 Rui Xu
Rui Xu Shaojun Du
Shaojun Du
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Cell Dev. Biol., 26 October 2021
Sec. Morphogenesis and Patterning
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.746818
Lifeact-GFP is a frequently used molecular probe to study F-actin structure and dynamic assembly in living cells. In this study, we generated transgenic zebrafish models expressing Lifeact-GFP specifically in cardiac muscles to investigate the effect of Lifeact-GFP on heart development and its application to study cardiomyopathy. The data showed that transgenic zebrafish with low to moderate levels of Lifeact-GFP expression could be used as a good model to study contractile dynamics of actin filaments in cardiac muscles in vivo. Using this model, we demonstrated that loss of Smyd1b, a lysine methyltransferase, disrupted F-actin filament organization in cardiomyocytes of zebrafish embryos. Our studies, however, also demonstrated that strong Lifeact-GFP expression in cardiomyocytes was detrimental to actin filament organization in cardiomyocytes that led to pericardial edema and early embryonic lethality of zebrafish embryos. Collectively, these data suggest that although Lifeact-GFP is a good probe for visualizing F-actin dynamics, transgenic models need to be carefully evaluated to avoid artifacts induced by Lifeact-GFP overexpression.
Actin is one of the most abundant proteins in eukaryotic cells and exists as either a free monomer called G-actin (globular) or a linear polymer microfilament called F-actin (filamentous). The F-actin is an important component of the cytoskeleton in eukaryotic cells and thin filaments in myofibrils of muscle cells. F-actin participates in many important cellular processes, including cell division, intracellular cargo transport, cell migration, cell morphogenesis, and muscle contraction (Pollard and Cooper, 2009; Dominguez and Holmes, 2011). A large number of illnesses and diseases are caused by genetic mutations in actin and its associated proteins. In muscle cells, actin thin filaments are an essential part of the sarcomere structure, the basic unit of muscle contraction. Actin thin filaments work together with myosin thick filaments to drive muscle contraction in heart and skeletal muscles. Defective thin filament assembly from actin mutations is associated with actin-accumulation myopathy, nemaline myopathy, and cardiomyopathy (Nowak et al., 1999; Costa et al., 2004; Despond and Dawson, 2018; Frustaci et al., 2018).
Visualization of actin filament structures and dynamic assembly in living cells is critical for the study of various cellular processes of cell division, migration, polarization, and muscle contraction. Several different techniques and approaches are currently applied to analyze and visualize cellular actin structures and their dynamic assembly and disassembly in various cell and model systems (Belin et al., 2014; Lemieux et al., 2014; Spracklen et al., 2014; Du et al., 2015). Phalloidin is the standard F-actin marker commonly used to label F-actin in fixed samples and tissues. However, phalloidin has a toxic side effect to living cells (Wehland et al., 1977; Cooper, 1987), thus limiting its application in live cell imaging. Live-cell analysis of actin filaments and their dynamic assembly largely relies on genetically modified GFP derivatives that tag actin directly or actin-binding domains (Melak et al., 2017). Tagging actin directly using GFP is a simple and popular technique, which has been successfully used for studying actin dynamics and turnover within a given cellular structure using the fluorescence recovery after photobleaching (FRAP) approach (Koestler et al., 2009; Clark et al., 2013). However, the relatively large size of GFP-tagged actin can give rise to problems in terms of the incorporation of Actin-GFP monomers into filaments. In addition, the Actin-GFP chimeras can interfere with the normal functionality of actin cytoskeleton resulting in experimental artifacts (Aizawa et al., 1997; Nagasaki et al., 2017).
To avoid the problem of steric clashes with actin from using large and bulky Actin-GFP chimeras, small fluorophore labeled peptides, such as Lifeact-GFP, were developed. Lifeact-GFP is the most popular and widely used actin probe for visualizing F-Actin structures in various cell types and model organisms. Lifeact-GFP is generated by fusing a short 17-amino-acid peptide (Lifeact) from yeast Abp140 with GFP (Riedl et al., 2008; Fraccaroli et al., 2012; Reischauer et al., 2014; Higuchi-Sanabria et al., 2018). The specific and selective binding of Lifeact with F-actin permits the study of intracellular actin in the native environment without significantly interfering with actin dynamics in vitro or in vivo. Thus, Lifeact-GFP surpasses other reagents like fluorescent phalloidin, actin-GFP, and anti-actin antibodies. It enables live imaging of the actin cytoskeleton and therefore the study of many fundamental biological processes in vivo.
Numerous studies have been published describing the application of Lifeact in studying cell motility (Duleh and Welch, 2012; Fiolka et al., 2012; Rullo et al., 2012; Grikscheit et al., 2015; Suarez et al., 2015). Cells expressing Lifeact-GFP showed no defects of actin dynamics, cell physiology, or tissue organization, suggesting that Lifeact-GFP expression does not interfere with cellular processes. Transgenic mice expressing Lifeact-GFP that were viable, phenotypically normal, and fertile have been successfully generated (Riedl et al., 2010), thus validating Lifeact-EGFP as a reporter that doesn’t interfere with physiological processes. Additionally, Lifeact-GFP has been successfully employed to generate other transgenic models including Arabidopsis (Vidali et al., 2009; Van Der Honing et al., 2011), Drosophila (Zanet et al., 2012; Huelsmann et al., 2013), and zebrafish (Phng et al., 2013; Reischauer et al., 2014; Xu et al., 2014; Fukuda et al., 2017). These Lifeact-EGFP transgenic models provide new opportunities to study actin remodeling in all cells, within native tissues.
Although it was believed that Lifeact-GFP does not interfere with actin dynamics in vitro or in vivo, several recent reports have raised concerns on Lifeact-associated artifacts at the cellular and whole organismal levels. It has been reported that overexpression of Lifeact caused infertility and severe actin defects and multiple nuclei in follicle cells in Drosophila (Spracklen et al., 2014). Similarly, strong Lifeact expression disturbed actin assembly in fission yeast (Courtemanche et al., 2016). Recent studies demonstrated that Lifeact altered cell cytoskeleton and morphology in mammalian cells (Flores et al., 2019). Most of the Lifeact-GFP induced artifacts were observed in actin cytoskeleton structures in non-muscle cells. It is not clear whether Lifeact-GFP can impede actin thin filaments in sarcomeres of skeletal and cardiac muscle cells.
In addition to Lifeact, actin binding domains of F-tractin, UtrCH and Fimbrin have been successfully used in constructing actin probes for live imaging (Melak et al., 2017). F-tractin contains a 43-amino-acid long peptide from the rat actin-binding inositol 1,4,5-trisphosphate 3-kinase A (Schell et al., 2001; Belin et al., 2014), whereas UtrCH comprises the first 261 amino acids of human utrophin, an actin-binding protein (Winder et al., 1995). Fimbrin on the other hand is an actin cross-linking protein, important in filopodia formation (Bretscher, 1981; Matsudaira, 1994). The actin binding domains in these proteins fused with GFP have been successfully used for imaging actin filaments in a wide range of organisms and cell types (Sheahan et al., 2004; Wang et al., 2004; Burkel et al., 2007; Holubcová et al., 2013).
SiR-actin and Actin-Chromobody are two recently introduced actin markers. SiR-actin is a cell-permeable chemically synthesized probe, a structural analog of F-actin-binding toxin from a marine sponge (Bubb et al., 1994; Lukinavičius et al., 2014; D’Este et al., 2015). Advantages of SiR-actin are its ease of use in live imaging as it avoids the time-consuming steps of cell transfection and protein overexpression. However, SiR-actin can cause F-actin stabilization or induce actin polymerization, making interpretations of observed F-actin structures more difficult. In comparison, it appears that high levels of Actin-Chromobody expression does not alter actin dynamics. Actin chromobodies are fluorescent-protein-tagged actin nanobodies that have been successfully used for imaging sub-organellar actin dynamics in cultured cells and live imaging of endogenous protein dynamics in zebrafish (Panza et al., 2015; Schiavon et al., 2020).
Zebrafish embryos, with their optical transparency, offer the potential to image contractile structures in skeletal and cardiac muscles in vivo. Transgenic zebrafish expressing Lifeact-GFP reporter have been generated and successfully used for a high-resolution imaging analysis of actin myofibril dynamics in the beating heart of zebrafish embryos and for studying cardiomyopathy from drug exposure and genetic mutations (Reischauer et al., 2014; Fukuda et al., 2017). The potential adverse effects of Lifeact-GFP overexpression on the heart have not been investigated. In this study, we generated Tg(myl7:Lifeact-GFP) transgenic zebrafish models to investigate the effect of Lifeact-GFP expression on heart development and the use of Lifeact-GFP to study cardiomyopathy. We showed that transgenic zebrafish with low to moderate levels of Lifeact-GFP expression could serve as a useful model to study contractile dynamics of actin filaments in cardiac muscles in vivo. Using this model, we found that loss of smyd1b function resulted in severe actin filament disassembly in cardiac myocytes leading to pericardial edema in zebrafish embryos. Moreover, our studies also demonstrated that strong cardiac expression of Lifeact-GFP was detrimental to actin filament organization in cardiomyocytes of zebrafish embryos resulting in pericardial edema and early embryonic lethality. Collectively, these data demonstrate that the use of Lifeact requires a more elaborate evaluation and optimization to avoid Lifeact-GFP induced artifacts.
This study was carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. To ease pain and facilitate animal handling, fish embryos over one day old were anaesthetized in 0.6 mM Tricaine (pH 7.0) before fixation in 4% paraformaldehyde (PFA) for whole mount observation and immunostaining.
All adult zebrafish were kept at the zebrafish facility at the Institute of Marine and Environmental Technology, University of Maryland. Zebrafish larvae and adult were maintained at 28.5°C in a recirculating aquatic system at a photoperiod of 14-h light and 10 h dark cycle. The smyd1bsa15678 mutant was obtained from ZIRC (Busch-Nentwich et al., 2013). The Tg(myl7:Lifeact-GFP) and Tg(acta1b:Lifeact-GFP) constructs used to generate the transgenic lines and transient expression assay were gifts from Dr. Didier Stainier (Reischauer et al., 2014). The pTol2-α-actin-EGFP plasmid was constructed in Tol2 vector using α-actin promoter to drive EGFP expression (Li et al., 2020).
