 Shan Liang
Shan Liang Yun-Shan Wu1†
Yun-Shan Wu1† Ji-Xin Tang
Ji-Xin Tang Hua-Feng Liu
Hua-Feng Liu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 15 October 2021
Sec. Cell Death and Survival
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.766142
This article is part of the Research Topic The Regulation of Autophagy Activity in Pathogenic Microorganism Infection View all 8 articles
As an evolutionarily conserved cellular process, autophagy plays an essential role in the cellular metabolism of eukaryotes as well as in viral infection and pathogenesis. Under physiological conditions, autophagy is able to meet cellular energy needs and maintain cellular homeostasis through degrading long-lived cellular proteins and recycling damaged organelles. Upon viral infection, host autophagy could degrade invading viruses and initial innate immune response and facilitate viral antigen presentation, all of which contribute to preventing viral infection and pathogenesis. However, viruses have evolved a variety of strategies during a long evolutionary process, by which they can hijack and subvert host autophagy for their own benefits. In this review, we highlight the function of host autophagy in the key regulatory steps during viral infections and pathogenesis and discuss how the viruses hijack the host autophagy for their life cycle and pathogenesis. Further understanding the function of host autophagy in viral infection and pathogenesis contributes to the development of more specific therapeutic strategies to fight various infectious diseases, such as the coronavirus disease 2019 epidemic.
Autophagy, or cellular self-digestion, is an evolutionarily conserved cellular process through which long-lived proteins, damaged organelles, or invading pathogens could be degraded by the lysosome (Levine et al., 2011; Deretic et al., 2013; Levine and Kroemer, 2019; Mizushima and Levine, 2020). According to the way that eukaryotic cells deliver cytoplasmic materials to lysosomes for degradation, autophagy can be divided into three major types: microautophagy, chaperone-mediated autophagy (CMA), and macroautophagy (Mizushima et al., 2008; Figure 1). Microautophagy engulfs cytoplasmic materials or large structures through non-selectively invaginating lysosomal membrane or selectively delivering soluble cytosolic proteins to the multivesicular bodies (MVBs) (Mijaljica et al., 2011; Sahu et al., 2011). CMA only degrades soluble proteins in a selective manner through the lysosomal LAMP2A receptor to recognize and translocate unfolding proteins with a specific signal sequence—KFERQ (Orenstein and Cuervo, 2010; Kaushik and Cuervo, 2012). Macroautophagy could both selectively or non-selectively engulf bulk cytoplasmic components by sequestering these cargoes to a specialized double-membrane vesicle (DMV) known as the autophagosome (Feng et al., 2014; Melia et al., 2020; Nakatogawa, 2020). Here, we focus on macroautophagy, and hereafter refer to “macroautophagy” simply as “autophagy.”
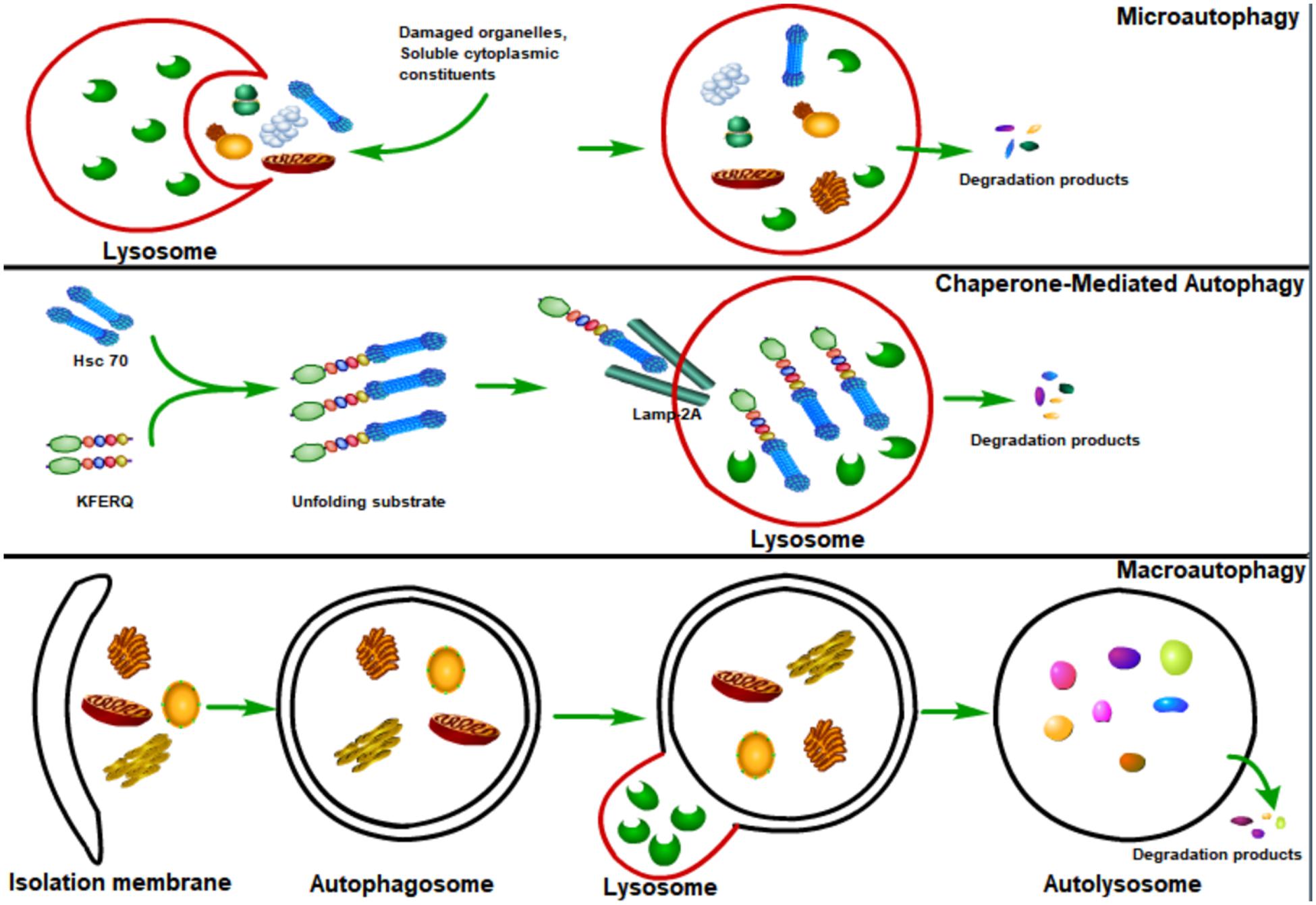
Figure 1. Major types of autophagy. According to the way that eukaryotic cells deliver cytoplasmic cargo to lysosomes for degradation, autophagy can be divided into three major types: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). Microautophagy refers to the lysosome itself engulfing cytoplasmic material or large structures by invading the lysosome membrane. The CMA only degrades soluble proteins in a selective manner through the LAMP2A receptor on the lysosome to recognize and translocate unfolding proteins with a specific signal sequence—KFERQ. Macroautophagy could both selectively and non-selectively engulf bulk cytoplasmic components by sequestering these cargoes to a specialized double-membrane vesicle known as the autophagosome; autophagosome is then fused with the lysosome, where the cargo is degraded and the resulting macromolecules are released into the cytosol for reuse.
Autophagy begins with the sequestration of cargoes into a cup-shaped double membrane known as the isolation membrane or phagophore, which stems from several cellular compartments (Shibutani and Yoshimori, 2014; Melia et al., 2020). The sequestration of cargoes could be either non-specific (such as the engulfment of bulk cytoplasm) or selective (such as specific engulfment of organelles or invading pathogens). The gradually expanding phagophore envelops the engulfed cargoes, forming the autophagosome, which then fuses with the lysosome, causing the formation of autolysosome (Hamasaki et al., 2013; Lamb et al., 2013). The lysosome provides hydrolases to the autolysosome, where the autophagosome inner membrane is lysed and the cargoes break down, and then the resulting macromolecules are released back into the cytosol for reuse through membrane permeases (Mizushima et al., 2008; Lamb et al., 2013).
Autophagy is involved in a variety of mammalian physiological processes such as the maintenance of energy homeostasis, cell differentiation and development, and innate immunity against invading pathogens (Kuma et al., 2004; Levine and Kroemer, 2008; Mizushima and Levine, 2010; Levine et al., 2011; Deretic et al., 2013). Given the powerful function to degrade intracellular substances, host autophagy is activated during the viral infection so as to degrade various invading viruses (Levine et al., 2011; Deretic et al., 2013). However, an increasing body of evidence suggests that viruses have developed various strategies to hijack and subvert the host autophagy for their life cycle and pathogenesis (Heaton and Randall, 2010; Levine et al., 2011; Deretic et al., 2013). In this review, we focus on the function of autophagy in the process of viral infection and pathogenesis and then discuss the mechanisms of how viruses usurp the host autophagy to facilitate their life cycle and pathogenesis.
As a multi-step and tightly regulated cellular process for maintaining eukaryotic cellular homeostasis, autophagy is the only pathway that is able to degrade whole cellular organelles (such as mitochondria, peroxisomes, endoplasmic reticulum, nucleus, and liposomes) and various invading pathogens (including viruses) in either a selective or a non-selective manner (Khaminets et al., 2015; Mochida et al., 2015; Ammanathan et al., 2020; Jo et al., 2020; Miceli et al., 2020; Onishi et al., 2021). Upon viral infection, the induction of autophagy by viruses (known as virophagy) could be either proviral or antiviral (Dong and Levine, 2013; Delorme-Axford and Klionsky, 2019; Mijaljica and Klionsky, 2020). Virophagy plays its antiviral function probably through (1) selectively targeting viral particles to the lysosome for degradation (Figure 2), (2) promoting interferon production by activating host innate immune response, or (3) coordinating adaptive immunity by promoting antigen presentation.
