 Wenjia Wang1,2
Wenjia Wang1,2 Jin Li1,2
Jin Li1,2 Lan Lan1,2Linyi Xie1,2Fen Xiong1,2Jing Guan1,2
Lan Lan1,2Linyi Xie1,2Fen Xiong1,2Jing Guan1,2 Hongyang Wang1,2*
Hongyang Wang1,2* Qiuju Wang1,2*
Qiuju Wang1,2*- 1College of Otolaryngology, Head and Neck Surgery, Institute of Otolaryngology, Chinese PLA General Hospital, Beijing, China
- 2National Clinical Research Center for Otolaryngologic Diseases, Beijing, China
Objective: The objective of this study is to analyze the genotype–phenotype correlation of patients with auditory neuropathy (AN), which is a clinical condition featuring normal cochlear responses and abnormal neural responses, and ATP1A3 c.2452 G > A (p.E818K), which has been generally recognized as a genetic cause of cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss (CAPOS) syndrome.
Methods: Four patients diagnosed as AN by clinical evaluation and otoacoustic emission and auditory brainstem responses were recruited and analyzed by next-generation sequencing to identify candidate disease-causing variants. Sanger sequencing was performed on the patients and their parents to verify the results, and short tandem repeat-based testing was conducted to confirm the biological relationship between the parents and the patients. Furthermore, cochlear implantation (CI) was performed in one AN patient to reconstruct hearing.
Results: Four subjects with AN were identified to share a de novo variant, p.E818K in the ATP1A3 gene. Except for the AN phenotype, patients 1 and 2 exhibited varying degrees of neurological symptoms, implying that they can be diagnosed as CAPOS syndrome. During the 15 years follow-up of patient 1, we observed delayed neurological events and progressive bilateral sensorineural hearing loss in pure tone threshold (pure tone audiometry, PTA). Patient 2 underwent CI on his left ear, and the result was poor. The other two patients (patient 3 and patient 4, who were 8 and 6 years old, respectively) denied any neurological symptoms.
Conclusion: ATP1A3 p.E818K has rarely been documented in the Chinese AN population. Our study confirms that p.E818K in the ATP1A3 gene is a multiethnic cause of AN in Chinese individuals. Our study further demonstrates the significance of genetic testing for this specific mutation for identifying the special subtype of AN with somewhat favorable CI outcome and offers a more accurate genetic counseling about the specific de novo mutation.
Introduction
Auditory neuropathy (AN) is a kind of hearing disorder involving different lesion sites beyond the outer hair cells (OHCs), ranging from the inner hair cells (IHCs) and synapses to auditory nerve and higher auditory centers (Rance and Starr, 2015; Moser and Starr, 2016). AN features impaired or absent response in auditory brainstem responses (ABR) and the presence of cochlear microphonics and/or detectable otoacoustic emission (OAE), showing evidence of intact outer hair cell function. Varying degrees of hearing loss are present in subjects with AN; however, their speech recognition rates are generally poor, disproportionate to the degree of hearing loss (Harrison et al., 2015; Rance and Starr, 2015; Moser and Starr, 2016). A variety of etiologies can trigger AN, generally considered to be hypoxia, infection, kernicterus, cytotoxic oncologic drugs, and genetic factors (Harrison et al., 2015).
Considering postlingual-onset AN, many syndromic forms that cause sensory and motor neuropathy have been documented, including Charcot-Marie-Tooth disease, Friedreich’s ataxia, deafness-dystonia-optic neuropathy (DDON) syndrome, autosomal dominant optic atrophy (ADOA), and AUNX1 due to AIFM1 gene mutations in apoptosis-inducing factor (Han et al., 2017). Han et al. (2017) explored the molecular etiology of three unrelated Korean subjects manifesting AN with postlingual onset, identified a mutation in c.2452 G > A (p.E818K) of ATP1A3 gene, and demonstrated ATP1A3 gene to be an important and prevalent causative gene for progressive AN with postlingual onset. ATP1A3 gene has thus been said to occur frequently de novo in both Caucasians and Koreans (Han et al., 2017). However, p.E818K in the ATP1A3 gene has rarely been documented in the Chinese population.
In this study, we reported four Chinese AN patients carrying a de novo variant, p.E818K mutation of ATP1A3 gene, identified through next generation sequencing (NGS). ATP1A3 p.E818K has been generally recognized as a genetic cause of cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss (CAPOS) syndrome. A genotype–phenotype correlation study of ATP1A3 was performed, and one AN patient underwent cochlear implantation (CI) to reconstruct hearing.
Materials and Methods
Ethics Statement
The study was approved by the Committee of Medical Ethics of Chinese PLA General Hospital. Written informed consent was obtained from participants.
Subject Recruitment and Clinical Evaluations
Four AN patients at the Institute of Otolaryngology, Chinese PLA General Hospital were recruited and underwent genetic testing. The diagnostic criteria were as follows: ABR had no obvious differentiation waveform and the OAE and/or the cochlear microphonic (CM) potential could be normally extracted. Medical evidence of hearing loss, tinnitus, and other clinical abnormalities of either affected or unaffected family members was identified. Pure tone threshold (pure tone audiometry, PTA), speech discrimination score (SDS), ABR, OAE, and electrocochleogram (ECochG) were carried out to evaluate auditory status. In general, low frequencies were primarily affected; thus, we focused on the low-frequency data and calculated the PTA as the average of the thresholds of 250–1,000 Hz to avoid bias in the assessment of the degree of AN hearing loss. High-resolution computed tomography (CT) scans of the temporal bone and MRI of the internal auditory canal were performed to exclude other possible neuropathic or anatomical disorders. For patients who exhibited neurological symptoms, we carried out neurological examinations to evaluate those symptoms.
Genetic Techniques
Next generation sequencing, including whole genome sequencing and targeted genes capture and sequencing, was performed on the four probands, Sanger sequencing was performed on the patients and their parents to verify any candidate gene variations as previously described. Variation interpretation (evaluation of the pathogenicity) was based on the standards and guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG and AMP). Short tandem repeat (STR)-based testing was conducted to confirm the biological relationship between the parents and the patients (Wang et al., 2018).
Results
General Clinical Information
A total of four AN cases with ATP1A3 variants were recruited in this study. Details on the patients in this report were collated. Clinical and audiological data were collected retrospectively. Audiological details were outlined in Figure 1 and Supplementary Figure 1.