DNA constructs of Tg(myl7:Lifeact-GFP), Tg(acta1b:Lifeact-GFP) and pTol2-α-actin-EGFP were dissolved in sterile water to a final concentration of 50 or 100 ng/μl. For transient expression analysis, approximately 1–2 nl of DNA construct (50–100 pg) was injected into each embryo at one- or two-cell stage. For the generation of transgenic zebrafish lines, the DNA constructs were mixed with Tol2 transposase mRNA (50 ng/μl). Approximately 1–2 nl of mixed DNA construct/Tol2 transposase mRNA was injected into each embryo at one- or two-cell stage. Transgenic zebrafish founders (P1) were screened by examining GFP expression in their F1 embryos at 24 hpf under a fluorescence microscope (Axioplan 2, Zeiss).
Immunostaining of zebrafish embryonic hearts was carried out at 3 and 14 days-post-fertilization (dpf) as described previously (Yang and Xu, 2012). In brief, the embryos were fixed with 4% formaldehyde. The hearts were dissected with forceps, and then stained with α-actinin (clone EA-53, A7811; MilliporeSigma) primary antibody, and visualized using goat anti-mouse Alexa Fluor-555 conjugated secondary antibody. To increase penetration, hearts from 2-weeks old larvae was treated with 0.1% CHAPS overnight before adding primary antibodies. Phalloidin staining was carried out on the dissected hearts by incubation with 20 ng/ml phalloidin-TRITC (P1951; MilliporeSigma) for 1 h at room temperature in the dark. The images were photographed using a confocal microscope (SP8; Leica Microsystems, Buffalo Grove, IL, United States).
Inverse PCR was performed as described (Kawakami et al., 2004; Kawakami, 2007) with some modifications. Genomic DNA was isolated from 30 transgenic embryos at 5 dpf using the DNeasy Blood & Tissue Kits (Qiagen). A 1 μg of genomic DNA was digested with Alul or HaeIII, and then circularized by ligation. The ligated circular DNA was purified and used for two rounds of nested PCR to isolate the sequence at the 5′ junction of the transgene. The first round of PCR was carried out using Tol2-5′/f1 (5′-AAGTACTTTTTACTCCTTACAA-3′) and Tol2-5′/r1 5′-TGATTTTTAATTGTACTCAAGT-3′) primers. The second round was performed using Tol2-5′/f2 (5′- TTACAGTCAAAAAGTACTTA-3′) and Tol2-5′/r2 (5′-CAAGTAAAGTAAAAATCCC-3′) primers. The PCR products were purified and cloned into pGEM-T easy for sequencing.
The standard control-MO and the Lifeact-GFP-MO (GATCAAATCTGCGACACCCATCCCC) targeted to the ATG start site of Lifeact were purchased from Gene Tools (Corvallis, OR, United States). The morpholino antisense oligos were dissolved in 1 × Danieau buffer (Nasevicius and Ekker, 2000) to a final concentration of 0.5 mM. Zebrafish embryos at the 1 or 2 cell stage were injected with approximately 2 nl (10 ng) of the MOs.
Total RNA was isolated from transgenic larvae at 5 days post-fertilization (dpf) using Trizol (Invitrogen, United States). Three biological replicates were carried out for each line. The RNA samples were treated with RNase-free DNase (Promega, United States). For each sample, 0.5 μg of RNA was used for reverse transcription using Maxima First Strand cDNA Synthesis Kit (Thermo Fisher, United States). qPCR was performed on the Applied Biosystems QuantStudio 3 (Thermo Fisher, United States) using the PowerUpTM SYBRTM Green Master Mix (Thermo Fisher, United States). The GFP and elongation factor 1α (EF1α) specific primers are: GFP-qpcr-F (5-CAAGCAGAAGAACGGCATCAAGGTG-3), GFP-qpcr-R (5-G GACTGGGTGCTCAGGTAGTGGTTG-3), EF1α-P3 (5′ – CTT CAACGCTCAGGTCATCAT – 3′) and EF1α-P4 (5′ – ACAGC AAAGCGACCAAGAGGA – 3′). The PCR was performed with the following parameters including 2 min initial activation at 50°C and followed by 40 cycles of PCR reaction. Each cycle includes 15 s denaturation at 95°C, 10 s annealing at 64°C, and 20 s elongation at 72°C. For each qPCR experiment, samples were run with three technical replicates. The relative amount of a given mRNA was calculated by using the formulae 2–ΔΔCt with EF1α as reference.
GFP fluorescent images were collected under a fluorescent microscopy (Leica Microsystems) with the same parameters. For each transgenic line, three embryos were randomly chosen for imaging at 24 h post-fertilization (hpf). ImageJ was used to manually measure GFP fluorescent intensity for each heart tube.
Data are presented as the mean ± sd. Statistical differences were analyzed by using a student’s t test. P value < 0.05 was set as the threshold for statistical significance.
Previous studies demonstrated that Smyd1 lysine methyltransferase plays a vital role in heart development and myofibril assembly in cardiac muscles (Gottlieb et al., 2002; Tan et al., 2006; Just et al., 2011; Li et al., 2013; Jiao et al., 2021). Genetic mutations in human SMYD1 are associated with congenital and hypertrophic cardiomyopathies in human (Coyan et al., 2019; Fan et al., 2019). To assess the application of Lifeact-GFP reporter in analysis of actin filament assembly in smyd1bsa15678 mutant with heart defects, we generated transgenic zebrafish expressing Lifeact-GFP specifically in cardiac myocytes using the Tg(myl7:Lifeact-GFP) transgene (Reischauer et al., 2014). Six transgenic lines were generated from eight transgenic founders (Figures 1A–D). Consistent with the previous report (Reischauer et al., 2014), Lifeact-GFP clearly revealed F-actin myofibril structures in cardiac myocytes of Tg(myl7:Lifeact-GFP)mb21 zebrafish embryos (Figure 1E). Co-staining with phalloidin-TRITC revealed that Lifeact-GFP co-localized with Phalloidin staining at the F-actin thin filaments of cardiomyocytes (Figures 1F,G).
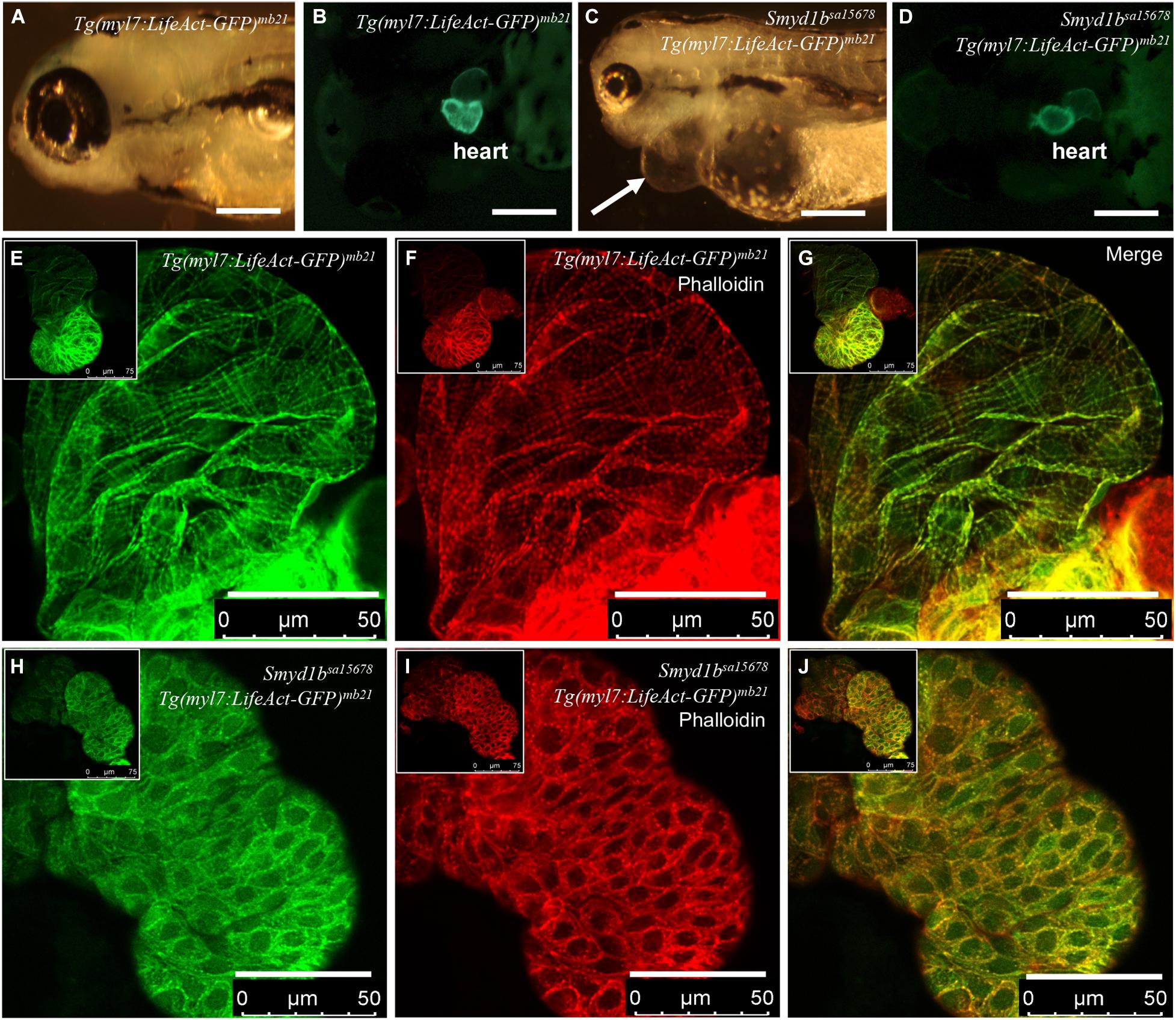
Figure 1. Analysis of congenital cardiomyopathy in smyd1bsa15678 mutant using Tg(myl7:Lifeact-GFP) transgenic zebrafish model. (A–D) Morphology and Lifeact-GFP expression in the heart region of WT Tg(myl7:Lifeact-GFP)mb21 (A,B), and smyd1bsa15678; Tg(myl7:Lifeact-GFP)mb21 mutant (C,D) transgenic larvae at 72 hpf. Pericardial edema is indicated by arrow in smyd1bsa15678; Tg(myl7:Lifeact-GFP)mb21 transgenic mutant larvae (C). Scale bars: 250 μm. (E–J) Actin think filaments in cardiomyocytes revealed by Lifeact-GFP (E,H) and phalloidin staining (F,I) in WT Tg(myl7:Lifeact-GFP)mb21 (E–G), and smyd1bsa15678; Tg(myl7:Lifeact-GFP)mb21 transgenic mutant (H–J) embryos at 72 hpf. (G,J) are merged pictures of (E,F,H,I), respectively. Scale bars: 50 μm.