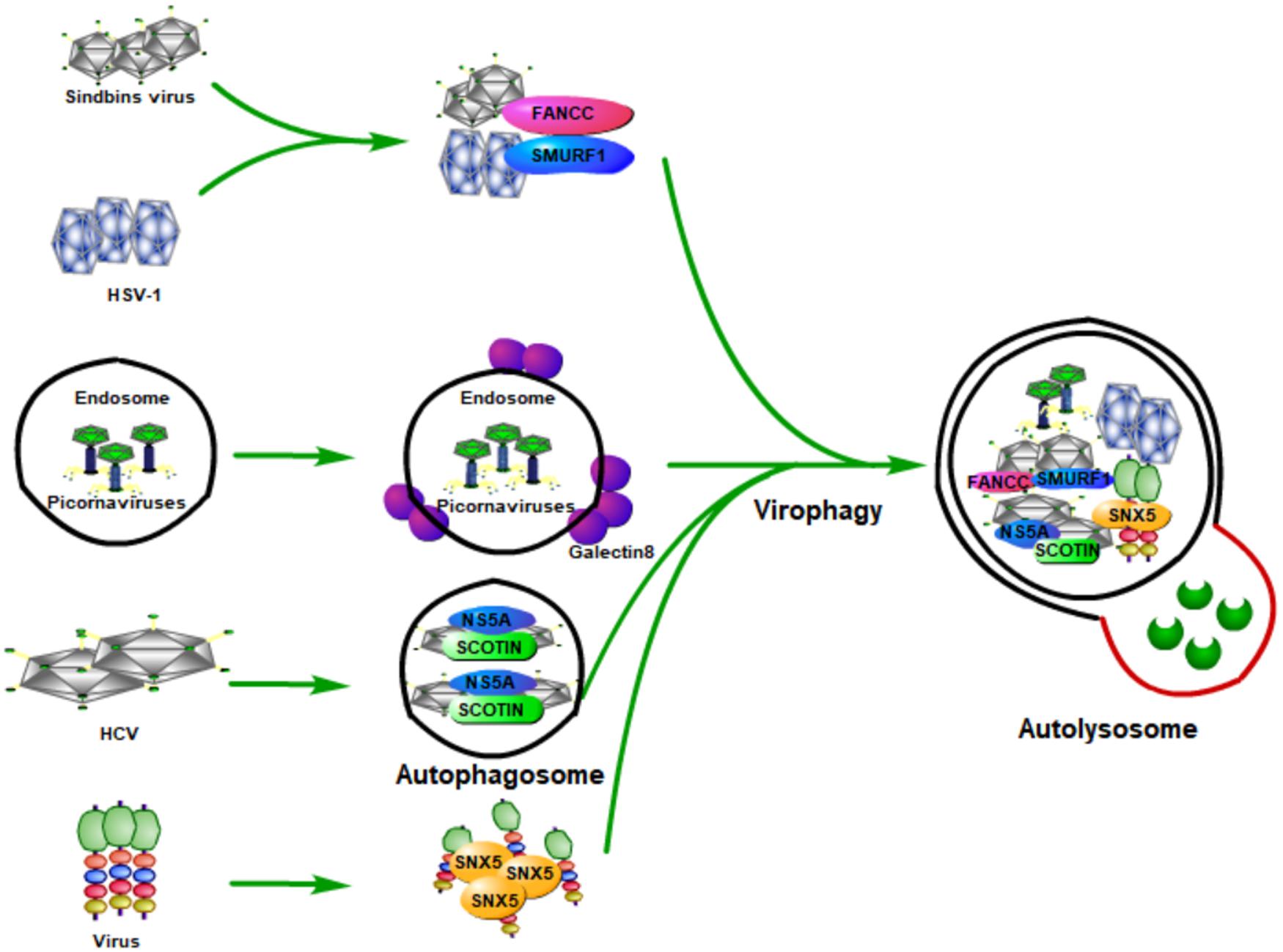
Figure 2. Host autophagy fights viral infection by selectively targeting viral particles to lysosome for degradation. During the viral infection, host autophagy can be inducted (known as virophagy), which can degrade viral particles, viral components, as well as host factors that are required by viruses for their replication. Some viruses are degraded by selective autophagy by being recognized by specific proteins of the host. Smad-Ubiquitin Regulatory Factor 1 (SMURF1), a HECT-domain ubiquitin ligase, and Fanconi anemia group C protein (FANCC) can target Sindbis capsid protein as well as the herpes simplex virus type 1 (HSV-1) to autophagosomes for virophagy, contributing to prevent viral infection. The invasion of the picornaviruses will puncture the endosomal membrane to release their genome into the host cytoplasm, causing the exposure of β-galactosides. Galectin-8 could specifically recognize β-galactosides and therefore mark the permeated endosomes for autophagic degradation. The endoplasmic reticulum (ER) protein SCOTIN can inhibit HCV replication by interacting with the HCV non-structural 5A (NS5A) protein, a critical factor for HCV replication, which can help to form autophagosomes for degradation and suppress infectious virion production in cells. Moreover, the endosomal protein sorting nexin 5 (SNX5) can target some viruses for virophagy, but not for basal level autophagy or stress-induced autophagy.
By delivering the invading pathogens to the lysosomes for degradation so as to discard them, autophagy is believed to be an important part of the host defense system. The process of degrading foreign microbial invaders by autophagy is known as xenophagy (Levine, 2005). The function of xenophagy to eliminate invading viruses, bacteria, fungi, or parasites, makes it an important immune player in pathogen infection. During viral infection, host autophagy can degrade viral particles, viral components, and the host factors required by viruses for their replication; therefore, host autophagy functions as a key innate antiviral response (Figure 2).
The first evidence of autophagy functions as antiviral means came from Sindbis viral infection. During its infection, overexpression of Beclin 1 (the mammalian Atg6 ortholog) in neurons could inhibit the Sindbis virus spread, reduce cellular apoptosis, and protect against fatal Sindbis virus encephalitis (Liang et al., 1998), whereas Atg5 (an essential component for the formation of autophagosomes in mammalian cells) deficiency in Sindbis-infected neurons leads to delayed clearance of viral proteins, accumulation of the p62 (also known as SQSTM1) adaptor protein, and increased cell death in neurons (Mizushima et al., 2001). Moreover, an in vitro study found that p62 could interact with Sindbis capsid protein and targets the viral capsid to the autophagosome (Orvedahl et al., 2010). Orvedahl et al. (2011) further found that Smad-Ubiquitin Regulatory Factor 1 (SMURF1), a HECT-domain ubiquitin ligase, is not required for general autophagy, but is needed by selective autophagy, such as virophagy and mitophagy. Moreover, they found that SMURF1 could interact with p62 and target Sindbis capsid to autophagosomes for virophagy. Fanconi anemia group C protein (FANCC) was also found to play an essential role in virophagy and mitophagy; its deficiency could inhibit the autophagic clearance of viruses (virophagy) and make mice more susceptible to lethal viruses (Sumpter et al., 2016). FANCC plays its role by interacting with the Sindbis capsid protein and therefore contributes to preventing viral infection (Minton, 2016; Sumpter et al., 2016). Besides Sindbis, SMURF1 and FANCC also inhibit HSV-1 infection through autophagy, indicating that these two proteins probably play virophagic factor functions during viral infection (Orvedahl et al., 2011; Sumpter et al., 2016).
Picornaviruses are archetypical non-enveloped viruses, which are a major cause of human and veterinary infections that lead to various diseases, such as polio and the common cold (Zhang et al., 2020). Host galectin 8 could detect picornaviruses and inhibit their infection through autophagy to degrade the viral genomic RNA (Staring et al., 2017). Specifically, the picornaviruses that enter the host will puncture the endosomal membrane and release their genome into the host cytoplasm, which causes the exposure of β-galactosides. Galectin-8 could specifically recognize β-galactosides and therefore marks the permeated endosomes for autophagic degradation.
As a small, enveloped RNA virus that mainly targets human hepatocytes, hepatitis C virus (HCV) is a major cause of liver cirrhosis and hepatocellular carcinoma worldwide (Zeisel et al., 2013; Kaplan, 2020). The prevention measures for HCV are absent and the current antiviral treatment for it is limited because of resistance, toxicity, and high costs (Zeisel et al., 2011; Hickman et al., 2015). Because of the discovery of HCV, the 2020 Nobel Prize in Physiology or Medicine has been awarded to Harvey J. Alter, Michael Houghton, and Charles M. Rice (Burki, 2020; Hoofnagle and Feinstone, 2020). Kim et al. (2016) found that the overexpression of endoplasmic reticulum (ER) protein SCOTIN inhibits HCV replication and infectious virion production in cells transfected with HCV. A further study found that SCOTIN could interact with HCV non-structural 5A (NS5A) protein, a critical factor for HCV replication, and target NS5A to autophagosomes for degradation. Moreover, inhibition of autophagy by silencing ATG7 or administering lysosomal inhibitors could relieve the suppressive effect of SCOTIN on HCV replication. They also showed that SCOTIN is merely a substrate for degradation of autophagy, but not affecting the whole process of autophagy; of note, the binding of transmembrane/proline-rich domain (TMPRD) of SCOTIN with Domain-II of NS5A is critically required for the trafficking of autophagosomal and NS5A degradation (Kim et al., 2016). These results suggest that autophagy restricts HCV replication through SCOTIN to target HCV NS5A protein to autophagosomes for degradation.
