 Fang Hao
Fang Hao Chen Wang†
Chen Wang† Xunlei Kang
Xunlei Kang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol., 19 August 2021
Sec. Signaling
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.730400
This article is part of the Research TopicThe Roles of Oncogenic Phosphatase/Kinase in TumorsView all 10 articles
Protein tyrosine phosphatases (PTPs) are modulators of cellular functions such as differentiation, metabolism, migration, and survival. PTPs antagonize tyrosine kinases by removing phosphate moieties from molecular signaling residues, thus inhibiting signal transduction. Two PTPs, SHP-1 and SHP-2 (SH2 domain-containing phosphatases 1 and 2, respectively) and another inhibitory phosphatase, SH2 domain-containing inositol phosphatase (SHIP), are essential for cell function, which is reflected in the defective phenotype of mutant mice. Interestingly, SHP-1, SHP-2, and SHIP mutations are identified in many cases of human leukemia. However, the impact of these phosphatases and their mutations regarding the onset and progression of leukemia is controversial. The ambiguity of the role of these phosphatases imposes challenges on the development of targeting therapies for leukemia. This fundamental problem, confronted by the expanding investigational field of leukemia, will be addressed in this review, which will include a discussion of the molecular mechanisms of SHP-1, SHP-2, and SHIP in normal hematopoiesis and their role in leukemia. Clinical development of leukemic therapies achieved by targeting these phosphatases will be addressed as well.
SH2 domain-containing phosphatase 1 (SHP-1), encoded by the PTPN6 gene, is expressed mainly in hemopoietic cellular systems. SHP-1, with a molecular weight of 68 kDa, is made up of three domains: the N-terminal Src homology-2 (SH2) domain, the C-terminal SH2 domain, and the C-terminal catalytic Protein tyrosine phosphatase (PTP) domain (Figure 1A; Lorenz, 2009). Two tyrosine residues at the C-terminus of SHP-1 (Y536 and Y564) are phosphorylated by various stimuli. The phosphorylation of tyrosine residues modifies the function and activity of SHP-1 depending on the property of the stimulus. SHP-1 exists in an auto-inhibited conformation (Yang et al., 2003). The N-SH2 domain binds tightly to the PTP domain and blocks substrate access to the catalytic domain, thus keeping the enzyme in its inactive conformation. On the contrary, when the phospho-peptide is bound to the C-SH2 domain, the N-terminal SH2 domain is released from the PTP domain, which relieves this autoinhibition and catalytically activates the enzyme (Figure 1B). The “moth-eaten” mice demonstrated a deficiency of SHP-1 and were affected by many hemopoietic disorders, including autoimmune hyperactivation of macrophages, which that suggested a defect in negative regulation (Shultz et al., 1997; Tsui et al., 2006). The SHP-1 gene has two promoters: the distal promoter, which is only present in epithelial cells, and the proximal promoter, which is active in both epithelial and hemopoietic cells. Therefore, under normal conditions, expression levels of SHP-1 vary between epithelial and hematopoietic cells. This concept is supported by an aberrant level of SHP-1 in cancers (Tsui et al., 2006). In hematopoietic cancers, such as myeloma, promoter methylation inhibited SHP-1 expression. Conversely, SHP-1 has been found to be overexpressed in epithelial cancer, such as breast cancer (Tsui et al., 2006).
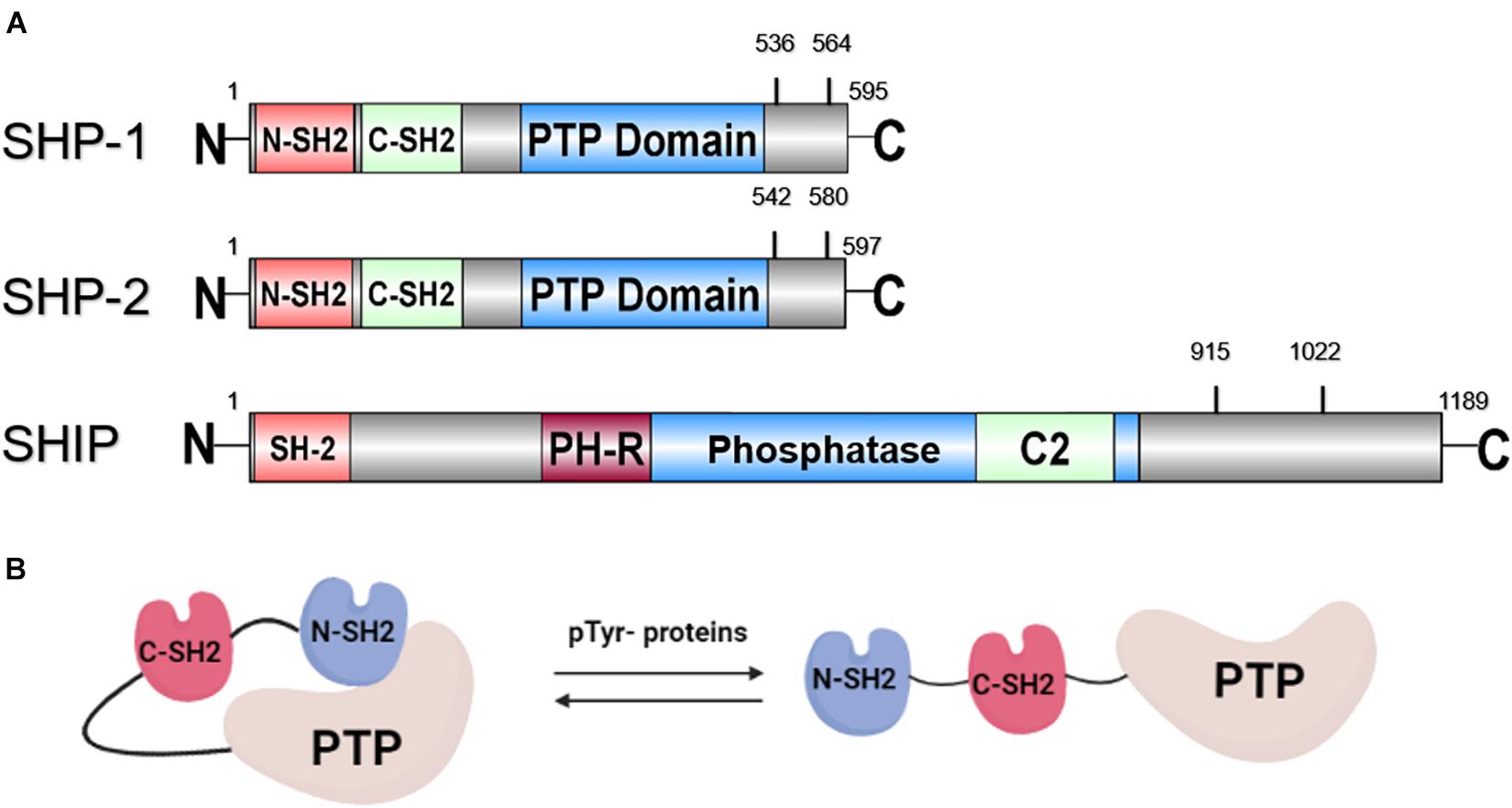
Figure 1. (A) Schematic representation of the structure of SHP-1, SHP-2, and SHIP with tyrosine phosphorylation sites. (B) Structure of SHP protein in auto-inhibited and activated conformation.
