 Jojanneke Stoof
Jojanneke Stoof Emily Harrold2,3
Emily Harrold2,3 Maeve A. Lowery
Maeve A. Lowery Naomi Walsh
Naomi Walsh
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 12 October 2021
Sec. Molecular and Cellular Pathology
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.749490
This article is part of the Research Topic Translational Insights into Pancreatic Ductal Adenocarcinoma View all 16 articles
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal cancers worldwide, and survival rates have barely improved in decades. In the era of precision medicine, treatment strategies tailored to disease mutations have revolutionized cancer therapy. Next generation sequencing has found that up to a third of all PDAC tumors contain deleterious mutations in DNA damage repair (DDR) genes, highlighting the importance of these genes in PDAC. The mechanisms by which DDR gene mutations promote tumorigenesis, therapeutic response, and subsequent resistance are still not fully understood. Therefore, an opportunity exists to elucidate these processes and to uncover relevant therapeutic drug combinations and strategies to target DDR deficiency in PDAC. However, a constraint to preclinical research is due to limitations in appropriate laboratory experimental models. Models that effectively recapitulate their original cancer tend to provide high levels of predictivity and effective translation of preclinical findings to the clinic. In this review, we outline the occurrence and role of DDR deficiency in PDAC and provide an overview of clinical trials that target these pathways and the preclinical models such as 2D cell lines, 3D organoids and mouse models [genetically engineered mouse model (GEMM), and patient-derived xenograft (PDX)] used in PDAC DDR deficiency research.
Pancreatic cancer is one of the most lethal cancers worldwide, accounting for 2.6% of all new cancer cases but causing 4.8% of all cancer deaths (Ferlay et al., 2019). Despite recent advances in personalized and targeted therapy, little progress has been made to improve overall survival (OS) and the 5-year survival rate is estimated at 9% (Siegel et al., 2020).
Currently, curative treatment is limited to low-stage, resectable disease but over 80% of patients present with advanced or metastatic disease (Bilimoria et al., 2007; Stathis and Moore, 2010; Ilic and Ilic, 2016). Current standard of care treatment for advanced pancreatic ductal adenocarcinoma (PDAC) consists of either combination treatment of nab-paclitaxel with gemcitabine or 5-fluorouracil, leucovorin, irinotecan, oxaliplatin (FOLFIRINOX) (Mohammed et al., 2014; Mohammad, 2018; Adel, 2019). In 2013, the MPACT trial showed that nab-paclitaxel with gemcitabine improved OS by 1.8 months compared to gemcitabine alone (8.5 vs. 6.7 months, p < 0.001) (Von Hoff et al., 2013). The PRODIGE trial found that FOLFIRINOX improved OS by 4.3 months compared to gemcitabine alone (11.1 vs. 6.8 months, p < 0.001) (Conroy et al., 2011). However, FOLFIRINOX is associated with higher toxicity profiles and is therefore generally reserved for patients with a good performance status and given as a modified regimen. Systemic treatment options for PDAC include gemcitabine, gemcitabine with erlotinib, FOLFIRINOX, gemcitabine with nab-paclitaxel, nano-liposomal irinotecan with 5-FU, pembrolizumab (patients with microsatellite instability), larotrectinib/entrectinib (patients with NTRK-fusion), and olaparib (patients with gBRCA mutation).
Genomic analyses have revealed a complex mutational landscape that is predominated by mutations in TP53, KRAS, SMAD4, and CDKN2A (Bailey et al., 2016; Aguirre et al., 2018). Despite extensive research, targeted therapies for these mutations have not reached clinical practice (Qian et al., 2020). In addition, PDAC is characterized by genome instability (Alexandrov et al., 2013). Genome instability has been described as one of the enabling hallmarks of cancer by Hanahan and Weinberg (2011) and can be attributed to multiple sources, including increased sensitivity to mutagenic agents, defects in the genomic maintenance machinery, loss of telomeric DNA, and aberrant surveillance mechanisms. While these aberrations can partly be contributed to these four commonly mutated genes, additional pathway deficiencies are also involved. The DNA damage response (DDR) pathway plays a central role in genome maintenance and repair. In contrast to TP53 and KRAS, DDR deficiency is targetable, with multiple drugs already available in the clinic for non-PDAC cancer types, such as breast and prostate cancer (Wengner et al., 2020).
This review briefly covers the role and definition of DDR deficiency in PDAC and provides an overview of clinical trials that investigate DDR targeting drugs. The main focus is on how cell lines, organoids, and mouse models are used to study DDR deficient pathways in PDAC.
To combat the development of mutations and the effects these may have on the cell a complex network of DNA damage repair (DDR) pathways exists (Giglia-Mari et al., 2011). At the core of this network are the pathways for base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), interstrand crosslink repair (ICL repair), and double strand break repair [both homologous recombination (HR) and non-homologous end-joining (NHEJ)] (Figure 1). Each pathway can roughly be divided into three phases or steps: recognition of the damage, excision or processing of the damaged strand(s), and the actual repair.
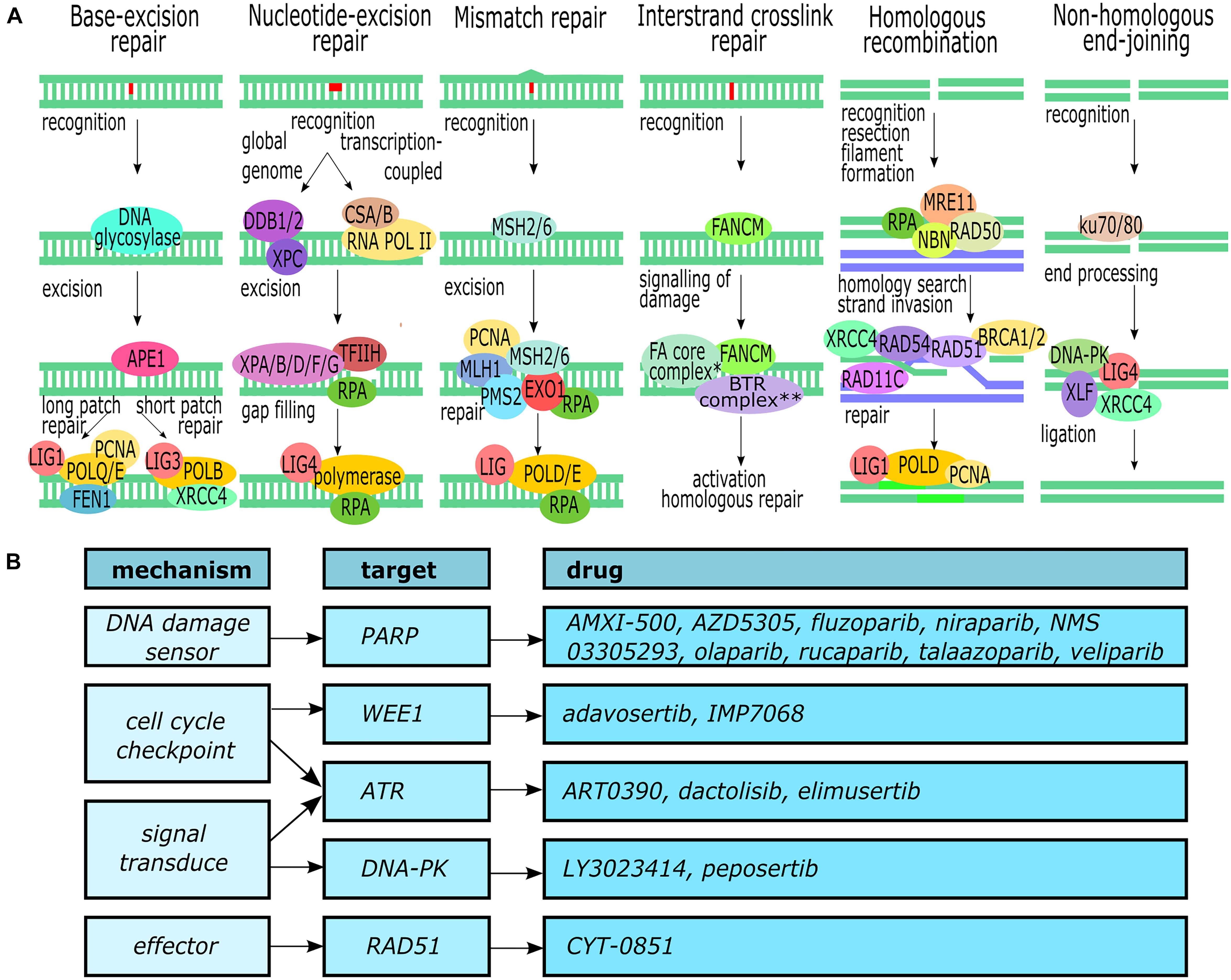
Figure 1. DNA damage repair. (A) Overview of the major DNA repair pathways (Deans and West, 2011; Yang et al., 2016; Ranjha et al., 2018; Gupta and Heinen, 2019; Lee and Kang, 2019). ∗The Fanconi anemia core complex consists of FANCA, FANCB, FANCC, FANCD, FANCE, FANCF, FANCG, and FAAP100. ∗∗The BTR complex consists of BLM, TOPOIII, RMI1, and RMI2. (B) Molecular targets within the DDR pathways and available inhibitors. This figure was created in Inkscape.
Base excision repair removes non-bulky single-base lesions such as oxidation or deamination damage (Lee and Kang, 2019). The damaged base is recognized and removed by one of multiple specific DNA glycosylases (such as UNG, SMUG1, or NEIL1), depending on the type of lesion. Next, the newly created abasic site is excised and processed by APE1 to generate a 3′-hydroxyl site. This 3′-hydroxyl is then used by DNA polymerase to fill the gap using the opposing strand as template.
Nucleotide excision repair is the main pathway for the removal of bulky lesions but can also remove intrastrand crosslinks and cyclobutene pyrimidine dimers that are produced by UV radiation (Schärer, 2013; Lee and Kang, 2019). While two subpathways can be distinguished – global genome NER (GG-NER) for the whole genome and transcription-coupled NER (TC-NER) for the transcribing strand of active genes, the general repair process is similar to BER. GG-NER recognizes distortions of the DNA helix through DDB1, DDB2, and XPC, whereas TC-NER CSA and CSB recognize blockage of the RNA polymerase. TFIIH opens up the DNA to enable XPD to verify the lesion upon which several other XP endonucleases and RPA are recruited to excise the lesion. Finally, the resulting 22–32 nt long gap is filled and ligated to the original DNA strand by DNA polymerases and ligases.
The MMR pathway removes single nucleotide mismatches and small insertions or deletions created by DNA polymerase during DNA synthesis (Gupta and Heinen, 2019). The lesions are recognized by the heterodimer MSH2/MSH6. The dimer recruits another heterodimer, MLH1–PMS2, and together they recruit several other proteins including Exo1 to excise the damage. Finally, polymerase eta or delta fills the newly created gap.
Interstrand crosslinks (ICLs) are caused by bifunctional alkylating agents that form covalent bonds between the two DNA strands (Deans and West, 2011; Hashimoto et al., 2016). In quiescent cells the lesion is recognized and repaired by the NER pathway, but during the S phase several steps take place to activate the HR pathway. When a DNA replication fork encounters an ICL the fork stalls and, through a complex containing FANCM, the lesion is recognized and the Fanconi anemia complex and BTR complex are recruited. These complex create a double-strand break (DSB) which is subsequently recognized and repaired by the HR pathway.
