 Yalan Jiang
Yalan Jiang Zehua Tao
Zehua Tao Hua Chen2
Hua Chen2- 1Department of Immunology, School of Medicine, Jiangsu University, Zhenjiang, China
- 2Department of Colorectal Surgery, Affiliated Kunshan Hospital of Jiangsu University, Kunshan, China
The endoplasmic reticulum quality control (ERQC) system, including endoplasmic reticulum-associated degradation (ERAD), the unfolded protein response (UPR), and autophagy, presides over cellular protein secretion and maintains proteostasis in mammalian cells. As part of the immune system, a variety of proteins are synthesized and assembled correctly for the development, activation, and differentiation of immune cells, such as dendritic cells (DCs), macrophages, myeloid-derived-suppressor cells (MDSCs), B lymphocytes, T lymphocytes, and natural killer (NK) cells. In this review, we emphasize the role of the ERQC in these immune cells, and also discuss how the imbalance of ER homeostasis affects the immune response, thereby suggesting new therapeutic targets for immunotherapy.
Introduction
The endoplasmic reticulum (ER) serves as a cell factory, playing a key role in protein synthesis, folding, and modification. However, an accumulation of misfolded or unfolded proteins in the ER leads to an ER stress response. Cells have evolved an endoplasmic reticulum quality control (ERQC) system to maintain ER homeostasis. As a complex monitoring system, the ERQC regulates ER homeostasis through three mechanisms: endoplasmic reticulum-associated degradation (ERAD), the unfolded protein response (UPR), and autophagy. ERAD and autophagy are responsible for the degradation of misfolded proteins in the ER, while constant ER stress induced by misfolded protein accumulation activates the inositol-requiring enzyme 1α (IRE1α), protein kinase RNA-like ER kinase (PERK), and activating transcription factor 6 (ATF6) signaling pathways to trigger the UPR response. Immune cells, including dendritic cells (DCs), macrophages, myeloid-derived-suppressor cells (MDSCs), B cells, T cells, and natural killer (NK) cells are key effectors in the immune system. In the development of these cells and even their activation and differentiation in the immune response, an amount of newly synthesized membrane and secretion proteins are indispensable, which first need correct intracellular folding. In this review, we summarized the recent work on the ERQC in immune cells and hope to demonstrate its role within the immune system.
The Endoplasmic Reticulum: Structure and Function
The ER is an intracellular membranous network that is present in all eukaryotic cells. It consists of interconnected, branching membranous tubules, vesicles, and cisternae, giving rise to two main dynamic and interconvertible structures: smooth endoplasmic reticulum (SER) and rough endoplasmic reticulum (RER), with membranes of the latter being decorated by ribosomes on its outer surface (English et al., 2009; Corazzari et al., 2017). Although some cells may have little SER, all eukaryotic cells have large amounts of RER, as these play a key role in the synthesis of many integral membrane proteins and several cytosolic proteins. RER is abundant in secretory cells, such as antibody-producing plasma cells, insulin-secreting beta cells, or cells of milk-producing glands (Burgoyne and Duncan, 1998; Harding and Ron, 2002; Oyadomari et al., 2002; van Anken et al., 2009). SER, which lacks ribosomes, is not involved in protein synthesis but is responsible for the synthesis of essential lipids, such as fatty acids and phospholipids, metabolism of carbohydrates, and regulation of calcium homeostasis (Schönthal, 2012). After synthesis, proteins are translocated into the ER lumen, where they are further folded, and then dispersed to their destination. Unfortunately, these new synthesized proteins sometimes do not fold or aggregate normally (Ellgaard and Helenius, 2003). The accumulation of unfolded, misfolded, or damaged proteins leads to ER stress, which further initiates ERAD, UPR, and autophagy responses to rescue the cell from cell destruction or death.
Endoplasmic Reticulum Stress Response and Protein Degradation
Protein folding is a complex process that depends on the interaction of chaperone proteins, folding enzymes, and glycosylases (Malhotra and Kaufman, 2007; Ron and Walter, 2007). When there is an imbalance between protein-folding capacity and protein-folding demand, this leads to an accumulation of misfolded proteins in the cell, known as ER stress (Hetz and Papa, 2018). Protein degradation is an important step in processing ER stress. The major protein degradation system includes the ubiquitin-proteasome system (UPS) and the autophagic-lysosomal system (ALS), which can selectively degrade aberrant protein fragments (Xia et al., 2020). In most cases, micromolecules and short-lived soluble proteins are retrotranslocated into the cytosol, ubiquitinated, and then degraded by the proteasomes in the ERAD system. However, when the misfolded proteins exceed ER degradation capacity, the UPR response will be activated. At the same time, aggregates of misfolded proteins that could not be processed by the proteasome are degraded through autophagy (Nam et al., 2017).
Endoplasmic Reticulum-Associated Degradation
Endoplasmic reticulum-associated degradation is a conserved quality control mechanism in the cell, responsible for retrotranslocation of misfolded proteins into the cytosol for proteasomal degradation (Zhou et al., 2020). The SEL1L-HRD1 protein complex comprises the most conserved members of ERAD from yeast to mammals. SEL1L is a type I transmembrane protein, which resides on the ER membrane and controls the stability of E3 ligase HRD1 (Sun et al., 2014; Jeong et al., 2016). ERAD acts in a multistep process involving recognition, extraction, polyubiquitination, and degradation (Christianson and Ye, 2014). In a simplified model of ERAD, molecular chaperones and lectins, including BiP, EDEM, ERdj, OS9, and XTP3B recognize unfolded nascent polypeptide substrates. Subsequently, unfolded proteins bind with these chaperones and are further delivered to ERAD adaptors, including SEL1L and Erlins (Qi et al., 2017). Finally, the polyubiquitinated substrates are transported to the cytosol through the retrotranslocation channel (candidates include HRD1, DER1, and DFM1) and degraded by 26S proteasome (Wu and Rapoport, 2018; Figure 1). Chronic ER stress may not necessarily trigger harmful changes, such as cell death (Hotamisligil, 2010). For example, DCs and developing B cells can tolerate ERAD deficiency and chronic ER stress, which may arise from other compensatory mechanisms, such as the UPR and autophagy (Yang et al., 2014; Ji et al., 2016). Therefore, the role of ERAD in cell development should be specifically considered according to different substrates and cell types.
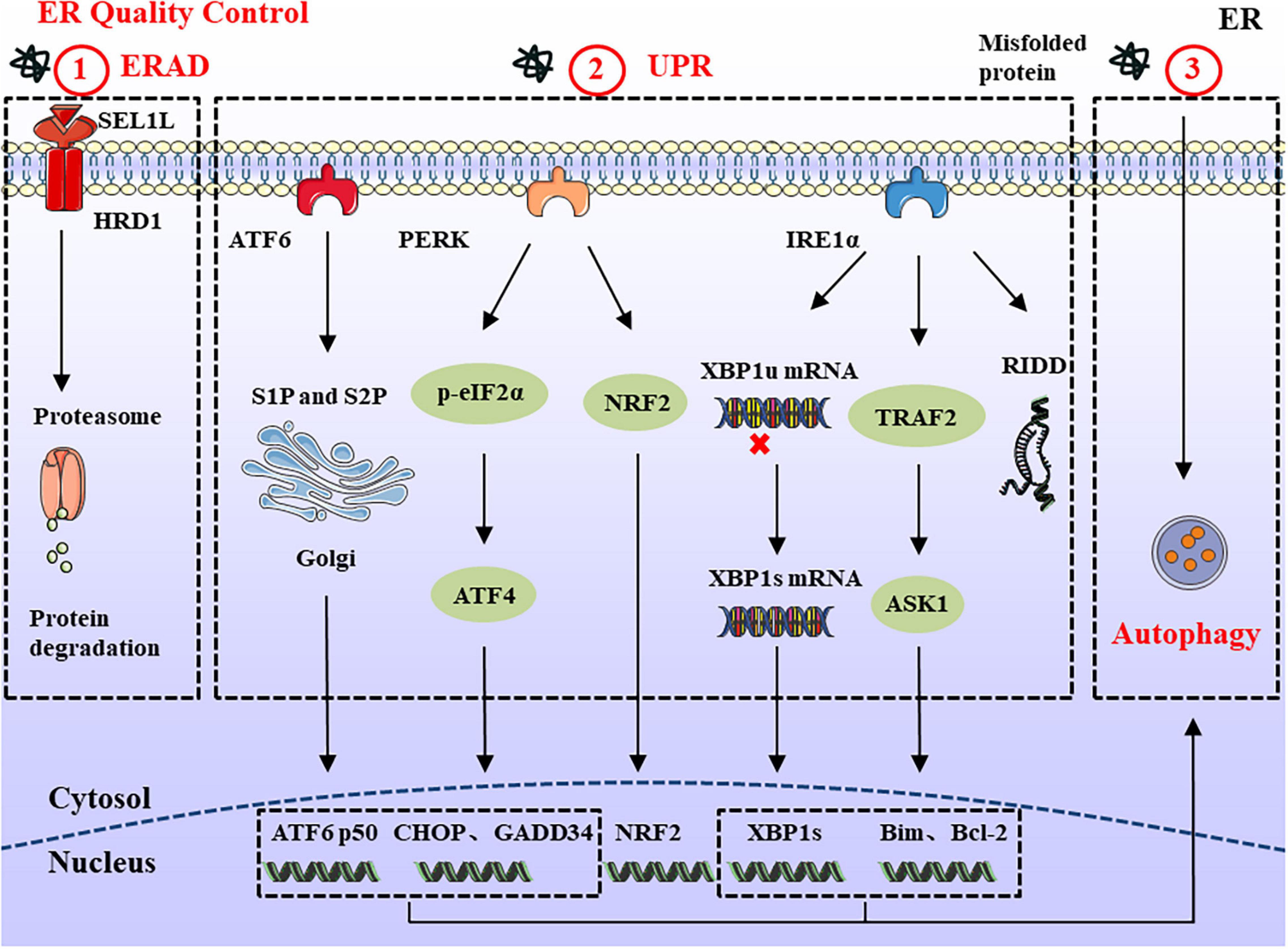
Figure 1. ERQC in Endoplasmic reticulum. ERQC monitors the correct folding of proteins via ERAD, UPR, and autophagy. (1) The soluble protein aggregates are retrotranslocated into cytosol through SEL1L-HRD1, where the ubiquitinated substrates are degraded by the proteasome. (2) When the misfolded proteins exceed ER degradation capacity, it leads to the ER stress response. In order to maintain ER homeostasis, the UPR response is activated which is composed of IRE1α, PERK, and ATF6 branches. The activated IRE1α catalyzes the splicing of XBP1 to generate XBP1s. In addition, IRE1α can also reduce protein synthesis through RIDD, and interacts with TRAF2 to regulate the expression of pro-/anti-apoptotic proteins. PERK reduces protein load by phosphorylating eIF2α and NRF2. Meanwhile, the upregulated ATF4 participates in cell apoptosis and protein synthesis via CHOP and GADD34. ER stress also causes the translocation of ATF6 to the Golgi apparatus, where it is spliced by S1P and S2P proteases, thereby mediating the expression of ERAD and ER chaperones. (3) In the ER stress response, large protein aggregates are degraded through the autophagy-lysosomal pathway.
Unfolded Protein Response
When the accumulated improperly folded proteins exceed a threshold, cells activate the UPR to restore ER homeostasis. The main purpose of the UPR is to eliminate misfolded proteins and reduce the synthesis of new proteins in the ER. The UPR slows down the transcription and translation of new mRNA by degrading existing mRNA, thereby reducing the flow of new proteins into the ER lumen (Grootjans et al., 2016). The production of genes encoding molecular chaperones and foldases also increases to process the large amounts of accumulated proteins.