Next, SH2 domain-containing phosphatase 2 (SHP-2), encoded by the PTPN11 gene, with a molecular weight of 68 kDa, is another member of the PTP family. SHP-2 shares a structure similar to SHP-1: both are composed of two SH2 domains at the N-terminus followed by a PTP domain and two tyrosine residues for phosphorylation by various stimuli at the C-terminus (Figure 1A). Comparable to SHP-1, SHP-2 is also auto-inhibited by its N-SH2 domain (Figure 1B). Activating SHP-2 mutations have been detected in many cancers, such as melanoma, acute myeloid leukemia (AML), lung cancer, colorectal cancer, etc. (Bentires-Alj et al., 2004; Miyamoto et al., 2008). This result suggests that PTPN11 may be a proto-oncogene. Despite its oncogenic potential, SHP-2 plays a role in tissue development. This is illustrated as SHP-2 expression was found to be present in neurons during brain development. A loss-of-function mutation of SHP-2 in mice led to inhibition of sympathetic neurite outgrowth (Chen et al., 2002). Furthermore, SHP-2 deficiency is related to autoimmune disorders with varying defectives in organ development, such as Noonan syndrome and Leopard syndrome. PTPN11 mutations were detected in 50 and 80% cases, respectively, in patients with these two syndromes (Table 1) (Shen et al., 2020).
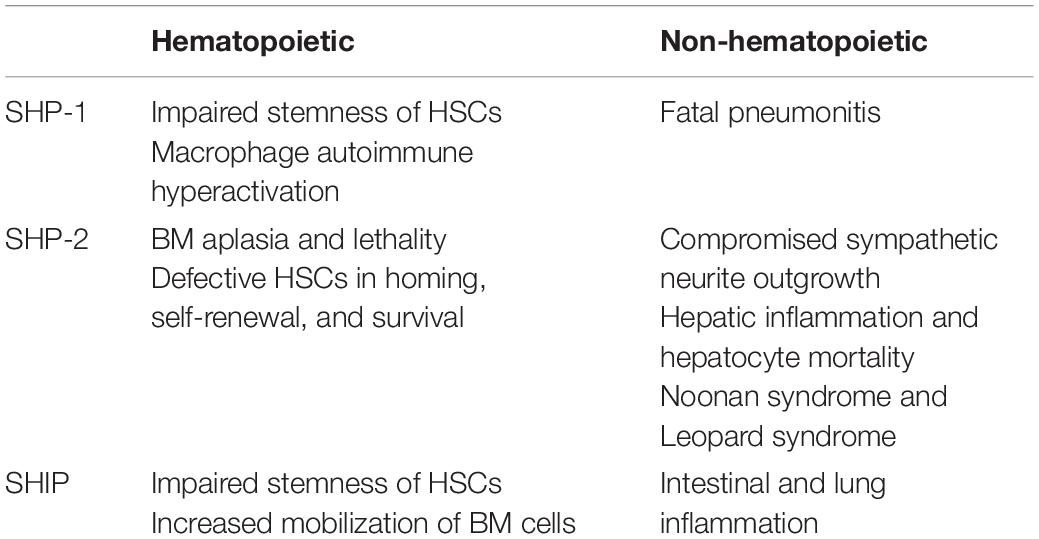
Table 1. Hematopoietic and non-hematopoietic abnormalities in vivo mediated by dysregulated SHP-1, SHP-2, and SHIP.
Lastly, SH2 domain-containing inositol phosphatase (SHIP) is a 145 kDa sized protein that is encoded by the INPP5 (inositol polyphosphate-5-phosphatase) gene. SHIP interferes with the PI3K (phosphatidylinositol 3-kinase)/Akt pathway by dephosphorylating the P13K product PI(3,4,5)P3 to PI(3,4)P2 (Blunt and Ward, 2012). In the presence of SHIP, a controlled level of PI(3,4,5)P3 is produced, which activates downstream Akt and induces cell proliferation. Alternate transcriptional splice variants of SHIP encoding different isoforms of the protein have been characterized. One isoform of SHIP, SHIP-1, is expressed primarily in the hematopoietic system and suppresses the proliferation of hematopoietic progenitor cells (Fu et al., 2019). As shown in Figure 1A, the SH2 domain of SHIP is not involved in autoinhibition, but instead is responsible for membrane translocation and recruitment to upstream kinases. SHIP is activated by phosphorylation of Ser440 in the phosphatase domain by PKA (cyclic AMP-dependent protein kinase) (Zhang et al., 2009), or it is allosterically activated by binding of PI(3,4)P2 to the C2 domain (Ong et al., 2007). The dephosphorylation activity of SHIP to produce PI (3,4,5)P3 is carried out by both the Pleckstrin homology-related (PH-R) and phosphatase domain (Pauls and Marshall, 2017). In addition, the tyrosine residues Y915 and Y1022, located on the C-terminal region of SHIP, regulate the binding of the adapter protein and the activation of subsequent signaling (Lamkin et al., 1997). Furthermore, deficient SHIP led to inflammatory disorders in vivo since SHIP serves to suppress production of inflammatory cytokine by innate immune cells, such as neutrophils and basophils (Kerr, 2011). Severe inflammatory lung disease has been observed in SHIP knockout mice (Lo et al., 2019). Crohn’s disease-like intestinal inflammation and fibrosis were revealed in SHIP knockout mice and irradiated wild-type mice reconstituted with splenocytes from SHIP knockout mice (Kerr, 2011; Maxwell et al., 2011; Lo et al., 2019) as well (Table 1).
In this review, we will interpret SHP-1, SHP-2, and SHIP in terms of their association with cell surface receptors, downstream signaling pathways, and roles in hematopoiesis. We will also focus on how SHP-1, SHP-2, and SHIP are involved in hematopoietic malignant diseases, particularly in AML, and the current development of leukemic therapy involving manipulation of their expression.
SH2 domain-containing phosphatase 1, SHP-2, and SHIP are recruited by and bound to multiple receptors on the cell surface through their SH2 domains. In myeloid cells, SHP-1 is associated with growth factor receptors, such as c-Kit (tyrosine protein kinase, CD117), and several immunoreceptor tyrosine-based inhibitory motif (ITIM) containing receptors, including paired immunoglobulin-like receptor B (PIR-B), leukocyte immunoglobulin-like receptor 1 (LIR-1), and leukocyte immunoglobulin-like receptor 2 (LIR-2; Zhang et al., 2000). In hematopoietic stem cells (HSCs), SHP-2 is activated by Kit, a mast/stem cell growth factor receptor (Kan et al., 2018). In addition, SHP-2 is expressed in stem cells of other tissues, whereas SHP-1 expression is mainly in hematopoietic cells. For example, in neural stem cells, SHP-2 is activated by fibroblast growth factor (FGF) receptor, Vascular endothelial growth factor (VEGF) receptor, and other receptor tyrosine kinases. SHP-2 is also present in mesenchymal stem/progenitor cells and satellite cells, where its activity is regulated by various tyrosine kinases on the cell membrane. SHIP, through its SH2 domain, binds to ITIM receptors such as FcγRIIB and killer cell immunoglobulin-like receptor (KIR) as well as immunoreceptor tyrosine-based activation motif (ITAM) receptors in hematopoietic cells (Dempke et al., 2018).
SH2 domain-containing phosphatase 1 negatively regulates hematopoietic cell proliferation through many intracellular signals. SHP-1 inhibits cytokine receptors, including Epo-R, IL3-R, and IL-2R, as well as growth factor receptors with intrinsic tyrosine kinase activity, such as CSF-1 and GM-CSF that reduce the proliferation of macrophages and granulocytes (Zhang et al., 2000). SHP-1 also suppresses the activation of the growth factor-induced signaling pathway of PI3K/Akt and nuclear factor-kappa B (NF-κB). SHP-1 regulates extracellular signal-related kinases (ERKs) and c-Jun-amino terminal kinases (JNKs) in a positive or negative manner. In addition, SHP-1 binds to erythropoietin (EPO) receptor and blocks subsequent activation of Janus kinase 2 (Jak2), resulting in downregulation of signal transducer and activator of transcription (STAT) (Chong and Maiese, 2007; Dempke et al., 2018). Most of the downstream factors of SHP-1 are also modulated by SHP-2, however, they are regulated in different ways. Upon induction by growth factor, SHP-2 promotes the PI3K/Akt pathway, ERK, and NF-κB through its association with signal regulatory protein α1 (SIRPα1) or Grb2-associated binder-1 (Gab1). Depending on the circumstance, SHP-2 can negatively or positively regulate STAT activation through Jak2 and JNK activation via Ras (Chong and Maiese, 2007; Dempke et al., 2018). Moreover, SHP-2 directly binds to and activates receptors for many growth factors and cytokines, such as IL-3 and GM-CSF (Tamir et al., 2000). Finally, SHIP dephosphorylates the product of its upstream kinase, P13K, and therefore has an inhibitory effect on downstream factors of P13K, including Akt, ERK, and NF-κB. SHIP also affects other kinases, including Burton tyrosine kinase (BTK) and phospholipase-C gamma (PLC-γ), and various transcription factors, such as nuclear factor of activated T cells (NFAT) (Blunt and Ward, 2012; Dempke et al., 2018).
