen-DR (HLA-DR). Recently, they renamed it as Mhem (Boyle et al., 2012). Mhem exists only in hemorrhagic plaques, and the elevated expression levels of CD163 and HO-1 suggests that their function is to efficiently uptake and decompose hemoglobin and heme. Perl staining results have shown the accumulation of ferritin in Mhem. In addition, results of in vitro experiments have indicated that IL-10 also induces CD163 expression, which may form a positive feedback loop that can polarize macrophages in the presence of the Hb/Hp complex (Boyle et al., 2009). In another study, Mhem is also referred to as M hemoglobin (Finn et al., 2012). Compared to foam cells, Mhem has no lipid retention (Finn et al., 2012) but has lower ROS levels and stronger tolerance to oxidative stress in vitro (Boyle et al., 2012). Moreover, the upregulation of ATF1 increases lipid transport-related protein expression, such as liver X receptor (LXR)-β, LXR-α, and ATP-binding cassette transporter (ABCA) in HA-mac, which promotes cholesterol efflux (Boyle et al., 2012). Moreover, Mhem is considered as a type of conductive cell, which eliminates hemoglobin and heme, reduces oxidative stress, and prevents macrophages from transforming into foam cells.
However, Guo et al. (2018) showed that the expression of CD163 in macrophages induces the progression of atherosclerotic plaques. Due to the increase of ferritin in Mhem and the upregulated expression of iron-export protein, Ferroportin (FPN), the content of free iron in cells decreases significantly. This leads to a decrease in iron-dependent prolyl hydroxylase domain protein 2 (PHD2) activity that leads to the activation of the hypoxia-inducible factor (HIF)1α/vascular endothelial growth factor (VEGF)-A pathway. This series of events results in endothelial VCAM expression, monocyte recruitment, angiogenesis, and vascular permeability in plaques (Guo et al., 2018). Thus far, the effect of Mhem on atherosclerosis remains contradictory.
Notably, Mhem has only been reported in atherosclerosis. Currently, the reported subtypes of macrophages in atherosclerosis include M1, M2a, M2b, M2c, Mox, MHem, and M4. Potor et al. explored the pathophysiologic role of oxidation of hemoglobin (Hb) to ferrylHb in macrophages of atherosclerosis recently. They reported that macrophages exposed to ferrylHb in atherosclerotic plaques exhibited a distinct transcriptomic profile affecting gene expressions associated with inflammation (IL-1β and TNF-α et), angiogenesis, tissue remodeling, iron metabolism, apoptosis, PI3K signaling, lipid transport, and calcification, thereby contributing to the transformation of atherosclerotic lesions toward aproatherogenic phenotype (Potor et al., 2021).
Many of the proinflammatory effects of heme have been associated with activation of TLR4 signaling in macrophages (Figueiredo et al., 2007). Amino acids W23 and Y34 on myeloid differentiation factor-2 (MD-2) has recently been identified as a heme binding site in activation of TLR4 signaling (Belcher et al., 2020). Belcher et al. reveal that heme toxicity triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in experimental SCD mouse models, which exhibited typical signs of vascular inflammation (Belcher et al., 2003). Dutra et al. found that heme triggered the processing and secretion of IL-1β dependently on NLRP3 inflammasome, inducing lethality caused by sterile hemolysis (Dutra et al., 2014).
3 MYOCARDIAL DAMAGE OF HEME
In the 1990s, Bhoite-Solomon et al. (1990), Alvarado et al. (2015) discovered an association between hemin and myosin in vitro. Heme-injured myocytes exhibit morphological changes, reduce beating rates, and increases enzymes losses (Jeney et al., 2002). Other studies have suggested that heme modifies cardiac contractile proteins through posttranslational protein modifications by consolidating with myosin light chain 1, which results in serious contractile heart dysfunction (Alvarado et al., 2015). Heme exposure alters cardiomyocyte morphology and evokes a decrease in Ca2+ sensitivity (pCa50) (Bunn and Jandl, 1968), thereby causing a decrease in the contractile function of the human heart. In hemolytic mice, free heme accumulates in the heart, which drives ROS to affect Ca2+ homeostasis, and impairs systolic function (Ingoglia et al., 2017). In vitro, heme-treated adult rat cardiomyocytes have shown a significant reduction in systolic Ca2+ transient amplitudes (de Back et al., 2014). A possible mechanism is that heme-derived ROS may have directly modified Ca2+-handling proteins, such as the ryanodine receptor2 (RyR2) and the sarcoendoplasmic reticulum Ca2+-ATPase 2a (SERCA2a) (Köhler et al., 2014), and may have activated intracellular stress kinases, such as calcium/calmodulin-dependent protein kinase II (CaMKII) (Erickson et al., 2008), which phosphorylates RyR2 and exacerbates Ca2+ mishandling. Heme also induces contractile dysfunction in human cardiomyocytes caused by the oxidation of myofilament proteins (Alvarado et al., 2015).
These pathological processes can be instrumental in cardiac dysfunction in the form of myocardial ischemia-reperfusion injury, heart failure, and hemolytic diseases. In a murine model for hemolytic diseases, application of the heme-binding protein, hemopexin, reduces ROS generation and restores myocardial function (Vinchi et al., 2013), which suggests that heme is associated with systolic and diastolic heart dysfunction in hemolytic diseases such as sickle cell disease and thalassemia (Fukuda et al., 2001; Sachdev et al., 2007; Gladwin and Sachdev, 2012; Stoyanova et al., 2012).
Khechaduri et al. found δ-aminolevulinic acid synthase 2 (ALAS2), the rate-limiting enzyme in heme production, is upregulated in human failing hearts, leading to increased heme levels in cardiac myoblasts (Khechaduri et al., 2013). Sansbury et al. (2014) conducted a transverse aortic constriction (TAC) or permanent coronary occlusion (MI) in male C57BL/6J mice and has found that after 1 week of TAC and 5 days after MI, a significant increase in ascorbate, heme, and other indices of oxidative stress are observed in a metabolomic analysis of infarcted mouse hearts. Further studies have shown that due to heme-induced mitochondrial oxidative stress, the present ALAS2 transgenic mice has exhibited an increase in heme accumulation, aggravation of cell death, and exacerbation of cardiac dysfunction after coronary ligation compared with the control littermates (Sawicki et al., 2015b). In addition, the knockdown of ALAS2 in cultured cardiomyoblasts exposed to hypoxia has reversed the increase in heme production and cell death (Fredenburgh et al., 2015).
4 HEME AND DEGENERATIVE AORTIC VALVE STENOSIS
Aortic valve stenosis is characterized by a slowly progressive fibrocalcific remodeling of the valve leaflets (Rajamannan et al., 2011; Lindman et al., 2016). In the early stage of the disease, known as aortic sclerosis (AS), the valve becomes thickened and mildly calcified. Nevertheless, there is no obstruction of blood flow. Over the years, the disease develops into severe valve calcification with abnormal leaflet motion, which severely obstructs blood flow. This corresponds to calcified aortic valve stenosis (Rajamannan et al., 2007; Lindman et al., 2016). Numerous studies have shown that DAVS and atherosclerotic disease share a similar pathophysiology, which includes lipid oxidation, angiogenesis, and internal hemorrhage (Zhong et al., 2019; de Oliveira Sá et al., 2020). Aortic valve leaflets obtained from aortic valve replacement in patients with DAVS, often revealed intraleaflet hemorrhage in the valve leaflets of AS. The area of intraleaflet hemorrhage is positively related to the depth of 4-Hydroxynonenal (4-HNE) staining, which suggests oxidative tissue damage that is conducive for the development of DAVS. Oxidation of LDL in the stenotic valve may also be aggravated by the presence of hemoglobin, heme, and iron (Olsson et al., 1999; Akahori et al., 2011). In addition to intraleaflet hemorrhage, another form of intravalvular hematoma has been discovered. Morvan et al. (2019) revealed that intraleaflet iron accumulation precedes calcium deposition. Furthermore, RBCs enter the fibrosa secondary to endothelial microfissuring. The contact of valvular interstitial cells (VICs) with RBCs contributes to the osteoblastic phenotype and systematic inflammation, which are manifestations of upregulated IL-6, IL-1β, osteoprotegerin, bone sialoprotein, NF-κB, bone morphogenetic proteins (BMP)2, and muscle segment homeobox 2 (Morvan et al., 2019). However, a recent study suggests that heme-mediated activation of the Nrf2/HO-1 pathway inhibits the calcification of VICs in vitro, a contradiction to an earlier report (Balogh et al., 2021).