As a sensor of cytosolic DNA that activates the type I interferon pathway, cyclic GMP-AMP synthase (cGAS) could bind to microbial or self-DNA in the cytoplasm and therefore supervises infections or tissue damage (Sun et al., 2013). Specifically, cGAS is activated through binding with cytosolic DNA, which could further catalyze GTP and ATP to be cyclic GMP-AMP (cGAMP) (Wu et al., 2013). As a second messenger of cell, cGAMP could bind to and activate the stimulator of interferon genes (STING) (Ishikawa and Barber, 2008; Saitoh et al., 2009; Burdette et al., 2011; Wu et al., 2013; Zhang et al., 2013), which then recruits and activates the tank-binding kinase 1 (TBK1) causing the phosphorylation of the transcription factor IRF3 to induce the production of type I interferons and other cytokines (Cai et al., 2014; Liu et al., 2015; Zhang et al., 2019). Besides the important function in activating the immune response, STING could also activate autophagy during viral infections (Gui et al., 2019; Liu et al., 2019). Gui et al. (2019) showed that the binding of cGAMP with STING leads to the interaction of STING with SEC24C, causing the budding of STING from the endoplasmic reticulum into the COP-II vesicles and forming the endoplasmic reticulum–Golgi intermediate compartment (ERGIC), which functions as the membrane source for WIPI2 recruitment and LC3 lipidation, and finally causing the formation of autophagosomes targeting cytosolic DNA or DNA viruses to the lysosome for degradation (Gui et al., 2019). STING induces autophagy that is dependent on WIPI2 and ATG5, whereas other regulators of autophagy such as Beclin 1, Atg9a, ULK1, and p62 are not required (Gui et al., 2019; Liu et al., 2019). Interestingly, Gui et al. (2019) also showed that STING from the sea anemone is also able to induce autophagy but not interferons in response to the stimulation of cGAMP, which suggests that the cGAS-STING pathway-induced autophagy is probably an ancient and highly conserved mechanism that predates the emergence of the type I interferon pathway in vertebrates to eliminate the invading viruses.
How is autophagy induced in mammalian cells during viral infection? By using genome-wide short interfering RNA screens, the endosomal protein sorting nexin 5 (SNX5) is found to be required merely for virus-induced autophagy, but not for basal level autophagy or stress-induced autophagy (Dong et al., 2021). Deletion of SNX5 makes cultured cells more susceptible to viral infection, and mice deficient in SNX5 have a high lethality after infection with several human viruses (Dong et al., 2021). Moreover, they found that SNX5 could interact with PI3KC3-C1 and promote its activation at endosomes and therefore contributes to the initiation of autophagy during viral infection.
Collectively, host cells detect the invading viruses and activate the autophagy, which targets viruses to the lysosomes for degradation through the interaction of host protein with the viral protein. Future studies should focus on exploring additional viruses that can be selectively degraded by autophagy and clarifying the specific mechanisms involved in this process.
As the first line of defense against virus infection, the host innate immune system plays essential roles in recognizing invading viruses and inducing anti-viral responses to prevent viral invasion and pathogenesis before the generation of more specific protection by the adaptive immune system (Takeuchi and Akira, 2007; Koyama et al., 2008; Figure 3). The host innate immune system recognizes invading viruses by several classes of germline-encoded pattern-recognition receptors (PRRs), which could specifically recognize the pathogen-associated molecular patterns (PAMPs), such as viral DNA, viral double-stranded RNA (dsRNA), viral single-stranded RNA (ssRNA), or viral surface glycoproteins (Akira et al., 2006; Chan and Gack, 2016). The recognition of viral components by PRRs will induce the infected cells and other immune cells to produce type I interferons (IFNs) to aid in eliminating the invading viruses (Medzhitov, 2007).
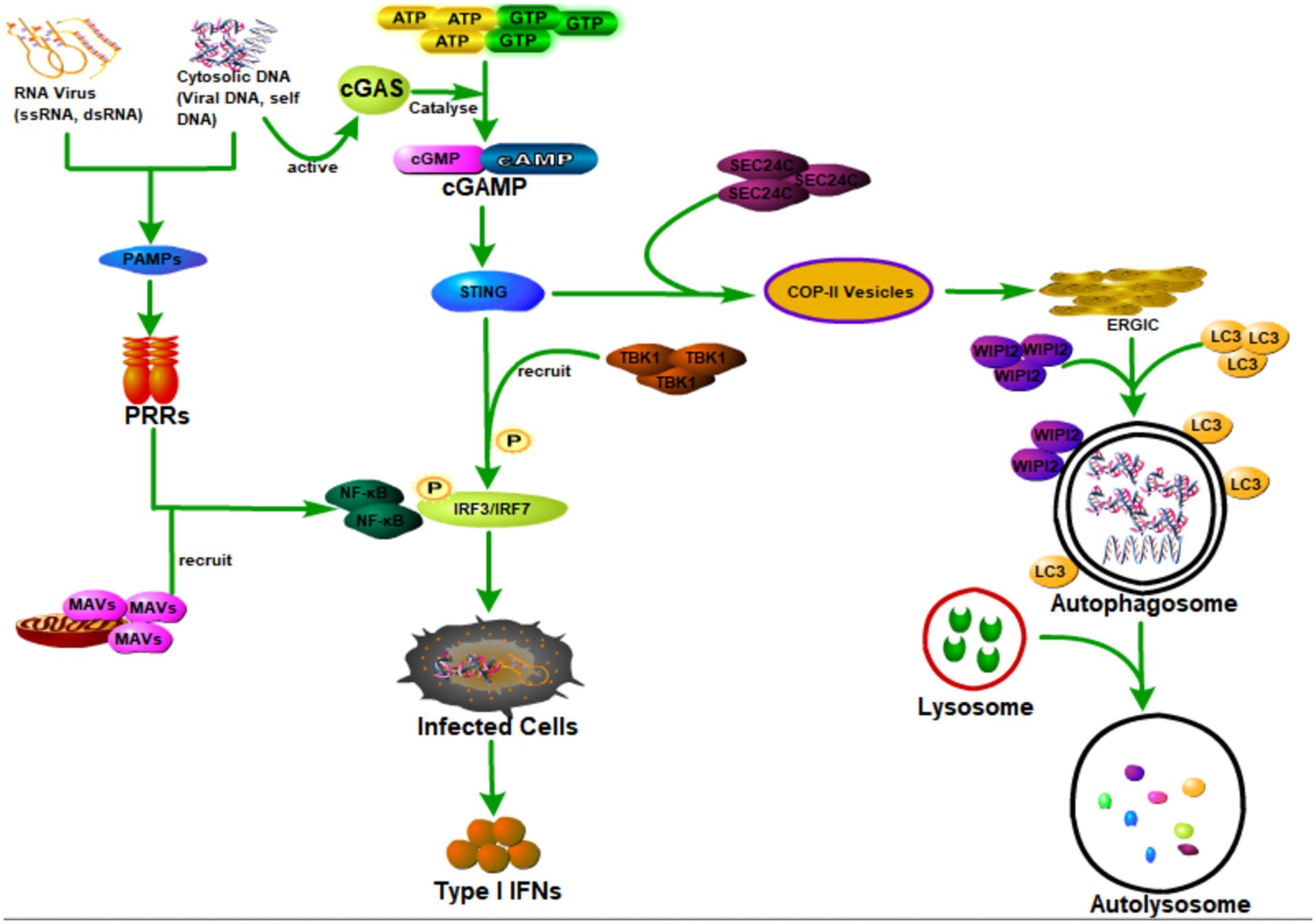
Figure 3. Host autophagy defense against viral infection by promoting interferon production through activating innate immune response. Cyclic GMP-AMP synthase (cGAS) can be activated when it is bound to the cytosolic DNA (microbial or self-DNA), then the activated cGAS can catalyze GTP and ATP to be cyclic GMP-AMP (cGAMP), which can bind to and activate the stimulator of interferon genes (STING). The binding of cGAMP with STING, on the one hand, can lead to the interaction of STING with SEC24C, causing the budding of STING from the ER into the COP-II vesicles and forming the endoplasmic reticulum–Golgi intermediate compartment (ERGIC), which functions as the membrane source for WIPI2 recruitment and LC3 lipidation, and finally causing the formation of autophagosomes targeting cytosolic DNA or DNA viruses to the lysosome for degradation. On the other hand, it can also recruit and activate the tank-binding kinase 1 (TBK1) causing the phosphorylation of the transcription factor IRF3 to induce the production of type I interferons (IFNs). In addition, viral DNA, viral double-stranded RNA (dsRNA), viral single-stranded RNA (ssRNA), or viral surface glycoproteins, express the pathogen-associated molecular patterns (PAMPs), which can be specifically recognized by several classes of germline-encoded pattern-recognition receptors (PRRs) of the host cells. Moreover, PRR could activate IRF3/IRF7 and NF-κB transcription factors by recruiting mitochondrial antiviral signaling protein (MAVS), leading to the activation of type I IFN responses and establishment of an antiviral state.