Mammalian cells have three ER-resident transmembrane proteins which work as sensors to detect the accumulation of unfolded or misfolded proteins: IRE1α, PERK, and ATF6 (Hetz et al., 2015; McQuiston and Diehl, 2017). Under physiological conditions, ER sensor proteins are maintained in an inactive state by interacting with glucose-regulated protein 78 (GRP78; also called BiP). As unfolded proteins accumulate, BiP dissociates from ER stress sensors and initiates the UPR. The IRE1α-X-box-binding protein 1 (XBP1) pathway is the most conserved UPR signaling branch. As a type I transmembrane protein with a cytosolic serine/threonine kinase domain, the activation of IRE1α causes conformational changes that trigger kinase activity and endoribonuclease activity to catalyze the excision of a 26-nt intron within the XBP1 mRNA, forming a stable and active transcription mRNA named XBP1s (Duwaerts et al., 2020). Furthermore, activated IRE1α forms a complex with tumor-necrosis factor-α (TNF-α)-receptor-associated factor 2 (TRAF2) at its cytosolic domain, which in turn engages apoptosis signal-regulating kinase 1 (ASK1) and JUN N-terminal kinase (JNK), leading to the activation of pro-apoptotic Bim and suppression of the anti-apoptotic activity of Bcl-2 (Schönthal, 2012; Corazzari et al., 2017). IRE1α also degrades ribosomal-associated mRNA through regulated IRE1-dependent decay (RIDD) to prevent the translation and accumulation of unfolded proteins (Coelho and Domingos, 2014). The RIDD substrate has a sequence CUGCAG similar to the stem-loop structure of XBP1 mRNA, and the activity of RIDD increased with the duration of ER stress (Maurel et al., 2014). So the basal activity of RIDD helps maintain ER homeostasis, but under prolonged ER stress, continuously increased RIDD induces apoptosis by degrading mRNAs.
The PERK arm phosphorylates eukaryotic initiation factor 2α (eIF2α) on Ser51, which reduces protein translation and lowers the protein load (Liu et al., 2015). Although the overall translation reduces after the phosphorylation of eIF2α, the expression of some key genes is upregulated, including ATF4 and its downstream products, which are related to ER homeostasis, amino acid metabolism, oxidative stress responses, and apoptosis (Rainbolt et al., 2014). Thus, ATF4 upregulates several key genes, including growth arrest and DNA damage-inducible protein 34 (GADD34) and pro-apoptotic C/EBP homologous protein (CHOP). As part of the negative feedback loop, GADD34 can dephosphorylate p-eIF2α and resume normal translation. Meanwhile, it can increase protein load during ER stress, thus aggravating stress and leading to cell death (Han et al., 2013). As the downstream product of ATF4, a large amount of CHOP is produced to trigger apoptosis after long-term ER stress (Nishitoh, 2012). Besides eIF2α, PERK can directly phosphorylate the transcription factor NRF2. Upon activation, NRF2 migrates to the nucleus, where it activates genes encoding detoxifying enzymes causing the antioxidant stress response (Cullinan and Diehl, 2004, 2006). Therefore, according to the severity and duration of ER stress, PERK activation can promote cell survival or induce cell apoptosis.
Endoplasmic reticulum stress also causes the translocation of ATF6 to the Golgi apparatus, where it is cleaved by site-1 protease (S1P) and site-2 protease (S2P). The released transcription factors activate the expression of genes related to protein folding and degradation whose products can promote protein folding, ER expansion, ERAD, and autophagy (Haze et al., 1999; Shen et al., 2002). Meanwhile, activated ATF6 can also induce the expression of XBP1 to optimize UPR (Figure 1).
Autophagy
Different from ERAD, which is limited to degrading proteins, ER stress-induced autophagy can be divided into ER stress-mediated autophagy and ER-phagy (Song et al., 2018). The former is characterized by the degradation of damaged proteins and organelles, while the latter selectively degrades part of the ER through ATG39 and ATG40 receptors. Previous studies have shown that three branches of the UPR (IRE1α, PERK, and ATF6) modulate the formation of autophagy (Song et al., 2017, 2018). IRE1α forms a complex with TRAF2 and ASK1 to activate JNK, thus releasing Beclin-1 to drive vesicle nucleation. Meanwhile, PERK upregulates the expression of ATF4 and CHOP to induce the formation of the ATG5-ATG12-ATG16L complex, which can promote autophagy elongation. By contrast, ATF6 indirectly affects autophagy by activating XBP1 and CHOP. A feature of autophagy is the production of autophagosomes, which are degraded in lysosomes. There are three main types of autophagy in the cell: macroautophagy, microautophagy, and chaperone-mediated autophagy (Yang and Klionsky, 2010; Nakatogawa, 2020). Among these three types, macroautophagy (hereafter simply autophagy) is the most thoroughly and widely studied autophagy pathway (Cybulsky, 2017). Autophagosomes sequester and degrade damaged organelles, unfolded proteins, and aggregates; and then fuse with lysosomes for degradation of the contents (Figure 1).
Although ER stress-mediated autophagy is mainly protective, the roles of autophagy in regulating cell survival remain controversial. Moderate ER stress-mediated autophagy can reduce ER stress and promote cell survival. However, autophagy induced by excessive ER stress can aggravate cell damage and promote cell apoptosis (Denton et al., 2012; Song et al., 2017). Thus, the ultimate fate of a cell is determined by the balance between autophagy and toxic protein accumulation. On the other hand, the loss of autophagy, such as the deletion of core autophagy genes, will also change the morphology and size of the ER and induce ER stress responses (Smith and Wilkinson, 2017). In consequence, autophagy can also regulate ER function which is essential for ER homeostasis.
Endoplasmic Reticulum Quality Control and Immune Cells
As a type of multipotent stem cell, bone marrow (BM) hematopoietic stem cells can be divided into common myeloid progenitor cells (CMPs) and common lymphoid progenitor cells (CLPs), which finally differentiate into diverse immune cells. Accumulating evidence shows that the ERQC system is essential for the development and the function of immune cells. Next, we will address the latest insights into the contribution of ERQC in the development, activation and differentiation of immune cells, although the regulation of immune response by ERQC is not limited to immune cells.
Dendritic Cell
As the professional antigen-presenting cells, DCs process and present antigens to prime T lymphocytes for activation and proliferation. Common DC progenitors in BM can differentiate into plasmacytoid DCs (pDCs) and conventional DCs (cDCs), which include cDC1 (CD8α+ DCs) and cDC2 cells (Macri et al., 2018). It has been reported that XBP1 plays an important role in the development of DCs (Iwakoshi et al., 2007). In the FLT3L induction system, XBP1–/– hematopoietic progenitor cells produced fewer DCs, and the apoptosis of XBP1–/– DCs was increased. However, after transfection of XBP1s, DC homeostasis was restored, and development was rescued. Compared with T cells and B cells, cDCs had increased expression of IRE1α, and the transcriptional activity of XBP1 in CD8α+ cDCs was higher. Osorio et al. (2014) showed that the absence of XBP1 in DCs affected the structure and morphology of the ER, but its function remained unchanged (Osorio et al., 2014). Additionally, there was no significant difference in the composition and number of DC subsets in the spleen of wild-type (WT) mice and Xbp1fl/flItgaxcre mice; however, the ability of CD8α+ cDCs to present MHC I antigens was impaired. Studies have found that the excessive activation of IRE1α in XBP1-deficient DCs led to an increase in RIDD activity but decreased the expression of Tapbp mRNA (the target of RIDD) (Osorio et al., 2014; Ramirez et al., 2019). After co-culture with OT-I T cells, the proliferation of CD8+ T cells was inhibited, and indicated that the cross-presentation function of CD8α+ cDCs was impaired. Although the expression of Tapbp is related to MHC class I antigen presentation, the specific mechanism by which RIDD affects this process remains unclear. Furthermore, it was reported that in ovarian cancer models, persistent activation of the IRE1α-XBP1 branch resulted in dysfunction of tumor-infiltrating dendritic cells (tDCs; Cubillos-Ruiz et al., 2015). Specifically, XBP1 signaling regulates the lipid metabolism and antigen presentation of tDCs, while silencing XBP1 in tDCs extends host survival by enhancing T cell anti-tumor immunity. Clearly, the IRE1α-XBP1 branch is very important for the biological functions of DC, which can further affect the activation of T cell.
Different from acute ER stress, the phosphorylation levels of eIF2α in the spleen and FLT3L BMDCs were significantly increased under steady-state conditions (Mendes et al., 2021). During the DC differentiation process, PERK was activated, which contributed to a high level of p-eIF2α. Meanwhile, cells could promote transcription through compensatory mechanisms, such as eIF2B and GADD34 (Perego et al., 2018; Kenner et al., 2019; Mendes et al., 2021). Therefore, although eIF2α negatively regulated translation, proteins were still continuously synthesized. GADD34 also coordinated with actin to regulate eIF2α phosphorylation levels, thereby affecting DC migration and cytokine secretion (Mendes et al., 2021). Furthermore, after lipopolysaccharide (LPS) stimulation, the secretion of IFN-β in PERK-deficient DCs was reduced, and the migration of DCs was impaired. However, the activation of T cells was not affected. These data indicate that although the PERK branch has no effect on DC functions and T cell activation, it is essential for type I interferon production and DC migration.
As glycoproteins on DCs, MHC class I and class II molecules bind to TCR receptors on the surface of T lymphocytes, presenting antigens to corresponding T cells (Adams and Luoma, 2013). The MHC class I heavy chain (MHC I HC) is the substrate for HRD1 degradation (Burr et al., 2011). Under normal circumstances, MHC I HC fails to bind to β2-microglobulin (β2m), which accumulates in the ER, eventually being recognized and degraded by ERAD (Hughes et al., 1997; Burr et al., 2011). Burr et al. (2013) showed that the SEL1L/HRD1/UBE2J1 complex could ubiquitinate abnormally folded MHC I HC to regulate the assembly of MHC class I molecules (Burr et al., 2013). Therefore, ERAD may be necessary for the expression of MHC class I molecules in DCs. Additionally, it was reported that B-lymphocyte-induced maturation protein 1 (BLIMP1) is the target of HRD1 ubiquitination and degradation, which can inhibit the transcription of CIITA and MHC-II genes (Yang et al., 2014). The deletion of HRD1 in DCs led to the accumulation of BLIMP1, which in turn inhibited the expression of MHC-II and CD4+ T cell proliferation. Interestingly, the deletion of HRD1 had no significant effect on the survival of DCs, indicating that it can specifically degrade substrates without affecting the final fate of cells. So, HRD1 appears to regulate the expression of the MHC complex through two different mechanisms. Although the specific role of ERAD in MHC-I antigen presentation remains unclear.
It is well known that DCs can process and load exogenous antigens with MHC-I molecules through cross-presentation pathways to activate naïve CD8+ T cells. The exogenous protein is internalized by the phagosome in DC and transported to the cytosol, where it is degraded by UPS. The generated antigen peptides are then translocated into the ER and combined with MHC-I molecules and finally loaded onto the surface of DC (Grotzke et al., 2017; Blander, 2018). Although studies have found that p97, a component of ERAD, played a role in cross-presentation retrotranslocation, HRD1 and BiP were not involved in this process (Grotzke and Cresswell, 2015). In addition, it remains unclear whether ERAD participates in the pruning and loading of peptide-MHC-I complexes (Table 1). Thus, based on these findings, determining the role of ERAD in DC cross-presentation will be an interesting future study.
