 Jianxin Deng
Jianxin Deng Yunxiu Liao
Yunxiu Liao Jianpin Liu2
Jianpin Liu2
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 24 December 2021
Sec. Epigenomics and Epigenetics
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.777258
This article is part of the Research TopicMetabolism and EpigeneticsView all 8 articles
Diabetic cardiomyopathy (DCM) is characterized by diastolic relaxation abnormalities in its initial stages and by clinical heart failure (HF) without dyslipidemia, hypertension, and coronary artery disease in its last stages. DCM contributes to the high mortality and morbidity rates observed in diabetic populations. Diabetes is a polygenic, heritable, and complex condition that is exacerbated by environmental factors. Recent studies have demonstrated that epigenetics directly or indirectly contribute to pathogenesis. While epigenetic mechanisms such as DNA methylation, histone modifications, and non-coding RNAs, have been recognized as key players in the pathogenesis of DCM, some of their impacts remain not well understood. Furthering our understanding of the roles played by epigenetics in DCM will provide novel avenues for DCM therapeutics and prevention strategies.
According to the World Health Organization, over 422 million people (∼6% of the global population) have been diagnosed with diabetes, a number that continues to increase each year (Sneha and Gangil, 2019). Diabetes that is accompanied by various other syndromes represents the leading cause of morbidity and mortality across the global. Clinical parameters such as poor glycemic control and insulin response to diabetes contribute to heart problems (Matheus et al., 2013). Recent studies have found that cardiovascular patients with diabetes have a worse prognosis than those without diabetes. The mortality due to cardiovascular disease in diabetic patients is s3-5 times higher than that in the general population (Rawshani et al., 2017; Fedeli et al., 2019). Diabetic cardiomyopathy (DCM), a cardiovascular complication associated with diabetes, is a severe form of cardiac dysfunction caused by changes in the structure and contractility of the myocardium (Dong et al., 2017). Approximately 12% of diabetic patients eventually develop severe heart failure (HF) and often die due to DCM (Trachanas et al., 2014). Additional comorbidities in this population of patients include hypertension, obesity, dyslipidemia, and vascular disease (Lorenzo-Almoros et al., 2017).
DCM is a serious complication of the myocardium of diabetic patients characterized by ventricular dilation and hypertrophy, diastolic dysfunction, decreased or preserved systolic function, and reduced ejection fraction, with no accompanying coronary artery disease or hypertension (Rubler et al., 1972). The diagnostic criteria for DCM include left ventricular diastolic dysfunction, left ventricular ejection fraction (EF) reduction, pathological left ventricular hypertrophy, and interstitial fibrosis (Fontes-Carvalho et al., 2015). Despite these devastating effects, there are still no effective and specific tools to diagnose DCM. Numerous studies over the past decades have highlighted the complexity of DCM pathogenesis, identifying that multiple molecular mechanisms synergistically damage cardiomyocytes and impair heart function. The metabolic environment associated with diabetes (e.g., high blood sugar, increased circulating fatty acids and triglycerides, hyperinsulinemia, and increased inflammatory cytokines, that activate transcription factors and change various molecular pathways in cardiomyocytes), reduces myocardial contractility and causes cardiomyocyte dysfunction, cell damage, and death. Thus, the mechanisms underlying DCM pathogenesis are extremely complex and involve changes in 1), signal transduction (insulin signal, renin-angiotensin signal); 2), metabolism (glucose and lipid metabolism), calcium homeostasis, and mitochondrial function; 3), gene regulation (activation of transcription factors and epigenetic mechanisms); 4), post-translational modification of signaling proteins; 5), homeostasis of cellular processes such as apoptosis, autophagy, and endoplasmic reticulum stress (Figure 1).
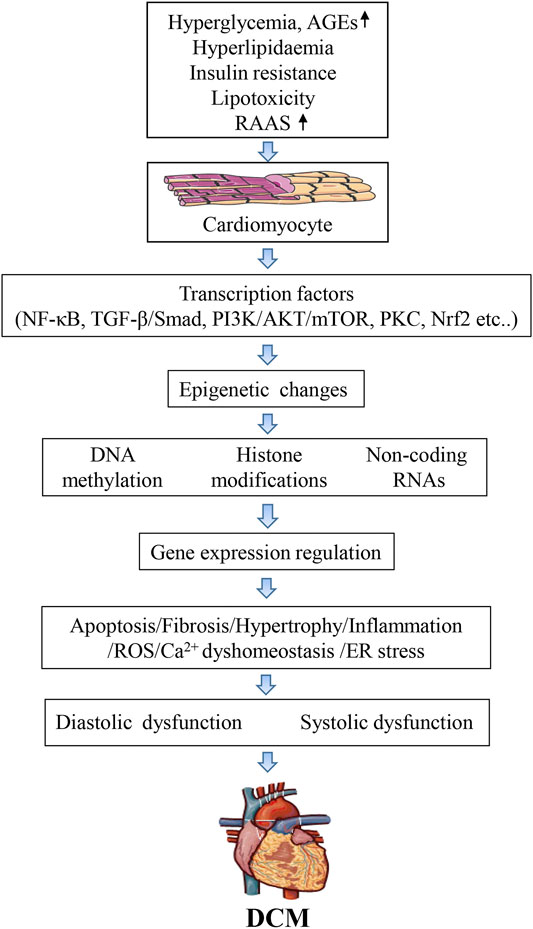
FIGURE 1. Mechanisms of epigenetic on diabetic cardiomyopathy.
Although DCM pathogenesis is multifactorial, hyperglycemia (Singh et al., 2018; Evangelista et al., 2019) is still considered a significant driver of myocardial damage (Seferovic and Paulus, 2015). Type 1 and type 2 diabetes(T2D) affect both systolic and diastolic function, and evidence from animal and the human study suggests that DCM occurs in T2D, affecting heart function and morphology and increasing the risk of HF (Holscher et al., 2016; Zhang et al., 2018; Yang et al., 2019). External stimuli or environmental factors often trigger this change. For example, changes in blood glucose levels can stress intracellular pathways that activate various transcription factors that alter global gene expression. Changes in the activity of transcription factors can ultimately lead to structural changes in the body (Lee et al., 2004). Dysregulation of coding and non-coding genes can lead to various cardiovascular diseases, including DCM (Dorn and Matkovich, 2008; Khraiwesh et al., 2010), and recent studies have implicated epigenetic and microRNAs (miRNAs) in this process. This review focuses on the role of epigenetic changes in DCM.
Epigenetics refers to heritable changes in gene expression that do not involve changes in nucleotide sequence. Epigenetic phenomena include DNA methylation, genomic imprinting, maternal effects, gene silencing, nucleolar dominance, dormant transposon activation, and RNA editing. Through these changes, the epigenome provides information about the structure of crucial functional elements that regulate gene expressions, such as methyl-labeled DNA and histones, and the interaction between the distal portions of chromatin (Romanoski et al., 2015). Epigenetic mechanisms such as DNA methylation, chromatin remodeling, and histone modifications regulate gene expression in response to change in the cellular microenvironment. These processes also play essential roles in HF (Egger et al., 2004; Mateo Leach et al., 2010), including HF that results from DCM. Although these epigenetic modifications can induce chronic disruptions in gene expression, recent studies have shown that complex interactions between genes and the environment may play an essential role in the pathogenesis of DCM and can be manipulated through diet, exercise, and drug interventions (Pepin and Wende, 2019).
DNA methylation is an important epigenetic mechanism and is one of the most widely studied epigenetic markers (Ambrosi et al., 2017). As a significant mechanism of epigenetic regulation in mammalian cells, DNA methylation is essential for mammalian development and plays a critical role in gene silencing, genomic stability, and parental imprinting during mitosis (Smith and Meissner, 2013). DNA methylation refers to the biochemical reaction of adding methyl groups to DNA nucleotides (cytosine or adenine) which is performed by DNA methyltransferases (DNMTs, Figure 2). The most frequently methylated nucleotides are cytosine residues located within CpG dinucleotides (Jin and Liu, 2018). They are usually found at the 5′ end of many gene regulatory regions but can extend into the exons (Deaton and Bird, 2011). CpG dinucleotides are not evenly distributed and can be categorized into dense CpG islands with CpG sequences or scattered out-of-island regions (Rodriguez-Rodero et al., 2017). In normal human somatic cells, 70%–90% of CpG dinucleotides are methylated (Ehrlich et al., 1982; Shaknovich et al., 2010). Cytosine methylation affects the expression of diabetes-related genes by changing the chromatin structure and altering the accessibility of transcription machinery. In addition to regulating the expression of various genes, methylation plays a vital role in cell differentiation and female X-chromosome inactivation (Shi et al., 2016). Repeating genomic sequences are extremely unstable, and their hypermethylation may prevent chromosomal instability, easy breakage, translocation, and gene disruption caused by the repeating DNA sequence, which can promote the expression of genes on that chromosome (Singh et al., 2011).