5 CARDIAC IRON OVERLOAD
Thalassemia, sickle cell anemia, primary hemochromatosis, and other diseases that require long-term repeated blood transfusions or iron supplements can lead to cardiac iron overload (Murphy and Oudit, 2010; Vaziri, 2012; Powell et al., 2016). Primary hemochromatosis is caused by mutations in genes that encode iron-transporting and iron-sensing proteins that disrupt the production of hepcidin. In the absence of the suppressive action of hepcidin, free iron in circulation continues to accumulate and eventually reaches the binding capacity of transferrin and ferritin. These result in the appearance of non-transferrin bound iron (NTBI) or free iron. In thalassemia and sickle cell anemia, a large number of RBCs and their metabolites in circulation are degraded by the liver and spleen. The degradative process releases excessive iron in the form of NTBI to circulation. NTBI is an unstable combination of excessive iron ions, various small molecules, such as citrate and acetate, and albumins in circulation (Grootveld et al., 1989). Therefore, the main cause of the decline in cardiac function in these patients is NTBI rather than hemoglobin. Fe2+ from NTBI is absorbed into cardiomyocytes through bivalent transporters, such as L-type Ca channels, ZIP8, and ZIP14 in the heart, thus inducing oxidative stress (Liuzzi et al., 2006; Kumfu et al., 2012; Wang et al., 2012; Kumfu et al., 2016). ROS production leads to suppressed SERCA2 function and contractive dysfunction. Lipid peroxidation also attacks the mitochondrial membrane, thereby leading to energy depletion, mitochondrial DNA damage and dysfunction (Das et al., 2015; Zhabyeyev and Oudit, 2017; Gordan et al., 2018). Theoretically, excessive intracellular Fe2+ may catalyze the cascade of lipid oxidation, which leads to ferroptosis (Shayeghi et al., 2005). However, there is still no precise evidence on the role of ferroptosis in cardiac iron overload.
A compared normo-ferremic ApoE−/− mice with iron-loaded ApoE−/− FPNwt/C326S mice reported that atherosclerosis can be exacerbated by iron-induced alterations in lipids, vascular permeabilization, sustained endothelial activation, elevated pro-atherogenic inflammatory mediators, and reduced nitric oxide availability. In vitro, NTBI induces ROS production and apoptosis in cultured vascular cells and stimulates MCP-1-mediated monocyte recruitment, which contributes to atherosclerosis (Vinchi et al., 2020). However, it has been reported that patients with hereditary hemochromatosis have a lower prevalence of cardiovascular disease (Demetz et al., 2020). This is because the deficiency of the haemochromatosis gene HFE increases LDL receptor expression in hepatocytes so that Kupffer cells are able to transfer more LDL-derived cholesterol to hepatocytes from circulation. Therefore, patients with hemochromatosis have a lower level of LDL-C and are less likely to develop atherosclerosis (Demetz et al., 2020).
Gbotosho et al. recently suggested that heme induced IL-6 expression and cardiac hypertrophy in patients and mice with sickle cell disease (SCD) (Gbotosho et al., 2020). Heme-laden erythrocyte membrane microparticles triggered rapid vaso-occlusions in kidneys and compromised microvascular dilation (Camus et al., 2015). Patrolling monocytes, which normally scavenge damaged cells and debris from the vasculature, express higher levels HO-1, protected SCD vasculature from vaso-occlusion (Liu et al., 2018). Although Hmox1 is usually regarded as a protective protein, Menon et al. revealed that increased heme in SCD causes upregulation of Hmox1 which consequently drives cardiomyopathy through ferroptosis (Menon et al., 2021).
6 Heme-related ferroptosis in cardiovascular diseases
Ferroptosis, an iron-dependent form of nonapoptotic cell death known as glutathione depletion and the amplification of lipid peroxidation, was first identified in 2012 (Dixon et al., 2012). To date, it has been reported in several cardiac pathologies, including doxorubicin-induced cardiomyopathy, acute I/R injury (Fang et al., 2019; Feng et al., 2019), post-myocardial infarction, heart failure (Park et al., 2019), atherosclerosis (Bai et al., 2020) and septic heart injury (Wang et al., 2020) (Figure 2). In acute I/R injury, post-myocardial infarction, early-stage heart failure, the presence of residual myocardial iron in the post-infarcted area, and GPX4 activity decrease, which results in ferroptosis in cardiac myocytes. Menon et al. revealed that increased heme in SCD caused upregulation of Hmox1 and free iron, which consequently drives cardiomyopathy through ferroptosis (Menon et al., 2021). Inhibition or induction of Hmox1 decreased or increased cardiac ferroptosis in SCD mice, respectively (Menon et al., 2021). In DOX-induced cardiomyopathy, the Nrf2/Hmox1 pathway mediates heme degradation and releases free iron, resulting in ferroptosis (Fang et al., 2019). Ferrastin-1 decreases iron content and lipid peroxidation and upregulates the levels of SLC7A11 and GPX4, which alleviates atherosclerotic lesions (Bai et al., 2020).
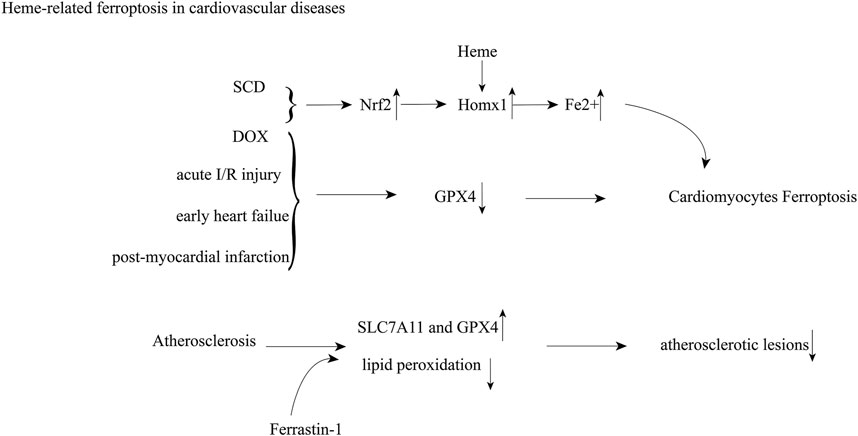
FIGURE 2. Heme-induced cardiovascular damage through ferroptosis; Abbreviations: sickle cell disease (SCD); doxorubicin (DOX); nuclear factor erythroid 2-related factor 2 (Nrf2); heme oxygenase 1 (Homx1); glutathione peroxidase 4 (GPX4); ischemia-reperfusion (I/R).
7 Conclusion
Heme is an ancient and indispensable biomolecule in aerobic organisms. It has evolved with a wide range of complex functions and metabolism in the human body (Celis and DuBois, 2019; Shimizu et al., 2019). Imbalance in heme metabolism plays an important role in cardiovascular diseases. Heme is a metabolic intermediate, of hemoglobin, red blood cells upstream in biosynthesis and carbon monoxide, biliverdin, and divalent iron downstream in the catabolism of RBC. Therefore, discussions on heme may not be comprehensive without considering these components. Heme leads to phenotypic changes of endothelial cells, macrophages, vascular smooth muscle cells and cardiomyocytes through oxidative stress, verification activation and other pathways. Most of these changes are harmful, but some studies have shown that prolonged exposure to a small amount of heme can induce antioxidant or anti-inflammatory phenotypes in cells. Heme binding proteins, like AIM and Hx, show their potential in treating heme-induced cardiovascular diseases, which may provide therapeutic opportunities to related cardiovascular diseases. The effects of heme on blood vessels, myocardium, and heart valves need to be further investigated.