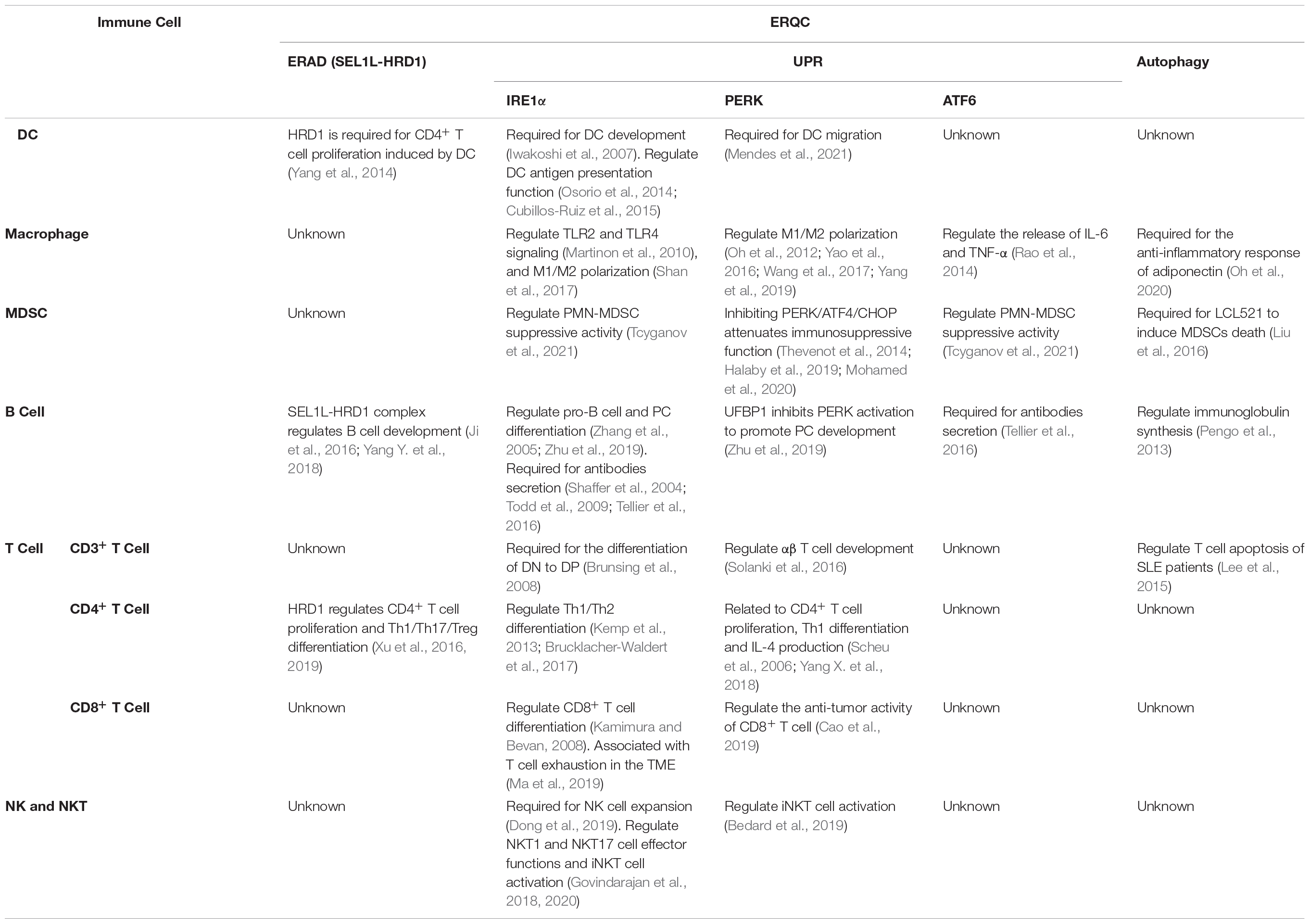
Table 1. ERQC regulates the differentiation and function of immune cells.
Macrophage
Macrophages are an important part of the innate immune system, which recognize and engulf pathogens through pattern recognition receptors (PRRs) to initiate cellular immune responses. Macrophages can be simply divided into two types, monocyte-derived macrophages and resident macrophages (Verschoor et al., 2012). Upon injury or infection, circulating monocytes migrate into the tissue and transform into mature macrophages, while resident macrophages can be divided into alveolar macrophages and liver Kupffer cells (KCs), etc. On the other hand, according to their activation status, macrophages can be divided into pro-inflammatory classical activated (M1) and anti-inflammatory alternate activated (M2) macrophages. XBP1 is a positive regulator of TLR2 and TLR4 signaling responses in macrophages (Martinon et al., 2010). TLRs promote the splicing of XBP1 and the production of pro-inflammatory cytokines through TNF receptor-related factor 6 (TRAF6) and NADPH oxidase NOX2, but the expression of ER stress markers do not increase. On the contrary, the PERK branch is inhibited and the expression of CHOP is reduced, which is conducive to the survival of macrophages and the initiation of immune responses. On the other hand, as a type of adipokine, accumulating evidence indicates that adiponectin exerts anti-inflammatory properties by inducing autophagy and ER stress in macrophages (Oh et al., 2020). After inhibition of ER stress by TUDCA, the induction of autophagy was impaired, and the expression of inflammatory cytokines increased. Therefore, ER stress-mediated autophagy plays a key role in the anti-inflammatory response and cytoprotection induced by adiponectin in macrophages.
It is well known that IL-4 is a key inducer of M2 macrophages and induces the activity of transcription factors such as c-Myc in macrophages, which in turn changes their migration and polarization (Brown et al., 2012; Sica and Mantovani, 2012; Li et al., 2018). As an ER membrane protein, homocysteine-inducible ER protein with ubiquitin-like domain 1 (Herpud1) is strongly upregulated in the ER stress response, and is induced by the three branches of UPR (Yamamoto et al., 2004). Moreover, Herpud1 can interact with SEL1L, HRD1, and other components of ERAD to degrade misfolded proteins (Leitman et al., 2014). A recent study has shown that IL-4 can induce the expression of the ER stress-related protein Herpud1 in M2 macrophages (Li et al., 2018). Inhibition of ER stress by 4-PBA or knockdown of the expression of Herpud1 suppressed the polarization and migration of M2 macrophages induced by IL-4. Clearly, Herpud1 plays an important role in IL-4 activated M2 polarization, but the potential mechanism remains undetermined.
As highly plastic and diverse cells, the polarized phenotype of macrophages can be transformed into each other under different microenvironments. In obesity, palmitic acid (PA) and LPS in the blood activate adipose tissue macrophages (ATMs) IRE1α branch, and then inhibit M2 and enhance M1 polarization, which ultimately lead to inflammation (Shan et al., 2017). However, the loss of IRE1α can reverse the imbalance of M1 and M2 polarization in ATMs and improve metabolism. Similarly in fatty liver, ER stress leads to the activation of the PERK branch in KCs, which induces the transformation of KCs into pro-inflammatory M1 (Yang et al., 2019). However, the absence of PERK can inhibit LPS induction of STAT1 activation and enhance IL-4 induction of STAT6 activation, thereby dampening the polarization of M1 and promoting the polarization of M2. In contrast, in patients with pulmonary fibrosis, diabetes, and allergic airway inflammation, the infiltration of M2 macrophages was increased, and the expression of ER stress markers, especially CHOP, was significantly upregulated (Oh et al., 2012; Yao et al., 2016; Wang et al., 2017). However, the loss of CHOP can inhibit the activation of STAT6 and then attenuate M2 polarization, thereby alleviating the inflammatory response. Furthermore, ATF6 activation was shown to amplify the pro-inflammatory response of TLR4 (Rao et al., 2014). Specifically, ATF6-mediated ER stress enhances the pro-inflammatory property of TLR4 by enhancing NF-κB signaling, while inhibiting ATF6 can alleviate ER stress and decrease the production of IL-6 and TNF-α. This indicates that there is a regulatory mechanism between ATF6 and TLR4 to prevent excessive inflammation.
Collectively, these findings indicate that the ER stress branches can regulate the polarization of M1 and M2, which can affect the progression of a variety of chronic diseases (Table 1). Further works are needed to explore the specific effect of macrophage ER stress in different diseases, which is expected to provide information for clinical targeted therapy.
Myeloid-Derived-Suppressor Cell
Myeloid-derived-suppressor cells are immature myeloid cells with immune suppressive activity, which can be divided into polymorphonuclear MDSC (PMN-MDSC) and monocytic MDSC (M-MDSC), and the large accumulation of MDSCs in cancer patients is the main factor limiting the efficacy of immunotherapy. Recently, apoptosis pathway was shown to play a key role in MDSCs homeostasis. Condamine et al. (2014) found that MDSCs in tumor-bearing mice had increased apoptosis and poor survival, which was associated with the increased expression of TRAIL receptors (TRAIL-Rs; Condamine et al., 2014). Subsequent further study showed that MDSCs from cancer patients exhibited higher ER stress levels, and the expression of TRAIL-Rs was upregulated, while targeting TRAIL-Rs improved the anti-tumor effect. In contrast, the acid ceramidase inhibitor LCL521 induced MDSCs death through an apoptosis-independent mechanism death (Liu et al., 2016). Specifically, LCL521 leads to the abnormal accumulation of autophagic vesicles and ER stress, which impaired autophagic flux and induced MDSCs death.
In addition, the immune function of MDSCs is also significantly affected by ER stress. The induction of ROS was impaired in CHOP-deficient MDSCs, which not only inhibited the immunosuppressive function, but also initiated T cell response to induce anti-tumor immunity (Thevenot et al., 2014). This transition was mediated through C/EBPβ and p-STAT-3 signaling. Although CHOP induction is mainly driven by ATF4, it is unclear whether ATF4 regulates MDSC function. A recent study showed that the GCN2-ATF4 axis drives the polarization of tumor-associated macrophages and the immunosuppressive functions of tumor-infiltrating MDSCs that attenuate anti-tumor immunity (Halaby et al., 2019). Likewise, Mohamed et al. (2020) found that in PERK-deficient tumor MDSCs, reduced NRF2 signaling elicited the accumulation of mtDNA and subsequently stimulated the STING pathway, which transformed MDSCs into cells that promote CD8+ T cell responses.
Of note, IRE1α-XBP1 and ATF6 also play an important role in altering the function of MDSCs. PMN-MDSCs in cancer patients exhibited higher expression of lectin-type oxidized LDL receptor-1 (LOX-1) and ER stress-related genes than neutrophils (PMNs) (Condamine et al., 2016). XBP1 inhibitor B-109 can suppress the upregulation of LOX-1 and the suppressive activity in PMN induced by ER stress. The function of PMN-MDSCs appears to be affected by IRE1α-XBP1. Supporting this observation, Tcyganov et al. (2021) found that IRE1α and ATF6 can regulate the suppressive activity of PMN-MDSC in tumors. The deletion of IRE1α and ATF6 abrogated PMN-MDSC suppressive activity and delayed tumor progression. Instead, the function of M-MDSC was controlled by IFN-γ signaling and was dispensable for ER stress. These findings indicate that UPR promotes immune evasion by regulating tumor MDSCs survival and immunosuppressive functions, which is related to cancer progression (Table 1).
B Cell
During the differentiation of B lymphocytes, pro-B cells differentiate into large pre-B cells expressing the pre-B cell receptor (pre-BCR). After several divisions, large pre-B cells differentiate into small resting pre-B cells and finally develop into immature B cells expressing BCR (Qi et al., 2017). Recent studies have shown that the SEL1L-HRD1 ERAD complex terminated pre-BCR signaling in a BiP-dependent way and indicated that ERAD regulated the development of B cells (Ji et al., 2016; Yang Y. et al., 2018). As an important node for B cell development, pre-BCR consists of IgH and the surrogate light chain (SLC), which contains λ5 and VpreB (Zhang et al., 2004). The silent expression of SLC in large pre-B cells could downregulate pre-BCR signaling and differentiate large pre-B cells into small pre-B cells. In the deletion of SEL1L-HRD1, the pre-BCR signaling complex was accumulated, leading to the strengthened proliferation of large pre-B cells and developmental defects in B cells. Eventually, the number of small pre-B cells decreased (Ji et al., 2016; Yang Y. et al., 2018). Unfortunately, it is unclear how ERAD differentiates between the pre-BCR and BCR complex expression on B cells.
