
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol., 26 November 2021
Sec. Cell Death and Survival
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.781365
 Chenqiang Jia1,2†
Chenqiang Jia1,2† Zhuqing Zhang1,3†
Zhuqing Zhang1,3† Jun Tang4†
Jun Tang4† Mei-Chun Cai1
Mei-Chun Cai1 Jingyu Zang1
Jingyu Zang1 Kaixuan Shi1Yunheng Sun1Jie Wu5Hailei Shi5Weiping Shi5
Kaixuan Shi1Yunheng Sun1Jie Wu5Hailei Shi5Weiping Shi5 Pengfei Ma4Xiaojing Zhao4*
Pengfei Ma4Xiaojing Zhao4* Zhuang Yu6*Yujie Fu4*
Zhuang Yu6*Yujie Fu4* Guanglei Zhuang1,3*
Guanglei Zhuang1,3*GSDME is a newly recognized executor of cellular pyroptosis, and has been recently implicated in tumor growth and immunity. However, knowledge about the molecular regulators underlying GSDME abundance remains limited. Here, we performed integrative bioinformatics analyses and identified that epithelial-mesenchymal transition (EMT) gene signatures exhibited positive correlation with GSDME levels across human cancers. A causal role was supported by the observation that EMT dictated GSDME reversible upregulation in multiple experimental models. Mechanistically, transcriptional activation of GSDME was directly driven by core EMT-activating transcription factors ZEB1/2, which bound to the GSDME promoter region. Of functional importance, elevated GSDME in mesenchymally transdifferentiated derivatives underwent proteolytic cleavage upon antineoplastic drug exposure, leading to pyroptotic cell death and consequent cytokine release. Taken together, our findings pinpointed a key transcriptional machinery controlling GSDME expression and indicated potential therapeutic avenues to exploit GSDME-mediated inflammatory pyroptosis for the treatment of mesenchymal malignancies.
Pyroptosis is a lytic type of programmed cell death characterized by cellular swelling and pore formation on cytomembrane, resulting in passive release of intracellular contents (He et al., 2015; Kayagaki et al., 2015; Shi et al., 2015; Liu et al., 2016; Strasser and Vaux, 2020). Several members of the gasdermin family (GSDMA, GSDMB, GSDMC, GSDMD, GSDME, and PJVK) have been recently identified to exert pyroptotic activity under different circumstances (Ding et al., 2016; Broz et al., 2020). For instance, GSDME-dependent pyroptosis can be triggered by chemotherapy or molecular targeted drugs in malignant cells (Rogers et al., 2017; Wang et al., 2017; Lu et al., 2018). During this process, GSDME is cleaved by activated caspase-3 to execute membrane permeabilization and proinflammatory cell killing. Indeed, antitumor immune responses during small-molecule therapy are reportedly enhanced by GSDME-mediated pyroptosis, which causes extracellular outflow of immunostimulatory factors and subsequent T cell propagation (Erkes et al., 2020; Rosenbaum et al., 2021; Zhang et al., 2021). Additionally, granzyme B from cytotoxic lymphocytes, by directly cleaving GSDME, induces caspase-independent pyroptosis and reinforces antineoplastic immunity (Zhang et al., 2020). Therefore, GSDME plays dual tumor-suppressive roles and lies in the intersection between pyroptotic cell death and rejuvenation of immune microenvironment, offering the possibility of harnessing its function to treat cancer.
However, tumor cells may delicately adopt various strategies to escape from GSDME-elicited suppression. Previous studies have extensively documented a molecular machinery involving epigenetic inactivation due to GSDME promoter hypermethylation in a range of human cancers (Akino et al., 2007; Kim et al., 2008a; Kim et al., 2008b; Yokomizo et al., 2012; Wang et al., 2013; Ibrahim et al., 2019; De Schutter et al., 2020). As a result, DNA methyltransferase inhibitors restore GSDME gene transcription and reinstate chemotherapy sensitivity (Wang et al., 2017; Fan et al., 2019). Nonetheless, we and others have observed ubiquitous expression of GSDME across multiple malignancies (Lu et al., 2018; Wu et al., 2019; Yu et al., 2019), suggesting that alternative regulatory mechanisms probably exist in certain context. Further understanding the determinants of GSDME levels holds the promise to therapeutically exploit disparate cell death modalities and deliberately produce pyroptosis-associated cytotoxicity and immune stimulation for cancer control.
In this study, we demonstrate that in addition to promoter methylation, epithelial-mesenchymal transition (EMT) dictates GSDME gene expression in diverse tumor models. We provide evidence that core EMT-activating transcription factors (EMT-TFs) ZEB1/2 are responsible for GSDME upregulation. Importantly, increased GSDME in mesenchymal cancer cells is functional to execute therapy-induced pyroptosis, leading to plasma membrane leakage and proinflammatory cytokine release.
All cell lines were obtained from ATCC, where cell characterization was verified via polymorphic short tandem repeat (STR) profiling. Cells were cultured in RPMI1640 (Invitrogen) supplemented with 10% fetal bovine serum (Gibco), 1% GlutaMAX, and 1% sodium pyruvate solution. TGFβ was purchased from Cell Signaling Technology, and used to treat cells at a final concentration of 5 nM. Decitabine, thioguanine, osimertinib, erlotinib, and salinomycin were purchased from Selleck Chemicals. All inhibitors were reconstituted in DMSO (Sigma-Aldrich) at a stock concentration of 10 mM, and used to treat cancer cells at a final concentration of 4 μM. Lactate dehydrogenase (LDH) assays were performed using CytoTox 96 Non-Radioactive Cytotoxicity Assay Kit (Promega).
Plasmids for gene overexpression were generated using the Gateway Cloning System (Invitrogen). All gene coding sequences were amplified using the PrimeSTAR GXL DNA Polymerase Kit (Takara) and authenticated by Sanger sequencing. The CRISPR-Cas9 system was applied to knock out genes of interest. The sequences of sgRNAs were provided in Supplementary Table S1. For virus packaging, 5 μg of lentiviral constructs, 5 μg of Δ8.9 plasmid, and 3 μg of VSVG plasmid were transfected into HEK293T cells with Lipofectamine 2000 (Invitrogen). Virus-containing supernatant was gathered 48 h after transfection and mixed with 8 μg/ml polybrene to infect target cells. Infected cells were selected with 5 μg/ml puromycin or blasticidin.
Cells were lysed in RIPA buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1% NP-40, 0.1% SDS, and 2 μM EDTA) supplemented with proteinase inhibitors (Roche). The cell lysates (20 μg) were subjected to SDS-PAGE and Western blot. Antibodies for the following proteins were used: E-cadherin (#3195), SLUG (#9585), SNAIL (#3879), Vimentin (#5741), TWIST (#46702), ZEB2 (#97885), and GAPDH (#8884, Cell Signaling Technology); ZEB1 (#21544-1-AP, Proteintech); GSDME (ab215191, Abcam).
Total RNA from cells was extracted using Trizol (Invitrogen) and subjected to reverse transcription with the HiScript III Q RT SuperMix Kit (Vazyme). The quantitative PCR (qPCR) was conducted on the Applied Biosystems ViiA7 machine. Relative mRNA expression of each gene was normalized to GAPDH as the endogenous control. At least three biological replicates were included for each condition.
The promoter region of human GSDME was cloned into the luciferase reporter vector (GeneCopoeia). HEK293T cells were transfected in 12-well plates with Lipofectamine 2000 (Invitrogen). Forty-eight hours after transfection, luciferase assays were conducted using Secrete-Pair Dual Luminescence Assay Kit (GeneCopoeia) following the manufacturer’s instructions. The gaussia luciferase activity was normalized to secreted alkaline phosphatase expression control. The normalized value was then normalized to the detection value of the control construct. Means and standard deviations were calculated in three independent replicates for each condition.