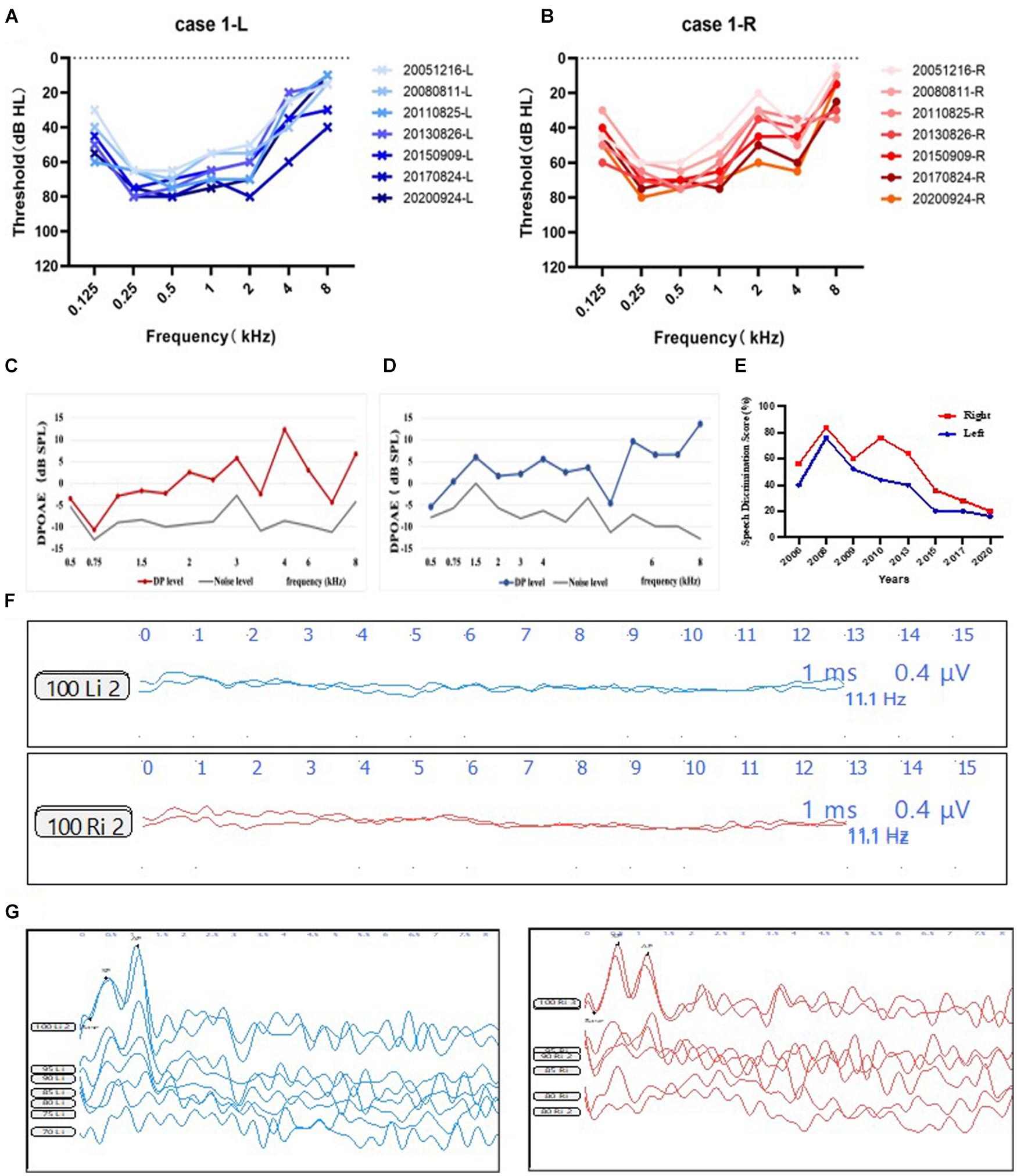
Figure 1. Audiological phenotype of subject 1. (A,B) PTA of the subject 1. Audiograms of patient 1 from 2005 to 2020 displaying moderate to severe sensorineural hearing loss, predominantly affecting the low frequencies and progressively deteriorated overtime. Blue, left ear; red, right ear. (C,D) Observed DPOAE responses from the right and the left ear. Red line, DPOAE level from the right ear; blue line, DPOAE level from the left ear; gray line, noise level. (E) SDS results from 2006 to 2020 showing decreased speech discrimination score. Blue, left ear; red, right ear. (F) Absent ABR waveforms at 90 dB nHL stimulus on both ears. (G) ECochG shows the mean ratios of -SP/AP were larger than 0.4 from both ears. Blue, left ear; red, right ear. Abbreviations: PTA, pure tone audiometry; SDS, speech discrimination score; DPOAE, distortion product optoacoustic emission; ABR, auditory brainstem response; CM, cochlear microphonic.
Detailed Medical History and Clinical Phenotype of Four Auditory Neuropathy Subjects
Patient 1 was a 24-year-old male (Figure 2A). He exhibited dystonia and ataxia at a very early age after having a fever. At age 5, he started to suffer from visual disturbance, and at age 7, his hearing level of both ears started to deteriorate. He was diagnosed as optic atrophy at Beijing Tongren Hospital at age 8. At age 9, he was diagnosed with AN at our hospital. His sensorineural hearing loss (SNHL) matched AN, with a detectable OAE response and disappearance of the ABR response (Figures 1C,D,F), and ratios of -SP/AP were more than 0.4 in both ears (Figure 1G). His pattern visual evoked potential (P-VEP) amplitude was already reduced, indicating amblyopia found in him. Imaging tests demonstrated anatomically intact auditory nerves (Figure 2G). Somatosensory evoked potentials (SSEPs) showed prolonged latency of the bilateral tibial nerve, suggesting abnormal tibial nerve conduction. Electromyographic (EMG) signals of the upper and lower limbs did not display abnormalities. The amplitude-integrated EEG (aEEG) detected minor dysfunction. Through 15 years of follow-up from 2005 to 2020, we observed that his eyesight deteriorated over time, as did his hearing level and speech discrimination score (Figures 1A,B,E).
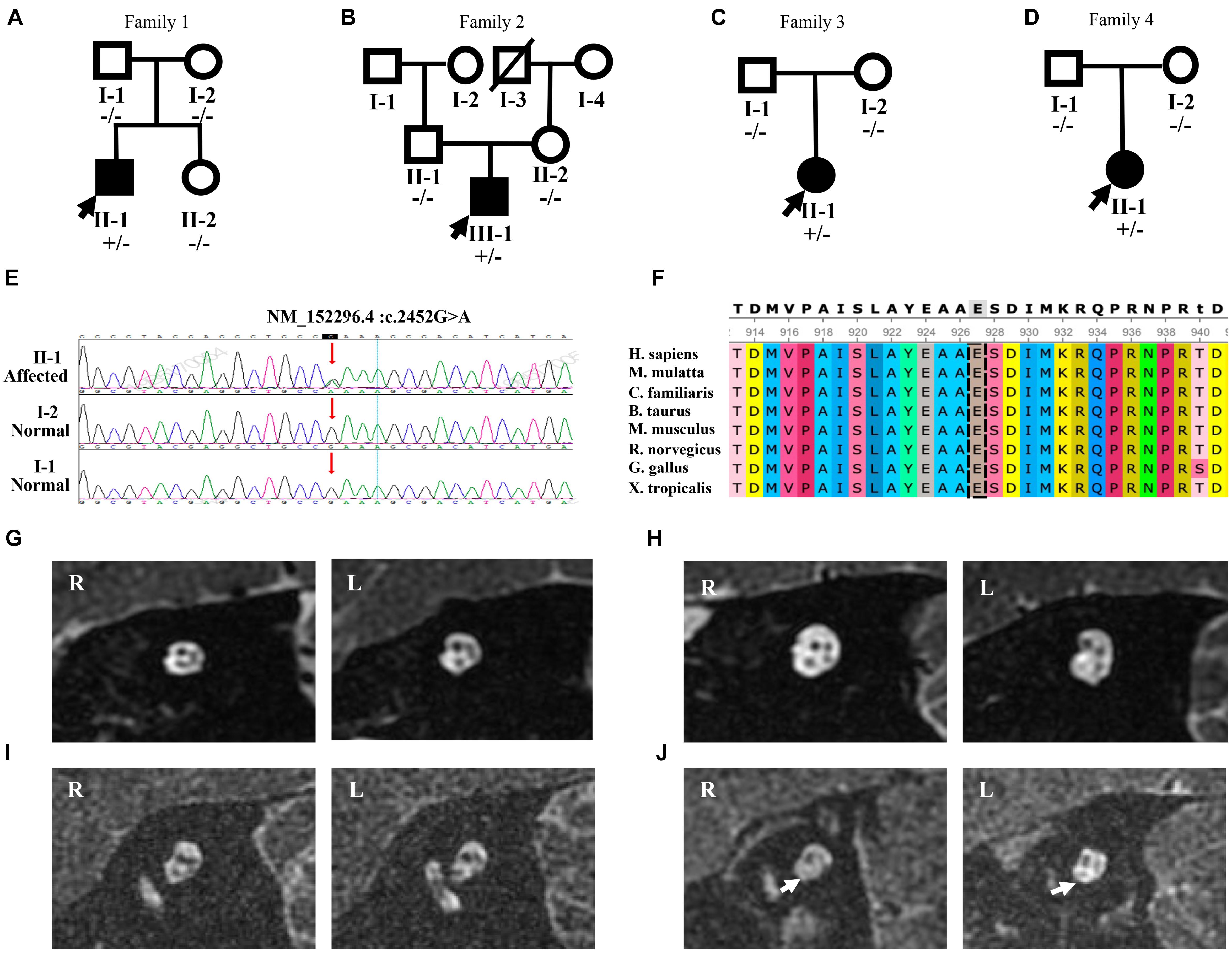
Figure 2. Mutation analysis and MRI results of four patients with the p.E818K mutation. (A–C), and (D) Pedigree of four families. Filled symbols denote affected individuals, whereas unfilled symbols denote the unaffected. A slash (/) indicates a deceased person. Minus sign (-/-) represents homozygosity for the wild-type allele, and plus-minus sign (±) represents heterozygosity for the mutant allele. Probands are indicated by arrows. (E) Sanger sequencing chromatograms of four pedigrees confirmed that all four subjects shared one common heterozygous mutation, c.2452 G > A (arrow) leading to p.E818K amino acid change, whereas their parents are normal. (F) Protein conservation of no. 818 amino acid among vertebrate species (H. sapiens, M. mulatta, C. familiaris, B. taurus, M. musculus, R. norvegicus, G. gallus, and X. tropicalis). No. 818 amino acid is highly conserved. (G–I), and (J) MRI results showing bilateral internal auditory canals (IACs) of patients 1, 2, 3, and 4, respectively. The oblique-sagittal image through the bilateral IACs of patient 4 (J) demonstrates absent bilateral cochlear nerves (white arrows).
