
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 21 September 2021
Sec. Molecular and Cellular Pathology
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.744838
This article is part of the Research Topic Hearing Loss: Mechanisms and Prevention View all 35 articles
 Tingting Dong1*†
Tingting Dong1*† Xuejie Zhang1†Yiqing Liu2
Xuejie Zhang1†Yiqing Liu2 Shan Xu3Haishuang Chang3Fengqiu Chen1Lulu Pan1Shaoru Hu1Min Wang1
Shan Xu3Haishuang Chang3Fengqiu Chen1Lulu Pan1Shaoru Hu1Min Wang1 Min Lu4*
Min Lu4*Optic atrophy1 (OPA1) is crucial for inner mitochondrial membrane (IMM) fusion and essential for maintaining crista structure and mitochondrial morphology. Optic atrophy and hearing impairment are the most prevalent clinical features associated with mutations in the OPA1 gene, but the function of OPA1 in hearing is still unknown. In this study, we examined the ability of Opa1 to protect against cisplatin-induced cochlear cell death in vitro and in vivo. Our results revealed that knockdown of Opa1 affects mitochondrial function in HEI-OC1 and Neuro 2a cells, as evidenced by an elevated reactive oxygen species (ROS) level and reduced mitochondrial membrane potential. The dysfunctional mitochondria release cytochrome c, which triggers apoptosis. Opa1 expression was found to be significantly reduced after cell exposed to cisplatin in HEI-OC1 and Neuro 2a cells. Loss of Opa1 aggravated the apoptosis and mitochondrial dysfunction induced by cisplatin treatment, whereas overexpression of Opa1 alleviated cisplatin-induced cochlear cell death in vitro and in explant. Our results demonstrate that overexpression of Opa1 prevented cisplatin-induced ototoxicity, suggesting that Opa1 may play a vital role in ototoxicity and/or mitochondria-associated cochlear damage.
Mitochondria are responsible for numerous vital cell functions, such as respiration, oxidative phosphorylation (OXPHOS), calcium homeostasis, and apoptotic signaling. These highly dynamic organelles continually modify their shape and undergo fusion and fission in order to maintain their morphology and activity (Hoppins et al., 2007; Rogers et al., 2017). Mitofusin 1 and 2 (Mfn1 and Mfn2) are crucial for outer mitochondrial membrane (OMM) fusion (Santel and Fuller, 2001; Chen et al., 2003), and the dynamin-related GTPase, OPA1, is essential for IMM fusion (Olichon et al., 2002). Dynamin-related protein 1 (Drp1) is a cytosolic protein that is required for mitochondrial fission. The recruitment of Drp1 is regulated by the OMM proteins, mitochondrial fission factor (Mff), mitochondrial division (Mid) 49, Mid51, and mitochondrial fission protein (Fis1) (Loson et al., 2013; Palmer et al., 2013). Mitochondrial fusion and fission are vital processes for cellular functions. Numerous studies have drawn connections between abnormal mitochondrial morphology and various diseases, including neurodegenerative diseases, cardiovascular diseases, and cancers (Mishra and Chan, 2014; Liu L. et al., 2016; He et al., 2017; Li A. et al., 2018; Gao et al., 2019; Zhang et al., 2019; Zhong et al., 2020; Fu et al., 2021b).
OPA1, which is localized at chromosome 3q28, has been identified as the causative gene of Dominant Optic Atrophy (DOA) (Alexander et al., 2000; Delettre et al., 2000). The OPA1 mRNA has eight alternatively spliced isoforms (Delettre et al., 2001), while the OPA1 protein exists in two forms, the membrane-bound long-OPA1 (L-OPA1) and the soluble short-OPA1 (S-OPA1). Both are required for mitochondrial fusion (Olichon et al., 2002; Ishihara et al., 2006). In addition to its function in IMM fusion, OPA1 is crucial for the maintenance of crista structure and mitochondrial morphology (Frezza et al., 2006; Patten et al., 2014). Opa1 depletion was associated with mitochondrial fragmentation, crista disorganization, and cytochrome c redistribution (Frezza et al., 2006). In contrast, Opa1 overexpression was associated with: normally shaped cristae; alterations in the responses of multiple tissues to apoptotic, necrotic, and atrophic stimuli; and phenotypic rescue of mitochondrial diseases in mice (Civiletto et al., 2015; Varanita et al., 2015). Opa1 also contributes to safeguarding mtDNA integrity, preserving mtDNA function in the face of mutations (Chen et al., 2010), and supporting cellular adaptation to metabolic demand (Patten et al., 2014). Opa1 dysfunction has been linked to ROS overproduction and unbalanced redox homeostasis (Yarosh et al., 2008; Chen et al., 2012).
In the clinic, about 60–70% of DOA cases are associated with pathogenic mutations of OPA1 (Alexander et al., 2000; Delettre et al., 2000). DOA is characterized by destruction of retinal ganglion cells (RGCs) and the optic nerve (Kjer, 1959), deafness, and chronic progressive external ophthalmoplegia (Treft et al., 1984; Meire et al., 1985). Hearing loss usually follows the onset of visual symptoms (Leruez et al., 2013). Although OPA1 is highly expressed in the retina, it is broadly expressed in multiple tissues. In cochlear tissue, OPA1 is expressed in both hair cells and ganglion cells. Unlike ganglion cells, which express OPA1 at birth, OPA1 expression in the organ of Corti increases after birth and approaches the mature expression level during the onset of hearing (Bette et al., 2007). This suggests that OPA1 is crucial for the function of hair cells and ganglion cells in the inner ear.
In this study, we investigated the potential effect of Opa1 to protect against cisplatin-induced apoptosis in vitro and in vivo. Our results indicated that loss of Opa1 caused mitochondrial dysfunction and thereby triggered apoptosis in HEI-OC1 and Neuro 2a cells, an auditory hair cell line and a neural cell line. Cisplatin treatment markedly reduced Opa1 expression in HEI-OC1 and Neuro 2a cells. In the two tested cell lines, Opa1 depletion aggravated the cisplatin-induced apoptosis and mitochondrial dysfunction, whereas Opa1 overexpression partially prevented cisplatin-induced apoptosis. Histological analyses were performed using cisplatin-treated organotypic cochlear cultures, and our results showed that Opa1 overexpression decreased cisplatin-induced cell death, indicating Opa1 is a potential therapeutics gene in cisplatin induced auditory impairment.
Murine HEI-OC1 cells were grown in DMEM supplemented with 10% heat-inactivated FBS and maintained at 33°C with 10% CO2. Murine Neuro 2a cells were grown in the same medium at 37°C with 5% CO2. HEI-OC1 and Neuro 2a cells were incubated in 96-well plates for cell viability assessment or 6-well plates for flow cytometry, transmission electron microscopy, and immunostaining. siRNA or plasmids were transfected after cells were grown to 60% confluence. siRNA was transfected using LipoRNAiMax (Invitrogen). Plasmids were transfected into Neuro 2a cells using Lipo2000 (Invitrogen, 11668-019) and into HEI-OC1 cells using Lipo3000 (Invitrogen, L30000015), with the relevant reagents diluted in Opti-MEM (Gibco). The cells were then treated with or without cisplatin for 24 h. For cell viability detection, HEI-OC1 or Neuro 2a cells were transfected with siRNA or plasmids, and then treated with or without cisplatin for 24 h, unless indicated the timepoint in the picture. For qRT-PCR analyses, HEI-OC1 or Neuro 2a cells were grown in a 6-well plate to 70–80% confluence, treated with siRNA, plasmids or cisplatin for 24 h. For Western blot, HEI-OC1 or Neuro 2a cells were grown in a 6-well plate to 70–80% confluence, treated with siRNA, plasmids or cisplatin for 48 h. For flow cytometry, transmission electron microscopy, and immunostaining, cells were transfected with siRNA or treated with or without cisplatin for 24 h. For almost all the experiments both HEI-OC1 and Neuro 2a cells were used. Several experiments (Mitochondria morphology detection after Opa1 knockdown and cisplatin treatment; Cytochrome C release analysis; TEM; Bcl-2 protein expression; and DCF level after cisplatin treatment) were only used HEI-OC1 cells.
HEI-OC1 and Neuro 2a cells were plated in 96-well plates (1 × 105 cells/well) and incubated with 100 μL drug-supplemented DMEM. After 24, 48, or 72 h, cell viability was measured using a Cell Counting Kit-8 (CCK-8; Dojindo) according to the manufacturer’s instructions. The optical density at 450 nm (OD450 nm) was measured using a Tecan Spark multimode microplate reader.
To test the ROS level and mitochondrial membrane potential (MMP), cells were collected, washed thrice with PBS, and stained using DCFH-A (Sigma, D6883; 10 mM in DMSO, 1:1,000) or TMRM (Invitrogen, 134361; 1:1,000) at 37°C for 30 min, and then counterstained with DAPI (Invitrogen,D357; 1:1,000). Apoptosis was assessed with apoptosis detection kit (BioLegend, San Diego, CA, United States). Briefly, cells were collected, washed thrice with PBS, suspended with 500 μL binding buffer, and stained with Annexin V-FITC and propidium iodide following the manufacturer’s instructions. Stained cells were analyzed using a flow cytometer and data were processed with the FlowJo software (FlowJo, LLC, Ashland, OR, United States).
Collected cells were lysed with RIPA buffer (Thermo Fisher Scientific, 89900) containing a complete protease inhibitor cocktail (Roche) at 4°C for 20 min with shaking, and then centrifuged at 4°C for 20 min at 13,000 rpm. The supernatant was collected, the total proteins were quantified, and 10 μg of total protein from each sample was boiled for 10 min in sample buffer (BioRad, #161-0737), resolved by SDS-PAGE, and transferred to a PVDF membrane. The membrane was blocked with TBST containing 5% skim milk, incubated with primary antibodies in TBST containing 5% skim milk, and incubated with HRP-conjugated secondary antibodies. The results were visualized with ECL reagents. The following primary antibodies were used: mouse anti-Opa1 (BD, 612606), rabbit anti-Hsp60 (CST, 12165), rabbit anti-Bcl2 (CST,3498), and rabbit anti-β-actin (CST, 4967S).