Several receptor tyrosine kinases (RTK) have been identified in hematopoietic cells and are critical mediators of cell signaling (Reilly, 2003). RTKs respond to chemokines, other cytokines, and numerous ligands. Ligand-induced phosphorylation of the RTK tyrosine residues invokes physiological actions, including cell growth, differentiation, metabolism, migration, and survival (Ruvolo, 2019). Phosphatases serve to regulate the actions of RTK through dephosphorylation of tyrosine residues and are required for maintenance of hematopoiesis in the hematopoietic microenvironment. Aberrant tyrosine phosphorylation induced by an imbalance between the activity of RTKs and phosphatases, such as SHP-1, SHP-2, and SHIP, can lead to abnormal cell signaling and hematopoietic defects (Table 1).
SHP-1 and its isoforms are widely expressed in all hematopoietic lineages and maturation stages. SHP-1 inhibits RTK pathways activated by various growth factors and cytokines (Neel et al., 2003; Lorenz, 2009; Abram and Lowell, 2017). Therefore, SHP-1 negatively affects hematopoietic differentiation of embryonic stem cells (Paling and Welham, 2005). The expression of dominant-negative SHP-1 in embryonic stem cells increased the formation of myeloid colonies during differentiation and was reduced by the expression of wild-type SHP-1. Mice lacking SHP-1 exhibited a plethora of perturbations in their hematopoietic and immune systems. Defective myelopoiesis has been found in mice with the SHP-1 inactivation mutation. SHP-1 mutant mice demonstrated an enlarged neutrophil and monocyte population in peripheral blood and increased macrophage proliferation, contributing to the development of fatal pneumonitis (Table 1). In lymphocytes, SHP-1 stimulated cell growth and suppressed their oncogenic capacity (Tibaldi et al., 2011). Mice with specific deletion of SHP-1 in B cells or dendritic cells exhibited increased differentiation and autoimmunity of B-1a and Th1 cells (Pao et al., 2007; Kaneko et al., 2012). Loss of SHP-1 expression in tumor-specific T cells or natural killer cells promoted immune response and antitumor function in a mouse model of disseminated leukemia (Stromnes et al., 2012; Viant et al., 2014). Our previous in vitro study reported that HSCs from SHP-1 knockout mice have attenuated quiescence and impaired long-term self-renewal (Jiang et al., 2018). Therefore, we identified SHP-1 as a regulatory PTP to maintain the microenvironmental homeostasis of HSCs.
SH2 domain-containing phosphatase 2 is also required to maintain hematopoietic growth and homeostasis. SHP-2 contributes to cytokine-mediated signaling through several tyrosine kinases, and therefore loss of SHP-2 results in many dysfunctions in hematopoiesis (Nabinger and Chan, 2012; Rehman et al., 2019). The absence of the PTPN11 gene in murine hematopoietic cells has been shown to result in BM (bone marrow) aplasia and lethality in vivo (Chan et al., 2011). Mice with PTPN11 deletion also exhibited a rapid loss of functional HSCs and myeloid progenitors as well as reduced cellularity in the BM, spleen, and peripheral blood (Chan et al., 2011; Zhu et al., 2011). PTPN11 knockout led to aberrant proliferation and promoted apoptosis of HSCs and progenitor cells in vivo, resulting in defects in homing, self-renewal, and survival (Zhu et al., 2011). Therefore, HSCs with deficient SHP-2 cannot reconstitute peripheral blood in lethally irradiated mouse recipients. SHP-2 is required not only for mouse hematopoiesis, but it is also necessary for human hematopoiesis. Upon SHP-2 knockout in human CD34+ cord blood cells, cell proliferation and colony formation decreased (Li et al., 2011). HSCs of a patient with a point mutation in PTPN11, eliminating the phosphatase activity of SHP-2 in human CD34+ cord blood cells, lost the ability to form colonies (Broxmeyer et al., 2013). These findings reveal a critical role for SHP-2 in the maintenance of functional HSCs and progenitors.
SH2 domain-containing inositol phosphatase is involved in the regulation of HSC proliferation and self-renewal. SHIP knockout induced an increase in the number of HSCs in the BM, spleen, and peripheral blood in vivo (Desponts et al., 2006). Mice with SHIP deletion demonstrated increased HSC cycling that was originally quiescent, which is justified by an increase in 5-FU sensitivity (Helgason et al., 2003). Although HSCs in SHIP knockout mice showed increased proliferation and reduced apoptosis, they also exhibited decreased homing ability to the BM after transplantation due to lower expression of CXCR4 and VCAM-1 receptors (Desponts et al., 2006). HSCs with SHIP knockout also revealed less self-renewal behavior in vivo as the expansion level of competitive repopulating cells (CRU) in the absence of SHIP was significantly lower than wild-type CRUs following transplantation (Helgason et al., 2003). Additionally, SHIP affects HSC adhesion in BM niches, and mobilization of primitive BM cells was expanded in SHIP knockout mice due to increased chemokine responsiveness (Helgason et al., 2003). These data demonstrate that SHIP plays a negative regulatory role in HSC proliferation and survival, and that SHIP is important in the maintenance of primitive hematopoietic cell homeostasis and regeneration.
Dysregulation of SHP-1, SHP-2, and SHIP is associated with uncontrolled cell growth and metabolism, which results in the activation of multiple pro-oncogenic cascades and eventually leads to leukemia. In fact, mutations of these phosphatases are identified in a fair percentage of patients with leukemia, and abnormal levels of SHP-1, SHP-2, and SHIP have been detected in mouse models of leukemia (Dempke et al., 2018). This suggests that they are associated with leukemia development. Therefore, agents that modify the levels or activity of these phosphatases are currently investigated for potential leukemic therapies.
Many studies have identified suppressive effects of SHP-1 on leukemia. For example, promotor methylation leading to silencing of SHP-1 has been reported in 10% of AML cases (Johan et al., 2005). Reexpression of SHP-1 by 5-Azacytidine, a DNA methyltransferase inhibitor, led to increased apoptosis of MV4-11 cells through down-regulation of STAT3 (Al-Jamal et al., 2015). In adult T cell leukemia/lymphoma, the SHP-1 protein has been shown to dephosphorylate and inactivate Sirtuin-1 (SIRT1) that repairs DNA of leukemia cells through homologous recombination (Yu et al., 2018). Upregulation of SHP-1 in Jurkat cells resulted in increased DNA damage, a higher incidence of apoptosis, and reduced colony formation in vitro. On the contrary, other studies indicate that the presence of SHP-1 is associated with the development of leukemia. In vivo myeloproliferative diseases induced by FMS like tyrosine kinase 3-internal tandem duplications (FLT3-ITD), a mutation present in approximately 30% of AML patients, were compromised by deletion of SHP-1 (Reich et al., 2020). Furthermore, our group has demonstrated that LAIR1-mediated SHP-1 activation recruited CAMK1 as an autonomous phosphatase signal adapter for downstream activation of CREB in AML cells, which contributes to the self-renewal of AML cells (Kang et al., 2015).