There are several classes of PRRs with specific functions; virus-derived nucleic acids with distinct features are recognized by these specific host transmembrane or cytosol PRRs (Medzhitov, 2007; Gürtler and Bowie, 2013; Wu and Chen, 2014). As an important group of cytoplasmic RNA sensors, the RIG-I-like receptors (RLRs) are composed of three proteins (RIG-I, MDA5, and LGP2) that are similar in their structure and function, and all of them are able to recognize viral nucleic acid signatures during the viral infections (Rehwinkel and e Sousa, 2010; Bruns and Horvath, 2014). Recently, Hou et al. (2021) have found a novel selective autophagy receptor, CCDC50, which could deliver K63 polyubiquitination-activated RIG-I/MDA5 for degradation by lysosomes during viral infection and therefore negatively regulate the IFNs signaling pathway initiated by RLRs. As the best characterized class of PRR, toll-like receptors (TLRs) are transmembrane receptors that recognize viral nucleic acids within endosomal compartments (Medzhitov, 2007). Both of these two classes of PRR could activate IRF3/IRF7 and NF-κB transcription factors by recruiting mitochondrial antiviral signaling protein (MAVS), leading to the activation of type I interferon (IFN) responses and establishment of antiviral state (Seth et al., 2005; Akira et al., 2006; Medzhitov, 2007).
Lee et al. (2007) first elucidated the key function of autophagy in promoting interferon secretion in plasmacytoid dendritic cells (pDCs). They found that autophagy was required for the transportation of cytosolic viral into the lysosomes and the recognition of ssRNA viruses such as vesicular stomatitis virus (VSV) or Sendai virus (SeV) by TLR7. Moreover, they also found that autophagy was critically needed by pDCs during the production of interferon-α.
As an essential DNA virus sensor, cGAS could prompt IFNs production by generating cGAMP, which binds to and activates an endoplasmic reticulum-associated adaptor protein STING (Sun et al., 2013; Wu et al., 2013). Chen et al. (2016) have found that TRIM14 could block cGAS degradation through selective autophagy and therefore promote innate immune responses during viral infections. Specifically, upon the viral infections, TRIM14 could recruit USP14 to cut the lysine 48 (K48)-linked ubiquitin chains of cGAS at K414, causing inhibition of p62-mediated autophagic degradation of cGAS, therefore promoting the activation of type I interferon signaling to aid the elimination of the invading viruses (Chen et al., 2016).
These studies suggest that host autophagy plays an essential role in activating the innate immune response to eliminate the invading viruses by promoting the infected cells and other immune cells to produce IFNs. However, there are also some studies reporting that autophagy or its components contributed to the negative regulation of host innate immune response (Jounai et al., 2007; Lei et al., 2012; Liang et al., 2014; Jin et al., 2017; Du et al., 2018; Prabakaran et al., 2018). Therefore, during viral infection, the interaction between PRRs and autophagy resulted in the activation and/or inhibition of various host innate immune responses, causing first-rank antiviral effects.
The efficient adaptive immune response is essential for the elimination of invading viruses. The first step of the adaptive immune response is the presentation of peptides of foreign or self-proteins on major histocompatibility complex (MHC) molecules at the cell surface of antigen-presenting cells (APCs) such as dendritic cells and macrophages, which then can be recognized by CD8+ or CD4+ T lymphocytes (Rammensee et al., 1993; Villadangos, 2001). In general, MHC class I (MHC-I) molecules specifically present antigenic peptides derived from intracellular proteins that have been digested by the proteasomal degradation system, whereas MHC class II (MHC-II) molecules specifically present antigenic peptides stemming from exogenous and membrane proteins that have been degraded by the endosomal/lysosomal system (Neefjes, 1999; Princiotta et al., 2003; Dengjel et al., 2005; Choi et al., 2018). However, there are certain situations where MHC-I molecules could present peptides stemming from exogenous antigens, which is a process called cross-presentation, mainly executed by a specific subset of dendritic cells (DCs) through endocytic and phagocytic pathways (Morón et al., 2004; Joffre et al., 2012; Blander, 2018; Muntjewerff et al., 2020). In addition, the peptides derived from intracellular proteins could also be loaded on MHC-II molecules through the process of autophagy (Nimmerjahn et al., 2003; Dörfel et al., 2005; Paludan et al., 2005; Münz, 2006).
Macrophages infected with the Herpes simplex type 1 virus could activate the process of autophagy, which plays an essential role in the targeting of viral proteins to lysosomes and then loaded on MHC class I molecules for presentation (English et al., 2009b). During this process, a novel type of autophagosomes is involved, which is formed by coiling of the viral proteins enriched with nuclear membrane (English et al., 2009a). With the help of this process, the peptide derived from HSV-1 glycoprotein B (gB) could be presented to CD8+ T cells aided by proteasome function and the secretory pathway.
An endogenous human cytomegalovirus (HCMV) latency-associated protein, pUL138, could be presented by MHC-I through both the conventional TAP-dependent and the non-conventional TAP-independent pathways (Tey and Khanna, 2012). The TAP-dependent process uses the proteasomal machinery and ER-resident proteases of the conventional MHC class I pathway, whereas the TAP-independent process uses the vacuolar pathway mediated by autophagy. Importantly, this autophagy-mediated pathway is not dependent on proteasomal processing and Golgi transport, but dependent on the alternate cross-presentation pathway that only occurs within the endovacuolar compartment. Although this autophagy-mediated pathway uses minimal components of the conventional MHC-I machinery, it could generate and present the same peptide epitope as the conventional pathway.
The MHC class II-positive cells such as dendritic, B, and epithelial cells constitutively form autophagosome, which then continuously fuse with multivesicular MHC class II-loading compartments to deliver cytoplasmic proteins for the presentation of MHC class II and antiviral immunity (Schmid et al., 2007). Endogenous Epstein–Barr virus nuclear antigen 1 (EBNA1) could be presented by MHC-II molecules and then recognized by CD4+ T cells (Paludan et al., 2005). Inhibition of lysosomal acidification led to the slow accumulation of EBNA1 in cytosolic autophagosomes. Moreover, blocking of autophagy using a PI3K inhibitor or by knockdown of ATG12 could decrease the presentation of EBNA1 to CD4+ T cells by MHC-II (Paludan et al., 2005).
Herpes simplex virus (HSV) infection resulted in the impairment of CD4+ T-cell priming in Atg5-deficient mice in DCs, which succumbed to HSV infection (Lee et al., 2010). Moreover, autophagy is essential for the presentation of various phagocytosed antigens with TLR agonists, whereas autophagy is not required for innate immune recognition, antigen maturation, or cytokine production, as most of these processes remained intact in DCs deficient in Atg5 (Lee et al., 2010). These results suggest that autophagy plays an essential part in the processing and presentation of extracellular viral antigens by MHC-II in DCs.
By presentation of extracellular, endocytosed antigens on MHC-I molecules, cross-presentation plays an essential role in the priming of CD8+ T-cell responses (Joffre et al., 2012). In vivo, subset-specific DCs are the major cells responsible for cross-presentation by adapting their endocytic and phagocytic pathways, which is essential for the immune defense against viruses and other intracellular pathogens (Blander, 2018; Theisen et al., 2018; Muntjewerff et al., 2020). On the one hand, autophagy could enhance cross-presentation by modulation of endosomes and assist the packaging of antigens released by donor cells, such as virus-infected cells, tumor cells, or dying cells, to neighbor DCs (Mintern et al., 2015; Dasari et al., 2016; Cruz et al., 2017); on the other hand, autophagy also contributed to the cross-presentation of viral antigens to CD8+ T cells during vaccination, which was facilitated through a stress-dependent initiation of autophagy in DCs (Ravindran et al., 2014). In addition, exocytosis mediated by autophagy could transfer extracellular antigens in LC3-coated autophagosome from the donor cells to DCs (Smed-Sörensen et al., 2012).
Thus, autophagy is involved in both the classical and the non-classical antigen presentation process, which is essential for the optimal processing and presentation of viral antigens. Moreover, the autophagy-mediated antigen presentation process is an important supplement to the conventional antigen presentation process; it may circumvent the various viral immune evasion strategies targeting the MHC-I or MHC-II machinery, therefore contributing to the elimination of invading viruses.
In general, host autophagy could inhibit viral replication, degrade viral particles, and activate host immune response, all of which contribute to the prevention of viral infection and pathogenesis. However, some viruses have developed various strategies to hijack and subvert host autophagy to aid their own infections and pathogenesis (Orvedahl and Levine, 2009a,b; Figure 4). These strategies include (1) directly inhibiting autophagy activation through blocking the function of host ATG proteins; (2) inhibiting autophagy downstream degradation pathway; and (3) subverting host autophagy to benefit for viral replication.