Double-strand breaks are repaired through two main pathways: HR and NHEJ (Giglia-Mari et al., 2011; Yang et al., 2016; Ranjha et al., 2018). HR can take place during the S- and G2-phase of the cell cycle when it can use the homologous sequence of a sister chromatid to accurately repair the break. The DSB is recognized by the MRN complex (consisting of MRE11, RAD50, and NBN), the ends of the break are resected, and RPA binds to and forms a filament between the newly resected single-stranded DNA section. Next, BRCA2 recruits RAD51 to replace the RPA-filament and, assisted by several other proteins, homology search and strand invasion of the sister chromatid takes place. Using the sister chromatid as template, polymerase delta synthesizes the missing nucleotides of the broken strand and the ends are ligated. NHEJ, in contrast, can take place during every phase of the cell cycle and is quicker than HR but is also error-prone and commonly results in small deletions. The break is recognized by the ku70/80 heterodimer which subsequently recruits DNA-PKcs, XLF, XRCC4, and Lig4 to process and ligate the broken ends of the DNA strands. In recent years, important progress has been made in deciphering the molecular underpinnings of PDAC due to the unparalleled power of next generation sequencing (NGS) technologies. Constitutive mutations of PDAC have been described as selective DDR pathways in PDAC, however, the main problem encountered is the heterogeneity of somatic alterations among patients outside of the four most frequently mutated genes (KRAS, CDKN2A, TP53, and SMAD4) which poses a challenge to the identification of potential predictive and prognostic biomarkers.
Approximately 10% of all PDAC cases are considered familial; defined as a family with at least two first-degree relatives with PDAC (Turati et al., 2013). While several germline pathogenic alterations that increase an individual’s lifetime risk of PDAC (e.g., hereditary pancreatitis and Lynch syndrome) have been characterized, the causative germline mutation of most familial cases remains unclear (Klein, 2012). The most commonly mutated genes in familial pancreatic cancer are BRCA2, CDKN2A, BRCA1, and PALB2 (Perkhofer et al., 2020). Pathogenic germline alterations have also been identified in patients who do not meet criteria for familial PDAC, and may involve genes beyond those previously associated with hereditary pancreatic cancer. These pathogenic germline alterations are therapeutically considered actionable in 5–10% of patients, and clinical guidelines now support routinely offering germline genetic testing with a broad panel of known hereditary cancer predisposition genes to all PDAC patients.
The presence of DDR gene mutations has been reported in 17–43% of all sporadic PDAC patients (Waddell et al., 2015; Aguirre et al., 2018). However, these papers focused on a limited selection of well-characterized DDR genes and potentially actionable DDR mutations may be more prevalent. We queried the GENIE cohort (The AACR Project GENIE Consortium, 2017) containing 3706 PDAC patients with somatic mutation profiling for the presence of mutations in any of the genes of the six major DDR pathways (BER, NER, HR, NHEJ, ICL repair, and MMR) (Deans and West, 2011; Yang et al., 2016; Ranjha et al., 2018; Gupta and Heinen, 2019; Lee and Kang, 2019). A comprehensive list of 352 genes was collated based on the gene lists of the respective pathways in the Gene Ontology database. Mutations were reported in 117 (33%) of the genes, with 46 (13%) and 14 (4.0%) genes being mutated in more than 1 and 2% of the patients, respectively. The most commonly mutated genes were TP53 (68.9%), BRCA2 (4.4%), ATM (4%), and PRKDC (3.9%). An overview of these genes and associated pathways can be found in Table 1, Figure 1, and Supplementary Table 1.
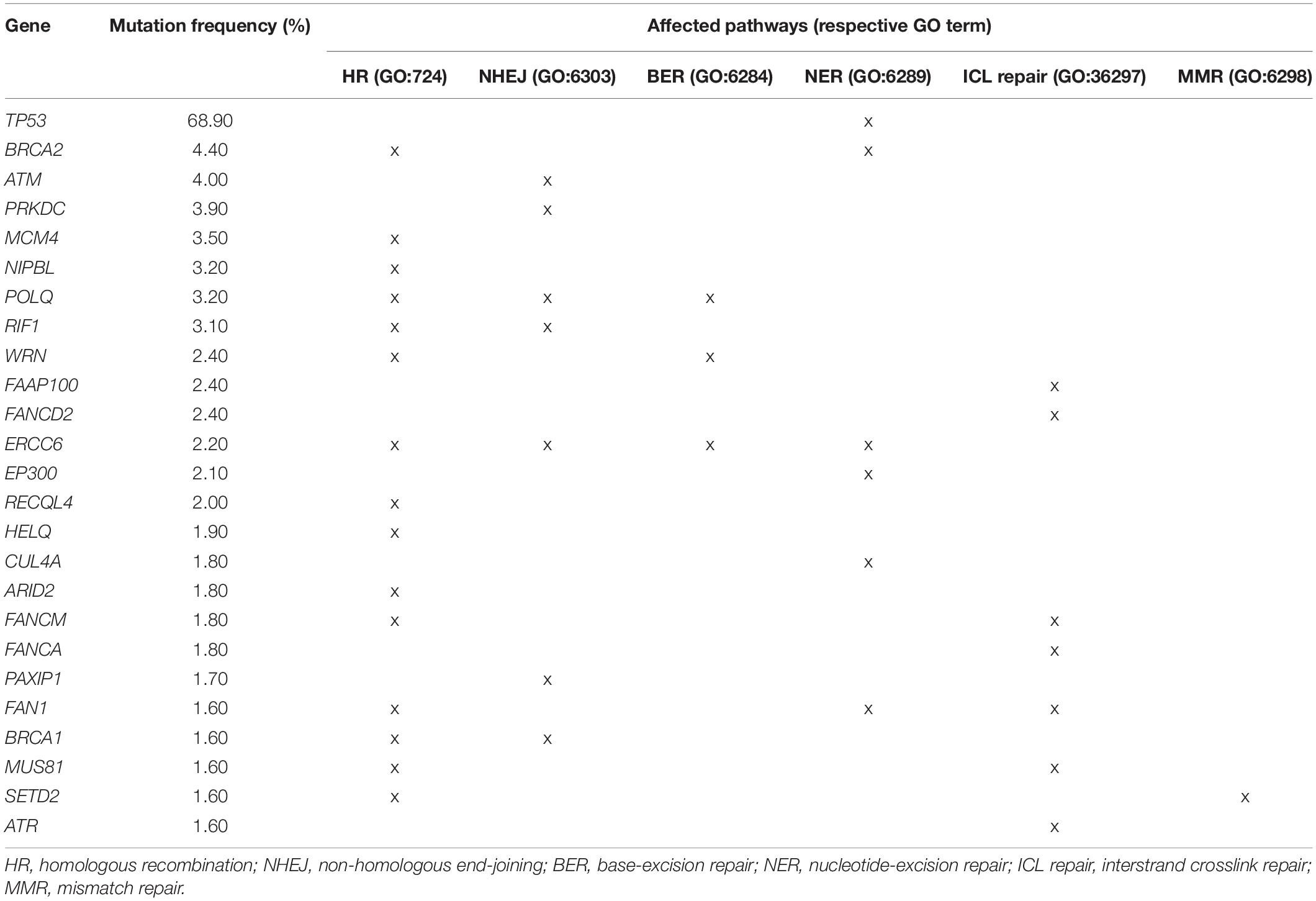
Table 1. Prevalence of the top 25 most frequently found somatic DDR gene mutations and associated pathways in a cohort of 3706 PDAC patients.
The relatively high prevalence of DDR gene mutations opens up opportunities for targeted therapies based on the synthetic lethality principle: tumors with a DDR pathway deficiency are more dependent on alternative DNA repair pathways to repair double-stranded DNA breaks (Guo et al., 2011; Topatana et al., 2020). Synthetic lethality has been applied successfully in cancers harboring BRCA1/2 mutations (homologous repair pathway) by treating them with PARP inhibitors (PARP is involved in the single-strand break repair pathway) (Lord and Ashworth, 2017). Unrepaired single-strand breaks will turn into DSBs during DNA replication which will accumulate to the point of cell death due to the HR deficiency.
Multiple research groups have performed next-generation sequencing and expression profiling to classify molecular PDAC subtypes that can be used to tailor therapies and guide clinical decision making (Collisson et al., 2011; Moffitt et al., 2015; Bailey et al., 2016; Puleo et al., 2018). At its simplest, a distinction is made between classical and basal-like subtypes, though most classifications include more specific subtypes as well (Figure 2). Bailey et al. (2016) defined four PDAC subtypes (immunogenic, pancreatic progenitor, ADEX, and squamous) based on 10 discriminatory gene programs found by transcriptional network analysis. Over 50 DDR genes were included in the gene program “proliferation” which is associated with the squamous subtype. Functionally, the squamous subtype is associated with histological adenosquamous carcinoma and a poor survival. The classifications by Collisson et al. (2011), Moffitt et al. (2015), and Puleo et al. (2018) found no associations with DDR deficiency.
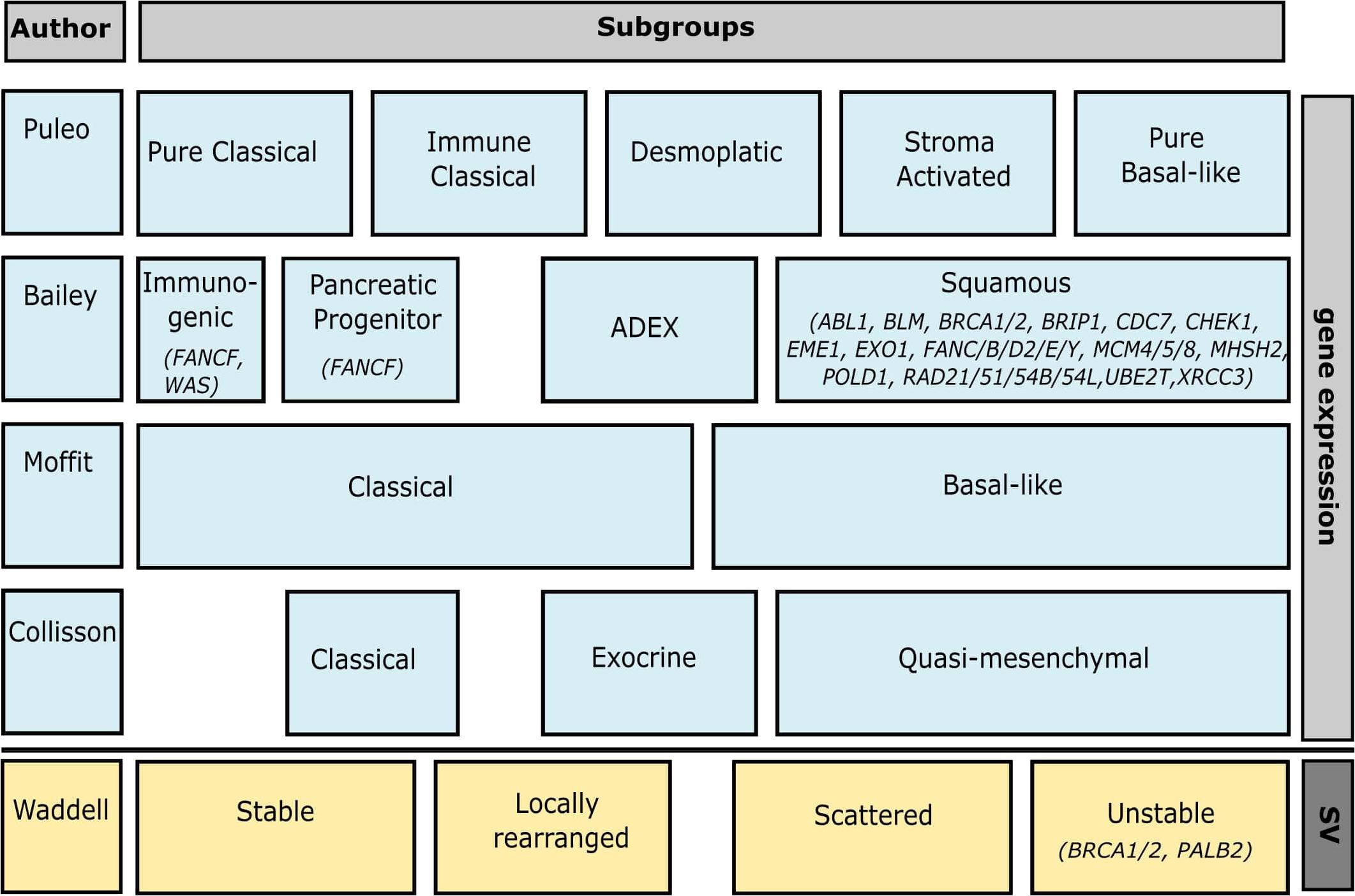
Figure 2. Overview of genomic pancreatic subtypes and how they overlap. Associated DDR genes of which mutations have been found in PDAC patients (see Supplementary Table 1) are included under their respective subtypes. The bar on the right indicates whether the classification is based on gene expression or structural variation (SV). Associated DDR genes not mutated in patients include CDC45, FEN1, GINS2/4, MAD2L2, MCM2/3/6/7, RMI2, RPA3, TIMELESS, HMGB2, POLA1, LIG1, DNA2, RDC2/3/4/5, PCNA, COPS5, BRIP1, HMGA2M, CETN2, UBC, TP73, PSMD14, POLR2D, and CDK7 for Bailey’s squamous subtype, and USP7 for Bailey’s Pancreatic Progenitor subtype. This figure was created in Inkscape.