Likely due to the undetermined role of SHP-1 in the pathogenesis of AML, treatments that alter the expression level of SHP-1 do not currently exist for AML patient therapy. From 2009 to 2018, sodium stibogluconate (SS), a SHP inhibitor with a potent inhibitory effect on SHP-1, underwent several clinical trials and was proven to treat Leishmaniasis (Table 2). On the other hand, the antileukemia effect of SS was reported in vitro in early 2002 (Pathak et al., 2002). This study showed that NB4, a human AML cell line, exhibited higher differentiation, with cell growth arrest in S phase, and increased apoptosis following SS treatment. SS has also been shown to inhibit in vitro growth and induced differentiation of HL-60 and U937 cells. However, no study has confirmed a significant SS-mediated anti-AML effect in vivo, and this may be due to various bodily microenvironments weakening the efficacy of the drug. In addition to the lack of in vivo studies of the SHP-1 inhibitor, the clinical trial of AML treatments involving alterations of SHP-1 activity does not show a significant result. An AML preclinical trial consisting of a therapy that combines azacytidine and gemtuzumab ozogamicin (GO) aimed to enhance the cytotoxicity of the CD33 antibody against AML blasts through epigenetic modifications of PTPN6 (Medeiros et al., 2018). Although 24% of participating patients obtained complete remission, an association between SHP-1 expression and clinical response was not found. The contradicting effects of SHP-1 in AML in vitro and the lack of significant findings in vivo indicate a lack of understanding of SHP-1 signaling pathways and mechanisms involving AML pathogenesis. This blind spot in knowledge must be explored more explicitly in order to better understand the therapeutic potential of targeting SHP-1 in AML.
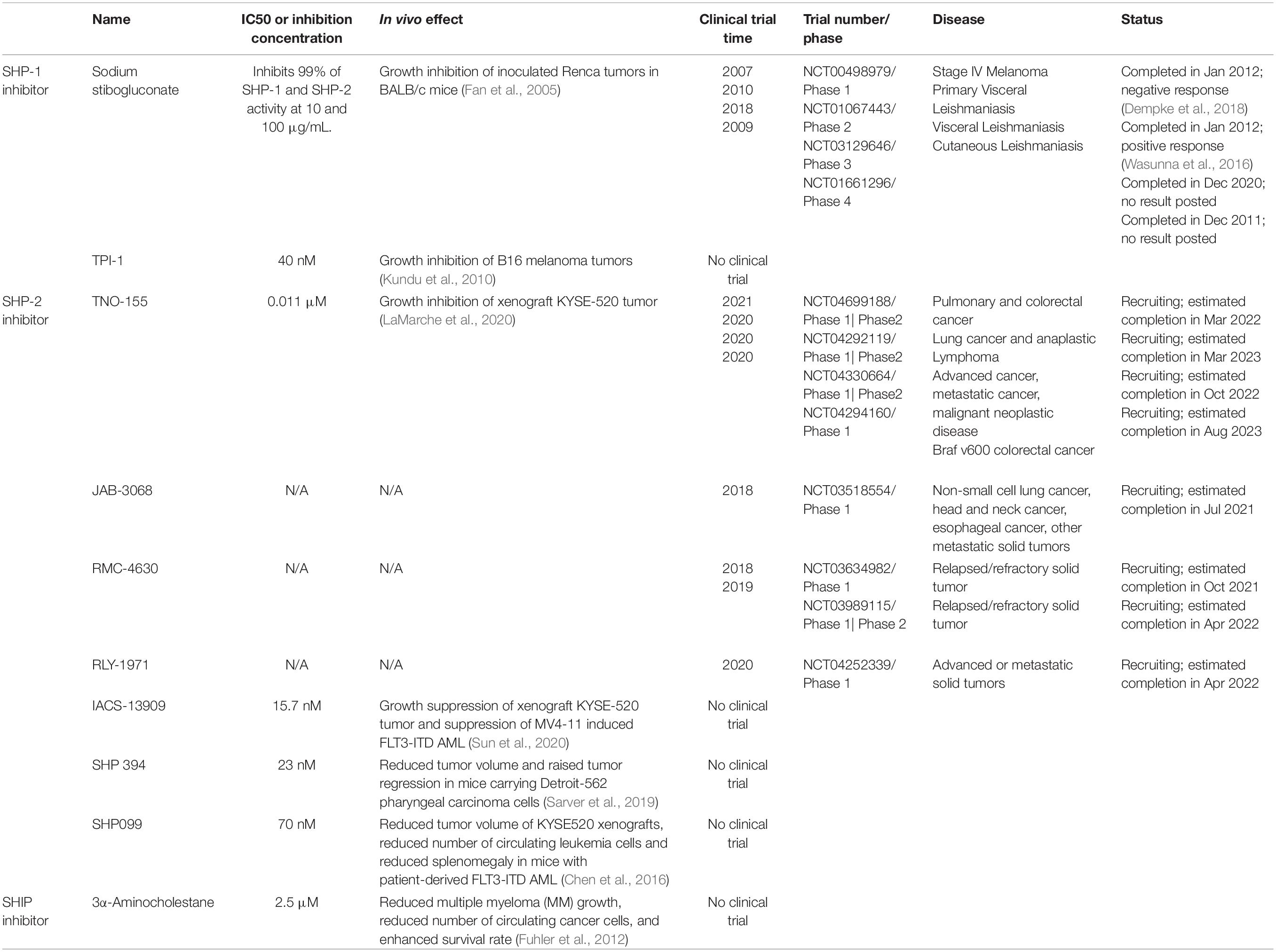
Table 2. SHP and SHIP inhibitors with clinical trial or with tumor suppression effect in vivo.
SH2 domain-containing phosphatase 1 has also demonstrated oncogenic properties in other leukemias. From the in vitro assay of chronic lymphocytic leukemia (CLL), SHP-1 underwent differential phosphorylation and, resultantly, exhibited differential functions and cellular localizations (Tibaldi et al., 2017). SHP-1 with phospho-S591 supported aberrant Lyn-dependent tyrosine phosphorylation of proteins in the cytosol of CLL cells and eventually formed a network of anti-apoptotic signaling. In B cell acute lymphoblastic leukemia (B-ALL), inducible ablation of SHP-1 reduced proliferation and stemness and increased cell cycle arrest in murine B-ALL cells. In vivo deletion of SHP-1 also extended the latency of leukemia and improved the survival rate of mice (Chen Z. et al., 2015). In patients with acute leukemia (AL), the differential expression of SHP-1 and the six-cytokine signaling suppressor (SOCS6) has been detected. SHP-1 and SOCS6 mRNA levels tended to be higher among patients in AL remission than in newly diagnosed patients. Therefore, the expression of SHP-1 and SOCS6 is associated with favorable outcomes, suggesting an anticancer property in AL and potential targets for gene therapy (Liu et al., 2017).
A SHP-1 inhibitor specific for leukemia treatment has not yet been developed. SS, a SHP-1 antagonist that showed effective suppression on inoculated tumors in vivo, underwent two Phase I clinical trials for malignant melanoma (Table 2). However, the outcome was poor, demonstrated by no objective response, life-threatening events in 68% of patients, and side effects such as pancreatitis, BM suppression, and nausea (Dempke et al., 2018). The pessimistic results of these clinical trials further imply an elusive role of SHP-1 in tumor development, and additional is needed to elucidate the potential of SHP-1 as a drug target for tumor treatment.