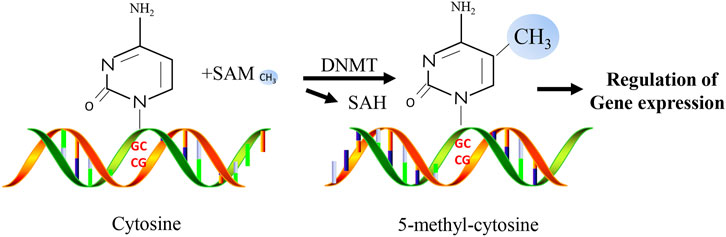
FIGURE 2. Simplified overview of molecular mechanisms of DNA methylation.
DNA methylation is an important epigenetic mechanism that controls cell differentiation and transcriptional potential in mammals. It regulates gene expression by inhibiting the binding of transcription factors to DNA. Many factors affect DNA methylation status, including the environment, diet, and aging. Recent studies have shown that abnormal DNA methylation is closely related to the occurrence and development of many cardiovascular diseases, such as coronary heart disease (Kim et al., 2010; Navas-Acien et al., 2021), atherosclerosis (AS) (Tao et al., 2021), hypertension (Dupriest et al., 2020; Amenyah et al., 2021), HF (Kao et al., 2010; Haas et al., 2013; Madsen et al., 2020), and DCM (Liu ZZ. et al., 2014; Guo et al., 2021).
Studies have shown that CpG islands are present in the promoter region of the sarcoplasmic reticulum Ca2+-ATPase (SERCA2a). In diabetic hyperglycemia, in vivo stimulation of cardiomyocytes with pro-inflammatory tumor necrosis factor-alpha (TNF-α: 50 ng/ml) increases methylation of the SERCA2a promoter region by increasing the level of DNA methyltransferase, which reduce the expression of SERCA2a (Kao et al., 2010). SERCA2a mediates the relaxation of the heart by transferring Ca2+ from the cells into the sarcoplasmic reticulum. Downregulation of SERCA2 expression can cause diastolic dysfunction and ultimately lead to the development of DCM. Changes in TNF-α expression in cardiomyocytes may underlie HF in diabetic patients since there is a positive correlation between HF severity and TNF-α (Asrih and Steffens, 2013). El-Osta et al. have shown that transient exposure of aortic endothelial cells to hyperglycemia induces sustained-onset epigenetic changes in the promoter of the NF-κB p65 subunit, which leads to increased expression of the p65 gene (El-Osta et al., 2008). Pirola et al. found that hyperglycemia can alter CpG methylation and, thus gene expression under diabetic conditions. A study reported that Keap1 protein expression was increased by demethylation of CpG islands in the promoter and reduced Nrf2 activity, thus inhibiting the transcription of various antioxidant genes and ultimately disrupting the redox balance in diabetes (Liu ZZ. et al., 2014). Activation of the renin-angiotensin-aldosterone system pathway plays a significant role in DCM. In DCM, genes in this pathway are typically upregulated, which eventually leads to myocardial hypertrophy. Bogdarina et al. showed that the proximal promoter of the angiotensin II type-1b (AT1b) gene is hypomethylated in the adrenal gland, and AT1b expression highly depends on promoter methylation in vitro (Bogdarina et al., 2007).
Hyperglycemia significantly affects human vascular chromatin and transcriptional upregulation of genes involved in metabolism and cardiovascular diseases (Pirola et al., 2011). The Epidemiology of Diabetes Interventions and Complications (EDIC) study examined DNA methylation profiles in whole blood isolated at baseline. The results indicate that changes in DNA methylation differences during diabetes persist at specific sites associated with blood glucose for several years. There was also evidence of persistent hypomethylation of the thioredoxin-interacting protein (TXNIP) gene associated with hyperglycemia and related complications (Chen et al., 2016). Recent studies identified that liver X receptor alpha (LXRα) is expressed in the myocardium of diabetic rats induced by streptozotocin (STZ). In addition, there are significant differences in the methylation status of the LXRα gene in the ventricles of control rats compared to the status in diabetic rats (Cheng et al., 2011).
A clinical study showed that age-related increases in methylation are negatively correlated with hepatic liver glucokinase (Gck) expression by studying the degree of Gck methylation and GcK expression in three age groups (Jiang et al., 2008). These results suggest that DNA methylation plays a significant role in increasing age-dependent insulin resistance and susceptibility to diabetes. Insulin resistance impairs heart contractility and increases oxidative stress, leading to cardiomyocyte apoptosis, myocardial fibrosis, remodeling, and cardiac hypertrophy, and thus resulting in DCM (Dobrin and Lebeche, 2010; Chemaly et al., 2011; Bobbert et al., 2012; Lebeche, 2015). Recent studies have shown that cardiac insulin resistance significantly contributes to the pathogenesis and progression of HF (Saotome et al., 2019).
JunD, a member of the activator protein 1 (AP-1) family transcription factors, stimulates or inhibits the expression of a variety of genes. JunD is under several layers of regulation, including transcriptional, post-transcriptional, protein post-translational modification, and protein-protein interactions. JunD is involved in the occurrence and development of DCM. Hussain et al. reported that the levels of JunD mRNA and protein are downregulated in the heart of patients with T2D and STZ-induced diabetic mice. JunD is epigenetically regulated by promoter hypermethylation, post-translational histone modifications, and miRNA-mediated translational repression by miR-673/menin axis. This indicates that multiple epigenetic mechanisms can synergize o alter gene expression rather than acting independently (Hussain et al., 2020). In addition, this cell-type-specific analysis revealed that gene programs associated with distinct biological processes are differentially regulated in diabetes.
Interestingly, despite these changes in gene expression, cell-type-specific DNA methylation signatures in genic and regulatory regions remain stable in diabetes. Analysis of heterocellular interactions in the diabetic heart suggests that the interplay between fibroblasts and monocytes is pivotal. A previous study showed that diabetes could change gene expression but not DNA methylation in cardiac cells (Lother et al., 2021).
Other regulatory factors are also involved in the occurrence and development of DCM. BRD4 is a member of the BET (bromodomain and extra-terminal domain) family of epigenetic regulators. High expression of BRD4 results in cardiac hypertrophy and plays an essential role in the pathogenesis of high glucose-induced cardiomyocyte hypertrophy through the AKT pathway in H9C2 cells and a diabetes rat model (Wang et al., 2019). In addition, JQ1 inhibits BRD4, improves mitochondrial function, and repairs cardiac structure and function by activating PINK1/Parkin-mediated mitophagy in high-fat diet-induced DCM (Mu et al., 2020).
In summary, these findings indicate that epigenetic marks such as DNA methylation play a significant role in DCM (Table 1).
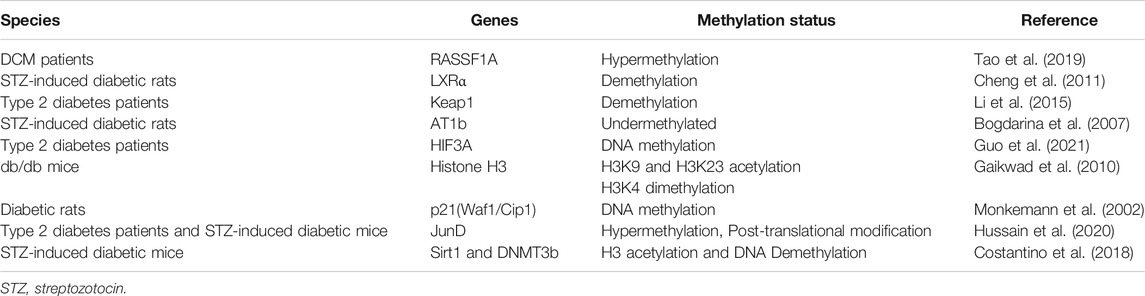
TABLE 1. Summary of studies examining DNA methylation in DCM.