Patient 2 was a 22-year-old male patient (Figure 2B). He had a fever and took gentamicin at the age of 2, and he presented with bilateral hearing loss at the age of 7. His SNHL deteriorated over time, accelerating from 13 to 14 years old. Along with bilateral tinnitus, he experienced alalia as well as fatigue. However, he denied other neurologic episodes and could even ride a bicycle, although his eyesight began to decline at the age of 12. His threshold of 40 Hz AERP was 90 dB HL in both ears. Both CT scan and MRI of the internal auditory canal showed only normal findings (Figure 2H). He underwent CI on the left ear at our hospital in 2013, yet his speech perception did not improve much in 1-year follow-up period. In addition, he exhibited facial muscle twitching when the cochlear implant was used in louder environment.
Patient 3 was an 8-year-old girl (Figure 2C). She did not exhibit abnormal hearing function until the age of 7, when she suffered from a fever. Three days after her fever, she experienced hearing loss accompanied by poor speech discrimination. Her symptoms did not improve with the administration of Ginaton as well as other neurotrophic drugs, such as vitamin B12. Her PTA showed an upward sloping configuration, with an average threshold being 70 dB HL in the left ear and 61 dB HL in the right ear (Supplementary Figure 1A). In contrast, her reduction in speech recognition was drastic, being 8% in the left ear and 4% in the right ear. Her SNHL was entirely compatible with AN, considering a detectable OAE response without ABR response (Supplementary Figures 1C,E). CT scan showed no abnormality, and MRI of the internal auditory canal confirmed that she had intact cochlear nerve (Figure 2I). With no obvious neurological condition exhibited, she was diagnosed with bilateral AN.
Patient 4 was a 6-year-old girl (Figure 2D). Her syndrome of hearing loss accompanied by tinnitus appeared at age 4. Unable to benefit from the hearing aids at age 5, she came to our clinic for help. Her PTA showed an upward sloping configuration (Supplementary Figure 1B). She was diagnosed with AN with no neurological symptoms found (Supplementary Figures 1D,F). MRI result showed bilateral cochlear nerve aplasia (Figure 2J).
All the four probands passed the newborn hearing screening test successfully, suggesting that their SNHL was not congenital. Furthermore, they presented with the presence of OAEs and/or CMs, indicating preserved cochlear outer hair cell activity. However, the ABR, a sign of neural dyssynchrony, was grossly abnormal. In addition, a poor SDS was observed in all four patients, especially in noisy environments, and was disproportionate to the level of hearing loss.
Molecular Genetic Testing and Delineation of the Causative Variant From the Four Subjects
We performed NGS, including targeted genes capture and whole genome capture, in four probands with sporadic AN and identified the most convincing causative variant that has previously been described (Kim et al., 2015). Candidate variants were validated by standard Sanger sequencing. All four AN subjects shared one heterozygous missense variant, c.2452 G > A (p.E818K) in the ATP1A3 gene (NM_152296.4, NP_689509.1, OMIM ∗182350), which was located on chromosome 19q13.2 and classified as ‘‘pathogenic’’ according to CLINVAR1 (Landrum et al., 2016). Unaffected parents showed no variation in ATP1A3, indicating a de novo occurrence of an autosomal dominant variant, p.E818K, in all four families (Figure 2E). Evolutionary conservation analysis revealed that this variant was highly conserved throughout evolution among species (Figure 2F). STR-based testing was conducted, confirming the biological relationship between the parents and the cases and indicating that the ATP1A3 mutation was de novo in nature, which strongly supports the pathogenicity. According to the ACMG guidelines, p. E818K variant met the PS2, PS3, PS4, PM2, PP1_Strong, and PP3 criteria and could be classified as pathogenic.
Discussion
Hearing loss is the most common sensorineural disorder. It is estimated that more than half of hearing loss cases are attributable to genetic factors (He Y. et al., 2017; Wang et al., 2017; Zhu et al., 2018; Fang et al., 2019; He Z. et al., 2019; Fu et al., 2021), while the other half of hearing loss could be caused by aging, chronic infections, infectious diseases, ototoxic drugs, and noise exposure (Liu L. et al., 2016; He Z. et al., 2017; Cheng et al., 2019; Gao et al., 2019; Tan et al., 2019; Zhang et al., 2019; Zhong et al., 2020). The functions of these hearing loss genes play an essential role in the development and function of hair cells and synaptic transmission of spiral ganglion neurons (Qi et al., 2020; Qian et al., 2020; Zhang et al., 2020, 2021; Cheng et al., 2021; Lv et al., 2021). Thus, hearing loss is often induced by loss of sensory hair cells (Liu Y. et al., 2019; Qi et al., 2019; Han et al., 2020; He et al., 2020, 2021; Chen et al., 2021) and spiral ganglion neurons (Guo et al., 2016, 2019, 2020, 2021; Liu W. et al., 2019; Hu et al., 2021; Liu W. et al., 2021) in the inner ear cochlea. AN is a hearing disorder with functional outer hair cells and disrupted function of inner hair cells and/or the auditory nerve itself. Han et al. (2017) first proposed that p.E818K in ATP1A3 gene, which encodes the NKAα3 subunit of Na, K-ATPases, can cause AN in Caucasians and Koreans, implying that ATP1A3 serves as a global contributor to AN (Supplementary Table 1). In our study, four AN cases were tested with p.E818K in the ATP1A3 gene. The incidence of p.E818K accounting for AN in Chinese individuals is still under investigation. Nevertheless, we found that the p.E818K mutation accounts for a high proportion of AN with postlingual onset, not only in Koreans and Caucasians but also in Chinese individuals.