Instead of mutational signatures, Waddell et al. (2015) based their classification on structural variation. Using a dataset of 75 primary samples and 25 patient-derived cell lines (PDCLs) they defined four subtypes: stable, locally rearranged, scattered, and unstable. The unstable subtype co-segregated with inactivation of BRCA1, BRCA2, and PALB2, as well as a mutational signature of DDR deficiency. Mutations of ATM and other genes involved in DNA maintenance (e.g., XRCC4/6 and FANCM) were also regularly found in these tumors.
Currently only gBRCA is used in the clinic as biomarker for sensitivity to PARP inhibition (PARPi) olaparib. Our query of somatic DDR mutations found that BRCA2 is mutated in 4% of all PDAC patients indicating that a larger group of patients may benefit from targeted therapy. In addition, multiple clinical trials are recruiting patients for treatment with DDR inhibitors based on a larger selection of DDR mutations, including but not limited to PALB2, CHEK2, ATM, and RAD51 (Supplementary Table 2). Their outcomes will show whether more DDR genes can be included as biomarkers for targeted therapy in the clinic.
Already, DDR deficiency has been associated with a significantly better patient survival compared to DDR proficiency independently of tumor subtype classification (Zimmermann et al., 2021). However, it should be noted that analysis of the interaction with precise treatment regimens is limited, in particular with regard to receipt of platinum based therapy, and this can be a considerable confounder. In addition, a small retrospective study in 36 patients treated with first-line FOLFIRINOX in a metastatic setting found that DDR deficiency, as based on a 14-gene panel, was significantly associated with improved survival (p = 0.04) (Sehdev et al., 2018). While the DDR deficient patient group also had a better median OS (14 vs. 5 months) this difference was not significant (p = 0.08). A similar retrospective study in 40 patients with metastatic PDAC treated with first-line platinum chemotherapy in combination with FOLFIRINOX was published a year later (Palacio et al., 2019). Based on a 35-gene panel, the patients with DDR deficiency had a significantly longer progression-free survival (PFS) (18.5 vs. 6.9 months, p = 0.003), with a trend toward superior median OS as well. Further research is needed to confirm these findings in a larger cohort and to investigate whether DDR deficiency is associated with response to FOLFIRINOX treatment or OS in general.
Despite the promising results for many targeted therapies in other solid tumors such as breast, lung, and colon, the use of targeted therapies in PDAC has had limited survival benefit in the clinic. Target discovery and successful development of targeted therapies is highly dependent on the relevance of the preclinical models used and therapies frequently fail at the transition to clinical trials. Multiple recent papers are available which review the preclinical models used in PDAC research (Moreira et al., 2018; Garcia et al., 2020; Swayden et al., 2020; Yu et al., 2021). This review will extend upon published literature by focusing on the application of these models to further target DDR pathways.
Cell lines remain the most commonly used preclinical model for cancer research. Their widespread use has ensured that they are readily available and most commercial cell lines are well-characterized (Deer et al., 2010). The main advantages of cell lines are that they are cheap, require little maintenance, and are easy to manipulate. In addition, cell lines are considered to be more homogenous than other preclinical models, thus contributing to a better reproducibility which makes them well-suited for high-throughput drug screening. However, this also means that cell lines lack the complexity and heterogeneity typical of tumors. At the same time, clonality and adaptation to 2D culturing conditions as well as immortalization and repeated passaging can all contribute to genomic drift which can significantly affect drug responses (Hughes et al., 2007; Monberg et al., 2021). Other disadvantages of 2D culture include loss of part of the normal 3D morphology, cell polarity, and cell–cell or cell–stroma interactions, especially the interaction with cancer-associated fibroblasts (CAFs) and immune cells (Feig et al., 2012). While some of these disadvantages can be resolved or diminished by adapting the culture methods (e.g., using early passage PDCLs instead of established cell lines, or co-culturing with fibroblasts) other disadvantages are inherent to the model system itself.
While the possible applications of cancer cell lines are diverse, ranging from biomarker discovery to functional studies, the use of cell lines to study the DDR pathways in PDAC has mainly been limited to drug sensitivity studies. Investigated drugs include PARP inhibitors (e.g., veliparib, olaparib, and rucaparib), WEE1 inhibitors, ATM inhibitors, ATR inhibitors, DNA protein kinase catalytic domain (PRKCD) inhibitors, and more.
DNA damage repair pathway deficiency has been shown to confer sensitivity to PARPi. Multiple studies found that the BRCA2-deficient cell line Capan-1 is significantly more sensitive to several PARP inhibitors and cisplatin, but not to gemcitabine, compared to the BRCA2-proficient cell lines MiaPaCa-2 and Panc-1 (Porcelli et al., 2013; Andrei et al., 2015; de Soto, 2020). In addition, restoration of BRCA2 expression in Capan-1 cell lines was shown to reduce sensitivity to olaparib and HYDAMTIQ (Mini et al., 2017; Sullivan-Reed et al., 2018). Similarly, shRNA-mediated knockdown of BRCA2 in Panc-1 cells impaired homology-directed repair and conferred sensitivity to BMN-673 (but not to veliparib) (Andrei et al., 2015). Furthermore, increased sensitivity to PARPi (olaparib, BMN-673, and rucaparib) and cisplatin has been found in DDR deficient PDCLs (Dreyer et al., 2021).
Acquired resistance is a problem in many cancer treatments. Likewise, long-term treatment of Capan-1 cells with low dose PARPi can induce resistance, including cross-resistance to other PARPi and cisplatin. Several mechanisms have been suggested for the development of resistance in Capan-1 BRCA2-deficient cell line, the simplest being the restoration of BRCA2 expression. Sakai et al. (2008) found that 7 out of 14 Capan-1 clones that developed resistance to cisplatin treatment had additional mutations in the BRCA2 gene which corrected the original frameshift mutation found in Capan-1. The truncated protein was also still present suggesting that these restorative mutations were preceded by gene duplication. However, the amplification of the truncated protein might in itself also contribute to resistance. Park et al. (2020) investigated PARPi resistant Capan-1 clones and did not detect reversion mutations, though several clones had additional copies of the mutant BRCA2 allele as well as an increased BRCA2 protein expression. Immunoprecipitation of BRCA2 followed by mass spectrometry showed enrichment of PALB2, RAD51, MLLT10, and DOT1L in the resistant clones, but not in the parent cells, which may contribute to resistance. Alternatively, Chen et al. (2020) also found additional mutations in BRCA2 in PARPi resistant Capan-1 clones, but these mutations resulted in truncated splice isoforms. In addition, they found overexpression of the anti-apoptotic proteins COX-2 and BIRC3. Depletion of either BRCA2, COX-2, or BIRC3 partially restored PARPi sensitivity. In contrast, combined depletion had no additive effect, suggesting that additional mechanisms contribute to PARPi resistance.
Sensitivity to the WEE1 inhibitor AZD-1775 has been evaluated in multiple studies, but due to contradicting findings its role in DDR deficiency remains unclear. Two studies found that Capan-1 is markedly more sensitive to AZD-1775 than other (PDAC) cell lines, suggesting that BRCA2 deficiency might play a role (Dréan et al., 2017; Parsels et al., 2018). However, while restoration of the BRCA2 open reading frame due to secondary mutations induced by CRISPR-Cas9 reduced the sensitivity to PARP inhibitors olaparib and BMN-673, it did not affect the sensitivity to AZD-1775. On the other hand, Lal et al. (2016) investigated sensitivity to AZD-1775 in a panel of nine PDAC cell lines and reported a medium sensitivity for Capan-1. In addition, they found that knockdown of BRCA2 by siRNA in MiaPaCa-2 and PL5 induced resistance to AZD-1775. These contradicting findings highlight the need for further investigation.
The application of ATR inhibitors in PDAC has been investigated in multiple in vitro studies in both human PDAC cell lines and mouse KPC and KPCB cell lines but so far drugs have shown limited potential and sensitivity to treatment does not correlate with the DDR status (Wallez et al., 2018; Elliott et al., 2019; Dreyer et al., 2021). However, multiple studies have found that ATRi (VE-821 and VE-822) sensitizes to gemcitabine and radiotherapy through impairment of the DNA repair (Fokas et al., 2012; Wallez et al., 2018). siRNA knockdown of another major signal transducer, ATM, in combination with ATRi in MiaPaCa-2 was able to prevent gemcitabine-induced activation of ATR completely (Wallez et al., 2018), suggesting that ATM-mutant tumors may be especially sensitive to this combination treatment. In addition, combination treatment with chloroquine, an autophagy inhibitor that is used in the treatment of malaria, significantly reduced proliferation in 24 or 17 out of 26 tested PDAC cell lines compared to VE-822 or chloroquine alone (Elliott et al., 2019). Azorsa et al. (2009) used an RNAi screen to identify which genes, when silenced, sensitized pancreatic cancer cells to gemcitabine. Silencing of CHK1 was found to be most effective and was further validated with additional siRNAs and two small molecule inhibitors (SB218078 and PD407824) in MiaPaCa-2 and BxPC3 cell lines (Azorsa et al., 2009).
A disadvantage of DDR targeted therapy is that it is not inherently cytotoxic. By inhibiting multiple DNA repair genes the cancer cells will accrue DNA damage, but whether this results in cell death or senescence depends on additional factors, such as the proliferation rate and how well the cells tolerate replicative stress. Combination treatment with chemo- or radiotherapy can increase the anti-tumor effect by inducing additional DNA damage (Porcelli et al., 2013). Perkhofer et al. (2017) generated stable mouse cell lines from tumors with pancreas-specific loss of Kras (KC), and Kras and Atm (AKC). Atm-deficient AKC cells showed a significant increase in DNA damage markers 53BP1 and γH2AFX upon treatment with 5 Gy of ionizing radiation compared to KC cells (p < 0.03), indicating impaired DSB repair, and had decreased proliferation. No significant differences were observed in sensitivity to cisplatin, 5-FU, or gemcitabine. Treatment with olaparib or niraparib reduced viability in an Atm-dependent manner and was potentiated by combination with gemcitabine or radiation (p < 0.01).
The cell lines used for DDR pathway studies in PDAC are mainly limited to Capan-1 as model for DDR/BRCA2-deficiency, and MiaPaCa-2, BxPC-3, and Panc-1 for DDR-proficiency. Studies in additional cell line models are required to analyze the role of the DDR pathways in more depth. Table 2 provides an overview of the mutations found in the 10 most frequently mutated DDR genes (excluding TP53) as per Table 1 for all PDAC cell lines found in the Cancer Cell Line Encyclopedia. Twenty of the 46 cell lines had a mutation in one or more of the investigated genes, of which three (Capan-1, PL18, and SNU-324) had a mutation annotated as pathogenic or likely pathogenic in the ClinVar or COSMIC database.