Currently, there are 5 classes of histones identified in mammals: H1, H2A, H2B, H3, and H4. Histone modification mechanism. Histone modification is an epigenetic event that occurs via methylation, acetylation, phosphorylation, adenylation, ubiquitination, and ADP ribosylation (Bauer and Martin, 2017). These modifications affect the transcriptional activity of associated genes.
Covalent post-translational modification of histones can alter genome stability in response to changes in the environment, resulting in alterations to gene expression in pathological states such as metabolic stress. Along with CpG methylation, histone modifications control the accessibility of nucleosomes for transcription. Histone modifications also influence the binding capacity of other proteins to histones through changes in local hydrophobicity, RNA polymerase status, and binding affinity to other transcription coactivators. Various post-translational modifications can occur at the N-terminus of histones, including phosphorylation, acetylation, methylation, and ADP-ribosylation. It is challenging to decode specific post-translational modifications for individual histones or nucleosomes (e.g., location of nucleosomes regarding the gene transcriptional start site). However, histone modifications can communicate with each other. Factors including sites, types, and degrees of histone modifications contribute to the complexity of the histone code.
Histone acetylation and deacetylation, mediated through coactivator complexes containing histone acetyltransferases (HATs) and co-repressor complexes containing HDACs, respectively, represent the primary machineries controlling gene expression. HAT-mediated histone acetylation disengages intra- and inter-nucleosomal interactions to loosen the chromatin structure and turn on gene transcription. Histone changes through acetylation (attaching an acetyl group to lysine residues to neutralize its basic charge) or deacetylation using HDACs modulates the chromatin state (euchromatin is accessible whereas heterochromatin is inaccessible (Pasquier et al., 2015).
HDAC1 plays a significant role in the prevention and treatment of cardiac dysfunction. Resveratrol activates HDAC1 to prevent cardiomyocyte apoptosis and endoplasmic reticulum stress, reducing heart dysfunction in diabetic rats. SIRT1-dependent H3 deacetylation is also important to the response to myocardial injury and involves PERK/eIF2α, ATF6/CHOP, and IRE1α/JNK (Guo et al., 2015). The downregulation of Sirt1 and DNMT3b induced by diabetes promotes H3 acetylation and DNA demethylation of the p66Shc promoter, leading to DCM (Costantino et al., 2018). In addition, a recent study on obese mice reported that the lack of cardiac mitochondrial acetaldehyde dehydrogenase 2 affected the epigenetic SUV39H-SIRT1 loop, resulting in changes in transcription, autophagy, and myocardial metabolism (Wang et al., 2018).
We often refer to this relationship between glycemic control and the development of organ dysfunction as “blood sugar memory.” Unstable blood glucose can lead to DCM (Pepin and Wende, 2019) due to a combination of the release of ROS from chronic exposure to hyperglycemia (Lebeche, 2015), histone H3K4 methylation, and epigenetic activation of NF-κB-p65 in aortic vascular endothelial cells (El-Osta et al., 2008), and eventually lead to DCM. El-Osta et al. further studied the expression of the p65 gene and found that in transient hyperglycemic conditions, glucose inhibited the H3K9me2 and H3K9me3 marks on the p65 promoter and promoted the H3K4me1 mark (Brasacchio et al., 2009). Furthermore, histone lysine methyltransferase, SET7/9 (a novel coactivator of NF-κB), can target histone H3K4, enhance its methylation, and increase in NF-κB expression through histone methylation. Studies have shown that NF-κB-p65 is involved in the pathogenesis of pathological cardiac hypertrophy (Xu et al., 2015). Moreover, telmisartan and esculetin attenuate increases in histone modifications, such as H3K9me2, H3K9Ac, H2AK119Ub, and H2BK120Ub in the heart of T2D rats and ameliorate type 2 DCM by reversing H3, H2A, and H2B histone modifications (Kadakol et al., 2017). This suggests that histone modifications induced by exposure to high blood glucose concentrations play an essential role in DCM.
Histone dysregulation caused by environmental factors can synergize with hyperglycemia to induce diabetic complications. Gaikwad et al. reported a metabolic abnormality associated with renal failure in diabetic nephropathy. Renal metabolic disorders alter histone H3 acetylation in diabetic mice, further damaging myocardial cells. Examining the cross-sections of the hearts of the diabetic mice demonstrated that renal failure increases myocardial disease-related gene expression and cardiomyocyte hypertrophy (one characteristic of DCM) rather than cardiomyocyte proliferation (Gaikwad et al., 2010). Cardiac histone H3 modification caused by diabetic nephropathy thus plays a significant role in driving DCM.
The role of HDACs in cardiac hypertrophy and failure is complex, with some displaying antihypertrophic properties, whereas others exhibit pro-hypertrophic features. Recent studies have shown that HDACs can improve myocardial function and inhibit cardiac remodeling in DCM, as HDACs can improve cardiac function and inhibit myocardial remodeling in diabetic hearts (Chen et al., 2015). Kronlage et al. showed that O-GlcNAcylation of HDAC4 alleviates HF in diabetes and found that O-GlcNAcylation of HDAC4 at serine (Ser)-642 is cardioprotective in DCM and inhibits Ca2+/calmodulin-dependent protein kinase II signaling (Kronlage et al., 2019).
Taken together, these studies suggest that histone acetylation plays a significant role in regulating gene expression associated with diabetic complications, including DCM. In diabetes, endothelial cells are exposed to hyperglycemia, and histone acetylation is increased in the promoters of crucial genes in the extracellular matrix, leading to increased human p300 (Mathiyalagan et al., 2010), vascular diabetic complications and cardiomyocyte hypertrophy.
In addition, histone modification affects blood glucose memory in cardiomyocytes, mainly through changes in the expression of important genes.
Accumulating evidence suggests that pathological hypertrophy and cardiac remodeling may contribute to DCM. Non-coding RNAs, including long non-coding RNAs (lncRNAs) and small non-coding RNAs (e.g., miRNAs, short-interfering RNA, and piwi-interacting RNAs), play important roles in DCM. MiRNAs are ubiquitous in eukaryotic cells and are about 21–23 nucleotides in length. As endogenous post-translational inhibitors, miRNAs bind to the 3’end of complementary mRNA to degrade the mRNA or inhibit translation, thereby reducing the expression of the target gene. lncRNA usually regulates protein through processes such as competitive inhibition and recruitment. They also participate in the entire transcription process and play important roles as scaffolds that provide platforms for the interaction between chromatin modification complexes and transcription complexes.
In recent years, small RNAs, especially miRNAs, have gained increasing attention in cardiovascular disease studies. Exploring the regulatory importance of small RNAs in DCM will facilitate the development of new DCM therapeutic.
Small RNAs negatively modulate gene expression, primarily through binding to the target mRNA and subsequently inducing their degradation or suppressing translation. In short, small RNAs can regulate DCM pathogenesis through the aggravation of myocardial fibrosis, oxidative stress, apoptosis, cardiac electrical remodeling, or epigenetic modification (Table 2). Recent studies have identified that miRNAs play a vital role in the etiology of DCM (Guo and Nair, 2017; Xia and Song, 2020). By upregulating or down-regulating different target genes, the same miRNA can play many roles in cardiomyocyte or myocardial fiber pathology (Xia and Song, 2020). Feng et al. reported that miR-133a was downregulated in hypertrophic cardiac tissue under high glucose conditions, and miR-133a overexpression prevented hypertrophic changes in cardiomyocytes (Feng et al., 2010). Different miRNAs can also synergistically regulate DCM by upregulating or down-regulating target genes (Xia and Song, 2020). According to previous reports, multiple miRNAs, including miR-1 (Ikeda et al., 2009), miR-133a (Zhu Y.-F. et al., 2021), miR-351 (Zhu Y.-F. et al., 2021), miR-199a (Li et al.,2017), and miR-451 (Kuwabara et al., 2015), regulate cardiac hypertrophy in DCM by upregulating or down-regulating target genes. In addition, some miRNAs only alter the levels of one target gene under high-glucose conditions. They achieve DCM pathological changes by singly up- or down-regulating target genes under high glucose conditions. Liang et al. revealed that knockdown of miR-451 attenuated cardiac fibrosis and improved cardiac function by suppressing the endothelial-to-mesenchymal transition (EndMT) in diabetic mouse hearts (Liang et al., 2019).