The Tg(myl7:Lifeact-GFP)mb21 transgenic line that showed normal development was selected to cross with smyd1bsa15678/+ mutant to generate smyd1bsa15678/+; Tg(myl7:Lifeact-GFP)mb21 heterozygous transgenic mutants. The smyd1bsa15678/+; Tg(myl7:Lifeact-GFP)mb21 heterozygous transgenic mutants were subsequently in-crossed to generate smyd1bsa15678; Tg(myl7:Lifeact-GFP)mb21 homozygous transgenic mutant embryos (Figure 1C,D). Lifeact-GFP expression was detected in cardiac muscle of wildtype (WT) and smyd1bsa15678 mutant transgenic embryos (Figures 1B,D). However, in contrast to WT Tg(myl7:Lifeact-GFP)mb21 transgenic embryos (Figures 1E–G), Lifeact-GFP and phalloidin staining showed little or no F-actin myofibrils in cardiomyocytes of smyd1bsa15678 homozygous mutant embryos (Figures 1H–J). Lifeact-GFP and phalloidin-TRITC staining appeared as punctate in cardiomyocytes of smyd1bsa15678; Tg(myl7:Lifeact-GFP)mb21 mutant transgenic fish embryos (Figures 1H–J). The homozygous transgenic mutant embryos died around 5 dpf. Together, these data indicate that Smyd1b is required for F-actin myofibril organization in cardiomyocyte, and Tg(myl7:Lifeact-GFP)mb21 transgenic zebrafish are a useful model to analyze F-actin myofibril defects in cardiomyopathy.
Tg(myl7:Lifeact-GFP) transgenic embryos from six out of the eight transgenic founders showed normal development (Figures 2A,B). They were able to grow into viable fertile adults. However, two transgenic founders, Tg(myl7:Lifeact-GFP)mb22 and Tg(myl7:Lifeact-GFP)mb23, gave F1 transgenic embryos with pericardial edema and died at early larval stages (Figures 2C,D). All transgenic embryos (n = 23) from the Tg(myl7:Lifeact-GFP)mb22 founder showed severe edema phenotype and were 100% embryonic lethal around 6 dpf. The non-transgenic sibling from the same Tg(myl7:Lifeact-GFP)mb22 transgenic founder were normal. In contrast, transgenic embryos from the Tg(myl7:Lifeact-GFP)mb23 founder showed normal cardiac morphology at early stage. However, they developed an edema phenotype around 14 dpf (Figure 2D). Visible blood clots began to form in the yolk near the swimming bladder (Figure 2D). All Tg(myl7:Lifeact-GFP)mb23 transgenic larvae died around 25–30 dpf. Together, these data suggest that expression of Lifeact-GFP might have an adverse effect on heart development.
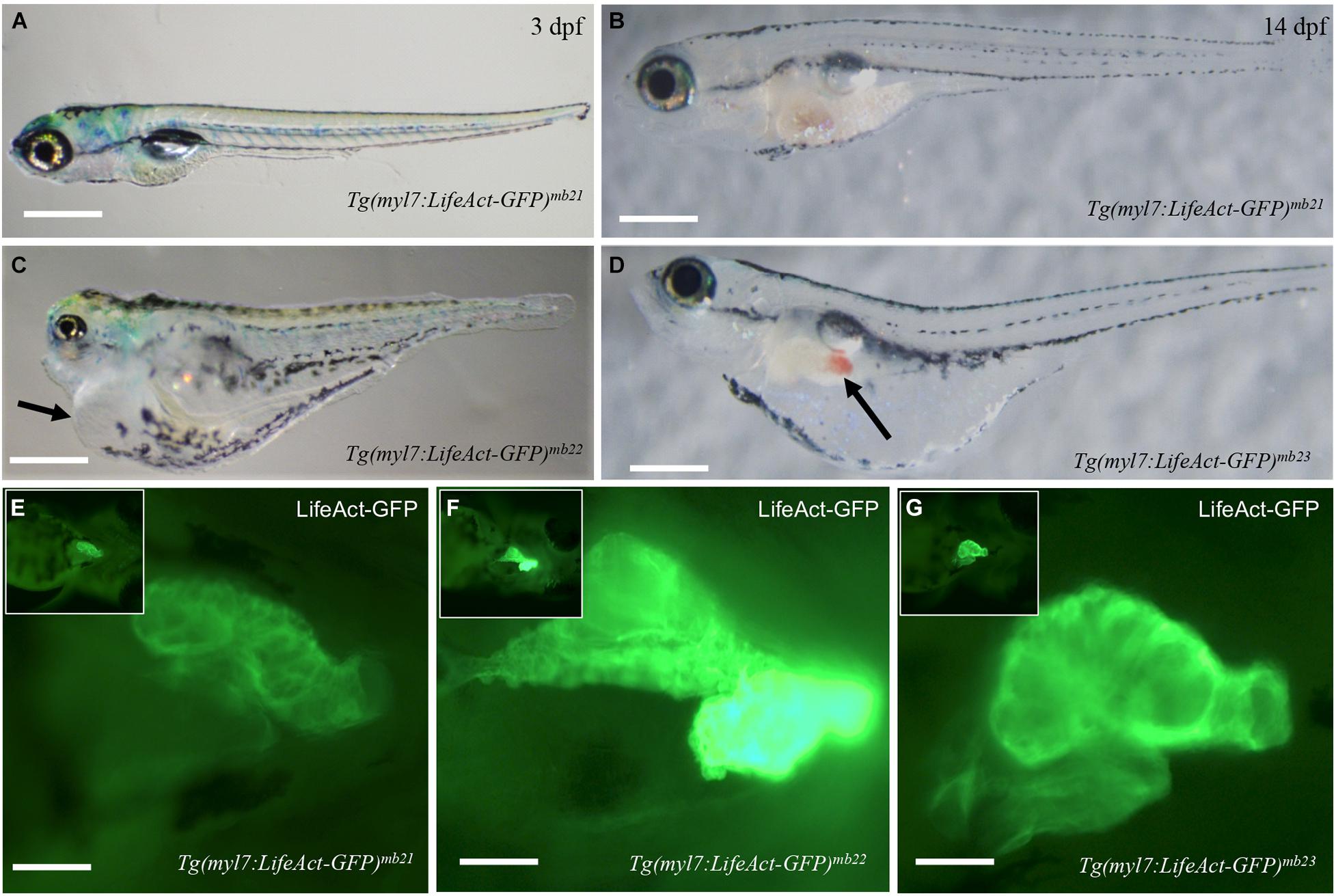
Figure 2. Development of pericardial edema in Tg(myl7:Lifeact-GFP)mb22 and Tg(myl7:Lifeact-GFP)mb23 transgenic embryos. (A–D) Morphology of representative transgenic embryos from Tg(myl7:Lifeact-GFP)mb21 line with no edema (A,B), and Tg(myl7:Lifeact-GFP)mb22 (C), and Tg(myl7:Lifeact-GFP)mb23 (D) transgenic lines with edema phenotypes at 3 (C) and 14 dpf (D), respectively. Pericardial edema is indicated by arrows in Tg(myl7:Lifeact-GFP)mb22 (C), and Tg(myl7:Lifeact-GFP)mb23 (D) transgenic larvae. Scale bars: 500 μm. (E–G) Lifeact-GFP expression in the hearts of Tg(myl7:Lifeact-GFP)mb21 (E), Tg(myl7:Lifeact-GFP)mb22 (F), and Tg(myl7:Lifeact-GFP)mb23 (G) transgenic embryos at 3 dpf. Scale bars: 100 μm.
To determine whether the pericardial edema phenotype correlated with higher levels of Lifeact-GFP expression, we compared the fluorescent intensity of Lifeact-GFP expression in Tg(myl7:Lifeact-GFP)mb22 and Tg(myl7:Lifeact-GFP)mb23 transgenic embryos that showed edema phenotypes (Figures 2C,D,F,G) with transgenic embryos from the Tg(myl7:Lifeact-GFP)mb21 line that showed no edema phenotype (Figures 2A,B,E). The data revealed a clear correlation between the severity of the edema phenotype and the increased levels of Lifeact-GFP expression in the heart. Tg(myl7:Lifeact-GFP)mb21 transgenic embryos with no edema phenotype had a lower Lifeact-GFP fluorescent intensity in the heart (Figure 2E). In contrast, Tg(myl7:Lifeact-GFP)mb22 transgenic embryos with a large pericardial edema showed a strong Lifeact-GFP expression (Figure 2F). Tg(myl7:Lifeact-GFP)mb22 transgenic embryos that displayed the edema phenotype at a later stage showed moderate levels of Lifeact-GFP fluorescent intensity (Figure 2G). To confirm that the Tg(myl7:Lifeact-GFP)mb22 transgenic line had higher levels of Lifeact-GFP expression, we compared the GFP fluorescence intensity and quantified the levels of GFP mRNA expression in cardiac muscles of three transgenic lines that showed the sever, moderate and no heart defects (Supplementary Figure 1). The data confirmed a strong correlation between the levels of Lifeact-GFP expression and the cardiac muscle defects (Supplementary Figures 1D,E). Collectively, these data indicate that high levels of Lifeact-GFP expression in cardiac myocytes could impede heart development and cardiac function, leading to pericardial edema and early lethality.