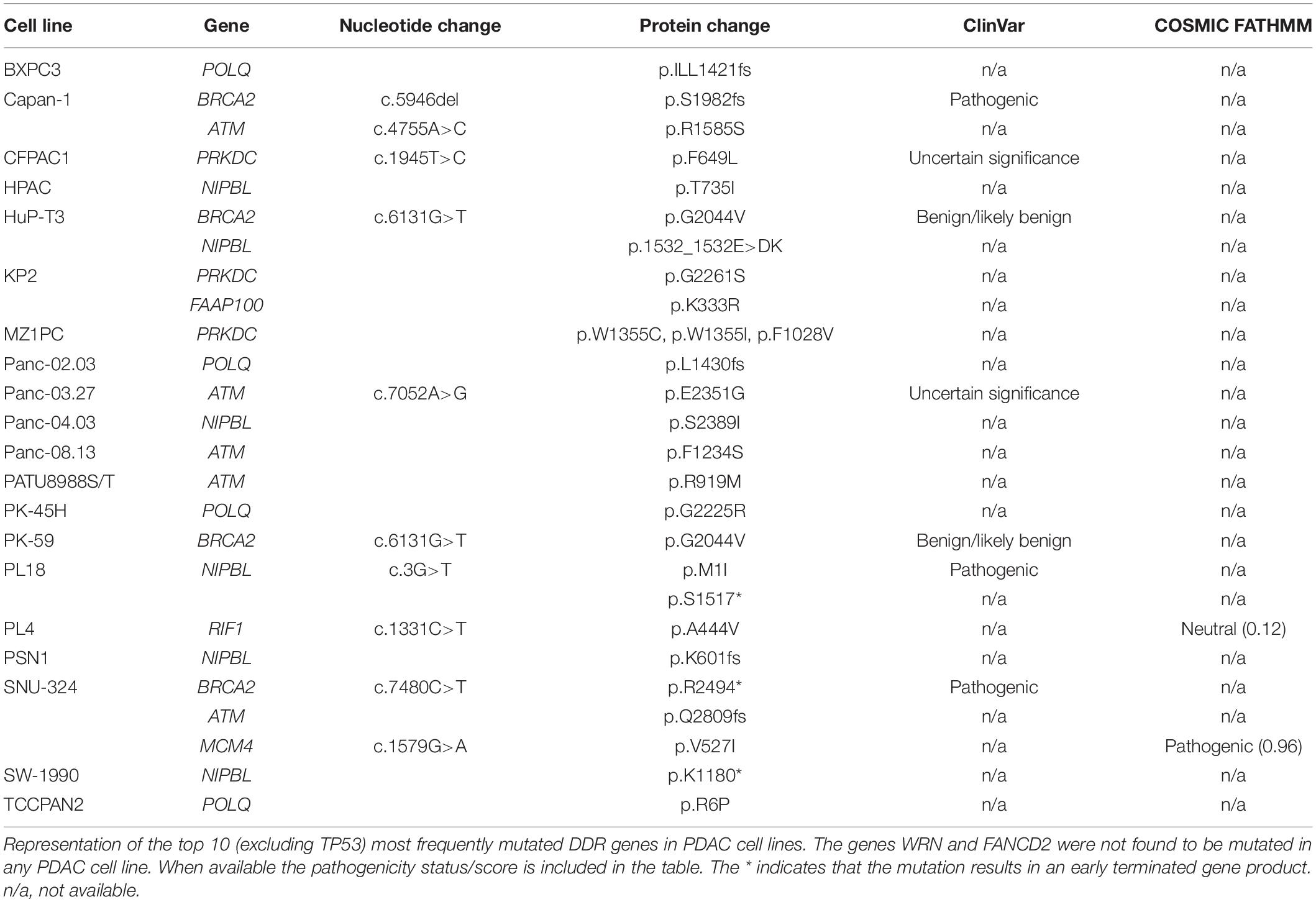
Table 2. Mutation status of DDR genes in cell lines.
Apart from Capan-1, SNU-324 is the only other available cell line with a suspected deleterious BRCA2 mutation (Ku et al., 2002). SNU-324, established in 2001, is derived from a poorly differentiated primary pancreatic tumor of a 50-year-old male. The cells are mainly adherent, but a fraction of the cells grow in suspension and frequently form aggregates. SNU-324 does not contain mutations in KRAS or TP53, but is microsatellite instable (Ku et al., 2002). Despite its usefulness for BRCA2-deficiency studies no other publications are available which have used this cell line. Therefore there is a need for additional well-defined BRCA2-deficient PDAC cell lines.
Patient-derived organoids are still a relatively new model for pancreatic cancer and there are few studies published that focused on the DDR of PDAC organoids. However, there is limited information on patient derived organoid (PDO) sensitivity to DDR-targeted drugs.
Driehuis et al. (2019) performed high-throughput drug screening of 76 drugs in 24 PDOs and found that, in general, PDOs have a similar response to agents that target the same biological process or molecular pathway. Drug response was found to be PDO-specific, thus reflecting patient heterogeneity. Of the 24 PDOs, 1 PDO had a BRCA2 indel and was among the most sensitive PDOs for most of the tested drugs.
Tiriac et al. (2018) performed pharmacotyping on 66 PDOs for the drugs gemcitabine, paclitaxel, irinotecan, 5-FU, and oxaliplatin. They found that PDO response reflected interpatient variability. For nine patients, the PDO response could be compared to patient response. Eight out of nine patients exhibited an outcome consistent with their matched PDO. Additionally, they investigated the correlation between AUC distribution and genotype for a range of drugs, including olaparib. The three samples with the lowest AUC had ATM loss, PALB2 loss, and ATM frameshift plus BRCA2 loss, respectively. The researchers observed a trend between olaparib sensitivity and complete loss of PALB2, but these data must be interpreted cautiously as there were just four PDOs with complete PALB2 loss. In addition, they state that single-copy losses of a range of genes involved in HR deficiency do not correspond with olaparib sensitivity. In line with these findings Dreyer et al. (2021) reported that they found no correlation between DDR status and the response to ATR or WEE1 inhibition in the six PDOs they investigated.
Based on genomic, transcriptomic, and histologic data, organoids are representative models of PDAC (Tiriac et al., 2018; Driehuis et al., 2019; Gendoo et al., 2019). Yet, limited studies have been published highlighting their use in the study of DDR deficiency in PDAC. However, drug screening and correlation to patient response studies are promising and suggest that organoids are good models to determine drug sensitivity for targeted therapies, and might also be used to identify biomarkers for drug sensitivity.
Patient-derived xenografts (PDXs) are well established cancer models and have been reviewed extensively (Garcia et al., 2020; Shi et al., 2020). PDAC xenografts can be established from resection, biopsy material, and ascites. Copy number alterations and gene expression profiling are largely maintained between primary samples and PDX and genomic signatures can be fitted to the Collisson, Moffitt, and Bailey subtypes (Golan et al., 2017; Nicolle et al., 2017; Pham et al., 2021). Interestingly, even though the mouse hosts are immune-deprived, PDX tumor models can reproduce the immune-related phenotype that is found in certain human primary tumors (Nicolle et al., 2017).
However, the application of PDX toward studying DDR deficiency in PDAC is still used infrequently. Golan et al. (2018) developed six PDXs from metastatic lesions of germline BRCA-mutated patients to recapitulate the clinical scenario of BRCA-associated PDAC in xenografts. Patient samples were taken before treatment and during progression to represent treatment naïve and resistant patients. Four models had bi-allelic inactivation of BRCA1/2 and demonstrated increased somatic mutational load compared to the two models that had retained one wild-type copy. Three PDX were treated with olaparib and cisplatin monotherapy, and PDX treatment response as well as HR deficiency profile were found to be associated with patient treatment response to platinum and PARPi.
In a similar study, Lohse et al. (2015) compared treatment sensitivity of four xenografts containing a germline mutation in BRCA1/2 resulting in heterozygous or homozygous loss to three xenografts with wild-type BRCA1/2. Mice were treated for 4 weeks with gemcitabine or cisplatin. The BRCA mutant xenografts were significantly more sensitive to both gemcitabine and cisplatin compared to the BRCA wild-type xenografts (p < 0.0001). In another study, using a BRCA2 mutant and a BRCA2 wild-type xenograft, Lohse et al. (2016) found no significant difference in sensitivity to radiation treatment or olaparib. Additionally, olaparib did not sensitize to radiation but instead reduced the induction of DNA damage in the BRCA mutant xenografts which was attributed to an increased repair of DSBs by the NHEJ pathway and activation of DNA-PK in the BRCA mutant xenograft.
Waddell et al. (2015) compared gemcitabine and cisplatin treatment sensitivity of three PDX with an unstable genome and/or high BRCA mutational signature burden to four PDX without. None of the DDR-proficient xenografts responded to cisplatin, while two out of three DDR-deficient xenografts did. The DDR-deficient xenograft that did not respond had a BRCA mutational signature burden but no unstable genome or mutation in the BRCA pathway. Response to gemcitabine was varied in both groups.
Roger et al. (2020) compared the efficacy of multi-DDR interference as maintenance therapy to continuous FOLFIRINOX treatment in a cell line derived xenograft model. Atm/Kras-deficient PDAC mouse cell lines (AKC) were orthotopically transplanted in mice and treated with four cycles of FOLFIRINOX to mimic a clinical setting, followed by either a combination of olaparib, VE-822 (ATRi), and CC-115 (DNA-PKi); FOLFIRINOX; or vehicle until an ethical endpoint was reached. OS was significantly longer in the multi-DDR group compared to the FOLFIRINOX or placebo groups (28.5 vs. 24.5 vs. 18.0 days, p < 0.02). In addition, the FOLFIRINOX treatment was shown to select for more aggressive subclones, which could partly be erased by multi-DDR treatment. The combination of multiple targeted drugs allowed for lower dosing than used in monotherapy which reduced side effects to a similar level as for the FOLFIRINOX treatment.
The advance of genetic manipulation has allowed for the development of genetically engineered mouse models (GEMMs). Carefully chosen germline mutations induce tumor formation at an early age and at a relatively high penetrance. In contrast to xenografts, tumors in GEMM develop progressively and can therefore also be used to study precancerous lesions and low grade tumors (Gopinathan et al., 2015).
The most used PDAC models are the KC and KPC mice (Hingorani et al., 2003, 2005; Gopinathan et al., 2015; Lee et al., 2016; Ariston Gabriel et al., 2020). The KC mice is characterized by a germline mutation in Kras (K-rasLSL.G12D/+) and the KPC mice has an additional mutation in Tp53 (p53LSL.R172H/+). The presence of Pdx1-Cre removes the floxed transcriptional STOP cassette which silenced the mutant alleles in the pancreas. Both models develop PanINs and eventually also PDAC although the onset and penetrance of PDAC is later and lower in KC mice.
The KC and KPC mouse models have been instrumental in understanding tumor development in DDR-proficient PDAC but have also been used as a basis for DDR-deficient GEMM models. The following section will describe DDR-deficient GEMM models (Brca2-deficient and Atm-deficient) that have been published in literature (Table 3).