Author Contributions
YG contributed manuscript preparation and wrote the manuscript and TY contributed to the revision of this manuscript. QZ contributed to the conception of the study and revised the manuscript provided guidance to the manuscript.
Funding
This work was partly supported by the National Natural Science Foundation of China (Grant Numbers 82070403 and 81770386), the Frontier Research Program of Guangzhou Regenerative Medicine and Health Guangdong Laboratory (Grant Number 2018GZR110105001), the Science and Technology Program of Guangdong Province (Grant Number 2021A0505030031) and the Youth Science and Technology Innovation Talent of Guangdong TeZhi Plan (Grant Number 2019TQ05Y136).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Reference
Aft, R. L., and Mueller, G. C. (1983). Hemin-mediated DNA Strand Scission. J. Biol. Chem. 258 (19), 12069–12072. doi:10.1016/s0021-9258(17)44341-9
Aft, R. L., and Mueller, G. C. (1984). Hemin-mediated Oxidative Degradation of Proteins. J. Biol. Chem. 259 (1), 301–305. doi:10.1016/s0021-9258(17)43657-x
Agarwal, A., Balla, J., Balla, G., Croatt, A. J., Vercellotti, G. M., and Nath, K. A. (1996). Renal Tubular Epithelial Cells Mimic Endothelial Cells upon Exposure to Oxidized LDL. Am. J. Physiol. 271 (4 Pt 2), F814–F823. doi:10.1152/ajprenal.1996.271.4.F814
Akahori, H., Tsujino, T., Naito, Y., Matsumoto, M., Lee-Kawabata, M., Ohyanagi, M., et al. (2011). Intraleaflet Haemorrhage Is Associated with Rapid Progression of Degenerative Aortic Valve Stenosis. Eur. Heart J. 32 (7), 888–896. doi:10.1093/eurheartj/ehq479
Almeida, A. S., Figueiredo-Pereira, C. u., and Vieira, H. L. A. (2015). Carbon Monoxide and Mitochondriaâ€"modulation of Cell Metabolism, Redox Response and Cell Death. Front. Physiol. 6, 33. doi:10.3389/fphys.2015.00033
Alvarado, G., Jeney, V., Tóth, A., Csősz, É., Kalló, G., Huynh, A. T., et al. (2015). Heme-induced Contractile Dysfunction in Human Cardiomyocytes Caused by Oxidant Damage to Thick Filament Proteins. Free Radic. Biol. Med. 89, 248–262. doi:10.1016/j.freeradbiomed.2015.07.158
Andrews, N. C. (2007). When Is a Heme Transporter Not a Heme Transporter? when It's a Folate Transporter. Cell Metab. 5 (1), 5–6. doi:10.1016/j.cmet.2006.12.004
Arruda, M. A., Barcellos-de-Souza, P., Sampaio, A. L. F., Rossi, A. G., Graça-Souza, A. V., and Barja-Fidalgo, C. (2006). NADPH Oxidase-Derived ROS: Key Modulators of Heme-Induced Mitochondrial Stability in Human Neutrophils. Exp. Cell Res. 312 (19), 3939–3948. doi:10.1016/j.yexcr.2006.08.022
Bai, T., Li, M., Liu, Y., Qiao, Z., and Wang, Z. (2020). Inhibition of Ferroptosis Alleviates Atherosclerosis through Attenuating Lipid Peroxidation and Endothelial Dysfunction in Mouse Aortic Endothelial Cell. Free Radic. Biol. Med. 160, 92–102. doi:10.1016/j.freeradbiomed.2020.07.026
Balla, G., Vercellotti, G. M., Muller-Eberhard, U., Eaton, J., and Jacob, H. S. (1991). Exposure of Endothelial Cells to Free Heme Potentiates Damage Mediated by Granulocytes and Toxic Oxygen Species. Lab. Invest. 64 (5), 648–655.
Balla, G., Jacob, H. S., Balla, J., Rosenberg, M., Nath, K., Apple, F., et al. (1992). Ferritin: a Cytoprotective Antioxidant Strategem of Endothelium. J. Biol. Chem. 267 (25), 18148–18153. doi:10.1016/s0021-9258(19)37165-0
Balla, G., Jacob, H. S., Eaton, J. W., Belcher, J. D., and Vercellotti, G. M. (1991). Hemin: a Possible Physiological Mediator of Low Density Lipoprotein Oxidation and Endothelial Injury. Arterioscler Thromb. 11 (6), 1700–1711. doi:10.1161/01.atv.11.6.1700
Balla, J., Nath, K. A., Balla, G., Juckett, M. B., Jacob, H. S., and Vercellotti, G. M. (1995). Endothelial Cell Heme Oxygenase and Ferritin Induction in Rat Lung by Hemoglobin In Vivo. Am. J. Physiol. 268 (2 Pt 1), L321–L327. doi:10.1152/ajplung.1995.268.2.L321
Balla, J., Balla, G., Jeney, V., Kakuk, G., Jacob, H. S., and Vercellotti, G. M. (2000). Ferriporphyrins and Endothelium: a 2-edged Sword-Promotion of Oxidation and Induction of Cytoprotectants. Blood 95 (11), 3442–3450. doi:10.1182/blood.v95.11.3442.011k51_3442_3450
Balla, J., Jacob, H. S., Balla, G., Nath, K., Eaton, J. W., and Vercellotti, G. M. (1993). Endothelial-cell Heme Uptake from Heme Proteins: Induction of Sensitization and Desensitization to Oxidant Damage. Proc. Natl. Acad. Sci. 90 (20), 9285–9289. doi:10.1073/pnas.90.20.9285
Balogh, E., Chowdhury, A., Ababneh, H., Csiki, D. M., Tóth, A., and Jeney, V. (2021). Heme-Mediated Activation of the Nrf2/HO-1 Axis Attenuates Calcification of Valve Interstitial Cells. Biomedicines 9 (4). doi:10.3390/biomedicines9040427
Barbagallo, I., Galvano, F., Frigiola, A., Cappello, F., Riccioni, G., Murabito, P., et al. (2013). Potential Therapeutic Effects of Natural Heme Oxygenase-1 Inducers in Cardiovascular Diseases. Antioxid. Redox Signaling 18 (5), 507–521. doi:10.1089/ars.2011.4360
Belcher, J. D., Bryant, C. J., Nguyen, J., Bowlin, P. R., Kielbik, M. C., Bischof, J. C., et al. (2003). Transgenic Sickle Mice Have Vascular Inflammation. Blood 101 (10), 3953–3959. doi:10.1182/blood-2002-10-3313
Belcher, J. D., Chen, C., Nguyen, J., Milbauer, L., Abdulla, F., Alayash, A. I., et al. (2014). Heme Triggers TLR4 Signaling Leading to Endothelial Cell Activation and Vaso-Occlusion in Murine Sickle Cell Disease. Blood 123 (3), 377–390. doi:10.1182/blood-2013-04-495887
Belcher, J. D., Zhang, P., Nguyen, J., Kiser, Z. M., Nath, K. A., Hu, J., et al. (2020). Identification of a Heme Activation Site on the MD-2/TLR4 Complex. Front. Immunol. 11, 1370. doi:10.3389/fimmu.2020.01370
Berggård, T., Thelin, N., Falkenberg, C., Enghild, J. J., and Akerström, B. (1997). Prothrombin, Albumin and Immunoglobulin A Form Covalent Complexes with Alpha1-Microglobulin in Human Plasma. Eur. J. Biochem. 245 (3), 676–683. doi:10.1111/j.1432-1033.1997.00676.x
Bhoite-Solomon, V., Kessler-Icekson, G., and Shaklai, N. (1990). Association of Iron-Protoporphyrin-IX (Hemin) with Myosins. FEBS Lett. 266 (1-2), 9–12. doi:10.1016/0014-5793(90)81493-8
Bohle, D. S., Dodd, E. L., and Stephens, P. W. (2012). Structure of Malaria Pigment and Related Propanoate-Linked Metalloporphyrin Dimers. Chem. Biodiversity 9 (9), 1891–1902. doi:10.1002/cbdv.201200033