Total RNA was extracted from cultured cells using a commercially available kit (Qiagen, #74136) with the optional DNase digestion step. The RNA was reverse transcribed with an Evo M-MLV RT kit with gDNA Clean for qPCR (Accurate Biotechnology, China) and qRT-PCR was performed using a Roche LightCycler 480 with a SYBR Green Premix Pro Taq HS qPCR kit (Accurate Biotechnology) and the following cycling conditions: 95°C for 5 min followed by 45 cycles of 95°C for 10 s, 60°C for 10 s, and 72°C for 10 s. The relative level of each target gene was normalized to that of endogenous Rpl19 and calculated using the comparative Ct (ΔΔCt) method. The sequences of the utilized primers were as follows: 5′- TGGAAAATGGTTCGAGAGTCAG-3′ (forward) and 5′- CATTCCGTCTCTAGGTTAAAGCG-3′ (reverse) for Opa1; 5′-ACGGAGGCTGGGATGCCTTTG-3′ (forward) and 5′-AGTGATGCAGGCCCCGACCA-3′ (reverse) for Bcl2; and 5′-ACCTGGATGAGAAGGATGAG-3′ (forward) and 5′-ACCTTCAGGTACAGGCTGTG-3′ (reverse) for Rpl19.
HEI-OC1 cells were collected, placed in 2.5% glutaraldehyde, and incubated at room temperature for 20 min and then at 4°C for 1 h. The cells were post-fixed with 1% osmium tetroxide for 2 h at room temperature, dehydrated in a graded alcohol series, and infiltrated in a graded alcohol series containing eopn812. The samples were double-stained with uranyl acetate followed by lead citrate and examined with a 120 kV transmission electron microscope (FEI Talos L120C).
Organotypic cochlear cultures were generated from the cochlea of postnatal day 1–2 (P1–P2) mouse pups of both sexes. Briefly, mouse pups were sacrificed by decapitation and dissected in HBSS supplemented with HEPES. The basal membrane was isolated, plated in a glass-bottom dish with DMEM/F12 medium containing 1 × N2 (Sigma, A1370701) and 1 × B27 (Sigma, 17504044), and incubated overnight at 37°C with 5% CO2. For immunostaining, the basal membrane cells were simultaneously transfected with Opa1 plasmid (pOpa1) or siRNA and treated with 60 μM cisplatin for 24 h. Control cultures were treated with cisplatin alone for 24 h or left untreated.
HEI-OC1 and Neuro 2a cells were collected and rinsed thrice in phosphate-buffered saline (PBS) for 10 min per wash. The cells were fixed in 4% paraformaldehyde (PFA) for 30 min and incubated overnight at 4°C with anti-cleaved caspase-3 (CST, 9661; rabbit, 1:400). The cells were rinsed thrice with PBS, incubated with Alexa Fluor 488 donkey anti-rabbit IgG antibody (Invitrogen, 1:400) for 60 min at room temperature, rinsed thrice in PBS, and stained with DAPI (YEASEN, 36308ES20). Alternatively, cells were incubated with MitoTracker (Invitrogen; M7510, 1:10,000) at 37°C for 30 min, rinsed thrice in PBS, fixed in 4% PFA for 30 min, and then either directly sealed with DAPI or incubated overnight at 4°C with anti-cytochrome c (CST, 11940; rabbit, 1:100), rinsed thrice in PBS, incubated with Alexa Fluor 488 donkey anti-rabbit IgG antibody (Invitrogen; 1:400) for 60 min at room temperature, rinsed thrice in PBS, and then sealed with DAPI.
The basal membrane cells were collected, rinsed thrice in PBS for 10 min per rinse, fixed in 4% PFA for 30 min, incubated overnight at 4°C with MYO 7A (proteus biosciences; 1:500), rinsed thrice in PBS, incubated with Alexa Fluor 488 donkey anti-rabbit IgG antibody (Invitrogen; 1:400) for 60 min at room temperature, rinsed thrice in PBS, and sealed with DAPI.
Data are presented as mean ± standard errors of the mean (SEM). Experiments were performed in triplicate and repeated three times. Statistical significance was assessed with t-test, one-way ANOVA, or two-way ANOVA, as applied using the GraphPad Prism 9.0 software (GraphPad Software). A value of p < 0.05 was considered statistically significant.
To study the role of Opa1 in the inner ear, we used siRNA transfection to assess the effect of Opa1 knockdown in HEI-OC1 and Neuro 2a cells, which were chosen to represent two important cell types of the inner ear: hair cells and spiral ganglion neurons. In the latter case, we selected the most common used neuronal cell line because there was no specific cell line available for spiral ganglion neurons. The transfection efficiency was monitored by transfecting cells with fluorescence-tagged sham siRNA. Almost all HEI-OC1 and Neuro 2a cells were fluorescence-positive at 24 h post-transfection (Figure 1A). We designed three different mouse Opa1 siRNA (siOpa1-260, siOpa1-1681, siOpa1-2361) and analyzed their effects in HEI-OC1 and Neuro 2a cells compared to those of a control scramble siRNA (Ctrl-siRNA). At 24 h after siOpa1 transfection, Opa1 mRNA expression was reduced by 70% in HEI-OC1 cells (Figure 1B) and by 90% in Neuro 2a cells (Figure 1C). To test whether there was a corresponding reduction in the protein expression of Opa1, we performed Western blot analysis at 48 h after siOpa1 transfection. All three of the Opa1 siRNA were found to significantly decrease the Opa1 protein levels in the two cell lines (Figures 1D,F). Quantification revealed that Neuro 2a cells displayed less Opa1 protein expression than HEI-OC1 cells (Figures 1E,G), which was consistent with the trends in their mRNA expression levels (Figures 1B,C). These results indicate that Opa1 could be successfully suppressed by the designed siRNA in HEI-OC1 and Neuro 2a cells. For the following experiments, siOpa1-260 was used for Opa1 knockdown subsequently.
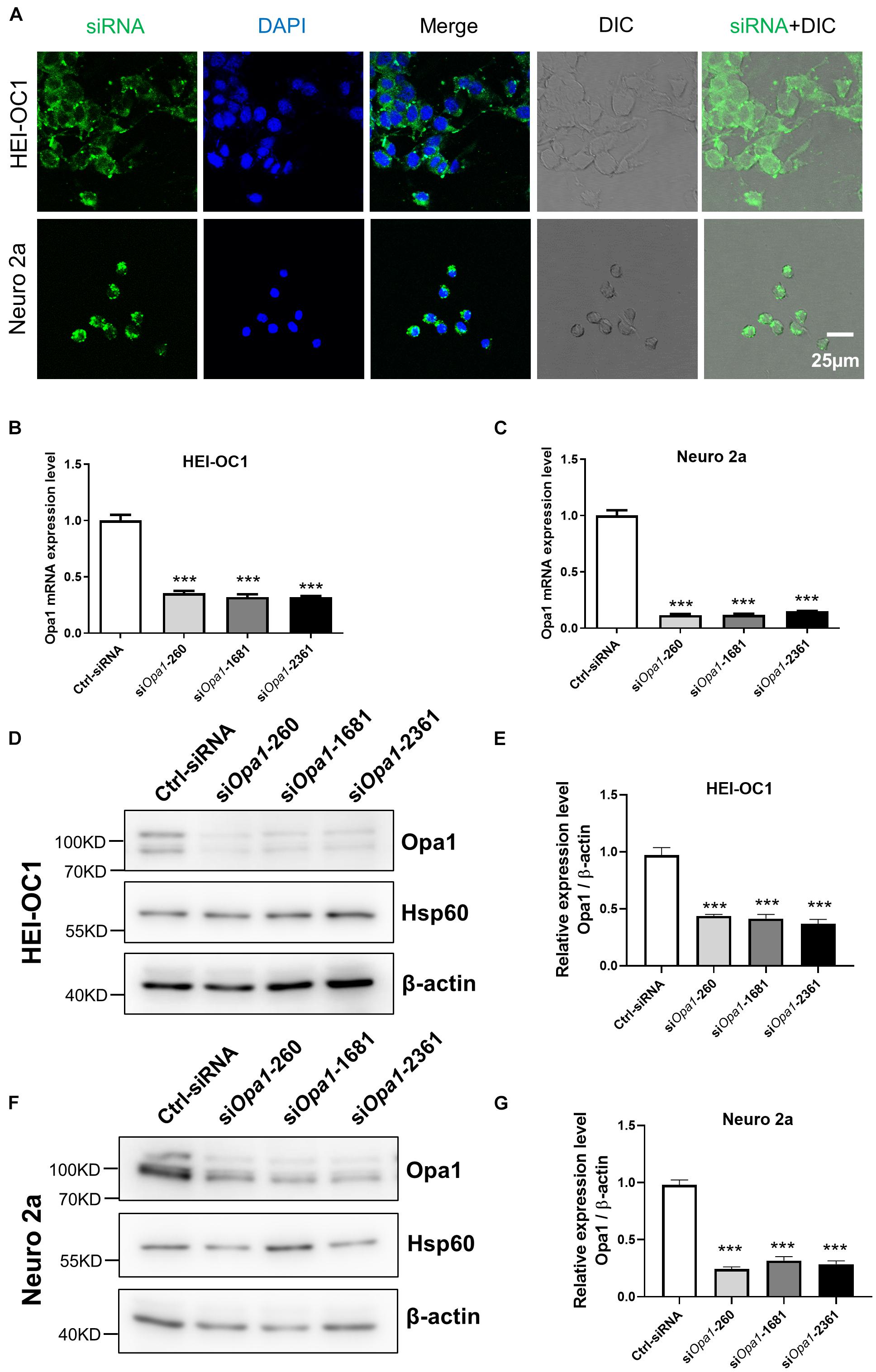
Figure 1. Depletion of Opa1 in HEI-OC1 and Neuro 2a cells. (A) Representative confocal images of HEI-OC1 and Neuro 2a cells after siRNA transfection for 24 h, showing transfection efficiency. DIC: Differential interference contrast. Quantification of Opa1 mRNA expression levels in HEI-OC1 (B) and Neuro 2a (C) cells transfected with siOpa1 for 24 h. In this and all following graphs: the levels of Opa1 mRNA were normalized to that of mouse rpl19; error bars represent standard error of the mean (SEM); and experiments were performed in triplicate and repeated three times. ***p < 0.001 (one-way ANOVA with Dunnett’s multiple comparisons test). Representative Western blot images (D) and quantification (E) of Opa1 expression in HEI-OC1 cells transfected with siOpa1 for 48 h. In this and all following graphs, the protein levels of Opa1 are normalized to that of β-actin. ***p < 0.001 (one-way ANOVA with Dunnett’s multiple comparisons test). Representative Western blot images (F) and quantification (G) of Opa1 expression in Neuro 2a cells transfected with siOpa1 for 48 h. ***p < 0.001 (one-way ANOVA with Dunnett’s multiple comparisons test).
