 Yi-Han Chiu1†
Yi-Han Chiu1† Chia-Jung Li
Chia-Jung Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 17 September 2021
Sec. Molecular and Cellular Pathology
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.743892
This article is part of the Research Topic Mitochondrial Dysfunction in Metabolic Diseases View all 6 articles
Mitochondria are double-membraned organelles that exhibit fluidity. They are the main site of cellular aerobic respiration, providing energy for cell proliferation, migration, and survival; hence, they are called “powerhouses.” Mitochondria play an important role in biological processes such as cell death, cell senescence, autophagy, lipid synthesis, calcium homeostasis, and iron balance. Fission and fusion are active processes that require many specialized proteins, including mechanical enzymes that physically alter mitochondrial membranes, and interface proteins that regulate the interaction of these mechanical proteins with organelles. This review discusses the molecular mechanisms of mitochondrial fusion, fission, and physiopathology, emphasizing the biological significance of mitochondrial morphology and dynamics. In particular, the regulatory mechanisms of mitochondria-related genes and proteins in animal cells are discussed, as well as research trends in mitochondrial dynamics, providing a theoretical reference for future mitochondrial research.
Mitochondria are organelles composed of an outer mitochondrial membrane (OMM) and an inner mitochondrial membrane (IMM), forming an inter-membrane space (IM) between. Reactions in the mitochondrial matrix provide energy for the normal functions of different cell types. Under physiological conditions, the OMM and IMM work together, continuously undergoing fusion and fission. While mitochondrial fusion and fission are essential for cellular homeostasis (Wu et al., 2018) and mitochondrial function, each physiological process plays a different role in mitochondrial function. Mitochondrial fusion can lead to an extension of mitochondrial structure, an increase in ATP content, and the transfer of various mitochondrial active substances to the newly fused mitochondria (Li et al., 2017). When cells are subjected to stress conditions (e.g., starvation or light stimulation), mitochondrial fusion reaches a maximum, producing enough ATP to counteract the stress (Silva Ramos et al., 2016). In contrast to fusion, mitochondrial fission produces new, smaller mitochondria that contribute to cell fission and mitosis. In addition, mitochondrial division, a process of mitochondrial proliferation, is ongoing and highly co-ordinated in eukaryotic cells and can also help maintain cellular constancy through mitophagy (Twig et al., 2008). Mitochondrial biogenesis and autophagy are highly regulated by fusion or fission, which also highlights the enormous impact of mitochondrial homeostasis on cellular function. As shown in Table 1, when mitochondrial fusion and/or fission are damaged, mitochondrial function is disrupted, which can eventually lead to neurodegenerative diseases, cardiovascular diseases, metabolic diseases, as well as many others (Archer, 2013, 2014).

Table 1. Summary of mitochondrial morphological effect proteins.
The structure of mitochondria and the synthesis of particular products are influenced by the needs of the cells in which they are located. Both the structure and metabolism of mitochondria play a key role in cell cycle progression, cell differentiation, development, the immune response, dynamic regulation of lipids and calcium, and apoptosis, in addition to influencing the generation of energy required by the cell (Nunnari and Suomalainen, 2012). Dynamin-related proteins (DRPs) mediate dynamic fusion and fission of mitochondria and remodel membrane shape, mainly through GTP-dependent self-assembly and GTP hydrolysis-mediated conformational changes (Lackner, 2013). Among different species, dynamin-1 protein (Dnm1, yeast) and dynamin-related protein 1 (Drp1, mammals) similarly lead to mitochondrial membrane fission, and mitofusins [Fzo1 (yeast)/Mfn1 and Mfn2 (mammals)] and the DRP Mgm1 (yeast)/Opa1 (mammals) mediate the fusion of outer the OMM and inner mitochondrial membranes the IMM, respectively (Gao and Hu, 2021).
Rapid mitochondrial fusion and fission are mechanisms for eliminating abnormal mitochondria from the cell (Yu et al., 2020). Mitochondrial fusion and fission are paired in a continuous progression, and cumulative probability analysis indicates that fusion triggers fission, but fission cannot affect the process of fusion that occurs later. Moreover, mitochondrial fusion is selective; after fission, depolarized daughter mitochondria are unlikely to participate in later fusions and will eventually be eliminated by autophagy (Chen Z. et al., 2018). As shown in Figure 1, mitochondrial fusion is mediated by the interaction of Mfn1 and Mfn2 at the OMM and Opa1 at the IMM; Fis1, Mff, and Mid49/51 drive the receptor-mediated enrichment of Drp1 from the cytoplasm to the OMM to the endoplasmic reticulum (ER)-labeled fission site, driving mitochondrial fission (Wai and Langer, 2016).
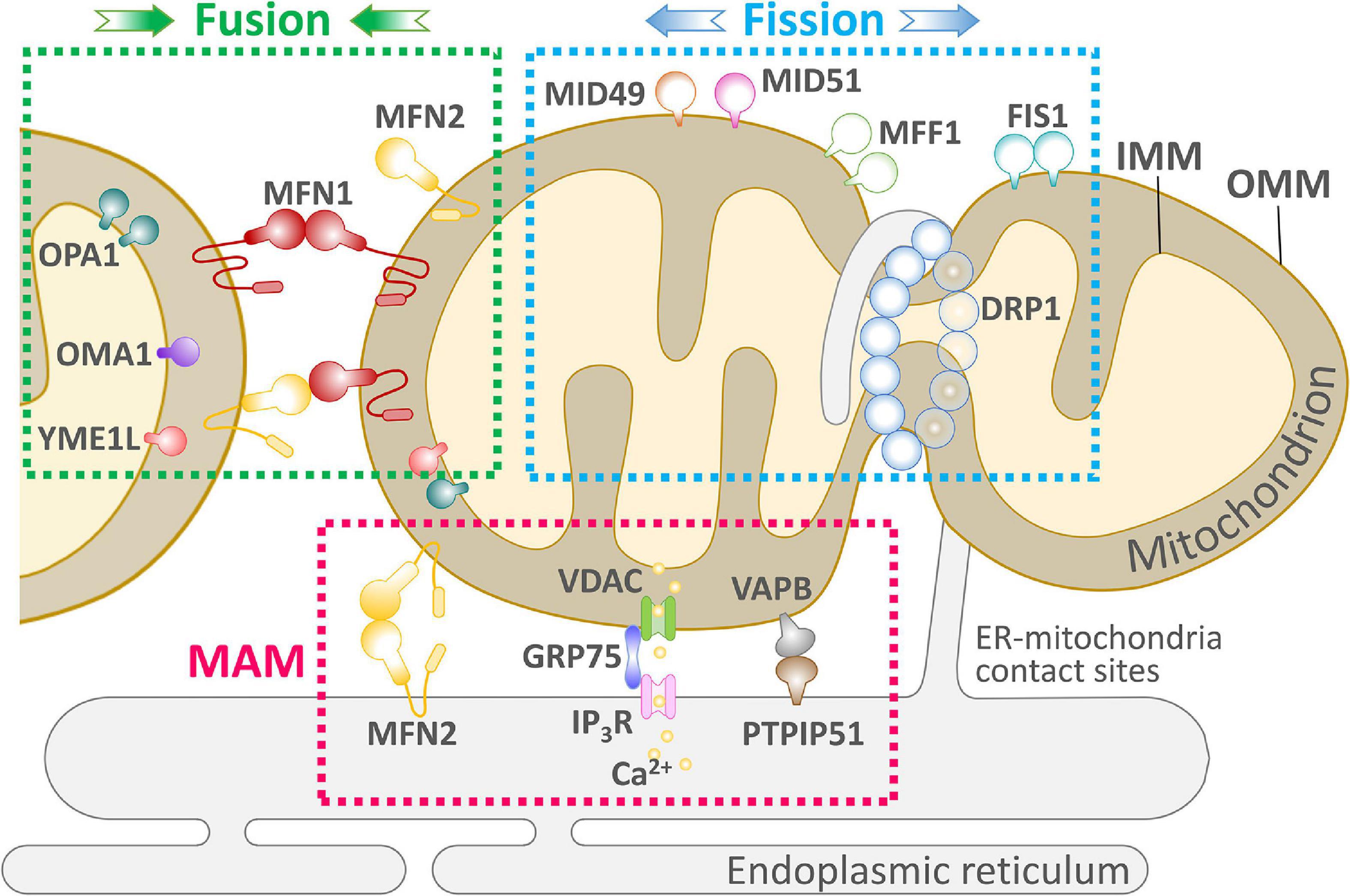
Figure 1. An overview of the key components of mitochondrial dynamics-ER communication and maintenance of cellular physiology. Mitochondria undergo two successive and opposite processes related to membranes: fission and fusion. Depending on the cellular environment and circumstances, the balance between fission and fusion can change, and the balance tends to favor one of these two processes. When cells are subjected to mild stress, mitochondria form an elongated and interconnected network to adapt to nutrient deficiencies while adapting to cellular stress. In contrast, under severe stress, mitochondria present a fragmented shape. Mitochondria and ER are in close proximity to each other and form the MAM. The MAM allows efficient Ca2+-stable exchange between the ER and mitochondria through the IP3R, VDAC mitochondrial calcium unidirectional transport protein (MCU) and PTP. Several proteins involved in mitochondrial fission, such as Mff and Fis1 mediate the localization of Drp1 on the ER, suggesting the existence of other unknown pathways to control ER-controlled mitochondrial dynamics.