SH2 domain-containing phosphatase 2 has been determined to have an oncogenic effect on cell proliferation and growth. Mutations in the N-SH2 domain of SHP-2 cause constitutive activation of the SHP-2 protein in the hematopoietic stem and progenitor compartment, resulting in the development of clinical leukemia. A PTPN11 mutation was common in diagnosed patients with secondary AML (Makishima et al., 2017), relapsed pediatric AML (Farrar et al., 2016), and acute lymphoblastic leukemia (ALL; Oshima et al., 2016). The activating mutation of SHP-2 has been identified in 10% of AML cases (Dempke et al., 2018). The prevalence of PTPN11 mutations has been found to be higher in patients over 60 years of age and associated with a poor prognosis (Tsai et al., 2016). Secondary AML patients with PTPN11 mutations tended to progress rapidly and have lower overall survival (Makishima et al., 2017). The PTPN11 mutation exists not only in older patients with AML but also in children with de novo AML. A study identified the substitution of trinucleotides at position 211–213 in 24 pediatric patients as well as other PTPN11 mutations in children with Juvenile myelomonocytic leukemia (JMML) and myelodysplastic syndrome (MDS; Tartaglia et al., 2003). Interestingly, almost all of the mutations identified in these leukemias are located in the N-SH2 domain, which confirms the essential role of the N-SH2 domain in regulating SHP-2 activity and thus the growth and proliferation of hematopoietic cells. The SHP-2 protein also plays an oncogenic role in the development of AML from a molecular perspective. SHP-2 inhibition negatively regulates the activation of downstream factors in FLT3-ITD pathways, which was illustrated in vitro as SHP-2 inhibition led to proliferation of blast cells with FLT3-ITD. Mutation of the FLT3-ITD residue that recruits SHP-2 also reduced FLT3-ITD-induced myeloproliferative disease in vivo (Richine et al., 2016). Furthermore, SHP-2 is involved in the signaling pathway of leukocyte immunoglobulin-like receptor B4 (LILRB4), an immunoreceptor tyrosine-based inhibition motif-containing receptor (Deng et al., 2018). LILRB4 inhibited T cell proliferation and improved the infiltration of AML cells into organs both in vitro and in vivo, and these effects were reversed by knockout of SHP-2 in human AML cell lines.
In addition, the mixed-lineage leukemia (MLL) translocation, MLL-AF10, is known to occur in patients with a G503A mutation in PTPN11. With the inclusion of both the MLL-AF10 and G503A mutations in a mouse model, there was an accelerated rate of disease development compared to the control group (Fu et al., 2017). PTPN11 E76K mutation resulted in increased proliferation of mouse HSCs in vitro. Transplantation of HSCs co-expressing PTPN11 E76K and MLL-AF9 fusion oncogenes induced a lower survival rate and more severe AML phenotype in vivo, such as splenomegaly (Chen L. et al., 2015). Given that SHP-2 imposes an oncogenic effect on hematopoietic cells and that its mutation has been identified in multiple malignancies, efforts have been devoted to discovering inhibitors of SHP-2 and examining their effects against cancers. For example, IACS-13909 (Sun et al., 2020) and SHP099 (Chen et al., 2016) inhibited the proliferation of human AML cell lines and reduced tumor burden derived from xenograft AML cells in vivo (Table 2). Importantly, there are several SHP-2 inhibitors, including TNO-155 (LaMarche et al., 2020; Liu et al., 2021), currently in clinical trials for multiple solid tumors and cancers that will be completed in spring 2022. Although results have yet to be announced, the potential of SHP-2 inhibitors as a treatment option for AML is expected given the strong oncogenicity of SHP-2 and its entanglement in AML oncogenic pathways.
SH2 domain-containing inositol phosphatase 1, a SHIP isoform, has been proposed to play a regulatory role in the development of AML. This is because approximately 50–70% of patients with AML demonstrated constitutive activation of the SHIP-1-regulated P13K/Akt pathway, and approximately 3% displayed a missense mutation in INPP5D (Täger et al., 2017). A higher level of SHIP-1 correlated with prolonged overall survival among 290 patients with AML as well. Furthermore, overexpression of SHIP-1 reduced the proliferation of CD34+ cells in AML patients (Metzner et al., 2009). Studies based on AML cells and the AML mouse model also supported that SHIP-1 acts as an AML suppressor. SHIP-1 transfection to THP-1, a human AML cell line lacking endogenous expression of SHIP-1, resulted in a higher proportion of apoptotic cells. Moreover, SHIP-1 was significantly down-regulated in patients with late-stage MDS, and SHIP-1 expression reduced the number of colonies formed by primary patient myeloid leukemia blasts (Lee et al., 2012). Furthermore, SHIP-1 overexpression extended the life span of the NSG mice model transplanted with human AML cells (Täger et al., 2017).
Considering the role of SHIP-1 as an AML suppressor, factors that suppress SHIP-1 have been investigated. SHIP-1 has been demonstrated to be targeted and down-regulated by miR-155. Compared to normal CD34+ cells, miR-155 expression was significantly higher in cells isolated from patients with late-stage MDS (Lee et al., 2012). Therefore, it is suggested that inhibition of miR-155 can restore suppression of AML by SHIP-1. One study identified Silvestrol as a miR-155 inhibitor and found it to down-regulate miR-155 levels in MV4-11 cells as well as inhibit the growth of MV4-11 and THP-1 cells (Brooks et al., 2010). Silvestrol treatment also improved the survival rate of AML mice in vivo and increased in vitro apoptosis of primary blasts from AML patients. Another study showed that MLN4924 decreased the miR-155 level in AML cells (Khalife et al., 2015). Treatment of MV4-11 cells with MLN4924 resulted in a reduced level of miR-155, upregulation of SHIP-1, suppression of the P13K/Akt pathway, and monocytic differentiation. MLN4924 also reduced the viability of MV4-11 cells and blasts from AML patients. Furthermore, the administration of MLN4924 to an AML mouse model prolonged the survival period, although all treated mice eventually died.
Based on these studies, SHIP-1 is recognized as an AML suppressor via attenuation of the P13K/Akt pathway and reduction of subsequent cell proliferation. However, some studies exhibit dual roles of SHIP-1. In some cases, SHIP-1 amplified survival or proliferative signals in neoplastic cells. The enzyme product of SHIP-1, PI(3,4) P2, has been shown to have higher affinity for Akt and led to more potent activation of the Akt pathway in vitro (Brooks et al., 2010). Treatment of the KG-1 AML cell line with 3α-aminocholestane (3-AC), a SHIP-1 inhibitor, resulted in reduced cell viability in vitro. Furthermore, SHIP-1 expression in patients with AML are largely variable and are not inversely associated with activated Akt level. Therefore, the role of SHIP-1 in the P13K/Akt pathway and AML cell proliferation needs to be clarified further. Considering that the antileukemic effect of 3-AC has not been tested in an AML mouse model, effects of SHIP-1 inhibition in vivo may provide more insight regarding the role and function of the phosphatase in AML.
The identification of SHP-1, SHP-2, and SHIP and their downstream signaling in hematopoietic cells provides new clues to the development and treatment of leukemia. The studies reviewed here demonstrate that an abnormal level of these phosphatases, instead of altered signaling pathways, is associated with aberrant proliferation of hematopoietic cells and leukemia development. Abnormal expression levels derive either from genetic mutations, such as a loss-of-function mutation of SHP-1 or a gain-of-function SHP-2 mutation, or dysregulated regulatory signals, such as upregulated miR-155 levels that inhibit transcription of the SHIP-1 gene in AML patients. Therefore, SHP-1, SHP-2, and SHIP can be potential targets for anti-leukemia therapy. However, scientists are still not in uniform agreement on the true role of these phosphatases in leukemia, which imposes challenges on the development of leukemia-treating reagents that manipulate expression levels of these proteins. Until now, most studies have supported the oncogenic property of SHP-2 and the tumor suppressor property of SHIP, but the effects of SHP-1 on leukemia are still controversial. Consequently, future studies must focus on confirming the effects of SHP-1 and its downstream pathways in different types of leukemia. In regard to SHP-2, it is necessary to refine its antagonists to suppress leukemia development in vivo for clinical trials. Furthermore, SHP-2 inhibitors, which have already undergone clinical trials to treat other tumors and cancers, need to be evaluated for their efficacy in the treatment of leukemia. Finally, miR-155 inhibitors, which upregulate SHIP-1, exhibit suppressive effects in AML in vivo and should also be refined for AML clinical trials.