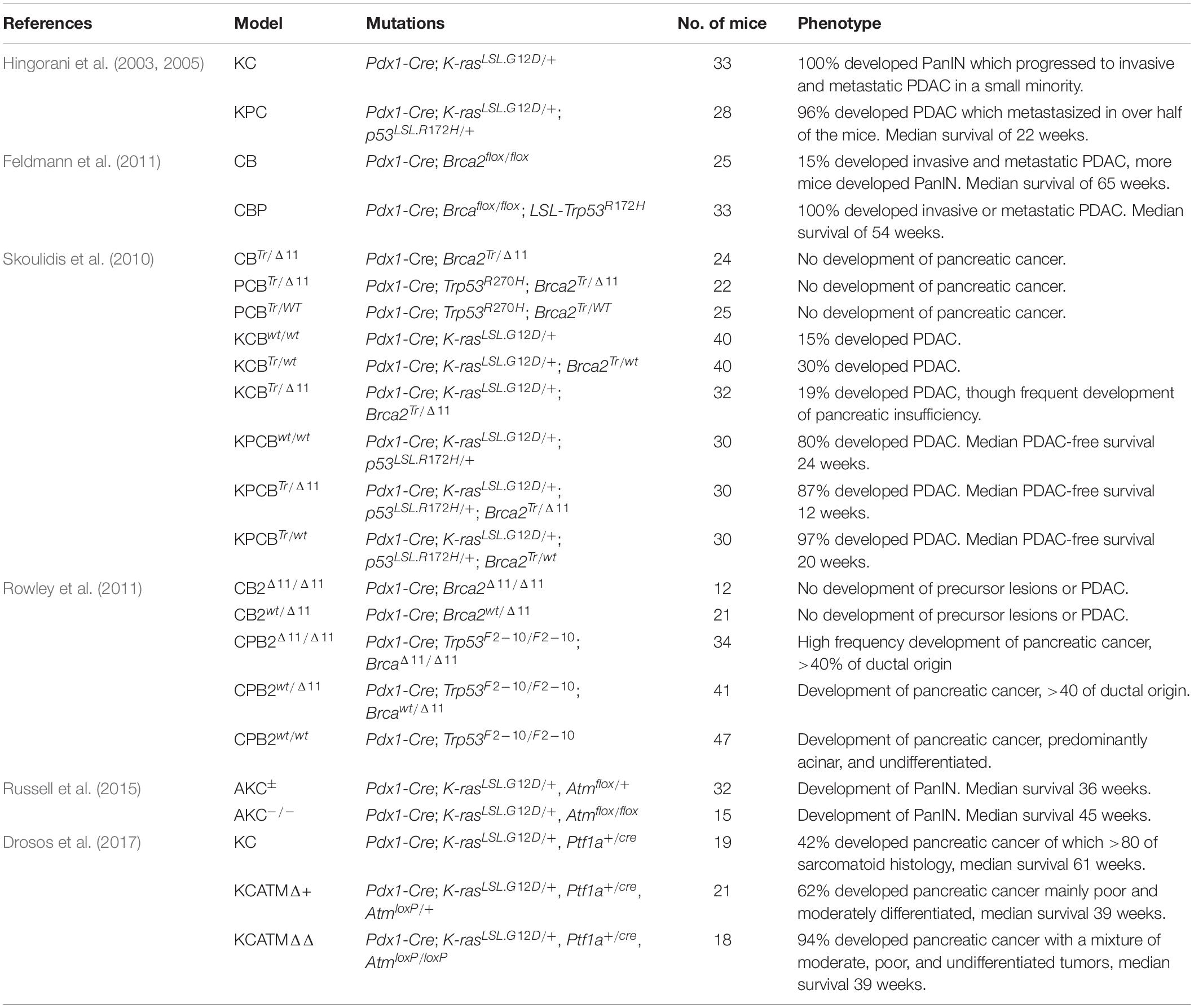
Table 3. Overview DDR deficient pancreatic cancer GEMMs studies.
Feldmann et al. (2011) established two BRCA2-mutated GEMM, abbreviated as CB (Pdx1-Cre; Brca2flox/flox) and CBP (Pdx1-Cre; Brcaflox/flox; LSL-Trp53R172H), and performed extensive histopathological characterization and survival analysis. Both models developed the full spectrum of PanIN lesions which replaced the pancreatic parenchyma and acinar tissue. The additional Trp53 mutation in the CBP cohort enhanced the frequency of invasive neoplasia and resulted in earlier mortality (375 vs. 454 days, p = 0.085). Five mice from the CBP cohort developed metastatic lesions (after 15 months), two were categorized as moderately differentiated adenocarcinomas, two as a combination of adenocarcinoma and sarcomatoid carcinoma, and one as anaplastic carcinoma with giant cells. Sequencing of metastatic PanIN (n = 2) and PDAC (n = 2) in CB and CBP mice found no secondary Kras mutations in any of the mutation hotspots (codon 12, 13, and 61) indicating that Kras mutation is not prerequisite for tumorigenesis in the presence of Brca2 mutation in mice.
These findings were contradicted by Skoulidis et al. (2010) who argued that Brca2-deficiency alone is not sufficient to induce carcinogenesis. They developed several mouse models with a combination of mutations in Brca2, Trp53, and Kras (Skoulidis et al., 2010). Of the resulting models, those with solely Brca2-deficiency (CBTr/Δ11: Pdx1-Cre; Brca2Tr/Δ11) had a longer survival than those with combined Brca2-deficiency and Trp53 loss (PCBTr/Δ11: Trp53R270H, Pdx1-Cre; Brca2Tr/Δ11 and PCBTr/WT: Trp53R270H, Pdx1-Cre; Brca2Tr/WT) which the researchers contribute to development of pancreatic insufficiency in a fraction of mice. Mice with triple mutation (KPCBTr/Δ11: KPC mouse with additional Pdx1-Cre; Brca2Tr/Δ11) nearly all developed tumors and had the worst survival. However, homozygous Brca2 inactivation did contribute to a significantly more aggressive disease with rapid clinical decline compared to wild-type or heterozygous loss in combination with Kras and Trp53 mutation (p < 0.002). Tumors in KCB and KPCB mice displayed a range of histological features that can also be found in human pancreatic cancers, ranging from PDAC to sarcomatoid tumors and acinar-cell carcinoma.
In line with this Rowley et al. (2011) found that loss of Brca2 alone is not enough to induce tumorigenesis, but that it can promote tumorigenesis in combination with Trp53 inactivation. They developed Brca2-deficient (CB2Δ11/Δ11: Pdx1-Cre; Brca2Δ11/Δ11; CB2wt/Δ11: Pdx1-Cre; Brca2wt/Δ11) and Trp53-null (CPB2Δ11/Δ11, CPB2wt/Δ11, and CPB2wt/wt) mice. The CB2 mice did not develop PDAC whereas the CPB2 mice did. Heterozygous and homozygous Brca2 loss significantly reduced pancreatic cancer-free survival (p < 0.0001), with the strongest effect seen in the homozygous-loss mice. The tumors observed in these mice were similar to several human pancreatic cancer types: 40% were of ductal origin, 35% high grade undifferentiated carcinomas, 20% were mucinous tumors, and the remaining 15% were acinar carcinomas. CPB2wt/wt mice presented mainly with acinar and undifferentiated tumors. Seventy-two percent of the CPB2Δ11/Δ11 mice were found to have PanIN lesions at the time of tumor resection or death, while this was less than 6% in CPB2wt/Δ11 and CPB2wt/wt mice. Remarkably, additional KrasG12D mutation (CKB2 mice) decreased pancreatic cancer formation; Tumors were found in 66% of CKB2wt/Δ11 and 61% of CKB2wt/wt mice, but in just 13% of CKB2Δ11/Δ11 mice. The majority of these tumors (>90%) were PDACs.
These studies established that bi-allelic loss of Brca2 in combination with Tp53 deregulation can induce a spectrum of pancreatic lesions. Whether bi-allelic loss of Brca2 alone can also induce pancreatic cancer remains unclear as Feldmann et al. (2011) did not investigate the mutation status of other genes besides Kras and Tp53.
Two studies have published a KC Atm-deficient mouse model. Russell et al. (2015) found that KC mice with floxed Atm (abbreviated as AKC) had developed more acinar-to-ductal metaplasia lesions and PanINs compared with KC mice at 10 weeks old. The higher tumorigenicity of KC Atm-deficient mice was confirmed by Drosos et al. (2017) who studied KC mice with either AtmloxP/+ or AtmloxP/loxP (abbreviated as KCATMΔ+ and KCATMΔΔ). Post-mortem analysis identified pancreatic cancers in 94 and 62% of the KCATMΔ+ and KCATMΔΔ mice compared to 42% in KC mice. In addition, the Atm-deficient mice had a comparable and significantly reduced median OS in both studies (p < 0.01).
Both studies performed subtype analysis. Russell et al. (2015) performed gene expression profiling of 10-week-old KC and AKC mice and compared this to PDAC subtypes as described by Collisson et al. (2011). Hierarchical clustering revealed that AKC pancreatic tumors were closer associated with the quasi-mesenchymal human PDAC subtype than with KC pancreatic tumors. In contrast, Drosos et al. (2017) used KC, KCATMΔ+ and KCATMΔΔ primary tumor cell lines for subtyping and concluded that their tumors were primarily of the pancreatic progenitor/classical phenotype based on the high expression of several progenitor marker genes (Pdx1, Hnf1β, and Lgals4) and a classical marker gene (Gata6). This discrepancy in subtyping may be explained by the sample material used. The subtyping as defined by Collisson et al. (2011) is based on FFPE material and therefore includes tumor cells, stroma, and normal tissue, by using cell lines the gene expression profile is altered compared to the primary tissue due to the lack of stroma which will likely affect the subtype definition, especially with respect to the quasi-mesenchymal subtype.
The past decade has seen a rising interest in the combination of cytotoxic chemotherapy with targeted approaches to exploit the synthetic lethality of this combinatorial approach. As of July 2021, there are 51 clinical trials registered on clinicaltrials.gov that investigate DDR targeted therapies alone or in combination with chemotherapy in PDAC (either in PDAC alone or as part of a larger cancer patient cohort). The majority of these trials (78%, n = 40) focus on PARP inhibitors, although ATM/ATR, CHK1, DNA-PK, and WEE1 inhibitors are also being investigated (Supplementary Table 2).1 Except for a single trial, all trials are either phase I or II, with limited numbers of patients and often single-arm treatment protocols which renders efficacy analysis more challenging.
The pivotal phase III trial leading to FDA approval for the use of PARPi in metastatic PDAC was conducted by Golan et al. (2019) and evaluated olaparib as maintenance therapy in metastatic PDAC patients with germline mutation of BRCA (NCT02184195). Patients were eligible if their tumor had not progressed on first-line platinum-based chemotherapy (e.g., cisplatin or oxaliplatin). Treatment with olaparib was compared to placebo and a significant increase in PFS was observed (7.4 vs. 3.8 months, p = 0.004), but at interim analysis (data maturity 46%) no significant difference was found in median OS (18.9 vs. 18.1 months, p = 0.68). The updated results of this study were presented in January 2021 (Golan et al., 2021). Disappointingly there was again no difference in median OS between the groups (OS 19.0 vs. 19.2 months, p = 0.35). Notably, however, PFS2, i.e., the time from randomization to second disease progression or death, was significantly longer in the olaparib-treated group (PFS2, 16.9 vs. 9.3 months, p = 0.0061).
A comparable phase I trial (NCT00515866) in PDAC patients with locally advanced or metastatic PDAC being treated in the first line setting included a comparison of olaparib combined with gemcitabine vs. gemcitabine alone in the expansion phase (n = 22) (Bendell et al., 2015). Patients were eligible for inclusion regardless of genetic/molecular status. No significant benefit was found regarding objective response rate (ORR), OS, or PFS for the combination treatment. While the researchers noted that nine patients had BRCA mutation status available, analysis of response by BRCA mutation status was not performed due to the small number of patients for whom this data was recorded.
O’Reilly et al.’s (2018) phase IB trial of the addition of another PARPi, veliparib, to first line chemotherapy (cisplatin, gemcitabine) demonstrated a striking ORR of 78% in patients with stage III/IV PDAC with BRCA1/2 germline mutations and an equally impressive median OS of 23.3 months. The investigators then proceeded to a phase II trial of this combination in patients with PDAC and germline BRCA or PALB2 mutations (O’Reilly et al., 2020). Response rate in the combination arm was 79 vs. 65.2% in the chemotherapy alone arm (p = 0.02). However, there was no statistically significant difference in PFS or OS between the groups (PFS 10.1 vs. 9.7 months, p = 0.73; OS 15.5 vs. 16.4 months, p = 0.6). The 2- and 3-year OS of 30.7 and 17.8%, respectively, in this study are the longest ever reported in a clinical trial in this cohort. Notably a phase II study of veliparib alone in the second line setting in patients with BRCA mutant PDAC reported no confirmed responses; 4 patients (4/16) had stable disease for 4 months (Lowery et al., 2018).