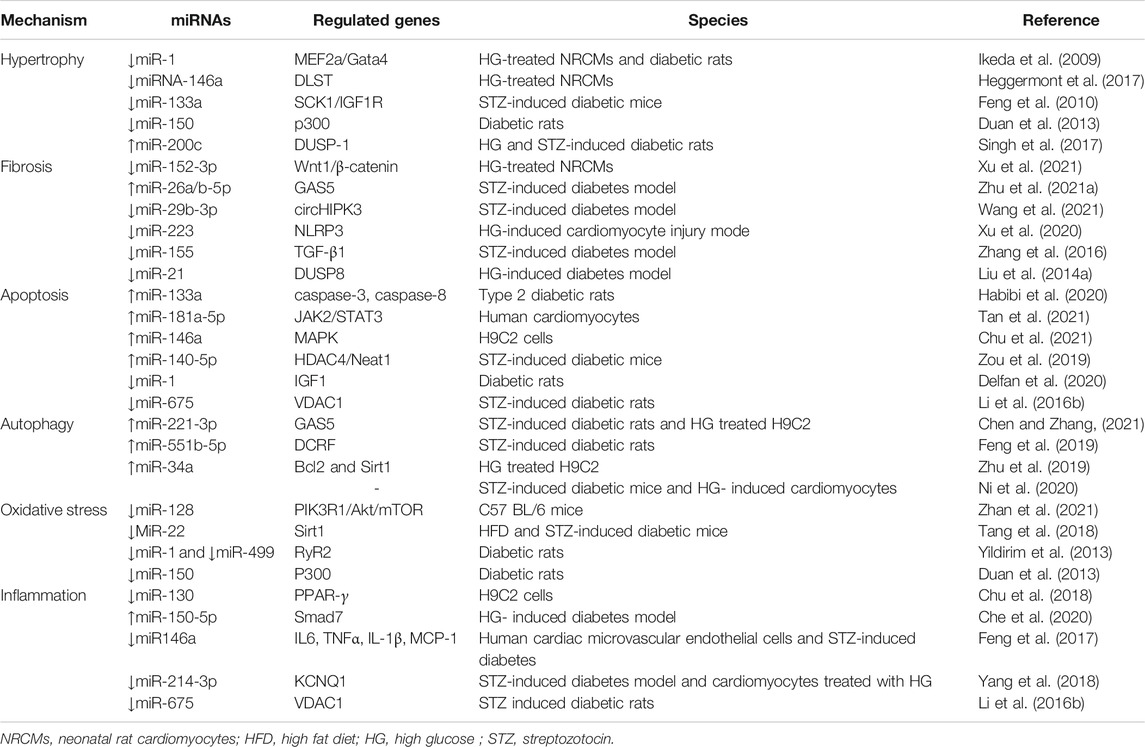
TABLE 2. Summary of miRNAs involved in the pathogenesis of DCM.
The above findings reveal a novel mechanism of DCM pathogenesis involving small RNAs, provide new strategies for the clinical diagnosis, treatment, or prevention of DCM, and highlight potential strategies for the development of small RNA-based therapies to treat diabetes-related cardiovascular complications. Li et al. demonstrated that inhibiting miR-320 could rescue DCM in diabetic mice (Li et al., 2019). This suggests that targeting miR-320 may represent a potential therapeutic strategy to treat diabetes-induced cardiac dysfunction. A recent study has identified miRNA-497 as a potential therapeutic agent for diabetic wound healing, owing to its repressive effect on pro-inflammatory cytokines (Ban et al., 2020). Furthermore, some studies indicate that as circulating miRNAs can be altered depending on the phase of the disease, they could also be used as potential biomarkers for assessing the development and progression of DCM. This also means that early intervention can prevent severe complications in DCM (Guo and Nair, 2017). In addition, other small RNAs, such as piwi-interacting RNAs, could provide therapeutic targets for the treatment of pathological hypertrophy and maladaptive cardiac remodeling (Gao et al., 2020).
In summary, epigenetics is an exciting and promising emerging research field in cardiovascular research and therapy. Achieving a better understanding of the roles played by epigenetics in DCM is an important strategy to improve early diagnosis and treatment of DCM, as well as the establishment of new therapeutics. In addition, drugs such as methylation and acetylation inhibitors represent promising candidates for targeted DCM therapies, and tissue-specific epigenetic modifier drugs may offer a fresh therapeutic perspective for DCM patients.
JD and YL wrote the article. JL and WL designed the graphs and tables; JD and DY revised and reviewed the article. All authors read and approved the final version of the article for publication.
This work was supported by the National Science Foundation of China (NO. 81670759 to JD), Basic Research Foundation of Shenzhen (NO. JCYJ20170306092910641 to JD), Bethune charitable foundation (NO. G-X-2019-056 to JD), and Sanming project of medicine in Shenzhen (NO.SZSM201612007 to DY)
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are particularly grateful to Dr. Long Chao for her professional revised and constructive suggestions.
AP-1, activator protein 1; AT1b, angiotensin II type 1b; ATF6: activating transcription factor 6; AKT/PKB, protein kinase B; BET, bromodomain and extra-terminal domain; BRD4, bromodomain-containing protein 4; CHOP, transcription factor C/EBP homologous protein; DCM, diabetic cardiomyopathy; DLST, dihydrolipoamide S-succinyltransferase; DNMTs, DNA methyltransferases; DNMT3b, DNA methyltransferase 3b; DUSP-1: dual specificity phosphatase 1; DUSP-8, dual specificity phosphatase 8; eIF2α, eukaryotic initiation factor 2 alpha; EndMT, endothelial-to-mesenchymal transition; GAS5, growth arrest-specific transcript 5; Gck, glucokinase; HAT, histone acetyltransferase; HDAC, histone deacetylase; HF, heart failure; HIF3A, hypoxia-inducible factor 3alpha; IRE1α, inositol-requiring protein 1alpha; IGF1R, insulin-like growth factor 1 receptor; JAK2, janus kinase 2; Keap1, Kelch-like ECH-associated protein 1; LXRα, liver X receptor α; MAPK, mitogen-activated protein kinases; Mef2a, myocyte enhancer factor 2a; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NLRP3: nod-like receptor protein 3; Nrf2, nuclear factor erythroid 2-related factor 2; PERK, Protein kinase RNA-like endoplasmic reticulum kinase; PPAR-γ, peroxisome proliferative activated receptor, gamma; RASSF1A, ras association domain family protein1 isoform A; RyR2, ryanodine receptor 2; SERCA2a, sarcoplasmic reticulum Ca2+-ATPase 2a; Sirt1, sirtuin 1; Smad7, SMAD family member 7; STAT3, signal transducer and activator of transcription 3; SUV39H1, suppresssor of variegation 3-9 homolog; T2D, type 2 diabetes; TNFα, tumor necrosis factor-alpha; TXNIP, thioredoxin-interacting protein; VDAC 1, voltage-dependent anion channel 1.