Bonkovsky, H. L., Guo, J. T., Hou, W., Li, T., Narang, T., and Thapar, M. (2013). Porphyrin and Heme Metabolism and the Porphyrias. Compr. Physiol. 3 (1), 365–401. doi:10.1002/cphy.c120006
Boyle, J. J., Harrington, H. A., Piper, E., Elderfield, K., Stark, J., Landis, R. C., et al. (2009). Coronary Intraplaque Hemorrhage Evokes a Novel Atheroprotective Macrophage Phenotype. Am. J. Pathol. 174 (3), 1097–1108. doi:10.2353/ajpath.2009.080431
Boyle, J. J., Johns, M., Kampfer, T., Nguyen, A. T., Game, L., Schaer, D. J., et al. (2012). Activating Transcription Factor 1 Directs Mhem Atheroprotective Macrophages through Coordinated Iron Handling and Foam Cell protection. Circ. Res. 110 (1), 20–33. doi:10.1161/circresaha.111.247577
Brown, S. B., Jones, P., and Lantzke, I. R. (1969). Infrared Evidence for an Oxo-Bridged (Fe-O-Fe) Haemin Dimer. Nature 223 (5209), 960–961. doi:10.1038/223960a0
Bunn, H. F., and Jandl, J. H. (1968). Exchange of Heme Among Hemoglobins and between Hemoglobin and Albumin. J. Biol. Chem. 243 (3), 465–475. doi:10.1016/s0021-9258(18)93628-8
Camus, S. M., De Moraes, J. A., Bonnin, P., Abbyad, P., Le Jeune, S., Lionnet, F., et al. (2015). Circulating Cell Membrane Microparticles Transfer Heme to Endothelial Cells and Trigger Vasoocclusions in Sickle Cell Disease. Blood 125 (24), 3805–3814. doi:10.1182/blood-2014-07-589283
Celis, A. I., and DuBois, J. L. (2019). Making and Breaking Heme. Curr. Opin. Struct. Biol. 59, 19–28. doi:10.1016/j.sbi.2019.01.006
Chang, T., Wu, L., and Wang, R. (2008). Inhibition of Vascular Smooth Muscle Cell Proliferation by Chronic Hemin Treatment. Am. J. Physiology-Heart Circulatory Physiol. 295 (3), H999–h1007. doi:10.1152/ajpheart.01289.2007
Cheng, S., Enserro, D., Xanthakis, V., Sullivan, L. M., Murabito, J. M., Benjamin, E. J., et al. (2014). Association of Exhaled Carbon Monoxide with Subclinical Cardiovascular Disease and Their Conjoint Impact on the Incidence of Cardiovascular Outcomes. Eur. Heart J. 35 (42), 2980–2987. doi:10.1093/eurheartj/ehu052
Chiabrando, D., Marro, S., Mercurio, S., Giorgi, C., Petrillo, S., Vinchi, F., et al. (2012). The Mitochondrial Heme Exporter FLVCR1b Mediates Erythroid Differentiation. J. Clin. Invest. 122 (12), 4569–4579. doi:10.1172/jci62422
Chiabrando, D., Vinchi, F., Fiorito, V., Mercurio, S., and Tolosano, E. (2014). Heme in Pathophysiology: a Matter of Scavenging, Metabolism and Trafficking across Cell Membranes. Front. Pharmacol. 5, 61. doi:10.3389/fphar.2014.00061
Chistiakov, D. A., Orekhov, A. N., and Bobryshev, Y. V. (2015). Contribution of Neovascularization and Intraplaque Haemorrhage to Atherosclerotic Plaque Progression and Instability. Acta Physiol. 213 (3), 539–553. doi:10.1111/apha.12438
Chung, J., Chen, C., and Paw, B. H. (2012). Heme Metabolism and Erythropoiesis. Curr. Opin. Hematol. 19 (3), 156–162. doi:10.1097/moh.0b013e328351c48b
Clegg, G. A., Fitton, J. E., Harrison, P. M., and Treffry, A. (1980). Ferritin: Molecular Structure and Iron-Storage Mechanisms. Prog. Biophys. Mol. Biol. 36 (2-3), 56–86.
Das, S. K., Wang, W., Zhabyeyev, P., Basu, R., McLean, B., Fan, D., et al. (2015). Iron-overload Injury and Cardiomyopathy in Acquired and Genetic Models Is Attenuated by Resveratrol Therapy. Sci. Rep. 5, 18132. doi:10.1038/srep18132
de Back, D. Z., Kostova, E. B., van Kraaij, M., van den Berg, T. K., and van Bruggen, R. (2014). Of Macrophages and Red Blood Cells; a Complex Love story. Front. Physiol. 5, 9. doi:10.3389/fphys.2014.00009
de Oliveira Sá, M. P. B., Cavalcanti, L. R. P., Perazzo, Á. M., Gomes, R. A. F., Clavel, M. A., Pibarot, P., et al. (2020). Calcific Aortic Valve Stenosis and Atherosclerotic Calcification. Curr. Atheroscler. Rep. 22 (2), 2. doi:10.1007/s11883-020-0821-7
Delaby, C., Rondeau, C., Pouzet, C., Willemetz, A., Pilard, N., Desjardins, M., et al. (2012). Subcellular Localization of Iron and Heme Metabolism Related Proteins at Early Stages of Erythrophagocytosis. PLoS One 7 (7), e42199. doi:10.1371/journal.pone.0042199
Demetz, E., Tymoszuk, P., Hilbe, R., Volani, C., Haschka, D., Heim, C., et al. (2020). The Haemochromatosis Gene Hfe and Kupffer Cells Control LDL Cholesterol Homeostasis and Impact on Atherosclerosis Development. Eur. Heart J. 41 (40), 3949–3959. doi:10.1093/eurheartj/ehaa140
Derbyshire, E. R., and Marletta, M. A. (2012). Structure and Regulation of Soluble Guanylate Cyclase. Annu. Rev. Biochem. 81, 533–559. doi:10.1146/annurev-biochem-050410-100030
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an Iron-dependent Form of Nonapoptotic Cell Death. Cell 149 (5), 1060–1072. doi:10.1016/j.cell.2012.03.042
Duffy, S. P., Shing, J., Saraon, P., Berger, L. C., Eiden, M. V., Wilde, A., et al. (2010). The Fowler Syndrome-Associated Protein FLVCR2 Is an Importer of Heme. Mol. Cell Biol 30 (22), 5318–5324. doi:10.1128/mcb.00690-10
Dutra, F. F., Alves, L. S., Rodrigues, D., Fernandez, P. L., de Oliveira, R. B., Golenbock, D. T., et al. (2014). Hemolysis-induced Lethality Involves Inflammasome Activation by Heme. Proc. Natl. Acad. Sci. USA 111 (39), E4110–E4118. doi:10.1073/pnas.1405023111
Erdei, J., Tóth, A., Balogh, E., Nyakundi, B. B., Bányai, E., Ryffel, B., et al. (2018). Induction of NLRP3 Inflammasome Activation by Heme in Human Endothelial Cells. Oxid Med. Cell Longev 2018, 4310816. doi:10.1155/2018/4310816
Erickson, J. R., Joiner, M.-l. A., Guan, X., Kutschke, W., Yang, J., Oddis, C. V., et al. (2008). A Dynamic Pathway for Calcium-independent Activation of CaMKII by Methionine Oxidation. Cell 133 (3), 462–474. doi:10.1016/j.cell.2008.02.048
Eskew, J. D., Vanacore, R. M., Sung, L., Morales, P. J., and Smith, A. (1999). Cellular Protection Mechanisms against Extracellular Heme. J. Biol. Chem. 274 (2), 638–648. doi:10.1074/jbc.274.2.638
Fang, X., Wang, H., Han, D., Xie, E., Yang, X., Wei, J., et al. (2019). Ferroptosis as a Target for protection against Cardiomyopathy. Proc. Natl. Acad. Sci. USA 116 (7), 2672–2680. doi:10.1073/pnas.1821022116