FH and CW wrote the manuscript and prepared figures. CS provided critical comments and proofreading the manuscript. MC provided comments and joined the discussion. XK organized the structure of the manuscript, provided critical comments and proofreading the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank the AHA (9CDA34770036) and NIH (R37CA241603) for generous support. We regret that we have been unable to cite many relevant primary references due to space limitations.
Abram, C. L., and Lowell, C. A. (2017). Shp1 function in myeloid cells. J. Leuk. Biol. 102, 657–675. doi: 10.1189/jlb.2mr0317-105r
Al-Jamal, H. A., Mat Jusoh, S. A., Hassan, R., and Johan, M. F. (2015). Enhancing SHP-1 expression with 5-azacytidine may inhibit STAT3 activation and confer sensitivity in lestaurtinib (CEP-701)-resistant FLT3-ITD positive acute myeloid leukemia. BMC Cancer 15:869. doi: 10.1186/s12885-015-1695-x
Bentires-Alj, M., Paez, J. G., David, F. S., Keilhack, H., Halmos, B., Naoki, K., et al. (2004). Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 64, 8816–8820. doi: 10.1158/0008-5472.can-04-1923
Blunt, M. D., and Ward, S. G. (2012). Targeting PI3K isoforms and SHIP in the immune system: new therapeutics for inflammation and leukemia. Curr. Opin. Pharmacol. 12, 444–451. doi: 10.1016/j.coph.2012.02.015
Brooks, R., Fuhler, G. M., Iyer, S., Smith, M. J., Park, M. Y., Paraiso, K. H., et al. (2010). SHIP1 inhibition increases immunoregulatory capacity and triggers apoptosis of hematopoietic cancer cells. J. Immunol. 184, 3582–3589. doi: 10.4049/jimmunol.0902844
Broxmeyer, H. E., Etienne-Julan, M., Gotoh, A., Braun, S. E., Lu, L., Cooper, S., et al. (2013). Hematopoietic colony formation from human growth factor-dependent TF1 cells and human cord blood myeloid progenitor cells depends on SHP2 phosphatase function. Stem Cells Dev. 22, 998–1006. doi: 10.1089/scd.2012.0478
Chan, G., Cheung, L. S., Yang, W., Milyavsky, M., Sanders, A. D., Gu, S., et al. (2011). Essential role for Ptpn11 in survival of hematopoietic stem and progenitor cells. Blood 117, 4253–4261. doi: 10.1182/blood-2010-11-319517
Chen, B., Hammonds-Odie, L., Perron, J., Masters, B. A., and Bixby, J. L. (2002). SHP-2 mediates target-regulated axonal termination and NGF-dependent neurite growth in sympathetic neurons. Dev. Biol. 252, 170–187. doi: 10.1006/dbio.2002.0847
Chen, L., Chen, W., Mysliwski, M., Serio, J., Ropa, J., Abulwerdi, F. A., et al. (2015). Mutated Ptpn11 alters leukemic stem cell frequency and reduces the sensitivity of acute myeloid leukemia cells to Mcl1 inhibition. Leukemia 29, 1290–1300. doi: 10.1038/leu.2015.18
Chen, Y. N., LaMarche, M. J., Chan, H. M., Fekkes, P., Garcia-Fortanet, J., Acker, M. G., et al. (2016). Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 535, 148–152.
Chen, Z., Shojaee, S., Buchner, M., Geng, H., Lee, J. W., Klemm, L., et al. (2015). Signalling thresholds and negative B-cell selection in acute lymphoblastic leukaemia. Nature 521, 357–361.
Chong, Z. Z., and Maiese, K. (2007). The Src homology 2 domain tyrosine phosphatases SHP-1 and SHP-2: diversified control of cell growth, inflammation, and injury. Histol. Histopathol. 22, 1251–1267.
Dempke, W. C. M., Uciechowski, P., Fenchel, K., and Chevassut, T. (2018). Targeting SHP-1, 2 and SHIP pathways: a novel strategy for cancer treatment? Oncology. 95, 257–269. doi: 10.1159/000490106
Deng, M., Gui, X., Kim, J., Xie, L., Chen, W., Li, Z., et al. (2018). LILRB4 signalling in leukaemia cells mediates T cell suppression and tumour infiltration. Nature 562, 605–609. doi: 10.1038/s41586-018-0615-z
Desponts, C., Hazen, A. L., Paraiso, K. H., and Kerr, W. G. (2006). SHIP deficiency enhances HSC proliferation and survival but compromises homing and repopulation. Blood 107, 4338–4345. doi: 10.1182/blood-2005-12-5021
Fan, K., Zhou, M., Pathak, M. K., Lindner, D. J., Altuntas, C. Z., Tuohy, V. K., et al. (2005). Sodium stibogluconate interacts with IL-2 in anti-Renca tumor action via a T cell-dependent mechanism in connection with induction of tumor-infiltrating macrophages. J. Immunol. 175, 7003–7008. doi: 10.4049/jimmunol.175.10.7003
Farrar, J. E., Schuback, H. L., Ries, R. E., Wai, D., Hampton, O. A., Trevino, L. R., et al. (2016). Genomic profiling of pediatric acute myeloid leukemia reveals a changing mutational landscape from disease diagnosis to relapse. Cancer Res. 76, 2197–2205. doi: 10.1158/0008-5472.can-15-1015
Fu, J. F., Liang, S. T., Huang, Y. J., Liang, K. H., Yen, T. H., Liang, D. C., et al. (2017). Cooperation of MLL/AF10(OM-LZ) with PTPN11 activating mutation induced monocytic leukemia with a shorter latency in a mouse bone marrow transplantation model. Int. J. Cancer 140, 1159–1172.
Fu, Q., Huang, Y., Ge, C., Li, Z., Tian, H., Li, Q., et al. (2019). SHIP1 inhibits cell growth, migration, and invasion in non-small cell lung cancer through the PI3K/AKT pathway. Oncol. Rep. 41, 2337–2350.
Fuhler, G. M., Brooks, R., Toms, B., Iyer, S., Gengo, E. A., Park, M. Y., et al. (2012). Therapeutic potential of SH2 domain-containing inositol-5’-phosphatase 1 (SHIP1) and SHIP2 inhibition in cancer. Mol. Med. 18, 65–75. doi: 10.2119/molmed.2011.00178
Helgason, C. D., Antonchuk, J., Bodner, C., and Humphries, R. K. (2003). Homeostasis and regeneration of the hematopoietic stem cell pool are altered in SHIP-deficient mice. Blood 102, 3541–3547. doi: 10.1182/blood-2002-12-3939
Jiang, L., Han, X., Wang, J., Wang, C., Sun, X., Xie, J., et al. (2018). SHP-1 regulates hematopoietic stem cell quiescence by coordinating TGF-beta signaling. J. Exp. Med. 215, 1337–1347. doi: 10.1084/jem.20171477
Johan, M. F., Bowen, D. T., Frew, M. E., Goodeve, A. C., and Reilly, J. T. (2005). Aberrant methylation of the negative regulators RASSFIA, SHP-1 and SOCS-1 in myelodysplastic syndromes and acute myeloid leukaemia. Br. J. Haematol. 129, 60–65. doi: 10.1111/j.1365-2141.2005.05412.x
Kan, C., Yang, F., and Wang, S. (2018). SHP2-mediated signal networks in stem cell homeostasis and dysfunction. Stem Cells Int. 2018:8351374.