To further characterize the effect of Lifeact-GFP overexpression on cardiac muscle differentiation, we compared F-actin thin filament organization in cardiomyocytes of Tg(myl7:Lifeact-GFP)mb21 and Tg(myl7:Lifeact-GFP)mb22 transgenic embryos by direct observation of Lifeact-GFP localization and phalloidin staining. F-actin thin filaments were clearly detected in cardiomyocytes of Tg(myl7:Lifeact-GFP)mb21 transgenic embryos that showed no edema phenotype (Figure 3A). Staining with phalloidin revealed a co-localization with Lifeact-GFP in the F-actin thin filaments of cardiomyocytes (Figures 3B,C). In contrast, Tg(myl7:Lifeact-GFP)mb22 transgenic embryos displayed a significant disruption of F-actin thin filaments in sarcomeres of cardiomyocytes (Figure 3D). Very few sarcomeres were detected in cardiomyocytes of Tg(myl7:Lifeact-GFP)mb22 transgenic embryos by Lifeact-GFP (Figure 3D) or phalloidin staining (Figure 3E,F). To better quantify the myofibril defect, we compared the number of sarcomeres in a defined area of cardiac muscles in Tg(myl7:Lifeact-GFP)mb21 and Tg(myl7:Lifeact-GFP)mb22 transgenic embryos. The data showed that the number of sarcomeres were dramatically reduced in cardiomyocytes of the Tg(myl7:Lifeact-GFP)mb22 transgenic larvae compared with the Tg(myl7:Lifeact-GFP)mb21 transgenic line (Supplementary Figure 2C). Together, these data demonstrate that Lifeact-GFP overexpression in Tg(myl7:Lifeact-GFP)mb22 disrupted F-actin filament organization in cardiomyocytes of zebrafish embryos.
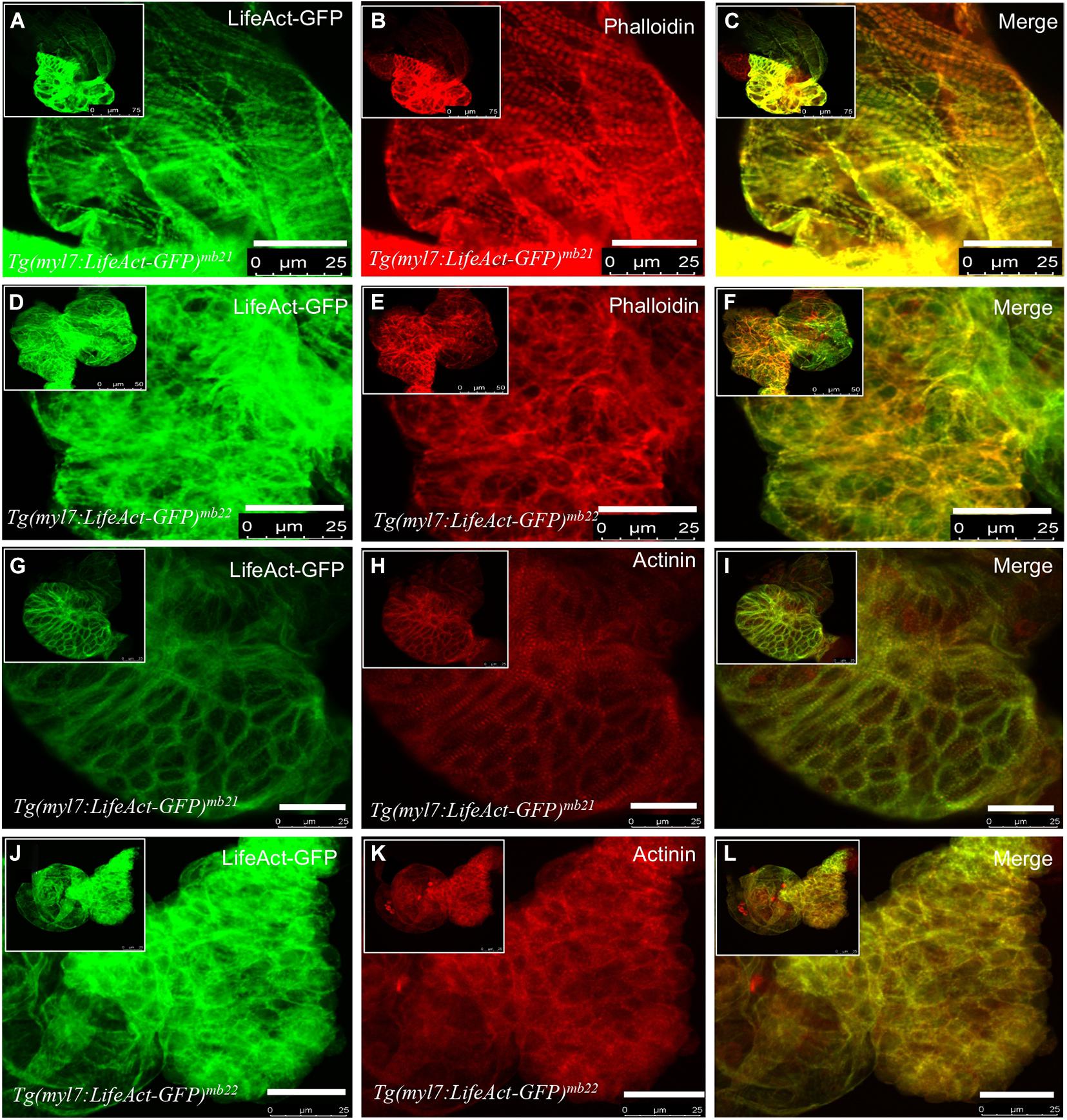
Figure 3. The effect of Lifeact-GFP expression on actin filament and sarcomere organization in cardiomyocytes of Tg(myl7:Lifeact-GFP)mb21 and Tg(myl7:Lifeact-GFP)mb22 transgenic embryos. (A–F) Actin think filaments revealed by Lifeact-GFP (A,D) and phalloidin staining (B,E) in Tg(myl7:Lifeact-GFP)mb21 (A–C), and Tg(myl7:Lifeact-GFP)mb22 (D–F) transgenic embryos at 72 hpf. (C,F) are merged pictures of A-B and D-E, respectively. (G–L) The sarcomeric organization of actin think filament and Z-lines revealed by Lifeact-GFP (G,J) anti-α-actinin antibody staining (H,K) in Tg(myl7:Lifeact-GFP)mb21 (G–I), and Tg(myl7:Lifeact-GFP)mb22 (J–L) transgenic embryos at 72 hpf. (I,L) are merged pictures of (G,H,J,K), respectively. Scale bars: 25 μm.
F-actin thin filaments are key structures in sarcomeres of skeletal and cardiac muscles. Actin thin filaments are anchored at the Z-discs by a protein called α-actinin. To test whether disruption of actin thin filaments by Lifeact-GFP overexpression could affect the Z-line organization in the sarcomere, we next examined the Z-discs integrity using anti-α-actinin antibody staining (Figures 3G–L). The data showed that cardiomyocytes in the normal Tg(myl7:Lifeact-GFP)mb21 transgenic line displayed striated Z-line structures (Figure 3H). However, the striated Z-lines were absent in cardiomyocytes of Tg(myl7:Lifeact-GFP)mb22 transgenic embryos with the edema phenotype (Figure 3K). Cardiac myocytes in Tg(myl7:Lifeact-GFP)mb22 embryos exhibited irregular F-actin structure and poor Z-line organization. Lifeact-GFP and α-actinin staining appeared as dispersed punctate within the cardiomyocytes (Figures 3J–L). Collectively, these data indicate that disruption of F-actin myofibril assembly by Lifeact-GFP overexpression could impede other sarcomere structures in cardiac myocytes.
It has been reported that overexpression of GFP could also induce cytotoxicity and apoptosis (Liu et al., 1999; Ansari et al., 2016). To test whether the defective actin filament organization could be caused by GFP overexpression, we compared the effects of GFP and Lifeact-GFP overexpression on actin filament and sarcomere organization in skeletal muscle fibers of zebrafish embryos. Zebrafish embryonic myofibers with their easy characterization provide a good model for analyzing protein cytotoxicity in vivo. DNA constructs expressing the Lifeact-GFP or GFP were microinjected into fertilized zebrafish embryos at 1–2 cell stages. Muscle specific expression of GFP or Lifeact-GFP was clearly detected in muscle fibers of injected embryos. Actin filament organization was characterized by phalloidin staining in the skeletal myofibers. The data showed that approximately 5% of the Lifeact-GFP expressing myofibers showed abnormal actin filament organization. In some of the injected embryos, almost half of the Lifeact-GFP expressing fibers showed actin filament defect (Supplementary Figures 3D–F). In contrast, myofibers expressing GFP appeared normal (Supplementary Figures 3A–C). These data indicate that the actin filament defect was likely caused by Lifeact rather than GFP.
Integration of Tg(myl7:Lifeact-GFP) transgenes into zebrafish genome could disrupt expression and function of genes at the integration sites (Clark et al., 2004; Kawakami et al., 2004; Kawakami, 2007). To assess this potential possibility in Tg(myl7:Lifeact-GFP)mb22 and Tg(myl7:Lifeact-GFP)mb23 transgenic lines that displayed the edema phenotype, we mapped the transgene integration sites by inverse PCR. The data revealed that in the Tg(myl7:Lifeact-GFP)mb22 transgenic line, the transgene was inserted into the third intron of the cadherin2 gene (Figure 4A), which encodes a transmembrane protein named N-cadherin that has been implicated in zebrafish cardiovascular development (Bagatto et al., 2006). In the Tg(myl7:Lifeact-GFP)mb23 transgenic line, the transgene was integrated into the last exon of traf4a (Figure 4B), which encodes tumor necrosis factor (TNF)-receptor–associated factor-4 (TRAF4). Previous studies showed that Cadherin2 or traf4a heterozygous mutants showed normal development and could survive to adult stage without any cardiac abnormality (Brusés, 2011; Hehr et al., 2018), indicating that the cardiac muscle defects observed in Tg(myl7:Lifeact-GFP)mb22 and Tg(myl7:Lifeact-GFP)mb23 hemizygous transgenic lines were unlikely caused by the transgene integration in these genes.