Feng, Y., Madungwe, N. B., Imam Aliagan, A. D., Tombo, N., and Bopassa, J. C. (2019). Liproxstatin-1 Protects the Mouse Myocardium against Ischemia/reperfusion Injury by Decreasing VDAC1 Levels and Restoring GPX4 Levels. Biochem. Biophysical Res. Commun. 520 (3), 606–611. doi:10.1016/j.bbrc.2019.10.006
Figueiredo, R. T., Fernandez, P. L., Mourao-Sa, D. S., Porto, B. N., Dutra, F. F., Alves, L. S., et al. (2007). Characterization of Heme as Activator of Toll-like Receptor 4. J. Biol. Chem. 282 (28), 20221–20229. doi:10.1074/jbc.m610737200
Finn, A. V., Nakano, M., Polavarapu, R., Karmali, V., Saeed, O., Zhao, X., et al. (2012). Hemoglobin Directs Macrophage Differentiation and Prevents Foam Cell Formation in Human Atherosclerotic Plaques. J. Am. Coll. Cardiol. 59 (2), 166–177. doi:10.1016/j.jacc.2011.10.852
Fredenburgh, L. E., Merz, A. A., and Cheng, S. (2015). Haeme Oxygenase Signalling Pathway: Implications for Cardiovascular Disease. Eur. Heart J. 36 (24), 1512–1518. doi:10.1093/eurheartj/ehv114
Fukuda, N., Sasaki, D., Ishiwata, S. i., and Kurihara, S. (2001). Length Dependence of Tension Generation in Rat Skinned Cardiac Muscle. Circulation 104 (14), 1639–1645. doi:10.1161/hc3901.095898
Gáll, T., Pethő, D., Nagy, A., Hendrik, Z., Méhes, G., Potor, L., et al. (2018). Heme Induces Endoplasmic Reticulum Stress (HIER Stress) in Human Aortic Smooth Muscle Cells. Front. Physiol. 9, 1595. doi:10.3389/fphys.2018.01595
Gbotosho, O. T., Kapetanaki, M. G., Ghosh, S., Villanueva, F. S., Ofori-Acquah, S. F., and Kato, G. J. (2020). Heme Induces IL-6 and Cardiac Hypertrophy Genes Transcripts in Sickle Cell Mice. Front. Immunol. 11, 1910. doi:10.3389/fimmu.2020.01910
Giulivi, C., and Davies, K. J. A. (1994). [30] Hydrogen Peroxide-Mediated Ferrylhemoglobin Generation In Vitro and in Red Blood Cells. Methods Enzymol. 231, 490–496. doi:10.1016/0076-6879(94)31032-7
Gladwin, M. T., and Sachdev, V. (2012). Cardiovascular Abnormalities in Sickle Cell Disease. J. Am. Coll. Cardiol. 59 (13), 1123–1133. doi:10.1016/j.jacc.2011.10.900
Gordan, R., Wongjaikam, S., Gwathmey, J. K., Chattipakorn, N., Chattipakorn, S. C., and Xie, L.-H. (2018). Involvement of Cytosolic and Mitochondrial Iron in Iron Overload Cardiomyopathy: an Update. Heart Fail. Rev. 23 (5), 801–816. doi:10.1007/s10741-018-9700-5
Grootveld, M., Bell, J. D., Halliwell, B., Aruoma, O. I., Bomford, A., and Sadler, P. J. (1989). Non-transferrin-bound Iron in Plasma or Serum from Patients with Idiopathic Hemochromatosis. J. Biol. Chem. 264 (8), 4417–4422. doi:10.1016/s0021-9258(18)83758-9
Guo, L., Akahori, H., Harari, E., Smith, S. L., Polavarapu, R., Karmali, V., et al. (2018). CD163+ Macrophages Promote Angiogenesis and Vascular Permeability Accompanied by Inflammation in Atherosclerosis. J. Clin. Invest. 128 (3), 1106–1124. doi:10.1172/jci93025
Hagen, W. R., Hagedoorn, P.-L., and Honarmand Ebrahimi, K. (2017). The Workings of Ferritin: a Crossroad of Opinions. Metallomics 9 (6), 595–605. doi:10.1039/c7mt00124j
Hanna, D. A., Harvey, R. M., Martinez-Guzman, O., Yuan, X., Chandrasekharan, B., Raju, G., et al. (2016). Heme Dynamics and Trafficking Factors Revealed by Genetically Encoded Fluorescent Heme Sensors. Proc. Natl. Acad. Sci. USA 113 (27), 7539–7544. doi:10.1073/pnas.1523802113
Hanna, D. A., Hu, R., Kim, H., Martinez-Guzman, O., Torres, M. P., and Reddi, A. R. (2018). Heme Bioavailability and Signaling in Response to Stress in Yeast Cells. J. Biol. Chem. 293 (32), 12378–12393. doi:10.1074/jbc.ra118.002125
Harrison, P. M., and Arosio, P. (1996). The Ferritins: Molecular Properties, Iron Storage Function and Cellular Regulation. Biochim. Biophys. Acta (BBA) - Bioenerg. 1275 (3), 161–203. doi:10.1016/0005-2728(96)00022-9
Hellings, W. E., Peeters, W., Moll, F. L., Piers, S. R. D., van Setten, J., Van der Spek, P. J., et al. (2010). Composition of Carotid Atherosclerotic Plaque Is Associated with Cardiovascular Outcome. Circulation 121 (17), 1941–1950. doi:10.1161/circulationaha.109.887497
Hill-Kapturczak, N., Voakes, C., Garcia, J., Visner, G., Nick, H. S., and Agarwal, A. (2003). A Cis -Acting Region Regulates Oxidized Lipid-Mediated Induction of the Human Heme Oxygenase-1 Gene in Endothelial Cells. Atvb 23 (8), 1416–1422. doi:10.1161/01.atv.0000081656.76378.a7
Hrkal, Z., Vodrazka, Z., and Kalousek, I. (1974). Transfer of Heme from Ferrihemoglobin and Ferrihemoglobin Isolated Chains to Hemopexin. Eur. J. Biochem. 43 (1), 73–78. doi:10.1111/j.1432-1033.1974.tb03386.x
Hvidberg, V., Maniecki, M. B., Jacobsen, C., Højrup, P., Møller, H. J., and Moestrup, S. K. (2005). Identification of the Receptor Scavenging Hemopexin-Heme Complexes. Blood 106 (7), 2572–2579. doi:10.1182/blood-2005-03-1185
Ingoglia, G., Sag, C. M., Rex, N., De Franceschi, L., Vinchi, F., Cimino, J., et al. (2017). Hemopexin Counteracts Systolic Dysfunction Induced by Heme-Driven Oxidative Stress. Free Radic. Biol. Med. 108, 452–464. doi:10.1016/j.freeradbiomed.2017.04.003
Ishikawa, K., Sugawara, D., Wang, X.-p., Suzuki, K., Itabe, H., Maruyama, Y., et al. (2001). Heme Oxygenase-1 Inhibits Atherosclerotic Lesion Formation in Ldl-Receptor Knockout Mice. Circ. Res. 88 (5), 506–512. doi:10.1161/01.res.88.5.506
Jansen, T., and Daiber, A. (2012). Direct Antioxidant Properties of Bilirubin and Biliverdin. Is There a Role for Biliverdin Reductase? Front. Pharmacol. 3, 30. doi:10.3389/fphar.2012.00030
Jansen, T., Hortmann, M., Oelze, M., Opitz, B., Steven, S., Schell, R., et al. (2010). Conversion of Biliverdin to Bilirubin by Biliverdin Reductase Contributes to Endothelial Cell protection by Heme Oxygenase-1-Evidence for Direct and Indirect Antioxidant Actions of Bilirubin. J. Mol. Cell Cardiol. 49 (2), 186–195. doi:10.1016/j.yjmcc.2010.04.011
Jeney, V., Balla, J., Yachie, A., Varga, Z., Vercellotti, G. M., Eaton, J. W., et al. (2002). Pro-oxidant and Cytotoxic Effects of Circulating Heme. Blood 100 (3), 879–887. doi:10.1182/blood.v100.3.879
Juckett, M. B., Balla, J., Balla, G., Jessurun, J., Jacob, H. S., and Vercellotti, G. M. (1995). Ferritin Protects Endothelial Cells from Oxidized Low Density Lipoprotein In Vitro. Am. J. Pathol. 147 (3), 782–789.