Plasma cells (PCs) are terminally differentiated B lymphocytes responsible for secreting antibodies, which are essential components of humoral immunity. To synthesize large amounts of immunoglobulins, new PCs must increase the size of the ER and potentially induce ER stress. Thus, the UPR, as a complex signal transduction pathway might play an important role in this process. Indeed, the lack of XBP1 in B cells severely impaired the synthesis and secretion of antibodies, but was dispensable for the development of PC and memory B cells (Todd et al., 2009). In addition, as an essential transcription factor for PC differentiation, BLIMP1 promoted the splicing of XBP1 by regulating ATF6 and IRE1α, which was required for antibody secretion (Shaffer et al., 2004; Tellier et al., 2016). However, beyond affecting the differentiation of PC, IRE1α can also regulate the differentiation of pro-B cells by promoting Ig genes rearrangements and the formation of BCRs (Zhang et al., 2005). Beyond the role of UPR in PCs, it is now recognized that autophagy is also essential for the differentiation of PCs (Sandoval et al., 2018). In a mouse model lacking the autophagy gene ATG5, PCs were shown to have an expanded ER and stronger ER stress (Pengo et al., 2013). Despite enhanced immunoglobulin synthesis, the antibody titers and survival of PCs decreased, indicating that autophagy is essential for PC homeostasis, which can limit immunoglobulin synthesis and optimize humoral immunity (Table 1).
As reported in most literature, the PERK pathway is not required for antibody secretion. Zhu et al. (2019) showed that UFBP1 can suppress the PERK pathway and promote the development of PCs (Zhu et al., 2019). Compared with WT B cells, the expression of ATF4 was significantly increased in UFBP1-deficient B cells (Zhu et al., 2019). Meanwhile, PC frequencies in the spleen and BM were comparable between WT, PerkF/FCD19cre, and PerkF/FUfbp1F/FCD19cre mice, but obviously lower in Ufbp1F/FCD19cre mice. This result confirmed the previous conclusion that the absence of PERK does not affect the differentiation of PCs. Moreover, the IRE1α-XBP1 branch was shown to regulate the expression of UFBP1 to promote ER expansion (Zhu et al., 2019). These findings indicate that UFBP1 affects the development and function of PCs by participating in different branches of the UPR pathway. However, the specific mechanism for UFBP1 regulating PERK remains unidentified. UFBP1 may inhibit PERK activity through a pathway unrelated to ER stress, thereby promoting the differentiation of naïve B cells into PCs.
Orosomucoid-like 3 (ORMDL3) is a new family of ER proteins, which was reported to play an important role in maintaining ER homeostasis by regulating the UPR response (Dang et al., 2017). Compared with WT mice, the level of B-cell activating factor (Baff) mRNA in spleen cells and serum in Ormdl3–/– mice were significantly decreased, which was crucial for B cell development and function (Parameswaran et al., 2010; Dang et al., 2017). The loss of Baff influenced the differentiation of T1 B cells to T2 B cells, thereby impairing B cell development, and ultimately decreasing the number of mature B cells (Dang et al., 2017). Beclin-1 is a core component of the class III PI3K complex and is essential for the initiation of autophagy. Experiments showed that ORMDL3 could induce autophagy and inhibit apoptosis through Beclin-1 to promote cell survival. In addition, Dang et al. (2017) found that ORMDL3 mediated ER stress mainly through the ATF6 pathway (Dang et al., 2017). Compared with HEK293 cells overexpressing ORMDL3, siRNA-mediated silencing of ATF6 decreased the expression of Beclin-1 and LC3-II. Collectively, ORMDL3 promotes autophagy and inhibits apoptosis through the ATF6-Beclin-1 autophagy regulatory pathway, and ORMDL3 may be important for B cell differentiation and maturation.
Of note, through the analysis of activated B cells, Gaudette et al. (2020) found that B cells upregulated many UPR-related genes before secreting robust antibodies (Gaudette et al., 2020). Instead of XBP1, mTORC1 drives the expression of these genes. Before the XBP1 arm is completely induced, activated B cells initiate the mTORC1 signal, independent of XBP1, upregulating many UPR components to lay the foundation for the early activation of UPR. For instance, by early upregulation of BiP expression, the ER function could be enhanced, reducing the early PCs ER stress caused by the synthesis of large amounts of Ig (Gaudette et al., 2020). This hypothesis reveals aspects that are overlooked in the early PC differentiation process. These results provide suggestions for the identification of early PCs and mature PCs.
Consistent with its critical role in normal PC biology, UPR aberrant activation is extensively related to multiple myeloma (MM). Studies have shown that overexpression of XBP1 appears to promote the initiation of MM. Compared with control mice, Eμ-XBP1s mice exhibited enhanced B cell proliferation and aberrant transformation of PCs (Carrasco et al., 2007). Transcriptome analysis showed that the expression of genes related to the pathogenesis of MM was generally upregulated, indicating the driving role of XBP in MM. Similarly, Patterson et al. (2008) found that the activities of XBP1 and ATF6 were abnormally increased in MM cells, but the PERK pathway inducing apoptosis was not activated. The role of PERK in MM appears to be unclear. Only recently has PERK been proven to play an important role in MM growth. The high expression of BLIMP1 in MM cells inhibited the activation of IRE1α-ASK1 and PERK pathways, therefore, MM cells can survive for a long term without apoptosis (Lin et al., 2012; Liu et al., 2020). These findings indicate that UPR appears to regulate MM differently. Based on these, many small molecule inhibitors or proteasome inhibitors (PIs) have been studied in MM. For example, as a potent PI, bortezomib triggers ER stress by blocking ERAD, and then upregulates CHOP to induce apoptosis (Obeng et al., 2006). However, attributed to the existence of PI resistance, MM still remains incurable. The reason for the failure of PI treatment appears to be related to IRE1α. Leung-Hagesteijn et al. (2013) found that the lack of IRE1α-XBP1 signaling blocked the maturation of PCs, which can promote PI resistance. Therefore, although IRE1α kinase inhibitors can reduce the viability of MM cells, its role in the treatment of MM is controversial.
On the other hand, as a pro-survival mechanism, autophagy promotes MM cells proliferation and survival by degrading protein aggregates (Milan et al., 2016). Based on this, the current researches of autophagy in anti-myeloma treatment mainly focus on two methods (Yun et al., 2017). The first is to induce persistent ER stress and trigger apoptosis by inhibiting autophagy or other protein degradation pathways. The second is to induce cell death by enhancing autophagy. Thus, based on the importance of autophagy in MM cells survival, targeting autophagy may become a new approach for MM treatment.
T Cell
In the thymus, immature T cells gradually differentiate into CD4–CD8– double negative (DN) cells, CD4+CD8+ double positive (DP) cells, and finally develop into CD4+, or CD8+ single positive (SP) cells with MHC restricted recognition and autoantigen tolerance (Brunsing et al., 2008). During these developmental stages, cells transition from quiescence to proliferation, accompanied with increased protein synthesis, which may trigger ER stress. Studies have shown that IRE1α was active in the DP stage, and the loss of IRE1α in BM pluripotent stem cells prevented the differentiation of DN to DP (Brunsing et al., 2008). Moreover, ribosomal protein L22 (RPL22) was shown to promote the development of αβ T cells by inhibiting ER stress responses (Anderson et al., 2007; Solanki et al., 2016; Kemp and Poe, 2019). The deletion of RPL22 aggravated ER stress in αβ T cells, leading to developmental arrest through the selective induction of p53 (Solanki et al., 2016). Downregulation of the PERK signaling pathway could inactivate p53 induction, thereby rescuing the developmental defects of αβ T cells. These results indicate that PERK appears to affect the development of αβ T cells by regulating the expression of p53, although the mechanism for the induction of p53 remains unclear. On the other hand, the activation of TCR induced ER stress, resulting in the upregulation of the expression of ER chaperones Gp96 and BiP (Thaxton et al., 2017). The lack of Gp96 hindered the differentiation of CD4+ T cells. While Gp96 is the target molecular chaperone of ATF6, it remains unknown whether ATF6 is involved in the differentiation of CD4+ T cells.
It is well known that disrupted T cell homeostasis is one of the pathogenic mechanisms of systemic lupus erythematosus (SLE). ER stress induced by ultraviolet irradiation or viral infection is responsible for SLE flare-ups (Hirabayashi et al., 2007). A previous study found that after TG induced ER stress, compared with healthy people, the expression of ER stress markers and autophagy decreased in T cells of SLE patients, while the expression of CHOP increased (Lee et al., 2015). Eventually the level of T cell apoptosis was higher and homeostasis was dysregulated. However, the effect of ER stress-induced autophagy and apoptosis on T cell function and subsets remains unclear, and further research is needed to provide more effective information for the clinical treatment of SLE (Table 1). Beyond affecting T cell development and survival, ERQC is also closely related to the differentiation and effector functions of T cells.
CD4+ T Cell
CD4+ T cells are at the center of the adaptive immune response which activation is strictly regulated. When stimulated with antigen, naïve CD4+ T cells are induced into activation, proliferation, and cytokine secretion and further differentiate into different subsets, including T helper type 1 (Th1), Th2, Th17, and regulatory T cells (Tregs). HRD1 was shown to regulate the development and differentiation of CD4+ T cells (Xu et al., 2016). Loss of HRD1 in T cells results in the accumulation of p27kip1 protein, which arrests the cell cycle at G1/G0, and then disrupts T cells development and inhibits the proliferation of CD4+ T cells (Xu et al., 2016). However, the deletion of p27kip1 in HRD1-deficient T cells does not rescue IL-2 production and Th1 or Th17 differentiation. Considering that E3 ubiquitin ligase usually targets a variety of substrates to achieve its physiological functions, HRD1 may modulate other substrates to regulate CD4+ T cell differentiation.
On the other hand, it has been confirmed that ATF4 plays a role in Th1 differentiation. During the activation of T cells, the increased level of glutathione promotes the expression of ATF4, thereby enhancing protein synthesis to ensure the demand for T cell proliferation (Yang X. et al., 2018). Compared with WT mice, the level of total glutathione in Atf4–/– mice was significantly reduced, resulting in impaired redox homeostasis and reduced CD4+ T cell proliferation (Yang X. et al., 2018). Also, under Th1 polarization conditions, the secretion of IFN-γ was reduced. But, IL-17 secretion remained unchanged under Th17 polarization conditions. In the experimental allergic encephalomyelitis (EAE) model, the Th1 response in Atf4–/– mice was impaired, while Th17 levels were increased, suggesting that ATF4 is necessary for Th1 differentiation. In addition, Brucklacher-Waldert et al. (2017) confirmed that XBP1 can affect Th17 differentiation efficiency. Mice lacking XBP1 showed impaired Th17 differentiation and slower EAE progression. Collectively, these findings indicate that ERQC can regulate the differentiation of Th1 and Th17, which may provide new insights for the development of autoimmune diseases.
Moreover, the IRE1α-XBP1 branch is also involved in Th2 differentiation. Through transcriptome analysis, it has been found that the expression of XBP1 is significantly upregulated when CD4+ T cells are activated and differentiate into Th2 (Kemp et al., 2013; Pramanik et al., 2018). After treating CD4+ T cells with IRE1α RNase inhibitor 4μ8c, the levels of IL-4, IL-5, and IL-13 decreased under Th2 polarization. Interestingly, the PERK-eIF2α branch appears to be related to the secretion of IL-4. TCR stimulation resulted in increased phosphorylation of eIF2α in Th2 cells, but IL-4 protein expression was not detected (Scheu et al., 2006). Unexpectedly, restimulation of Th2 cells showed increased dephosphorylation of eIF2α, and the translation block was relieved, which promoted the production of IL-4. This indicates that the promotion or inhibition of ER stress affect IL-4 secretion, and UPR appears to regulate Th2 effector function in different ways.
Expression of the transcription factor Foxp3 controls the differentiation and function of Tregs. Under inflammatory conditions, ER stress downregulates Foxp3 expression. Eventually, Tregs fail to maintain stable expression, and their function is weakened (Rudensky, 2005; Xu and Fang, 2020). A recent study reported that IRE1α expression was increased in Hrd1fl/fl-Foxp3cre mice, and Foxp3 expression was impaired. After treatment with IRE1α inhibitor, the expression of Foxp3 was rescued, and the function of Tregs was restored (Xu et al., 2019). Therefore, inhibiting ER stress responses mediated by IRE1α or HRD1 might maintain Tregs homeostasis (Table 1).