Two additional phase II trials have published results on the addition of veliparib to 5-FU based chemotherapy. The first study (NCT02890355) compared the combination of veliparib with modified FOLFIRI vs. FOLFIRI alone as second line treatment in metastatic pancreatic cancer patients (Chiorean et al., 2019). In total, 108 patients were included in the analysis. The addition of veliparib to FOLFIRI treatment was shown to increase toxicity. Moreover, veliparib did not improve either OS (5.1 vs. 5.9 months in combination vs. monotherapy arm, respectively, HR 1.3, p = 0.21) or PFS (2.1 vs. 2.9 months, HR 1.5, p = 0.05). Additionally, blood and tumor biopsies were collected at baseline to explore HR or DDR biomarkers. Nine percent of the tumors had HR deficiency (BRCA1/2, ATM, PALB2, ATM, or CDK12 mutation), and an additional 20% had mutations in other DDR genes (FANC, BLM, SLX4, CHEK2, POLD1, RIF1, and MSH2/6). Correlative analysis of HR or DDR deficiency with treatment response is still ongoing.
Pishvaian et al. (2020) performed a phase I/II clinical trial (NCT01489865) to evaluate the safety and response of PDAC patients to combination treatment of veliparib with modified FOLFOX. For the phase I portion of the study patients were not selected based on genetic history; however, for the phase II part of the trial, patients were selected based on the presence of HR-DDR deficiency or family history suggesting breast or ovarian cancer syndrome, and a distinction was made between previously treated and untreated patients. The ORR was 20% in the phase I unselected cohort (n = 23) and 31% in the phase II cohort (n = 33) selected for HR-DDR deficiency. Further analysis of the phase II cohort showed that treatment-naïve patients had a better ORR and OS than previously treated patients (40 vs. 22%, and 13.0 vs. 4.5 months, respectively). In the treatment-naïve HR-DDR patients, the ORR was 57%. However, due to the lack of a placebo or control arm the magnitude of benefit attributable to the addition of veliparib is difficult to quantify. Previously reported OS of metastatic PDAC patients receiving FOLFOX as second-line treatment are in a similar range (3.3 and 6.3 months) (Yoo et al., 2009; Hecht et al., 2020).
Multiple trials for PARP inhibitors are currently recruiting or preparing for recruitment of patients with DDR deficiency (either for BRCA mutations specifically, or for a panel of DDR genes). This includes phase I and II trials for niraparib both as monotherapy and in combination with other drugs (NCT04673448, NCT04764084, NCT03601923, and NCT04493060), Olaparib (NCT04548752), rucaparib (NCT04171700), talazoparib (NCT04550494), and NMS-03305293 (NCT04182516).
Laquente et al. (2017) performed a phase I/II clinical trial for the CHK1 inhibitor rabusertib (LY2603618) combined with gemcitabine vs. gemcitabine alone in patients with locally advanced or metastatic PDAC (NCT00839332). Although no significant differences were found in the number and severity of adverse events, no significant differences in OS, PFS, ORR, or duration of response were found either.
A phase II trial on the safety and efficacy of the combination of the DNA-PK inhibitor LY3023414 with abemaciclib in previously treated metastatic PDAC patients compared to standard-of-care gemcitabine or capecitabine found that the combination treatment had a significantly worse PFS (1.81 vs. 3.25 months, p = 0.012) (NCT02981342).
Preclinical models have shown that several targeted therapies are more effective in models that defects in complementary DNA repair pathways and this principle of synthetic lethality has been adapted by clinical trials. Multiple trials are currently running which select patients based on the presence BRCA mutations. While the application of targeted therapy in patients with of BRCA mutations has had success in other cancer types (most notably in breast and ovarian cancer), the fraction of patients with BRCA mutations is relatively small and patients with other DDR gene mutations may also benefit from these therapies. Several studies have included additional mutations in their selection criteria, but based on Table 1 these panels could be extended upon. However, preclinical trials might be needed to warrant this.
Alternatively, DDR deficiency might serve as biomarker for response to non-targeted therapies. One study that is currently investigating this is the Precision Panc trial which recruits PDAC patients for molecular profiling and allows patients to enroll in a PRIMUS trial (NCT0461417), one of which aims to test a DDR deficiency biomarker for response to FOLFOX-A treatment (NCT04176952).
The dismal survival rate of PDAC underscores the urgent need to develop new and more effective preventative and therapeutic strategies. So far, clinical trials for DDR targeting drugs have shown limited results beyond the approval of Olaparib in the maintenance setting for patients with germline BRCA1/2 mutations. To improve treatment, representative models are needed, especially those that model DDR deficiency, to test drugs and to develop biomarkers that predict patient response. Clinical rationale exists to expand the use of DDR targeting agents, however, to date there are no validated predictors of treatment response with these agents for patients with DDR deficient tumors beyond BRCA1/2.
Therefore, in order to expand the impact of targeting DDR pathway genes, we performed a comprehensive review of published preclinical models which can potentially be used for DDR deficiency targeted drug screening. Using a 352 DDR gene panel we queried the GENIE database and found multiple frequently mutated DDR genes, some of which are not commonly used in DDR panels. This suggests that DDR deficiency might occur more frequently than previously thought but also provides additional options for biomarkers or targeted therapies.
Cell lines are by far the most frequently used model for PDAC. However, DDR deficiency studies are mainly limited to the BRCA2-mutant cell line Capan-1. Our query of the PDAC cell lines in the CCLE database for the top 10 most commonly mutated DDR genes (excluding TP53) found that 20 cell lines contain one or multiple DDR gene mutations. However, for the majority of these mutations the pathogenicity is unknown and functional characterization of the DDR status is warranted.
The application of organoids toward PDAC studies is still relatively new and at the time of writing no studies in PDAC organoids on DDR-deficiency specifically have been published. We therefore stress the need for characterization of DDR status in existing organoids based on NGS and functional profiling as well as the development and characterization of DDR deficiency in existing organoids through gene editing.
In contrast to cell lines and organoids, several DDR deficient mouse models have been published. So far the application of these models has mainly focused on the contribution of DDR deficiency on tumorigenesis and tumor progression, but there is opportunity for their application in drug sensitivity studies.
The process from biomarker/drug discovery to clinical practice is a long and often unsuccessful path, with 95% of the drugs failing at the translation from preclinical model to clinical trials (Seyhan, 2019). Preclinical models that closely recapitulate the primary tumor have a higher translational value and thus improve the chance of success.
Interest in DDR deficiency as a target for personalized therapy is rising, indicated by the high number of clinical trials currently open in this area. Of the 51 trials running, 29 phase I or II trials are currently recruiting or not yet recruiting. More evidence of the efficacy of these therapies in preclinical PDAC models are required to support their clinical rationale, as many studies have not achieved their desired endpoint. While limitations exist to preclinical in vitro and in vivo models (Nelson and Walsh, 2020), appropriate preclinical models can reflect histopathological subtypes, assist in early prioritization of promising therapies, be used in high-throughput screening to identify ineffective therapies earlier, and thus prevent the excessive time and money resources of a failed clinical trial.
NW and ML conceived the study and provided the significant guidance. NW drew up the outline of the review. JS wrote the manuscript and generated the illustrations. EH contributed with the section on clinical trials. SM contributed to the writing of early drafts. All authors approved the final manuscript.
JS and SM were funded under the EU Horizon-2020 Program “PancREatic Cancer OrganoiDs rEsearch Network” (grant agreement ID: 861196). ML and NW were funded by the Pancreatic Cancer Research Fund (award round 2018).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to acknowledge the American Association for Cancer Research and its financial and material support in the development of the AACR Project GENIE registry, as well as members of the consortium for their commitment to data sharing. Interpretations are the responsibility of study authors.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.749490/full#supplementary-material
Adel, N. (2019). Current treatment landscape and emerging therapies for pancreatic cancer. Am. J. Manag. Care 25, S3–S10.
Aguirre, A. J., Nowak, J. A., Camarda, N. D., Moffitt, R. A., Ghazani, A. A., Hazar-Rethinam, M., et al. (2018). Real-time genomic characterization of advanced pancreatic cancer to enable precision medicine. Cancer Discov. 8, 1096–1111. doi: 10.1158/2159-8290.CD-18-0275
Alexandrov, L. B., Nik-Zainal, S., Wedge, D. C., Aparicio, S. A. J. R., Behjati, S., Biankin, A. V., et al. (2013). Signatures of mutational processes in human cancer. Nature 500, 415–421. doi: 10.1038/nature12477
Andrei, A.-Z., Hall, A., Smith, A. L., Bascuñana, C., Malina, A., Connor, A., et al. (2015). Increased in vitro and in vivo sensitivity of BRCA2-associated pancreatic cancer to the poly(ADP-ribose) polymerase-1/2 inhibitor BMN 673. Cancer Lett. 364, 8–16. doi: 10.1016/j.canlet.2015.04.003
Ariston Gabriel, A. N., Jiao, Q., Yvette, U., Yang, X., Al-Ameri, S. A., Du, L., et al. (2020). Differences between KC and KPC pancreatic ductal adenocarcinoma mice models, in terms of their modeling biology and their clinical relevance. Pancreatology 20, 79–88. doi: 10.1016/j.pan.2019.11.006
Azorsa, D. O., Gonzales, I. M., Basu, G. D., Choudhary, A., Arora, S., Bisanz, K. M., et al. (2009). Synthetic lethal RNAi screening identifies sensitizing targets for gemcitabine therapy in pancreatic cancer. J. Transl. Med. 7:43. doi: 10.1186/1479-5876-7-43
Bailey, P., Chang, D. K., Nones, K., Johns, A. L., Patch, A. M., Gingras, M. C., et al. (2016). Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52. doi: 10.1038/nature16965
Bendell, J., O’Reilly, E. M., Middleton, M. R., Chau, I., Hochster, H., Fielding, A., et al. (2015). Phase I study of olaparib plus gemcitabine in patients with advanced solid tumours and comparison with gemcitabine alone in patients with locally advanced/metastatic pancreatic cancer. Ann. Oncol. 26, 804–811. doi: 10.1093/annonc/mdu581
Bilimoria, K. Y., Bentrem, D. J., Ko, C. Y., Ritchey, J., Stewart, A. K., Winchester, D. P., et al. (2007). Validation of the 6th edition AJCC pancreatic cancer staging system. Cancer 110, 738–744. doi: 10.1002/cncr.22852
Chen, H.-D., Guo, N., Song, S.-S., Chen, C.-H., Miao, Z.-H., and He, J.-X. (2020). Novel mutations in BRCA2 intron 11 and overexpression of COX-2 and BIRC3 mediate cellular resistance to PARP inhibitors. Am. J. Cancer Res. 10, 2813–2831.