To study the cellular function of Opa1 in HEI-OC1 and Neuro 2a cells, we first monitored mitochondrial morphology by electron microscope and MitoTracker staining. MitoTracker staining and Electron microscopic analysis of HEI-OC1 cells transfected with Ctrl-siRNA revealed mainly elongated mitochondria (Figure 2A), with some tubular mitochondria (Figure 2B). HEI-OC1 cells transfected with Opa1 siRNA for 24 h exhibited highly fragmented, dot-like mitochondria, as assessed by MitoTracker staining (Figure 2A) and electron microscopy (Figure 2B). In contrast, mitochondria of Neuro 2a cells displayed fragmented shapes prior to siRNA transfection (data not shown), and there was no further difference of mitochondria shape between Ctrl-siRNA- and siOpa1-transfected Neuro 2a cells under MitoTracker staining (data not shown). To assess whether Opa1 depletion affected mitochondrial function, we transfected the cells with siRNA for 24 h and measured the ROS level and mitochondrial membrane potential (MMP, ΔΨm) using DCFH-DA and TMRM, respectively. HEI-OC1 cells lacking Opa1 exhibited an increased ROS level (Figures 2C,D) and reduced MMP (Figures 2G,H) compared with control cells. Despite displaying no alteration of mitochondrial shape, Opa1-silenced Neuro 2a cells displayed a higher ROS level (Figures 2E,F) and lower MMP (Figures 2I,J) compared with control cells. These findings indicate that Opa1 depletion affects mitochondrial function in HEI-OC1 and Neuro 2a cells, independent of its contribution to mitochondrial formation.
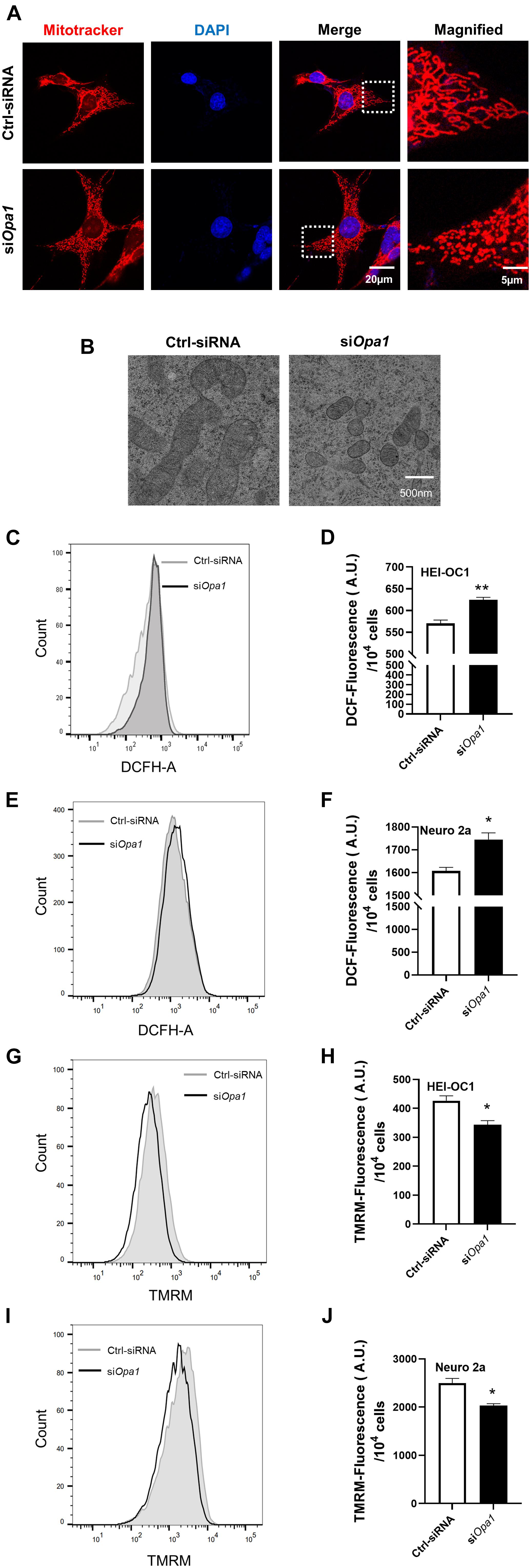
Figure 2. Loss of Opa1 leads to mitochondrial dysfunction in HEI-OC1 and Neuro 2a cells. (A) Representative confocal microscopic images of MitoTracker staining at 24 h after siRNA transfection of HEI-OC1 cells. Right panels show magnified views of the areas marked in the left panels. (B) HEI-OC1 cells were transfected with scramble (Ctrl) siRNA or siOpa1 for 24 h and fixed, and thin sections of cells were visualized by transmission electron microscopy. (C) Intracellular ROS level was measured by flow cytometric analysis using DCFH-DA in HEI-OC1 cells transfected with siRNA for 24 h. (D) Quantitative histograms of the mean fluorescence intensity of DCFH-DA in HEI-OC1 cells. **p < 0.01 (non-parametric Mann-Whitney t-test). (E,F) Representative flow cytometric plot (E) and quantitative histograms (F) of the mean fluorescence intensity of DCFH-DA in Neuro 2a cells transfected with siRNA for 24 h. *p < 0.05 (non-parametric Mann-Whitney t-test). (G) The mitochondrial membrane potential of HEI-OC1 cells was measured by flow cytometric analysis using TMRM fluorescence at 24 h after siRNA transfection. (H) Bar graph shows the relative TMRM mean intensity in HEI-OC1 cells. *p < 0.05 (non-parametric Mann-Whitney t-test). Representative flow cytometric plot (I) and quantitative histograms (J) of the relative TMRM mean intensity in Neuro 2a cells. *p < 0.05 (non-parametric Mann-Whitney t-test).
Next we sought to identify the cellular changes that arise from Opa1 silencing. At 24 h post-transfection, the cell numbers were comparable between siOpa1- and Ctrl-siRNA-transfected HEI-OC1 and Neuro 2a cells (Figures 3A,B, left). At 48 h post-transfection, Opa1-depleted Neuro 2a cells displayed a significant increase in cell death, whereas Opa1-depleted HEI-OC1 cells did not (Figures 3A,B, middle). At 72 h post-transfection, Opa1-depleted Neuro 2a and HEI-OC1 cells both exhibited significant increases in cell death (Figures 3A,B, right). To assess whether cells underwent apoptosis upon Opa1 knockdown, we stained siRNA-transfected cells for cleaved caspase-3. Cleaved caspase-3 staining was evident in HEI-OC1 and Neuro 2a cells transfected with siOpa1 (Figure 3C), indicating that apoptosis was activated upon Opa1 depletion. In agreement with this increased caspase-3 activation, transmission electron microscopy revealed the presence of apoptotic cells in cultures of Opa1-depleted HEI-OC1 cells (Figure 3C). We postulated that loss of Opa1 decreased the MMP (Figures 2E,I) and led to cytochrome c leakage, thereby increasing caspase-3 activation. In support of this proposal, staining experiments revealed that cytochrome c and mitochondria were colocalized in HEI-OC1 cells transfected with Ctrl-siRNA, as reflected by an orange color representing the merging of green (cytochrome c) and red (MitoTracker) fluorescence signals (Figure 3D). In Opa1-knockdown cells, in contrast, we observed largely non-overlapping green and red signals that suggested the cytochrome c had been released from the mitochondria (Figure 3D). To further confirm that cells underwent apoptosis following siOpa1 transfection, we preformed Annexin V/PI staining and flow cytometric analysis. Annexin V staining showed that knockdown of Opa1 induced more early apoptotic cells in HEI-OC1 cultures (Figures 3E,F) and more late apoptotic cells in Neuro 2a cultures (Figures 3G,H). This finding is consistent with our observation that Opa1 depletion led to earlier cell death in Neuro 2a cells than in HEI-OC1 cells (Figures 3A,B). Bcl family proteins, such as Bcl-2, regulate the release of cytochrome c through the OMM (Adams and Cory, 2007). To assess whether Bcl2 was involved in the apoptosis induced by Opa1 knockdown, we detected Bcl2 expression in cells transfected with siRNA for 24 h. After Opa1 depletion, the mRNA level of Bcl2 was markedly reduced in both HEI-OC1 and Neuro 2a cells (Figures 3I,J), and the protein level of Bcl2 was reduced in Neuro 2a cells (Figure 3K).