ATP1A3 p.E818K has been regarded as a causal mutation of CAPOS syndrome since identified in 2014 (Carecchio et al., 2018), long after the initial description in 1996 (Nicolaides et al., 1996). CAPOS syndrome is a rare disorder, with fewer than 100 CAPOS patients worldwide, and is sporadically reported (Demos et al., 2014; Rosewich et al., 2014; Heimer et al., 2015; Potic et al., 2015; Maas et al., 2016; Duat Rodriguez et al., 2017). Growing evidence shows that sensorineural hearing loss in CAPOS syndrome is AN, which may progress slowly over time. The typical CAPOS phenotype is characterized by acute neurological deterioration manifesting in infancy and triggered by stressful episodes, such as a febrile illness (Paquay et al., 2018). Meticulous history as well as comprehensive neurologic examination should be performed on postlingual-onset AN subjects, to detect any episode of visual disturbance and ataxia associated with CAPOS syndrome triggered by a fever that might otherwise go unnoticed. CAPOS syndrome patients initially have cochlear apical turns, manifested in a low-frequency loss in PTA. However, neuronal distribution of NKAα3 is also observed in middle and basal turns of the cochlea (Paquay et al., 2018), indicating that mid and high frequencies may also be involved. Through 15 years follow-up, we detected progressing hearing defects involving all frequencies, predominantly the low frequencies in patient 1. It is worth continuing the follow-up periods for patients with p.E818K to detect changes in PTA over time, to clarify the significance of the p.E818K mutation in AN.
In previous study, Demos et al. (2014) reported follow-up of three patients with ATP1A3 p.E818K. All the patients showed slow progression of all symptoms with no further acute episodes. Demos et al. (2014) further reported two Caucasian families with ATP1A3 p.E818K. In the first family, all affected suffered from acute episodic ataxic encephalopathy and/or weakness triggered by a febrile illness onset in infancy or early childhood. Recovery was not as desirable after these episodes, when variable neurologic abnormalities was lingering. All affected individuals eventually had optic atrophy and sensorineural hearing loss. One patient exhibited mild cognitive dysfunction at age 10 years. In the second family, the affected mother and her two children presented with recurrent acute episodes of neurologic disorders associated with febrile illnesses beginning in the first years of life. Features included weakness, ataxia, and a progressive decline in hearing and vision. In our study, patient 1 exhibited dystonia and ataxia at a very early age after having a fever. He exhibited optic atrophy and auditory neuropathy at an early age, which were slowly progressive over time. Follow-up period was needed for every patient to further clarify whether there was a slow progression of all symptoms, be it neurological disorders or visual and hearing impairment.
The ATP1A3 gene has also been reported in two other clinical entities, alternating hemiplegia of childhood (AHC, AHC2, OMIM #614820) and rapid-onset dystonia Parkinsonism (RDP, DYT12, OMIM #128235) (Blanco and Mercer, 1998; Starr et al., 2005; Rodacker et al., 2006; Blanco-Arias et al., 2009). Mutation sites are distributed across the entire coding region of ATP1A3 in RDP and AHC (Supplementary Table 1; Clausen et al., 2017). Interestingly, all CAPOS patients reported without exception share a common mutation, p.E818K in the ATP1A3 gene. Also, p.E818K is seemingly tightly coupled with CAPOS syndrome and has never been reported in other two disorders (Han et al., 2017; Figure 3). There is no report of hearing or visual impairment in AHC and RDP, which is vastly different from CAPOS syndrome (Figure 4; Tranebjærg et al., 2018a). However, those three entities present with an expanding phenotypic spectrum and are more often reported to share somewhat overlapping neurological phenotypes. In addition, atypical phenotypes due to ATP1A3 gene mutations have recently been identified (Gropman et al., 2020; Guo et al., 2020). It becomes gradually clear that these overlapping phenotypes may be a series of ATP1A3-related symptoms, rather than allelic disorders (Carecchio et al., 2018). Moreover, the expression timing of neurological deficits of the mutation varies and has still not been fully characterized until now (Han et al., 2017). In our study, it was not very hard to observe the latency of occurrence of numerous symptoms in patients 1 and 2. Notably, patients 3 and 4 denied all neurological episodes. Continuation of the follow-up periods for these four patients is therefore necessary for the discovery of other delayed neurological symptoms. Nevertheless, whether AN is simply a symptom of CAPOS or a completely new phenotype of the ATP1A3 gene remains to be seen.
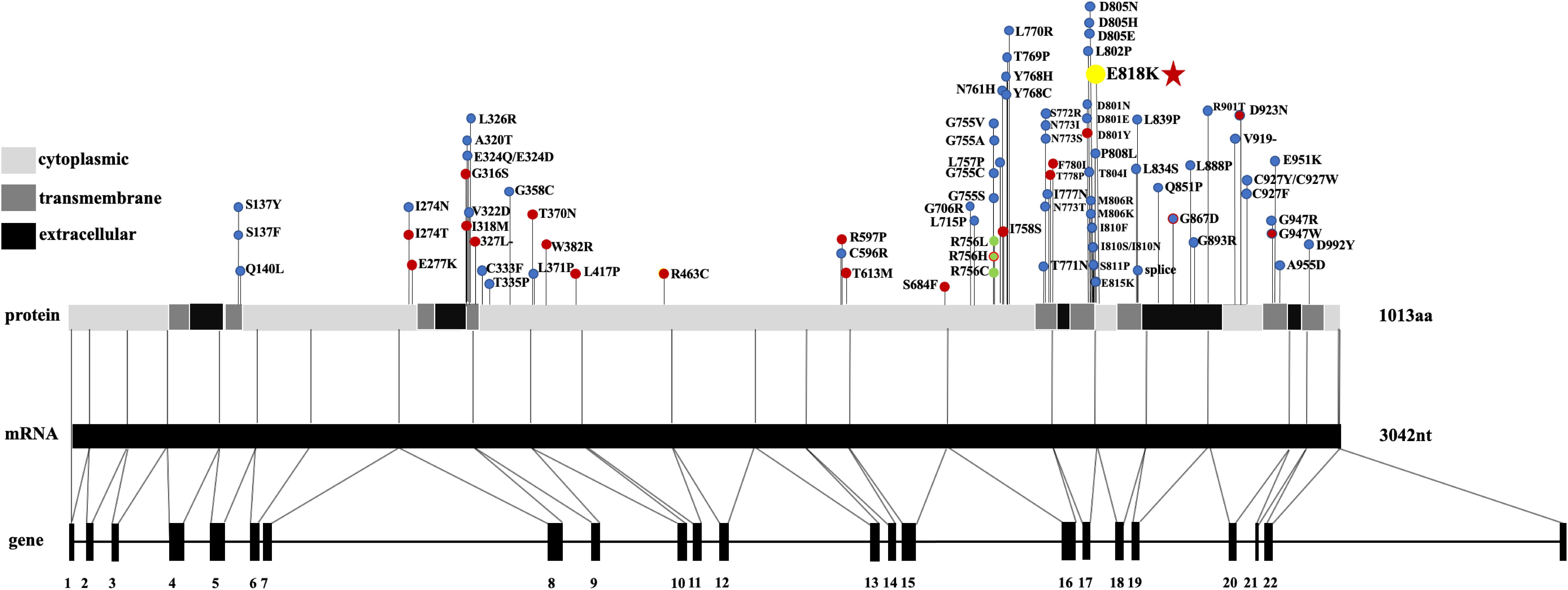
Figure 3. The location of AHC-causing (blue dots), RDP-causing (red dots), and CAPOS-causing (yellow dots) mutations in ATP1A3, mRNA, and protein. Three rare polymorphisms identified are indicated by the green dots. Mutation shared between different phenotypes is located at D923N (blue dot with a red dot inside), G867N (red dot with a blue dot inside), and R756H (red dot with a green dot inside). Different regions (cytoplasmic, transmembrane, and extracellular) of the protein are listed on the left. Abbreviations: AHC, alternating hemiplegia of childhood; CAPOS, cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss; RDP, rapid-onset dystonia Parkinsonism.