Kaneko, T., Saito, Y., Kotani, T., Okazawa, H., Iwamura, H., Sato-Hashimoto, M., et al. (2012). Dendritic cell-specific ablation of the protein tyrosine phosphatase Shp1 promotes Th1 cell differentiation and induces autoimmunity. J. Immunol. 188, 5397–5407. doi: 10.4049/jimmunol.1103210
Kang, X., Lu, Z., Cui, C., Deng, M., Fan, Y., Dong, B., et al. (2015). The ITIM-containing receptor LAIR1 is essential for acute myeloid leukaemia development. Nat. Cell Biol. 17, 665–677. doi: 10.1038/ncb3158
Kerr, W. G. (2011). Inhibitor and activator: dual functions for SHIP in immunity and cancer. Ann. N. Y. Acad. Sci. 1217, 1–17. doi: 10.1111/j.1749-6632.2010.05869.x
Khalife, J., Radomska, H. S., Santhanam, R., Huang, X., Neviani, P., Saultz, J., et al. (2015). Pharmacological targeting of miR-155 via the NEDD8-activating enzyme inhibitor MLN4924 (Pevonedistat) in FLT3-ITD acute myeloid leukemia. Leukemia 29, 1981–1992.
Kundu, S., Fan, K., Cao, M., Lindner, D. J., Zhao, Z. J., Borden, E., et al. (2010). Novel SHP-1 inhibitors tyrosine phosphatase inhibitor-1 and analogs with preclinical anti-tumor activities as tolerated oral agents. J. Immunol. 184, 6529–6536. doi: 10.4049/jimmunol.0903562
LaMarche, M. J., Acker, M., Argintaru, A., Bauer, D., Boisclair, J., Chan, H., et al. (2020). Identification of TNO155, an allosteric SHP2 inhibitor for the treatment of cancer. J. Med. Chem. 63, 13578–13594.
Lamkin, T. D., Walk, S. F., Liu, L., Damen, J. E., Krystal, G., and Ravichandran, K. S. (1997). Shc interaction with Src homology 2 domain containing inositol phosphatase (SHIP) in vivo requires the Shc-phosphotyrosine binding domain and two specific phosphotyrosines on SHIP. J. Biol. Chem. 272, 10396–10401. doi: 10.1074/jbc.272.16.10396
Lee, D. W., Futami, M., Carroll, M., Feng, Y., Wang, Z., Fernandez, M., et al. (2012). Loss of SHIP-1 protein expression in high-risk myelodysplastic syndromes is associated with miR-210 and miR-155. Oncogene 31, 4085–4094. doi: 10.1038/onc.2011.579
Li, L., Modi, H., McDonald, T., Rossi, J., Yee, J. K., and Bhatia, R. (2011). A critical role for SHP2 in STAT5 activation and growth factor-mediated proliferation, survival, and differentiation of human CD34+ cells. Blood 118, 1504–1515. doi: 10.1182/blood-2010-06-288910
Liu, C., Lu, H., Wang, H., Loo, A., Zhang, X., Yang, G., et al. (2021). Combinations with allosteric SHP2 inhibitor TNO155 to block receptor tyrosine kinase signaling. Clin. Cancer Res. 27, 342–354. doi: 10.1158/1078-0432.ccr-20-2718
Liu, J., Zheng, Y., Gao, J., Zhu, G., Gao, K., Zhang, W., et al. (2017). Expression of SHP-1 and SOCS6 in patients with acute leukemia and their clinical implication. OncoTargets Ther. 10, 1915–1920. doi: 10.2147/ott.s131537
Lo, Y., Sauve, J. P., Menzies, S. C., Steiner, T. S., and Sly, L. M. (2019). Phosphatidylinositol 3-kinase p110δ drives intestinal fibrosis in SHIP deficiency. Mucosal Immunol. 12, 1187–1200. doi: 10.1038/s41385-019-0191-z
Lorenz, U. (2009). SHP-1 and SHP-2 in T cells: two phosphatases functioning at many levels. Immunol. Rev. 228, 342–359. doi: 10.1111/j.1600-065x.2008.00760.x
Makishima, H., Yoshizato, T., Yoshida, K., Sekeres, M. A., Radivoyevitch, T., Suzuki, H., et al. (2017). Dynamics of clonal evolution in myelodysplastic syndromes. Nat. Genet. 49, 204–212.
Maxwell, M. J., Duan, M., Armes, J. E., Anderson, G. P., Tarlinton, D. M., and Hibbs, M. L. (2011). Genetic segregation of inflammatory lung disease and autoimmune disease severity in SHIP-1-/- mice. J. Immunol. 186, 7164–7175. doi: 10.4049/jimmunol.1004185
Medeiros, B. C., Tanaka, T. N., Balaian, L., Bashey, A., Guzdar, A., Li, H., et al. (2018). A Phase I/II trial of the combination of Azacitidine and Gemtuzumab Ozogamicin for treatment of relapsed acute myeloid leukemia. Clin. Lymph. Myeloma Leukemia 18, 346–352.e5.
Metzner, A., Precht, C., Fehse, B., Fiedler, W., Stocking, C., Günther, A., et al. (2009). Reduced proliferation of CD34(+) cells from patients with acute myeloid leukemia after gene transfer of INPP5D. Gene Ther. 16, 570–573.
Miyamoto, D., Miyamoto, M., Takahashi, A., Yomogita, Y., Higashi, H., Kondo, S., et al. (2008). Isolation of a distinct class of gain-of-function SHP-2 mutants with oncogenic RAS-like transforming activity from solid tumors. Oncogene 27, 3508–3515. doi: 10.1038/sj.onc.1211019
Nabinger, S. C., and Chan, R. J. (2012). Shp2 function in hematopoietic stem cell biology and leukemogenesis. Curr. Opin. Hematol. 19, 273–279. doi: 10.1097/moh.0b013e328353c6bf
Neel, B. G., Gu, H., and Pao, L. (2003). The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 28, 284–293. doi: 10.1016/s0968-0004(03)00091-4
Ong, C. J., Ming-Lum, A., Nodwell, M., Ghanipour, A., Yang, L., Williams, D. E., et al. (2007). Small-molecule agonists of SHIP1 inhibit the phosphoinositide 3-kinase pathway in hematopoietic cells. Blood 110, 1942–1949. doi: 10.1182/blood-2007-03-079699
Oshima, K., Khiabanian, H., da Silva-Almeida, A. C., Tzoneva, G., Abate, F., Ambesi-Impiombato, A., et al. (2016). Mutational landscape, clonal evolution patterns, and role of RAS mutations in relapsed acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. U.S.A. 113, 11306–11311. doi: 10.1073/pnas.1608420113
Paling, N. R. D., and Welham, M. J. (2005). Tyrosine phosphatase SHP-1 acts at different stages of development to regulate hematopoiesis. Blood 105, 4290–4297. doi: 10.1182/blood-2004-08-3271
Pao, L. I., Lam, K. P., Henderson, J. M., Kutok, J. L., Alimzhanov, M., Nitschke, L., et al. (2007). B cell-specific deletion of protein-tyrosine phosphatase Shp1 promotes B-1a cell development and causes systemic autoimmunity. Immunity 27, 35–48. doi: 10.1016/j.immuni.2007.04.016
Pathak, M. K., Hu, X., and Yi, T. (2002). Effects of sodium stibogluconate on differentiation and proliferation of human myeloid leukemia cell lines in vitro. Leukemia 16, 2285–2291. doi: 10.1038/sj.leu.2402692
Pauls, S. D., and Marshall, A. J. (2017). Regulation of immune cell signaling by SHIP1: a phosphatase, scaffold protein, and potential therapeutic target. Eur. J. Immunol. 47, 932–945. doi: 10.1002/eji.201646795
Rehman, A. U., Rafiq, H., Rahman, M. U., Li, J., Liu, H., Luo, S., et al. (2019). Gain-of-Function SHP2 E76Q mutant rescuing autoinhibition mechanism associated with juvenile Myelomonocytic leukemia. J. Chem. Inform. Model. 59, 3229–3239. doi: 10.1021/acs.jcim.9b00353
Reich, D., Kresinsky, A., Müller, J. P., Bauer, R., Kallenbach, J., Schnoeder, T. M., et al. (2020). SHP1 regulates a STAT6-ITGB3 axis in FLT3ITD-positive AML cells. Leukemia 34, 1444–1449. doi: 10.1038/s41375-019-0676-5
Reilly, J. T. (2003). Receptor tyrosine kinases in normal and malignant haematopoiesis. Blood Rev. 17, 241–248. doi: 10.1016/s0268-960x(03)00024-9
Richine, B. M., Virts, E. L., Bowling, J. D., Ramdas, B., Mali, R., Naoye, R., et al. (2016). Syk kinase and Shp2 phosphatase inhibition cooperate to reduce FLT3-ITD-induced STAT5 activation and proliferation of acute myeloid leukemia. Leukemia 30, 2094–2097. doi: 10.1038/leu.2016.131
Ruvolo, P. P. (2019). Role of protein phosphatases in the cancer microenvironment. Biochim. Biophys. Acta Mol. Cell Res. 1866, 144–152. doi: 10.1016/j.bbamcr.2018.07.006
Sarver, P., Acker, M., Bagdanoff, J. T., Chen, Z., Chen, Y. N., Chan, H., et al. (2019). 6-Amino-3-methylpyrimidinones as potent, selective, and orally efficacious SHP2 inhibitors. J. Med. Chem. 62, 1793–1802.