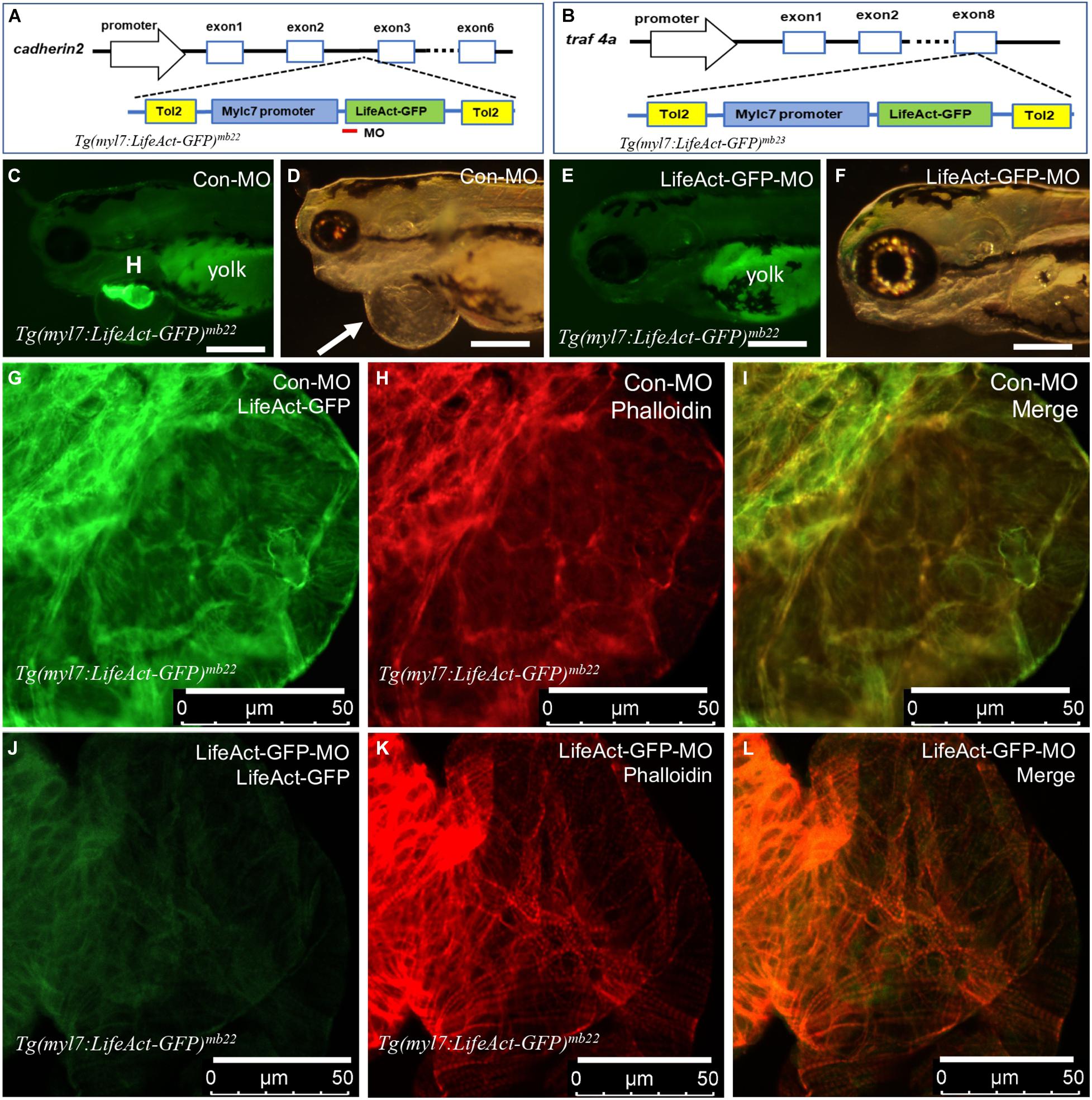
Figure 4. Identification of transgene integration sites in Tg(myl7:Lifeact-GFP)mb22 and Tg(myl7:Lifeact-GFP)mb23 transgenic embryos and the rescue of heart defect by knockdown of Lifeact-GFP expression. (A,B) Mapping the transgene integration sites in Tg(myl7:Lifeact-GFP)mb22 and Tg(myl7:Lifeact-GFP)mb23 transgenic embryos at cadherin2 and traf4a genes, respectively. The Lifeact-GFP-MO is indicated by the red line. (C–F) Morphology of heart region of Tg(myl7:Lifeact-GFP)mb22 transgenic embryos at 72 hpf injected with control-MO (C,D) and Lifeact-GFP-MO (E,F), respectively. Pericardial edema is indicated by the arrow in control-MO injected Tg(myl7:Lifeact-GFP)mb22 (D) transgenic mutant larvae. H, heart; Yolk shows autofluorescence. Scale bars: 250 μm. (G–L) Lifeact-GFP expression (G,J) and phalloidin staining (H,K) showing actin think filaments in cardiomyocytes of Tg(myl7:Lifeact-GFP)mb22 transgenic embryos injected with control-MO (G–I) or Lifeact-GFP-MO (J–L) at 72 hpf transgenic embryos. (I,L) are merged pictures of (G,H,J,K), respectively. Scale bars: 50 μm.
To confirm the heart defect in Tg(myl7:Lifeact-GFP)mb22 transgenic zebrafish embryos was indeed caused by Lifeact-GFP expression, we decided to knock down Lifeact-GFP expression in the transgenic embryos and assess the effect on heart development. A Lifeact-GFP specific antisense morpholino (Lifeact-GFP-MO) was designed that targets the translational start site of Lifeact-GFP (Figure 4A). The MO was microinjected into fertilized eggs of Tg(myl7:Lifeact-GFP)mb22 transgenic fish. Compared with the control MO (Con-MO) injected embryos that showed normal Lifeact-GFP expression and a strong edema phenotype (Figures 4C,D), Lifeact-GFP-MO injection dramatically reduced the Lifeact-GFP expression (Figure 4E). While the control-MO injected embryos had a pronounced edema, strikingly, knockdown of Lifeact-GFP expression completely rescued the pericardiac edema phenotype in the Lifeact-GFP-MO injected transgenic embryos (Figure 4F), suggesting that edema phenotype was indeed caused by Lifeact-GFP overexpression.
To better characterize the heart phenotype and rescue, we analyzed F-actin thin filaments in cardiomyocytes of MO injected embryos by Lifeact-GFP localization and phalloidin staining. As shown in Figure 4G, a heart from control-MO injected embryos showed strong Lifeact-GFP expression. However, no F-actin myofibrils could be detected in cardiomyocytes of the control-MO injected embryos (Figures 4H,I). In contrast, in Lifeact-GFP-MO injected embryos, Lifeact-GFP expression was dramatically reduced (Figure 4J). Phalloidin staining revealed highly organized thin filaments in cardiomyocytes of the Lifeact-GFP knockdown embryos (Figures 4K,L). To quantify the myofibril recovery, we compared the number of sarcomeres in cardiomyocytes of Tg(myl7:Lifeact-GFP)mb22 transgenic embryos injected with Con-MO and Lifeact-GFP-MO. Compared with the Con-MO injected embryos, the number of sarcomeres was dramatically increased in cardiomyocytes of Lifeact-GFP-MO injected embryos (Supplementary Figure 4). Collectively, these data indicate that Lifeact-GFP overexpression could disrupt F-actin filament organization in cardiomyocytes of zebrafish embryos, and thus the use of Lifeact-GFP requires caution and evaluation of the transgenic model to avoid artifacts.
In this study, we generated Tg(myl7:Lifeact-GFP) transgenic zebrafish models to study cardiomyopathy in smyd1b mutants and to investigate the effect of Lifeact-GFP overexpression on heart development. We showed that Lifeact-GFP transgenic zebrafish is a useful model for studying contractile dynamics of F-actin filaments in cardiomyocytes in vivo. Using this model, we found that loss of smyd1b resulted in poor actin filament organization in cardiac myocytes. Moreover, our studies also demonstrated that strong Lifeact-GFP expression was detrimental to F-actin filament organization in cardiomyocytes, causing pericardial edema and early embryonic lethality of the transgenic embryos. Collectively, these data indicate that although Lifeact-GFP is a powerful probe for studying actin dynamic in cardiomyocytes in vivo, Lifeact-GFP could alter actin filament arrangement and dynamics in cardiomyocytes. Lifeact-GFP mediated artifacts are concentration-dependent and thus the use of Lifeact requires a closer examination and optimization to minimize the Lifeact-GFP induced adverse effects.
Using Lifeact-GFP, we found that smyd1b is required for actin filament assembly in cardiac myocytes of zebrafish embryos. This finding is consistent with data from previous studies using phalloidin and immunostaining that showed that Smyd1 is required for sarcomere assembly in skeletal and cardiac muscles (Tan et al., 2006; Just et al., 2011; Li et al., 2013; Prill et al., 2015; Jiao et al., 2021). The molecular mechanism of Smyd1 function in sarcomere assembly is not well known. Actin thin filament is a key component of the sarcomere. Thin filaments work together with thick filaments for generation and propagation of mechanical force. It has been reported that Smyd1 is a myosin heavy chain binding protein (Just et al., 2011). Biochemical analysis revealed that myosin heavy chain (MHC) and several other muscle proteins are methylated at lysine residues (Huszar and Elzinga, 1969; Hardy et al., 1970; Huszar, 1972; Tong and Elzinga, 1983; Iwabata et al., 2005). Given that Smyd1b is a lysine methyltransferase, Smyd1 may directly methylate MHC and possibly other muscle proteins critical to protein folding and stability. However, the methylation targets of Smyd1 have yet to be identified. Our previous studies have demonstrated that Smyd1 is required for MHC protein expression and stability in skeletal and cardiac muscle cells (Li et al., 2013; Jiao et al., 2021), and that loss of myosin heavy chain abolished sarcomere assembly, including actin thin filaments in zebrafish embryonic skeletal muscles (Xu et al., 2012; Li et al., 2020). We cannot rule out the possibility that the defective F-actin assembly in cardiomyocytes of smyd1b mutant mutants was a secondary defect from loss of MHC in smyd1b mutants.