Mitochondria change their morphological structure (elongating, shortening, bifurcating, bending, and swelling) by fusion and fission, maintaining the physiological functions of the cell. Mitochondrial fusion is mainly involved in the synthesis of new mitochondria and the repair of damaged mitochondria [e.g., mitochondrial DNA (mtDNA) mutations, decrease in membrane potential]. When damaged mitochondria fuse with normal mitochondria, mtDNA is reintegrated, repaired, and the membrane potential adjusts to normal levels (Gaziev et al., 2014). When an organism is under stress (e.g., disease, starvation, etc.), mitochondrial fusion leads to maximal ATP production, which supports the energy needs of the organism (Silva Ramos et al., 2016). Mitochondrial fusions include OMM fusions and IMM fusions. OMM fusions are mainly mediated by functionally similar Mfn1 and Mfn2 proteins (Table 1; Sloat et al., 2019).
Homologous mammalian genes (Mfn1 and Mfn2) regulate the fusion of the OMM and maintain the dynamic needs of reticulate mitochondria in the cell (Mishra and Chan, 2014). Both Mfn1 and Mfn2 are OMM transmembrane GTPases containing several conserved regions, including the amino-terminal GTP-binding structural domain, two coiled-coil structural domains, and the carboxy-terminus of a transmembrane structural domain (Chandhok et al., 2018). They are anchored to the OMM via a C-terminal transmembrane structural domain and mediate OMM fusion through homo- and heterotypic interactions with GTP hydrolysis. The impairment of either Mfn2 or GTPase activity prevents normal mitochondrial fusion, resulting in a small mitochondrial network in cells that fracture during mitochondrial fission (Escobar-Henriques and Joaquim, 2019).
Although Mfn1 and Mfn2 have similar functions and are even functionally complementary in specific situations, only mutations in Mfn2 cause significant physiological changes leading to neurodegenerative diseases, such as type 2A peroneal muscular atrophy neuropathy (Sloat et al., 2019). The two proteins play different functions in mitochondrial fusion, e.g., mitochondrial elongation under hypoxia is mainly regulated by sirt1-mediated Mfn1 deacetylation (Oanh et al., 2017), whereas Mfn1 mediates the formation of mitochondrial and ER contacts (Basso et al., 2018). The difference in functions of Mfn1 and Mfn2 may be due to the lack of the N-terminal RAS binding domain in Mfn1 (Chen et al., 2004). The membrane proteins responsible for mitochondrial fusion mediate fusion on adjacent mitochondria, indicating that the fusion complex needs to be between adjacent mitochondria to function. That is, the second coiled-coil domain of each protein forms an anti-parallel coiled-coil structure after forming a homo- or heterodimer, thereby restraining and regulating mitochondrial fusion. Therefore, mitochondrial fusion proteins promote fusion mainly through the action of molecular dimers on neighboring mitochondria (Koshiba et al., 2004).
Two forms of Opa1 have been identified in mammalian cells and tissues as a result of selective splicing and proteolytic cleavage by two endosomal peptidases: Oma1 and the i-AAA protease Yme1l. The Opa1 form obtained by selective splicing is the long-chain Opa1 (L-Opa1), whereas proteolytic cleavage generates the short-chain Opa1 (S-Opa1). L-Opa1 is anchored to the IMM through the N-terminal transmembrane structural domain and is released by proteolytic cleavage, producing the soluble form of S-Opa1 (Ishihara et al., 2006). Thus, L-Opa1 processing helps maintain the balance between mitochondrial fusion and cleavage, an important regulatory mechanism of the mitochondrial network structure (Anand et al., 2013). In contrast, IMM fusion is mainly mediated by Opa1, which is responsible for joining and integrating the two IMM systems and forming a complete IMM system when the OMM is fused (Figure 1). In mitochondria, Opa1 is proteolytically cleaved into two isoforms, long and short, but the two proteins alone are not physiologically active and only interact while mediating mitochondrial fusion (Del Dotto et al., 2018). The relative concentrations of the two isoforms are nearly equal in the cell, which is necessary for proper function in IMM fusion in normal cellular physiology (Sanchis-Gomar and Derbre, 2014). Mitochondrial fusion is closely related to cellular physiological function, and fusion transfers mitochondrial proteins and mtDNA to newly synthesized mitochondria, which helps prevent the accumulation of damaged mtDNA (Chan, 2020). In contrast, the rate of mitochondrial fusion can be affected by the physicochemical properties of mitochondria, such as changes in mitochondrial membrane potential. In a study of pancreatic cancer cells in G1 phase with high/low metabolism to alter the cellular mitochondrial membrane potential, the mitochondrial membrane potential increased; oxidative phosphorylation (OxPhos) as well as mitochondrial respiration and fusion rates were correspondingly elevated (Little et al., 2020).
Mitochondrial fission plays a balancing role with mitochondrial fusion in mitochondrial dynamics. There are two main modes of mitochondrial fission: (1) “Inside-out,” in which the IMM fractures first, eventually leading to fracture of the OMM, and (2) “Extrusion,” in which the mitochondria accumulate from the fission point inward, leading to mitochondrial fission. Currently, most of the proteins known to be associated with mitochondrial fission are GTPase family proteins, including Drp1, Fis1, Mff, and Mid49/51 (Chen K. H. et al., 2018; Kamerkar et al., 2018; Yu et al., 2019a,b). Also present are dynamin protein 2 (Dyn2/Dnm2), and Bax interacting factor 1 (Bif1), which assist Drp1 in regulating mitochondrial fission (Mahecic et al., 2021), and ganglioside-induced differentiation-associated protein 1 (GDAP1) (Rzepnikowska and Kochanski, 2018), death-associated protein 3 (DAP3) (Xiao et al., 2015), and mitochondria protein 18 (MTP18) (Aung et al., 2019). Maintaining physiological constancy consumes energy, and the energy required for mitochondrial fission is mainly provided by the hydrolysis of GTP by GTPases. However, the fission process requires the involvement of the ER, which helps initiate mitochondrial fission before GTPase hydrolysis. As shown in Figure 1, mitochondrial fission begins with “tagging” of the mitochondrial membrane by the ER and the ER-mitochondrial contact sites (ERMCSs), resulting in the recruitment of Drp1 by the Drp1 receptor. Drp1 then forms oligomers around the fission site, further constricting membrane GTPase activity, tightening the mitochondria, and leading to Dyn1/Dyn2 replenishment, while allowing additional GTPase activity. Dyn2 replenishment allows for additional GTP hydrolysis to complete the cleavage process, producing two separate mitochondria. Mitochondrial fission is essential for intracellular mitochondrial remodeling and rearrangement, and for the transfer of healthy mtDNA and other active material to daughter cells after mitotic fission (Pagliuso et al., 2018). Drp1 performs OMM fission, mediating mitochondrial fission, and in response to specific cellular signals, Drp1 moves from the cytoplasm to the OMM, where it clusters into a loop at the fission site. Several Drp1 receptor recruitment elements at the OMM have been identified, including Fis1, Mff, Mid49, and Mid51 (Civiletto et al., 2015). The mechanism of inner membrane fission is still unclear. Two IMM proteins, S-Opa1 and MTP18, have been suggested to have an important role in mitochondrial fission and may be part of the endosomal fission mechanism (Wai and Langer, 2016).