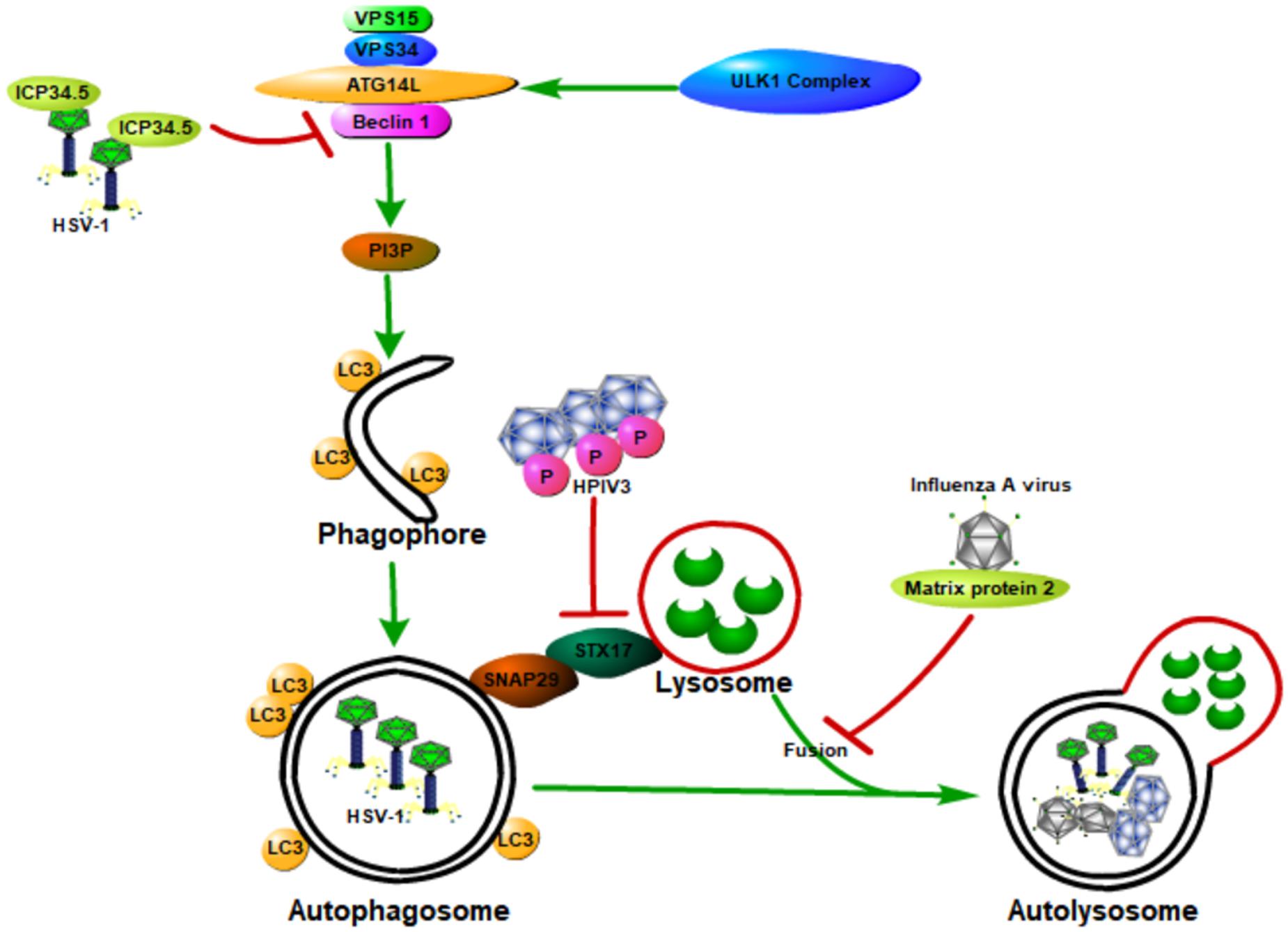
Figure 4. Viruses hijack and subvert host autophagy to aid their own infections and pathogenesis. Some viruses have developed various strategies to hijack and subvert host autophagy to aid their own infections and pathogenesis. These strategies include (1) directly inhibiting autophagy activation through blocking the function of host ATG proteins; (2) inhibiting autophagy downstream degradation pathway; and (3) subverting host autophagy to benefit viral replication. Class III PI3K complex I (PI3KC3-C1), which can produce PI3P, is necessary for the nucleation of autophagosomes and is composed of VPS34, VPS15, Beclin 1, and ATG14L. ICP34.5 expressed by the HSV-1 could inhibit host autophagy activation through binding with the host autophagy protein Beclin 1. The matrix protein 2 of influenza A virus could inhibit autophagy function by blocking the autophagosome–lysosome fusion, causing the enhanced virus-induced cell death of infected cells and elevated viral antigen release. The human parainfluenza virus type 3 (HPIV3) phosphoprotein (P) binds to SNAP29, blocking its binding to syntaxin 17, therefore inhibiting the fusion of autophagosome-lysosome mediated by these two host SNARE proteins. All of these will contribute to the viral infection and pathogenesis.
Neurovirulence protein ICP34.5, which is expressed by the herpes simplex virus type 1 (HSV-1), could inhibit host autophagy activation through binding with the host autophagy protein Beclin 1 (Orvedahl et al., 2007). HSV-1 virus deficient in the Beclin 1-binding domain of ICP34.5 is unable to block autophagy in neurons and shows a decrease in the ability to cause lethal encephalitis in mice. Deletion of PKR, an autophagy-inducing signaling molecule, could restore the neurovirulence of this Beclin 1-binding mutant virus (Orvedahl et al., 2007). These results suggest that the binding of Beclin 1 with ICP34.5 could inhibit host autophagy and contribute to viral neurovirulence, and the PKR is a Beclin 1 upstream effector during host defense against HSV-1. Moreover, the Us11 protein of HSV-1 could also inhibit host autophagy function by directly interacting with the PKR protein kinase, therefore contributing to the HSV-1 infection and pathogenesis (Lussignol et al., 2013).
Matrix protein 2 of influenza A virus could inhibit autophagy function through blocking the autophagosome–lysosome fusion, causing the enhanced virus-induced cell death of infected cells and elevated viral antigen release (Gannagé et al., 2009; Rossman and Lamb, 2009). Human parainfluenza virus type 3 (HPIV3) could also lead to the incomplete autophagy of host cells through blocking autophagosome fusion with the lysosome, causing an increase in virus production (Ding et al., 2014). Specifically, the viral phosphoprotein (P) binds to SNAP29, blocking its binding to syntaxin 17 (STX17), therefore inhibiting autophagosome–lysosome fusion mediated by these two host SNARE proteins. Autophagosome accumulation due to incomplete autophagy could increase extracellular viral production but does not affect viral protein synthesis, therefore contributing to the viral infection and pathogenesis.
Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2), an enveloped, single-stranded and positive-sense RNA β-coronavirus, is the cause of the COVID-19 pandemic (Wang et al., 2020; Wu et al., 2020; Zhou et al., 2020). ORF3a, an accessory protein of SARS-CoV-2, could strongly inhibit autophagy activity by blocking the fusion of autophagosomes with lysosomes (Hayn et al., 2021; Miao et al., 2021; Qu et al., 2021; Zhang et al., 2021). Specifically, ORF3a is localized in the late endosome and directly interacts with and sequestrates VPS39, an essential component of the homotypic fusion and protein sorting (HOPS) complex, thereby preventing the HOPS complex from interacting with the STX17 or RAB7, which prevented the fusion machinery packaging, leading to an abnormal autophagosome–lysosome fusion. Moreover, SARS-CoV-2-expressed ORF3a and ORF7a can directly induce lysosomes injury and impair their function, such as inhibiting their acidification (Hayn et al., 2021; Koepke et al., 2021; Miao et al., 2021; Shroff and Nazarko, 2021). By doing this, SARS-CoV-2 could escape host lysosome degradation. Besides, the SARS-CoV-2 spike could hijack host autophagy to promote host cell inflammation and apoptosis probably through the ROS-suppressed PI3K/AKT/mTOR signaling (Li et al., 2021).
As a DMV formed during autophagy, autophagosome provides a perfect place for the RNA viral replication through concentration of essential intermediates for viral package and protection of viral RNAs away from the detection by innate immune supervision and degradation. The host autophagy has been required by several viruses such as Coxsackieviruses (CVB) 3, CVB 4, foot and mouth disease virus, HCV, and poliovirus for their own replication, as genetic or pharmacological inhibition of autophagy could decrease viral yields (Taylor and Kirkegaard, 2008; Wong et al., 2008; Yoon et al., 2008; Dreux et al., 2009; O’Donnell et al., 2011). Moreover, some viruses such as dengue virus (DENV), usurp autophagy to enhance their replication by regulating cellular lipid metabolism (Heaton and Randall, 2010). Specifically, DENV infection induces the activation of lipophagy to release free fatty acids, causing an increase of cellular β-oxidation and more ATP generation, which contributes to the replication of DENV.
In summary, host autophagy is a double-edged sword during viral infection and pathogenesis. On the one hand, hosts could utilize their own autophagy to prevent viral infection and pathogenesis; on the other hand, viruses have evolved various strategies by which they hijack and subvert host autophagy to aid their infection and pathogenesis.
As an evolutionarily conserved cellular process, autophagy is essential for both hosts and invading viruses. On the one hand, it could prevent viral infections and pathogenesis mainly by degrading viruses, initiating innate immune response, and facilitating antigen presentation. On the other hand, a mass of viruses have evolved strategies to hijack host autophagy for their own benefits. Although the function of host autophagy in viral infection and pathogenesis has been widely studied in the past two decades, current knowledge of autophagy in viral infections is still in its infancy and many important questions remain. For example, why some viruses have evolved strategies to evade host autophagy, whereas others have not? What is the driving force for this evolution? What roles do a variety of selective autophagy (such as mitophagy and lysophagy) play in viral infection and pathogenesis? What roles do microautophagy and chaperone-mediated autophagy play in viral infection and pathogenesis? Moreover, the specific function of autophagic proteins and the mechanisms controlling autophagy during viral infection and pathogenesis are still unclear. Further research is needed to elucidate the specific functions of different types of autophagy and the specific function in viral infections and pathogenesis, to develop more specific targeted drugs to combat epidemic viral infections, such as COVID-19.