Smyd1 function in cardiac muscles is conserved during evolution from fish to human. Recent studies demonstrated that genetic mutations in human SMYD1 are associated with dilated cardiomyopathy and hypertrophic cardiomyopathy (Coyan et al., 2019; Fan et al., 2019). An infant patient carrying a SMYD1 mutation suffered from dilated cardiomyopathy and needed heart transplantation at the infant stage (Coyan et al., 2019). A peripheral muscle biopsy showed disorganized muscle fibers with minimal mitochondria (Coyan et al., 2019). The sarcomere and mitochondria defects resemble the heart defects from zebrafish and mouse Smyd1 mutant models (Tan et al., 2006; Just et al., 2011; Rasmussen et al., 2015; Warren et al., 2018). In mice, knockout of the Smyd1 gene results in death of the fetus or embryos and various failures in the development of the cardiac and vascular system (Gottlieb et al., 2002; Rasmussen et al., 2015; Nagandla et al., 2016; Stewart et al., 2016). In zebrafish, loss of smyd1 causes edema and no muscle contraction (Just et al., 2011; Cai et al., 2019; Jiao et al., 2021). Tg(myl7:Lifeact-GFP) transgenic zebrafish could be a useful model for future studies of cardiomyopathy from Smyd1 deficiency.
Data from this study showed that overexpression of Lifeact-GFP disrupted actin filament organization in cardiac myocytes, leading to pericardial edema and early larval lethality. This adverse effect was not observed in a previous study using the same Tg(myl7:Lifeact-GFP) transgene (Reischauer et al., 2014). Reischauer and colleagues showed that expression of Lifeact-GFP in zebrafish heart did not interfere with cardiac muscle development. We speculate that the difference might be due to varied levels of Lifeact-GFP expression in different transgenic lines. Indeed, we observed in the same study here that six of the eight Tg(myl7:Lifeact-GFP) transgenic founders could produce healthy and viable transgenic offspring. All these six transgenic lines had lower levels of Lifeact-GFP expression. The other two lines with higher levels of Lifeact-GFP expression developed edema and were early embryonic lethal. The varied levels of gene expression were likely due to the positional effect of transgene integration. We have mapped the integration sites in Tg(myl7:Lifeact-GFP)mb22 and Tg(myl7:Lifeact-GFP)mb23 to the cadherin2 and traf4a genes, respectively. We do not think the heart phenotype in these lines was caused by disruption of these host genes at the integration sites because the heterozygous mutant of cadherin2 or traf4a in zebrafish showed no edema phenotypes (Brusés, 2011; Hehr et al., 2018), and moreover, the edema phenotype in Tg(myl7:Lifeact-GFP)mb22 could be rescued by knockdown of Lifeact-GFP expression.
It has been reported that higher levels of Lifeact-GFP expression could influence the assembly of actin skeleton in cell culture, yeast, Drosophila, and plant cells (Van Der Honing et al., 2011; Spracklen et al., 2014; Courtemanche et al., 2016). Strong expression of Lifeact-GFP in Drosophila disrupted F-actin remodeling, and resulted in female fertility defects (Spracklen et al., 2014). In fission yeast, it was shown that Lifeact altered actin assembly during endocytosis and cytokinesis (Courtemanche et al., 2016). Recent studies showed that Lifeact also induced dose-response effects on F-actin assembly in human Mesenchymal Stem Cells (Flores et al., 2019), and transient transfection of Lifeact-GFP was toxic to C2C12 myoblast cells (Yu, 2013). Most of these previous studies were primarily focused on Lifeact-GFP induced artifacts in cytoskeleton structures of non-muscle cells. Data from this study demonstrate that Lifeact can induce defective actin myofibril organization in the sarcomeres of skeletal and cardiac muscle cells.
The molecular structure of Lifeact and its basis for inducing actin filament artifacts have been recently investigated (Belyy et al., 2020; Kumari et al., 2020). Structural analysis of the Lifeact–F-actin complex showed that Lifeact interacts with a hydrophobic binding pocket on F-actin (Belyy et al., 2020; Kumari et al., 2020). This hydrophobic binding pocket is required for myosin binding. Lifeact could compete with myosin for binding with actin, thus providing a mechanistic explanation for the adverse effects of Lifeact on cell morphology and muscle cell differentiation in vivo. However, previous studies have shown that excessive Lifeact stabilizes actin filaments and bundles even in the absence of Myosins (Courtemanche et al., 2016). Structural analysis revealed that Lifeact interacts with a hydrophobic binding pocket on F-actin and stretches over 2 adjacent actin subunits, stabilizing the DNase I-binding loop (D-loop) of actin in the closed conformation (Belyy et al., 2020; Kumari et al., 2020). D-loop plasticity has been implicated in filament formation and stability (Durer et al., 2012; Grintsevich et al., 2017; Das et al., 2020). Thus, an alternative possibility of Lifeact action on actin filament disruption could be contributed by locking D-loop in the closed conformation.
In addition to Lifeact, several other actin probes have been developed using actin binding domains from other proteins or actin nanobody for live imaging. It has been reported that in contrast to Lifeact, F-tractin does not alter actin rearrangement during Drosophila follicle development (Spracklen et al., 2014). However, F-tractin expression could perturb actin cytoskeleton organization in a Xenopus cell line (Belin et al., 2014). Compared with Lifeact, F-tractin is larger in size, it might interfere with actin binding with other regulatory or accessory proteins. Actin-Chromobody represents another novel approach to visualize actin dynamic assembly in living cells. Chromobodies combine the specificity of antibodies with the convenience of live fluorescence imaging, which can be easily expressed by a DNA construct (Panza et al., 2015). It appears that high levels of actin-Chromobody expression does not alter actin dynamics (Schiavon et al., 2020). Chromobodies have been successfully applied to visualize actin in mammalian nuclei (Plessner et al., 2015), in tobacco leaf cells (Rocchetti et al., 2014) and zebrafish (Panza et al., 2015). These alternative actin probes could be evaluated in zebrafish embryonic cardiac and skeletal muscles to determine their cytotoxicity in vivo.
In summary, our studies show that Tg(myl7:Lifeact-GFP) transgenic zebrafish is a useful model to study contractile dynamics of F-actin filaments in cardiomyocytes in vivo and to investigate cardiomyopathy from defective thin filament organization. Data from this study also demonstrated that strong Lifeact-GFP expression in cardiomyocytes could alter actin filament arrangement and dynamics in cardiomyocytes causing heart development defect and embryonic lethality. The Lifeact-GFP induced artifacts are concentration-dependent and thus the use of Lifeact requires careful evaluation of the transgenic model to minimize the adverse effects.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee of University of Maryland Baltimore.
RX and SD conceived and designed the research project and analyzed the data. RX performed the experiments, data collection, and wrote the first draft of the manuscript. SD revised the manuscript. Both authors read and approved the final manuscript.
This research was supported by grant funding from the National Institutes of Health (R01AR072703 to SD). RX was supported by a fellowship from the Chinese Scholarship Council.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Didier Y. R. Stainier from Max Planck Institute for Heart and Lung Research for sharing the Tg(myl7:Lifeact-GFP) and Tg(acta1b:Lifeact-GFP) constructs with us.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.746818/full#supplementary-material
Aizawa, H., Sameshima, M., and Yahara, I. A. (1997). green fluorescent protein-actin fusion protein dominantly inhibits cytokinesis, cell spreading, and locomotion in Dictyostelium. Cell Struct. Funct. 22, 335–345. doi: 10.1247/csf.22.335
Ansari, A. M., Ahmed, A. K., Matsangos, A. E., Lay, F., Born, L. J., Marti, G., et al. (2016). Cellular GFP Toxicity and Immunogenicity: Potential Confounders in in Vivo Cell Tracking Experiments. Stem. Cell Rev. Rep. 12, 553–559. doi: 10.1007/s12015-016-9670-8
Bagatto, B., Francl, J., Liu, B., and Liu, Q. (2006). Cadherin2 (N-cadherin) plays an essential role in zebrafish cardiovascular development. BMC Dev. Biol. 6:23. doi: 10.1186/1471-213X-6-23
Belin, B. J., Goins, L. M., and Mullins, R. D. (2014). Comparative analysis of tools for live cell imaging of actin network architecture. Bioarchitecture 4, 189–202. doi: 10.1080/19490992.2014.1047714
Belyy, A., Merino, F., Sitsel, O., and Raunser, S. (2020). Structure of the Lifeact-F-actin complex. PLoS Biol. 18:e3000925. doi: 10.1371/journal.pbio.3000925
Bretscher, A. (1981). Fimbrin is a cytoskeletal protein that crosslinks F-actin in vitro. Proc. Natl. Acad. Sci. U.S.A. 78, 6849–6853.
Brusés, J. L. N. (2011). -cadherin regulates primary motor axon growth and branching during zebrafish embryonic development. J. Comparat. Neurol. 519, 1797–1815.
Bubb, M. R., Senderowicz, A. M., Sausville, E. A., Duncan, K. L., and Korn, E. D. (1994). Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J. Biol. Chem. 269, 14869–14871.
Burkel, B. M., von Dassow, G., and Bement, W. M. (2007). Versatile fluorescent probes for actin filaments based on the actin-binding domain of utrophin. Cell Motil. Cytoskeleton. 64, 822–832. doi: 10.1002/cm.20226
Busch-Nentwich, E., Kettleborough, R., Dooley, C. M., Scahill, C., Sealy, I., White, R., et al. (2013). Sanger Institute Zebrafish Mutation Project mutant data submission. Eugene: ZFIN Direct Data Submission.