CD8+ T Cell
CD8+ cytotoxic T cells are the main effector cells of anti-tumor immunity. Adverse conditions in the tumor microenvironment (TME), such as nutrient deprivation, hypoxia, low pH, and ROS, disrupt protein folding, and aggravate ER stress (Urra et al., 2016; Oakes, 2020). Studies have shown that the splicing of XBP1 was increased in CD8+ T cells during acute infection, which can promote the expression of KLRG1 and enhance effector function (Kamimura and Bevan, 2008). However, the loss of XBP1 led to a decrease in the number of KLRG1high effector cells, and the terminal differentiation of CD8+ T cells was hindered. In contrast, cholesterol in tumor tissues induced the expression of XBP1 in tumor-infiltrating CD8+ T cells (Ma et al., 2019). Subsequently, XBP1 induced T cell exhaustion by upregulating the expression of a variety of inhibitory receptors (such as PD-1, 2B4). Therefore, although XBP1 can promote the differentiation of CD8+ cytotoxic T cells, prolonged ER stress is harmful to T cells in the TME.
Contrary to the role of UPR in the development and differentiation of T cells, CHOP is responsible for the impaired tumor immunity. In CD8+ T lymphocytes of patients with advanced ovarian cancer, CHOP expression was elevated, and associated with its poor clinical response (Cao et al., 2019). The accumulation of ROS in the TME could activate PERK and induce the expression of ATF4, which resulted in the upregulation of CHOP. A further study found that the pro-apoptotic factor CHOP can bind and inhibit the transcriptional activity of Tbx21, thereby downregulating the expression of T-bet protein, leading to a decrease in the cellular activity of effector CD8+ T cells (Cao et al., 2019). CD8+ T cells lacking CHOP showed enhanced anti-tumor activity and a delayed tumor growth. Although the loss of CHOP weakens the immunosuppressive function of tumor MDSCs and enhances the activity of tumor infiltrating T cells, ER stress can also modulate tumor cell survival. Therefore, the efficacy of the clinical application of CHOP inhibition needs to be further tested.
On the other hand, in viral infection condition, naïve CD8+ T cells recognize the specific antigen presented by DCs, and then activate and divide into effector T cells. After the pathogen is cleared in vivo, most effector CD8+ T cells die, and only a few differentiate into memory T cells (Puleston et al., 2014). During the transition from effector to memory cells, excess proteins accumulate in cytosol, and ER load dramatically increases. Studies have found that autophagy activity increases during this contraction phase, which affects the development of memory CD8+ T cells through lipid metabolism (Xu et al., 2014). Given that autophagy modulates ER homeostasis of T lymphocytes, it may also regulate the differentiation of memory CD8+ T cells through ER stress; a point which deserves further study (Table 1).
Natural Killer and Natural Killer T Cell
Natural killer cells are special innate lymphocytes derived from CLPs, which have cytotoxic functions similar to cytotoxic T lymphocytes (CTLs), but lack TCR receptors. To date, NK cells play an indispensable role in viral infection and anti-tumor immunity. It is reported that mouse cytomegalovirus (MCMV) infection activates IRE1α-XBP1 in NK cells and drives cell proliferation through the c-Myc signaling pathway (Dong et al., 2019). Although the loss of IRE1α has no effect on NK cell activation and apoptosis, the proliferation and expansion of these cells are impaired. Moreover, XBP1 can promote the survival and enhance the cytotoxic activity of IL-15-driven NK cells (Wang et al., 2019). This indicates that ER stress is an important factor in the expansion of NK cells and suggests that IRE1α-XBP1 may be a potential target for improving the therapeutic effect of expanded NK cells in vitro (Table 1).
Natural killer T (NKT) cells are a special type of T lymphocyte that express TCR and the NK cell receptors. Present studies divide NKT cells into two subsets, most of which are type I NKT cells, also known as invariant NKT (iNKT) cells (Kumar and Delovitch, 2014). Unlike conventional T cells, NKT cells are activated by the lipid antigen presented by CD1d and release cytokines rapidly (Wu and Van Kaer, 2011). Current data indicate that iNKT cells can differentiate into NKT1, NKT2, and NKT17, which play different roles in immunity (Hogquist and Georgiev, 2020). Govindarajan et al. (2018) found that TCR-dependent stimulation of iNKT sublineages resulted in the increased expression of XBP1 in NKT1 and NKT17 (Govindarajan et al., 2018). Although IRE1α had no effect on the development and maturation of resting iNKT cells, the deletion of IRE1α could inhibit cytokine production of NKT1 and NKT17 subsets, which suggested that IRE1α can modulate NKT1 and NKT17 cell effector functions. In contrast to the pro-inflammatory effect of PA in macrophages, a recent study found that PA enhanced the degradation of RIDD on T-bet and gata-3 mRNA by inducing ER stress in iNKT cells, thereby suppressing the production of IL-4 and IFN-γ and attenuating arthritis (Ko et al., 2017). In addition, ER stress induced IRE1α and PERK-dependent branches can increase lipid antigens bound to CD1d molecules, thereby enhancing iNKT cell activation (Bedard et al., 2019; Govindarajan et al., 2020). This indicates that ER-stressed APCs can affect iNKT cell activation via CD1d. Given that self-lipids participate in the positive selection of thymic iNKT cells, it is possible that ER stress in thymocytes may impact self-lipids presented by CD1d to further affect the positive selection and development of iNKT cells (Table 1).
Pharmacological Regulation of Endoplasmic Reticulum Stress
Drugs that regulate ER stress have been reviewed in detail elsewhere (Hetz et al., 2019; Chen and Cubillos-Ruiz, 2021). Therefore, this section will briefly introduce these compounds. The acute activation of the IRE1α branch under ER stress has a protective effect, but its chronic activation is related to diseases such as cancer. There are two kinds of IRE1α inhibitors, RNase inhibitor and kinase inhibitor (Hetz et al., 2019). RNase inhibitors include 4μ8c, MKC3946, and STF-083010, which can block the splicing of XBP1, and the kinase inhibitors include KIRA6 and AMG18. Studies have shown that the use of IRE1α inhibitors may become a potential treatment strategy. For example, STF-083010 and AMG18 inhibit the proliferation of tumor cells in MM and pancreatic neuroendocrine tumors, respectively (Papandreou et al., 2011; Moore et al., 2019). In addition, B-I09 (4μ8C analogs) can induce apoptosis of B-cell leukemia cells (Tang et al., 2014). However, there are also reports showing that 4μ8c exhibits off-target effects (Sato et al., 2017). 4μ8c treatment of pancreatic β-cells blocked the secretion of insulin, indicating that 4μ8c has other side effects unrelated to the inhibition of IRE1α activity.
Unlike GSK2606414 and GSK2656157, AMG44 and AMG52 are highly selective PERK inhibitors without pancreatic toxicity (Smith et al., 2015). PERK kinase inhibitors are widely used in a variety of diseases, which can suppress the growth of human tumor xenografts and improve the progression of neurodegenerative diseases including frontotemporal dementia (Atkins et al., 2013; Radford et al., 2015). However, these compounds have a dose-dependent defect and further clinical studies are needed to evaluate their potential side effects. Apart from directly inhibiting PERK signaling, ISRIB is a potent eIF2α inhibitor, which significantly inhibits the progression of prostate cancer by blocking the phosphorylation signaling of eIF2α (Nguyen et al., 2018). Notably, activation of PERK and phosphorylation of eIF2α can also play a protective role. CCT020312 and its derivative MK-28 activated the PERK pathway and improved the symptoms of Huntington’s disease in mice (Ganz et al., 2020). However, the activation mechanism remains undefined and needs further exploration. Moreover, similar to Salubrinal in preventing eIF2α dephosphorylation, guanabenz and its analog Sephin1 target GADD34 to delay translation recovery and relieve amyotrophic lateral sclerosis (Das et al., 2015). Paradoxically, studies showed that guanabenz and Sephin1 do not inhibit GADD34 activity, and it is necessary to further explain how these compounds induce sustained eIF2α phosphorylation (Crespillo-Casado et al., 2017, 2018). Although the PERK activators have been shown to have potential therapeutic value, prolonged phosphorylation of eIF2α may induce the expression of pro-apoptotic genes. Therefore, in order to obtain the desired therapeutic effect, these factors must be carefully considered.
Contrary to the role of IRE1α and PERK in diseases, ATF6 appears to have less effect on organism functions. Recently, with the development of ATF6 compounds, the unique therapeutic potential has gradually emerged. Ceapin-A7 is a highly effective ATF6 inhibitor, which prevents the movement of ATF6 by inducing the accumulation of ATF6 in the ER, but the exact target is still unclear (Gallagher et al., 2016). Alternatively, compounds 147 and 263 have been shown to specifically activate ATF6, of which 147 can protect the heart from ischemia/reperfusion injury (Blackwood et al., 2019).
Thus, although regulators of the three UPR branches have been developed, each method faces the problem of targeted toxicity and mechanism of action. In the future, it is necessary to define the role of UPR signaling in different physiological and pathological conditions and optimize the dosing regimen to promote the clinical application of UPR modulators.
Conclusion and Perspective
Protein homeostasis is essential for the normal survival of immune cells. However, the accumulation of misfolded proteins above a threshold will induce ER stress. To restore homeostasis, cells reduce protein synthesis through ERQC, including activating the UPR response, ERAD and autophagy. Although an increasing number of data recognize that ERQC plays an important role in the pathogenesis of tumors and inflammatory disease, its exact role in the biology of specific immune cells still waits for further exploration.
Moreover, the application of small molecule inhibitors has confirmed that UPR is involved in various diseases, there are still many problems to be solved. For example, UPR targeted therapy will have off-target effects, and the UPR in a state of over-activation or inhibition can also cause harmful effects. In the future, it may be possible to adopt short-term treatment or local delivery strategies to reduce organ toxicity.
Taken together, regulation of the ERQC system may constitute a new method for promoting immunity, but more investigation is needed.
Author Contributions
SX conceived the idea and designed the work. YJ designed and drafted the manuscript. ZT perfected the literature search and edited the manuscript. HC helped in the concept discussion. All authors contributed to the manuscript revision and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation, China (grant nos. 81871234, 81901579, and 82172763) and the Jiangsu Provincial Key Research and Development Program (grant no. BE2017696).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ERQC, Endoplasmic reticulum quality control; ERAD, Endoplasmic reticulum-associated degradation; UPR, Unfolded protein response; DCs, Dendritic cells; MDSCs, Myeloid-derived-suppressor cells; NK, Natural killer; ER, Endoplasmic reticulum; IRE1α, Inositol-requiring enzyme 1α; PERK, Protein kinase RNA-like ER kinase; ATF6, Activating transcription factor 6; SER, Smooth endoplasmic reticulum; RER, Rough endoplasmic reticulum; UPS, Ubiquitin-proteasome system; ALS, Autophagic-lysosomal system; GRP78/Bip, Glucose-regulated protein 78; XBP1, X-box-binding protein 1; TRAF2, Tumor-necrosis factor-α-receptor-associated factor 2; ASK1, Apoptosis signal-regulating kinase 1; JNK, JUN N-terminal kinase; RIDD, Regulated IRE1-dependent decay; eIF2α, Eukaryotic initiation factor 2α; GADD34, Growth arrest and DNA damage-inducible protein 34; CHOP, C/EBP homologous protein; BM, Bone marrow; CMPs, Common myeloid progenitor cells; CLPs, Common lymphoid progenitor cells; BLIMP1, B-lymphocyte-induced maturation protein 1; KCs, Kupffer cells; PA, Palmitic acid; ATMs, Adipose tissue macrophages; PMN-MDSC, Polymorphonuclear MDSC; M-MDSC, Monocytic MDSC; TRAIL-Rs, TRAIL receptors; LOX-1, Lectin-type oxidized LDL receptor-1; Pre-BCR, Pre-B cell receptor; PCs, Plasma cells; UFBP1, UFM1 binding protein 1; ORMDL3; Orosomucoid-like 3; Baff, B-cell activating factor; MM, Multiple myeloma; PIs, Proteasome inhibitors; RPL22, Ribosomal protein L22; SLE, Systemic lupus erythematosus; ROS, Reactive oxygen species; EAE, Experimental allergic encephalomyelitis; TME, The tumor microenvironment; MCMV, Mouse cytomegalovirus; iNKT, Invariant NKT.