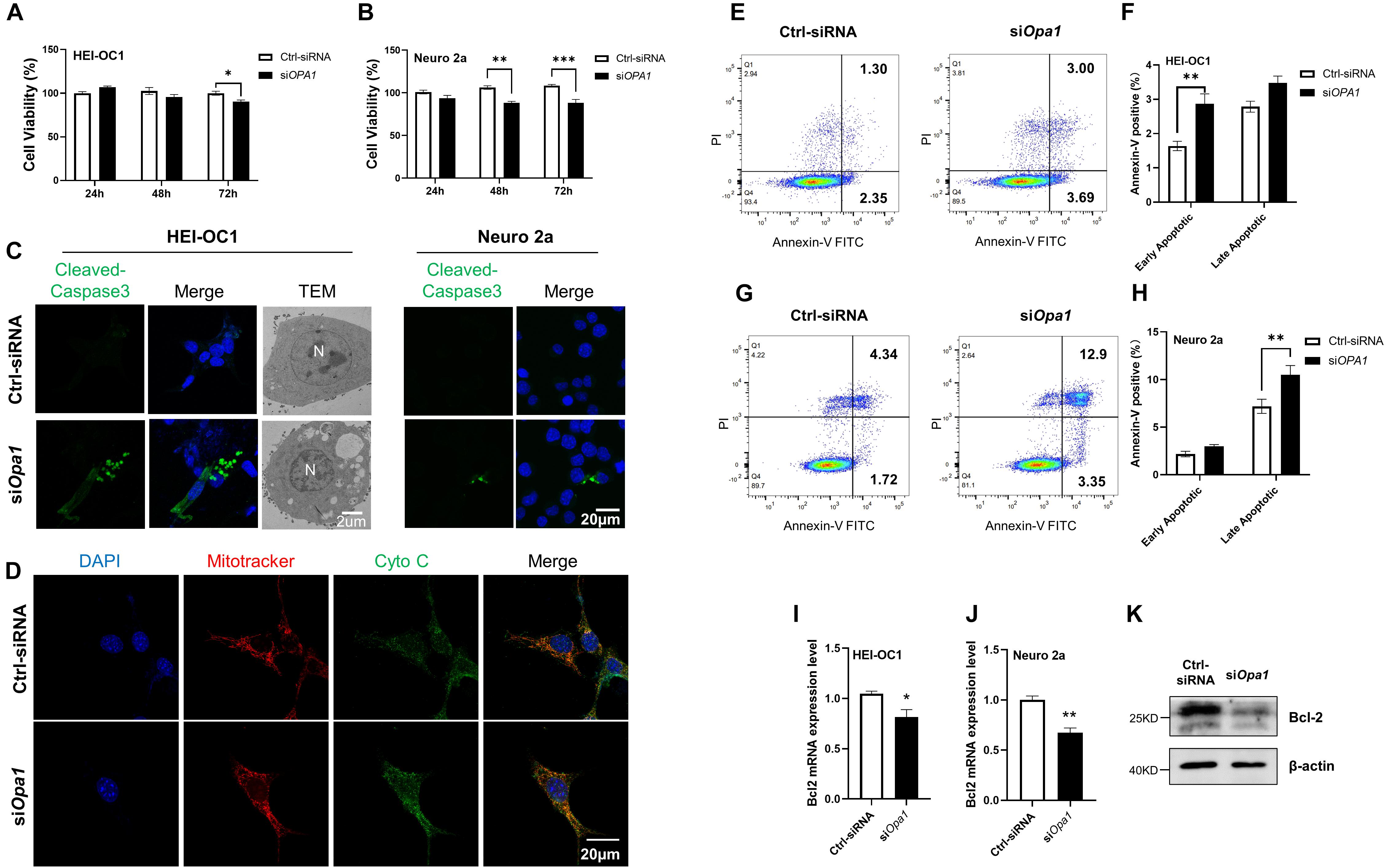
Figure 3. Lacking Opa1 triggers apoptosis in HEI-OC1 and Neuro 2a cells. (A,B) CCK-8 was used to assess the cell viability of HEI-OC1 (A) and Neuro 2a (B) cells cultured for 24, 48, and 72 h after transfection with scramble siRNA (Ctrl) or siOpa1. *p < 0.05, **p < 0.01, ***p < 0.001 (two-way ANOVA). (C) Representative confocal microscopic images of cleaved caspase-3 staining at 24 h after siRNA transfection of HEI-OC1 and Neuro 2a cells. Representative transmission electron microscopic images of HEI-OC1 cells at 24 h after siRNA transfection. (D) Representative confocal images of HEI-OC1 cells co-stained for mitochondria (MitoTracker) and cytochrome c at 24 h after siRNA transfection. (E) Representative flow cytometric plots generated from HEI-OC1 cells stained with Annexin V-FITC/PI to detect apoptosis at 24 h after transfection with scramble siRNA (Ctrl) or siOpa1. (F) The percentages of early and late apoptotic cells were compared between Ctrl-siRNA- and siOPA1-transfected HEI-OC1 cells. **p < 0.01 (two-way ANOVA). FITC, fluorescein isothiocyanate; PI, propidium iodide. (G) Representative flow cytometric plots generated from Neuro 2a cells stained with Annexin V-FITC/PI to detect apoptosis at 24 h after transfection with scramble siRNA (Ctrl) or siOpa1. (H) The percentages of early and late apoptotic cells were compared between Ctrl-siRNA- and siOPA1-transfected Neuro 2a cells. **p < 0.01 (two-way ANOVA). (I,J) Quantification of Bcl2 mRNA expression levels in HEI-OC1 (I) and Neuro 2a (J) cells transfected with siRNA for 24 h. *p < 0.05, **p < 0.01 (non-parametric Mann-Whitney t-test). (K) Representative Western blot images showing Bcl2 protein levels in Neuro 2a cells transfected with siRNA for 48 h.
Serious side effects, such as ototoxicity, are associated with the use of cisplatin. Several studies demonstrated that cisplatin-induced cytotoxicity is closely related to mitochondrial dysfunction such as that signaled by ROS generation (van Gisbergen et al., 2015; de Sa Junior et al., 2017). Increased ROS alters the MMP and induces damage in the respiratory chain, thereby triggering apoptosis. To identify a suitable concentration of cisplatin that could induce cellular damage in our system, we exposed HEI-OC1 and Neuro 2a cells to various concentrations of cisplatin. After incubation with 60 μM cisplatin for 24 h, cell death was observed in 60% of HEI-OC1 cells and 50% of Neuro 2a cells (Figures 4A,B). Interestingly, Opa1 mRNA expression was reduced in HEI-OC1 and Neuro 2a cells exposed to cisplatin for 24 h (Figure 4C). Consistent with mRNA level, Opa1 protein level also decreased after cisplatin treatment in the two cell lines (Figure 4D). According to MitoTracker staining, HEI-OC1 cells treated with cisplatin for 24 h exhibited highly fragmented, dot-like mitochondria (Figure 4E), which is similar to the morphology of mitochondrion in HEI-OC1 cells with Opa1 knockdown (Figure 2A). Treatment with siOpa1 for 24 h did not cause cell death in HEI-OC1 or Neuro 2a cells, but loss of Opa1 significantly worsened cell survival upon cisplatin treatment (Figures 4F,G). To confirm these cell viability results, we performed Annexin V/PI staining followed by flow cytometric analysis. Annexin V staining showed that cisplatin treatment for 24 h induced ∼12% late apoptotic cells in HEI-OC1 cells and ∼3.5% late apoptotic cells in Neuro 2a cells (Figures 4H–K). Silencing of Opa1 additively enhanced the proportions of late apoptotic cells in cisplatin-treated HEI-OC1 and Neuro 2a cell cultures (Figures 4I,K). Moreover, the mitochondrial dysfunction caused by cisplatin treatment was worsened by Opa1 depletion, as evidenced by increased ROS generation in HEI-OC1 cells (Figures 4L,M). These results indicate that Opa1 depletion exacerbates the mitochondrial dysfunction and apoptosis induced by cisplatin in both HEI-OC1 and Neuro 2a cells.
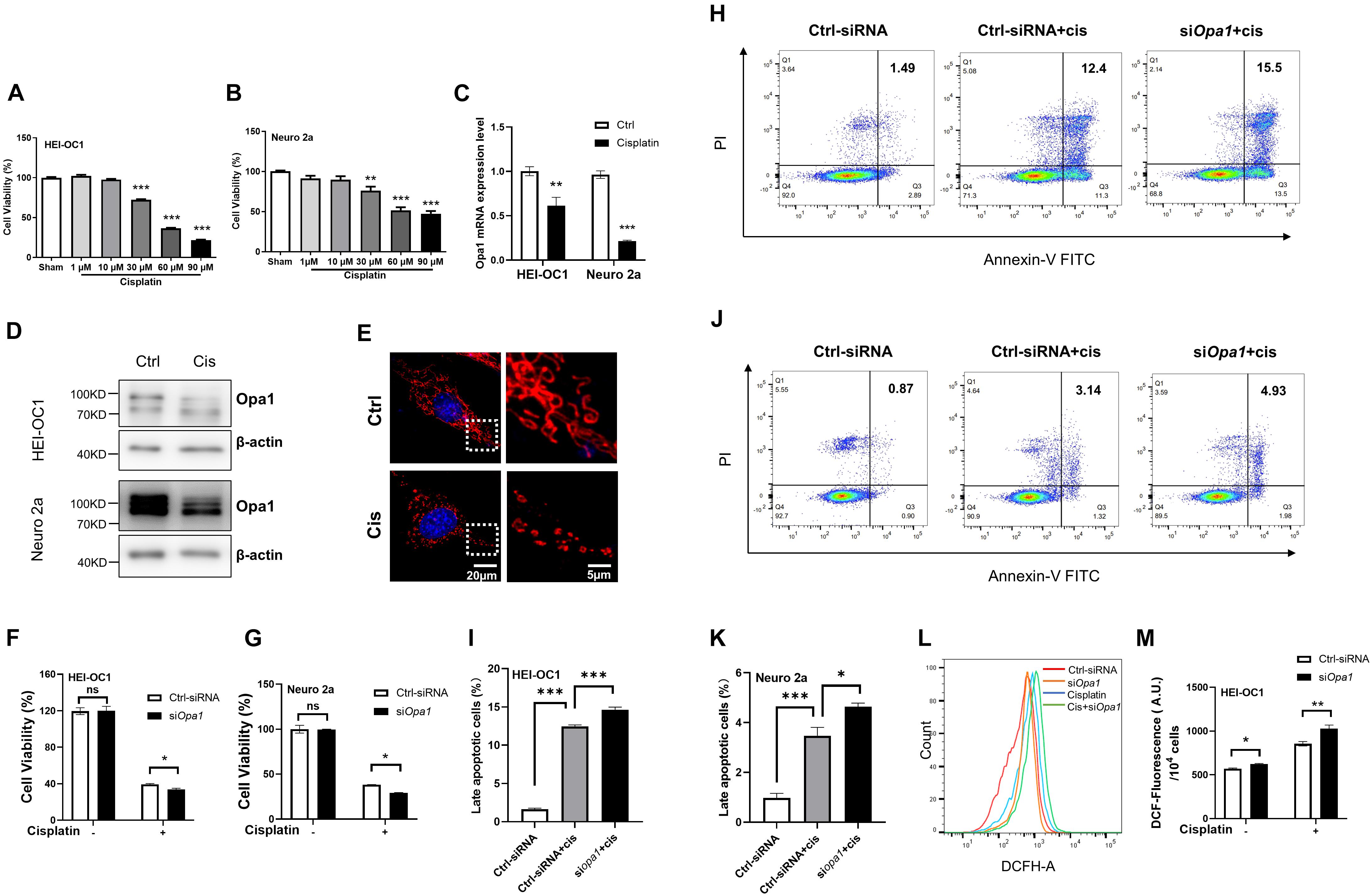
Figure 4. Knockdown of Opa1 aggravates cisplatin-induced apoptosis and mitochondrial dysfunction in HEI-OC1 and Neuro 2a cells (A,B) CCK-8 was used to assess cell viability in HEI-OC1 (A) and Neuro 2a (B) cells treated with the indicated doses of cisplatin for 24 h. **p < 0.01, ***p < 0.001 (one-way ANOVA with Dunnett’s multiple comparisons test). (C) Quantification of Opa1 mRNA expression levels in HEI-OC1 and Neuro 2a cells treated with cisplatin for 24 h. **p < 0.01, ***p < 0.001 (two-way ANOVA). (D) Representative Western blot images of Opa1 expression in HEI-OC1 and Neuro 2a cells treated with cisplatin for 24 h. (E) Representative confocal microscopic images of MitoTracker staining at 24 h after cisplatin treatment of HEI-OC1 cells. Right panels show magnified views of the areas marked in the left panels. (F,G) CCK-8 was used to assess cell viability in HEI-OC1 (F) and Neuro 2a (G) cells at 24 h after siRNA transfection with or without cisplatin treatment. N.S., non-significant, *p < 0.05 (two-way ANOVA). (H) Representative flow cytometric plots were generated from HEI-OC1 cells stained with Annexin V-FITC/PI to detect apoptosis at 24 h after siRNA transfection with or without cisplatin treatment. (I) The percentage of late apoptotic cells in each group. ***p < 0.001 (one-way ANOVA with Dunnett’s multiple comparisons test). (J) Representative flow cytometric plots were generated from Neuro 2a cells stained with Annexin V-FITC/PI to detect apoptosis at 24 h after siRNA transfection with or without cisplatin treatment. (K) The percentage of late apoptotic cells in each group. *p < 0.05, ***p < 0.001 (one-way ANOVA with Dunnett’s multiple comparisons test). (L) Intracellular ROS levels were measured by flow cytometric analysis of HEI-OC1 cells stained with DCFH-DA at 24 h after siRNA transfection with or without cisplatin treatment. (M) Quantitative histograms of the mean fluorescence intensity of DCFH-DA in HEI-OC1 cells. *p < 0.05, **p < 0.01 (two-way ANOVA).