Cells were crosslinked with RPM1640 medium supplemented with 1% formaldehyde for 10 min at room temperature, and terminated with 1/20 volume of 2.5 M glycine. The cells were collected and washed by cold PBS. Cell samples were suspended and rocked in three lysis buffers in sequence as previously described (Lee et al., 2006). The lysates were sonicated with an ultrasonic processor VC505 (Sonics and Materials) for obtaining 200–500 bp fragments. Subsequently, 50 μl lysates were reserved as whole cell extract (WCE). The remaining lysates were incubated with antibody-conjugated magnetic beads overnight. For preparing beads, 100 μl of magnetic beads (Invitrogen) were incubated with 10 μg ZEB1 (#21544-1-AP, Proteintech), normal rabbit IgG (#2729, Cell Signaling Technology) or ZEB2 (sc-271984, Santacruz) antibodies overnight, respectively. The fragments-attached beads were washed several times, and eluted by elution buffer (50 mmol/L Tris-HCl pH 8.0, 10 mmol/L EDTA, 1% SDS) at 65°C for 15 min. Both the elution samples and WCE samples were incubated overnight at 65°C for crosslink reversal. These samples were treated with RNase A and Proteinase K, and then purified with QIAquick PCR Purification Kit (Qiagen). Purified DNA samples were employed to confirm the enrichment of target DNA fragments by quantitative PCR. The enrichment of target sequences in samples from specific immunoprecipitation was normalized to their amplification in the sample from IgG immunoprecipitation.
The Bio-Plex Pro Human Cytokine 27-plex assay (Bio-Rad) was performed for quantifying released cytokines using Luminex liquid suspension chip detection technology according to the manufacturer’s instructions. Briefly, culture supernatants were collected from cells treated with erlotinib for 72 h, and incubated with specific capture beads. Subsequently, detection antibodies and Phycoerythrin (PE)-conjugated streptavidin were added into each well in sequence. The fluorescent signal analysis was carried out with a Bio-Plex MAGPIX Multiplex Reader.
The RNA sequencing data of 33 TCGA cancer types were provided by the Pan-Cancer Atlas (Hoadley et al., 2018; Liu et al., 2018; Sanchez-Vega et al., 2018) and downloaded from the UCSC Xena Explorer (cohort: TCGA Pan-Cancer). The transcriptomic data of the gasdermin family and EMT transcription factors in cancer cell lines were obtained from Broad Institute Cancer Cell Line Encyclopedia (CCLE) (Ghandi et al., 2019). All datasets were processed as described previously (Cai et al., 2019). The methylation data were obtained from TCGA Pan-Cancer Atlas and CCLE, and then analyzed as described previously (Du et al., 2010; Ghandi et al., 2019). The β average value of all CpG sites between TSS −2000–+1000 was identified as the methylation value of the indicated gene in each sample. The methylation statuses of promoters were assessed as hypomethylation, partial methylation, and hypermethylation as described previously (Maekawa et al., 2019).
The gene signatures were derived from the Molecular Signatures Database (MSigDB) (Subramanian et al., 2005). We quantified the relative level of each gene signature in each sample by performing the single-sample gene set enrichment analysis (ssGSEA). The GSVA package in R was employed to calculate ssGSEA scores (Hänzelmann et al., 2013).
Statistical analysis was performed using the GraphPad Prism software. In all experiments, comparisons between two groups were based on two-sided Student’s t-test. P-values of <0.05 were considered statistically significant. The Pearson correlation analysis was used to evaluate the correlation between continuous factors. The geneset network was constructed using the Cytoscape software.
In line with our prior observation that GSDME was readily detectable in non-small cell lung cancer (NSCLC) (Lu et al., 2018), a panel of 30 cell lines exhibited a range of GSDME and GSDMD protein levels in immunoblotting analysis (Figure 1A). To assess whether GSDME abundance was regulated by gene methylation as reported, 12 models were selected and treated with two distinct DNA methyltransferase (DNMT) inhibitors, including a pan-DNMT inhibitor decitabine (Figure 1B) and a specific DNMT1 inhibitor thioguanine (Figure 1C). In a subset of NSCLC cell lines, GSDME expression, rather than GSDMD expression, was indeed restored by decitabine and thioguanine to various extent. In contrast, other NSCLC cell lines, e.g., NCI-H596, NCI-H358, and HCC4006, were not evidently impacted by either compound. Moreover, the promoter region of GSDME gene was mostly hypomethylated or partially methylated in cancer cell lines (Supplementary Figure S1A) and primary tumors (Supplementary Figure S1B). For comparison, the promoter region of SOX10 gene was universally hypermethylated. These results suggested that GSDME gene methylation only partly accounted for its modulation and additional regulatory mechanisms might play an important role in human cancer.
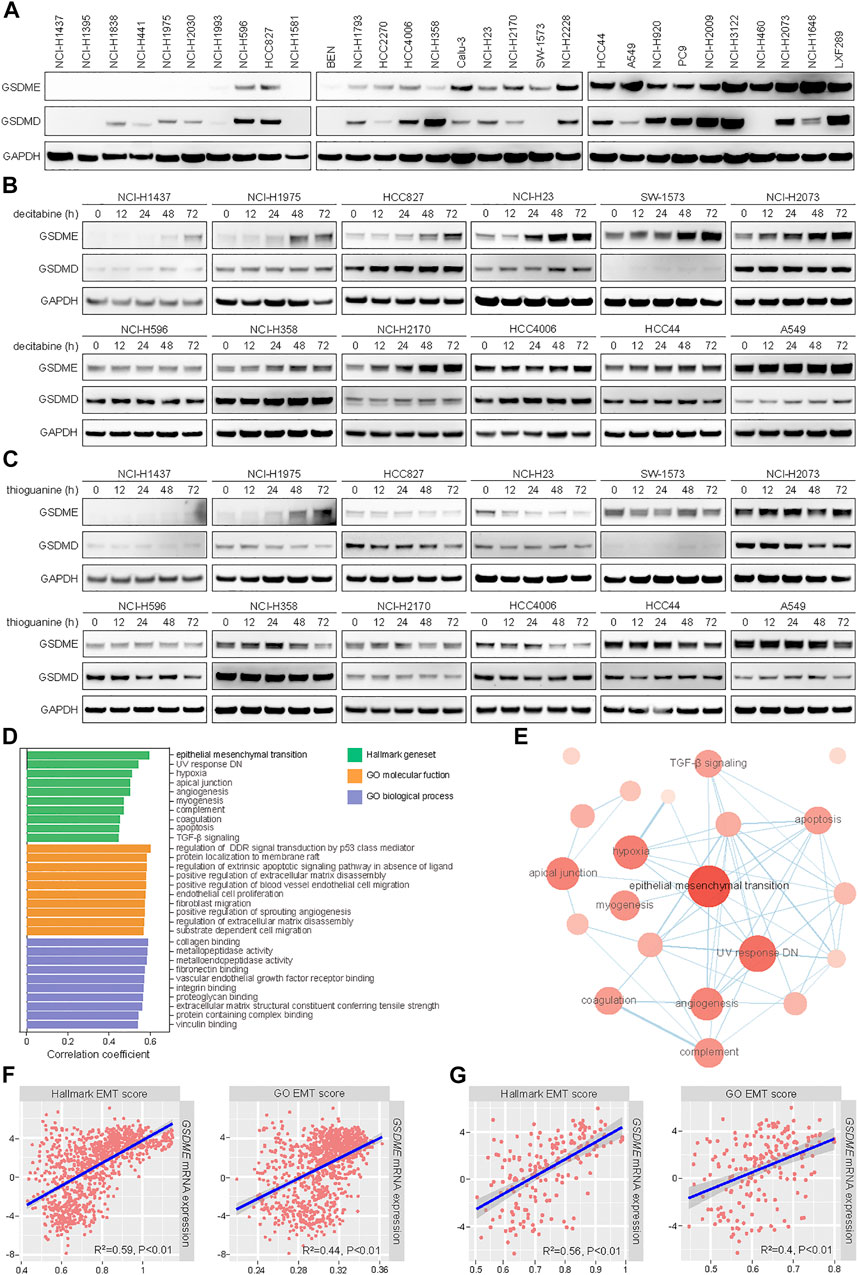
FIGURE 1. Positive correlation of expression levels between GSDME gene and EMT markers in human cancer. (A) The protein expression of GSDME and GSDMD was analyzed by Western blot in 30 lung cancer cell lines. (B) Indicated lung cancer cell lines with variable GSDME levels were treated with decitabine (4 μM) at a time course manner and analyzed by Western blot for GSDME and GSDMD protein expression. (C) Indicated lung cancer cell lines with variable GSDME levels were treated with thioguanine (4 μM) at a time course manner and analyzed by Western blot for GSDME and GSDMD protein expression. (D) Hallmark gene sets and gene ontology terms with ssGSEA scores that positively correlated with GSDME transcript expression in CCLE cancer cell lines. (E) The interaction network of hallmark gene sets with ssGSEA scores that positively correlated with GSDME transcript expression in CCLE cancer cell lines. (F) Scatterplots with linear regression line and shaded 95% confidence region for correlation estimation of GSDME transcript expression and EMT ssGSEA scores in CCLE cancer cell lines. R indicated Pearson correlation coefficient. (G) Scatterplots with linear regression line and shaded 95% confidence region for correlation estimation of GSDME transcript expression and EMT ssGSEA scores in CCLE lung cancer cell lines. R indicated Pearson correlation coefficient.