Kakhlon, O., and Cabantchik, Z. I. (2002). The Labile Iron Pool: Characterization, Measurement, and Participation in Cellular Processes1 1This Article Is Part of a Series of Reviews on "Iron and Cellular Redox Status." the Full List of Papers May Be Found on the Homepage of the Journal. Free Radic. Biol. Med. 33 (8), 1037–1046. doi:10.1016/s0891-5849(02)01006-7
Khan, A. A., and Quigley, J. G. (2013). Heme and FLVCR-Related Transporter Families SLC48 and SLC49. Mol. Aspects Med. 34 (2-3), 669–682. doi:10.1016/j.mam.2012.07.013
Khechaduri, A., Bayeva, M., Chang, H.-C., and Ardehali, H. (2013). Heme Levels Are Increased in Human Failing Hearts. J. Am. Coll. Cardiol. 61 (18), 1884–1893. doi:10.1016/j.jacc.2013.02.012
Köhler, A. C., Sag, C. M., and Maier, L. S. (2014). Reactive Oxygen Species and Excitation-Contraction Coupling in the Context of Cardiac Pathology. J. Mol. Cell Cardiol 73, 92–102.
Kolodgie, F. D., Gold, H. K., Burke, A. P., Fowler, D. R., Kruth, H. S., Weber, D. K., et al. (2003). Intraplaque Hemorrhage and Progression of Coronary Atheroma. N. Engl. J. Med. 349 (24), 2316–2325. doi:10.1056/nejmoa035655
Kumfu, S., Chattipakorn, S. C., Fucharoen, S., and Chattipakorn, N. (2016). Dual T-type and L-type Calcium Channel Blocker Exerts Beneficial Effects in Attenuating Cardiovascular Dysfunction in Iron-Overloaded Thalassaemic Mice. Exp. Physiol. 101 (4), 521–539. doi:10.1113/ep085517
Kumfu, S., Chattipakorn, S., Chinda, K., Fucharoen, S., and Chattipakorn, N. (2012). T-type Calcium Channel Blockade Improves Survival and Cardiovascular Function in Thalassemic Mice. Eur. J. Haematol. 88 (6), 535–548. doi:10.1111/j.1600-0609.2012.01779.x
Lane, D. J. R., Merlot, A. M., Huang, M. L.-H., Bae, D.-H., Jansson, P. J., Sahni, S., et al. (2015). Cellular Iron Uptake, Trafficking and Metabolism: Key Molecules and Mechanisms and Their Roles in Disease. Biochim. Biophys. Acta (BBA) - Mol. Cell Res. 1853 (5), 1130–1144. doi:10.1016/j.bbamcr.2015.01.021
Lawson, D. M., Treffry, A., Artymiuk, P. J., Harrison, P. M., Yewdall, S. J., Luzzago, A., et al. (1989). Identification of the Ferroxidase centre in Ferritin. FEBS Lett. 254 (1-2), 207–210. doi:10.1016/0014-5793(89)81040-3
Li, W., Östblom, M., Xu, L. H., Hellsten, A., Leanderson, P., Liedberg, B., et al. (2006). Cytocidal Effects of Atheromatous Plaque Components: the Death Zone Revisited. FASEB j. 20 (13), 2281–2290. doi:10.1096/fj.06-6114com
Lindman, B. R., Clavel, M.-A., Mathieu, P., Iung, B., Lancellotti, P., Otto, C. M., et al. (2016). Calcific Aortic Stenosis. Nat. Rev. Dis. Primers 2, 16006. doi:10.1038/nrdp.2016.6
Liu, S. C., Zhai, S., Lawler, J., and Palek, J. (1985). Hemin-mediated Dissociation of Erythrocyte Membrane Skeletal Proteins. J. Biol. Chem. 260 (22), 12234–12239. doi:10.1016/s0021-9258(17)39015-4
Liu, Y., Jing, F., Yi, W., Mendelson, A., Shi, P., Walsh, R., et al. (2018). HO-1hi Patrolling Monocytes Protect against Vaso-Occlusion in Sickle Cell Disease. Blood 131 (14), 1600–1610. doi:10.1182/blood-2017-12-819870
Liuzzi, J. P., Aydemir, F., Nam, H., Knutson, M. D., and Cousins, R. J. (2006). Zip14 (Slc39a14) Mediates Non-transferrin-bound Iron Uptake into Cells. Proc. Natl. Acad. Sci. 103 (37), 13612–13617. doi:10.1073/pnas.0606424103
Menon, A. V., Liu, J., Tsai, H. P., Zeng, L., Yang, S., Asnani, A., et al. (2021). Excess Heme Upregulates Heme Oxygenase 1 and Promotes Cardiac Ferroptosis in Mice with Sickle Cell Disease. Blood, 2020008455.
Moestrup, S. r. K., Gliemann, J. r., and Pallesen, G. (1992). Distribution of the ?2-macroglobulin Receptor/low Density Lipoprotein Receptor-Related Protein in Human Tissues. Cell Tissue Res 269 (3), 375–382. doi:10.1007/bf00353892
Moraes, J. A., Barcellos-de-Souza, P., Rodrigues, G., Nascimento-Silva, V., Silva, S. V., Assreuy, J., et al. (2012). Heme Modulates Smooth Muscle Cell Proliferation and Migration via NADPH Oxidase: a Counter-regulatory Role for Heme Oxygenase System. Atherosclerosis 224 (2), 394–400. doi:10.1016/j.atherosclerosis.2012.07.043
Morvan, M., Arangalage, D., Franck, G., Perez, F., Cattan-Levy, L., Codogno, I., et al. (2019). Relationship of Iron Deposition to Calcium Deposition in Human Aortic Valve Leaflets. J. Am. Coll. Cardiol. 73 (9), 1043–1054. doi:10.1016/j.jacc.2018.12.042
Muckenthaler, M. U., Rivella, S., Hentze, M. W., and Galy, B. (2017). A Red Carpet for Iron Metabolism. Cell 168 (3), 344–361. doi:10.1016/j.cell.2016.12.034
Murphy, C. J., and Oudit, G. Y. (2010). Iron-overload Cardiomyopathy: Pathophysiology, Diagnosis, and Treatment. J. Card. Fail. 16 (11), 888–900. doi:10.1016/j.cardfail.2010.05.009
Nagy, E., Jeney, V., Yachie, A., Szabó, R. P., Wagner, O., Vercellotti, G. M., et al. (2005). Oxidation of Hemoglobin by Lipid Hydroperoxide Associated with Low-Density Lipoprotein (LDL) and Increased Cytotoxic Effect by LDL Oxidation in Heme Oxygenase-1 (HO-1) Deficiency. Cell Mol Biol (Noisy-le-grand) 51 (4), 377–385.