Shen, D., Chen, W., Zhu, J., Wu, G., Shen, R., Xi, M., et al. (2020). Therapeutic potential of targeting SHP2 in human developmental disorders and cancers. Eur. J. Med. Chem. 190:112117. doi: 10.1016/j.ejmech.2020.112117
Shultz, L. D., Rajan, T. V., and Greiner, D. L. (1997). Severe defects in immunity and hematopoiesis caused by SHP-1 protein-tyrosine-phosphatase deficiency. Trends Biotechnol. 15, 302–307. doi: 10.1016/s0167-7799(97)01060-3
Stromnes, I. M., Fowler, C., Casamina, C. C., Georgopolos, C. M., McAfee, M. S., Schmitt, T. M., et al. (2012). Abrogation of SRC homology region 2 domain-containing phosphatase 1 in tumor-specific T cells improves efficacy of adoptive immunotherapy by enhancing the effector function and accumulation of short-lived effector T cells in vivo. J. Immunol. 189, 1812–1825. doi: 10.4049/jimmunol.1200552
Sun, Y., Meyers, B. A., Czako, B., Leonard, P., Mseeh, F., Harris, A. L., et al. (2020). Allosteric SHP2 Inhibitor, IACS-13909, overcomes EGFR-dependent and EGFR-independent resistance mechanisms toward Osimertinib. Cancer Res. 80, 4840–4853. doi: 10.1158/0008-5472.can-20-1634
Täger, M., Horn, S., Latuske, E., Ehm, P., Schaks, M., Nalaskowski, M., et al. (2017). SHIP1, but not an AML-derived SHIP1 mutant, suppresses myeloid leukemia growth in a xenotransplantation mouse model. Gene Ther. 24, 749–753. doi: 10.1038/gt.2017.88
Tamir, I., Dal Porto, J. M., and Cambier, J. C. (2000). Cytoplasmic protein tyrosine phosphatases SHP-1 and SHP-2: regulators of B cell signal transduction. Curr. Opin. Immunol. 12, 307–315. doi: 10.1016/s0952-7915(00)00092-3
Tartaglia, M., Niemeyer, C. M., Fragale, A., Song, X., Buechner, J., Jung, A., et al. (2003). Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat. Genet. 34, 148–150. doi: 10.1038/ng1156
Tibaldi, E., Brunati, A. M., Zonta, F., Frezzato, F., Gattazzo, C., Zambello, R., et al. (2011). Lyn-mediated SHP-1 recruitment to CD5 contributes to resistance to apoptosis of B-cell chronic lymphocytic leukemia cells. Leukemia 25, 1768–1781. doi: 10.1038/leu.2011.152
Tibaldi, E., Pagano, M. A., Frezzato, F., Trimarco, V., Facco, M., Zagotto, G., et al. (2017). Targeted activation of the SHP-1/PP2A signaling axis elicits apoptosis of chronic lymphocytic leukemia cells. Haematologica 102, 1401–1412. doi: 10.3324/haematol.2016.155747
Tsai, C. H., Hou, H. A., Tang, J. L., Liu, C. Y., Lin, C. C., Chou, W. C., et al. (2016). Genetic alterations and their clinical implications in older patients with acute myeloid leukemia. Leukemia 30, 1485–1492.
Tsui, F. W., Martin, A., Wang, J., and Tsui, H. W. (2006). Investigations into the regulation and function of the SH2 domain-containing protein-tyrosine phosphatase, SHP-1. Immunol. Res. 35, 127–136. doi: 10.1385/ir:35:1:127
Viant, C., Fenis, A., Chicanne, G., Payrastre, B., Ugolini, S., and Vivier, E. (2014). SHP-1-mediated inhibitory signals promote responsiveness and anti-tumour functions of natural killer cells. Nat. Commun. 5:5108.
Wasunna, M., Njenga, S., Balasegaram, M., Alexander, N., Omollo, R., Edwards, T., et al. (2016). Efficacy and safety of AmBisome in combination with sodium Stibogluconate or Miltefosine and Miltefosine monotherapy for African Visceral Leishmaniasis: phase II randomized trial. PLoS Negl. Trop. Dis. 10:e0004880. doi: 10.1371/journal.pntd.0004880
Yang, J., Liu, L., He, D., Song, X., Liang, X., Zhao, Z. J., et al. (2003). Crystal structure of human protein-tyrosine phosphatase SHP-1. J. Biol. Chem. 278, 6516–6520.
Yu, W., Li, L., Wang, G., Zhang, W., Xu, J., and Liang, A. (2018). KU70 inhibition impairs both non-homologous end joining and homologous recombination DNA damage repair through SHP-1 induced dephosphorylation of SIRT1 in adult T-cell leukemia-lymphoma cells. Cell. Physiol. Biochem. 49, 2111–2123. doi: 10.1159/000493815
Zhang, J., Somani, A. K., and Siminovitch, K. A. (2000). Roles of the SHP-1 tyrosine phosphatase in the negative regulation of cell signalling. Semin. Immunol. 12, 361–378. doi: 10.1006/smim.2000.0223
Zhang, J., Walk, S. F., Ravichandran, K. S., and Garrison, J. C. (2009). Regulation of the Src homology 2 domain-containing inositol 5’-phosphatase (SHIP1) by the cyclic AMP-dependent protein kinase. J. Biol. Chem. 284, 20070–20078.
Keywords: SHP-1, SHP-2, SHIP, leukemia, AML, PTP inhibitor, signaling pathway, thrapeutic target
Citation: Hao F, Wang C, Sholy C, Cao M and Kang X (2021) Strategy for Leukemia Treatment Targeting SHP-1,2 and SHIP. Front. Cell Dev. Biol. 9:730400. doi: 10.3389/fcell.2021.730400
Received: 24 June 2021; Accepted: 28 July 2021;
Published: 19 August 2021.
Edited by:
Mirco Galiè, University of Verona, ItalyReviewed by:
Chengcheng “Alec” Zhang, University of Texas Southwestern Medical Center, United StatesCopyright © 2021 Hao, Wang, Sholy, Cao and Kang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xunlei Kang, a2FuZ3h1QGhlYWx0aC5taXNzb3VyaS5lZHU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.