Ambrosi, C., Manzo, M., and Baubec, T. (2017). Dynamics and Context-Dependent Roles of DNA Methylation. J. Mol. Biol. 429, 1459–1475. doi:10.1016/j.jmb.2017.02.008
Amenyah, S. D., Ward, M., Mcmahon, A., Deane, J., Mcnulty, H., Hughes, C., et al. (2021). DNA Methylation of Hypertension-Related Genes and Effect of Riboflavin Supplementation in Adults Stratified by Genotype for the MTHFR C677T Polymorphism. Int. J. Cardiol. 322, 233–239. doi:10.1016/j.ijcard.2020.09.011
Asrih, M., and Steffens, S. (2013). Emerging Role of Epigenetics and miRNA in Diabetic Cardiomyopathy. Cardiovasc. Pathol. 22, 117–125. doi:10.1016/j.carpath.2012.07.004
Ban, E., Jeong, S., Park, M., Kwon, H., Park, J., Song, E. J., et al. (2020). Accelerated Wound Healing in Diabetic Mice by miRNA-497 and its Anti-Inflammatory Activity. Biomed. Pharmacother. 121, 109613. doi:10.1016/j.biopha.2019.109613
Bauer, A. J., and Martin, K. A. (2017). Coordinating Regulation of Gene Expression in Cardiovascular Disease: Interactions between Chromatin Modifiers and Transcription Factors. Front. Cardiovasc. Med. 4, 19. doi:10.3389/fcvm.2017.00019
Bobbert, P., Jenke, A., Bobbert, T., Kühl, U., Rauch, U., Lassner, D., et al. (2012). High Leptin and Resistin Expression in Chronic Heart Failure: Adverse Outcome in Patients with Dilated and Inflammatory Cardiomyopathy. Eur. J. Heart Fail. 14, 1265–1275. doi:10.1093/eurjhf/hfs111
Bogdarina, I., Welham, S., King, P. J., Burns, S. P., and Clark, A. J. L. (2007). Epigenetic Modification of the Renin-Angiotensin System in the Fetal Programming of Hypertension. Circ. Res. 100, 520–526. doi:10.1161/01.RES.0000258855.60637.58
Brasacchio, D., Okabe, J., Tikellis, C., Balcerczyk, A., George, P., Baker, E. K., et al. (2009). Hyperglycemia Induces a Dynamic Cooperativity of Histone Methylase and Demethylase Enzymes Associated with Gene-Activating Epigenetic marks that Coexist on the Lysine Tail. Diabetes 58, 1229–1236. doi:10.2337/db08-1666
Che, H., Wang, Y., Li, Y., Lv, J., Li, H., Liu, Y., et al. (2020). Inhibition of microRNA‐150‐5p Alleviates Cardiac Inflammation and Fibrosis via Targeting Smad7 in High Glucose‐Treated Cardiac Fibroblasts. J. Cel Physiol. 235, 7769–7779. doi:10.1002/jcp.29386
Chemaly, E. R., Hadri, L., Zhang, S., Kim, M., Kohlbrenner, E., Sheng, J., et al. (2011). Long-Term In Vivo Resistin Overexpression Induces Myocardial Dysfunction and Remodeling in Rats. J. Mol. Cell Cardiol. 51, 144–155. doi:10.1016/j.yjmcc.2011.04.006
Chen, D., and Zhang, M. (2021). GAS5 Regulates Diabetic Cardiomyopathy via miR-221-3p/p27 A-xis-associated A-utophagy. Mol. Med. Rep. 23. doi:10.3892/mmr.2020.11774
Chen, Y., Du, J., Zhao, Y. T., Zhang, L., Lv, G., Zhuang, S., et al. (2015). Histone Deacetylase (HDAC) Inhibition Improves Myocardial Function and Prevents Cardiac Remodeling in Diabetic Mice. Cardiovasc. Diabetol. 14, 99. doi:10.1186/s12933-015-0262-8
Chen, Z., Miao, F., Paterson, A. D., Lachin, J. M., Zhang, L., Schones, D. E., et al. (2016). Epigenomic Profiling Reveals an Association between Persistence of DNA Methylation and Metabolic Memory in the DCCT/EDIC Type 1 Diabetes Cohort. Proc. Natl. Acad. Sci. USA. 113, E3002–E3011. doi:10.1073/pnas.1603712113
Cheng, Y., Liu, G., Pan, Q., Guo, S., and Yang, X. (2011). Elevated Expression of Liver X Receptor Alpha (LXRα) in Myocardium of Streptozotocin-Induced Diabetic Rats. Inflammation 34, 698–706. doi:10.1007/s10753-010-9281-5
Chu, X., Wang, Y., Pang, L., Huang, J., Sun, X., and Chen, X. (2018). miR‐130 Aggravates Acute Myocardial Infarction‐induced Myocardial Injury by Targeting PPAR‐γ. J. Cel Biochem. 119, 7235–7244. doi:10.1002/jcb.26903
Chu, Y., Teng, J., Feng, P., Liu, H., Wang, F., and Wang, H. (2021). Dexmedetomidine Attenuates Hypoxia/reoxygenation Injury of H9C2 Myocardial Cells by Upregulating miR-146a Expression via the MAPK Signal Pathway. Pharmacology, 1–14. doi:10.1159/000506814
Costantino, S., Paneni, F., Mitchell, K., Mohammed, S. A., Hussain, S., Gkolfos, C., et al. (2018). Hyperglycaemia-Induced Epigenetic Changes Drive Persistent Cardiac Dysfunction via the Adaptor P66 Shc. Int. J. Cardiol. 268, 179–186. doi:10.1016/j.ijcard.2018.04.082
Deaton, A. M., and Bird, A. (2011). CpG Islands and the Regulation of Transcription. Genes Dev. 25, 1010–1022. doi:10.1101/gad.2037511
Delfan, M., Delphan, M., Kordi, M. R., Ravasi, A. A., Safa, M., Gorgani-Firuzjaee, S., et al. (2020). High Intensity Interval Training Improves Diabetic Cardiomyopathy via miR-1 Dependent Suppression of Cardiomyocyte Apoptosis in Diabetic Rats. J. Diabetes Metab. Disord. 19, 145–152. doi:10.1007/s40200-019-00485-0
Dobrin, J. S., and Lebeche, D. (2010). Diabetic Cardiomyopathy: Signaling Defects and Therapeutic Approaches. Expert Rev. Cardiovasc. Ther. 8, 373–391. doi:10.1586/erc.10.17
Dong, S., Zhang, R., Liang, Y., Shi, J., Li, J., Shang, F., et al. (2017). Changes of Myocardial Lipidomics Profiling in a Rat Model of Diabetic Cardiomyopathy Using UPLC/Q-TOF/MS Analysis. Diabetol. Metab. Syndr. 9, 56. doi:10.1186/s13098-017-0249-6
Dorn, G. W., and Matkovich, S. J. (2008). Put Your Chips on Transcriptomics. Circulation 118, 216–218. doi:10.1161/CIRCULATIONAHA.108.789933
Duan, Y., Zhou, B., Su, H., Liu, Y., and Du, C. (2013). miR-150 Regulates High Glucose-Induced Cardiomyocyte Hypertrophy by Targeting the Transcriptional Co-Activator P300. Exp. Cel Res. 319, 173–184. doi:10.1016/j.yexcr.2012.11.015
Dupriest, E., Hebert, J., Morita, M., Marek, N., Meserve, E. E. K., Andeen, N., et al. (2020). Fetal Renal DNA Methylation and Developmental Programming of Stress-Induced Hypertension in Growth-Restricted Male Mice. Reprod. Sci. 27, 1110–1120. doi:10.1007/s43032-019-00121-5
Egger, G., Liang, G., Aparicio, A., and Jones, P. A. (2004). Epigenetics in Human Disease and Prospects for Epigenetic Therapy. Nature 429, 457–463. doi:10.1038/nature02625
Ehrlich, M., Gama-Sosa, M. A., Huang, L.-H., Midgett, R. M., Kuo, K. C., Mccune, R. A., et al. (1982). Amount and Distribution of 5-methylcytosine in Human DNA from Different Types of Tissues or Cells. Nucl. Acids Res. 10, 2709–2721. doi:10.1093/nar/10.8.2709
El-Osta, A., Brasacchio, D., Yao, D., Pocai, A., Jones, P. L., Roeder, R. G., et al. (2008). Transient High Glucose Causes Persistent Epigenetic Changes and Altered Gene Expression during Subsequent Normoglycemia. J. Exp. Med. 205, 2409–2417. doi:10.1084/jem.20081188
Evangelista, I., Nuti, R., Picchioni, T., Dotta, F., and Palazzuoli, A. (2019). Molecular Dysfunction and Phenotypic Derangement in Diabetic Cardiomyopathy. Int. J. Mol. Sci. 20, 3264. doi:10.3390/ijms20133264
Fedeli, U., Schievano, E., Targher, G., Bonora, E., Corti, M. C., and Zoppini, G. (2019). Estimating the Real burden of Cardiovascular Mortality in Diabetes. Eur. Rev. Med. Pharmacol. Sci. 23, 6700–6706. doi:10.26355/eurrev_201908_18561
Feng, B., Chen, S., George, B., Feng, Q., and Chakrabarti, S. (2010). miR133a Regulates Cardiomyocyte Hypertrophy in Diabetes. Diabetes Metab. Res. Rev. 26, 40–49. doi:10.1002/dmrr.1054
Feng, B., Chen, S., Gordon, A. D., and Chakrabarti, S. (2017). miR-146a Mediates Inflammatory Changes and Fibrosis in the Heart in Diabetes. J. Mol. Cell Cardiol. 105, 70–76. doi:10.1016/j.yjmcc.2017.03.002