Chiorean, E. G., Guthrie, K. A., Philip, P. A., Swisher, E. M., Jalikis, F., Pishvaian, M. J., et al. (2019). Randomized phase II study of second-line modified FOLFIRI with PARP inhibitor ABT-888 (Veliparib) (NSC-737664) versus FOLFIRI in metastatic pancreatic cancer (mPC): SWOG S1513. J. Clin. Oncol. 37:4014. doi: 10.1200/jco.2019.37.15_suppl.4014
Collisson, E. A., Sadanandam, A., Olson, P., Gibb, W. J., Truitt, M., Gu, S., et al. (2011). Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 17, 500–503. doi: 10.1038/nm.2344
Conroy, T., Desseigne, F., Ychou, M., Bouché, O., Guimbaud, R., Bécouarn, Y., et al. (2011). FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 364, 1817–1825. doi: 10.1056/nejmoa1011923
de Soto, J. A. (2020). Evaluating the sensitivity of sporadic pancreatic cancer to poly(ADP-ribose) polymerase (PARP) inhibition (Velaparib, Olaparib, AG14361) as single agents and as chemo-sensitizers. bioRxiv [Preprint] doi: 10.1101/2020.03.28.013763
Deans, A. J., and West, S. C. (2011). DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 11, 467–480. doi: 10.1038/nrc3088
Deer, E. L., González-Hernández, J., Coursen, J. D., Shea, J. E., Ngatia, J., Scaife, C. L., et al. (2010). Phenotype and genotype of pancreatic cancer cell lines. Pancreas 39, 425–435. doi: 10.1097/MPA.0b013e3181c15963
Dréan, A., Williamson, C. T., Brough, R., Brandsma, I., Menon, M., Konde, A., et al. (2017). Models and technologies modeling therapy resistance in BRCA1/2-mutant cancers. Mol. Cancer Ther. 16, 2022–2034. doi: 10.1158/1535-7163.MCT-17-0098
Dreyer, S. B., Upstill-Goddard, R., Paulus-Hock, V., Paris, C., Lampraki, E. M., Dray, E., et al. (2021). Targeting DNA damage response and replication stress in pancreatic cancer. Gastroenterology 160, 362–377.e13. doi: 10.1053/j.gastro.2020.09.043
Driehuis, E., Van Hoeck, A., Moore, K., Kolders, S., Francies, H. E., Gulersonmez, M. C., et al. (2019). Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad. Sci. U.S.A. 116, 26580–26590. doi: 10.1073/pnas.1911273116
Drosos, Y., Escobar, D., Chiang, M. Y., Roys, K., Valentine, V., Valentine, M. B., et al. (2017). ATM-deficiency increases genomic instability and metastatic potential in a mouse model of pancreatic cancer. Sci. Rep. 7:11144. doi: 10.1038/s41598-017-11661-8
Elliott, I. A., Dann, A. M., Xu, S., Kim, S. S., Abt, E. R., Kim, W., et al. (2019). Lysosome inhibition sensitizes pancreatic cancer to replication stress by aspartate depletion. Proc. Natl. Acad. Sci. U.S.A. 116, 6842–6847. doi: 10.1073/PNAS.1812410116
Feig, C., Gopinathan, A., Neesse, A., Chan, D. S., Cook, N., and Tuveson, D. A. (2012). The pancreas cancer microenvironment. Clin. Cancer Res. 18, 4266–4276. doi: 10.1158/1078-0432.CCR-11-3114
Feldmann, G., Karikari, C., Dal Molin, M., Duringer, S., Volkmann, P., Bartsch, D. K., et al. (2011). Inactivation of Brca2 cooperates with Trp53R172H to induce invasive pancreatic ductal adenocarcinomas in mice: a mouse model of familial pancreatic cancer. Cancer Biol. Ther. 11, 959–968. doi: 10.4161/cbt.11.11.15534
Ferlay, J., Colombet, M., Soerjomataram, I., Mathers, C., Parkin, D. M., Piñeros, M., et al. (2019). Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 144, 1941–1953. doi: 10.1002/ijc.31937
Fokas, E., Prevo, R., Pollard, J. R., Reaper, P. M., Charlton, P. A., Cornelissen, B., et al. (2012). Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 3:e441. doi: 10.1038/cddis.2012.181
Garcia, P. L., Miller, A. L., and Yoon, K. J. (2020). Patient-derived xenograft models of pancreatic cancer: overview and comparison with other types of models. Cancers 12:1327. doi: 10.3390/cancers12051327
Gendoo, D. M. A., Denroche, R. E., Zhang, A., Radulovich, N., Jang, G. H., Lemire, M., et al. (2019). Whole genomes define concordance of matched primary, xenograft, and organoid models of pancreas cancer. PLoS Comput. Biol. 15:e1006596. doi: 10.1371/journal.pcbi.1006596
Giglia-Mari, G., Zotter, A., and Vermeulen, W. (2011). DNA damage response. Cold Spring Harb. Perspect. Biol. 3:a000745. doi: 10.1101/cshperspect.a000745
Golan, T., Hammel, P., Reni, M., Van Cutsem, E., Macarulla, T., Hall, M. J., et al. (2021). Overall survival from the phase 3 POLO trial: maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. J. Clin. Oncol. 39:378. doi: 10.1200/JCO.2021.39.3_SUPPL.378
Golan, T., Hammel, P., Reni, M., Van Cutsem, E., Macarulla, T., Hall, M. J., et al. (2019). Maintenance olaparib for germline BRCA -mutated metastatic pancreatic cancer. N. Engl. J. Med. 381, 317–327. doi: 10.1056/NEJMoa1903387
Golan, T., Stossel, C., Atias, D., Buzhor, E., Halperin, S., Cohen, K., et al. (2018). Recapitulating the clinical scenario of BRCA-associated pancreatic cancer in pre-clinical models. Int. J. Cancer 143, 179–183. doi: 10.1002/ijc.31292
Golan, T., Stossel, C., Schvimer, M., Atias, D., Halperin, S., Buzhor, E., et al. (2017). Pancreatic cancer ascites xenograft-an expeditious model mirroring advanced therapeutic resistant disease. Oncotarget 8, 40778–40790. doi: 10.18632/oncotarget.17253
Gopinathan, A., Morton, J. P., Jodrell, D. I., and Sansom, O. J. (2015). GEMMs as preclinical models for testing pancreatic cancer therapies. DMM Dis. Model. Mech. 8, 1185–1200. doi: 10.1242/dmm.021055
Guo, G., Zhang, F., Gao, R., Delsite, R., Feng, Z., and Powell, S. N. (2011). DNA repair and synthetic lethality. Int. J. Oral Sci. 3, 176–179. doi: 10.4248/IJOS11064
Gupta, D., and Heinen, C. D. (2019). The mismatch repair-dependent DNA damage response: Mechanisms and implications. DNA Repair 78, 60–69. doi: 10.1016/j.dnarep.2019.03.009
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Hashimoto, S., Anai, H., and Hanada, K. (2016). Mechanisms of interstrand DNA crosslink repair and human disorders. Genes Environ. 38:9. doi: 10.1186/s41021-016-0037-9
Hecht, J. R., Lonardi, S., Bendell, J. C., Sim, H.-W., Macarulla, T., Lopez, C. D., et al. (2020). Randomized phase III study of FOLFOX alone and with pegilodecakin as second-line therapy in patients with metastatic pancreatic cancer (SEQUOIA). J. Clin. Oncol. 38:637. doi: 10.1200/jco.2020.38.4_suppl.637
Hingorani, S. R., Petricoin, E. F., Maitra, A., Rajapakse, V., King, C., Jacobetz, M. A., et al. (2003). Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450. doi: 10.1016/S1535-6108(03)00309-X
Hingorani, S. R., Wang, L., Multani, A. S., Combs, C., Deramaudt, T. B., Hruban, R. H., et al. (2005). Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483. doi: 10.1016/j.ccr.2005.04.023
Hughes, P., Marshall, D., Reid, Y., Parkes, H., and Gelber, C. (2007). The costs of using unauthenticated, over-passaged cell lines: how much more data do we need? Biotechniques 43, 575–586. doi: 10.2144/000112598
Ilic, M., and Ilic, I. (2016). Epidemiology of pancreatic cancer. World J. Gastroenterol. 22, 9694–9705. doi: 10.3748/wjg.v22.i44.9694
Klein, A. P. (2012). Genetic susceptibility to pancreatic cancer. Mol. Carcinog. 51, 14–24. doi: 10.1002/mc.20855
Ku, J. L., Yoon, K. A., Kim, W. H., Jang, J., Suh, K. S., Kim, S. W., et al. (2002). Establishment and characterization of four human pancreatic carcinoma cell lines: genetic alterations in the TGFBR2 gene but not in the MADH4 gene. Cell Tissue Res. 308, 205–214. doi: 10.1007/s00441-001-0510-y
Lal, S., Zarei, M., Chand, S. N., Dylgjeri, E., Mambelli-Lisboa, N. C., Pishvaian, M. J., et al. (2016). WEE1 inhibition in pancreatic cancer cells is dependent on DNA repair status in a context dependent manner. Sci. Rep. 6:33323. doi: 10.1038/srep33323
Laquente, B., Lopez-Martin, J., Richards, D., Illerhaus, G., Chang, D. Z., Kim, G., et al. (2017). A phase II study to evaluate LY2603618 in combination with gemcitabine in pancreatic cancer patients. BMC Cancer 17:137. doi: 10.1186/s12885-017-3131-x
Lee, J. W., Komar, C. A., Bengsch, F., Graham, K., and Beatty, G. L. (2016). Genetically engineered mouse models of pancreatic cancer: the KPC model (LSL-KrasG12D/+;LSL-Trp53R172H/+;Pdx-1-Cre), its variants, and their application in immuno-oncology drug discovery. Curr. Protoc. Pharmacol. 73, 14.39.1–14.39.20. doi: 10.1002/cpph.2
Lee, T. H., and Kang, T. H. (2019). DNA oxidation and excision repair pathways. Int. J. Mol. Sci. 20:6092. doi: 10.3390/ijms20236092
Lohse, I., Borgida, A., Cao, P., Cheung, M., Pintilie, M., Bianco, T., et al. (2015). BRCA1 and BRCA2 mutations sensitize to chemotherapy in patient-derived pancreatic cancer xenografts. Br. J. Cancer 113, 425–432. doi: 10.1038/bjc.2015.220
Lohse, I., Kumareswaran, R., Cao, P., Pitcher, B., Gallinger, S., Bristow, R. G., et al. (2016). Effects of combined treatment with ionizing radiation and the PARP inhibitor olaparib in BRCA mutant and wild type patient-derived pancreatic cancer xenografts. PLoS One 11:e0167272. doi: 10.1371/journal.pone.0167272
Lord, C. J., and Ashworth, A. (2017). PARP inhibitors: synthetic lethality in the clinic. Science 355, 1152–1158. doi: 10.1126/science.aam7344
Lowery, M. A., Kelsen, D. P., Capanu, M., Smith, S. C., Lee, J. W., Stadler, Z. K., et al. (2018). Phase II trial of veliparib in patients with previously-treated BRCA-mutated pancreas ductal adenocarcinoma. Eur. J. Cancer 89:19. doi: 10.1016/J.EJCA.2017.11.004
Mini, E., Landini, I., Lucarini, L., Lapucci, A., Napoli, C., Perrone, G., et al. (2017). The inhibitory effects of HYDAMTIQ, a novel PARP inhibitor, on growth in human tumor cell lines with defective DNA damage response pathways. Oncol. Res. 25, 1441–1451. doi: 10.3727/096504017X14926854178616
Moffitt, R. A., Marayati, R., Flate, E. L., Volmar, K. E., Loeza, S. G. H., Hoadley, K. A., et al. (2015). Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 47, 1168–1178. doi: 10.1038/ng.3398
Mohammad, A. A. (2018). Advanced pancreatic cancer: the standard of care and new opportunities. Oncol. Rev. 12, 98–104. doi: 10.4081/oncol.2018.370
Mohammed, S., Van Buren, G., and Fisher, W. E. (2014). Pancreatic cancer: advances in treatment. World J. Gastroenterol. 20, 9354–9360. doi: 10.3748/wjg.v20.i28.9354
Monberg, M. E., Geiger, H., Lee, J. J., Sharma, R., Semaan, A., Bernard, V., et al. (2021). Occult polyclonality of preclinical pancreatic cancer models drives in vitro evolution. Brief title: intratumoral heterogeneity of PDAC preclinical models. bioRxiv [Preprint] doi: 10.1101/2021.04.13.439717
Moreira, L., Bakir, B., Chatterji, P., Dantes, Z., Reichert, M., and Rustgi, A. K. (2018). Pancreas 3D organoids: current and future aspects as a research platform for personalized medicine in pancreatic cancer. Cell. Mol. Gastroenterol. Hepatol. 5, 289–298. doi: 10.1016/j.jcmgh.2017.12.004