We sought to systematically explore the biological determinants of GSDME levels in neoplastic cells. To this end, ssGSEA scores were calculated for a total of 9,243 hallmark gene sets and gene ontology terms (Subramanian et al., 2005) using CCLE RNA sequencing data (Ghandi et al., 2019), followed by correlation estimation with GSDME transcript quantity (Figure 1D). Interestingly, the analysis revealed that epithelial-mesenchymal transition was among the top hallmark pathways which were positively associated with GSDME expression (Figure 1E). The significant correlation was consistently observed with two different EMT-related transcriptional signatures in pan-cancer (Figure 1F) and lung cancer cell lines (Figure 1G). We further corroborated the finding of EMT-GSDME interconnection across 33 TCGA cancer types (Supplementary Figures S1C,D). Of note, the remaining five members of gasdermin family (GSDMA, GSDMB, GSDMC, GSDMD, and PJVK) did not show an obvious correlation with EMT ssGSEA scores in either cancer cell lines (Supplementary Figure S2) or primary tumors (Supplementary Figure S3). Together, our comprehensive investigations pinpointed that the EMT process might be specifically involved in GSDME regulation in lung and other human malignancies.
To establish the causal role of EMT in GSDME upregulation, we first attempted to induce mesenchymal transdifferentiation with transforming growth factor-β (TGFβ) in a variety of cancer cell lines. As anticipated, elevated GSDME protein was detected at a time-dependent manner in lung cancer cells, such as NCI-H1975, HCC827, and NCI-H1437 (Figure 2A). Confirming previous reports (Miettinen et al., 1994; Zavadil and Böttinger, 2005; Thiery et al., 2009), TGFβ incited a gradual decrease of epithelial marker E-cadherin, a progressive increase of mesenchymal marker vimentin, and a corresponding acquisition of spindle-shaped morphology (Figure 2B). Similar GSDME upregulation along with EMT was noted in ovarian cancer cells including OVCAR3, OVCAR4, and COV 318 (Figures 2C,D). Furthermore, a larger panel of NSCLC cell lines verified that GSDME levels were augmented by TGFβ stimulation in most cases with just a few exceptions (Supplementary Figures S4A,B). In addition to TGFβ-mediated EMT, we introduced a distinct model (Sequist et al., 2011; Zhang et al., 2012) and validated GSDME upregulation by generating a serial of osimertinib-resistant NCI-H1975 and erlotinib-resistant HCC827 subclones (Figure 2E), which expectedly developed fibroblast-like features manifested as vimentin activation. Finally, upon TGFβ withdrawal from the NCI-H1975 and HCC827 mesenchymal derivatives, GSDME molecules demonstrated an analogous reversibility to the EMT program (Figure 2F). Therefore, EMT seemed to dictate GSDME expression in tumor cells.
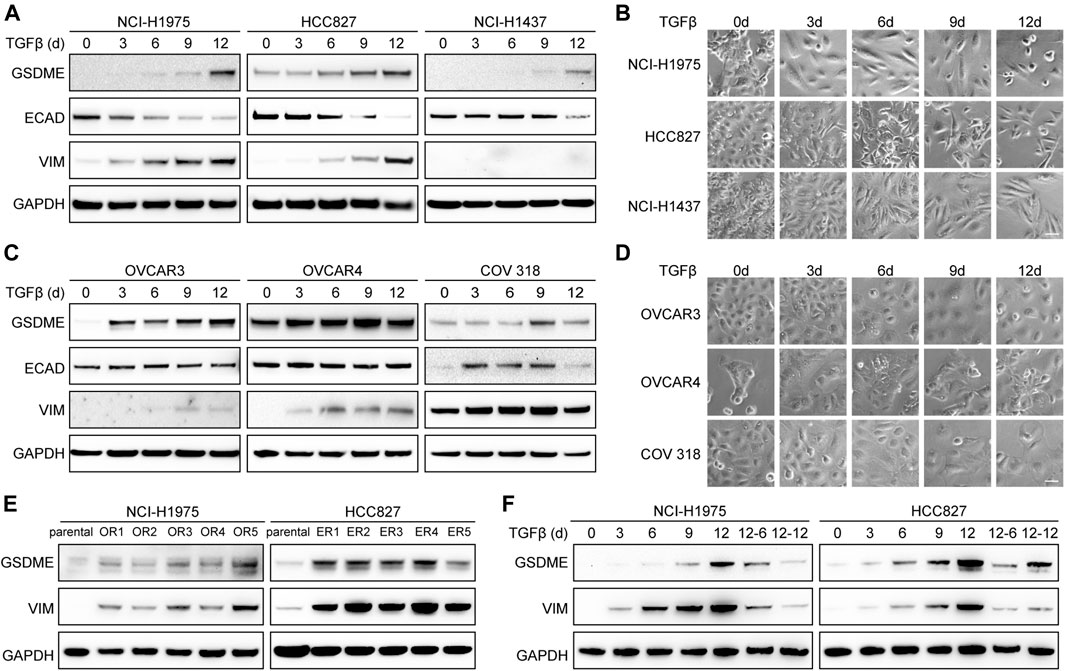
FIGURE 2. Reversible GSDME upregulation during EMT. (A) Three indicated lung cancer cell lines were treated with TGFβ (5 ng/μl) at a time course manner and analyzed by Western blot for GSDME protein expression. (B) Imaging analysis of lung cancer cells showed EMT morphology upon TGFβ treatment (scale bar = 50 μm). (C) Three indicated ovarian cancer cell lines were treated with TGFβ (5 ng/μl) at a time course manner and analyzed by Western blot for GSDME protein expression. (D) Imaging analysis of ovarian cancer cells showed EMT morphology upon TGFβ treatment (scale bar = 50 μm). (E) Osimertinib-resistant NCI-H1975 (OR) and erlotinib-resistant HCC827 (ER) cells were generated by long-term culture and analyzed by Western blot for GSDME protein expression. (F) NCI-H1975 and HCC827 cells were treated with TGFβ (5 ng/μl) to induce EMT. TGFβ was withdrawn as indicated and cells were analyzed by Western blot for GSDME protein expression.
We further dissected the molecular mechanism underlying EMT-associated GSDME regulation. Initially, quantitative PCR was performed in NCI-H1975, HCC827, and NCI-H1437 cells, and uncovered that GSDME, but not other gasdermins (Supplementary Figure S5A), was uniquely upregulated by TGFβ at the transcriptional level (Figure 3A). Likewise, five core EMT-inciting transcription factors (Stemmler et al., 2019), i.e., ZEB1, ZEB2, SNAIL, SLUG, and TWIST, displayed general elevation in response to TGFβ treatment (Supplementary Figure S5B), and thus were individually tested using a luciferase reporter fused with the GSDME promoter. Remarkably, overexpressed ZEB1/2 significantly enhanced the luciferase activity (Figure 3B) and consistently, endogenous ZEB1/2 directly bound to a consensus motif located in the GSDME promoter (Figure 3C), implying a crucial role of the classical ZEB family EMT-TFs. Indeed, ZEB1 and ZEB2 exhibited positive correlation with GSDME transcript across 188 CCLE lung cancer cell lines (Supplementary Figure S6A). Constitutive ectopic expression of ZEB1/2 (Supplementary Figure S6B) was competent to drive GSDME gene (Figure 3D) and protein upregulation (Figure 3E). Notably, GSDME abundance was also increased in selected cell lines at the gene (Supplementary Figure S7A) and protein level (Supplementary Figure S7B) in the presence of exogenous SNAIL, SLUG or TWIST (Supplementary Figure S7C), implicating functional interplay and/or redundancy of different EMT-TFs. Nevertheless, synchronous ZEB1 and ZEB2 deficiency substantially attenuated TGFβ-conferred GSDME production in HCC827 cells (Figure 3F). These results collectively proved that core EMT-activating transcription factors, in particular ZEB1/2, were both sufficient and required for GSDME transcriptional induction.