Feng, Y., Xu, W., Zhang, W., Wang, W., Liu, T., and Zhou, X. (2019). LncRNA DCRF Regulates Cardiomyocyte Autophagy by Targeting miR-551b-5p in Diabetic Cardiomyopathy. Theranostics 9, 4558–4566. doi:10.7150/thno.31052
Fontes-Carvalho, R., Ladeiras-Lopes, R., Bettencourt, P., Leite-Moreira, A., and Azevedo, A. (2015). Diastolic Dysfunction in the Diabetic Continuum: Association with Insulin Resistance, Metabolic Syndrome and Type 2 Diabetes. Cardiovasc. Diabetol. 14, 4. doi:10.1186/s12933-014-0168-x
Gaikwad, A. B., Sayyed, S. G., Lichtnekert, J., Tikoo, K., and Anders, H.-J. (2010). Renal Failure Increases Cardiac Histone H3 Acetylation, Dimethylation, and Phosphorylation and the Induction of Cardiomyopathy-Related Genes in Type 2 Diabetes. Am. J. Pathol. 176, 1079–1083. doi:10.2353/ajpath.2010.090528
Gao, X.-Q., Zhang, Y.-H., Liu, F., Ponnusamy, M., Zhao, X.-M., Zhou, L.-Y., et al. (2020). The piRNA CHAPIR Regulates Cardiac Hypertrophy by Controlling METTL3-dependent N6-Methyladenosine Methylation of Parp10 mRNA. Nat. Cel Biol. 22, 1319–1331. doi:10.1038/s41556-020-0576-y
Guo, R., Liu, W., Liu, B., Zhang, B., Li, W., and Xu, Y. (2015). SIRT1 Suppresses Cardiomyocyte Apoptosis in Diabetic Cardiomyopathy: An Insight into Endoplasmic Reticulum Stress Response Mechanism. Int. J. Cardiol. 191, 36–45. doi:10.1016/j.ijcard.2015.04.245
Guo, R., and Nair, S. (2017). Role of microRNA in Diabetic Cardiomyopathy: From Mechanism to Intervention. Biochim. Biophys. Acta (Bba) - Mol. Basis Dis. 1863, 2070–2077. doi:10.1016/j.bbadis.2017.03.013
Guo, Y., Zou, J., Xu, X., Zhou, H., Sun, X., Wu, L., et al. (2021). Short-Chain Fatty Acids Combined with Intronic DNA Methylation of HIF3A: Potential Predictors for Diabetic Cardiomyopathy. Clin. Nutr. 40, 3708–3717. doi:10.1016/j.clnu.2021.04.007
Haas, J., Frese, K. S., Park, Y. J., Keller, A., Vogel, B., Lindroth, A. M., et al. (2013). Alterations in Cardiac DNA Methylation in Human Dilated Cardiomyopathy. EMBO Mol. Med. 5, 413–429. doi:10.1002/emmm.201201553
Habibi, P., Alihemmati, A., Ahmadiasl, N., Fateh, A., and Anvari, E. (2020). Exercise Training Attenuates Diabetes-Induced Cardiac Injury through Increasing miR-133a and Improving Pro-apoptosis/anti-apoptosis Balance in Ovariectomized Rats. Iran J. Basic Med. Sci. 23, 79–85. doi:10.22038/IJBMS.2019.36731.8750
Heggermont, W. A., Papageorgiou, A.-P., Quaegebeur, A., Deckx, S., Carai, P., Verhesen, W., et al. (2017). Inhibition of microRNA-146a and Overexpression of its Target Dihydrolipoyl Succinyltransferase Protect against Pressure Overload-Induced Cardiac Hypertrophy and Dysfunction. Circulation 136, 747–761. doi:10.1161/CIRCULATIONAHA.116.024171
Hölscher, M., Bode, C., and Bugger, H. (2016). Diabetic Cardiomyopathy: Does the Type of Diabetes Matter. Int. J. Mol. Sci. 17, 2136. doi:10.3390/ijms17122136
Hussain, S., Khan, A. W., Akhmedov, A., Suades, R., Costantino, S., Paneni, F., et al. (2020). Hyperglycemia Induces Myocardial Dysfunction via Epigenetic Regulation of JunD. Circ. Res. 127, 1261–1273. doi:10.1161/CIRCRESAHA.120.317132
Ikeda, S., He, A., Kong, S. W., Lu, J., Bejar, R., Bodyak, N., et al. (2009). MicroRNA-1 Negatively Regulates Expression of the Hypertrophy-Associated Calmodulin and Mef2a Genes. Mol. Cel Biol 29, 2193–2204. doi:10.1128/MCB.01222-08
Jiang, M. H., Fei, J., Lan, M. S., Lu, Z. P., Liu, M., Fan, W. W., et al. (2008). Hypermethylation of Hepatic Gck Promoter in Ageing Rats Contributes to Diabetogenic Potential. Diabetologia 51, 1525–1533. doi:10.1007/s00125-008-1034-8
Jin, Z., and Liu, Y. (2018). DNA Methylation in Human Diseases. Genes Dis. 5, 1–8. doi:10.1016/j.gendis.2018.01.002
Kadakol, A., Malek, V., Goru, S. K., Pandey, A., and Gaikwad, A. B. (2017). Telmisartan and Esculetin Combination Ameliorates Type 2 Diabetic Cardiomyopathy by Reversal of H3, H2A, and H2B Histone Modifications. Indian J. Pharmacol. 49, 348–356. doi:10.4103/ijp.IJP_710_16
Kao, Y.-H., Chen, Y.-C., Cheng, C.-C., Lee, T.-I., Chen, Y.-J., and Chen, S.-A. (2010). Tumor Necrosis Factor-α Decreases Sarcoplasmic Reticulum Ca2+-ATPase Expressions via the Promoter Methylation in Cardiomyocytes*. Crit. Care Med. 38, 217–222. doi:10.1097/CCM.0b013e3181b4a854
Khraiwesh, B., Arif, M. A., Seumel, G. I., Ossowski, S., Weigel, D., Reski, R., et al. (2010). Transcriptional Control of Gene Expression by microRNAs. Cell 140, 111–122. doi:10.1016/j.cell.2009.12.023
Kim, M., Long, T. I., Arakawa, K., Wang, R., Yu, M. C., and Laird, P. W. (2010). DNA Methylation as a Biomarker for Cardiovascular Disease Risk. PLoS One 5, e9692. doi:10.1371/journal.pone.0009692
Kronlage, M., Dewenter, M., Grosso, J., Fleming, T., Oehl, U., Lehmann, L. H., et al. (2019). O-GlcNAcylation of Histone Deacetylase 4 Protects the Diabetic Heart from Failure. Circulation 140, 580–594. doi:10.1161/CIRCULATIONAHA.117.031942
Kuwabara, Y., Horie, T., Baba, O., Watanabe, S., Nishiga, M., Usami, S., et al. (2015). MicroRNA-451 Exacerbates Lipotoxicity in Cardiac Myocytes and High-Fat Diet-Induced Cardiac Hypertrophy in Mice through Suppression of the LKB1/AMPK Pathway. Circ. Res. 116, 279–288. doi:10.1161/CIRCRESAHA.116.304707
Lebeche, D. (2015). Diabetic Cardiomyopathy: Is Resistin a Culprit. Cardiovasc. Diagn. Ther. 5, 387–393. doi:10.3978/j.issn.2223-3652.2015.05.04
Lee, Y., Kim, M., Han, J., Yeom, K.-H., Lee, S., Baek, S. H., et al. (2004). MicroRNA Genes Are Transcribed by RNA Polymerase II. EMBO J. 23, 4051–4060. doi:10.1038/sj.emboj.7600385
Li, H., Fan, J., Zhao, Y., Zhang, X., Dai, B., Zhan, J., et al. (2019). Nuclear miR-320 Mediates Diabetes-Induced Cardiac Dysfunction by Activating Transcription of Fatty Acid Metabolic Genes to Cause Lipotoxicity in the Heart. Circ. Res. 125, 1106–1120. doi:10.1161/CIRCRESAHA.119.314898
Li, K. K., Lau, K. M., and Ng, H. K. (2015). Signaling Pathway and Molecular Subgroups of Medulloblastoma. Int J Clin Exp Pathol. 2013; 6(7): 1211-22. Int. J. Clin. Exp. Patholint J. Clin. Exp. Pathol. 8 (9), 11945. doi:10.4103/0377-4929.116164
Li, X., Wang, H., Yao, B., Xu, W., Chen, J., and Zhou, X. (2016b). lncRNA H19/miR-675 axis Regulates Cardiomyocyte Apoptosis by Targeting VDAC1 in Diabetic Cardiomyopathy. Sci. Rep. 6, 36340. doi:10.1038/srep36340
Li, Z., Song, Y., Liu, L., Hou, N., An, X., Zhan, D., et al. (2017). miR-199a Impairs Autophagy and Induces Cardiac Hypertrophy through mTOR Activation. Cell Death Differ 24, 1205–1213. doi:10.1038/cdd.2015.95
Liang, C., Gao, L., Liu, Y., Liu, Y., Yao, R., Li, Y., et al. (2019). MiR-451 Antagonist Protects against Cardiac Fibrosis in Streptozotocin-Induced Diabetic Mouse Heart. Life Sci. 224, 12–22. doi:10.1016/j.lfs.2019.02.059
Liu, S., Li, W., Xu, M., Huang, H., Wang, J., and Chen, X. (2014a). Micro-RNA 21Targets Dual Specific Phosphatase 8 to Promote Collagen Synthesis in High Glucose-Treated Primary Cardiac Fibroblasts. Can. J. Cardiol. 30, 1689–1699. doi:10.1016/j.cjca.2014.07.747
Liu, Z. Z., Zhao, X. Z., Zhang, X. S., and Zhang, M. (2014b). Promoter DNA Demethylation of Keap1 Gene in Diabetic Cardiomyopathy. Int. J. Clin. Exp. Pathol. 7, 8756–8762.