Contact sites exist between mitochondria and the ER and these sites are critical for phospholipid synthesis, Ca2+ homeostasis, and labeling of cleavage sites (Friedman et al., 2011). In mammals, the function of the contact sites is mainly regulated by Mfn2 (de Brito and Scorrano, 2008). Although mitochondria are dynamic, the location of mitochondrial and ER contact sites is kept relatively constant; the exact mechanism of regulation needs to be further investigated (Friedman et al., 2010). Many researchers have suggested that the role of the ER is to mark the initiation site of mitochondrial fusion and fission (Friedman et al., 2011). IFN2 located in the ER induces the polymerization of actin at the interface between the ER and mitochondrial contact sites, driving contraction of mitochondrial fission sites, probably because Drp1 oligomers cannot wrap the mitochondrial membrane and induce fission in the absence of receptors (Korobova et al., 2013). Depending on the cell type, mitochondria are generally greater than 200 nm in diameter, and the Drp1 oligomers recognize and bind mitochondrial membranes with diameters of 110–130 nm before further contraction of the membrane structure. In contrast, ER contraction sites are approximately 138–146 nm in diameter, suggesting that ER-initiated contraction precedes the formation of Drp1 oligomers at mitochondrial membrane contraction sites (Friedman et al., 2011). This suggests that the ER plays an important role in initiating contraction prior to mitochondrial contraction, and it also demonstrates that the ER is the initiation site for labeling mitochondrial contraction and fission (Korobova et al., 2013).
Mitochondrial fission is mainly regulated by Drp1, Fis1, Dyn1/2, Mff, and other related genes and proteins. Drp1 is primarily localized to the cytoplasmic matrix, but Drp1 does not possess a lipid-binding pleckstrin homology domain and cannot bind directly to the mitochondrial membrane. Therefore, its mediation of mitochondrial fission requires recruitment to the mitochondria (Mears et al., 2011). As shown in Figure 1, mitochondrial proteins must act as Drp1 receptors to cluster Drp1 into the OMM. In mammalian cells, Drp1 receptors identified include Fis1, Mff, and Mid49/51 (Otera et al., 2010; Palmer et al., 2011; Zhao et al., 2011; Loson et al., 2013); all three can recruit Drp1 to the OMM with Mff having the strongest binding effect (Loson et al., 2013). Once Drp1 is recruited to the OMM, it forms a cyclic oligomer and uses its GTPase activity to further contract the mitochondria, but it cannot complete the fission process due to the limitation of contraction strength (Frohlich et al., 2013). Dyn2/Dnm2 is a mitochondrial contraction and fission protein (Scholtes and Giguere, 2021) and, similar to Drp1, can form a cyclic oligomer around the membrane, further contracting the mitochondria and completing fission with the hydrolysis of GTP (Ferguson and De Camilli, 2012). In mammals, the two fission proteins must work in concert without one another.
In addition, the two GTPase proteins Dyn2/Dnm2 and Drp1 are depleted during mitochondrial fission, and when either protein is depleted, the mitochondria become elongated and display a finer tubular network. Mitochondrial fission begins when Drp1 is activated and is subsequently translocated from the cytoplasmic matrix to the OMM where it binds to the receptor, thereby initiating fission (Lee et al., 2016). However, the rate of fission depends on the amount of Drp1 binding and the level of phosphorylation modifications. For example, in neuronal cell, Ca2+ flux inward through voltage-dependent Ca2+ channels leads to a rapid arrest of mitochondrial motility and induces mitochondrial fission. The Ca2+ channels activate Ca2+/calmodulin-dependent protein kinase (CaMK)-alpha which in turn stimulates Drp1 serine (s600) phosphorylation, resulting in an increased affinity for Fis1 and promoting mitochondrial fission (Portz and Lee, 2021).
During the transition from the G1/S phase in rat kidney cells, when large amounts of ATP are required by cells for nucleic acid and protein synthesis, the rate of mitochondrial fusion is significantly enhanced and the level of ATP in cells is much higher than during other periods (Mitra et al., 2009). However, there are many factors that alter the mitochondrial membrane potential, such as aging, apoptosis, and disease. Thus, whether there is a positive correlation between the mitochondrial fusion rate and membrane potential needs further investigation. When mice were exposed to high-fat diets, Mfn1 and Mfn2 expression was significantly decreased, accompanied by mitochondrial respiratory dysfunction and decreased skeletal muscle ATP levels (Liu et al., 2014). After swimming training in obese mice, ER stress was initiated and mitochondria-associated membrane content was increased, which in turn led to enhanced mitochondrial function and increased skeletal muscle ATP (Zhang et al., 2020). In severely starved mice, the rate of mitochondrial fusion and the mitochondrial network were significantly increased compared to well-fed mice; this resulted in elevated cellular AMP levels and activation of protein kinase A (PKA). In turn, PKA phosphorylated Drp1, resulting in a slowing of the mitochondrial fission rate, elevated fusion rate and OxPhos levels, and high expression of ATP synthase, maintaining the cellular ATP supply (Gomes et al., 2011). While starvation and stress can lead to the generation of more energy to maintain cellular function, they also increase the risk of mitochondrial reactive oxygen species (ROS) production and oxidative damage (Lee et al., 2014). Experiments using UV light and actinomycin D stimulation of mouse fibroblasts found that with enhanced mitochondrial fusion, ATP content and OxPhos levels were elevated and ROS levels were significantly elevated with increased mitochondrial damage and diminished function (Tondera et al., 2009). From the above, it is clear that there is a linear relationship between mitochondrial function and fusion rate, i.e., the rate of mitochondrial fusion is strongest when the fusion rate reaches some critical point, but at the same time, the risk of oxidative damage is greatest (Farmer et al., 2018).
In fibroblast studies, ionizing radiation triggered CaM-K alpha and activated Drp1 (s616) phosphorylation, thus accelerating the rate of lysis. However, when CaM-K alpha activity was inhibited, both Drp1 (s616) phosphorylation and lysis rates were significantly suppressed (Salaciak et al., 2021). Meanwhile, PKA mediated mouse fibroblast Drp1 (s637) phosphorylation allowing it to interact with Mff and be recruited to the OMM, resulting in an accelerated rate of mitochondrial fission (Yu et al., 2019b). Similarly, differences in nutritional status affect the rate of mitochondrial fission and ATP content in cells. Islet β-cells under high glucose and high fat nutritional conditions exhibit an increased mitochondrial fission rate, decreased FIS1 expression, reduced fusion rate, and reduced ROS production, triggering a mechanism that may be protective of normal cellular function by reducing excessive cellular ATP consumption (Molina et al., 2009). In skeletal muscle cells of high-fat dietary mice, Fis1 and Drp1 expression increased and fusion protein Mfn 1/2 expression decreased compared with control mice (Liu et al., 2014). Taken together, it is clear that when the organism (cell) contains high lipid levels, the rate of mitochondrial ATP synthesis is accelerated, which is also accompanied by an increase in ROS synthesis, increasing the risk of mitochondrial oxidative damage. Mitochondrial fission also removes damaged mitochondria, and healthy mtDNA and active substances are passed to the offspring mitochondria.
Mitochondrial hyper fission leads to mitochondrial fragmentation and damage, decreased mitochondrial membrane potential, increased permeability, and decreased ATP production. At the same time, the disruption of mitochondrial autophagy leads to an excessive accumulation of intracellular mitochondria with abnormal function and high levels of oxygen radical production, producing large amounts of neurotoxic substances and eventually causing neurodegeneration disease (Portz and Lee, 2021; Yang et al., 2021). Parkinson’s disease, Alzheimer’s disease, and Huntington’s chorea, several neurodegenerative diseases are associated with mitochondrial dysfunction (Stanga et al., 2020; Table 2). Mendelian genetics seems to play an important role in the link between mitochondrial dysfunction and Parkinson’s disease (Buneeva et al., 2020). All three gene products responsible for familial autosomal recessive Parkinson’s disease can be found in mitochondria, including the Pink1 protein (a protein that maintains free radical metabolism, calcium homeostasis, and mtDNA in mitochondria) (Schapira et al., 2009; Schapira, 2012). It is also highly likely that Alzheimer’s disease is associated with polymorphisms in the OMM protein Tomm40 gene (Devi et al., 2006), which may be associated with age as an important risk factor for triggering Alzheimer’s disease (Roses et al., 2010). Similarly, Huntington’s chorea is associated with abnormal calcium handling due to mitochondrial defects, increased calcium-induced sensitivity to opening of mitochondrial permeability pores, and reduced mitochondrial respiration (Lim et al., 2008). Mutant Huntington proteins bind to the mitochondrial membrane, impairing axonal transport in mitochondria and reducing the synaptic ATP concentration (Orr et al., 2008). Mutant Huntington proteins also interact with and increase the sensitivity of inositol 1,4,5-trisphosphate receptors on mitochondria-associated membranes, thereby promoting calcium dysregulation in Huntington’s disease (Kaltenbach et al., 2007).