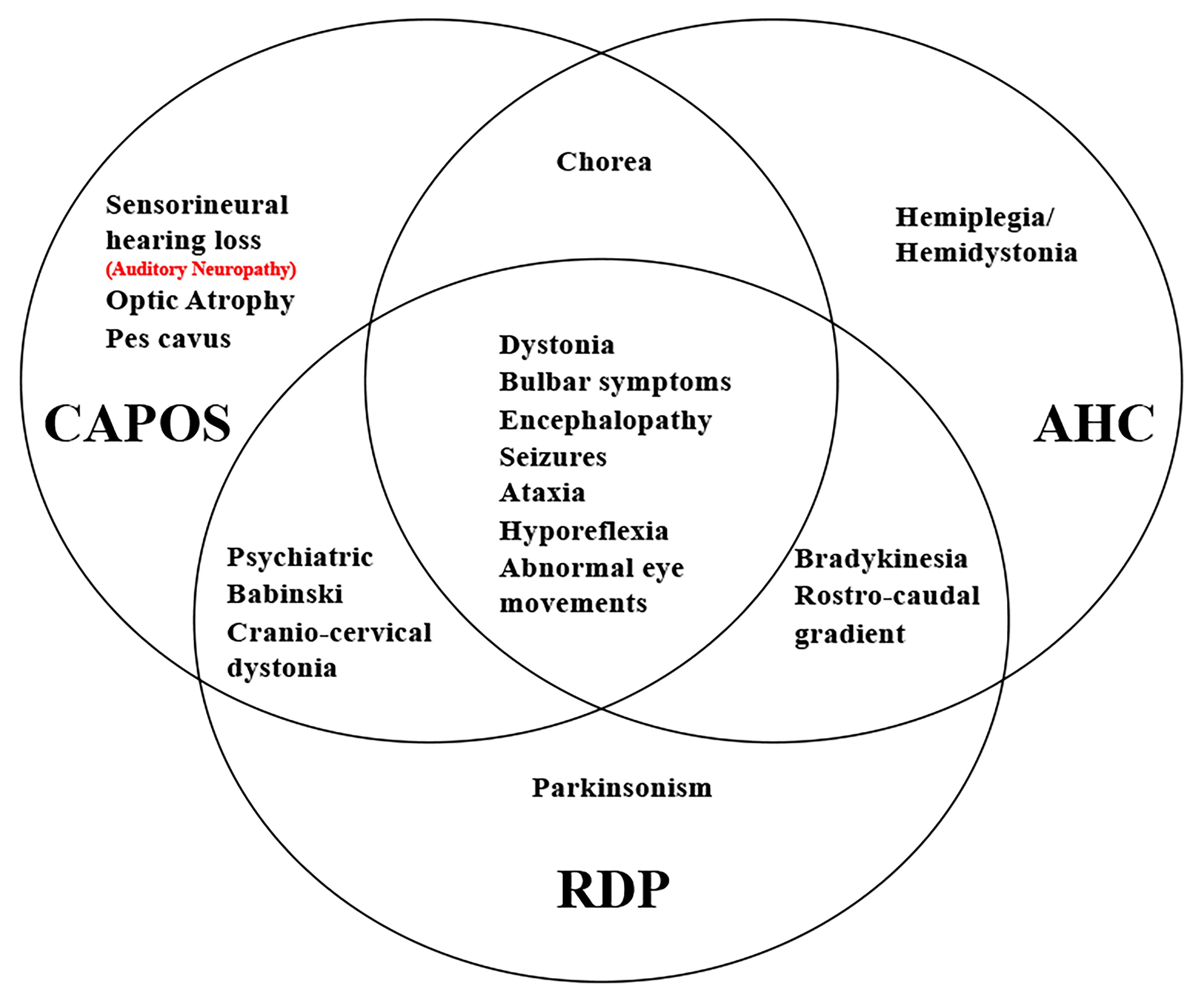
Figure 4. Unique and shared features between AHC, RDP, and CAPOS phenotypes. Auditory neuropathy is either a symptom of CAPOS or a new phenotype of ATP1A3 gene.
Early diagnosis of hearing loss and timely prevention of sensory deprivation are of vital importance and confer lifelong benefits to CAPOS patients, especially for those with visual interference. The 818 position is previously confirmed located at the cytoplasmic end of transmembrane helix 6 of NKAα3, forming a salt bridge with the backbone carbonyl of Arg930, which is a residue known to stabilize the C-terminus. Tranebjærg et al. (2018b) have evaluated functional impact of Glu818Lys to the C-terminal structure and demonstrated that Glu818Lys leads to low affinity for extracellular sodium and a high rate of sodium release from the third ion-binding portion; therefore, it disrupts the function of the NKAα3 subunit of Na, K-ATPases, which is a key regulator of regaining the resting potential of the membrane after excitatory activity in the auditory pathway. In the auditory pathway, NKAα3 is expressed in the membranes of type I afferent terminals contacting the inner hair cells, spiral ganglion, and medial efferent terminals that contact the outer hair cells (Erichsen et al., 1996; McLean et al., 2009; Bottger et al., 2011), providing evidence that CI in those patients could generate likely preferable outcomes. One example of CI having an ideal outcome is CI in AN together with ADOA (OMIM #165500) owing to OPA1 variations (Santarelli et al., 2008; Huang et al., 2009; Moser and Starr, 2016). The expression site of OPA1 significantly overlaps with that of NKAα3, being it in the hair cells as well as in the neuronal fibers innervating the inner hair cells, ganglion cells of the cochlea, and vestibular organs (Bette et al., 2007).
Auditory neuropathy with ATP1A3 mutation are generally considered to have ideal CI results. However, CI produces dubious outcomes in some reported cases. Tranebjaerg et al. (Fang et al., 2019) stated that four CAPOS patients received CI, two of whom gained significant benefits and the other two was relatively poor. In contrast, Atilgan et al. (2019) documented a much more beneficial effect of CI in a patient with CAPOS syndrome as compared to patients with SNHL. These findings could be explained by the fact that the ATP1A3 gene is expressed not only in synapse but also in postsynaptic spiral ganglion cells (SGNs). If the lesion is mainly at the synapse, good CI results can be expected, similar to those in OPA1 variants (Han et al., 2017), whereas CI is less useful with lesions dominantly at the SGNs. In fact, in most postsynaptic AN, CI has a dubious outcome (Supplementary Table 2). An example is CI in the SGNs (Fasquelle et al., 2011; Eppsteiner et al., 2012; Atilgan et al., 2019). CI performed earlier in TMPRSS3 generated a preferable outcome, while the outcomes in later implantees were not as favorable (Chung et al., 2014). Patients who benefit most from CI were reported to be relatively younger (Han et al., 2017; Tranebjærg et al., 2018b), yet an accurate CI efficacy estimation with age and other possible impact factors taken into consideration is still awaiting discussion. Chaudhry et al. (2020) summarized 25 postsynaptic CI results, with 22 displaying modest to significant benefit, and demonstrated that CI behaves variably but generally good in postsynaptic AN. Moreover, the best functional outcome of CI observed at the short-term follow-up may decrease over time. Although earlier electrical stimulation of SGNs may attenuate the degeneration of the neurons, progressive degeneration of SGNs could possibly occur due to a disrupted function of NKAα3 (Han et al., 2017). In our study, patient 2 underwent CI on his left ear, but the result was hardly desirable. One possible explanation is that his age by the time he received surgery and the duration of his hearing loss are too long to ensure SGN activities. Moreover, the result of CI may be in relation with the characteristic of the PTA of the patients. Therefore, a continuous follow-up period for CI performance in those patients is warranted, to better determine if the current benefits from CI decreases over time.