Lorenzo-Almorós, A., Tuñón, J., Orejas, M., Cortés, M., Egido, J., and Lorenzo, Ó. (2017). Diagnostic Approaches for Diabetic Cardiomyopathy. Cardiovasc. Diabetol. 16, 28. doi:10.1186/s12933-017-0506-x
Lother, A., Bondareva, O., Saadatmand, A. R., Pollmeier, L., Härdtner, C., Hilgendorf, I., et al. (2021). Diabetes Changes Gene Expression but Not DNA Methylation in Cardiac Cells. J. Mol. Cell Cardiol. 151, 74–87. doi:10.1016/j.yjmcc.2020.11.004
Madsen, A., Höppner, G., Krause, J., Hirt, M. N., Laufer, S. D., Schweizer, M., et al. (2020). An Important Role for DNMT3A-Mediated DNA Methylation in Cardiomyocyte Metabolism and Contractility. Circulation 142, 1562–1578. doi:10.1161/CIRCULATIONAHA.119.044444
Mateo Leach, I., Van Der Harst, P., and De Boer, R. A. (2010). Pharmacoepigenetics in Heart Failure. Curr. Heart Fail. Rep. 7, 83–90. doi:10.1007/s11897-010-0011-y
Matheus, A. S. d. M., Tannus, L. R. M., Cobas, R. A., Palma, C. C. S., Negrato, C. A., and Gomes, M. d. B. (2013). Impact of Diabetes on Cardiovascular Disease: an Update. Int. J. Hypertens. 2013, 1–15. doi:10.1155/2013/653789
Mathiyalagan, P., Chang, L., Du, X.-J., and El-Osta, A. (2010). Cardiac Ventricular chambers Are Epigenetically Distinguishable. Cell Cycle 9, 612–617. doi:10.4161/cc.9.3.10612
Mönkemann, H., De Vriese, A. S., Blom, H. J., Kluijtmans, L. A. J., Heil, S. G., Schild, H. H., et al. (2002). Early Molecular Events in the Development of the Diabetic Cardiomyopathy. Amino Acids 23, 331–336. doi:10.1007/s00726-001-0146-y
Mu, J., Zhang, D., Tian, Y., Xie, Z., and Zou, M.-h. (2020). BRD4 Inhibition by JQ1 Prevents High-Fat Diet-Induced Diabetic Cardiomyopathy by Activating PINK1/Parkin-Mediated Mitophagy In Vivo. J. Mol. Cell Cardiol. 149, 1–14. doi:10.1016/j.yjmcc.2020.09.003
Navas-Acien, A., Domingo-Relloso, A., Subedi, P., Riffo-Campos, A. L., Xia, R., Gomez, L., et al. (2021). Blood DNA Methylation and Incident Coronary Heart Disease. JAMA Cardiol. 6, 1237. doi:10.1001/jamacardio.2021.2704
Ni, T., Lin, N., Lu, W., Sun, Z., Lin, H., Chi, J., et al. (2020). Dihydromyricetin Prevents Diabetic Cardiomyopathy via miR-34a Suppression by Activating Autophagy. Cardiovasc. Drugs Ther. 34, 291–301. doi:10.1007/s10557-020-06968-0
Pasquier, J., Fakhro, K., Rafii, A., and Abi Khalil, C. (2015). Epigenetics and Cardiovascular Disease in Diabetes. Curr. Diab. Rep. 15, 108. doi:10.1007/s11892-015-0677-3
Pepin, M. E., and Wende, A. R. (2019). Epigenetics in the Development of Diabetic Cardiomyopathy. Epigenomics 11, 469–472. doi:10.2217/epi-2019-0027
Pirola, L., Balcerczyk, A., Tothill, R. W., Haviv, I., Kaspi, A., Lunke, S., et al. (2011). Genome-wide Analysis Distinguishes Hyperglycemia Regulated Epigenetic Signatures of Primary Vascular Cells. Genome Res. 21, 1601–1615. doi:10.1101/gr.116095.110
Rawshani, A., Rawshani, A., Franzén, S., Eliasson, B., Svensson, A.-M., Miftaraj, M., et al. (2017). Mortality and Cardiovascular Disease in Type 1 and Type 2 Diabetes. N. Engl. J. Med. 376, 1407–1418. doi:10.1056/NEJMoa1608664
Rodríguez-Rodero, S., Delgado-Álvarez, E., Díaz-Naya, L., Martín Nieto, A., and Menéndez Torre, E. (2017). Epigenetic Modulators of Thyroid Cancer. Endocrinología, Diabetes y Nutrición 64, 44–56. doi:10.1016/j.endinu.2016.09.006
Romanoski, C. E., Glass, C. K., Stunnenberg, H. G., Wilson, L., and Almouzni, G. (2015). Roadmap for Regulation. Nature 518, 314–316. doi:10.1038/518314a
Rubler, S., Dlugash, J., Yuceoglu, Y. Z., Kumral, T., Branwood, A. W., and Grishman, A. (1972). New Type of Cardiomyopathy Associated with Diabetic Glomerulosclerosis. Am. J. Cardiol. 30, 595–602. doi:10.1016/0002-9149(72)90595-4
Saotome, M., Ikoma, T., Hasan, P., and Maekawa, Y. (2019). Cardiac Insulin Resistance in Heart Failure: the Role of Mitochondrial Dynamics. Int. J. Mol. Sci. 20, 3552. doi:10.3390/ijms20143552
Seferović, P. M., and Paulus, W. J. (2015). Clinical Diabetic Cardiomyopathy: a Two-Faced Disease with Restrictive and Dilated Phenotypes. Eur. Heart J. 36, 17181727a–17271727c. doi:10.1093/eurheartj/ehv134
Shaknovich, R., Figueroa, M. E., and Melnick, A. (2010). HELP (HpaII Tiny Fragment Enrichment by Ligation-Mediated PCR) Assay for DNA Methylation Profiling of Primary normal and Malignant B Lymphocytes. Methods Mol. Biol. 632, 191–201. doi:10.1007/978-1-60761-663-4_12
Shi, Y. J., Li, Q. H., and Liu, X. H. (2016). Progress in Studies of DNA Methylation and Gene Expression Regulation. China Biotechnol. 33, 90–96.