J-XT and H-FL formulated and conceived the study. Y-SW, SL, D-YL, J-XT, and H-FL wrote the manuscript. All authors helped to interpret results and approved the final version of the manuscript.
This work was supported by grants from the National Natural Science Foundation of China (81974095) and Natural Science Foundation of Guangdong Province (2019A1515110152).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Akira, S., Uematsu, S., and Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell 124, 783–801. doi: 10.1016/j.cell.2006.02.015
Ammanathan, V., Mishra, P., Chavalmane, A. K., Muthusamy, S., Jadhav, V., Siddamadappa, C., et al. (2020). Restriction of intracellular Salmonella replication by restoring TFEB-mediated xenophagy. Autophagy 16, 1584–1597. doi: 10.1080/15548627.2019.1689770
Blander, J. M. (2018). Regulation of the cell biology of antigen cross-presentation. Annu. Rev. Immunol. 36, 717–753. doi: 10.1146/annurev-immunol-041015-055523
Bruns, A. M., and Horvath, C. M. (2014). Antiviral RNA recognition and assembly by RLR family innate immune sensors. Cytokine Growth Factor Rev. 25, 507–512. doi: 10.1016/j.cytogfr.2014.07.006
Burdette, D. L., Monroe, K. M., Sotelo-Troha, K., Iwig, J. S., Eckert, B., Hyodo, M., et al. (2011). STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518. doi: 10.1038/nature10429
Burki, T. (2020). Nobel prize for hepatitis C virus discoverers. Lancet 396:1058. doi: 10.1016/S0140-6736(20)32111-5
Cai, X., Chiu, Y.-H., and Chen, Z. J. (2014). The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol. Cell 54, 289–296. doi: 10.1016/j.molcel.2014.03.040
Chan, Y. K., and Gack, M. U. (2016). Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol. 14, 360–373. doi: 10.1038/nrmicro.2016.45
Chen, M., Meng, Q., Qin, Y., Liang, P., Tan, P., He, L., et al. (2016). TRIM14 inhibits cGAS degradation mediated by selective autophagy receptor p62 to promote innate immune responses. Mol. Cell 64, 105–119. doi: 10.1016/j.molcel.2016.08.025
Choi, Y., Bowman, J. W., and Jung, J. U. (2018). Autophagy during viral infection—a double-edged sword. Nat. Rev. Microbiol. 16, 341–354. doi: 10.1038/s41579-018-0003-6
Cruz, F. M., Colbert, J. D., Merino, E., Kriegsman, B. A., and Rock, K. L. (2017). The biology and underlying mechanisms of cross-presentation of exogenous antigens on MHC-I molecules. Annu. Rev. Immunol. 35, 149–176. doi: 10.1146/annurev-immunol-041015-055254
Dasari, V., Rehan, S., Tey, S. K., Smyth, M. J., Smith, C., and Khanna, R. (2016). Autophagy and proteasome interconnect to coordinate cross-presentation through MHC class I pathway in B cells. Immunol. Cell Biol. 94, 964–974. doi: 10.1038/icb.2016.59
Delorme-Axford, E., and Klionsky, D. J. (2019). Inflammatory-Dependent Sting Activation Induces Antiviral Autophagy to Limit Zika Virus in the Drosophila Brain. Milton Park: Taylor & Francis. doi: 10.1080/15548627.2018.1539585
Dengjel, J., Schoor, O., Fischer, R., Reich, M., Kraus, M., Müller, M., et al. (2005). Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc. Natl. Acad. Sci. U.S.A. 102, 7922–7927. doi: 10.1073/pnas.0501190102
Deretic, V., Saitoh, T., and Akira, S. (2013). Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 13, 722–737. doi: 10.1038/nri3532
Ding, B., Zhang, G., Yang, X., Zhang, S., Chen, L., Yan, Q., et al. (2014). Phosphoprotein of human parainfluenza virus type 3 blocks autophagosome-lysosome fusion to increase virus production. Cell Host Microbe 15, 564–577. doi: 10.1016/j.chom.2014.04.004
Dong, X., and Levine, B. (2013). Autophagy and viruses: adversaries or allies? J. Innate Immun. 5, 480–493. doi: 10.1159/000346388
Dong, X., Yang, Y., Zou, Z., Zhao, Y., Ci, B., Zhong, L., et al. (2021). Sorting nexin 5 mediates virus-induced autophagy and immunity. Nature 589, 456–461. doi: 10.1038/s41586-020-03056-z
Dörfel, D., Appel, S., Grünebach, F., Weck, M. M., Müller, M. R., Heine, A., et al. (2005). Processing and presentation of HLA class I and II epitopes by dendritic cells after transfection with in vitro–transcribed MUC1 RNA. Blood 105, 3199–3205. doi: 10.1182/blood-2004-09-3556
Dreux, M., Gastaminza, P., Wieland, S. F., and Chisari, F. V. (2009). The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. U.S.A. 106, 14046–14051. doi: 10.1073/pnas.0907344106
Du, Y., Duan, T., Feng, Y., Liu, Q., Lin, M., Cui, J., et al. (2018). LRRC25 inhibits type I IFN signaling by targeting ISG15-associated RIG-I for autophagic degradation. EMBO J. 37, 351–366. doi: 10.15252/embj.201796781
English, L., Chemali, M., Duron, J., Rondeau, C., Laplante, A., Gingras, D., et al. (2009b). Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 10, 480–487. doi: 10.1038/ni.1720
English, L., Chemali, M., and Desjardins, M. (2009a). Nuclear membrane-derived autophagy, a novel process that participates in the presentation of endogenous viral antigens during HSV-1 infection. Autophagy 5, 1026–1029. doi: 10.4161/auto.5.7.9163
Feng, Y., He, D., Yao, Z., and Klionsky, D. J. (2014). The machinery of macroautophagy. Cell Res. 24, 24–41. doi: 10.1038/cr.2013.168
Gannagé, M., Dormann, D., Albrecht, R., Dengjel, J., Torossi, T., Rämer, P. C., et al. (2009). Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 6, 367–380. doi: 10.1016/j.chom.2009.09.005
Gui, X., Yang, H., Li, T., Tan, X., Shi, P., Li, M., et al. (2019). Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 567, 262–266. doi: 10.1038/s41586-019-1006-9
Gürtler, C., and Bowie, A. G. (2013). Innate immune detection of microbial nucleic acids. Trends Microbiol. 21, 413–420. doi: 10.1016/j.tim.2013.04.004
Hamasaki, M., Furuta, N., Matsuda, A., Nezu, A., Yamamoto, A., Fujita, N., et al. (2013). Autophagosomes form at ER–mitochondria contact sites. Nature 495, 389–393. doi: 10.1038/nature11910
Hayn, M., Hirschenberger, M., Koepke, L., Nchioua, R., Straub, J. H., Klute, S., et al. (2021). Systematic functional analysis of SARS-CoV-2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep. 35:109126. doi: 10.1016/j.celrep.2021.109126
Heaton, N. S., and Randall, G. (2010). Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe 8, 422–432. doi: 10.1016/j.chom.2010.10.006
Hickman, M., De Angelis, D., Vickerman, P., Hutchinson, S., and Martin, N. (2015). Hcv treatment as prevention in people who inject drugs–testing the evidence. Curr. Opin. Infect Dis. 28:576. doi: 10.1097/QCO.0000000000000216
Hoofnagle, J. H., and Feinstone, S. M. (2020). The discovery of Hepatitis C—the 2020 Nobel prize in physiology or medicine. N. Engl. J. Med. 383, 2297–2299. doi: 10.1056/NEJMp2031110
Hou, P., Yang, K., Jia, P., Liu, L., Lin, Y., Li, Z., et al. (2021). A novel selective autophagy receptor, CCDC50, delivers K63 polyubiquitination-activated RIG-I/MDA5 for degradation during viral infection. Cell Res. 31, 62–79. doi: 10.1038/s41422-020-0362-1
Ishikawa, H., and Barber, G. N. (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678. doi: 10.1038/nature07317
Jin, S., Tian, S., Luo, M., Xie, W., Liu, T., Duan, T., et al. (2017). Tetherin suppresses type I interferon signaling by targeting MAVS for NDP52-mediated selective autophagic degradation in human cells. Mol. Cell 68, 308–322. e304. doi: 10.1016/j.molcel.2017.09.005
Jo, D. S., Park, S. J., Kim, A.-K., Park, N. Y., Kim, J. B., Bae, J.-E., et al. (2020). Loss of HSPA9 induces peroxisomal degradation by increasing pexophagy. Autophagy 16, 1989–2003. doi: 10.1080/15548627.2020.1712812
Joffre, O. P., Segura, E., Savina, A., and Amigorena, S. (2012). Cross-presentation by dendritic cells. Nat. Rev. Immunol. 12, 557–569. doi: 10.1038/nri3254
Jounai, N., Takeshita, F., Kobiyama, K., Sawano, A., Miyawaki, A., Xin, K.-Q., et al. (2007). The Atg5–Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. U.S.A. 104, 14050–14055. doi: 10.1073/pnas.0704014104
Kaplan, D. E. (2020). Hepatitis C virus. Ann. Intern. Med. 173, ITC33–ITC48. doi: 10.7326/AITC202009010
Kaushik, S., and Cuervo, A. M. (2012). Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 22, 407–417. doi: 10.1016/j.tcb.2012.05.006
Khaminets, A., Heinrich, T., Mari, M., Grumati, P., Huebner, A. K., Akutsu, M., et al. (2015). Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522, 354–358. doi: 10.1038/nature14498
Kim, N., Kim, M.-J., Sung, P. S., Bae, Y. C., Shin, E.-C., and Yoo, J.-Y. (2016). Interferon-inducible protein SCOTIN interferes with HCV replication through the autolysosomal degradation of NS5A. Nat. Commun. 7:10631. doi: 10.1038/ncomms10631
Koepke, L., Hirschenberger, M., Hayn, M., Kirchhoff, F., and Sparrer, K. M. J. A. (2021). Manipulation of autophagy by SARS-CoV-2 proteins. Autophagy 1–3. doi: 10.1080/15548627.2021.1953847 [Epub ahead of print].