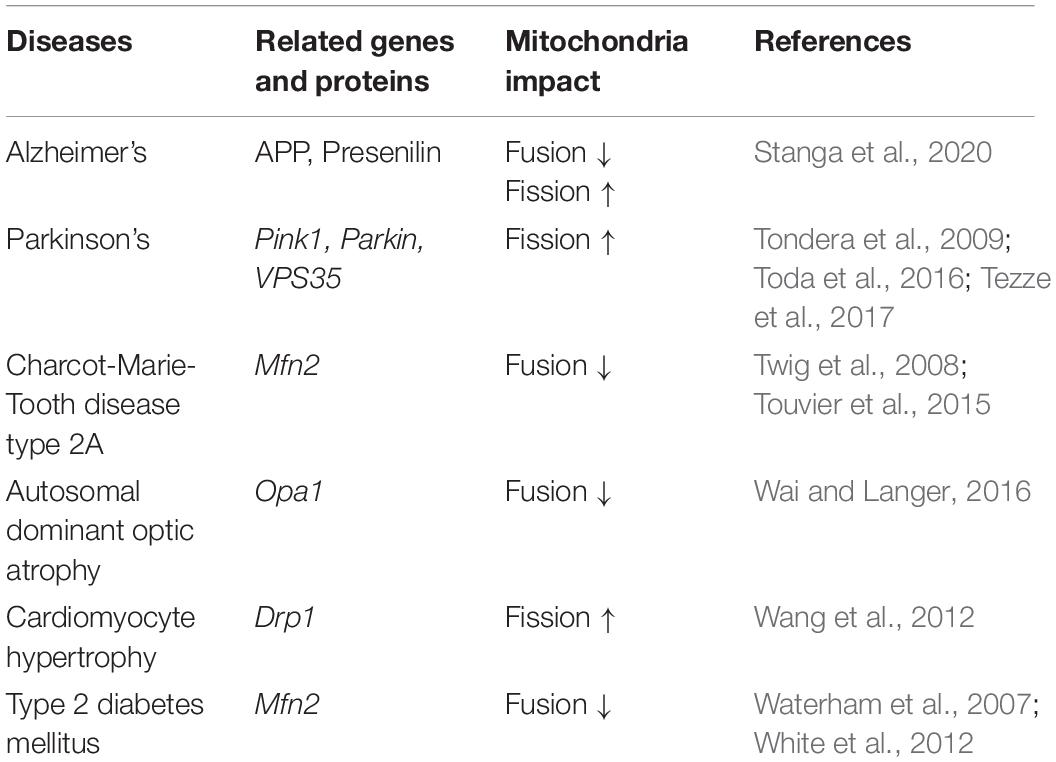
Table 2. Common diseases related to mitochondria dynamics.
Mitochondrial DNA damage and malfunction are closely related to OxPhos and further affect oxidative stress. Diabetes associated with mitochondrial DNA abnormalities accounts for up to 1% of all cases of diabetes (Murphy et al., 2008), referred to as “mitochondrial diabetes.” Glucose induces neuronal activation and reduces ROS production in the ventral medial nucleus of the hypothalamus owing to the key role of uncoupling protein 2 (UCP2) (Toda et al., 2016), which regulates neuronal activity by controlling ROS production (Coppola et al., 2007; Andrews et al., 2008; Diano and Horvath, 2012). Furthermore, mitochondria in the ventral medial nucleus of the hypothalamus depend on UCP2 for regulation of the fission process (Toda et al., 2016). In summary, blood glucose concentration can regulate UCP2 secretion in the ventral medial nucleus of the hypothalamus, which in turn affects mitochondrial fission.
An imbalance in mitochondrial dynamics can cause pancreatic β-cell dysfunction and insulin resistance, thus inducing diabetes. An imbalance leads to a decrease in the level of mitochondrial OxPhos in the cell, proton efflux, mitochondrial membrane potential, and an increase in the production of ROS, which puts the cell in a state of oxidative stress. The inflammatory effect of the stress response reduces the sensitivity of the insulin signaling pathway (Gonzalez-Franquesa and Patti, 2017). First the expression level of Mfn2 affects the insulin signaling pathway (Mahdaviani et al., 2017); individuals with low Mfn2 expression in the liver are more likely to develop insulin resistance (Sebastian et al., 2012). Second, the expression level of Drp1 affects insulin resistance, and the mitochondrial Drp1 content is significantly increased in a mouse model of hyperinsulinemia. Inhibition of mitochondrial fission has been shown to improve obesity-induced insulin resistance in skeletal muscle. Myocardial biopsies from diabetic or hypoglycemic patients showed significantly higher mitochondrial deletion in myocytes than that in non-diabetic patients, and the number of deletions was higher and the sizes differed. Examination of skeletal muscle in diabetic patients revealed reduced levels of mitochondrial OxPhos (Kim et al., 2011). Electron microscopic observation of mitochondrial morphology in pancreatic islet β-cells of diabetic and obese patients revealed abnormal mitochondrial morphology, including fragmentation of mitochondria, destruction of mitochondrial cristae morphology, and a significantly lower volume and number of mitochondria than that of normal subjects. This is a more intuitive representation of the imbalance of mitochondrial fusion in the insulin-resistant state (Gonzalez-Franquesa and Patti, 2017).
This may be due to mitochondrial fragmentation caused by increased mitochondrial fission or decreased fusion. Therefore, mitochondrial dynamics are closely associated with the development of diabetes. It is hypothesized that an imbalance in mitochondrial dynamics under any circumstances can lead to β-cell dysfunction, manifested by decreased insulin secretion, β-cell failure, and eventually β-cell apoptosis, inducing the development of diabetes mellitus. One of the effective ways to improve the function of pancreatic β-cells is through regulation of Mfn2, Opa1, and other related mitochondrial dynamics proteins to maintain the balance of mitochondrial dynamics.
When vascular endothelial cells (VEC) are stimulated by oxidative stress, their mitochondrial morphology becomes abnormal, as evidenced by increases in mitochondrial fission, abnormal mitochondrial structure, mitochondrial ROS production, and free radicals, and a decrease in OxPhos levels; VEC dysfunction occurs. Interference with mitochondrial fission is associated with myocardial ischemia/reperfusion injury; in animal models, inhibition of Drp1 expression in cardiomyocytes reduced myocardial infarct size and thus myocardial protection (Sharp, 2015). A significant decrease in Mfn2 and Drp1 expression was reported in heart failure. By interfering with mitochondrial fusion- and fission-related proteins, mitochondrial dynamics are affected, which in turn disrupts the normal physiological function of the VEC and induces many cardiovascular diseases (Givvimani et al., 2014). In addition, Mfn2 prevents cardiovascular I/R damage by enhancing mitochondrial fusion and activating the AMPK/Sirt3 signaling pathway (Liu et al., 2020). Furthermore, inhibition of Fis1 or Drp1 expression prevents high-glucose-induced ROS generation and mitochondrial fragmentation in venous endothelial cells with elevated levels of Fis1 protein in patients with atherosclerosis and increased abundance of Fis1 and Drp1 proteins in human aortic endothelial cells cultured in high-glucose medium (Mai et al., 2010).