To determine if the overexpression of Opa1 suppresses the apoptosis induced by cisplatin, we first transiently transfected HEI-OC1 and Neuro 2a cells with Opa1 and examined Opa1 mRNA and protein expression levels. At 24 h after Opa1 transfected to HEI-OC1 and Neuro 2a cells, the expression levels of the Opa1 mRNA (Figures 5A,C) and protein (Figures 5B,D) were significantly elevated. Treatment of both cell lines with 60 μM cisplatin for 24 h caused almost half of the cells to die (Figures 5E,F), but these death rates were diminished significantly in cells transfected with Opa1 (Figures 5E,F). Furthermore, flow cytometric analysis showed that upregulation of Opa1 reduced the number of early and late apoptotic cells in HEI-OC1 cells (Figures 5G,H). Opa1 was also found to protect Neuro 2a cells from cisplatin-induced apoptosis (Figures 5I,J).
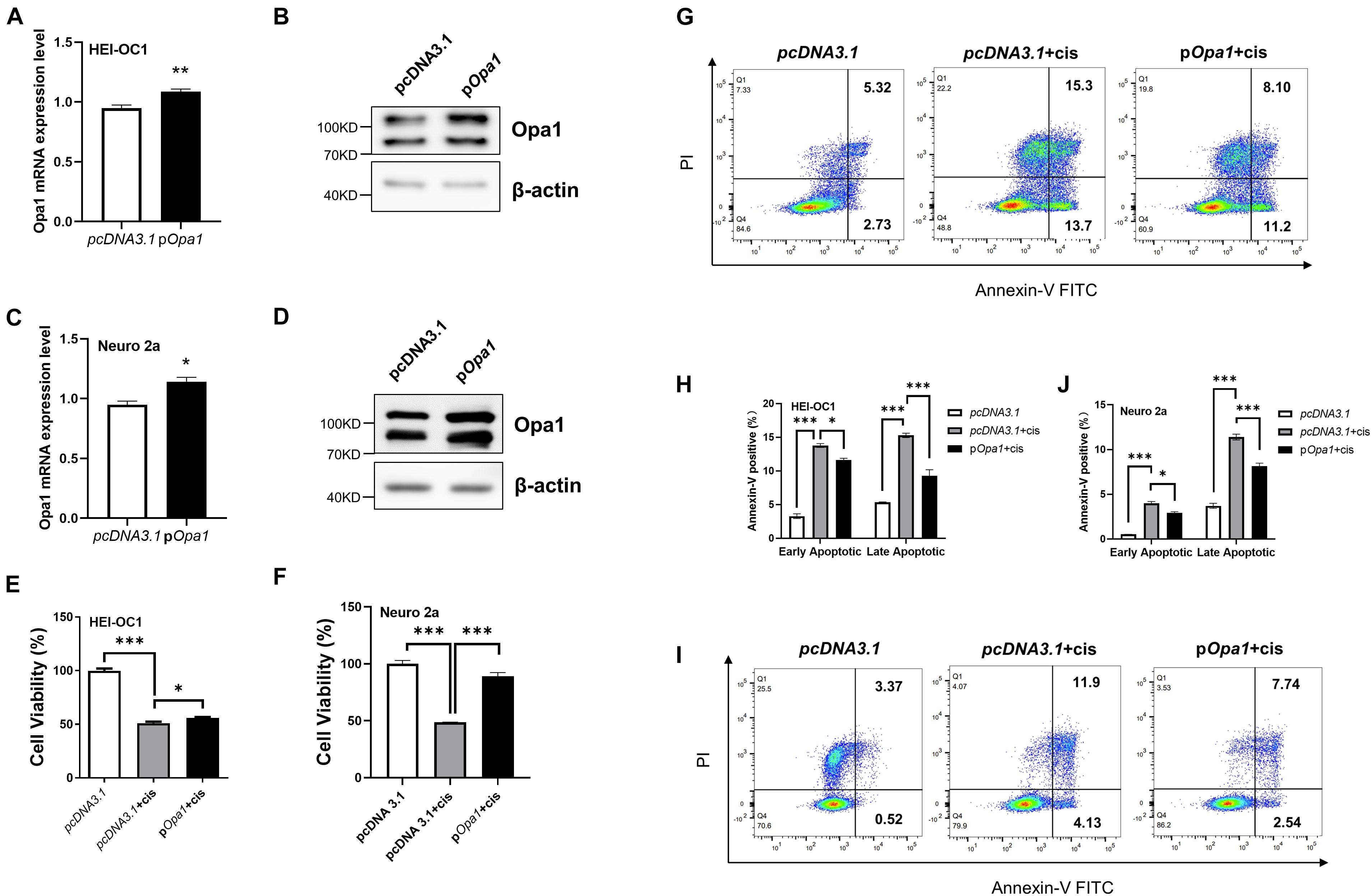
Figure 5. Overexpression of Opa1 partially restores cisplatin-induced apoptosis in HEI-OC1 and Neuro 2a cells (A) Quantification of Opa1 mRNA expression in HEI-OC1 cells transfected with Opa1 for 24 h. **p < 0.01 (non-parametric Mann-Whitney t-test). (B) Representative Western blot images showing the Opa1 protein expression level in HEI-OC1 cells transfected with Opa1 for 48 h. (C) Quantification of Opa1 mRNA expression in Neuro 2a cells transfected with Opa1 for 24 h. *p < 0.05 (non-parametric Mann-Whitney t-test). (D) Representative Western blot images showing the Opa1 protein expression level in Neuro 2a cells transfected with Opa1 for 48 h. (E,F) CCK-8 was used to assess cell viability in HEI-OC1 (E) and Neuro 2a (F) cells at 24 h after cisplatin treatment with or without Opa1 transfection. *p < 0.05, ***p < 0.001 (one-way ANOVA with Dunnett’s multiple comparisons test). (G) Representative flow cytometric plots were generated from HEI-OC1 cells stained with Annexin V-FITC/PI to detect apoptosis at 24 h after cisplatin treatment with or without Opa1 transfection. (H) The percentages of early and late apoptotic cells in each group. *p < 0.05, ***p < 0.001 (two-way ANOVA). (I) Representative flow cytometric plots were generated from Neuro 2a cells stained with Annexin V-FITC/PI to detect apoptosis at 24 h after cisplatin treatment with or without Opa1 transfection. (J) The percentages of early and late apoptotic cells in each group. *p < 0.05, ***p < 0.001 (two-way ANOVA).
Next, we tested the otoprotective potential of Opa1 in vivo. Organotypic cochlear cultures were treated with 60 μM cisplatin for 24 h, and hair cell damage was observed in whole-organ inner ear explants. Hair cell morphology and structure were evaluated by immunolabeling of the hair cell marker, myosin 7a. As shown in the Figure 6A, knockdown or overexpressed Opa1 didn’t affect cochlea morphology. Under cisplatin treatment, the numbers of OHCs were significantly reduced in the apical, middle, and basal turns of the cochlea, but there was no change in the number of IHCs (Figures 6B,C). Opa1 knockdown followed by cisplatin treatment resulted in more severe cochlear lesions: There were 30% fewer OHCs and 20% fewer IHCs compared to the cisplatin treatment groups (Figures 6B,C). On the contrary, the overexpression of Opa1 was modestly effective in preventing OHCs death in the middle turn of explants exposed to cisplatin (Figures 6B,C).