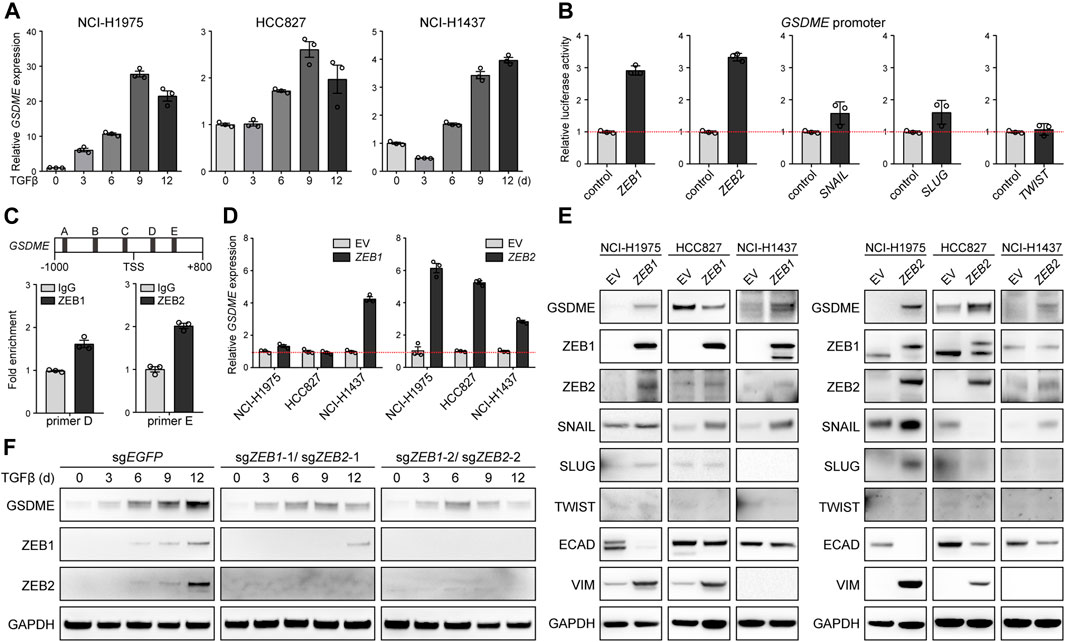
FIGURE 3. Direct induction of GSDME transcription by the EMT activator ZEB1/2. (A) Three indicated lung cancer cell lines were treated with TGFβ (5 ng/μl) at a time course manner and analyzed by qPCR for GSDME gene expression. Data are means ± SEM pooled from three independent experiments. (B) Five core EMT-TFs (ZEB1, ZEB2, SNAIL, SLUG, and TWIST) were co-transfected with GSDME promoter fused with dual luciferase reporter system and luciferase activity was assayed against the vector control. Data are means ± SEM pooled from three independent experiments. (C) ZEB1 or ZEB2 was overexpressed in NCI-H1975 cells and ChIP-qPCR analysis was performed with primer sets flanking five predicted ZEB1/2-binding consensus sequence proximal to the GSDME TSS (transcription start site). ZEB1 and ZEB2 was observed to bind to (D,E) locus at the GSDME promoter region, respectively. Data are means ± SEM pooled from three independent experiments. (D) EMT-TFs ZEB1 or ZEB2 was overexpressed in three indicated lung cancer cell lines and analyzed by qPCR for GSDME gene expression. Data are means ± SEM pooled from three independent experiments. (E) EMT-TFs ZEB1 or ZEB2 was overexpressed in three indicated lung cancer cell lines and analyzed by Western blot for GSDME protein expression. EV, empty vector. (F) ZEB1 and ZEB2 were simultaneously knocked out in HCC827 cells using the CRISPR-Cas9 system. Cells were treated with TGFβ (5 ng/μl) at a time course manner and analyzed by Western blot for GSDME protein expression.
A key question emerged whether the elevated GSDME in mesenchymal cells underwent proteolytic cleavage to trigger cellular pyroptosis upon therapeutic treatment. To address this pivot biological relevance, alteration-matched EGFR tyrosine kinase inhibitors (erlotinib or osimertinib) and salinomycin, a potassium ionophore with EMT-specific toxicity (Gupta et al., 2009), were respectively assessed in parental and TGFβ-stimulated lung cancer models. In the mesenchymally transdifferentiated HCC827 (Figure 4A) and NCI-H1975 (Figure 4B) cell lines, these compounds invariably yielded more GSDME N-terminal fragments at a time-dependent manner, presumably unleashing its intrinsic pore-forming activity in response to drug exposure. Conversely, GSDME gene depletion using the CRISPR-Cas9 system was sufficient to eliminate therapy-induced GSDME cleavage products in TGFβ-mediated (Figure 4C) or ZEB2-overexpressing (Figure 4D) mesenchymal HCC827 cells. Consequently, GSDME knockout resulted in significantly decreased lactate dehydrogenase (LDH) release into the culture supernatant following erlotinib or salinomycin inhibition (Figures 4E,F). These data indicated that gained GSDME protein during EMT retained the capacity to execute cell-lytic pyroptosis.
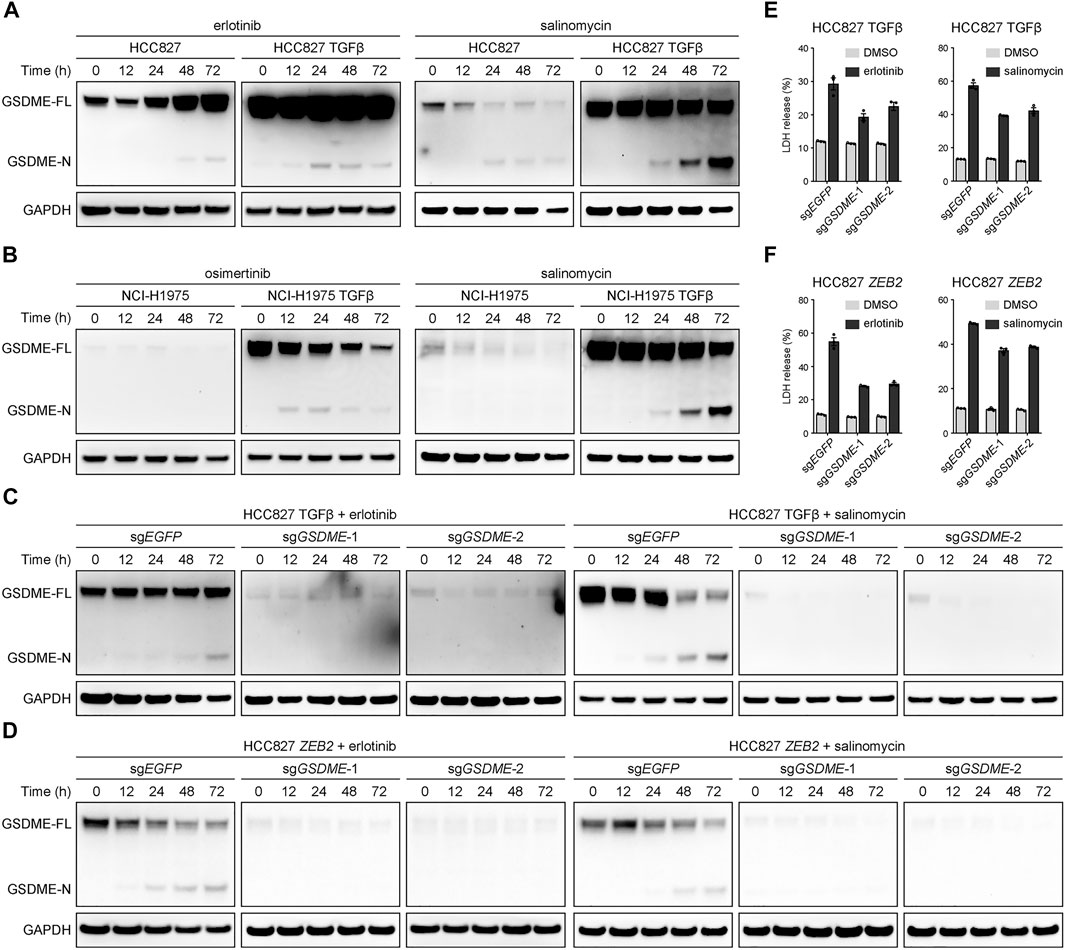
FIGURE 4. GSDME-dependent pyroptosis in mesenchymal cells upon drug treatment. (A) Parental and TGFβ-stimulated HCC827 cell lines were treated with erlotinib (4 μM) or salinomycin (4 μM) at a time course manner and analyzed by Western blot for GSDME protein cleavage. GSDME-FL, full-length GSDME; GSDME-N, GSDME N-terminal domain. (B) Parental and TGFβ-stimulated NCI-H1975 cell lines were treated with osimertinib (4 μM) or salinomycin (4 μM) at a time course manner and analyzed by Western blot for GSDME protein cleavage. (C) GSDME was knocked out in HCC827 cells using the CRISPR-Cas9 system. Cells were stimulated with TGFβ (5 ng/μl) to induce EMT, treated with erlotinib (4 μM) or salinomycin (4 μM), and analyzed by Western blot for GSDME protein cleavage. (D) GSDME was knocked out in HCC827 cells using the CRISPR-Cas9 system. Cells were transfected with exogenous ZEB2 to induce EMT, treated with erlotinib (4 μM) or salinomycin (4 μM), and analyzed by Western blot for GSDME protein cleavage. (E) GSDME was knocked out in HCC827 cells using the CRISPR-Cas9 system. Cells were stimulated with TGFβ (5 ng/μl) to induce EMT, and LDH release in the presence of DMSO or indicated inhibitors was measured. Data are means ± SEM pooled from three independent experiments. (F) GSDME was knocked out in HCC827 cells using the CRISPR-Cas9 system. Cells were transfected with exogenous ZEB2 to induce EMT, and LDH release in the presence of DMSO or indicated inhibitors was measured. Data are means ± SEM pooled from three independent experiments.