The identification of p.E818K in ATP1A3 has implications for future risk calculation and genetic counseling. In CAPOS families with an autosomal dominant inheritance pattern, the chance of offspring carrying pathogenic mutations is 50%, with varying degrees of penetrance. In addition, all reported CAPOS patients had unaffected siblings without exception, revealing that all pedigrees of CAPOS syndrome are sporadic cases. It is still unclear why de novo variants frequently occur in ATP1A3. The age of the father has been reported to be a strong impactor of de novo mutations (Niederberger, 2013). However, that study failed to explain all de novo cases. A genetic context in ATP1A3 that favors the occurrence of de novo variants may account for this phenomenon, awaiting further clarification.
Here, four AN patients were identified with p.E818K mutation in the gene ATP1A3, causing progressive AN with postlingual onset and varying degrees of syndromic features. Our study confirms that ATP1A3 in Chinese individuals is undoubtedly an important genetic cause of progressive AN with postlingual onset. Genetic testing for this specific mutation site allows for identification of a special subtype of AN with somewhat preferable CI results, as well as accurate genetic counseling.
Data Availability Statement
The data presented in the study are deposited in the ClinVar repository, accession number VCV000156238.
Ethics Statement
The studies involving human participants were reviewed and approved by Committee of Medical Ethics of Chinese PLA General Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
HW and WW conceived and designed the experiments and wrote the manuscript. HW, WW, JL, LX, LL, and FX performed the experiments. HW analyzed the data. JL, LL, LX, and FX contributed to the reagents, materials, and analysis tools. HW, JG, and QW critically read and discussed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the grants of the National Natural Science Foundation of China (Major Project No. 81830028; Youth Projects 81900950 and 81900951). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.749484/full#supplementary-material
Supplementary Figure 1 | Audiological phenotype of subject 3 and 4. (A,B) PTA of subjects 3 and 4. Blue, left ear; red, right ear. (C,D) Observed DPOAE responses from the right and the left ear. Red line, DPOAE level from the right ear; blue line, DPOAE level from the left ear; gray line, noise level from both ears. (E,F) Absent ABR waveforms at 90 dB nHL stimulus on both ears of subjects 3 and 4. Blue, left ear; red, right ear.
Supplementary Table 1 | ATP1A3 genotypes as they correlate with RDP, AHC, and CAPOS phenotypes.
Supplementary Table 2 | Postsynaptic auditory neuropathies and their CI outcomes.
Footnotes
References
Atilgan, A., Yuksel, M., and Ciprut, A. (2019). Cochlear implantation in a case of auditory neuropathy spectrum disorder with CAPOS syndrome. Med. Med. J. 34, 318–323. doi: 10.5222/MMJ.2019.53503
Bette, S., Zimmermann, U., Wissinger, B., and Knipper, M. (2007). OPA1, the disease gene for optic atrophy type Kjer, is expressed in the inner ear. Histochem. Cell Biol. 128, 421–430. doi: 10.1007/s00418-007-0321-7
Blanco, G., and Mercer, R. (1998). Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am. J. Physiol. 275, F633–F650. doi: 10.1152/ajprenal.1998.275.5.F633
Blanco-Arias, P., Einholm, A., Mamsa, H., Concheiro, C., Gutiérrez-de-Terán, H., Romero, J., et al. (2009). Mutation of ATP1A3 underscores the crucial role of sodium affinity in the pathophysiology of rapid-onset dystonia-parkinsonism. Hum. Mol. Genet. 18, 2370–2377. doi: 10.1093/hmg/ddp170
Bottger, P., Tracz, Z., Heuck, A., Nissen, P., Romero-Ramos, M., and Lykke-Hartmann, K. (2011). Distribution of Na/K-ATPase alpha 3 isoform, a sodium-potassium P-type pump associated with rapid-onset of dystonia parkinsonism (RDP) in the adult mouse brain. J. Comp. Neurol. 519, 376–404. doi: 10.1002/cne.22524
Carecchio, M., Zorzi, G., Ragona, F., Zibordi, F., and Nardocci, N. (2018). ATP1A3-related disorders: an update. Eur. J. Paediatr. Neurol. 22, 257–263. doi: 10.1016/j.ejpn.2017.12.009
Chaudhry, D., Chaudhry, A., Muzaffar, J., Monksfield, P., and Bance, M. (2020). Cochlear implantation outcomes in post synaptic auditory neuropathies: a systematic review and narrative synthesis. J. Int. Adv. Otol. 16, 411–431. doi: 10.5152/iao.2020.9035
Chen, Y., Gu, Y., Li, Y., Li, G. L., Chai, R., Li, W., et al. (2021). Generation of mature and functional hair cells by co-expression of Gfi1, Pou4f3, and Atoh1 in the postnatal mouse cochlea. Cell Rep. 35:109016. doi: 10.1016/j.celrep.2021.109016
Cheng, C., Hou, Y., Zhang, Z., Wang, Y., Lu, L., Zhang, L., et al. (2021). Disruption of the autism-related gene Pak1 causes stereocilia disorganization, hair cell loss, and deafness in mice. J. Genet. Genomics 48, 324–332. doi: 10.1016/j.jgg.2021.03.010
Cheng, C., Wang, Y., Guo, L., Lu, X., Zhu, W., Muhammad, W., et al. (2019). Age-related transcriptome changes in Sox2+ supporting cells in the mouse cochlea. Stem Cell Res. Ther. 10:365. doi: 10.1186/s13287-019-1437-0
Chung, J., Park, S., Chang, S., Chung, T., Lee, K., Kim, A., et al. (2014). A novel mutation of TMPRSS3 related to milder auditory phenotype in Korean postlingual deafness: a possible future implication for a personalized auditory rehabilitation. J. Mol. Med. (Berlin, Germany) 92, 651–663. doi: 10.1007/s00109-014-1128-3
Clausen, M., Hilbers, F., and Poulsen, H. (2017). The structure and function of the Na,K-ATPase isoforms in health and disease. Front. Physiol. 8:371. doi: 10.3389/fphys.2017.00371
Demos, M., van Karnebeek, C., Ross, C., Adam, S., Shen, Y., Zhan, S., et al. (2014). A novel recurrent mutation in ATP1A3 causes CAPOS syndrome. Orphanet J. Rare Dis. 9:15. doi: 10.1186/1750-1172-9-15
Duat Rodriguez, A., Prochazkova, M., Santos Santos, S., Rubio Cabezas, O., Cantarin Extremera, V., and Gonzalez-Gutierrez-Solana, L. (2017). Early diagnosis of CAPOS syndrome before acute-onset ataxia-review of the literature and a new family. Pediatr. Neurol. 71, 60–64. doi: 10.1016/j.pediatrneurol.2017.01.009
Eppsteiner, R., Shearer, A., Hildebrand, M., Deluca, A., Ji, H., Dunn, C., et al. (2012). Prediction of cochlear implant performance by genetic mutation: the spiral ganglion hypothesis. Hear. Res. 292, 51–58. doi: 10.1016/j.heares.2012.08.007
Erichsen, S., Zuo, J., Curtis, L., Rarey, K., and Hultcrantz, M. (1996). Na,K-ATPase alpha- and beta-isoforms in the developing cochlea of the mouse. Hear. Res. 100, 143–149. doi: 10.1016/0378-5955(96)00105-0
Fang, Q., Zhang, Y., Da, P., Shao, B., Pan, H., He, Z., et al. (2019). Deletion of Limk1 and Limk2 in mice does not alter cochlear development or auditory function. Sci. Rep. 9:3357. doi: 10.1038/s41598-019-39769-z
Fasquelle, L., Scott, H., Lenoir, M., Wang, J., Rebillard, G., Gaboyard, S., et al. (2011). Tmprss3, a transmembrane serine protease deficient in human DFNB8/10 deafness, is critical for cochlear hair cell survival at the onset of hearing. J. Biol. Chem. 286, 17383–17397. doi: 10.1074/jbc.M110.190652
Fu, X., An, Y., Wang, H., Li, P., Lin, J., Yuan, J., et al. (2021). Deficiency of Klc2 induces low-frequency sensorineural hearing loss in C57BL/6 J mice and human. Mol. Neurobiol. doi: 10.21203/rs.3.rs-277106/v1
Gao, S., Cheng, C., Wang, M., Jiang, P., Zhang, L., Wang, Y., et al. (2019). Blebbistatin inhibits neomycin-induced apoptosis in hair cell-like HEI-OC-1 cells and in cochlear hair cells. Front. Cell Neurosci. 13:590. doi: 10.3389/fncel.2019.00590