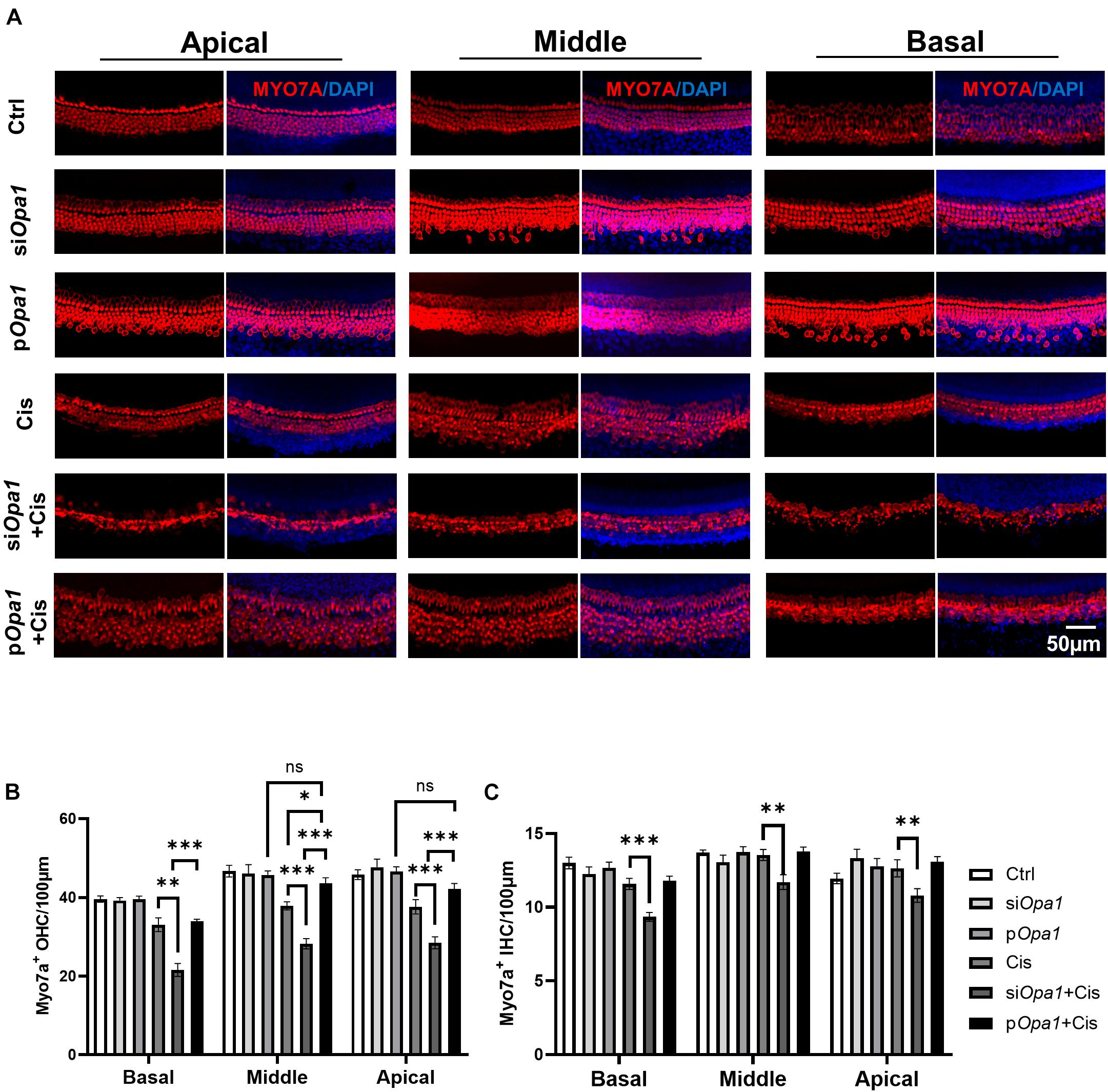
Figure 6. Opa1 knockdown or overexpression respectively aggravates or partially rescues cisplatin induced hair cell damage in organotypic cochlear cultures. (A) Representative confocal images of the apical, middle, and basal regions of organotypic cochlear cultures transfected with siOpa1 or Opa1 and treated with 60 μM cisplatin. Hair cells are indicated by MYO7A staining (red) and nuclei are indicated by DAPI staining (blue). (B,C) Quantification of IHCs (B) and OHCs (C) in the apical, middle, and basal turns. N = 3–5 per groups. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (two-way ANOVA).
Mitochondrial fission and fusion are counterbalancing mechanisms that control the shape, size, and number of organelles. These dynamic processes are also critical in regulating cell death, mitophagy, and organelle distribution (Itoh et al., 2013). In many adherent cell types, such as Hela cells, mitochondria form elongated tubules that continually divide and fuse to form a dynamic interconnecting network (Suen et al., 2008). As shown in Figures 2A,B, we observed elongated and tubular mitochondria in HEI-OC1 cells, then they became highly fragmented upon the loss of Opa1. In Neuro 2a cells, in contrast, fragmented mitochondria were observed by MitoTracker staining even in untreated control cells (data not shown), which is consistent with a previous report that only 30% of mitochondria in this cell line had a length at least twice their width when observed by transmission electron microscopy (Qiao et al., 2017). Because of this, we were unable to observe a change in mitochondrial shape following Opa1 knockdown in Neuro 2a cells, as assessed by MitoTracker staining. In addition to this difference in mitochondrial shape in the effect of Opa1 knockdown, HEI-OC1 cells and Neuro 2a cells exhibited differences in the progression of cell death following Opa1 knockdown: Opa1 downregulation was associated with significant cell death at 48 h in Neuro 2a cells and at 72 h in HEI-OC1 cells (Figures 3A,B). Moreover, at 24 h after Opa1 depletion, more early apoptotic cells were seen in HEI-OC1 cells, whereas more late apoptotic cells were found in Neuro 2a cells (Figures 3G,H). This difference appeared to reflect the knockdown efficiencies of the two cell lines, indicating that Opa1 knockdown level is correlated to apoptosis.
Cisplatin is an effective chemotherapeutic agent that is widely used to treat a variety of malignant tumors. In recent years, cisplatin has been reported to affect many different pathways, including cell cycle arrest, apoptosis, proliferation, DNA repair, the TCA cycle, and glycolysis (Pabla and Dong, 2008; Li et al., 2016; Li H. et al., 2018; Gentilin et al., 2019; Liu W. et al., 2019; Zhang et al., 2020; Liu et al., 2021). The anti-tumor effect of cisplatin is mainly due to its ability to interfere with tumor cell proliferation (Wang and Lippard, 2005). Soon after its administration, cisplatin binds to nuclear DNA, where it blocks transcription and induces double-strand breaks leading to cell cycle arrest (Siddik, 2003). As cells from the cochlea are not proliferative, it is thought that mtDNA damage is a more likely cause of cisplatin-induced hearing loss than nuclear DNA damage (Hutchin and Cortopassi, 2000). Hair cells, spiral ganglion neurons, and the stria vascularis have been considered the three major targets of cisplatin ototoxicity (Schacht et al., 2012; He et al., 2019, 2020, 2021; Liu Y. et al., 2019; Zhou et al., 2020; Fu et al., 2021b). Several molecular mechanisms have been proposed as mediators of cisplatin-induced ototoxicity. Cisplatin was reported to stimulate ROS, which in turn triggers inflammatory pathways in the cochlea and promotes apoptotic and necrotic cell death (Rybak et al., 2009; Mukhopadhyay et al., 2012; Qi et al., 2019; Ding et al., 2020; Cheng et al., 2021; Fu et al., 2021a; Lv et al., 2021). As shown in Figure 2 of the present work, Opa1 deletion significantly elevated ROS generation and reduced MMP in both hair cells and neural cells. Cisplatin was reported to decrease Opa1 mRNA expression in the HK-2 kidney cell line (Choi et al., 2015). We therefore speculated that Opa1 expression might involve in cisplatin induced hair cell damage. Furthermore, our observation that HEI-OC1 and Neuro2a cells treated with cisplatin exhibited downregulation of Opa1 transcription and translation level (Figures 4C,D). Meanwhile fragmented mitochondria were observed when HEI-OC1 cells treated with cisplatin (Figure 4E), which may due to the reduction of Opa1. These results indicated Opa1 depletion would exacerbate cisplatin-induced damage.
OPA1 is essential for IMM fusion and plays a prominent role in maintaining the membrane ultrastructure of cristae and the function of mitochondria (Chan, 2020). OPA1 is ubiquitously expressed in all human tissue (Alexander et al., 2000; Delettre et al., 2000). The highest transcript level is found in retina, followed by the brain, testis, heart and skeletal muscle (Alexander et al., 2000).
Though OPA1 is expressed in multiple systems, OPA1 dysfunction diseases are all related to peripheric neuropathy. Furthermore, the rich expression of OPA1 is found in both white and brown adipocytes, and it’s expression elevated during lipid accumulation in adipocytes (Chu et al., 2017). OPA1 has been reported as a dual-specificity A-kinase anchoring protein associated with lipid droplets (Pidoux et al., 2011). At the meantime, many studies reported that Opa1 dysfunction has been involved in muscle mass and cardiac function (Wai et al., 2015; Tezze et al., 2017). Many OPA1 disease mutations have been identified, some of which result in OPA1 haploinsufficiency (Chun and Rizzo, 2017). Mutations in OPA1, particularly truncating mutations, most often manifest as DOA (Cipolat et al., 2004). DOA is characterized by progressive bilateral vision loss, along with hearing loss, myopathy, and peripheral neuropathy (Amati-Bonneau et al., 2008; Hudson et al., 2008). Opa1 dysfunction have been known resulted in RGC neuropathy, increasing the susceptibility of RGCs to apoptosis and vulnerability to oxidative stress (Williams et al., 2010). Given the crucial functions of OPA1, many studies have sought to increase Opa1 levels in vivo and in vitro as a potential therapeutic approach (Del Dotto et al., 2018). In a transgenic mouse model, a mild increase of the Opa1 isoform 1 protein level was found to protect mice from denervation-induced muscular atrophy, ischemic heart and brain damage, and hepatocellular apoptosis (Varanita et al., 2015). Moreover, Opa1 overexpression efficiently rescued the phenotypes of two mouse models of defective mitochondrial bioenergetics: Ndufs4–/– and Cox15sm/sm (Civiletto et al., 2015). Overexpression of each of the eight Opa1 isoforms or different constructs encoding isoform 1 of Opa1 in Opa1–/– MEFs revealed that any isoform could restore the crista structure, mtDNA abundance, and energetic efficiency independent of the mitochondrial network morphology (Del Dotto et al., 2017). Although it is known that mutation of OPA1 often leads to hearing loss, it remains unclear how OPA1 is involved in cochlear dysfunction. Our present results indicate that Opa1 plays key roles in maintaining mitochondrial function and preventing apoptosis induced by cisplatin damage. Future work is warranted to assess the potential of OPA1 as a treatment target for cochlear protection and/or repair.