Nelson, S. R., and Walsh, N. (2020). Genetic alterations featuring biological models to tailor clinical management of pancreatic cancer patients. Cancers 12:1233. doi: 10.3390/CANCERS12051233
Nicolle, R., Blum, Y., Marisa, L., Loncle, C., Gayet, O., Moutardier, V., et al. (2017). Pancreatic adenocarcinoma therapeutic targets revealed by tumor-stroma cross-talk analyses in patient-derived xenografts. Cell Rep. 21, 2458–2470. doi: 10.1016/j.celrep.2017.11.003
O’Reilly, E. M., Lee, J. W., Lowery, M. A., Capanu, M., Stadler, Z. K., Moore, M. J., et al. (2018). Phase 1 trial evaluating cisplatin, gemcitabine, and veliparib in 2 patient cohorts: germline BRCA mutation carriers and wild-type BRCA pancreatic ductal adenocarcinoma. Cancer 124, 1374–1382. doi: 10.1002/CNCR.31218
O’Reilly, E. M., Lee, J. W., Zalupski, M., Capanu, M., Park, J., Golan, T., et al. (2020). Randomized, multicenter, phase II trial of gemcitabine and cisplatin with or without veliparib in patients with pancreas adenocarcinoma and a germline BRCA/PALB2 mutation. J. Clin. Oncol. 38, 1378–1388. doi: 10.1200/JCO.19.02931
Palacio, S., McMurry, H. S., Ali, R., Donenberg, T., Silva-Smith, R., Wideroff, G., et al. (2019). DNA damage repair deficiency as a predictive biomarker for FOLFIRINOX efficacy in metastatic pancreatic cancer. J. Gastrointest. Oncol. 10, 1133–1139. doi: 10.21037/jgo.2019.09.12
Park, P. H., Yamamoto, T. M., Li, H., Alcivar, A. L., Xia, B., Wang, Y., et al. (2020). Amplification of the mutation-carrying BRCA2 allele promotes RAD51 loading and PARP inhibitor resistance in the absence of reversion mutations. Mol. Cancer Ther. 19, 602–613. doi: 10.1158/1535-7163.MCT-17-0256
Parsels, L. A., Parsels, J. D., Tanska, D. M., Maybaum, J., Lawrence, T. S., and Morgan, M. A. (2018). The contribution of DNA replication stress marked by high-intensity, pan-nuclear γH2AX staining to chemosensitization by CHK1 and WEE1 inhibitors. Cell Cycle 17, 1076–1086. doi: 10.1080/15384101.2018.1475827
Perkhofer, L., Gout, J., Roger, E., Kude De Almeida, F., Baptista Simões, C., Wiesmüller, L., et al. (2020). DNA damage repair as a target in pancreatic cancer: state-of-the-art and future perspectives. Gut 70, 606–617. doi: 10.1136/gutjnl-2019-319984
Perkhofer, L., Schmitt, A., Romero Carrasco, M. C., Ihle, M., Hampp, S., Ruess, D. A., et al. (2017). ATM deficiency generating genomic instability sensitizes pancreatic ductal adenocarcinoma cells to therapy-induced DNA damage. Cancer Res. 77, 5576–5590. doi: 10.1158/0008-5472.CAN-17-0634
Pham, N.-A., Radulovich, N., Ibrahimov, E., Martins-Filho, S. N., Li, Q., Pintilie, M., et al. (2021). Patient-derived tumor xenograft and organoid models established from resected pancreatic, duodenal and biliary cancers. Sci. Rep. 11:10619. doi: 10.1038/s41598-021-90049-1
Pishvaian, M. J., Wang, H., He, A. R., Hwang, J. J., Smaglo, B. G., Kim, S. S., et al. (2020). A phase I/II study of veliparib (ABT-888) in combination with 5-fluorouracil and oxaliplatin in patients with metastatic pancreatic cancer. Clin. Cancer Res. 26, 5092–5101. doi: 10.1158/1078-0432.CCR-20-1301
Porcelli, L., Quatrale, A. E., Mantuano, P., Leo, M. G., Silvestris, N., Rolland, J. F., et al. (2013). Optimize radiochemotherapy in pancreatic cancer: PARP inhibitors a new therapeutic opportunity. Mol. Oncol. 7, 308–322. doi: 10.1016/j.molonc.2012.10.002
Puleo, F., Nicolle, R., Blum, Y., Cros, J., Marisa, L., Demetter, P., et al. (2018). Stratification of pancreatic ductal adenocarcinomas based on tumor and microenvironment features. Gastroenterology 155, 1999–2013. doi: 10.1053/j.gastro.2018.08.033
Qian, Y., Gong, Y., Fan, Z., Luo, G., Huang, Q., Deng, S., et al. (2020). Molecular alterations and targeted therapy in pancreatic ductal adenocarcinoma. J. Hematol. Oncol. 13:130. doi: 10.1186/s13045-020-00958-3
Ranjha, L., Howard, S. M., and Cejka, P. (2018). Main steps in DNA double-strand break repair: an introduction to homologous recombination and related processes. Chromosoma 127, 187–214. doi: 10.1007/s00412-017-0658-1
Roger, E., Gout, J., Arnold, F., Beutel, A. K., Müller, M., Abaei, A., et al. (2020). Maintenance therapy for ATM-deficient pancreatic cancer by multiple DNA damage response interferences after platinum-based chemotherapy. Cells 9:2110. doi: 10.3390/cells9092110
Rowley, M., Ohashi, A., Mondal, G., Mills, L., Yang, L., Zhang, L., et al. (2011). Inactivation of Brca2 promotes Trp53-associated but inhibits KrasG12D-dependent pancreatic cancer development in mice. Gastroenterology 140, 1303–1313.e1-3. doi: 10.1053/j.gastro.2010.12.039
Russell, R., Perkhofer, L., Liebau, S., Lin, Q., Lechel, A., Feld, F. M., et al. (2015). Loss of ATM accelerates pancreatic cancer formation and epithelial-mesenchymal transition. Nat. Commun. 6:7677. doi: 10.1038/ncomms8677
Sakai, W., Swisher, E. M., Karlan, B. Y., Agarwal, M. K., Higgins, J., Friedman, C., et al. (2008). Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 451, 1116–1120. doi: 10.1038/nature06633
Schärer, O. D. (2013). Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 5:a012609. doi: 10.1101/cshperspect.a012609
Sehdev, A., Gbolahan, O., Hancock, B. A., Stanley, M., Shahda, S., Wan, J., et al. (2018). Germline and somatic DNA damage repair gene mutations and overall survival in metastatic pancreatic adenocarcinoma patients treated with FOLFIRINOX. Clin. Cancer Res. 24, 6204–6211. doi: 10.1158/1078-0432.CCR-18-1472
Seyhan, A. A. (2019). Lost in translation: the valley of death across preclinical and clinical divide – identification of problems and overcoming obstacles. Transl. Med. Commun. 4:18. doi: 10.1186/S41231-019-0050-7
Shi, J., Li, Y., Jia, R., and Fan, X. (2020). The fidelity of cancer cells in PDX models: characteristics, mechanism and clinical significance. Int. J. Cancer 146, 2078–2088. doi: 10.1002/ijc.32662
Siegel, R. L., Miller, K. D., and Jemal, A. (2020). Cancer statistics, 2020. CA Cancer J. Clin. 70, 7–30. doi: 10.3322/caac.21590
Skoulidis, F., Cassidy, L. D., Pisupati, V., Jonasson, J. G., Bjarnason, H., Eyfjord, J. E., et al. (2010). Germline Brca2 heterozygosity promotes KrasG12D -driven carcinogenesis in a murine model of familial pancreatic cancer. Cancer Cell 18, 499–509. doi: 10.1016/j.ccr.2010.10.015
Stathis, A., and Moore, M. J. (2010). Advanced pancreatic carcinoma: current treatment and future challenges. Nat. Rev. Clin. Oncol. 7, 163–172. doi: 10.1038/nrclinonc.2009.236
Sullivan-Reed, K., Bolton-Gillespie, E., Wasik, M. A., Mazin, A. V., and Correspondence, T. S. (2018). Simultaneous targeting of PARP1 and RAD52 triggers dual synthetic lethality in BRCA-deficient tumor cells. CellReports 23, 3127–3136. doi: 10.1016/j.celrep.2018.05.034
Swayden, M., Soubeyran, P., and Iovanna, J. (2020). Upcoming revolutionary paths in preclinical modeling of pancreatic adenocarcinoma. Front. Oncol. 9:1443. doi: 10.3389/fonc.2019.01443
The AACR Project GENIE Consortium (2017). AACR project GENIE: powering precision medicine through an international consortium. Cancer Discov. 7, 818–831.
Tiriac, H., Belleau, P., Engle, D. D., Plenker, D., Deschênes, A., Somerville, T. D. D., et al. (2018). Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discov. 8, 1112–1129. doi: 10.1158/2159-8290.CD-18-0349
Topatana, W., Juengpanich, S., Li, S., Cao, J., Hu, J., Lee, J., et al. (2020). Advances in synthetic lethality for cancer therapy: cellular mechanism and clinical translation. J. Hematol. Oncol. 13:118. doi: 10.1186/s13045-020-00956-5
Turati, F., Edefonti, V., Bosetti, C., Ferraroni, M., Malvezzi, M., Franceschi, S., et al. (2013). Family history of cancer and the risk of cancer: a network of case-control studies. Ann. Oncol. 24, 2651–2656. doi: 10.1093/annonc/mdt280
Von Hoff, D. D., Ervin, T., Arena, F. P., Chiorean, E. G., Infante, J., Moore, M., et al. (2013). Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 369, 1691–1703. doi: 10.1056/NEJMoa1304369
Waddell, N., Pajic, M., Patch, A. M., Chang, D. K., Kassahn, K. S., Bailey, P., et al. (2015). Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 518, 495–501. doi: 10.1038/nature14169
Wallez, Y., Dunlop, C. R., Johnson, T. I., Koh, S.-B., Fornari, C., Yates, J. W. T., et al. (2018). The ATR inhibitor AZD6738 synergizes with gemcitabine in vitro and in vivo to induce pancreatic ductal adenocarcinoma regression. Mol. Cancer Ther. 17, 1670–1682. doi: 10.1158/1535-7163.MCT-18-0010
Wengner, A. M., Scholz, A., and Haendler, B. (2020). Targeting DNA damage response in prostate and breast cancer. Int. J. Mol. Sci. 21:8273. doi: 10.3390/ijms21218273
Yang, K., Guo, R., and Xu, D. (2016). Non-homologous end joining: advances and frontiers. Acta Biochim. Biophys. Sin. 48, 632–640. doi: 10.1093/abbs/gmw046
Yoo, C., Hwang, J. Y., Kim, J. E., Kim, T. W., Lee, J. S., Park, D. H., et al. (2009). A randomised phase II study of modified FOLFIRI.3 vs modified FOLFOX as second-line therapy in patients with gemcitabine-refractory advanced pancreatic cancer. Br. J. Cancer 101, 1658–1663. doi: 10.1038/sj.bjc.6605374
Yu, Y., Yang, G., Huang, H., Fu, Z., Cao, Z., Zheng, L., et al. (2021). Preclinical models of pancreatic ductal adenocarcinoma: challenges and opportunities in the era of precision medicine. J. Exp. Clin. Cancer Res. 40:8. doi: 10.1186/s13046-020-01787-5
Zimmermann, M. T., Mathison, A. J., Stodola, T., Evans, D. B., Abrudan, J. L., Demos, W., et al. (2021). Interpreting sequence variation in PDAC-predisposing genes using a multi-tier annotation approach performed at the gene, patient, and cohort level. Front. Oncol. 11:606820. doi: 10.3389/fonc.2021.606820
Keywords: DNA damage response (DDR), pancreatic duct adenocarcinoma (PDAC), preclinical model, cell line, organoid, genetically engineered mouse model (GEMM), xenograft, targeted therapy
Citation: Stoof J, Harrold E, Mariottino S, Lowery MA and Walsh N (2021) DNA Damage Repair Deficiency in Pancreatic Ductal Adenocarcinoma: Preclinical Models and Clinical Perspectives. Front. Cell Dev. Biol. 9:749490. doi: 10.3389/fcell.2021.749490
Received: 29 July 2021; Accepted: 22 September 2021;
Published: 12 October 2021.
Edited by:
Maarten Fokke Bijlsma, Academic Medical Center, NetherlandsReviewed by:
P. M. Krawczyk, Academic Medical Center, NetherlandsCopyright © 2021 Stoof, Harrold, Mariottino, Lowery and Walsh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maeve A. Lowery, bWxvd2VyeUB0Y2QuaWU=; Naomi Walsh, bmFvbWkud2Fsc2hAZGN1Lmll
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.