Considering that pyroptosis represented a form of proinflammatory cell death, it was plausible to speculate that tumor immune milieu might be broadly altered by GSDME upregulation in transformed mesenchymal cells as compared to the epithelial counterparts. To experimentally test this hypothesis, Luminex liquid chip technology was adopted to probe a set of 27 common inflammatory and chemotactic cytokines in the conditional media of ZEB2-overexpressing HCC827 cells treated with erlotinib (Figure 5A). The multiplex assay was performed in triplicates and we found that the analyte concentrations were predominantly reduced following genetic GSDME ablation, as exemplified by eight representative cytokines displaying the most dramatic changes (Figure 5B). It was worth noting that gene expression at the mRNA level did not convincingly explain the observed differences of secreted proteins (Figure 5C). Based on these results, we concluded that GSDME-elicited pyroptosis would reshape the tumor immune attributes most likely by facilitating direct release of proinflammatory cytokines and potentially danger-associated molecular patterns in mesenchymally transdifferentiated cancers subjected to physiological stresses or therapeutic challenges.
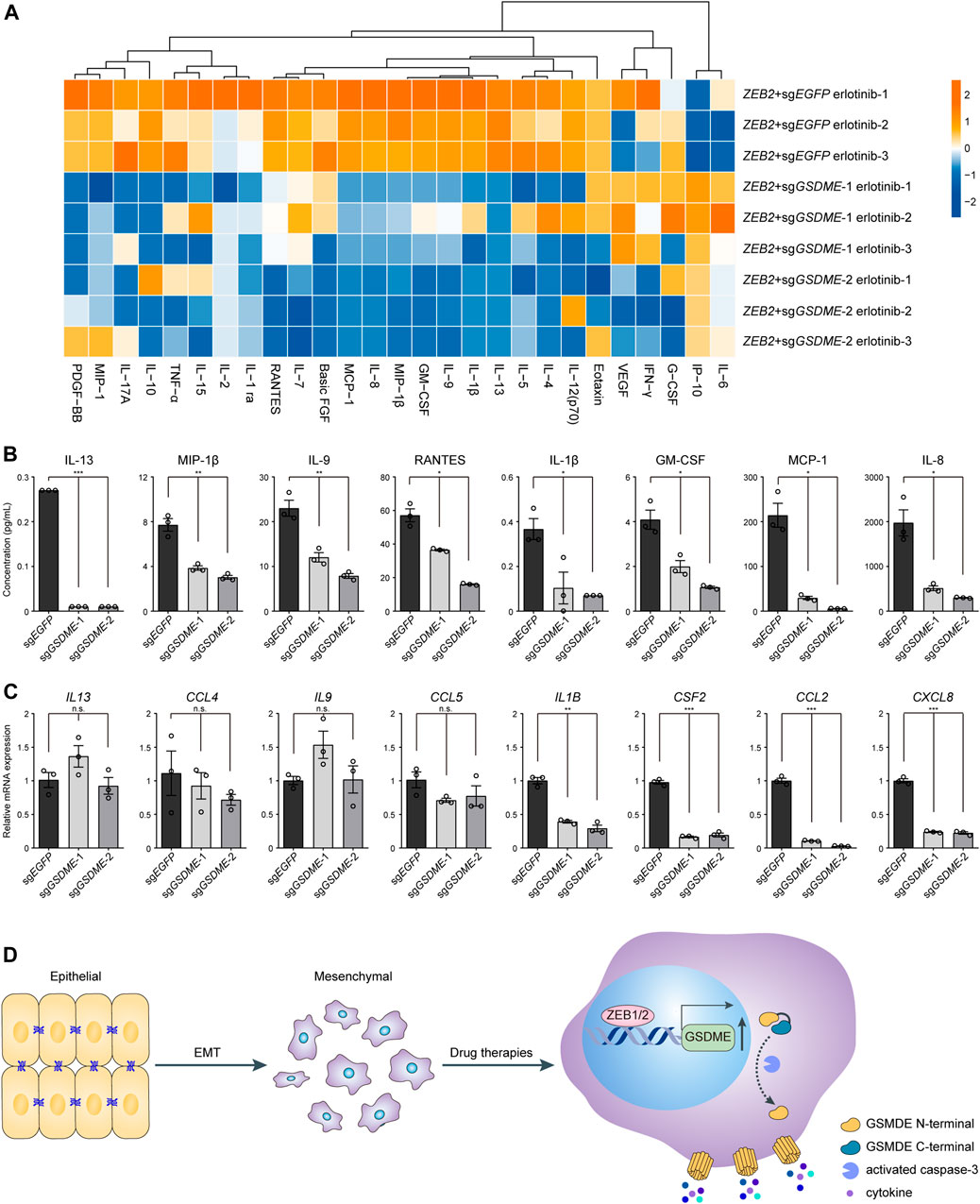
FIGURE 5. GSDME-mediated cytokine release from mesenchymal cells. (A) GSDME was knocked out in HCC827 cells using the CRISPR-Cas9 system. Cells were transfected with exogenous ZEB2 to induce EMT, and treated with erlotinib (4 μM) for 72 h. The secretion of 27 cytokines was analyzed by Luminex liquid suspension chip. (B) Concentrations of eight representative cytokines as measured by Luminex liquid suspension chip (*p < 0.05; **p < 0.01; ***p < 0.001). Data are means ± SEM pooled from three independent experiments. (C) Gene expression levels of eight representative cytokines were analyzed by qPCR (n.s. not significant; *p < 0.05; **p < 0.01; ***p < 0.001). Data are means ± SEM pooled from three independent experiments. (D) A schematic summary of the study showing that EMT-TFs ZEB1 and ZEB2 induce GSDME transcriptional activation in mesenchymal tumor cells, leading to inflammatory pyroptosis with cytokine release in response to drug therapies.
While published work heavily focuses on the epigenetic modifications of GSDME gene, its transcriptional regulatory programs and their implications for therapeutic development remain understudied. Our findings build on prior literature and identify epithelial-mesenchymal transition with a previously unrecognized role in controlling GSDME expression. Mechanistically, core EMT-TFs ZEB1/2 bind to the GSDME promoter and directly drive gene transcription for the execution of pyroptotic cell death, leading to extracellular release of inflammatory cytokines (please refer to the schematic model in Figure 5D). These data shed light on the molecular determinants of GSDME abundance and predict unique susceptibility to GSDME-mediated pyroptosis for malignant cells of mesenchymal state or lineage.