Gropman, A., Uittenbogaard, M., Brantner, C., Wang, Y., Wong, L., and Chiaramello, A. (2020). Molecular genetic and mitochondrial metabolic analyses confirm the suspected mitochondrial etiology in a pediatric patient with an atypical form of alternating hemiplegia of childhood. Mol. Genet. Metab. Rep. 24:100609. doi: 10.1016/j.ymgmr.2020.100609
Guo, R., Li, J., Chen, C., Xiao, M., Liao, M., Hu, Y., et al. (2021). Biomimetic 3D bacterial cellulose-graphene foam hybrid scaffold regulates neural stem cell proliferation and differentiation. Colloids Surf. B Biointerfaces 200:111590. doi: 10.1016/j.colsurfb.2021.111590
Guo, R., Ma, X., Liao, M., Liu, Y., Hu, Y., Qian, X., et al. (2019). Development and application of cochlear implant-based electric-acoustic stimulation of spiral ganglion neurons. ACS Biomater. Sci. Eng. 5, 6735–6741. doi: 10.1021/acsbiomaterials.9b01265
Guo, R., Xiao, M., Zhao, W., Zhou, S., Hu, Y., Liao, M., et al. (2020). 2D Ti3C2TxMXene couples electrical stimulation to promote proliferation and neural differentiation of neural stem cells. Acta Biomater. S1742-7061(20)30749-2. doi: 10.1016/j.actbio.2020.12.035
Guo, R., Zhang, S., Xiao, M., Qian, F., He, Z., Li, D., et al. (2016). Accelerating bioelectric functional development of neural stem cells by graphene coupling: implications for neural interfacing with conductive materials. Biomaterials 106, 193–204. doi: 10.1016/j.biomaterials.2016.08.019
Han, K., Oh, D., Lee, S., Lee, C., Han, J., Kim, M., et al. (2017). ATP1A3 mutations can cause progressive auditory neuropathy: a new gene of auditory synaptopathy. Sci. Rep. 7:16504. doi: 10.1038/s41598-017-16676-9
Han, S., Xu, Y., Sun, J., Liu, Y., Zhao, Y., Tao, W., et al. (2020). Isolation and analysis of extracellular vesicles in a Morpho butterfly wing-integrated microvortex biochip. Biosens. Bioelectron. 154:112073. doi: 10.1016/j.bios.2020.112073
Harrison, R. V., Gordon, K. A., Papsin, B. C., Negandhi, J., and James, A. L. (2015). Auditory neuropathy spectrum disorder (ANSD) and cochlear implantation. Int. J. Pediatr. Otorhinolaryngol. 79, 1980–1987. doi: 10.1016/j.ijporl.2015.10.006
He, Y., Lu, X., Qian, F., Liu, D., Chai, R., and Li, H. (2017). Insm1a is required for zebrafish posterior lateral line development. Front. Mol. Neurosci. 10:241. doi: 10.3389/fnmol.2017.00241
He, Z., Fang, Q., Li, H., Shao, B., Zhang, Y., Zhang, Y., et al. (2019). The role of FOXG1 in the postnatal development and survival of mouse cochlear hair cells. Neuropharmacology 144, 43–57. doi: 10.1016/j.neuropharm.2018.10.021
He, Z., Guo, L., Shu, Y., Fang, Q., Zhou, H., Liu, Y., et al. (2017). Autophagy protects auditory hair cells against neomycin-induced damage. Autophagy 13, 1884–1904. doi: 10.1080/15548627.2017.1359449
He, Z. H., Li, M., Fang, Q. J., Liao, F. L., Zou, S. Y., Wu, X., et al. (2021). FOXG1 promotes aging inner ear hair cell survival through activation of the autophagy pathway. Autophagy 1–22. doi: 10.1080/15548627.2021.1916194
He, Z. H., Zou, S. Y., Li, M., Liao, F. L., Wu, X., Sun, H. Y., et al. (2020). The nuclear transcription factor FoxG1 affects the sensitivity of mimetic aging hair cells to inflammation by regulating autophagy pathways. Redox Biol. 28:101364. doi: 10.1016/j.redox.2019.101364
Heimer, G., Sadaka, Y., Israelian, L., Feiglin, A., Ruggieri, A., Marshall, C., et al. (2015). CAOS-episodic cerebellar ataxia, areflexia, optic atrophy, and sensorineural hearing loss: a third allelic disorder of the ATP1A3 gene. J. Child Neurol. 30, 1749–1756. doi: 10.1177/0883073815579708
Hu, Y., Li, D., Wei, H., Zhou, S., Chen, W., Yan, X., et al. (2021). Neurite extension and orientation of spiral ganglion neurons can be directed by superparamagnetic iron oxide nanoparticles in a magnetic field. Int. J. Nanomed. 16, 4515–4526. doi: 10.2147/IJN.S313673
Huang, T., Santarelli, R., and Starr, A. (2009). Mutation of OPA1 gene causes deafness by affecting function of auditory nerve terminals. Brain Res. 1300, 97–104. doi: 10.1016/j.brainres.2009.08.083
Kim, N., Kim, A., Park, K., Kim, S., Kim, M., Nam, J., et al. (2015). Whole-exome sequencing reveals diverse modes of inheritance in sporadic mild to moderate sensorineural hearing loss in a pediatric population. Genet. Med. 17, 901–911. doi: 10.1038/gim.2014.213
Landrum, M., Lee, J., Benson, M., Brown, G., Chao, C., Chitipiralla, S., et al. (2016). ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44, D862–D868. doi: 10.1093/nar/gkv1222
Liu, L., Chen, Y., Qi, J., Zhang, Y., He, Y., Ni, W., et al. (2016). Wnt activation protects against neomycin-induced hair cell damage in the mouse cochlea. Cell Death Dis. 7:e2136. doi: 10.1038/cddis.2016.35
Liu, W., Xu, L., Wang, X., Zhang, D., Sun, G., Wang, M., et al. (2021). PRDX1 activates autophagy via the PTEN-AKT signaling pathway to protect against cisplatin-induced spiral ganglion neuron damage. Autophagy 5, 1–23. doi: 10.1080/15548627.2021.1905466
Liu, W., Xu, X., Fan, Z., Sun, G., Han, Y., Zhang, D., et al. (2019). Wnt signaling activates TP53-induced glycolysis and apoptosis regulator and protects against cisplatin-induced spiral ganglion neuron damage in the mouse cochlea. Antioxid. Redox Signal. 30, 1389–1410. doi: 10.1089/ars.2017.7288
Liu, Y., Qi, J., Chen, X., Tang, M., Chu, C., Zhu, W., et al. (2019). Critical role of spectrin in hearing development and deafness. Sci. Adv. 5:eaav7803. doi: 10.1126/sciadv.aav7803
Lv, J., Fu, X., Li, Y., Hong, G., Li, P., Lin, J., et al. (2021). Deletion of Kcnj16 in mice does not alter auditory function. Front. Cell Dev. Biol. 9:630361. doi: 10.3389/fcell.2021.630361
Maas, R., Schieving, J., Schouten, M., Kamsteeg, E., and van de Warrenburg, B. (2016). The genetic homogeneity of CAPOS syndrome: four new patients with the c.2452G>A (p.Glu818Lys) mutation in the ATP1A3 gene. Pediatr. Neurol. 59, 71–75.e1. doi: 10.1016/j.pediatrneurol.2016.02.010
McLean, W., Smith, K., Glowatzki, E., and Pyott, S. (2009). Distribution of the Na,K-ATPase alpha subunit in the rat spiral ganglion and organ of corti. J. Assoc. Res. Otolaryngol. 10, 37–49. doi: 10.1007/s10162-008-0152-9
Moser, T., and Starr, A. (2016). Auditory neuropathy–neural and synaptic mechanisms. Nat. Rev. Neurol. 12, 135–149. doi: 10.1038/nrneurol.2016.10
Nicolaides, P., Appleton, R., and Fryer, A. (1996). Cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss (CAPOS): a new syndrome. J. Med. Genet. 33, 419–421. doi: 10.1136/jmg.33.5.419
Niederberger, C. (2013). Re: rate of De Novo mutations and the importance of father’s age to disease risk. J. Urol. 189:655. doi: 10.1016/j.juro.2012.10.098
Paquay, S., Wiame, E., Deggouj, N., Boschi, A., De Siati, R., Sznajer, Y., et al. (2018). Childhood hearing loss is a key feature of CAPOS syndrome: a case report. Int. J. Pediatr. Otorhinolaryngol. 104, 191–194. doi: 10.1016/j.ijporl.2017.11.022
Potic, A., Nmezi, B., and Padiath, Q. (2015). CAPOS syndrome and hemiplegic migraine in a novel pedigree with the specific ATP1A3 mutation. J. Neurol. Sci. 358, 453–456. doi: 10.1016/j.jns.2015.10.002
Qi, J., Liu, Y., Chu, C., Chen, X., Zhu, W., Shu, Y., et al. (2019). A cytoskeleton structure revealed by super-resolution fluorescence imaging in inner ear hair cells. Cell Discov. 5:12. doi: 10.1038/s41421-018-0076-4
Qi, J., Zhang, L., Tan, F., Liu, Y., Chu, C., Zhu, W., et al. (2020). Espin distribution as revealed by super-resolution microscopy of stereocilia. Am. J. Transl. Res. 12, 130–141.