Mitochondrial dynamics imbalance leads to increased production of ROS in mitochondria, which has the effect of inducing Drp1 activation and fragmentation of mitochondria; the addition of ROS scavengers can prevent this process. Recently, it was found that the addition of mitochondrial division inhibitor 1 (Mdivi-1) or the knockdown of Drp1 in microglia inhibited ROS production (Maimaitijiang et al., 2016). In addition, ROS can lead to the release of Opa1 isoforms and cytochrome C from mitochondria into the cytoplasm, reduce apoptosis, and increase mitochondrial and cellular morphological stability (McBride and Soubannier, 2010; Huttemann et al., 2011). Interleukin-6 reduces mitochondrial Mfn2 protein expression by upregulating Fis1 expression and downregulating PGC-1α, biasing the mitochondrial dynamics balance toward fission (White et al., 2012). In the presence of tumor necrosis factor α (TNF-α), adipocytes 3T3-L1 exhibit abnormal mitochondrial morphology, increased Mfn2, and significantly increased Drp1 (Chen et al., 2010). However, TNF-α activates nuclear factor-κB (NF-κB) in pancreatic β-cells, and the activated NF-κB increases Opa1 expression, promotes mitochondrial fusion, and increases the level of mitochondrial OsPhos and ATP production, meeting the energy demand of mitochondrial fusion (Chan et al., 2012; Baltrusch, 2016). To avoid a critical reduction in total mitochondria, the TNF-α/NF-κB/Opa1 pathway is necessary to rebalance the system by increasing fusion proteins and enhancing the structure of mitochondrial ridges and the efficiency of the respiratory chain.
Previous studies have demonstrated the important role of mitochondrial dynamics in maintaining muscle and mitochondrial physiological functions. Mice deficient in both mitochondrial fusion proteins Mfn-1, -2 in skeletal muscle exhibit severe mitochondrial dysfunction, mitochondrial DNA damage and severe developmental defects, which in turn affect a significant reduction in motor performance (Bell et al., 2019). In addition, Opa1 deficiency leads to mitochondrial kinetic imbalance, oxidative stress and inflammation (Tezze et al., 2017; Rodriguez-Nuevo et al., 2018), and Opa1 deficiency also promotes the secretion of FGF21 in skeletal muscle, leading to altered lipid homeostasis, inflammation and aging of different tissues (Tezze et al., 2017). Furthermore, inhibition of mitochondrial fission by gene silencing of Fis1 and Drp1 in skeletal muscle has been shown to reduce muscle atrophy caused by overexpression of transcription factor FoxO3a (Romanello et al., 2010). Interestingly, both low and high Drp1 levels impair skeletal muscle growth and development, and Drp1 overexpression late in life triggers mild muscle atrophy and decreased mitochondrial mass (Touvier et al., 2015; Giovarelli et al., 2020; Leduc-Gaudet et al., 2021). This also suggests that abnormalities in mitochondrial fission are also a factor in muscle disease.
Mitochondrial diseases are challenging to diagnose and treat clinically because of the high genetic and clinical heterogeneity of the disease. Previous preclinical studies have reported that benzofibrate improves mitochondrial function in Drp1-deficient cells with abnormal mitochondrial fission and function (Douiev et al., 2020). Benzofibrate is a small molecule activator of peroxisome proliferator-activated receptor alpha (PPARα), which upon activation increases the expression of many transcriptional regulators and thus affects the expression of downstream groups (Djouadi and Bastin, 2019). Genetic abnormalities in mitochondrial dynamics also include mutations in the Mfn2 or Opa1 genes, manifesting as Charcot-Marie-Tooth type 2A and autosomal dominant optic atrophy, respectively (Alexander et al., 2000; Zuchner et al., 2004), as well as by affecting Drp1 and Mff (Waterham et al., 2007). Therefore, specific inhibitors affecting mitochondrial fusion and fission have been studied to improve the pathophysiology (Cassidy-Stone et al., 2008; Wang et al., 2012; Qi et al., 2013). In contrast, P110, a peptide inhibitor with reduced Drp1 enzyme activity, can prevent Drp1/Fis1 interaction between neurons, which can effectively improve mitochondrial morphology and reduce mitochondrial fission, thus reducing neuronal axon loss in Parkinson’s disease patients and thus acting as a neuroprotective agent (Qi et al., 2013). This effect may be related to the reduction of apoptotic signaling pathway in neuronal cells mediated by Drp1-dependent p53 mitochondrial translocation. S3 acts on the mitochondrial deubiquitinating enzyme USP30, which regulates mitochondrial morphology by modulating Mfn1 and Mfn2 deubiquitination, and this action increases Mfn1 and Mfn2 activity and induces mitochondrial fusion, suggesting its potential therapeutic value for diseases such as insulin resistance (Yue et al., 2014). In addition, resveratrol, mitochondria-targeted antioxidants, and caloric restriction may improve insulin sensitivity and mitochondrial function in individuals with type 2 diabetes (Cao et al., 2018). Metformin and resveratrol protect mitochondrial integrity by inhibiting Drp1 activity and protect cellular function in hyperglycemic conditions (Maresca et al., 2013). Mitochondria-targeted antioxidants such as SS-31 protect normal cellular function in hyperglycemic environments by regulating mitochondrial membrane potential, inhibiting NADPH oxidase-4 and transforming growth factor-β1 expression, and activating p38MAPK. In addition, heat shock protein 70 can improve mitochondrial bioenergetic metabolism and reverse diabetic sensory neuropathy. All of the above drugs could be potential agents for the treatment of diabetic neuropathies, among others.
While fragmented mitochondrial networks are a component of disease pathophysiology, increasing mitochondrial fusion or reducing mitochondrial fission are both enhancements to the mitochondrial network in these cells and are expected to prevent functional deterioration. However, genetic and chemical approaches implemented in model systems that rebalance mitochondrial kinetics, restored mitochondrial structure and function alleviate disease-related symptoms. Research focused on dissecting the mechanisms of mitochondrial fusion and division and understanding the integration of these processes with other cellular pathways will be an important aspect of developing effective therapies, but enhancing the regulation of mitochondrial transport to improve cellular function in disease is a different idea and therapeutic strategy.
A variety of factors and physiological and biochemical processes affect the degree of mitochondrial fusion or fission by regulating relevant factors that influence morphological changes in mitochondria, resulting in mitochondria showing different morphological characteristics. For example, increasing the degree of mitochondrial fusion or decreasing the degree of mitochondrial fission leads to the formation of larger, but fewer mitochondria in the cell. Conversely, decreasing the degree of mitochondrial fusion or increasing the degree of mitochondrial fission leads to the formation of smaller, but a large number of mitochondria. Thus, it can be speculated that when a disease or factor is associated with morphological changes in mitochondria, the process that is influenced can be identified. In summary, the role of mitochondrial fusion and fission proteins in the morphological structure and function of mitochondria has been elucidated. However, how breakthroughs in understanding mitochondrial dynamics can be better applied to existing research studies in the intervention of cell growth, aging, apoptosis, and other physiological and pathological processes needs to be studied more deeply.
Y-HC and C-JL: writing—original draft preparation. S-CL, C-HK, and C-JL: writing—review and editing. C-JL: supervision and funding acquisition. All authors have read and agreed to the published version of the manuscript.
This research was funded by the Ministry of Science Technology (MOST 109-2314-B-075B-002 and MOST 110-2314-B-075B-011) and the Kaohsiung Veterans General Hospital (VGHKS109-106 and KSVGH110-142).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alexander, C., Votruba, M., Pesch, U. E., Thiselton, D. L., Mayer, S., Moore, A., et al. (2000). OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 26, 211–215. doi: 10.1038/79944
Anand, R., Langer, T., and Baker, M. J. (2013). Proteolytic control of mitochondrial function and morphogenesis. Biochim. Biophys. Acta 1833, 195–204. doi: 10.1016/j.bbamcr.2012.06.025
Andrews, Z. B., Liu, Z. W., Walllingford, N., Erion, D. M., Borok, E., Friedman, J. M., et al. (2008). UCP2 mediates ghrelin’s action on NPY/AgRP neurons by lowering free radicals. Nature 14, 846–851. doi: 10.1038/nature07181
Archer, S. L. (2013). Mitochondrial dynamics–mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 5, 2236–2251. doi: 10.1056/nejmra1215233
Aung, L. H. H., Li, Y. Z., Yu, H., Chen, X., Yu, Z., Gao, J., et al. (2019). Mitochondrial protein 18 is a positive apoptotic regulator in cardiomyocytes under oxidative stress. Clin. Sci. 15, 1067–1084. doi: 10.1042/cs20190125
Baltrusch, S. (2016). Mitochondrial network regulation and its potential interference with inflammatory signals in pancreatic beta cells. Diabetologia 59, 683–687. doi: 10.1007/s00125-016-3891-x
Basso, V., Marchesan, E., Peggion, C., Chakraborty, J., von Stockum, S., Giacomello, M., et al. (2018). Regulation of ER-mitochondria contacts by Parkin via Mfn2. Pharmacol. Res. 138, 43–56. doi: 10.1016/j.phrs.2018.09.006
Bell, M. B., Bush, Z., McGinnis, G. R., and Rowe, G. C. (2019). Adult skeletal muscle deletion of Mitofusin 1 and 2 impedes exercise performance and training capacity. J. Appl. Physiol. (1985) 126, 341–353. doi: 10.1152/japplphysiol.00719.2018
Buneeva, O., Fedchenko, V., Kopylov, A., and Medvedev, A. (2020). Mitochondrial dysfunction in Parkinson’s disease: focus on mitochondrial DNA. Biomedicines 8:591. doi: 10.3390/biomedicines8120591
Cao, M. M., Lu, X., Liu, G. D., Su, Y., Li, Y. B., and Zhou, J. (2018). Resveratrol attenuates type 2 diabetes mellitus by mediating mitochondrial biogenesis and lipid metabolism via Sirtuin type 1. Exp. Ther. Med. 15, 576–584.