Accumulative evidence highlights GSDME as a candidate tumor suppressor and a critical factor influencing treatment responses (Wang et al., 2017; Lu et al., 2018; Rogers et al., 2019). In addition, GSDME aggravates adverse effects of certain anticancer regimens by causing pyroptosis-based normal tissue damage or cytokine release syndrome (Huang et al., 2020; Liu et al., 2020). However, the intricate machinery governing its expression has not been fully elucidated, precluding further clinical translation to improve therapeutic efficacy and reduce therapy-related toxicity. In order to thoroughly understand the mechanisms underlying GSDME regulation, we performed integrative bioinformatics analyses on publicly available pan-cancer datasets and discovered that EMT gene signatures were among the top correlates of GSDME levels. Subsequently, the novel link between EMT and GSDME was experimentally confirmed with multiple models in different tumor types, exemplifying its validity and generalizability. Thus, as epithelial lesions acquire mesenchymal properties via EMT, a well-defined cellular process often triggered by microenvironmental signals, there may be a growing tendency for neoplasms to undergo pyroptotic rather than apoptotic cell death accordingly. Reinforcing this notion, our study also captured pronounced plasticity of GSDME-marked pathway during the reverse process, known as mesenchymal-epithelial transition (MET). It is important to mention the new view in the field that EMT does not act as binary switches and can be activated to various degrees (Grigore et al., 2016; Pastushenko et al., 2018; Yuan et al., 2019). Therefore, the modes and dynamics of GSDME modulation within a continuum of EMT intermediate states desire future investigations.
We found that core EMT-TFs ZEB1/2 were probably responsible for GSDME gene activation. The observation was reminiscent of previous findings on other gasdermin members illustrating molecular regulation at the transcriptional level. For example, transcription factor IRF2 was identified as a direct driver of GSDMD expression in a spectrum of cell types (Kayagaki et al., 2019), and GSDMC transcription was enhanced by nuclear PD-L1/p-STAT3 co-activators under hypoxia (Hou et al., 2020). Despite that ZEB1 and ZEB2 normally serve as transcriptional suppressors of epithelial markers, they are able to exert pleiotropic functions depending on the cooperation with diverse co-factors (Zheng and Kang, 2014; Hegarty et al., 2015). Although we showed that ZEB1 and ZEB2 directly bound to the E-box motifs proximal to the GSDME transcription start site, whether ZEB1/2 acted individually or as a part of protein complexes to guide gene expression would require detailed exploration. In addition to transcriptional regulation, posttranslational mechanisms to fine-tune GSDME activity also exist and GSDME phosphorylation by an unknown kinase has been described to inhibit its pore-forming capacity (Rogers et al., 2019).
EMT has been linked to metastatic spread, cancer stemness and drug resistance, posing serious challenges to effective treatment (Thiery et al., 2009; Fischer et al., 2015; Zheng et al., 2015; Gainor et al., 2016; Shibue and Weinberg, 2017; Dongre and Weinberg, 2019; Williams et al., 2019). As such, our study promises to have profound therapeutic implications. First, because GSDME is upregulated along with the EMT process and remains functionally intact, pharmaceutical strategies activating GSDME-dependent pyroptosis present a specific opportunity for targeting human neoplasms with mesenchymal characteristics. Intriguingly, it is lately reported that high-mesenchymal state tumor cells, while often resistant to common therapeutics, are exquisitely vulnerable to ferroptosis, another lytic form of regulated cell death (Viswanathan et al., 2017; Sang et al., 2019). Hence, selected necrosis inducers can hold great potential for the inhibition of these typically unresponsive therapy-persisters (Hangauer et al., 2017). Second, pro-pyroptotic agents may simultaneously rejuvenate the immune microenvironment and render mesenchymally transdifferentiated cancers more susceptible to immunologic treatment by converting “cold” tumors into “hot” ones. Compared to the immunologically silent apoptosis, gasdermin-mediated pyroptosis is considered highly proinflammatory due to passive discharge of cytosolic contents that are enriched in cytokines and alarmins (Place and Kanneganti, 2019; Bedoui et al., 2020). Indeed, recent evidence has indicated that GSDME promotes lymphocyte infiltration and activation spontaneously or upon targeted inhibitors (Erkes et al., 2020; Wang et al., 2020; Zhang et al., 2020). In keeping with these reports, our data demonstrated that increased GSDME in mesenchymal cells facilitated drug-triggered cytokine release, though caution should be advised that some of them could also be detrimental to antitumor immunity. Continued fundamental research on GSDME regulation and biology to unravel its full immunostimulatory potential may ultimately improve clinical responses to current cancer immunotherapies.
Publicly available datasets were analyzed in this study. This data can be found here: The Cancer Genome Atlas (TCGA) and Cancer Cell Line Encyclopedia (CCLE).
Conception and design: CJ, YF, XZ, ZY, and GZ. Development of methodology: ZZ and JT. Acquisition of data (provided technical and material support): CJ, ZZ, JT, YF, KS, YS, HS, WS, and PM. Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): CJ, YF, M-CC, JZ, and JW. Writing, review, or revision of the article: CJ, XZ, ZY, and GZ.
This work was supported by the National Natural Science Foundation of China (81922047 and 82172596 to GZ; 82072560 to XZ; 81802809 to M-CC; 81802734 to PM), Shandong Provincial Natural Science Foundation (ZR202102240880 to ZY), Shanghai Municipal Education Commission-Gaofeng Clinical Medicine Grant Support (20161313 to GZ), Shanghai Collaborative Innovation Center for Translational Medicine (TM202005 to GZ), Science and Technology Commission of Shanghai Municipality (21Y11913900 to YF).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.781365/full#supplementary-material
Akino, K., Toyota, M., Suzuki, H., Imai, T., Maruyama, R., Kusano, M., et al. (2007). Identification of DFNA5 as a Target of Epigenetic Inactivation in Gastric Cancer. Cancer Sci. 98 (1), 88–95. doi:10.1111/j.1349-7006.2006.00351.x
Bedoui, S., Herold, M. J., and Strasser, A. (2020). Emerging Connectivity of Programmed Cell Death Pathways and its Physiological Implications. Nat. Rev. Mol. Cel Biol 21 (11), 678–695. doi:10.1038/s41580-020-0270-8
Broz, P., Pelegrín, P., and Shao, F. (2020). The Gasdermins, a Protein Family Executing Cell Death and Inflammation. Nat. Rev. Immunol. 20 (3), 143–157. doi:10.1038/s41577-019-0228-2