Qian, F., Wang, X., Yin, Z., Xie, G., Yuan, H., Liu, D., et al. (2020). The slc4a2b gene is required for hair cell development in zebrafish. Aging (Albany NY) 12, 18804–18821. doi: 10.18632/aging.103840
Rance, G., and Starr, A. (2015). Pathophysiological mechanisms and functional hearing consequences of auditory neuropathy. Brain 138, 3141–3158. doi: 10.1093/brain/awv270
Rodacker, V., Toustrup-Jensen, M., and Vilsen, B. (2006). Mutations Phe785Leu and Thr618Met in Na+,K+-ATPase, associated with familial rapid-onset dystonia parkinsonism, interfere with Na+ interaction by distinct mechanisms. J. Biol. Chem. 281, 18539–18548. doi: 10.1074/jbc.M601780200
Rosewich, H., Weise, D., Ohlenbusch, A., Gärtner, J., and Brockmann, K. (2014). Phenotypic overlap of alternating hemiplegia of childhood and CAPOS syndrome. Neurology 83, 861–863. doi: 10.1212/WNL.0000000000000735
Santarelli, R., Cama, E., Scimemi, P., Dal Monte, E., Genovese, E., and Arslan, E. (2008). Audiological and electrocochleography findings in hearing-impaired children with connexin 26 mutations and otoacoustic emissions. Eur. Arch. Otorhinolaryngol. 265, 43–51. doi: 10.1007/s00405-007-0412-z
Starr, P., Rau, G., Davis, V., Marks, W., Ostrem, J., Simmons, D., et al. (2005). Spontaneous pallidal neuronal activity in human dystonia: comparison with Parkinson’s disease and normal macaque. J. Neurophysiol. 93, 3165–3176. doi: 10.1152/jn.00971.2004
Tan, F., Chu, C., Qi, J., Li, W., You, D., Li, K., et al. (2019). AAV-ie enables safe and efficient gene transfer to inner ear cells. Nat. Commun. 10:3733. doi: 10.1038/s41467-019-11687-8
Tranebjærg, L., Strenzke, N., Lindholm, S., Rendtorff, N., Poulsen, H., Khandelia, H., et al. (2018a). The CAPOS mutation in ATP1A3 alters Na/K-ATPase function and results in auditory neuropathy which has implications for management. Hum. Genet. 137, 279–280. doi: 10.1007/s00439-018-1870-7
Tranebjærg, L., Strenzke, N., Lindholm, S., Rendtorff, N., Poulsen, H., Khandelia, H., et al. (2018b). The CAPOS mutation in ATP1A3 alters Na/K-ATPase function and results in auditory neuropathy which has implications for management. Hum. Genet. 137, 111–127. doi: 10.1007/s00439-017-1862-z
Wang, H., Guan, J., Lan, L., Yu, L., Xie, L., Liu, X., et al. (2018). A novel de novo mutation of ACTG1 in two sporadic non-syndromic hearing loss cases. Sci. China Life Sci. 61, 729–732. doi: 10.1007/s11427-017-9165-2
Wang, Y., Li, J., Yao, X., Li, W., Du, H., Tang, M., et al. (2017). Loss of CIB2 causes profound hearing loss and abolishes mechanoelectrical transduction in mice. Front. Mol. Neurosci. 10:401. doi: 10.3389/fnmol.2017.00401
Zhang, S., Dong, Y., Qiang, R., Zhang, Y., Zhang, X., Chen, Y., et al. (2021). Characterization of Strip1 expression in mouse cochlear hair cells. Front. Genet. 12:625867. doi: 10.3389/fgene.2021.625867
Zhang, S., Zhang, Y., Dong, Y., Guo, L., Zhang, Z., Shao, B., et al. (2020). Knockdown of Foxg1 in supporting cells increases the trans-differentiation of supporting cells into hair cells in the neonatal mouse cochlea. Cell. Mol. Life Sci. 77, 1401–1419. doi: 10.1007/s00018-019-03291-2
Zhang, Y., Li, W., He, Z., Wang, Y., Shao, B., Cheng, C., et al. (2019). Pre-treatment with Fasudil prevents neomycin-induced hair cell damage by reducing the accumulation of reactive oxygen species. Front. Mol. Neurosci. 12:264. doi: 10.3389/fnmol.2019.00264
Zhong, Z., Fu, X., Li, H., Chen, J., Wang, M., Gao, S., et al. (2020). Citicoline protects auditory hair cells against neomycin-induced damage. Front. Cell Dev. Biol. 8:712. doi: 10.3389/fcell.2020.00712
Keywords: auditory neuropathy (AN), ATP1A3, de novo, CAPOS syndrome, cochlear implantation (CI)
Citation: Wang W, Li J, Lan L, Xie L, Xiong F, Guan J, Wang H and Wang Q (2021) Auditory Neuropathy as the Initial Phenotype for Patients With ATP1A3 c.2452 G > A: Genotype–Phenotype Study and CI Management. Front. Cell Dev. Biol. 9:749484. doi: 10.3389/fcell.2021.749484
Received: 29 July 2021; Accepted: 31 August 2021;
Published: 08 October 2021.
Edited by:
Renjie Chai, Southeast University, ChinaReviewed by:
Guoqiang Wan, Nanjing University, ChinaYu Sun, Huazhong University of Science and Technology, China
Copyright © 2021 Wang, Li, Lan, Xie, Xiong, Guan, Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongyang Wang, whyx301@foxmail.com; Qiuju Wang, wqcr301@vip.sina.com