Koyama, S., Ishii, K. J., Coban, C., and Akira, S. (2008). Innate immune response to viral infection. Cytokine 43, 336–341. doi: 10.1016/j.cyto.2008.07.009
Kuma, A., Hatano, M., Matsui, M., Yamamoto, A., Nakaya, H., Yoshimori, T., et al. (2004). The role of autophagy during the early neonatal starvation period. Nature 432, 1032–1036. doi: 10.1038/nature03029
Lamb, C. A., Yoshimori, T., and Tooze, S. A. (2013). The autophagosome: origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 14, 759–774. doi: 10.1038/nrm3696
Lee, H. K., Lund, J. M., Ramanathan, B., Mizushima, N., and Iwasaki, A. (2007). Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 315, 1398–1401. doi: 10.1126/science.1136880
Lee, H. K., Mattei, L. M., Steinberg, B. E., Alberts, P., Lee, Y. H., Chervonsky, A., et al. (2010). In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity 32, 227–239. doi: 10.1016/j.immuni.2009.12.006
Lei, Y., Wen, H., Yu, Y., Taxman, D. J., Zhang, L., Widman, D. G., et al. (2012). The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity 36, 933–946. doi: 10.1016/j.immuni.2012.03.025
Levine, B. (2005). Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell 120, 159–162. doi: 10.1016/S0092-8674(05)00043-7
Levine, B., and Kroemer, G. (2008). Autophagy in the pathogenesis of disease. Cell 132, 27–42. doi: 10.1016/j.cell.2007.12.018
Levine, B., and Kroemer, G. (2019). Biological functions of autophagy genes: a disease perspective. Cell 176, 11–42. doi: 10.1016/j.cell.2018.09.048
Levine, B., Mizushima, N., and Virgin, H. W. (2011). Autophagy in immunity and inflammation. Nature 469, 323–335. doi: 10.1038/nature09782
Li, F., Li, J., Wang, P.-H., Yang, N., Huang, J., Ou, J., et al. (2021). SARS-CoV-2 spike promotes inflammation and apoptosis through autophagy by ROS-suppressed PI3K/AKT/mTOR signaling. Biochim. Biophys. Acta Mol. Basis Dis. 1867:166260. doi: 10.1016/j.bbadis.2021.166260
Liang, Q., Seo, G. J., Choi, Y. J., Kwak, M.-J., Ge, J., Rodgers, M. A., et al. (2014). Crosstalk between the cGAS DNA sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe 15, 228–238. doi: 10.1016/j.chom.2014.01.009
Liang, X. H., Kleeman, L. K., Jiang, H. H., Gordon, G., Goldman, J. E., Berry, G., et al. (1998). Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 72, 8586–8596. doi: 10.1128/JVI.72.11.8586-8596.1998
Liu, D., Wu, H., Wang, C., Li, Y., Tian, H., Siraj, S., et al. (2019). STING directly activates autophagy to tune the innate immune response. Cell Death Differ. 26, 1735–1749. doi: 10.1038/s41418-018-0251-z
Liu, S., Cai, X., Wu, J., Cong, Q., Chen, X., Li, T., et al. (2015). Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347:aaa2630. doi: 10.1126/science.aaa2630
Lussignol, M., Queval, C., Bernet-Camard, M.-F., Cotte-Laffitte, J., Beau, I., Codogno, P., et al. (2013). The herpes simplex virus 1 Us11 protein inhibits autophagy through its interaction with the protein kinase PKR. J. Virol. 87, 859–871. doi: 10.1128/JVI.01158-12
Medzhitov, R. (2007). Recognition of microorganisms and activation of the immune response. Nature 449, 819–826. doi: 10.1038/nature06246
Melia, T. J., Lystad, A. H., and Simonsen, A. (2020). Autophagosome biogenesis: from membrane growth to closure. J. Cell Biol. 219:e202002085. doi: 10.1083/jcb.202002085
Miao, G., Zhao, H., Li, Y., Ji, M., Chen, Y., Shi, Y., et al. (2021). ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev. Cell 56, 427–442. e425. doi: 10.1016/j.devcel.2020.12.010
Miceli, C., Roccio, F., Penalva-Mousset, L., Burtin, M., Leroy, C., Nemazanyy, I., et al. (2020). The primary cilium and lipophagy translate mechanical forces to direct metabolic adaptation of kidney epithelial cells. Nat. Cell Biol. 22, 1091–1102. doi: 10.1038/s41556-020-0566-0
Mijaljica, D., and Klionsky, D. J. (2020). Autophagy/Virophagy: A “Disposal Strategy” to Combat COVID-19. Milton Park: Taylor & Francis. doi: 10.1080/15548627.2020.1782022
Mijaljica, D., Prescott, M., and Devenish, R. J. (2011). Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy 7, 673–682. doi: 10.4161/auto.7.7.14733
Mintern, J. D., Macri, C., Chin, W. J., Panozza, S. E., Segura, E., Patterson, N. L., et al. (2015). Differential use of autophagy by primary dendritic cells specialized in cross-presentation. Autophagy 11, 906–917. doi: 10.1080/15548627.2015.1045178
Minton, K. (2016). Autophagy: inflammatory pathology of Fanconi anaemia. Nat. Rev. Mol. Cell Biol. 17, 330–331. doi: 10.1038/nrm.2016.64
Mizushima, N., and Levine, B. (2010). Autophagy in mammalian development and differentiation. Nat. Cell Biol. 12, 823–830. doi: 10.1038/ncb0910-823
Mizushima, N., and Levine, B. (2020). Autophagy in human diseases. N. Engl. J. Med. 383, 1564–1576. doi: 10.1056/NEJMra2022774
Mizushima, N., Levine, B., Cuervo, A. M., and Klionsky, D. J. (2008). Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075. doi: 10.1038/nature06639
Mizushima, N., Yamamoto, A., Hatano, M., Kobayashi, Y., Kabeya, Y., Suzuki, K., et al. (2001). Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 152, 657–668. doi: 10.1083/jcb.152.4.657
Mochida, K., Oikawa, Y., Kimura, Y., Kirisako, H., Hirano, H., Ohsumi, Y., et al. (2015). Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 522, 359–362. doi: 10.1038/nature14506
Morón, G., Dadaglio, G., and Leclerc, C. (2004). New tools for antigen delivery to the MHC class I pathway. Trends Immunol. 25, 92–97. doi: 10.1016/j.it.2003.11.008
Muntjewerff, E. M., Meesters, L. D., and Van Den Bogaart, G. (2020). Antigen cross-presentation by macrophages. Front. Immunol. 11:1276. doi: 10.3389/fimmu.2020.01276
Münz, C. (2006). Autophagy and antigen presentation. Cell. Microbiol. 8, 891–898. doi: 10.1111/j.1462-5822.2006.00714.x
Nakatogawa, H. (2020). Mechanisms governing autophagosome biogenesis. Nat. Rev. Mol. Cell Biol. 21, 439–458. doi: 10.1038/s41580-020-0241-0
Neefjes, J. (1999). CIIV, MIIC and other compartments for MHC class II loading. Eur. J. Immunol. 29, 1421–1425. doi: 10.1002/(SICI)1521-4141(199905)29:05<1421::AID-IMMU1421>3.0.CO;2-C
Nimmerjahn, F., Milosevic, S., Behrends, U., Jaffee, E. M., Pardoll, D. M., Bornkamm, G. W., et al. (2003). Major histocompatibility complex class II-restricted presentation of a cytosolic antigen by autophagy. Eur. J. Immunol. 33, 1250–1259. doi: 10.1002/eji.200323730
O’Donnell, V., Pacheco, J. M., LaRocco, M., Burrage, T., Jackson, W., Rodriguez, L. L., et al. (2011). Foot-and-mouth disease virus utilizes an autophagic pathway during viral replication. Virology 410, 142–150. doi: 10.1016/j.virol.2010.10.042
Onishi, M., Yamano, K., Sato, M., Matsuda, N., and Okamoto, K. (2021). Molecular mechanisms and physiological functions of mitophagy. EMBO J. 40:e104705. doi: 10.15252/embj.2020104705
Orenstein, S. J., and Cuervo, A. M. (2010). Chaperone-mediated autophagy: molecular mechanisms and physiological relevance. Semin. Cell Dev. Biol. 21, 719–26. doi: 10.1016/j.semcdb.2010.02.005
Orvedahl, A., Alexander, D., Tallóczy, Z., Sun, Q., Wei, Y., Zhang, W., et al. (2007). HSV-1 ICP34. 5 confers neurovirulence by targeting the beclin 1 autophagy protein. Cell Host Microbe 1, 23–35. doi: 10.1016/j.chom.2006.12.001
Orvedahl, A., and Levine, B. (2009a). Autophagy in mammalian antiviral immunity. Curr. Top Microbiol. Immunol. 335, 267–285. doi: 10.1007/978-3-642-00302-8_13
Orvedahl, A., and Levine, B. (2009b). Eating the enemy within: autophagy in infectious diseases. Cell Death Differ. 16, 57–69. doi: 10.1038/cdd.2008.130
Orvedahl, A., MacPherson, S., Sumpter, R. Jr., Tallóczy, Z., Zou, Z., and Levine, B. (2010). Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 7, 115–127. doi: 10.1016/j.chom.2010.01.007