Nagy, E., Eaton, J. W., Jeney, V., Soares, M. P., Varga, Z., Galajda, Z., et al. (2010). Red Cells, Hemoglobin, Heme, Iron, and Atherogenesis. Atvb 30 (7), 1347–1353. doi:10.1161/atvbaha.110.206433
Natarajan, R., Fisher, B. J., and Fowler, A. A. (2007). Hypoxia Inducible Factor-1 Modulates Hemin-Induced IL-8 Secretion in Microvascular Endothelium. Microvasc. Res. 73 (3), 163–172. doi:10.1016/j.mvr.2007.01.002
Nath, K. A., Balla, J., Croatt, A. J., and Vercellotti, G. M. (1995). Heme Protein-Mediated Renal Injury: a Protective Role for 21-aminosteroids In Vitro and In Vivo. Kidney Int. 47 (2), 592–602. doi:10.1038/ki.1995.75
Nielsen, M. J., Møller, H. J., and Moestrup, S. K. (2010). Hemoglobin and Heme Scavenger Receptors. Antioxid. Redox Signaling 12 (2), 261–273. doi:10.1089/ars.2009.2792
Olsson, M., Thyberg, J., and Nilsson, J. (1999). Presence of Oxidized Low Density Lipoprotein in Nonrheumatic Stenotic Aortic Valves. Atvb 19 (5), 1218–1222. doi:10.1161/01.atv.19.5.1218
Ortiz de Montellano, P. (2000). The Mechanism of Heme Oxygenase. Curr. Opin. Chem. Biol. 4 (2), 221–227. doi:10.1016/s1367-5931(99)00079-4
Pantopoulos, K., Porwal, S. K., Tartakoff, A., and Devireddy, L. (2012). Mechanisms of Mammalian Iron Homeostasis. Biochemistry 51 (29), 5705–5724. doi:10.1021/bi300752r
Park, T.-J., Park, J. H., Lee, G. S., Lee, J.-Y., Shin, J. H., Kim, M. W., et al. (2019). Quantitative Proteomic Analyses Reveal that GPX4 Downregulation during Myocardial Infarction Contributes to Ferroptosis in Cardiomyocytes. Cell Death Dis 10 (11), 835. doi:10.1038/s41419-019-2061-8
Pethő, D., Gáll, T., Hendrik, Z., Nagy, A., Beke, L., Gergely, A. P., et al. (2021). Ferryl Hemoglobin and Heme Induce A(1)-Microglobulin in Hemorrhaged Atherosclerotic Lesions with Inhibitory Function against Hemoglobin and Lipid Oxidation. Int. J. Mol. Sci. 22 (13).
Ponka, P., Sheftel, A. D., English, A. M., Scott Bohle, D., and Garcia-Santos, D. (2017). Do Mammalian Cells Really Need to Export and Import Heme? Trends Biochem. Sci. 42 (5), 395–406. doi:10.1016/j.tibs.2017.01.006
Posta, N., Csősz, É., Oros, M., Pethő, D., Potor, L., Kalló, G., et al. (2020). Hemoglobin Oxidation Generates Globin-Derived Peptides in Atherosclerotic Lesions and Intraventricular Hemorrhage of the Brain, Provoking Endothelial Dysfunction. Lab. Invest. 100 (7), 986–1002. doi:10.1038/s41374-020-0403-x
Potor, L., Bányai, E., Becs, G., Soares, M. P., Balla, G., Balla, J., et al. (2013). Atherogenesis May Involve the Prooxidant and Proinflammatory Effects of Ferryl Hemoglobin. Oxid Med. Cell Longev 2013, 676425. doi:10.1155/2013/676425
Potor, L., Hendrik, Z., Patsalos, A., Katona, É., Méhes, G., Póliska, S., et al. (2021). Oxidation of Hemoglobin Drives a Proatherogenic Polarization of Macrophages in Human Atherosclerosis. Antioxid. Redox Signaling 35 (12), 917–950. doi:10.1089/ars.2020.8234
Powell, L. W., Seckington, R. C., and Deugnier, Y. (2016). Haemochromatosis. The Lancet 388 (10045), 706–716. doi:10.1016/s0140-6736(15)01315-x
Qiu, A., Jansen, M., Sakaris, A., Min, S. H., Chattopadhyay, S., Tsai, E., et al. (2006). Identification of an Intestinal Folate Transporter and the Molecular Basis for Hereditary Folate Malabsorption. Cell 127 (5), 917–928. doi:10.1016/j.cell.2006.09.041
Rajagopal, A., Rao, A. U., Amigo, J., Tian, M., Upadhyay, S. K., Hall, C., et al. (2008). Haem Homeostasis Is Regulated by the Conserved and Concerted Functions of HRG-1 Proteins. Nature 453 (7198), 1127–1131. doi:10.1038/nature06934
Rajamannan, N. M., Bonow, R. O., and Rahimtoola, S. H. (2007). Calcific Aortic Stenosis: an Update. Nat. Rev. Cardiol. 4 (5), 254–262. doi:10.1038/ncpcardio0827
Rajamannan, N. M., Evans, F. J., Aikawa, E., Grande-Allen, K. J., Demer, L. L., Heistad, D. D., et al. (2011). Calcific Aortic Valve Disease: Not Simply a Degenerative Process. Circulation 124 (16), 1783–1791. doi:10.1161/circulationaha.110.006767
Roumenina, L. T., Rayes, J., Lacroix-Desmazes, S., and Dimitrov, J. D. (2016). Heme: Modulator of Plasma Systems in Hemolytic Diseases. Trends Mol. Med. 22 (3), 200–213. doi:10.1016/j.molmed.2016.01.004
Sachdev, V., Machado, R. F., Shizukuda, Y., Rao, Y. N., Sidenko, S., Ernst, I., et al. (2007). Diastolic Dysfunction Is an Independent Risk Factor for Death in Patients with Sickle Cell Disease. J. Am. Coll. Cardiol. 49 (4), 472–479. doi:10.1016/j.jacc.2006.09.038
Sadrzadeh, S. M., Graf, E., Panter, S. S., Hallaway, P. E., and Eaton, J. W. (1984). Hemoglobin. A Biologic fenton Reagent. J. Biol. Chem. 259 (23), 14354–14356. doi:10.1016/s0021-9258(17)42604-4
Sansbury, B. E., DeMartino, A. M., Xie, Z., Brooks, A. C., Brainard, R. E., Watson, L. J., et al. (2014). Metabolomic Analysis of Pressure-Overloaded and Infarcted Mouse Hearts. Circ. Heart Fail. 7 (4), 634–642. doi:10.1161/circheartfailure.114.001151
Sassa, S. (2004). Why Heme Needs to Be Degraded to Iron, Biliverdin IXα, and Carbon Monoxide? Antioxid. Redox Signal. 6 (5), 819–824. doi:10.1089/1523086041798006
Sawicki, K. T., Chang, H. C., and Ardehali, H. (2015). Role of Heme in Cardiovascular Physiology and Disease. J. Am. Heart Assoc. 4 (1), e001138. doi:10.1161/JAHA.114.001138
Sawicki, K. T., Shang, M., Wu, R., Chang, H. C., Khechaduri, A., Sato, T., et al. (2015). Increased Heme Levels in the Heart Lead to Exacerbated Ischemic Injury. J. Am. Heart Assoc. 4 (8), e002272. doi:10.1161/JAHA.115.002272
Schmitt, T. H., Frezzatti, W. A., and Schreier, S. (1993). Hemin-induced Lipid Membrane Disorder and Increased Permeability: a Molecular Model for the Mechanism of Cell Lysis. Arch. Biochem. Biophys. 307 (1), 96–103. doi:10.1006/abbi.1993.1566
Shayeghi, M., Latunde-Dada, G. O., Oakhill, J. S., Laftah, A. H., Takeuchi, K., Halliday, N., et al. (2005). Identification of an Intestinal Heme Transporter. Cell 122 (5), 789–801. doi:10.1016/j.cell.2005.06.025