Cai, M., Han, L., Liu, L., He, F., Chu, W., Zhang, J., et al. (2019). Defective sarcomere assembly in smyd1a and smyd1b zebrafish mutants. FASEB J. 33, 6209–6225. doi: 10.1096/fj.201801578R
Clark, A. G., Dierkes, K., and Paluch, E. K. (2013). Monitoring actin cortex thickness in live cells. Biophys. J. 105, 570–580. doi: 10.1016/j.bpj.2013.05.057
Clark, K. J., Geurts, A. M., Bell, J. B., and Hackett, P. B. (2004). Transposon vectors for gene-trap insertional mutagenesis in vertebrates. Genesis 39, 225–233. doi: 10.1002/gene.20049
Cooper, J. A. (1987). Effects of cytochalasin and phalloidin on actin. J. Cell Biol. 105, 1473–1478.
Costa, C. F., Rommelaere, H., Waterschoot, D., Sethi, K. K., Nowak, K. J., Laing, N. G., et al. (2004). Myopathy mutations in alpha-skeletal-muscle actin cause a range of molecular defects. J. Cell Sci. 117(Pt 15), 3367–3377. doi: 10.1242/jcs.01172
Courtemanche, N., Pollard, T. D., and Chen, Q. (2016). Avoiding artifacts when counting polymerized actin in live cells with Lifeact fused to fluorescent proteins. Nat. Cell Biol. 18, 676–683.
Coyan, G. N., Zinn, M. D., West, S. C., and Sharma, M. S. (2019). Heart transplantation from biventricular support in infant with novel smyd1 mutation. Pediatr. Cardiol 40, 1745–1747.
Das, S., Ge, P., Oztug Durer, Z. A., Grintsevich, E. E., Zhou, Z. H., Reisler, E., et al. (2020). D-loop Dynamics and Near-Atomic-Resolution Cryo-EM Structure of Phalloidin-Bound F-Actin. Structure 28, 586.e–593.e. doi: 10.1016/j.str.2020.04.004
Despond, E. A., and Dawson, J. F. (2018). Classifying Cardiac Actin Mutations Associated With Hypertrophic Cardiomyopathy. Front. Physiol. 9:405. doi: 10.3389/fphys.2018.00405
D’Este, E., Kamin, D., Göttfert, F., El-Hady, A., and Hell, S. W. (2015). STED nanoscopy reveals the ubiquity of subcortical cytoskeleton periodicity in living neurons. Cell Rep. 10, 1246–1251. doi: 10.1016/j.celrep.2015.02.007
Dominguez, R., and Holmes, K. C. (2011). Actin structure and function. Annu Rev Biophys 40, 169–186.
Du, J., Fan, Y. L., Chen, T. L., and Feng, X. Q. (2015). Lifeact and Utr230 induce distinct actin assemblies in cell nuclei. Cytoskeleton 72, 570–575. doi: 10.1002/cm.21262
Duleh, S. N., and Welch, M. D. (2012). Regulation of integrin trafficking, cell adhesion, and cell migration by WASH and the Arp2/3 complex. Cytoskeleton 69, 1047–1058. doi: 10.1002/cm.21069
Durer, Z. A., Kudryashov, D. S., Sawaya, M. R., Altenbach, C., Hubbell, W., and Reisler, E. (2012). Structural states and dynamics of the D-loop in actin. Biophys. J. 103, 930–939. doi: 10.1016/j.bpj.2012.07.030
Fan, L. L., Ding, D. B., Huang, H., Chen, Y. Q., Jin, J. Y., Xia, K., et al. (2019). A de novo mutation of SMYD1 (p.F272L) is responsible for hypertrophic cardiomyopathy in a Chinese patient. Clin. Chem. Lab. Med. 57, 532–539. doi: 10.1515/cclm-2018-0578
Fiolka, R., Shao, L., Rego, E. H., Davidson, M. W., and Gustafsson, M. G. (2012). Time-lapse two-color 3D imaging of live cells with doubled resolution using structured illumination. Proc. Natl. Acad. Sci. U S A. 109, 5311–5315. doi: 10.1073/pnas.1119262109
Flores, L. R., Keeling, M. C., Zhang, X., Sliogeryte, K., and Gavara, N. (2019). Lifeact-TagGFP2 alters F-actin organization, cellular morphology and biophysical behaviour[J]. Sci. Rep. 9, 1–13.
Fraccaroli, A., Franco, C. A., Rognoni, E., Neto, F., Rehberg, M., Aszodi, A., et al. (2012). Visualization of endothelial actin cytoskeleton in the mouse retina. PLoS One 7:e47488.
Frustaci, A., De Luca, A., Guida, V., Biagini, T., Mazza, T., Gaudio, C., et al. (2018). Novel α-Actin Gene Mutation p.(Ala21Val) Causing Familial Hypertrophic Cardiomyopathy, Myocardial Noncompaction, and Transmural Crypts. Clinical-Pathologic Correlation. J. Am. Heart Assoc. 7:e008068. doi: 10.1161/JAHA.117.008068
Fukuda, R., Gunawan, F., Beisaw, A., Jimenez-Amilburu, V., Maischein, H. M., Kostin, S., et al. (2017). Proteolysis regulates cardiomyocyte maturation and tissue integration. Nat. Commun. 8:14495.
Gottlieb, P. D., Pierce, S. A., Sims, R. J., Yamagishi, H., Weihe, E. K., Harriss, J. V., et al. (2002). Bop encodes a muscle-restricted protein containing MYND and SET domains and is essential for cardiac differentiation and morphogenesis. Nat. Genet. 31, 25–32.
Grikscheit, K., Frank, T., Wang, Y., and Grosse, R. (2015). Junctional actin assembly is mediated by Formin-like 2 downstream of Rac1. J. Cell Biol. 209, 367–376. doi: 10.1083/jcb.201412015
Grintsevich, E. E., Ge, P., Sawaya, M. R., Yesilyurt, H. M., Terman, J. R., Zhou, H., et al. (2017). Catastrophic disassembly of actin filaments via Mical-mediated oxidation. Nat. Commun. 8:2183. doi: 10.1038/s41467-017-02357-8
Hardy, M., Harris, I., Perry, S. V., and Stone, D. (1970). Epsilon-N-monomethyl-lysine and trimethyl-lysine in myosin. Biochem. J. 117, 44–45.
Hehr, C. L., Halabi, R., and McFarlane, S. (2018). Polarity and morphogenesis of the eye epithelium requires the adhesion junction associated adaptor protein Traf4[J]. Cell Adhes. Migr. 12, 489–502.
Higuchi-Sanabria, R., Paul, J. W., Durieux, J., Benitez, C., Frankino, P. A., Tronnes, S. U., et al. (2018). Spatial regulation of the actin cytoskeleton by HSF-1 during aging. Mole. Biol. Cell 29, 2522–2527.
Holubcová, Z., Howard, G., and Schuh, M. (2013). Vesicles modulate an actin network for asymmetric spindle positioning. Nat. Cell Biol. 15, 937–947. doi: 10.1038/ncb2802
Huelsmann, S., Ylänne, J., and Brown, N. H. (2013). Filopodia-like actin cables position nuclei in association with perinuclear actin in Drosophila nurse cells. Dev. Cell. 26, 604–615. doi: 10.1016/j.devcel.2013.08.014
Huszar, G. (1972). Amino acid sequences around the two -N-trimethyllysine residues in rabbit skeletal muscle myosin. J. Biol. Chem. 247, 4057–4062.
Iwabata, H., Yoshida, M., and Komatsu, Y. (2005). Proteomic analysis of organ-specific post-translational lysine-acetylation and -methylation in mice by use of anti-acetyllysine and -methyllysine mouse monoclonal antibodies. Proteomics 5, 4653–4664.
Jiao, S., Xu, R., and Du, S. (2021). Smyd1 is essential for myosin expression and sarcomere organization in craniofacial, extraocular, and cardiac muscles. J. Genet. Genomics. 48, 208–218. doi: 10.1016/j.jgg.2021.03.004
Just, S., Meder, B., Berger, I. M., Etard, C., Trano, N., Patzel, E., et al. (2011). The myosin-interacting protein SMYD1 is essential for sarcomere organization. J. Cell Sci. 124, 3127–3136.
Kawakami, K., Takeda, H., Kawakami, N., Kobayashi, M., Matsuda, N., and Mishina, M. A. (2004). transposon-mediated gene trap approach identifies developmentally regulated genes in zebrafish. Dev. Cell. 7, 133–144. doi: 10.1016/j.devcel.2004.06.005
Koestler, S. A., Rottner, K., Lai, F., Block, J., Vinzenz, M., and Small, J. V. F. (2009). - and G-actin concentrations in lamellipodia of moving cells. PLoS One. 4:e4810. doi: 10.1371/journal.pone.0004810
Kumari, A., Kesarwani, S., Javoor, M. G., Vinothkumar, K. R., and Sirajuddin, M. (2020). Structural insights into actin filament recognition by commonly used cellular actin markers. EMBO J. 39:e104006. doi: 10.15252/embj.2019104006
Lemieux, M. G., Janzen, D., Hwang, R., Roldan, J., Jarchum, I., and Knecht, D. A. (2014). Visualization of the actin cytoskeleton: different F-actin-binding probes tell different stories. Cytoskeleton 71, 157–169. doi: 10.1002/cm.21160
Li, H. Q., Zhong, Y. W., Wang, Z. F., Gao, J., Xu, J., Chu, W. Y., et al. (2013). Smyd1b is required for skeletal and cardiac muscle function in zebrafish. Mol. Biol. Cell 24, 3511–3521.
Li, S., Wen, H., and Du, S. J. (2020). Defective sarcomere organization and reduced larval locomotion and fish survival in slow muscle heavy chain 1 (smyhc1) mutants. FASEB J. 34, 1378–1397.
Liu, H. S., Jan, M. S., Chou, C. K., Chen, P. H., and Ke, N. J. (1999). Is green fluorescent protein toxic to the living cells? Biochem. Biophys. Res. Commun. 260, 712–717. doi: 10.1006/bbrc.1999.0954
Lukinavičius, G., Reymond, L., D’Este, E., Masharina, A., Göttfert, F., Ta, H., et al. (2014). Fluorogenic probes for live-cell imaging of the cytoskeleton. Nat. Methods 11, 731–733.