References
Adams, E., and Luoma, A. (2013). The adaptable major histocompatibility complex (MHC) fold: structure and function of nonclassical and MHC class I-like molecules. Annu. Rev. Immunol. 31, 529–561. doi: 10.1146/annurev-immunol-032712-095912
Anderson, S. J., Lauritsen, J. P., Hartman, M. G., Foushee, A. M., Lefebvre, J. M., Shinton, S. A., et al. (2007). Ablation of ribosomal protein L22 selectively impairs alphabeta T cell development by activation of a p53-dependent checkpoint. Immunity 26, 759–772.
Atkins, C., Liu, Q., Minthorn, E., Zhang, S., Figueroa, D., Moss, K., et al. (2013). Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 73, 1993–2002. doi: 10.1158/0008-5472.Can-12-3109
Bedard, M., Shrestha, D., Priestman, D., Wang, Y., Schneider, F., Matute, J., et al. (2019). Sterile activation of invariant natural killer T cells by ER-stressed antigen-presenting cells. Proc. Natl. Acad. Sci. U.S.A. 116, 23671–23681. doi: 10.1073/pnas.1910097116
Blackwood, E., Azizi, K., Thuerauf, D., Paxman, R., Plate, L., Kelly, J., et al. (2019). Pharmacologic ATF6 activation confers global protection in widespread disease models by reprograming cellular proteostasis. Nat. Commun. 10:187. doi: 10.1038/s41467-018-08129-2
Blander, J. (2018). Regulation of the cell biology of antigen cross-presentation. Annu. Rev. Immunol. 36, 717–753. doi: 10.1146/annurev-immunol-041015-055523
Brown, B., Ratner, B., Goodman, S., Amar, S., and Badylak, S. (2012). Macrophage polarization: an opportunity for improved outcomes in biomaterials and regenerative medicine. Biomaterials 33, 3792–3802. doi: 10.1016/j.biomaterials.2012.02.034
Brucklacher-Waldert, V., Ferreira, C., Stebegg, M., Fesneau, O., Innocentin, S., Marie, J., et al. (2017). Cellular stress in the context of an inflammatory environment supports TGF-β-independent T helper-17 differentiation. Cell Rep. 19, 2357–2370. doi: 10.1016/j.celrep.2017.05.052
Brunsing, R., Omori, S., Weber, F., Bicknell, A., Friend, L., Rickert, R., et al. (2008). B- and T-cell development both involve activity of the unfolded protein response pathway. J. Biol. Chem. 283, 17954–17961. doi: 10.1074/jbc.M801395200
Burgoyne, R. D., and Duncan, J. S. (1998). Secretion of milk proteins. J. Mammary Gland Biol. Neoplasia 3, 275–286.
Burr, M., Cano, F., Svobodova, S., Boyle, L., Boname, J., and Lehner, P. (2011). HRD1 and UBE2J1 target misfolded MHC class I heavy chains for endoplasmic reticulum-associated degradation. Proc. Natl. Acad. Sci. U.S.A. 108, 2034–2039. doi: 10.1073/pnas.1016229108
Burr, M., van den Boomen, D., Bye, H., Antrobus, R., Wiertz, E., and Lehner, P. (2013). MHC class I molecules are preferentially ubiquitinated on endoplasmic reticulum luminal residues during HRD1 ubiquitin E3 ligase-mediated dislocation. Proc. Natl. Acad. Sci. U.S.A. 110, 14290–14295. doi: 10.1073/pnas.1303380110
Cao, Y., Trillo-Tinoco, J., Sierra, R., Anadon, C., Dai, W., Mohamed, E., et al. (2019). ER stress-induced mediator C/EBP homologous protein thwarts effector T cell activity in tumors through T-bet repression. Nat. Commun. 10:1280. doi: 10.1038/s41467-019-09263-1
Carrasco, D., Sukhdeo, K., Protopopova, M., Sinha, R., Enos, M., Carrasco, D., et al. (2007). The differentiation and stress response factor XBP-1 drives multiple myeloma pathogenesis. Cancer Cell 11, 349–360. doi: 10.1016/j.ccr.2007.02.015
Chen, X., and Cubillos-Ruiz, J. (2021). Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 21, 71–88. doi: 10.1038/s41568-020-00312-2
Christianson, J. C., and Ye, Y. (2014). Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat. Struct. Mol. Biol. 21, 325–335.
Coelho, D., and Domingos, P. (2014). Physiological roles of regulated Ire1 dependent decay. Front. Genet. 5:76. doi: 10.3389/fgene.2014.00076
Condamine, T., Dominguez, G., Youn, J., Kossenkov, A., Mony, S., Alicea-Torres, K., et al. (2016). Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci. Immunol. 1:aaf8943. doi: 10.1126/sciimmunol.aaf8943
Condamine, T., Kumar, V., Ramachandran, I., Youn, J., Celis, E., Finnberg, N., et al. (2014). ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J. Clin. Invest. 124, 2626–2639. doi: 10.1172/jci74056
Corazzari, M., Gagliardi, M., Fimia, G., and Piacentini, M. (2017). Endoplasmic reticulum stress, unfolded protein response, and cancer cell fate. Front. Oncol. 7:78. doi: 10.3389/fonc.2017.00078
Crespillo-Casado, A., Chambers, J., Fischer, P., Marciniak, S., and Ron, D. (2017). PPP1R15A-mediated dephosphorylation of eIF2α is unaffected by Sephin1 or Guanabenz. eLife 6:e26109. doi: 10.7554/eLife.26109
Crespillo-Casado, A., Claes, Z., Choy, M., Peti, W., Bollen, M., and Ron, D. A. (2018). Sephin1-insensitive tripartite holophosphatase dephosphorylates translation initiation factor 2α. J. Biol. Chem. 293, 7766–7776. doi: 10.1074/jbc.RA118.002325
Cubillos-Ruiz, J., Silberman, P., Rutkowski, M., Chopra, S., Perales-Puchalt, A., Song, M., et al. (2015). ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell 161, 1527–1538. doi: 10.1016/j.cell.2015.05.025
Cullinan, S. B., and Diehl, J. A. (2004). PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 279, 20108–20117.
Cullinan, S. B., and Diehl, J. A. (2006). Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 38, 317–332.
Cybulsky, A. (2017). Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat. Rev. Nephrol. 13, 681–696. doi: 10.1038/nrneph.2017.129
Dang, J., Bian, X., Ma, X., Li, J., Long, F., Shan, S., et al. (2017). ORMDL3 facilitates the survival of splenic B cells via an ATF6α-endoplasmic reticulum stress-beclin1 autophagy regulatory pathway. J. Immunol. (Baltimore, Md : 1950) 199, 1647–1659. doi: 10.4049/jimmunol.1602124
Das, I., Krzyzosiak, A., Schneider, K., Wrabetz, L., D’Antonio, M., Barry, N., et al. (2015). Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science (New York, NY) 348, 239–242. doi: 10.1126/science.aaa4484
Denton, D., Nicolson, S., and Kumar, S. (2012). Cell death by autophagy: facts and apparent artefacts. Cell Death Differ. 19, 87–95.
Dong, H., Adams, N., Xu, Y., Cao, J., Allan, D., Carlyle, J., et al. (2019). The IRE1 endoplasmic reticulum stress sensor activates natural killer cell immunity in part by regulating c-Myc. Nat. Immunol. 20, 865–878. doi: 10.1038/s41590-019-0388-z
Duwaerts, C., Siao, K., Soon, R., Her, C., Iwawaki, T., Kohno, K., et al. (2020). Hepatocyte-specific deletion of XBP1 sensitizes mice to liver injury through hyperactivation of IRE1α. Cell Death Differ. 28, 1455–1465. doi: 10.1038/s41418-020-00671-1
Ellgaard, L., and Helenius, A. (2003). Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 4, 181–191.
English, A., Zurek, N., and Voeltz, G. (2009). Peripheral ER structure and function. Curr. Opin. Cell Biol. 21, 596–602. doi: 10.1016/j.ceb.2009.04.004
Gallagher, C., Garri, C., Cain, E., Ang, K., Wilson, C., Chen, S., et al. (2016). Ceapins are a new class of unfolded protein response inhibitors, selectively targeting the ATF6α branch. eLife 5:e11878. doi: 10.7554/eLife.11878
Ganz, J., Shacham, T., Kramer, M., Shenkman, M., Eiger, H., Weinberg, N., et al. (2020). A novel specific PERK activator reduces toxicity and extends survival in Huntington’s disease models. Sci. Rep. 10:6875. doi: 10.1038/s41598-020-63899-4
Gaudette, B. T., Jones, D. D., Bortnick, A., Argon, Y., and Allman, D. (2020). mTORC1 coordinates an immediate unfolded protein response-related transcriptome in activated B cells preceding antibody secretion. Nat. Commun. 11:723.
Govindarajan, S., Gaublomme, D., Van der Cruyssen, R., Verheugen, E., Van Gassen, S., Saeys, Y., et al. (2018). Stabilization of cytokine mRNAs in iNKT cells requires the serine-threonine kinase IRE1alpha. Nat. Commun. 9:5340. doi: 10.1038/s41467-018-07758-x
Govindarajan, S., Verheugen, E., Venken, K., Gaublomme, D., Maelegheer, M., Cloots, E., et al. (2020). ER stress in antigen-presenting cells promotes NKT cell activation through endogenous neutral lipids. EMBO Rep. 21, e48927. doi: 10.15252/embr.201948927
Grootjans, J., Kaser, A., Kaufman, R., and Blumberg, R. (2016). The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 16, 469–484. doi: 10.1038/nri.2016.62
Grotzke, J., and Cresswell, P. (2015). Are ERAD components involved in cross-presentation? Mol. Immunol. 68, 112–115. doi: 10.1016/j.molimm.2015.05.002
Grotzke, J., Sengupta, D., Lu, Q., and Cresswell, P. (2017). The ongoing saga of the mechanism(s) of MHC class I-restricted cross-presentation. Curr. Opin. Immunol. 46, 89–96. doi: 10.1016/j.coi.2017.03.015
Halaby, M., Hezaveh, K., Lamorte, S., Ciudad, M., Kloetgen, A., MacLeod, B., et al. (2019). GCN2 drives macrophage and MDSC function and immunosuppression in the tumor microenvironment. Sci. Immunol. 4:eaax8189. doi: 10.1126/sciimmunol.aax8189
Han, J., Back, S. H., Hur, J., Lin, Y. H., Gildersleeve, R., Shan, J., et al. (2013). ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 15, 481–490.
Harding, H., and Ron, D. (2002). Endoplasmic reticulum stress and the development of diabetes: a review. Diabetes 51 Suppl 3, S455–S461. doi: 10.2337/diabetes.51.2007.s455
Haze, K., Yoshida, H., Yanagi, H., Yura, T., and Mori, K. (1999). Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787–3799.
Hetz, C., and Papa, F. R. (2018). The unfolded protein response and cell fate control. Mol. Cell 69, 169–181.
Hetz, C., Axten, J., and Patterson, J. (2019). Pharmacological targeting of the unfolded protein response for disease intervention. Nat. Chem. Biol. 15, 764–775. doi: 10.1038/s41589-019-0326-2
Hetz, C., Chevet, E., and Oakes, S. A. (2015). Proteostasis control by the unfolded protein response. Nat. Cell Biol. 17, 829–838.
Hirabayashi, Y., Oka, Y., Tada, M., Takahashi, R., and Ishii, T. (2007). A potential trigger of nephritogenic anti-DNA antibodies in lupus nephritis. Ann. N. Y. Acad. Sci. 1108, 92–95. doi: 10.1196/annals.1422.010
Hogquist, K., and Georgiev, H. (2020). Recent advances in iNKT cell development. F1000Research 9, doi: 10.12688/f1000research.21378.1
Hotamisligil, G. S. (2010). Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140, 900–917.