Singh, G. B., Raut, S. K., Khanna, S., Kumar, A., Sharma, S., Prasad, R., et al. (2017). MicroRNA-200c Modulates DUSP-1 Expression in Diabetes-Induced Cardiac Hypertrophy. Mol. Cel Biochem 424, 1–11. doi:10.1007/s11010-016-2838-3
Singh, G. B., Sharma, R., and Khullar, M. (2011). Epigenetics and Diabetic Cardiomyopathy. Diabetes Res. Clin. Pract. 94, 14–21. doi:10.1016/j.diabres.2011.05.033
Singh, R. M., Waqar, T., Howarth, F. C., Adeghate, E., Bidasee, K., and Singh, J. (2018). Hyperglycemia-Induced Cardiac Contractile Dysfunction in the Diabetic Heart. Heart Fail. Rev. 23, 37–54. doi:10.1007/s10741-017-9663-y
Smith, Z. D., and Meissner, A. (2013). DNA Methylation: Roles in Mammalian Development. Nat. Rev. Genet. 14, 204–220. doi:10.1038/nrg3354
Sneha, N., and Gangil, T. (2019). Analysis of Diabetes Mellitus for Early Prediction Using Optimal Features Selection. J. Big Data. 6, 13. doi:10.1186/s40537-019-0175-6
Tan, J.-K., Ma, X.-F., Wang, G.-N., Jiang, C.-R., Gong, H.-Q., and Liu, H. (2021). LncRNA MIAT Knockdown Alleviates Oxygen-Glucose Deprivation-induced C-ardiomyocyte I-njury by R-egulating JAK2/STAT3 P-athway via miR-181a-5p. J. Cardiol. 78, 586–597. doi:10.1016/j.jjcc.2021.08.018
Tang, Q., Len, Q., Liu, Z., and Wang, W. (2018). Overexpression of miR-22 Attenuates Oxidative Stress Injury in Diabetic Cardiomyopathy via Sirt 1. Cardiovasc. Ther. 36, e12318. doi:10.1111/1755-5922.12318
Tao, H., Tao, J.-Y., Song, Z.-Y., Shi, P., Wang, Q., Deng, Z.-Y., et al. (2019). MeCP2 Triggers Diabetic Cardiomyopathy and Cardiac Fibroblast Proliferation by Inhibiting RASSF1A. Cell Signal. 63, 109387. doi:10.1016/j.cellsig.2019.109387
Tao, J., Xia, L., Cai, Z., Liang, L., Chen, Y., Meng, J., et al. (2021). Interaction between microRNA and DNA Methylation in Atherosclerosis. DNA Cel Biol. 40, 101–115. doi:10.1089/dna.2020.6138
Trachanas, K., Sideris, S., Aggeli, C., Poulidakis, E., Gatzoulis, K., Tousoulis, D., et al. (2014). Diabetic Cardiomyopathy: from Pathophysiology to Treatment. Hellenic J. Cardiol. 55, 411–421.
Wang, Q., Sun, Y., Li, T., Liu, L., Zhao, Y., Li, L., et al. (2019). Function of BRD4 in the Pathogenesis of High Glucose-induced C-ardiac H-ypertrophy. Mol. Med. Rep. 19, 499–507. doi:10.3892/mmr.2018.9681
Wang, S., Wang, C., Turdi, S., Richmond, K. L., Zhang, Y., and Ren, J. (2018). ALDH2 Protects against High Fat Diet-Induced Obesity Cardiomyopathy and Defective Autophagy: Role of CaM Kinase II, Histone H3K9 Methyltransferase SUV39H, Sirt1, and PGC-1α Deacetylation. Int. J. Obes. 42, 1073–1087. doi:10.1038/s41366-018-0030-4
Wang, W., Zhang, S., Xu, L., Feng, Y., Wu, X., Zhang, M., et al. (2021). Involvement of circHIPK3 in the Pathogenesis of Diabetic Cardiomyopathy in Mice. Diabetologia 64, 681–692. doi:10.1007/s00125-020-05353-8
Xia, L., and Song, M. (2020). Role of Non-coding RNA in Diabetic Cardiomyopathy. Adv. Exp. Med. Biol. 1229, 181–195. doi:10.1007/978-981-15-1671-9_10
Xu, D., Zhang, X., Chen, X., Yang, S., and Chen, H. (2020). Inhibition of miR-223 Attenuates the NLRP3 Inflammasome Activation, Fibrosis, and Apoptosis in Diabetic Cardiomyopathy. Life Sci. 256, 117980. doi:10.1016/j.lfs.2020.117980
Xu, S.-S., Ding, J.-F., Shi, P., Shi, K.-H., and Tao, H. (2021). DNMT1-induced miR-152-3p Suppression Facilitates Cardiac Fibroblast Activation in Cardiac Fibrosis. Cardiovasc. Toxicol. 21, 984–999. doi:10.1007/s12012-021-09690-x
Xu, T., Zhang, B., Yang, F., Cai, C., Wang, G., Han, Q., et al. (2015). HSF1 and NF-Κb P65 Participate in the Process of Exercise Preconditioning Attenuating Pressure Overload-Induced Pathological Cardiac Hypertrophy. Biochem. Biophysical Res. Commun. 460, 622–627. doi:10.1016/j.bbrc.2015.03.079
Yang, F., Qin, Y., Lv, J., Wang, Y., Che, H., Chen, X., et al. (2018). Silencing Long Non-Coding RNA Kcnq1ot1 Alleviates Pyroptosis and Fibrosis in Diabetic Cardiomyopathy. Cell Death Dis. 9, 1000. doi:10.1038/s41419-018-1029-4
Yang, F., Qin, Y., Wang, Y., Meng, S., Xian, H., Che, H., et al. (2019). Metformin Inhibits the NLRP3 Inflammasome via AMPK/mTOR-dependent Effects in Diabetic Cardiomyopathy. Int. J. Biol. Sci. 15, 1010–1019. doi:10.7150/ijbs.29680
Yildirim, S. S., Akman, D., Catalucci, D., and Turan, B. (2013). Relationship between Downregulation of miRNAs and Increase of Oxidative Stress in the Development of Diabetic Cardiac Dysfunction: Junctin as a Target Protein of miR-1. Cell Biochem Biophys. 67, 1397–1408. doi:10.1007/s12013-013-9672-y
Zhan, H., Huang, F., Niu, Q., Jiao, M., Han, X., Zhang, K., et al. (2021). Downregulation of miR-128 Ameliorates Ang II-Induced Cardiac Remodeling via SIRT1/PIK3R1 Multiple Targets. Oxidative Med. Cell Longevity 2021, 1–17. doi:10.1155/2021/8889195
Zhang, D., Cui, Y., Li, B., Luo, X., Li, B., and Tang, Y. (2016). miR-155 Regulates High Glucose-Induced Cardiac Fibrosis via the TGF-β Signaling Pathway. Mol. Biosyst. 13, 215–224. doi:10.1039/c6mb00649c
Zhang, M., Zhang, H., Liu, C., Li, X., Ling, M., Wang, Z., et al. (2018). Myocardial Protective Effects of Nicorandil on Rats with Type 2 Diabetic Cardiomyopathy. Med. Sci. Monit. Basic Res. 24, 141–145. doi:10.12659/MSMBR.910974
Zhu, C., Zhang, H., Wei, D., and Sun, Z. (2021a). Silencing lncRNA GAS5 Alleviates Apoptosis and Fibrosis in Diabetic Cardiomyopathy by Targeting miR-26a/b-5p. Acta Diabetol. 58, 1491–1501. doi:10.1007/s00592-021-01745-3
Zhu, Y.-F., Wang, R., Chen, W., Cao, Y.-D., Li, L.-P., and Chen, X. (2021b). miR-133a-3p Attenuates Cardiomyocyte Hypertrophy through Inhibiting Pyroptosis Activation by Targeting IKKε. Acta Histochem. 123, 151653. doi:10.1016/j.acthis.2020.151653
Zhu, Y., Qian, X., Li, J., Lin, X., Luo, J., Huang, J., et al. (2019). Astragaloside-IV Protects H9C2(2-1) Cardiomyocytes from High Glucose-Induced Injury via miR-34a-Mediated Autophagy Pathway. Artif. Cell Nanomedicine, Biotechnol. 47, 4172–4181. doi:10.1080/21691401.2019.1687492
Keywords: diabetes, diabetic cardiomyopathy, epigenetics, hyperglycemia, methylation
Citation: Deng J, Liao Y, Liu J, Liu W and Yan D (2021) Research Progress on Epigenetics of Diabetic Cardiomyopathy in Type 2 Diabetes. Front. Cell Dev. Biol. 9:777258. doi: 10.3389/fcell.2021.777258
Received: 15 September 2021; Accepted: 06 December 2021;
Published: 24 December 2021.
Edited by:
Joaquim SL Vong, The Chinese University of Hong Kong SAR, ChinaReviewed by:
Jun Ren, University of Washington, United StatesCopyright © 2021 Deng, Liao, Liu, Liu and Yan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianxin Deng, Y2hlZWhzaW5AMTI2LmNvbQ==; Dewen Yan, eWFuZHc5NjNAMTI2LmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.