Matsudaira, P. (1994). Actin crosslinking proteins at the leading edge. Semin. Cell Biol. 5, 165–174.
Melak, M., Plessner, M., and Grosse, R. (2017). Actin visualization at a glance. J. Cell Sci. 130, 525–530. doi: 10.1242/jcs.189068
Nagandla, H., Lopez, S., Yu, W., Rasmussen, T. L., Tucker, H. O., Schwartz, R. J., et al. (2016). Defective myogenesis in the absence of the muscle-specific lysine methyltransferase SMYD1. Dev. Biol. 410, 86–97.
Nagasaki, A., Kijima, T., Yumoto, T., Imaizumi, M., Yamagishi, A., Kim, H., et al. (2017). The Position of the GFP Tag on Actin Affects the Filament Formation in Mammalian Cells. Cell Struct. Funct. 42, 131–140. doi: 10.1247/csf.17016
Nasevicius, A., and Ekker, S. C. (2000). Effective targeted gene ‘knockdown’ in zebrafish. Nat. Genet. 26, 216–220. doi: 10.1038/79951
Nowak, K. J., Wattanasirichaigoon, D., Goebel, H. H., Wilce, M., Pelin, K., Donner, K., et al. (1999). Mutations in the skeletal muscle alpha-actin gene in patients with actin myopathy and nemaline myopathy. Nat. Genet. 23, 208–212. doi: 10.1038/13837
Panza, P., Maier, J., Schmees, C., Rothbauer, U., and Söllner, C. (2015). Live imaging of endogenous protein dynamics in zebrafish using chromobodies. Development 142, 1879–1884. doi: 10.1242/dev.118943
Phng, L. K., Stanchi, F., and Gerhardt, H. (2013). Filopodia are dispensable for endothelial tip cell guidance. Development 140, 4031–4040. doi: 10.1242/dev.097352
Plessner, M., Melak, M., Chinchilla, P., Baarlink, C., and Grosse, R. (2015). Nuclear F-actin formation and reorganization upon cell spreading. J. Biol. Chem. 290, 11209–11216. doi: 10.1074/jbc.M114.627166
Pollard, T. D., and Cooper, J. A. (2009). Actin, a Central Player in Cell Shape and Movement. Science 326, 1208–1212.
Prill, K., Windsor Reid, P., Wohlgemuth, S. L., and Pilgrim, D. B. (2015). Still Heart Encodes a Structural HMT, SMYD1b, with Chaperone-Like Function during Fast Muscle Sarcomere Assembly. PLoS One 10, e0142528.
Rasmussen, T. L., Ma, Y., Park, C. Y., Harriss, J., Pierce, S. A., Dekker, J. D., et al. (2015). Smyd1 facilitates heart development by antagonizing oxidative and ER stress responses. PLoS One 10, e0121765.
Reischauer, S., Arnaout, R., Ramadass, R., Tainier, D. Y., and Reischauer, S. (2014). Actin binding GFP allows 4D in vivo imaging of myofilament dynamics in the zebrafish heart and the identification of Erbb2 signaling as a remodeling factor of myofibril architecture[J]. Circulat. Res. 115, 845–856.
Riedl, J., Crevenna, A. H., Kessenbrock, K., Yu, J. H., Neukirchen, D., Bista, M., et al. (2008). Lifeact: a versatile marker to visualize F-actin. Nat. Methods 5, 605–607.
Riedl, J., Flynn, K. C., Raducanu, A., Gärtner, F., Beck, G., Bösl, M., et al. (2010). Lifeact mice for studying F-actin dynamics. Nat. Methods. 7, 168–169. doi: 10.1038/nmeth0310-168
Rocchetti, A., Hawes, C., and Kriechbaumer, V. (2014). Fluorescent labelling of the actin cytoskeleton in plants using a cameloid antibody. Plant Methods 10:12. doi: 10.1186/1746-4811-10-12
Rullo, J., Becker, H., Hyduk, S. J., Wong, J. C., Digby, G., Arora, P. D., et al. (2012). Actin polymerization stabilizes α4β1 integrin anchors that mediate monocyte adhesion. J. Cell Biol. 197, 115–129. doi: 10.1083/jcb.201107140
Schell, M. J., Erneux, C., and Irvine, R. F. (2001). Inositol 1,4,5-trisphosphate 3-kinase A associates with F-actin and dendritic spines via its N terminus. J. Biol. Chem. 276, 37537–37546. doi: 10.1074/jbc.M104101200
Schiavon, C. R., Zhang, T., Zhao, B., Moore, A. S., Wales, P., Andrade, L. R., et al. (2020). Actin chromobody imaging reveals sub-organellar actin dynamics. Nat. Methods 17, 917–921. doi: 10.1038/s41592-020-0926-5
Sheahan, M. B., Staiger, C. J., Rose, R. J., and McCurdy, D. W. A. (2004). green fluorescent protein fusion to actin-binding domain 2 of Arabidopsis fimbrin highlights new features of a dynamic actin cytoskeleton in live plant cells. Plant Physiol. 136, 3968–3978. doi: 10.1104/pp.104.049411
Spracklen, A. J., Fagan, T. N., Lovander, K. E., Tootle, T. L., and Spracklen, A. J. (2014). The pros and cons of common actin labeling tools for visualizing actin dynamics during Drosophila oogenesis. Dev. Biol. 393, 209–226.
Stewart, M. D., Lopez, S., Nagandla, H., Soibam, B., Benham, A., Nguyen, J., et al. (2016). Mouse myofibers lacking the SMYD1 methyltransferase are susceptible to atrophy, internalization of nuclei and myofibrillar disarray. Dis. Model Mech. 9, 347–359.
Suarez, C., Carroll, R. T., Burke, T. A., Christensen, J. R., Bestul, A. J., Sees, J. A., et al. (2015). Profilin regulates F-actin network homeostasis by favoring formin over Arp2/3 complex. Dev. Cell. 32, 43–53. doi: 10.1016/j.devcel.2014.10.027
Tan, X. G., Rotllant, J., Li, H. Q., DeDeyne, P., and Du, S. J. (2006). SmyD1, a histone methyltransferase, is required for myofibril organization and muscle contraction in zebrafish embryos. Proc. Natl. Acad. Sci. U S A 103, 2713–2718.
Tong, S. W., and Elzinga, M. (1983). The sequence of the NH2-terminal 204-residue fragment of the heavy chain of rabbit skeletal muscle myosin. J. Biol. Chem. 258, 13100–13110.
Van Der Honing, H. S., Van Bezouwen, L. S., Emons, A. M. C., and Ketelaar, T. (2011). High expression of Lifeact in Arabidopsis thaliana reduces dynamic reorganization of actin filaments but does not affect plant development. Cytoskeleton 68, 578–587.
Vidali, L., Rounds, C. M., Hepler, P. K., and Bezanilla, M. (2009). Lifeact-mEGFP reveals a dynamic apical F-actin network in tip growing plant cells. PLoS One 4:e5744. doi: 10.1371/journal.pone.0005744
Wang, Y. S., Motes, C. M., Mohamalawari, D. R., and Blancaflor, E. B. (2004). Green fluorescent protein fusions to Arabidopsis fimbrin 1 for spatio-temporal imaging of F-actin dynamics in roots. Cell Motil. Cytoskeleton 59, 79–93. doi: 10.1002/cm.20024
Warren, J. S., Tracy, C. M., Miller, M. R., Makaju, A., Szulik, M. W., Oka, S. I., et al. (2018). Histone methyltransferase Smyd1 regulates mitochondrial energetics in the heart. Proc. Natl. Acad. Sci. U S A. 115, E7871–E7880. doi: 10.1073/pnas.1800680115
Wehland, J., Osborn, M., and Weber, K. (1977). Phalloidin-induced actin polymerization in the cytoplasm of cultured cells interferes with cell locomotion and growth[J]. Proc. Natl. Acad. Sci. 74, 5613–5617.
Winder, S. J., Hemmings, L., Maciver, S. K., Bolton, S. J., Tinsley, J. M., Davies, K. E., et al. (1995). Utrophin actin binding domain: analysis of actin binding and cellular targeting. J. Cell Sci. 108(Pt 1), 63–71.
Xu, H., Ye, D., Behra, M., Burgess, S., Chen, S., and Lin, F. G. (2014). β1 controls collective cell migration by regulating the protrusive activity of leader cells in the posterior lateral line primordium. Dev. Biol. 385, 316–327. doi: 10.1016/j.ydbio.2013.10.027
Xu, J., Gao, J., Li, J., Xue, L., Clark, K. J., Ekker, S. C., et al. (2012). Functional analysis of slow myosin heavy chain 1 and myomesin-3 in sarcomere organization in zebrafish embryonic slow muscles. J. Genet. Genomics 39, 69–80.
Yang, J., and Xu, X. (2012). Immunostaining of dissected zebrafish embryonic heart. J. Vis. Exp. 2012:3510. doi: 10.3791/3510
Yu, S. F. (2013). The Role of the Actin Cytoskeleton During Muscle Development in Drosophila and Mouse. Ph.D. thesis. New York, NY: Gerstner Sloan Kettering Graduate school of Biomedical Sciences.
Keywords: F-actin filament, Lifeact-GFP, sarcomere, cardiomyocyte, Smyd1
Citation: Xu R and Du S (2021) Overexpression of Lifeact-GFP Disrupts F-Actin Organization in Cardiomyocytes and Impairs Cardiac Function. Front. Cell Dev. Biol. 9:746818. doi: 10.3389/fcell.2021.746818
Received: 24 July 2021; Accepted: 07 October 2021;
Published: 26 October 2021.
Edited by:
Enrique Martin-Blanco, Instituto de Biología Molecular de Barcelona (IBMB), SpainReviewed by:
Roland Wedlich-Söldner, University of Münster, GermanyCopyright © 2021 Xu and Du. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shaojun Du, c2R1QHNvbS51bWFyeWxhbmQuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.