Cassidy-Stone, A., Chipuk, J. E., Ingerman, E., Song, C., Yoo, C., Kuwana, T., et al. (2008). Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 14, 193–204. doi: 10.1016/j.devcel.2007.11.019
Chan, D. C. (2020). Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol. 24, 235–259. doi: 10.1146/annurev-pathmechdis-012419-032711
Chan, J. Y., Biden, T. J., and Laybutt, D. R. (2012). Cross-talk between the unfolded protein response and nuclear factor-kappaB signalling pathways regulates cytokine-mediated beta cell death in MIN6 cells and isolated mouse islets. Diabetologia 55, 2999–3009. doi: 10.1007/s00125-012-2657-3
Chandhok, G., Lazarou, M., and Neumann, B. (2018). Structure, function, and regulation of mitofusin-2 in health and disease. Biol. Rev. Camb. Philos. Soc. 93, 933–949. doi: 10.1111/brv.12378
Chen, K. H., Dasgupta, A., Lin, J., Potus, F., Bonnet, S., Iremonger, J., et al. (2018). Epigenetic dysregulation of the dynamin-related protein 1 binding partners MiD49 and MiD51 increases mitotic mitochondrial fission and promotes pulmonary arterial hypertension: mechanistic and therapeutic implications. Circulation 138, 287–304. doi: 10.1161/circulationaha.117.031258
Chen, K. H., Guo, X., Ma, D., Guo, Y., Li, Q., Yang, D., et al. (2004). Dysregulation of HSG triggers vascular proliferative disorders. Nat. Cell Biol. 6, 872–883. doi: 10.1038/ncb1161
Chen, X. H., Zhao, Y. P., Xue, M., Ji, C. B., Gao, C. L., Zhu, J. G., et al. (2010). TNF-alpha induces mitochondrial dysfunction in 3T3-L1 adipocytes. Mol. Cell. Endocrinol. 328, 63–69. doi: 10.1016/j.mce.2010.07.005
Chen, Z., Tong, L. J., Tang, B. Y., Liu, H. Y., Wang, X., Zhang, T., et al. (2018). C11, a novel fibroblast growth factor receptor 1 (FGFR1) inhibitor, suppresses breast cancer metastasis and angiogenesis. Acta Pharmacol. Sin. 40, 823–832. doi: 10.1038/s41401-018-0191-7
Civiletto, G., Varanita, T., Cerutti, R., Gorletta, T., Barbaro, S., Marchet, S., et al. (2015). Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Metab. 21, 845–854. doi: 10.1016/j.cmet.2015.04.016
Coppola, A., Liu, Z. W., Andrews, Z. B., Paradis, E., Roy, M. C., Friedman, J. M., et al. (2007). A central thermogenic-like mechanism in feeding regulation: an interplay between arcuate nucleus T3 and UCP2. Cell Metab. 5, 21–33. doi: 10.1016/j.cmet.2006.12.002
de Brito, O. M., and Scorrano, L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. doi: 10.1038/nature07534
Del Dotto, V., Fogazza, M., Carelli, V., Rugolo, M., and Zanna, C. (2018). Eight human OPA1 isoforms, long and short: what are they for? Biochim. Biophys. Acta Bioenerg. 1859, 263–269. doi: 10.1016/j.bbabio.2018.01.005
Devi, L., Prabhu, B. M., Galati, D. F., Avadhani, N. G., and Anandatheerthavarada, H. K. (2006). Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 26, 9057–9068. doi: 10.1523/jneurosci.1469-06.2006
Diano, S., and Horvath, T. L. (2012). Mitochondrial uncoupling protein 2 (UCP2) in glucose and lipid metabolism. Trends Mol. Med. 18, 52–58. doi: 10.1016/j.molmed.2011.08.003
Djouadi, F., and Bastin, J. (2019). Mitochondrial genetic disorders: cell signaling and pharmacological therapies. Cells 8:289. doi: 10.3390/cells8040289
Douiev, L., Sheffer, R., Horvath, G., and Saada, A. (2020). Bezafibrate improves mitochondrial fission and function in DNM1L-deficient patient cells. Cells 9:301. doi: 10.3390/cells9020301
Escobar-Henriques, M., and Joaquim, M. (2019). Mitofusins: disease gatekeepers and hubs in mitochondrial quality control by E3 ligases. Front. Physiol. 10:517. doi: 10.3389/fphys.2019.00517
Farmer, T., Naslavsky, N., and Caplan, S. (2018). Tying trafficking to fusion and fission at the mighty mitochondria. Traffic 19, 569–577. doi: 10.1111/tra.12573
Ferguson, S. M., and De Camilli, P. (2012). Dynamin, a membrane-remodelling GTPase. Nat. Rev. Mol. Cell Biol. 13, 75–88. doi: 10.1038/nrm3266
Friedman, J. R., Lackner, L. L., West, M., DiBenedetto, J. R., Nunnari, J., and Voeltz, G. K. (2011). ER tubules mark sites of mitochondrial division. Science. 334, 358–362. doi: 10.1126/science.1207385
Friedman, J. R., Webster, B. M., Mastronarde, D. N., Verhey, K. J., and Voeltz, G. K. (2010). ER sliding dynamics and ER-mitochondrial contacts occur on acetylated microtubules. J. Cell Biol. 190, 363–375. doi: 10.1083/jcb.200911024
Frohlich, C., Grabiger, S., Schwefel, D., Faelber, K., Rosenbaum, E., Mears, J., et al. (2013). Structural insights into oligomerization and mitochondrial remodelling of dynamin 1-like protein. EMBO J. 32, 1280–1292. doi: 10.1038/emboj.2013.74
Gao, S., and Hu, J. (2021). Mitochondrial fusion: the machineries in and out. Trends Cell Biol. 31, 62–74. doi: 10.1016/j.tcb.2020.09.008
Gaziev, A. I., Abdullaev, S., and Podlutsky, A. (2014). Mitochondrial function and mitochondrial DNA maintenance with advancing age. Biogerontology 15, 417–438. doi: 10.1007/s10522-014-9515-2
Giovarelli, M., Zecchini, S., Martini, E., Garre, M., Barozzi, S., Ripolone, M., et al. (2020). Drp1 overexpression induces desmin disassembling and drives kinesin-1 activation promoting mitochondrial trafficking in skeletal muscle. Cell Death Differ. 27, 2383–2401. doi: 10.1038/s41418-020-0510-7
Givvimani, S., Pushpakumar, S., Veeranki, S., and Tyagi, S. C. (2014). Dysregulation of Mfn2 and Drp-1 proteins in heart failure. Can. J. Physiol. Pharmacol. 92, 583–591. doi: 10.1139/cjpp-2014-0060
Gomes, L. C., Di Benedetto, G., and Scorrano, L. (2011). During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 13, 589–598. doi: 10.1038/ncb2220
Gonzalez-Franquesa, A., and Patti, M. E. (2017). Insulin resistance and mitochondrial dysfunction. Adv. Exp. Med. Biol. 982, 465–520.
Huttemann, M., Pecina, P., Rainbolt, M., Sanderson, T. H., Kagan, V. E., Samavati, L., et al. (2011). The multiple functions of cytochrome c and their regulation in life and death decisions of the mammalian cell: from respiration to apoptosis. Mitochondrion 11, 369–381.