Cai, M.-C., Chen, M., Ma, P., Wu, J., Lu, H., Zhang, S., et al. (2019). Clinicopathological, Microenvironmental and Genetic Determinants of Molecular Subtypes in KEAP1/NRF2-Mutant Lung Cancer. Int. J. Cancer 144 (4), 788–801. doi:10.1002/ijc.31975
De Schutter, E., Croes, L., Ibrahim, J., Pauwels, P., Op de Beeck, K., Vandenabeele, P., et al. (2020). GSDME and its Role in Cancer: From behind the Scenes to the Front of the Stage. Int. J. Cancer 148, 2872–2883. doi:10.1002/ijc.33390
Ding, J., Wang, K., Liu, W., She, Y., Sun, Q., Shi, J., et al. (2016). Pore-forming Activity and Structural Autoinhibition of the Gasdermin Family. Nature 535 (7610), 111–116. doi:10.1038/nature18590
Dongre, A., and Weinberg, R. A. (2019). New Insights into the Mechanisms of Epithelial-Mesenchymal Transition and Implications for Cancer. Nat. Rev. Mol. Cel Biol 20 (2), 69–84. doi:10.1038/s41580-018-0080-4
Du, P., Zhang, X., Huang, C.-C., Jafari, N., Kibbe, W. A., Hou, L., et al. (2010). Comparison of Beta-Value and M-Value Methods for Quantifying Methylation Levels by Microarray Analysis. BMC bioinformatics 11, 587. doi:10.1186/1471-2105-11-587
Erkes, D. A., Cai, W., Sanchez, I. M., Purwin, T. J., Rogers, C., Field, C. O., et al. (2020). Mutant BRAF and MEK Inhibitors Regulate the Tumor Immune Microenvironment via Pyroptosis. Cancer Discov. 10 (2), 254–269. doi:10.1158/2159-8290.CD-19-0672
Fan, J.-X., Deng, R.-H., Wang, H., Liu, X.-H., Wang, X.-N., Qin, R., et al. (2019). Epigenetics-Based Tumor Cells Pyroptosis for Enhancing the Immunological Effect of Chemotherapeutic Nanocarriers. Nano Lett. 19 (11), 8049–8058. doi:10.1021/acs.nanolett.9b03245
Fischer, K. R., Durrans, A., Lee, S., Sheng, J., Li, F., Wong, S. T. C., et al. (2015). Epithelial-to-mesenchymal Transition Is Not Required for Lung Metastasis but Contributes to Chemoresistance. Nature 527 (7579), 472–476. doi:10.1038/nature15748
Gainor, J. F., Dardaei, L., Yoda, S., Friboulet, L., Leshchiner, I., Katayama, R., et al. (2016). Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov. 6 (10), 1118–1133. doi:10.1158/2159-8290.CD-16-0596
Ghandi, M., Huang, F. W., Jané-Valbuena, J., Kryukov, G. V., Lo, C. C., McDonald, E. R., et al. (2019). Next-generation Characterization of the Cancer Cell Line Encyclopedia. Nature 569 (7757), 503–508. doi:10.1038/s41586-019-1186-3
Grigore, A., Jolly, M., Jia, D., Farach-Carson, M., and Levine, H. (2016). Tumor Budding: The Name Is EMT. Partial EMT. Jcm 5 (5), 51. doi:10.3390/jcm5050051
Gupta, P. B., Onder, T. T., Jiang, G., Tao, K., Kuperwasser, C., Weinberg, R. A., et al. (2009). Identification of Selective Inhibitors of Cancer Stem Cells by High-Throughput Screening. Cell 138 (4), 645–659. doi:10.1016/j.cell.2009.06.034
Hangauer, M. J., Viswanathan, V. S., Ryan, M. J., Bole, D., Eaton, J. K., Matov, A., et al. (2017). Drug-tolerant Persister Cancer Cells Are Vulnerable to GPX4 Inhibition. Nature 551 (7679), 247–250. doi:10.1038/nature24297
Hänzelmann, S., Castelo, R., and Guinney, J. (2013). GSVA: Gene Set Variation Analysis for Microarray and RNA-Seq Data. BMC bioinformatics 14, 7. doi:10.1186/1471-2105-14-7
He, W.-t., Wan, H., Hu, L., Chen, P., Wang, X., Huang, Z., et al. (2015). Gasdermin D Is an Executor of Pyroptosis and Required for Interleukin-1β Secretion. Cell Res 25 (12), 1285–1298. doi:10.1038/cr.2015.139
Hegarty, S. V., Sullivan, A. M., and O’Keeffe, G. W. (2015). Zeb2: A Multifunctional Regulator of Nervous System Development. Prog. Neurobiol. 132, 81–95. doi:10.1016/j.pneurobio.2015.07.001
Hoadley, K. A., Yau, C., Hinoue, T., Wolf, D. M., Lazar, A. J., Drill, E., et al. (2018). Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 173 (2), 291–e6. doi:10.1016/j.cell.2018.03.022
Hou, J., Zhao, R., Xia, W., Chang, C.-W., You, Y., Hsu, J.-M., et al. (2020). PD-L1-mediated Gasdermin C Expression Switches Apoptosis to Pyroptosis in Cancer Cells and Facilitates Tumour Necrosis. Nat. Cel Biol 22 (10), 1264–1275. doi:10.1038/s41556-020-0575-z
Huang, Z., Zhang, Q., Wang, Y., Chen, R., Wang, Y., Huang, Z., et al. (2020). Inhibition of Caspase-3-Mediated GSDME-Derived Pyroptosis Aids in Noncancerous Tissue protection of Squamous Cell Carcinoma Patients during Cisplatin-Based Chemotherapy. Am. J. Cancer Res. 10 (12), 4287–4307.
Ibrahim, J., Op de Beeck, K., Fransen, E., Peeters, M., and Van Camp, G. (2019). The Gasdermin E Gene Potential as a Pan-Cancer Biomarker, while Discriminating between Different Tumor Types. Cancers 11 (11), 1810. doi:10.3390/cancers11111810
Kayagaki, N., Lee, B. L., Stowe, I. B., Kornfeld, O. S., O'Rourke, K., Mirrashidi, K. M., et al. (2019). IRF2 Transcriptionally Induces GSDMD Expression for Pyroptosis. Sci. Signal. 12 (582), 12. doi:10.1126/scisignal.aax4917
Kayagaki, N., Stowe, I. B., Lee, B. L., O’Rourke, K., Anderson, K., Warming, S., et al. (2015). Caspase-11 Cleaves Gasdermin D for Non-canonical Inflammasome Signalling. Nature 526 (7575), 666–671. doi:10.1038/nature15541
Kim, M. S., Chang, X., Yamashita, K., Nagpal, J. K., Baek, J. H., Wu, G., et al. (2008). Aberrant Promoter Methylation and Tumor Suppressive Activity of the DFNA5 Gene in Colorectal Carcinoma. Oncogene 27 (25), 3624–3634. doi:10.1038/sj.onc.1211021
Kim, M. S., Lebron, C., Nagpal, J. K., Chae, Y. K., Chang, X., Huang, Y., et al. (2008). Methylation of the DFNA5 Increases Risk of Lymph Node Metastasis in Human Breast Cancer. Biochem. biophysical Res. Commun. 370 (1), 38–43. doi:10.1016/j.bbrc.2008.03.026
Lee, T. I., Johnstone, S. E., and Young, R. A. (2006). Chromatin Immunoprecipitation and Microarray-Based Analysis of Protein Location. Nat. Protoc. 1 (2), 729–748. doi:10.1038/nprot.2006.98
Liu, J., Lichtenberg, T., Hoadley, K. A., Poisson, L. M., Lazar, A. J., Cherniack, A. D., et al. (2018). An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 173 (2), 400–e11. doi:10.1016/j.cell.2018.02.052
Liu, X., Zhang, Z., Ruan, J., Pan, Y., Magupalli, V. G., Wu, H., et al. (2016). Inflammasome-activated Gasdermin D Causes Pyroptosis by Forming Membrane Pores. Nature 535 (7610), 153–158. doi:10.1038/nature18629
Liu, Y., Fang, Y., Chen, X., Wang, Z., Liang, X., Zhang, T., et al. (2020). Gasdermin E-Mediated Target Cell Pyroptosis by CAR T Cells Triggers Cytokine Release Syndrome. Sci. Immunol. 5 (43). doi:10.1126/sciimmunol.aax7969
Lu, H., Zhang, S., Wu, J., Chen, M., Cai, M.-C., Fu, Y., et al. (2018). Molecular Targeted Therapies Elicit Concurrent Apoptotic and GSDME-dependent Pyroptotic Tumor Cell Death. Clin. Cancer Res. 24 (23), 6066–6077. doi:10.1158/1078-0432.CCR-18-1478
Maekawa, R., Tamura, I., Shinagawa, M., Mihara, Y., Sato, S., Okada, M., et al. (2019). Genome-wide DNA Methylation Analysis Revealed Stable DNA Methylation Status during Decidualization in Human Endometrial Stromal Cells. BMC genomics 20 (1), 324. doi:10.1186/s12864-019-5695-0
Miettinen, P. J., Ebner, R., Lopez, A. R., and Derynck, R. (1994). TGF-beta Induced Transdifferentiation of Mammary Epithelial Cells to Mesenchymal Cells: Involvement of Type I Receptors. J. Cel Biol 127 (6 Pt 2), 2021–2036. doi:10.1083/jcb.127.6.2021