Shimizu, T., Lengalova, A., Martínek, V., and Martínková, M. (2019). Heme: Emergent Roles of Heme in Signal Transduction, Functional Regulation and as Catalytic Centres. Chem. Soc. Rev. 48 (24), 5624–5657. doi:10.1039/c9cs00268e
Silva, G., Jeney, V., Chora, Â., Larsen, R., Balla, J., and Soares, M. P. (2009). Oxidized Hemoglobin Is an Endogenous Proinflammatory Agonist that Targets Vascular Endothelial Cells. J. Biol. Chem. 284 (43), 29582–29595. doi:10.1074/jbc.m109.045344
Silver, J., and Lukas, B. (1983). Mössbauer Studies on Protoporphyrin IX Iron(III) Solutions. Inorg. Chim. Acta 78, 219–224. doi:10.1016/s0020-1693(00)86516-0
Stocker, R., Yamamoto, Y., McDonagh, A. F., Glazer, A. N., and Ames, B. N. (1987). Bilirubin Is an Antioxidant of Possible Physiological Importance. Science 235 (4792), 1043–1046. doi:10.1126/science.3029864
Stoyanova, E., Cloutier, G., Felfly, H., Lemsaddek, W., Ah-Son, N., and Trudel, M. (2012). Evidence for a Novel Mechanism Independent of Myocardial Iron in β-Thalassemia Cardiac Pathogenesis. PLoS ONE 7 (12), e52128. doi:10.1371/journal.pone.0052128
Tappel, A. L. (1953). The Mechanism of the Oxidation of Unsaturated Fatty Acids Catalyzed by Hematin Compounds. Arch. Biochem. Biophys. 44 (2), 378–395. doi:10.1016/0003-9861(53)90056-3
Valentijn, K. M., Sadler, J. E., Valentijn, J. A., Voorberg, J., and Eikenboom, J. (2011). Functional Architecture of Weibel-Palade Bodies. Blood 117 (19), 5033–5043. doi:10.1182/blood-2010-09-267492
Van der Zee, J., Barr, D. P., and Mason, R. P. (1996). ESR Spin Trapping Investigation of Radical Formation from the Reaction between Hematin and Tert-Butyl Hydroperoxide. Free Radic. Biol. Med. 20 (2), 199–206. doi:10.1016/0891-5849(95)02031-4
Vaziri, N. D. (2012). Epidemic of Iron Overload in Dialysis Population Caused by Intravenous Iron Products: a Plea for Moderation. Am. J. Med. 125 (10), 951–952. doi:10.1016/j.amjmed.2012.02.009
Vincent, S. H. (1989). Oxidative Effects of Heme and Porphyrins on Proteins and Lipids. Semin. Hematol. 26 (2), 105–113.
Vinchi, F., De Franceschi, L., Ghigo, A., Townes, T., Cimino, J., Silengo, L., et al. (2013). Hemopexin Therapy Improves Cardiovascular Function by Preventing Heme-Induced Endothelial Toxicity in Mouse Models of Hemolytic Diseases. Circulation 127 (12), 1317–1329. doi:10.1161/circulationaha.112.130179
Vinchi, F., Porto, G., Simmelbauer, A., Altamura, S., Passos, S. T., Garbowski, M., et al. (2020). Atherosclerosis Is Aggravated by Iron Overload and Ameliorated by Dietary and Pharmacological Iron Restriction. Eur. Heart J. 41 (28), 2681–2695. doi:10.1093/eurheartj/ehz112
Virmani, R., Narula, J., and Farb, A. (1998). When Neoangiogenesis Ricochets. Am. Heart J. 136 (6), 937–939. doi:10.1016/s0002-8703(98)70144-9
Wagener, F. A. D. T. G., Feldman, E., de Witte, T., and Abraham, N. G. (1997). Heme Induces the Expression of Adhesion Molecules ICAM-1, VCAM-1, and E Selectin in Vascular Endothelial Cells. Exp. Biol. Med. 216 (3), 456–463. doi:10.3181/00379727-216-44197
Wang, C.-Y., Jenkitkasemwong, S., Duarte, S., Sparkman, B. K., Shawki, A., Mackenzie, B., et al. (2012). ZIP8 Is an Iron and Zinc Transporter Whose Cell-Surface Expression Is Up-Regulated by Cellular Iron Loading. J. Biol. Chem. 287 (41), 34032–34043. doi:10.1074/jbc.m112.367284
Wang, C., Yuan, W., Hu, A., Lin, J., Xia, Z., Yang, C., et al. (2020). Dexmedetomidine Alleviated Sepsis-induced M-yocardial F-erroptosis and S-eptic H-eart I-njury. Mol. Med. Rep. 22 (1), 175–184. doi:10.3892/mmr.2020.11114
Wang, L. J., Lee, T. S., Lee, F. Y., Pai, R. C., and Chau, L. Y. (1998). Expression of Heme Oxygenase-1 in Atherosclerotic Lesions. Am. J. Pathol. 152 (3), 711–720.
Wu, M.-L., Ho, Y.-C., and Yet, S.-F. (2011). A central Role of Heme Oxygenase-1 in Cardiovascular protection. Antioxid. Redox Signaling 15 (7), 1835–1846. doi:10.1089/ars.2010.3726
Yang, Z., Philips, J. D., Doty, R. T., Giraudi, P., Ostrow, J. D., Tiribelli, C., et al. (2010). Kinetics and Specificity of Feline Leukemia Virus Subgroup C Receptor (FLVCR) export Function and its Dependence on Hemopexin. J. Biol. Chem. 285 (37), 28874–28882. doi:10.1074/jbc.m110.119131
Yuan, X., Cong, Y., Hao, J., Shan, Y., Zhao, Z., Wang, S., et al. (2004). Regulation of LIP Level and ROS Formation through Interaction of H-Ferritin with G-CSF Receptor. J. Mol. Biol. 339 (1), 131–144. doi:10.1016/j.jmb.2004.03.027
Yuan, X., Protchenko, O., Philpott, C. C., and Hamza, I. (2012). Topologically Conserved Residues Direct Heme Transport in HRG-1-Related Proteins. J. Biol. Chem. 287 (7), 4914–4924. doi:10.1074/jbc.m111.326785
Zhabyeyev, P., and Oudit, G. Y. (2017). Hemochromatosis Protein (HFE) Knockout Mice as a Novel Model of Hemochromatosis: Implications for Study and Management of Iron-Overload Cardiomyopathy. Can. J. Cardiol. 33 (7), 835–837. doi:10.1016/j.cjca.2017.04.013
Keywords: heme, cardiovascular diseases, atherosclerosis, aortic valve stenosis, heart failure, ferroptosis, iron overload
Citation: Guo Y, Zhao H, Lin Z, Ye T, Xu D and Zeng Q (2022) Heme in Cardiovascular Diseases: A Ubiquitous Dangerous Molecule Worthy of Vigilance. Front. Cell Dev. Biol. 9:781839. doi: 10.3389/fcell.2021.781839
Received: 23 September 2021; Accepted: 13 December 2021;
Published: 19 January 2022.
Edited by:
Gladys Oluyemisi Latunde-Dada, King’s College London, United KingdomReviewed by:
Stefan W. Ryter, Harvard Medical School, United StatesJózsef Balla, University of Debrecen, Hungary
Copyright © 2022 Guo, Zhao, Lin, Ye, Xu and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingchun Zeng, cWluZ2NodW56ZW5nQHNtdS5lZHUuY24=; Dingli Xu, ZGluZ2xpeHVAc211LmVkdS5jbg==