Orvedahl, A., Sumpter, R., Xiao, G., Ng, A., Zou, Z., Tang, Y., et al. (2011). Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 480, 113–117. doi: 10.1038/nature10546
Paludan, C., Schmid, D., Landthaler, M., Vockerodt, M., Kube, D., Tuschl, T., et al. (2005). Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 307, 593–596. doi: 10.1126/science.1104904
Prabakaran, T., Bodda, C., Krapp, C., Zhang, B. C., Christensen, M. H., Sun, C., et al. (2018). Attenuation of c GAS-STING signaling is mediated by a p62/SQSTM 1-dependent autophagy pathway activated by TBK1. EMBO J. 37:e97858. doi: 10.15252/embj.201797858
Princiotta, M. F., Finzi, D., Qian, S.-B., Gibbs, J., Schuchmann, S., Buttgereit, F., et al. (2003). Quantitating protein synthesis, degradation, and endogenous antigen processing. Immunity 18, 343–354. doi: 10.1016/S1074-7613(03)00051-7
Qu, Y., Wang, X., Zhu, Y., Wang, W., Wang, Y., Hu, G., et al. (2021). ORF3a-mediated incomplete autophagy facilitates severe acute respiratory syndrome coronavirus-2 replication. Front. Cell Dev. Biol. 9:716208. doi: 10.3389/fcell.2021.716208
Rammensee, H.-G., Falk, K., and Rötzschke, O. (1993). MHC molecules as peptide receptors. Curr. Opin. Immunol. 5, 35–44. doi: 10.1016/0952-7915(93)90078-7
Ravindran, R., Khan, N., Nakaya, H. I., Li, S., Loebbermann, J., Maddur, M. S., et al. (2014). Vaccine activation of the nutrient sensor GCN2 in dendritic cells enhances antigen presentation. Science 343, 313–317. doi: 10.1126/science.1246829
Rehwinkel, J., and e Sousa, C. R. (2010). RIGorous detection: exposing virus through RNA sensing. Science 327, 284–286. doi: 10.1126/science.1185068
Rossman, J. S., and Lamb, R. A. (2009). Autophagy, apoptosis, and the influenza virus M2 protein. Cell Host Microbe 6, 299–300. doi: 10.1016/j.chom.2009.09.009
Sahu, R., Kaushik, S., Clement, C. C., Cannizzo, E. S., Scharf, B., Follenzi, A., et al. (2011). Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 20, 131–139. doi: 10.1016/j.devcel.2010.12.003
Saitoh, T., Fujita, N., Hayashi, T., Takahara, K., Satoh, T., Lee, H., et al. (2009). Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl. Acad. Sci. U.S.A. 106, 20842–20846. doi: 10.1073/pnas.0911267106
Schmid, D., Pypaert, M., and Münz, C. (2007). Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 26, 79–92. doi: 10.1016/j.immuni.2006.10.018
Seth, R. B., Sun, L., Ea, C.-K., and Chen, Z. J. (2005). Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell 122, 669–682. doi: 10.1016/j.cell.2005.08.012
Shibutani, S. T., and Yoshimori, T. (2014). A current perspective of autophagosome biogenesis. Cell Res. 24, 58–68. doi: 10.1038/cr.2013.159
Shroff, A., and Nazarko, T. Y. J. C. (2021). The molecular interplay between human coronaviruses and autophagy. Cells 10:2022. doi: 10.3390/cells10082022
Smed-Sörensen, A., Chalouni, C., Chatterjee, B., Cohn, L., Blattmann, P., Nakamura, N., et al. (2012). Influenza A virus infection of human primary dendritic cells impairs their ability to cross-present antigen to CD8 T cells. PLoS Pathog. 8:e1002572. doi: 10.1371/journal.ppat.1002572
Staring, J., von Castelmur, E., Blomen, V. A., van den Hengel, L. G., Brockmann, M., Baggen, J., et al. (2017). PLA2G16 represents a switch between entry and clearance of picornaviridae. Nature 541, 412–416. doi: 10.1038/nature21032
Sumpter, R. Jr., Sirasanagandla, S., Fernández, ÁF., Wei, Y., Dong, X., Franco, L., et al. (2016). Fanconi anemia proteins function in mitophagy and immunity. Cell 165, 867–881. doi: 10.1016/j.cell.2016.04.006
Sun, L., Wu, J., Du, F., Chen, X., and Chen, Z. J. (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791. doi: 10.1126/science.1232458
Takeuchi, O., and Akira, S. (2007). Recognition of viruses by innate immunity. Immunol. Rev. 220, 214–224. doi: 10.1111/j.1600-065X.2007.00562.x
Taylor, M. P., and Kirkegaard, K. (2008). Potential subversion of autophagosomal pathway by picornaviruses. Autophagy 4, 286–289. doi: 10.4161/auto.5377
Tey, S.-K., and Khanna, R. (2012). Autophagy mediates transporter associated with antigen processing-independent presentation of viral epitopes through MHC class I pathway. Blood 120, 994–1004. doi: 10.1182/blood-2012-01-402404
Theisen, D. J., Davidson, J. T., Briseño, C. G., Gargaro, M., Lauron, E. J., Wang, Q., et al. (2018). WDFY4 is required for cross-presentation in response to viral and tumor antigens. Science 362, 694–699. doi: 10.1126/science.aat5030
Villadangos, J. A. (2001). Presentation of antigens by MHC class II molecules: getting the most out of them. Mol. Immunol. 38, 329–346. doi: 10.1016/S0161-5890(01)00069-4
Wang, C., Horby, P. W., Hayden, F. G., and Gao, G. F. J. T. (2020). A novel coronavirus outbreak of global health concern. Lancet 395, 470–473. doi: 10.1016/S0140-6736(20)30185-9
Wong, J., Zhang, J., Si, X., Gao, G., Mao, I., McManus, B. M., et al. (2008). Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 82, 9143–9153. doi: 10.1128/JVI.00641-08
Wu, F., Zhao, S., Yu, B., Chen, Y.-M., Wang, W., Song, Z.-G., et al. (2020). A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269. doi: 10.1038/s41586-020-2008-3
Wu, J., and Chen, Z. J. (2014). Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 32, 461–488. doi: 10.1146/annurev-immunol-032713-120156
Wu, J., Sun, L., Chen, X., Du, F., Shi, H., Chen, C., et al. (2013). Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830. doi: 10.1126/science.1229963
Yoon, S. Y., Ha, Y. E., Choi, J. E., Ahn, J., Lee, H., Kweon, H.-S., et al. (2008). Coxsackievirus B4 uses autophagy for replication after calpain activation in rat primary neurons. J. Virol. 82, 11976–11978. doi: 10.1128/JVI.01028-08
Zeisel, M. B., Felmlee, D. J., and Baumert, T. F. (2013). Hepatitis C virus entry. Hepatitis C virus: from molecular virology to antiviral therapy. Curr. Top Microbiol. Immunol. 369, 87–112. doi: 10.1007/978-3-642-27340-7_4
Zeisel, M. B., Fofana, I., Fafi-Kremer, S., and Baumert, T. F. (2011). Hepatitis C virus entry into hepatocytes: molecular mechanisms and targets for antiviral therapies. J. Hepatol. 54, 566–576. doi: 10.1016/j.jhep.2010.10.014
Zhang, C., Shang, G., Gui, X., Zhang, X., Bai, X.-C., and Chen, Z. J. (2019). Structural basis of STING binding with and phosphorylation by TBK1. Nature 567, 394–398. doi: 10.1038/s41586-019-1000-2
Zhang, X., Paget, M., Wang, C., Zhu, Z., and Zheng, H. (2020). Innate immune evasion by picornaviruses. Eur. J. Immunol. 50, 1268–1282. doi: 10.1002/eji.202048785
Zhang, X., Shi, H., Wu, J., Zhang, X., Sun, L., Chen, C., et al. (2013). Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell 51, 226–235. doi: 10.1016/j.molcel.2013.05.022
Zhang, Y., Sun, H., Pei, R., Mao, B., Zhao, Z., Li, H., et al. (2021). The SARS-CoV-2 protein ORF3a inhibits fusion of autophagosomes with lysosomes. Cell Discov. 7:31. doi: 10.1038/s41421-021-00268-z
Keywords: autophagy, xenophagy, virophagy, viral infection, innate immune response, antigen presentation, inflammation and immunity
Citation: Liang S, Wu Y-S, Li D-Y, Tang J-X and Liu H-F (2021) Autophagy in Viral Infection and Pathogenesis. Front. Cell Dev. Biol. 9:766142. doi: 10.3389/fcell.2021.766142
Received: 28 August 2021; Accepted: 17 September 2021;
Published: 15 October 2021.
Edited by:
Shou-Long Deng, Institute of Laboratory Animal Sciences, Chinese Academy of Medical Sciences, Peking Union Medical College, ChinaReviewed by:
Wenting Li, Henan Agricultural University, ChinaCopyright © 2021 Liang, Wu, Li, Tang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ji-Xin Tang, dGp4OTg2QDE2My5jb20=; Hua-Feng Liu, aGYtbGl1QDI2My5uZXQ=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.