Hughes, E., Hammond, C., and Cresswell, P. (1997). Misfolded major histocompatibility complex class I heavy chains are translocated into the cytoplasm and degraded by the proteasome. Proc. Natl. Acad. Sci. U.S.A. 94, 1896–1901. doi: 10.1073/pnas.94.5.1896
Iwakoshi, N., Pypaert, M., and Glimcher, L. (2007). The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J. Exp. Med. 204, 2267–2275.
Jeong, H., Sim, H. J., Song, E. K., Lee, H., Ha, S. C., Jun, Y., et al. (2016). Crystal structure of SEL1L: Insight into the roles of SLR motifs in ERAD pathway. Sci. Rep. 6:20261.
Ji, Y., Kim, H., Yang, L., Sha, H., Roman, C. A., Long, Q., et al. (2016). The sel1l-hrd1 endoplasmic reticulum-associated degradation complex manages a key checkpoint in B cell development. Cell Rep. 16, 2630–2640.
Kamimura, D., and Bevan, M. (2008). Endoplasmic reticulum stress regulator XBP-1 contributes to effector CD8+ T cell differentiation during acute infection. J. Immunol. (Baltimore, Md : 1950) 181, 5433–5441.
Kemp, K., and Poe, C. (2019). Stressed: the unfolded protein response in T cell development, activation, and function. Int. J. Mol. Sci. 20:1792.
Kemp, K., Lin, Z., Zhao, F., Gao, B., Song, J., Zhang, K., et al. (2013). The serine-threonine kinase inositol-requiring enzyme 1α (IRE1α) promotes IL-4 production in T helper cells. J. Biol. Chem. 288, 33272–33282. doi: 10.1074/jbc.M113.493171
Kenner, L., Anand, A., Nguyen, H., Myasnikov, A., Klose, C., McGeever, L., et al. (2019). eIF2B-catalyzed nucleotide exchange and phosphoregulation by the integrated stress response. Science (New York, NY) 364, 491–495. doi: 10.1126/science.aaw2922
Ko, J., Koh, J., So, J., Jeon, Y., Kim, H., and Chung, D. (2017). Palmitate inhibits arthritis by inducing t-bet and gata-3 mRNA degradation in iNKT cells via IRE1α-dependent decay. Sci. Rep. 7:14940. doi: 10.1038/s41598-017-14780-4
Kumar, V., and Delovitch, T. (2014). Different subsets of natural killer T cells may vary in their roles in health and disease. Immunology 142, 321–336. doi: 10.1111/imm.12247
Lee, W., Sung, M., Lee, E., Yoo, H., Cheon, Y., Chae, H., et al. (2015). A pathogenic role for ER stress-induced autophagy and ER chaperone GRP78/BiP in T lymphocyte systemic lupus erythematosus. J. Leukocyte Biol. 97, 425–433. doi: 10.1189/jlb.6A0214-097R
Leitman, J., Shenkman, M., Gofman, Y., Shtern, N., Ben-Tal, N., Hendershot, L., et al. (2014). Herp coordinates compartmentalization and recruitment of HRD1 and misfolded proteins for ERAD. Mol. Biol. Cell 25, 1050–1060. doi: 10.1091/mbc.E13-06-0350
Leung-Hagesteijn, C., Erdmann, N., Cheung, G., Keats, J., Stewart, A., Reece, D., et al. (2013). Xbp1s-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell 24, 289–304. doi: 10.1016/j.ccr.2013.08.009
Li, Y., Xie, Y., Hao, J., Liu, J., Ning, Y., Tang, Q., et al. (2018). ER-localized protein-Herpud1 is a new mediator of IL-4-induced macrophage polarization and migration. Exp. Cell Res. 368, 167–173. doi: 10.1016/j.yexcr.2018.04.023
Lin, F., Huang, S., Hung, K., Su, S., Chung, C., Matsuzawa, A., et al. (2012). ASK1 promotes apoptosis of normal and malignant plasma cells. Blood 120, 1039–1047. doi: 10.1182/blood-2011-12-399808
Liu, F., Li, X., Lu, C., Bai, A., Bielawski, J., Bielawska, A., et al. (2016). Ceramide activates lysosomal cathepsin B and cathepsin D to attenuate autophagy and induces ER stress to suppress myeloid-derived suppressor cells. Oncotarget 7, 83907–83925. doi: 10.18632/oncotarget.13438
Liu, H., Xiong, C., Liu, J., Sun, T., Ren, Z., Li, Y., et al. (2020). Aspirin exerts anti-tumor effect through inhibiting Blimp1 and activating ATF4/CHOP pathway in multiple myeloma. Biomed. Pharmacother. 125:110005. doi: 10.1016/j.biopha.2020.110005
Liu, Z., Lv, Y., Zhao, N., Guan, G., and Wang, J. (2015). Protein kinase R-like ER kinase and its role in endoplasmic reticulum stress-decided cell fate. Cell Death Dis. 6:e1822.
Ma, X., Bi, E., Lu, Y., Su, P., Huang, C., Liu, L., et al. (2019). Cholesterol induces CD8 T cell exhaustion in the tumor microenvironment. Cell Metab. 30, 143–156.e5.
Macri, C., Pang, E., Patton, T., and O’Keeffe, M. (2018). Dendritic cell subsets. Semin. Cell Dev. Biol. 84, 11–21. doi: 10.1016/j.semcdb.2017.12.009
Malhotra, J. D., and Kaufman, R. J. (2007). The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 18, 716–731.
Martinon, F., Chen, X., Lee, A., and Glimcher, L. (2010). TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 11, 411–418. doi: 10.1038/ni.1857
Maurel, M., Chevet, E., Tavernier, J., and Gerlo, S. (2014). Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 39, 245–254.
McQuiston, A., and Diehl, J. A. (2017). Recent insights into PERK-dependent signaling from the stressed endoplasmic reticulum. F1000Research 6:1897.
Mendes, A., Gigan, J., Rodriguez Rodrigues, C., Choteau, S., Sanseau, D., Barros, D., et al. (2021). Proteostasis in dendritic cells is controlled by the PERK signaling axis independently of ATF4. Life Sci. Alliance 4:e202000865. doi: 10.26508/lsa.202000865
Milan, E., Fabbri, M., and Cenci, S. (2016). Autophagy in plasma cell ontogeny and malignancy. J. Clin. Immunol. 36 Suppl 1, 18–24. doi: 10.1007/s10875-016-0254-9
Mohamed, E., Sierra, R., Trillo-Tinoco, J., Cao, Y., Innamarato, P., Payne, K., et al. (2020). The unfolded protein response mediator perk governs myeloid cell-driven immunosuppression in tumors through inhibition of STING signaling. Immunity 52, 668–682.e7. doi: 10.1016/j.immuni.2020.03.004
Moore, P., Qi, J., Thamsen, M., Ghosh, R., Peng, J., Gliedt, M., et al. (2019). Parallel signaling through IRE1α and PERK regulates pancreatic neuroendocrine tumor growth and survival. Cancer Res. 79, 6190–6203. doi: 10.1158/0008-5472.Can-19-1116
Nakatogawa, H. (2020). Autophagic degradation of the endoplasmic reticulum. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 96, 1–9.
Nam, T., Han, J. H., Devkota, S., and Lee, H. W. (2017). Emerging paradigm of crosstalk between autophagy and the ubiquitin-proteasome system. Mol. Cells 40, 897–905.
Nguyen, H., Conn, C., Kye, Y., Xue, L., Forester, C., Cowan, J., et al. (2018). Development of a stress response therapy targeting aggressive prostate cancer. Sci. Transl. Med. 10:eaar2036. doi: 10.1126/scitranslmed.aar2036
Oakes, S. A. (2020). Endoplasmic reticulum stress signaling in cancer cells. Am. J. Pathol. 190, 934–946.
Obeng, E., Carlson, L., Gutman, D., Harrington, W., Lee, K., and Boise, L. (2006). Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 107, 4907–4916. doi: 10.1182/blood-2005-08-3531
Oh, H., Lee, S., and Park, P. (2020). ER stress contributes to autophagy induction by adiponectin in macrophages: Implication in cell survival and suppression of inflammatory response. Cytokine 127:154959. doi: 10.1016/j.cyto.2019.154959
Oh, J., Riek, A., Weng, S., Petty, M., Kim, D., Colonna, M., et al. (2012). Endoplasmic reticulum stress controls M2 macrophage differentiation and foam cell formation. J. Biol. Chem. 287, 11629–11641. doi: 10.1074/jbc.M111.338673
Osorio, F., Tavernier, S., Hoffmann, E., Saeys, Y., Martens, L., Vetters, J., et al. (2014). The unfolded-protein-response sensor IRE-1α regulates the function of CD8α+ dendritic cells. Nat. Immunol. 15, 248–257. doi: 10.1038/ni.2808
Oyadomari, S., Araki, E., and Mori, M. (2002). Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis 7, 335–345. doi: 10.1023/a:1016175429877
Papandreou, I., Denko, N., Olson, M., Van Melckebeke, H., Lust, S., Tam, A., et al. (2011). Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 117, 1311–1314. doi: 10.1182/blood-2010-08-303099
Parameswaran, R., Müschen, M., Kim, Y., Groffen, J., and Heisterkamp, N. (2010). A functional receptor for B-cell-activating factor is expressed on human acute lymphoblastic leukemias. Cancer Res. 70, 4346–4356. doi: 10.1158/0008-5472.Can-10-0300
Patterson, J., Palombella, V., Fritz, C., and Normant, E. (2008). IPI-504, a novel and soluble HSP-90 inhibitor, blocks the unfolded protein response in multiple myeloma cells. Cancer Chemother. Pharmacol. 61, 923–932. doi: 10.1007/s00280-007-0546-0
Pengo, N., Scolari, M., Oliva, L., Milan, E., Mainoldi, F., Raimondi, A., et al. (2013). Plasma cells require autophagy for sustainable immunoglobulin production. Nat. Immunol. 14, 298–305. doi: 10.1038/ni.2524
Perego, J., Mendes, A., Bourbon, C., Camosseto, V., Combes, A., Liu, H., et al. (2018). Guanabenz inhibits TLR9 signaling through a pathway that is independent of eIF2α dephosphorylation by the GADD34/PP1c complex. Sci. Signal. 11:eaam8104. doi: 10.1126/scisignal.aam8104
Pramanik, J., Chen, X., Kar, G., Henriksson, J., Gomes, T., Park, J. E., et al. (2018). Genome-wide analyses reveal the IRE1a-XBP1 pathway promotes T helper cell differentiation by resolving secretory stress and accelerating proliferation. Genome Med. 10:76.
Puleston, D., Zhang, H., Powell, T., Lipina, E., Sims, S., Panse, I., et al. (2014). Autophagy is a critical regulator of memory CD8(+) T cell formation. Elife 3:e03706. doi: 10.7554/eLife.03706
Qi, L., Tsai, B., and Arvan, P. (2017). New insights into the physiological role of endoplasmic reticulum-associated degradation. Trends Cell Biol. 27, 430–440. doi: 10.1016/j.tcb.2016.12.002
Radford, H., Moreno, J., Verity, N., Halliday, M., and Mallucci, G. (2015). PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. 130, 633–642. doi: 10.1007/s00401-015-1487-z
Rainbolt, T. K., Saunders, J. M., and Wiseman, R. L. (2014). Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trends Endocrinol. Metab. 25, 528–537.
Ramirez, M., Hernandez, S., Soto-Pantoja, D., and Cook, K. (2019). Endoplasmic reticulum stress pathway, the unfolded protein response, modulates immune function in the tumor microenvironment to impact tumor progression and therapeutic response. Int. J. Mol. Sci. 21:169. doi: 10.3390/ijms21010169
Rao, J., Yue, S., Fu, Y., Zhu, J., Wang, X., Busuttil, R., et al. (2014). ATF6 mediates a pro-inflammatory synergy between ER stress and TLR activation in the pathogenesis of liver ischemia-reperfusion injury. Am. J. Transpl. 14, 1552–1561. doi: 10.1111/ajt.12711
Ron, D., and Walter, P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529.