Ishihara, N., Fujita, Y., Oka, T., and Mihara, K. (2006). Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 25, 2966–2977. doi: 10.1038/sj.emboj.7601184
Kaltenbach, L. S., Romero, E., Becklin, R. R., Chettier, R., Bell, R., Phansalkar, A., et al. (2007). Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet. 3:e82. doi: 10.1371/journal.pgen.0030082
Kamerkar, S. C., Kraus, F., Sharpe, A. J., Pucadyil, T. J., and Ryan, M. T. (2018). Dynamin-related protein 1 has membrane constricting and severing abilities sufficient for mitochondrial and peroxisomal fission. Nat. Commun. 9:5239.
Kim, B., McLean, L. L., Philip, S. S., and Feldman, E. L. (2011). Hyperinsulinemia induces insulin resistance in dorsal root ganglion neurons. Endocrinology 152, 3638–3647. doi: 10.1210/en.2011-0029
Korobova, F., Ramabhadran, V., and Higgs, H. N. (2013). An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 339, 464–467. doi: 10.1126/science.1228360
Koshiba, T., Detmer, S. A., Kaiser, J. T., Chen, H., McCaffery, J. M., and Chan, D. C. (2004). Structural basis of mitochondrial tethering by mitofusin complexes. Science 305, 858–862. doi: 10.1126/science.1099793
Lackner, L. L. (2013). Determining the shape and cellular distribution of mitochondria: the integration of multiple activities. Curr. Opin. Cell Biol. 25, 471–476. doi: 10.1016/j.ceb.2013.02.011
Leduc-Gaudet, J. P., Hussain, S. N. A., Barreiro, E., and Gouspillou, G. (2021). Mitochondrial dynamics and mitophagy in skeletal muscle health and aging. Int. J. Mol. Sci. 22:8179. doi: 10.3390/ijms22158179
Lee, J. E., Westrate, L. M., Wu, H., Page, C., and Voeltz, G. K. (2016). Multiple dynamin family members collaborate to drive mitochondrial division. Nature 540, 139–143. doi: 10.1038/nature20555
Lee, J. Y., Kapur, M., Li, M., Choi, M. C., Choi, S., Kim, H. J., et al. (2014). MFN1 deacetylation activates adaptive mitochondrial fusion and protects metabolically challenged mitochondria. J. Cell Sci. 127(Pt 22), 4954–4963.
Li, C. J., Chen, P. K., Sun, L. Y., and Pang, C. Y. (2017). Enhancement of mitochondrial transfer by antioxidants in human mesenchymal stem cells. Oxid. Med. Cell. Longev. 2017:8510805.
Lim, D., Fedrizzi, L., Tartari, M., Zuccato, C., Cattaneo, E., Brini, M., et al. (2008). Calcium homeostasis and mitochondrial dysfunction in striatal neurons of Huntington disease. J. Biol. Chem. 283, 5780–5789. doi: 10.1074/jbc.m704704200
Little, A. C., Kovalenko, I., Goo, L. E., Hong, H. S., Kerk, S. A., Yates, J. A., et al. (2020). High-content fluorescence imaging with the metabolic flux assay reveals insights into mitochondrial properties and functions. Commun. Biol. 3:271.
Liu, M., Li, X., and Huang, D. (2020). Mfn2 overexpression attenuates cardio-cerebrovascular ischemia-reperfusion injury through mitochondrial fusion and activation of the AMPK/Sirt3 signaling. Front. Cell Dev. Biol. 8:598078. doi: 10.3389/fcell.2020.598078
Liu, R., Jin, P., Yu, L., Wang, Y., Han, L., Shi, T., et al. (2014). Impaired mitochondrial dynamics and bioenergetics in diabetic skeletal muscle. PLoS One 9:e92810. doi: 10.1371/journal.pone.0092810
Loson, O. C., Song, Z., Chen, H., and Chan, D. C. (2013). Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 24, 659–667. doi: 10.1091/mbc.e12-10-0721
Mahdaviani, K., Benador, I. Y., Su, S., Gharakhanian, R. A., Stiles, L., Trudeau, K. M., et al. (2017). Mfn2 deletion in brown adipose tissue protects from insulin resistance and impairs thermogenes. EMBO Rep. 18, 1123–1138. doi: 10.15252/embr.201643827
Mahecic, D., Carlini, L., Kleele, T., Colom, A., Goujon, A., Matile, S., et al. (2021). Mitochondrial membrane tension governs fission. Cell Rep. 35:108947. doi: 10.1016/j.celrep.2021.108947
Mai, S., Klinkenberg, M., Auburger, G., Bereiter-Hahn, J., and Jendrach, M. (2010). Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. J. Cell Sci. 123(Pt 6), 917–926.
Maimaitijiang, A., Zhuang, X., Jiang, X., and Li, Y. (2016). Dynamin-related protein inhibitor downregulates reactive oxygen species levels to indirectly suppress high glucose-induced hyperproliferation of vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 471, 474–478. doi: 10.1016/j.bbrc.2016.02.051
Maresca, A., la Morgia, C., Caporali, L., Valentino, M. L., and Carelli, V. (2013). The optic nerve: a “mito-window” on mitochondrial neurodegeneration. Mol. Cell. Neurosci. 55, 62–76. doi: 10.1016/j.mcn.2012.08.004
McBride, H., and Soubannier, V. (2010). Mitochondrial function: OMA1 and OPA1, the grandmasters of mitochondrial health. Curr. Biol. 20, R274–R276.
Mears, J. A., Lackner, L. L., Fang, S., Ingerman, E., Nunnari, J., and Hinshaw, J. E. (2011). Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nat. Struct. Mol. Biol. 18, 20–26. doi: 10.1038/nsmb.1949
Mishra, P., and Chan, D. C. (2014). Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 15, 634–646. doi: 10.1038/nrm3877
Mitra, K., Wunder, C., Roysam, B., Lin, G., and Lippincott-Schwartz, J. (2009). A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc. Natl. Acad. Sci. U.S.A. 106, 11960–11965. doi: 10.1073/pnas.0904875106
Molina, A. J., Wikstrom, J. D., Stiles, L., Las, G., Mohamed, H., Elorza, A., et al. (2009). Mitochondrial networking protects beta-cells from nutrient-induced apoptosis. Diabetes 58, 2303–2315. doi: 10.2337/db07-1781
Murphy, R., Turnbull, D. M., Walker, M., and Hattersley, A. T. (2008). Clinical features, diagnosis and management of maternally inherited diabetes and deafness (MIDD) associated with the 3243A>G mitochondrial point mutation. Diabet. Med. 25, 383–399. doi: 10.1111/j.1464-5491.2008.02359.x
Nunnari, J., and Suomalainen, A. (2012). Mitochondria: in sickness and in health. Cell 148, 1145–1159. doi: 10.1016/j.cell.2012.02.035
Oanh, N. T. K., Park, Y. Y., and Cho, H. (2017). Mitochondria elongation is mediated through SIRT1-mediated MFN1 stabilization. Cell. Signal. 38, 67–75. doi: 10.1016/j.cellsig.2017.06.019
Orr, A. L., Li, S., Wang, C. E., Li, H., Wang, J., Rong, J., et al. (2008). N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J. Neurosci. 26, 2783–2792. doi: 10.1523/jneurosci.0106-08.2008
Otera, H., Wang, C., Cleland, M. M., Setoguchi, K., Yokota, S., Youle, R. J., et al. (2010). Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 191, 1141–1158. doi: 10.1083/jcb.201007152
Pagliuso, A., Cossart, P., and Stavru, F. (2018). The ever-growing complexity of the mitochondrial fission machinery. Cell. Mol. Life Sci. 75, 355–374. doi: 10.1007/s00018-017-2603-0
Palmer, C. S., Osellame, L. D., Laine, D., Koutsopoulos, O. S., Frazier, A. E., and Ryan, M. T. (2011). MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 12, 565–573. doi: 10.1038/embor.2011.54
Portz, P., and Lee, M. K. (2021). Changes in Drp1 function and mitochondrial morphology are associated with the alpha-synuclein pathology in a transgenic mouse model of Parkinson’s disease. Cells 10:885. doi: 10.3390/cells10040885
Qi, X., Qvit, N., Su, Y. C., and Mochly-Rosen, D. (2013). A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J. Cell Sci. 126(Pt 3), 789–802.