Pastushenko, I., Brisebarre, A., Sifrim, A., Fioramonti, M., Revenco, T., Boumahdi, S., et al. (2018). Identification of the Tumour Transition States Occurring during EMT. Nature 556 (7702), 463–468. doi:10.1038/s41586-018-0040-3
Place, D. E., and Kanneganti, T.-D. (2019). Cell Death-Mediated Cytokine Release and its Therapeutic Implications. J. Exp. Med. 216 (7), 1474–1486. doi:10.1084/jem.20181892
Rogers, C., Erkes, D. A., Nardone, A., Aplin, A. E., Fernandes-Alnemri, T., and Alnemri, E. S. (2019). Gasdermin Pores Permeabilize Mitochondria to Augment Caspase-3 Activation during Apoptosis and Inflammasome Activation. Nat. Commun. 10 (1), 1689. doi:10.1038/s41467-019-09397-2
Rogers, C., Fernandes-Alnemri, T., Mayes, L., Alnemri, D., Cingolani, G., and Alnemri, E. S. (2017). Cleavage of DFNA5 by Caspase-3 during Apoptosis Mediates Progression to Secondary Necrotic/pyroptotic Cell Death. Nat. Commun. 8, 14128. doi:10.1038/ncomms14128
Rosenbaum, S. R., Wilski, N. A., and Aplin, A. E. (2021). Fueling the Fire: Inflammatory Forms of Cell Death and Implications for Cancer Immunotherapy. Cancer Discov. 11 (2), 266–281. doi:10.1158/2159-8290.CD-20-0805
Sanchez-Vega, F., Mina, M., Armenia, J., Chatila, W. K., Luna, A., La, K. C., et al. (2018). Oncogenic Signaling Pathways in the Cancer Genome Atlas. Cell 173 (2), 321–e10. doi:10.1016/j.cell.2018.03.035
Sang, M., Luo, R., Bai, Y., Dou, J., Zhang, Z., Liu, F., et al. (2019). Mitochondrial Membrane Anchored Photosensitive Nano-Device for Lipid Hydroperoxides Burst and Inducing Ferroptosis to Surmount Therapy-Resistant Cancer. Theranostics 9 (21), 6209–6223. doi:10.7150/thno.36283
Sequist, L. V., Waltman, B. A., Dias-Santagata, D., Digumarthy, S., Turke, A. B., Fidias, P., et al. (2011). Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. Sci. Transl. Med. 3 (75). doi:10.1126/scitranslmed.3002003
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature 526 (7575), 660–665. doi:10.1038/nature15514
Shibue, T., and Weinberg, R. A. (2017). EMT, CSCs, and Drug Resistance: the Mechanistic Link and Clinical Implications. Nat. Rev. Clin. Oncol. 14 (10), 611–629. doi:10.1038/nrclinonc.2017.44
Stemmler, M. P., Eccles, R. L., Brabletz, S., and Brabletz, T. (2019). Non-redundant Functions of EMT Transcription Factors. Nat. Cel Biol 21 (1), 102–112. doi:10.1038/s41556-018-0196-y
Strasser, A., and Vaux, D. L. (2020). Cell Death in the Origin and Treatment of Cancer. Mol. Cel. 78 (6), 1045–1054. doi:10.1016/j.molcel.2020.05.014
Subramanian, A., Tamayo, P., Mootha, V. K., Mukherjee, S., Ebert, B. L., Gillette, M. A., et al. (2005). Gene Set Enrichment Analysis: a Knowledge-Based Approach for Interpreting Genome-wide Expression Profiles. Proc. Natl. Acad. Sci. 102 (43), 15545–15550. doi:10.1073/pnas.0506580102
Thiery, J. P., Acloque, H., Huang, R. Y. J., and Nieto, M. A. (2009). Epithelial-mesenchymal Transitions in Development and Disease. Cell 139 (5), 871–890. doi:10.1016/j.cell.2009.11.007
Viswanathan, V. S., Ryan, M. J., Dhruv, H. D., Gill, S., Eichhoff, O. M., Seashore-Ludlow, B., et al. (2017). Dependency of a Therapy-Resistant State of Cancer Cells on a Lipid Peroxidase Pathway. Nature 547 (7664), 453–457. doi:10.1038/nature23007
Wang, C.-J., Tang, L., Shen, D.-W., Wang, C., Yuan, Q.-Y., Gao, W., et al. (2013). The Expression and Regulation of DFNA5 in Human Hepatocellular Carcinoma DFNA5 in Hepatocellular Carcinoma. Mol. Biol. Rep. 40 (12), 6525–6531. doi:10.1007/s11033-013-2581-8
Wang, Q., Wang, Y., Ding, J., Wang, C., Zhou, X., Gao, W., et al. (2020). A Bioorthogonal System Reveals Antitumour Immune Function of Pyroptosis. Nature 579 (7799), 421–426. doi:10.1038/s41586-020-2079-1
Wang, Y., Gao, W., Shi, X., Ding, J., Liu, W., He, H., et al. (2017). Chemotherapy Drugs Induce Pyroptosis through Caspase-3 Cleavage of a Gasdermin. Nature 547 (7661), 99–103. doi:10.1038/nature22393
Williams, E. D., Gao, D., Redfern, A., and Thompson, E. W. (2019). Controversies Around Epithelial-Mesenchymal Plasticity in Cancer Metastasis. Nat. Rev. Cancer 19 (12), 716–732. doi:10.1038/s41568-019-0213-x
Wu, M., Wang, Y., Yang, D., Gong, Y., Rao, F., Liu, R., et al. (2019). A PLK1 Kinase Inhibitor Enhances the Chemosensitivity of Cisplatin by Inducing Pyroptosis in Oesophageal Squamous Cell Carcinoma. EBioMedicine 41, 244–255. doi:10.1016/j.ebiom.2019.02.012
Yokomizo, K., Harada, Y., Kijima, K., Shinmura, K., Sakata, M., Sakuraba, K., et al. (2012). Methylation of the DFNA5 Gene Is Frequently Detected in Colorectal Cancer. Anticancer Res. 32 (4), 1319–1322.
Yu, P., Wang, H.-y., Tian, M., Li, A.-x., Chen, X.-s., Wang, X.-l., et al. (2019). Eukaryotic Elongation Factor-2 Kinase Regulates the Cross-Talk between Autophagy and Pyroptosis in Doxorubicin-Treated Human Melanoma Cells In Vitro. Acta Pharmacol. Sin 40 (9), 1237–1244. doi:10.1038/s41401-019-0222-z
Yuan, S., Norgard, R. J., and Stanger, B. Z. (2019). Cellular Plasticity in Cancer. Cancer Discov. 9 (7), 837–851. doi:10.1158/2159-8290.CD-19-0015
Zavadil, J., and Böttinger, E. P. (2005). TGF-β and Epithelial-To-Mesenchymal Transitions. Oncogene 24 (37), 5764–5774. doi:10.1038/sj.onc.1208927
Zhang, Z., Lee, J. C., Lin, L., Olivas, V., Au, V., LaFramboise, T., et al. (2012). Activation of the AXL Kinase Causes Resistance to EGFR-Targeted Therapy in Lung Cancer. Nat. Genet. 44 (8), 852–860. doi:10.1038/ng.2330
Zhang, Z., Zhang, Y., and Lieberman, J. (2021). Lighting a Fire: Can We Harness Pyroptosis to Ignite Antitumor Immunity. Cancer Immunol. Res. 9 (1), 2–7. doi:10.1158/2326-6066.CIR-20-0525
Zhang, Z., Zhang, Y., Xia, S., Kong, Q., Li, S., Liu, X., et al. (2020). Gasdermin E Suppresses Tumour Growth by Activating Anti-tumour Immunity. Nature 579 (7799), 415–420. doi:10.1038/s41586-020-2071-9
Zheng, H., and Kang, Y. (2014). Multilayer Control of the EMT Master Regulators. Oncogene 33 (14), 1755–1763. doi:10.1038/onc.2013.128
Keywords: epithelial-mesenchymal transition, GSDME, transcription activation, pyroptosis, ZEB1/2
Citation: Jia C, Zhang Z, Tang J, Cai M-C, Zang J, Shi K, Sun Y, Wu J, Shi H, Shi W, Ma P, Zhao X, Yu Z, Fu Y and Zhuang G (2021) Epithelial-Mesenchymal Transition Induces GSDME Transcriptional Activation for Inflammatory Pyroptosis. Front. Cell Dev. Biol. 9:781365. doi: 10.3389/fcell.2021.781365
Received: 22 September 2021; Accepted: 08 November 2021;
Published: 26 November 2021.
Edited by:
Jiyan Zhang, Independent researcher, Beijing, ChinaReviewed by:
Bhesh Raj Sharma, St. Jude Children’s Research Hospital, United StatesCopyright © 2021 Jia, Zhang, Tang, Cai, Zang, Shi, Sun, Wu, Shi, Shi, Ma, Zhao, Yu, Fu and Zhuang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaojing Zhao, emhhb3hpYW9qaW5nQHJlbmppLmNvbQ==; Zhuang Yu, eXV6aHVhbmcyMDAyQDE2My5jb20=; Yujie Fu, cmljaGFyZGppZWppZUBob3RtYWlsLmNvbQ==; Guanglei Zhuang, emh1YW5nZ3VhbmdsZWlAZ21haWwuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.