Our results show that depletion of Opa1 affects the mitochondrial function and cell survival of cochlear cells. We herein report that Opa1 depletion is detrimental to the survival of cisplatin-treated cochlear cells both in vitro and in vivo, and that Opa1 overexpression protects cochlear cells against cisplatin-induced ototoxicity in cell lines and in organotypic tissue cultures. Our findings collectively suggest that Opa1 modulates mitochondrial function and is essential for the survival of cisplatin-exposed cochlear cells. In conclusion, our study provides strong cell biological evidence that Opa1 can protect against cisplatin-induced cochlear cell death by enhance mitochondrial function.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
The animal study was reviewed and approved by the Ethics Committee of the Shanghai Jiao Tong University School of Medicine.
TD designed, supervised the research, analyzed, and interpreted the data. TD and XZ performed the research. TD and ML wrote the manuscript. All authors reviewed the manuscript and discussed the work.
This study was funded by the Fundamental Research Program Funding of Ninth People’s Hospital affiliated to Shanghai Jiao Tong University School of Medicine (JYZZ096), and Cross Disciplinary Research Fund of Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine (JYJC202104).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the Electron Microscopy Center of Shanghai Institute of Precision Medicine, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, for their technical support and assistance in the electron microscopy. We also thank the Bioimaging Facility of Shanghai Institute of Precision Medicine, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, for their technical support and assistance in flow cytometry.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.744838/full#supplementary-material
Supplementary Figure 1 | The uncropped immunoblots for data presented in the Main Figures.
Adams, J. M., and Cory, S. (2007). The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 26, 1324–1337. doi: 10.1038/sj.onc.1210220
Alexander, C., Votruba, M., Pesch, U. E., Thiselton, D. L., Mayer, S., Moore, A., et al. (2000). OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 26, 211–215. doi: 10.1038/79944
Amati-Bonneau, P., Valentino, M. L., Reynier, P., Gallardo, M. E., Bornstein, B., Boissiere, A., et al. (2008). OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain 131(Pt 2), 338–351. doi: 10.1093/brain/awm298
Bette, S., Zimmermann, U., Wissinger, B., and Knipper, M. (2007). OPA1, the disease gene for optic atrophy type Kjer, is expressed in the inner ear. Histochem. Cell Biol. 128, 421–430. doi: 10.1007/s00418-007-0321-7
Chan, D. C. (2020). Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol. 15, 235–259. doi: 10.1146/annurev-pathmechdis-012419-032711
Chen, H., Detmer, S. A., Ewald, A. J., Griffin, E. E., Fraser, S. E., and Chan, D. C. (2003). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 160, 189–200. doi: 10.1083/jcb.200211046
Chen, H., Vermulst, M., Wang, Y. E., Chomyn, A., Prolla, T. A., McCaffery, J. M., et al. (2010). Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141, 280–289. doi: 10.1016/j.cell.2010.02.026
Chen, L., Liu, T., Tran, A., Lu, X., Tomilov, A. A., Davies, V., et al. (2012). OPA1 mutation and late-onset cardiomyopathy: mitochondrial dysfunction and mtDNA instability. J. Am. Heart Assoc. 1:e003012. doi: 10.1161/JAHA.112.003012
Cheng, C., Hou, Y., Zhang, Z., Wang, Y., Lu, L., Zhang, L., et al. (2021). Disruption of the autism-related gene Pak1 causes stereocilia disorganization, hair cell loss, and deafness in mice. J. Genet. Genomics. doi: 10.1016/j.jgg.2021.03.010
Choi, Y. M., Kim, H. K., Shim, W., Anwar, M. A., Kwon, J. W., Kwon, H. K., et al. (2015). Mechanism of Cisplatin-Induced cytotoxicity is correlated to impaired metabolism due to mitochondrial ROS generation. PLoS One 10:e0135083. doi: 10.1371/journal.pone.0135083
Chu, D.-T., Tao, Y., and Taskén, K. (2017). OPA1 in lipid metabolism: function of OPA1 in Lipolysis and Thermogenesis of Adipocytes. Hormone Metab. Res. 49, 276–285. doi: 10.1055/s-0043-100384
Chun, B. Y., and Rizzo, J. F. III (2017). Dominant Optic Atrophy and Leber’s hereditary optic neuropathy: update on clinical features and current therapeutic approaches. Semin. Pediatr. Neurol. 24, 129–134. doi: 10.1016/j.spen.2017.06.001
Cipolat, S., Martins, de Brito, O., Dal Zilio, B., and Scorrano, L. (2004). OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. U.S.A. 101, 15927–15932. doi: 10.1073/pnas.0407043101
Civiletto, G., Varanita, T., Cerutti, R., Gorletta, T., Barbaro, S., Marchet, S., et al. (2015). Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Metab. 21, 845–854. doi: 10.1016/j.cmet.2015.04.016
de Sa Junior, P. L., Camara, D. A. D., Porcacchia, A. S., Fonseca, P. M. M., Jorge, S. D., Araldi, R. P., et al. (2017). The roles of ROS in cancer heterogeneity and therapy. Oxid. Med. Cell. Longev. 2017:2467940. doi: 10.1155/2017/2467940
Del Dotto, V., Fogazza, M., Lenaers, G., Rugolo, M., Carelli, V., and Zanna, C. (2018). OPA1: How much do we know to approach therapy? Pharmacol. Res. 131, 199–210. doi: 10.1016/j.phrs.2018.02.018
Del Dotto, V., Mishra, P., Vidoni, S., Fogazza, M., Maresca, A., Caporali, L., et al. (2017). OPA1 isoforms in the hierarchical organization of mitochondrial functions. Cell Rep. 19, 2557–2571. doi: 10.1016/j.celrep.2017.05.073
Delettre, C., Griffoin, J. M., Kaplan, J., Dollfus, H., Lorenz, B., Faivre, L., et al. (2001). Mutation spectrum and splicing variants in the OPA1 gene. Hum. Genet. 109, 584–591. doi: 10.1007/s00439-001-0633-y
Delettre, C., Lenaers, G., Griffoin, J. M., Gigarel, N., Lorenzo, C., Belenguer, P., et al. (2000). Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 26, 207–210. doi: 10.1038/79936
Ding, Y., Meng, W., Kong, W., He, Z., and Chai, R. (2020). The Role of FoxG1 in the Inner Ear. Front. Cell Dev. Biol. 8:614954. doi: 10.3389/fcell.2020.614954
Frezza, C., Cipolat, S., Martins, de Brito, O., Micaroni, M., Beznoussenko, G. V., et al. (2006). OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126, 177–189. doi: 10.1016/j.cell.2006.06.025
Fu, X., An, Y., Wang, H., Li, P., Lin, J., Yuan, J., et al. (2021a). Deficiency of Klc2 induces low-frequency sensorineural hearing loss in C57BL/6 J mice and human. Mol. Neurobiol. doi: 10.1007/s12035-021-02422-w
Fu, X., Wan, P., Li, P., Wang, J., Guo, S., Zhang, Y., et al. (2021b). Mechanism and prevention of ototoxicity induced by aminoglycosides. Front. Cell. Neurosci. 15:692762. doi: 10.3389/fncel.2021.692762
Gao, S., Cheng, C., Wang, M., Jiang, P., Zhang, L., Wang, Y., et al. (2019). Blebbistatin inhibits neomycin-induced apoptosis in hair cell-like HEI-OC-1 cells and in cochlear hair cells. Front. Cell. Neurosci. 13:590. doi: 10.3389/fncel.2019.00590
Gentilin, E., Simoni, E., Candito, M., Cazzador, D., and Astolfi, L. (2019). Cisplatin-induced ototoxicity: updates on molecular targets. Trends Mol. Med. 25, 1123–1132. doi: 10.1016/j.molmed.2019.08.002
He, Z., Fang, Q., Li, H., Shao, B., Zhang, Y., Zhang, Y., et al. (2019). The role of FOXG1 in the postnatal development and survival of mouse cochlear hair cells. Neuropharmacology 144, 43–57. doi: 10.1016/j.neuropharm.2018.10.021
He, Z., Guo, L., Shu, Y., Fang, Q., Zhou, H., Liu, Y., et al. (2017). Autophagy protects auditory hair cells against neomycin-induced damage. Autophagy 13, 1884–1904. doi: 10.1080/15548627.2017.1359449
He, Z.-H., Li, M., Fang, Q.-J., Liao, F.-L., Zou, S.-Y., Wu, X., et al. (2021). FOXG1 promotes aging inner ear hair cell survival through activation of the autophagy pathway. Autophagy. doi: 10.1080/15548627.2021.1916194
He, Z.-H., Zou, S.-Y., Li, M., Liao, F.-L., Wu, X., Sun, H.-Y., et al. (2020). The nuclear transcription factor FoxG1 affects the sensitivity of mimetic aging hair cells to inflammation by regulating autophagy pathways. Redox Biol. 28:101364. doi: 10.1016/j.redox.2019.101364
Hoppins, S., Lackner, L., and Nunnari, J. (2007). The machines that divide and fuse mitochondria. Annu. Rev. Biochem. 76, 751–780. doi: 10.1146/annurev.biochem.76.071905.090048
Hudson, G., Amati-Bonneau, P., Blakely, E. L., Stewart, J. D., He, L., Schaefer, A. M., et al. (2008). Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain 131(Pt 2), 329–337. doi: 10.1093/brain/awm272
Hutchin, T. P., and Cortopassi, G. A. (2000). Mitochondrial defects and hearing loss. Cell. Mol. Life Sci. 57, 1927–1937. doi: 10.1007/PL00000673
Ishihara, N., Fujita, Y., Oka, T., and Mihara, K. (2006). Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 25, 2966–2977. doi: 10.1038/sj.emboj.7601184
Itoh, K., Nakamura, K., Iijima, M., and Sesaki, H. (2013). Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 23, 64–71. doi: 10.1016/j.tcb.2012.10.006
Kjer, P. (1959). Infantile optic atrophy with dominant mode of inheritance: a clinical and genetic study of 19 Danish families. Acta Ophthalmol. Suppl. 164(Suppl. 54), 1–147.