Rudensky, A. (2005). Foxp3 and dominant tolerance. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 360, 1645–1646.
Sandoval, H., Kodali, S., and Wang, J. (2018). Regulation of B cell fate, survival, and function by mitochondria and autophagy. Mitochondrion 41, 58–65. doi: 10.1016/j.mito.2017.11.005
Sato, H., Shiba, Y., Tsuchiya, Y., Saito, M., and Kohno, K. (2017). 4μ8C inhibits insulin secretion independent of IRE1α RNase Activity. Cell Struct. Funct. 42, 61–70. doi: 10.1247/csf.17002
Scheu, S., Stetson, D., Reinhardt, R., Leber, J., Mohrs, M., and Locksley, R. (2006). Activation of the integrated stress response during T helper cell differentiation. Nat. Immunol. 7, 644–651. doi: 10.1038/ni1338
Schönthal, A. H. (2012). Endoplasmic reticulum stress: its role in disease and novel prospects for therapy. Scientifica 2012:857516.
Shaffer, A., Shapiro-Shelef, M., Iwakoshi, N., Lee, A., Qian, S., Zhao, H., et al. (2004). XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 21, 81–93. doi: 10.1016/j.immuni.2004.06.010
Shan, B., Wang, X., Wu, Y., Xu, C., Xia, Z., Dai, J., et al. (2017). The metabolic ER stress sensor IRE1α suppresses alternative activation of macrophages and impairs energy expenditure in obesity. Nat. Immunol. 18, 519–529. doi: 10.1038/ni.3709
Shen, J., Chen, X., Hendershot, L., and Prywes, R. (2002). ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 3, 99–111.
Sica, A., and Mantovani, A. (2012). Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 122, 787–795. doi: 10.1172/jci59643
Smith, A., Andrews, K., Beckmann, H., Bellon, S., Beltran, P., Booker, S., et al. (2015). Discovery of 1H-pyrazol-3(2H)-ones as potent and selective inhibitors of protein kinase R-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 58, 1426–1441. doi: 10.1021/jm5017494
Solanki, N., Stadanlick, J., Zhang, Y., Duc, A., Lee, S., Lauritsen, J., et al. (2016). Rpl22 loss selectively impairs αβ T cell development by dysregulating endoplasmic reticulum stress signaling. J. Immunol. (Baltimore, Md : 1950) 197, 2280–2289. doi: 10.4049/jimmunol.1600815
Song, S., Tan, J., Miao, Y., and Zhang, Q. (2018). Crosstalk of ER stress-mediated autophagy and ER-phagy: involvement of UPR and the core autophagy machinery. J. Cell. Physiol. 233, 3867–3874. doi: 10.1002/jcp.26137
Song, S., Tan, J., Miao, Y., Li, M., and Zhang, Q. (2017). Crosstalk of autophagy and apoptosis: Involvement of the dual role of autophagy under ER stress. J. Cell. Physiol. 232, 2977–2984. doi: 10.1002/jcp.25785
Sun, S., Shi, G., Han, X., Francisco, A. B., Ji, Y., Mendonça, N., et al. (2014). Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc. Natl. Acad. Sci. U.S.A. 111, E582–E591.
Tang, C., Ranatunga, S., Kriss, C., Cubitt, C., Tao, J., Pinilla-Ibarz, J., et al. (2014). Inhibition of ER stress-associated IRE-1/XBP-1 pathway reduces leukemic cell survival. J.Clin. Invest. 124, 2585–2598. doi: 10.1172/jci73448
Tcyganov, E., Hanabuchi, S., Hashimoto, A., Campbell, D., Kar, G., Slidel, T., et al. (2021). Distinct mechanisms govern populations of myeloid-derived suppressor cells in chronic viral infection and cancer. J. Clin. Invest. 131:e145971. doi: 10.1172/jci145971
Tellier, J., Shi, W., Minnich, M., Liao, Y., Crawford, S., Smyth, G., et al. (2016). Blimp-1 controls plasma cell function through the regulation of immunoglobulin secretion and the unfolded protein response. Nat. Immunol. 17, 323–330. doi: 10.1038/ni.3348
Thaxton, J., Wallace, C., Riesenberg, B., Zhang, Y., Paulos, C., Beeson, C., et al. (2017). Modulation of endoplasmic reticulum stress controls CD4 T-cell activation and antitumor function. Cancer Immunol. Res. 5, 666–675. doi: 10.1158/2326-6066.Cir-17-0081
Thevenot, P., Sierra, R., Raber, P., Al-Khami, A., Trillo-Tinoco, J., Zarreii, P., et al. (2014). The stress-response sensor chop regulates the function and accumulation of myeloid-derived suppressor cells in tumors. Immunity 41, 389–401. doi: 10.1016/j.immuni.2014.08.015
Todd, D., McHeyzer-Williams, L., Kowal, C., Lee, A., Volpe, B., Diamond, B., et al. (2009). XBP1 governs late events in plasma cell differentiation and is not required for antigen-specific memory B cell development. J. Exp. Med. 206, 2151–2159. doi: 10.1084/jem.20090738
Urra, H., Dufey, E., Avril, T., Chevet, E., and Hetz, C. (2016). Endoplasmic reticulum stress and the hallmarks of cancer. Trends Cancer 2, 252–262.
van Anken, E., Pena, F., Hafkemeijer, N., Christis, C., Romijn, E., Grauschopf, U., et al. (2009). Efficient IgM assembly and secretion require the plasma cell induced endoplasmic reticulum protein pERp1. Proc. Natl. Acad. Sci. U.S.A. 106, 17019–17024. doi: 10.1073/pnas.0903036106
Verschoor, C., Puchta, A., and Bowdish, D. (2012). The macrophage. Methods Mol. Biol. (Clifton, NJ) 844, 139–156. doi: 10.1007/978-1-61779-527-5_10
Wang, Y., Zhang, Y., Yi, P., Dong, W., Nalin, A., Zhang, J., et al. (2019). The IL-15-AKT-XBP1s signaling pathway contributes to effector functions and survival in human NK cells. Nat. Immunol. 20, 10–17. doi: 10.1038/s41590-018-0265-1
Wang, Y., Zhu, J., Zhang, L., Zhang, Z., He, L., Mou, Y., et al. (2017). Role of C/EBP homologous protein and endoplasmic reticulum stress in asthma exacerbation by regulating the IL-4/signal transducer and activator of transcription 6/transcription factor EC/IL-4 receptor α positive feedback loop in M2 macrophages. J. Allergy Clin. Immunol. 140, 1550–1561.e8. doi: 10.1016/j.jaci.2017.01.024
Wu, L., and Van Kaer, L. (2011). Natural killer T cells in health and disease. Front. Biosci. (Scholar edition) 3:236–251. doi: 10.2741/s148
Wu, X., and Rapoport, T. (2018). Mechanistic insights into ER-associated protein degradation. Curr. Opin. Cell Biol. 53, 22–28. doi: 10.1016/j.ceb.2018.04.004
Xia, S., Wang, Z., Sun, S., Su, Y., Li, Z., Shao, J., et al. (2020). Endoplasmic reticulum stress and protein degradation in chronic liver disease. Pharmacol. Res. 161:105218. doi: 10.1016/j.phrs.2020.105218
Xu, X., Araki, K., Li, S., Han, J., Ye, L., Tan, W., et al. (2014). Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat. Immunol. 15, 1152–1161. doi: 10.1038/ni.3025
Xu, Y., and Fang, D. (2020). Endoplasmic reticulum-associated degradation and beyond: The multitasking roles for HRD1 in immune regulation and autoimmunity. J. Autoimmun. 109:102423.
Xu, Y., Melo-Cardenas, J., Zhang, Y., Gau, I., Wei, J., Montauti, E., et al. (2019). The E3 ligase Hrd1 stabilizes Tregs by antagonizing inflammatory cytokine-induced ER stress response. JCI Insight 4:e121887. doi: 10.1172/jci.insight.121887
Xu, Y., Zhao, F., Qiu, Q., Chen, K., Wei, J., Kong, Q., et al. (2016). The ER membrane-anchored ubiquitin ligase Hrd1 is a positive regulator of T-cell immunity. Nat. Commun. 7:12073. doi: 10.1038/ncomms12073
Yamamoto, K., Yoshida, H., Kokame, K., Kaufman, R., and Mori, K. (2004). Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J. Biochem. 136, 343–350. doi: 10.1093/jb/mvh122
Yang, F., Liu, Y., Ren, H., Zhou, G., Yuan, X., and Shi, X. (2019). ER-stress regulates macrophage polarization through pancreatic EIF-2alpha kinase. Cell. Immunol. 336, 40–47. doi: 10.1016/j.cellimm.2018.12.008
Yang, H., Qiu, Q., Gao, B., Kong, S., Lin, Z., and Fang, D. (2014). Hrd1-mediated BLIMP-1 ubiquitination promotes dendritic cell MHCII expression for CD4 T cell priming during inflammation. J. Exp. Med. 211, 2467–2479.
Yang, X., Xia, R., Yue, C., Zhai, W., Du, W., Yang, Q., et al. (2018). ATF4 regulates CD4 T cell immune responses through metabolic reprogramming. Cell Rep. 23, 1754–1766. doi: 10.1016/j.celrep.2018.04.032
Yang, Y., Kong, S., Zhang, Y., Melo-Cardenas, J., Gao, B., Zhang, Y., et al. (2018). The endoplasmic reticulum-resident E3 ubiquitin ligase Hrd1 controls a critical checkpoint in B cell development in mice. J. Biol. Chem. 293, 12934–12944. doi: 10.1074/jbc.RA117.001267
Yang, Z., and Klionsky, D. J. (2010). Eaten alive: a history of macroautophagy. Nat. Cell Biol. 12, 814–822.
Yao, Y., Wang, Y., Zhang, Z., He, L., Zhu, J., Zhang, M., et al. (2016). Chop deficiency protects mice against bleomycin-induced pulmonary fibrosis by attenuating M2 macrophage production. Mol. Ther. 24, 915–925. doi: 10.1038/mt.2016.36
Yun, Z., Zhichao, J., Hao, Y., Ou, J., Ran, Y., Wen, D., et al. (2017). Targeting autophagy in multiple myeloma. Leukemia Res. 59, 97–104. doi: 10.1016/j.leukres.2017.06.002
Zhang, K., Wong, H., Song, B., Miller, C., Scheuner, D., and Kaufman, R. (2005). The unfolded protein response sensor IRE1alpha is required at 2 distinct steps in B cell lymphopoiesis. J. Clin. Invest. 115, 268–281. doi: 10.1172/jci21848
Zhang, M., Srivastava, G., and Lu, L. (2004). The pre-B cell receptor and its function during B cell development. Cell. Mol. Immunol. 1, 89–94.
Zhou, Z., Torres, M., Sha, H., Halbrook, C. J., Van den Bergh, F., Reinert, R. B., et al. (2020). Endoplasmic reticulum-associated degradation regulates mitochondrial dynamics in brown adipocytes. Science 368, 54–60.
Keywords: ERQC, UPR, ERAD, autophagy, immune cell
Citation: Jiang Y, Tao Z, Chen H and Xia S (2021) Endoplasmic Reticulum Quality Control in Immune Cells. Front. Cell Dev. Biol. 9:740653. doi: 10.3389/fcell.2021.740653
Received: 14 July 2021; Accepted: 07 September 2021;
Published: 29 September 2021.
Edited by:
Lawrence H. Boise, Emory University, United StatesReviewed by:
Holger W. Auner, Imperial College London, United KingdomChong Wu, Sun Yat-sen University, China
Copyright © 2021 Jiang, Tao, Chen and Xia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sheng Xia, eGlhc2hlbmcxNTE5QDE2My5jb20=
†These authors have contributed equally to this work