Rodriguez-Nuevo, A., Diaz-Ramos, A., Noguera, E., Diaz-Saez, F., Duran, X., Munoz, J. P., et al. (2018). Mitochondrial DNA and TLR9 drive muscle inflammation upon Opa1 deficiency. EMBO J. 37:e96553.
Romanello, V., Guadagnin, E., Gomes, L., Roder, I., Sandri, C., Petersen, Y., et al. (2010). Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 29, 1774–1785. doi: 10.1038/emboj.2010.60
Roses, A. D., Lutz, M. W., Amrine-Madsen, H., Saunders, A. M., Crenshaw, D. G., Sundseth, S. S., et al. (2010). A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharmacogenomics J. 10, 375–384. doi: 10.1038/tpj.2009.69
Rzepnikowska, W., and Kochanski, A. (2018). A role for the GDAP1 gene in the molecular pathogenesis of CharcotMarieTooth disease. Acta Neurobiol. Exp. 78, 1–13. doi: 10.21307/ane-2018-002
Salaciak, K., Koszalka, A., Zmudzka, E., and Pytka, K. (2021). The calcium/calmodulin-dependent kinases II and IV as therapeutic targets in neurodegenerative and neuropsychiatric disorders. Int. J. Mol. Sci. 22:4307. doi: 10.3390/ijms22094307
Sanchis-Gomar, F., and Derbre, F. (2014). Mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 13, 1073–1074. doi: 10.1056/nejmc1316254
Schapira, A. H., Agid, Y., Barone, P., Jenner, P., Lemke, M. R., Poewe, W., et al. (2009). Perspectives on recent advances in the understanding and treatment of Parkinson’s disease. Eur. J. Neurol. 16, 1090–1099.
Scholtes, C., and Giguere, V. (2021). Transcriptional regulation of ROS homeostasis by the ERR subfamily of nuclear receptors. Antioxidants 10:437. doi: 10.3390/antiox10030437
Sebastian, D., Hernandez-Alvarez, M. I., Segales, J., Sorianello, E., Munoz, J. P., Sala, D., et al. (2012). Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc. Natl. Acad. Sci. U.S.A. 109, 5523–5528. doi: 10.1073/pnas.1108220109
Sharp, W. W. (2015). Dynamin-related protein 1 as a therapeutic target in cardiac arrest. J. Mol. Med. 93, 243–252. doi: 10.1007/s00109-015-1257-3
Silva Ramos, E., Larsson, N. G., and Mourier, A. (2016). Bioenergetic roles of mitochondrial fusion. Biochim. Biophys. Acta 1857, 1277–1283. doi: 10.1016/j.bbabio.2016.04.002
Sloat, S. R., Whitley, B. N., Engelhart, E. A., and Hoppins, S. (2019). Identification of a mitofusin specificity region that confers unique activities to Mfn1 and Mfn2. Mol. Biol. Cell. 130, 2309–2319. doi: 10.1091/mbc.e19-05-0291
Stanga, S., Caretto, A., Boido, M., and Vercelli, A. (2020). Mitochondrial dysfunctions: a red thread across neurodegenerative diseases. Int. J. Mol. Sci. 21:3719. doi: 10.3390/ijms21103719
Tezze, C., Romanello, V., Desbats, M. A., Fadini, G. P., Albiero, M., Favaro, G., et al. (2017). Age-associated loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metab. 25, 1374–1389.e6.
Toda, C., Kim, J. D., Impellizzeri, D., Cuzzocrea, S., Liu, Z. W., and Diano, S. (2016). UCP2 regulates mitochondrial fission and ventromedial nucleus control of glucose responsiveness. Cell 164, 872–883. doi: 10.1016/j.cell.2016.02.010
Tondera, D., Grandemange, S., Jourdain, A., Karbowski, M., Mattenberger, Y., Herzig, S., et al. (2009). SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 28, 1589–1600. doi: 10.1038/emboj.2009.89
Touvier, T., De Palma, C., Rigamonti, E., Scagliola, A., Incerti, E., Mazelin, L., et al. (2015). Muscle-specific Drp1 overexpression impairs skeletal muscle growth via translational attenuation. Cell Death Dis. 6:e1663. doi: 10.1038/cddis.2014.595
Twig, G., Elorza, A., Molina, A. J., Mohamed, H., Wikstrom, J. D., Walzer, G., et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446. doi: 10.1038/sj.emboj.7601963
Wai, T., and Langer, T. (2016). Mitochondrial dynamics and metabolic regulation. Trends Endocrinol. Metab. 27, 105–117. doi: 10.1016/j.tem.2015.12.001
Wang, D., Wang, J., Bonamy, G. M., Meeusen, S., Brusch, R. G., Turk, C., et al. (2012). A small molecule promotes mitochondrial fusion in mammalian cells. Angew. Chem. Int. Ed. Engl. 51, 9302–9305. doi: 10.1002/anie.201204589
Waterham, H. R., Koster, J., van Roermund, C. W., Mooyer, P. A., Wanders, R. J., and Leonard, J. V. (2007). A lethal defect of mitochondrial and peroxisomal fission. N. Engl. J. Med. 26, 1736–1741. doi: 10.1056/nejmoa064436
White, J. P., Puppa, M. J., Sato, S., Gao, S., Price, R. L., Baynes, J. W., et al. (2012). IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skelet. Muscle 2, 14.
Wu, M. Y., Yiang, G. T., Lai, T. T., and Li, C. J. (2018). The oxidative stress and mitochondrial dysfunction during the pathogenesis of diabetic retinopathy. Oxid. Med. Cell. Longev. 2018:3420187.
Xiao, L., Xian, H., Lee, K. Y., Xiao, B., Wang, H., Yu, F., et al. (2015). Death-associated protein 3 regulates mitochondrial-encoded protein synthesis and mitochondrial dynamics. J. Biol. Chem. 290, 24961–24974. doi: 10.1074/jbc.m115.673343
Yang, D., Ying, J., Wang, X., Zhao, T., Yoon, S., Fang, Y., et al. (2021). Mitochondrial dynamics: a key role in neurodegeneration and a potential target for neurodegenerative disease. Front. Neurosci. 15:654785. doi: 10.3389/fnins.2021.654785
Yu, R., Jin, S. B., Lendahl, U., Nister, M., and Zhao, J. (2019a). Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J. 38:e99748.
Yu, R., Lendahl, U., Nister, M., and Zhao, J. (2020). Regulation of mammalian mitochondrial dynamics: opportunities and challenges. Front. Endocrinol. 11:374. doi: 10.3389/fendo.2020.00374
Yu, R., Liu, T., Ning, C., Tan, F., Jin, S. B., Lendahl, U., et al. (2019b). The phosphorylation status of Ser-637 in dynamin-related protein 1 (Drp1) does not determine Drp1 recruitment to mitochondria. J. Biol. Chem. 296, 17262–17277. doi: 10.1074/jbc.ra119.008202
Yue, W., Chen, Z., Liu, H., Yan, C., Chen, M., Feng, D., et al. (2014). A small natural molecule promotes mitochondrial fusion through inhibition of the deubiquitinase USP30. Cell Res. 24, 482–496. doi: 10.1038/cr.2014.20
Zhang, Z., Cui, D., Zhang, T., Sun, Y., and Ding, S. (2020). Swimming differentially affects T2DM-induced skeletal muscle ER stress and mitochondrial dysfunction related to MAM. Diabetes Metab. Syndr. Obes. 30, 1417–1428. doi: 10.2147/dmso.s243024
Zhao, J., Liu, T., Jin, S., Wang, X., Qu, M., Uhlen, P., et al. (2011). Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. 30, 2762–2778. doi: 10.1038/emboj.2011.198
Keywords: mitochondrial dynamics, fusion, fission, pathophysiology, machinery
Citation: Chiu Y-H, Lin S-CA, Kuo C-H and Li C-J (2021) Molecular Machinery and Pathophysiology of Mitochondrial Dynamics. Front. Cell Dev. Biol. 9:743892. doi: 10.3389/fcell.2021.743892
Received: 19 July 2021; Accepted: 31 August 2021;
Published: 17 September 2021.
Edited by:
Kanchan Phadwal, University of Edinburgh, United KingdomReviewed by:
Jianquan Chen, Soochow University, ChinaCopyright © 2021 Chiu, Lin, Kuo and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chia-Jung Li, bmlnZWw2NzYxQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.