Leruez, S., Milea, D., Defoort-Dhellemmes, S., Colin, E., Crochet, M., Procaccio, V., et al. (2013). Sensorineural hearing loss in OPA1-linked disorders. Brain 136(Pt 7):e236. doi: 10.1093/brain/aws340
Li, A., You, D., Li, W., Cui, Y., He, Y., Li, W., et al. (2018). Novel compounds protect auditory hair cells against gentamycin-induced apoptosis by maintaining the expression level of H3K4me2. Drug Deliv. 25, 1033–1043. doi: 10.1080/10717544.2018.1461277
Li, H., Song, Y., He, Z., Chen, X., Wu, X., Li, X., et al. (2018). Meclofenamic acid reduces reactive oxygen species accumulation and apoptosis, inhibits excessive autophagy, and protects hair cell-Like HEI-OC1 cells from cisplatin-induced damage. Front. Cell. Neurosci. 12:139. doi: 10.3389/fncel.2018.00139
Li, Y., Li, A., Wu, J., He, Y., Yu, H., Chai, R., et al. (2016). MiR-182-5p protects inner ear hair cells from cisplatin-induced apoptosis by inhibiting FOXO3a. Cell Death Dis. 7:e2362. doi: 10.1038/cddis.2016.246
Liu, L., Chen, Y., Qi, J., Zhang, Y., He, Y., Ni, W., et al. (2016). Wnt activation protects against neomycin-induced hair cell damage in the mouse cochlea. Cell Death Dis. 7:e2136. doi: 10.1038/cddis.2016.35
Liu, W., Xu, L., Wang, X., Zhang, D., Sun, G., Wang, M., et al. (2021). PRDX1 activates autophagy via the PTEN-AKT signaling pathway to protect against cisplatin-induced spiral ganglion neuron damage. Autophagy. doi: 10.1080/15548627.2021.1905466
Liu, W., Xu, X., Fan, Z., Sun, G., Han, Y., Zhang, D., et al. (2019). Wnt Signaling Activates TP53-induced glycolysis and apoptosis regulator and protects against cisplatin-induced spiral ganglion neuron damage in the mouse cochlea. Antioxid. Redox Signal. 30, 1389–1410. doi: 10.1089/ars.2017.7288
Liu, Y., Qi, J., Chen, X., Tang, M., Chu, C., Zhu, W., et al. (2019). Critical role of spectrin in hearing development and deafness. Sci. Adv. 5:eaav7803. doi: 10.1126/sciadv.aav7803
Loson, O. C., Song, Z., Chen, H., and Chan, D. C. (2013). Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 24, 659–667. doi: 10.1091/mbc.E12-10-0721
Lv, J., Fu, X., Li, Y., Hong, G., Li, P., Lin, J., et al. (2021). Deletion of in mice does not alter auditory function. Front. Cell Dev. Biol. 9:630361. doi: 10.3389/fcell.2021.630361
Meire, F., De Laey, J. J., de Bie, S., van Staey, M., and Matton, M. T. (1985). Dominant optic nerve atrophy with progressive hearing loss and chronic progressive external ophthalmoplegia (CPEO). Ophthalmic Paediatr. Genet. 5, 91–97. doi: 10.3109/13816818509007861
Mishra, P., and Chan, D. C. (2014). Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 15, 634–646. doi: 10.1038/nrm3877
Mukhopadhyay, P., Horvath, B., Zsengeller, Z., Zielonka, J., Tanchian, G., Holovac, E., et al. (2012). Mitochondrial-targeted antioxidants represent a promising approach for prevention of cisplatin-induced nephropathy. Free Radic. Biol. Med. 52, 497–506. doi: 10.1016/j.freeradbiomed.2011.11.001
Olichon, A., Emorine, L. J., Descoins, E., Pelloquin, L., Brichese, L., Gas, N., et al. (2002). The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett. 523, 171–176. doi: 10.1016/s0014-5793(02)02985-x
Pabla, N., and Dong, Z. (2008). Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int. 73, 994–1007. doi: 10.1038/sj.ki.5002786
Palmer, C. S., Elgass, K. D., Parton, R. G., Osellame, L. D., Stojanovski, D., and Ryan, M. T. (2013). Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J. Biol. Chem. 288, 27584–27593. doi: 10.1074/jbc.M113.479873
Patten, D. A., Wong, J., Khacho, M., Soubannier, V., Mailloux, R. J., Pilon-Larose, K., et al. (2014). OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO J. 33, 2676–2691. doi: 10.15252/embj.201488349
Pidoux, G., Witczak, O., Jarnaess, E., Myrvold, L., Urlaub, H., Stokka, A. J., et al. (2011). Optic atrophy 1 is an A-kinase anchoring protein on lipid droplets that mediates adrenergic control of lipolysis. EMBO J. 30, 4371–4386. doi: 10.1038/emboj.2011.365
Qi, J., Liu, Y., Chu, C., Chen, X., Zhu, W., Shu, Y., et al. (2019). A cytoskeleton structure revealed by super-resolution fluorescence imaging in inner ear hair cells. Cell Discov. 5:12. doi: 10.1038/s41421-018-0076-4
Qiao, P., Zhao, F., Liu, M., Gao, D., Zhang, H., and Yan, Y. (2017). Hydrogen sulfide inhibits mitochondrial fission in neuroblastoma N2a cells through the Drp1/ERK1/2 signaling pathway. Mol. Med. Rep. 16, 971–977. doi: 10.3892/mmr.2017.6627
Rogers, C., Fernandes-Alnemri, T., Mayes, L., Alnemri, D., Cingolani, G., and Alnemri, E. S. (2017). Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 8:14128. doi: 10.1038/ncomms14128
Rybak, L. P., Mukherjea, D., Jajoo, S., and Ramkumar, V. (2009). Cisplatin ototoxicity and protection: clinical and experimental studies. Tohoku J. Exp. Med. 219, 177–186. doi: 10.1620/tjem.219.177
Santel, A., and Fuller, M. T. (2001). Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 114(Pt 5), 867–874.
Schacht, J., Talaska, A. E., and Rybak, L. P. (2012). Cisplatin and aminoglycoside antibiotics: hearing loss and its prevention. Anat. Rec. 295, 1837–1850. doi: 10.1002/ar.22578
Siddik, Z. H. (2003). Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene 22, 7265–7279. doi: 10.1038/sj.onc.1206933
Suen, D. F., Norris, K. L., and Youle, R. J. (2008). Mitochondrial dynamics and apoptosis. Genes Dev. 22, 1577–1590. doi: 10.1101/gad.1658508
Tezze, C., Romanello, V., Desbats, M. A., Fadini, G. P., Albiero, M., Favaro, G., et al. (2017). Age-Associated Loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metab. 25, 1374–1389.e6. doi: 10.1016/j.cmet.2017.04.021
Treft, R. L., Sanborn, G. E., Carey, J., Swartz, M., Crisp, D., Wester, D. C., et al. (1984). Dominant optic atrophy, deafness, ptosis, ophthalmoplegia, dystaxia, and myopathy. A new syndrome. Ophthalmology 91, 908–915. doi: 10.1016/s0161-6420(84)34214-2
van Gisbergen, M. W., Voets, A. M., Starmans, M. H., de Coo, I. F., Yadak, R., Hoffmann, R. F., et al. (2015). How do changes in the mtDNA and mitochondrial dysfunction influence cancer and cancer therapy? Challenges, opportunities and models. Mutat. Res. Rev. Mutat. Res. 764, 16–30. doi: 10.1016/j.mrrev.2015.01.001
Varanita, T., Soriano, M. E., Romanello, V., Zaglia, T., Quintana-Cabrera, R., Semenzato, M., et al. (2015). The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 21, 834–844. doi: 10.1016/j.cmet.2015.05.007
Wai, T., García-Prieto, J., Baker, M. J., Merkwirth, C., Benit, P., Rustin, P., et al. (2015). Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 350:aad0116. doi: 10.1126/science.aad0116
Wang, D., and Lippard, S. J. (2005). Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 4, 307–320. doi: 10.1038/nrd1691
Williams, P. A., Morgan, J. E., and Votruba, M. (2010). Opa1 deficiency in a mouse model of dominant optic atrophy leads to retinal ganglion cell dendropathy. Brain 133, 2942–2951. doi: 10.1093/brain/awq218
Yarosh, W., Monserrate, J., Tong, J. J., Tse, S., Le, P. K., Nguyen, K., et al. (2008). The molecular mechanisms of OPA1-mediated optic atrophy in Drosophila model and prospects for antioxidant treatment. PLoS Genet. 4:e6. doi: 10.1371/journal.pgen.0040006
Zhang, S., Zhang, Y., Dong, Y., Guo, L., Zhang, Z., Shao, B., et al. (2020). Knockdown of Foxg1 in supporting cells increases the trans-differentiation of supporting cells into hair cells in the neonatal mouse cochlea. Cell. Mol. Life Sci. 77, 1401–1419. doi: 10.1007/s00018-019-03291-2
Zhang, Y., Li, W., He, Z., Wang, Y., Shao, B., Cheng, C., et al. (2019). Pre-treatment With Fasudil prevents neomycin-induced hair cell damage by reducing the accumulation of reactive oxygen Species. Front. Mol. Neurosci. 12:264. doi: 10.3389/fnmol.2019.00264
Zhong, Z., Fu, X., Li, H., Chen, J., Wang, M., Gao, S., et al. (2020). Citicoline protects auditory hair cells against neomycin-induced damage. Front. Cell Dev. Biol. 8:712. doi: 10.3389/fcell.2020.00712
Keywords: OPA1, mitochondria, cisplatin, apoptosis, ototoxicity
Citation: Dong T, Zhang X, Liu Y, Xu S, Chang H, Chen F, Pan L, Hu S, Wang M and Lu M (2021) Opa1 Prevents Apoptosis and Cisplatin-Induced Ototoxicity in Murine Cochleae. Front. Cell Dev. Biol. 9:744838. doi: 10.3389/fcell.2021.744838
Received: 21 July 2021; Accepted: 30 August 2021;
Published: 21 September 2021.
Edited by:
Renjie Chai, Southeast University, ChinaReviewed by:
Sofia Waissbluth, Pontificia Universidad Católica de Chile, ChileCopyright © 2021 Dong, Zhang, Liu, Xu, Chang, Chen, Pan, Hu, Wang and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tingting Dong, ZHR0NjIzQGZveG1haWwuY29t; Min Lu, bHVtaW5zanR1QDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.