 Amir Bahat1†
Amir Bahat1† Thomas MacVicar
Thomas MacVicar Thomas Langer
Thomas Langer
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 27 July 2021
Sec. Signaling
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.720490
This article is part of the Research Topic Mitochondria as a Hub in Cellular Signaling View all 8 articles
Mitochondria are master regulators of metabolism and have emerged as key signalling organelles of the innate immune system. Each mitochondrion harbours potent agonists of inflammation, including mitochondrial DNA (mtDNA), which are normally shielded from the rest of the cell and extracellular environment and therefore do not elicit detrimental inflammatory cascades. Mitochondrial damage and dysfunction can lead to the cytosolic and extracellular exposure of mtDNA, which triggers inflammation in a number of diseases including autoimmune neurodegenerative disorders. However, recent research has revealed that the extra-mitochondrial exposure of mtDNA is not solely a negative consequence of mitochondrial damage and pointed to an active role of mitochondria in innate immunity. Metabolic cues including nucleotide imbalance can stimulate the release of mtDNA from mitochondria in order to drive a type I interferon response. Moreover, important effectors of the innate immune response to pathogen infection, such as the mitochondrial antiviral signalling protein (MAVS), are located at the mitochondrial surface and modulated by the cellular metabolic status and mitochondrial dynamics. In this review, we explore how and why metabolism and innate immunity converge at the mitochondria and describe how mitochondria orchestrate innate immune signalling pathways in different metabolic scenarios. Understanding how cellular metabolism and metabolic programming of mitochondria are translated into innate immune responses bears relevance to a broad range of human diseases including cancer.
Mitochondria are dynamic double-membrane organelles responsible for ATP production, the biosynthesis of macromolecules including lipids, proteins, and nucleotides and for cellular redox status. They serve as metabolic hubs and respond to intrinsic cues and environmental stressors with an incredible degree of plasticity, which enables them to participate in key signalling pathways such as programmed cell death and redox homoeostasis (Bahat and Gross, 2019; Chakrabarty and Chandel, 2021). Apoptotic signals trigger the oligomerisation of pro-apoptotic effector proteins BAX and BAK on the mitochondrial surface, leading to mitochondrial outer membrane (OM) permeabilisation and the release of pro-apoptotic signals into the cytosol to activate caspases and trigger cell death (Flores-Romero et al., 2020). Mitochondria thus play a central role in programmed cell death and this places them as important regulators of cellular responses to pathogen infection. Mitochondrial cell death pathways are directly and indirectly manipulated by viruses to combat innate immune responses and promote infection (Imre, 2020). However, the interplay between mitochondria and innate immunity goes well beyond the control of host cell death or survival. Recent work identified numerous innate immune responses that are controlled by mitochondria and revealed reciprocal relationships that exist between innate immunity and cellular metabolism.
The innate immune system is poised on the front line of defence against infection. Invading pathogens are recognised by a number of pattern recognition receptors (PRRs) that have evolved to sense a wide array of pathogen-associated molecular patterns (PAMPs) such as bacterial lipopolysaccharide (LPS) and viral nucleic acids (Brubaker et al., 2015). The majority of PRRs belong to four protein families: Toll-like receptors (TLRs), C-type lectin receptors (CLRs), NOD-like receptors (NLRs), and retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs). PRR activation triggers a variety of innate immune responses such as the type I interferon (IFN) pathway and expression of pro-inflammatory cytokines and interferon stimulated genes (ISGs), in order to concoct an anti-microbial environment and prime the adaptive immune system (Brubaker et al., 2015). A well described way by which mitochondria support the transduction of innate immune signals occurs downstream of RLR activation during RNA virus infection (Rehwinkel and Gack, 2020). Detection of cytosolic dsRNA by the RLR proteins, RIG-I, or melanoma differentiation-associated gene 5 (MDA5), causes a conformational change in these proteins, which leads them to the RLR adaptor protein called mitochondrial antiviral signalling protein (MAVS) (Kawai et al., 2005; Meylan et al., 2005; Seth et al., 2005; Xu et al., 2005). RIG-I and MDA5 utilise homotypic caspase activation and recruitment domain (CARD)-binding to interact with MAVS, which is anchored on the OM surface (Seth et al., 2005) as well as mitochondrial associated membranes (Horner et al., 2011) and peroxisomes (Dixit et al., 2010). This interaction leads to MAVS aggregation and subsequent activation of NF-kB and interferon regulatory factor (IRF) signalling pathways (Hou et al., 2011).
Innate immune responses are also triggered in the absence of pathogen infection, commonly referred to as sterile inflammation, whereby PRRs and non-PRR receptors sense damage-associated molecular patterns (DAMPs) that originate from within the cell itself (Gong et al., 2020). Besides harbouring pro-apoptotic signals, mitochondria contain numerous potent immunostimulatory DAMPs, including their own genome, that engage the innate immune system upon exposure to the cytosol or release into the extracellular environment. Mitochondria house circular molecules of mitochondrial DNA (mtDNA) within the matrix, which encode 13 subunits of the respiratory chain, 22 transfer RNAs (tRNA), and 2 ribosomal RNAs (rRNA). The exposure of mtDNA has long been known to be immunostimulatory likely owing to its bacterial origin and the release of mtDNA into the cytosol can activate a number of PRRs to trigger a variety of innate immune responses (Collins et al., 2004).
The DNA sensor cyclic GMP-AMP (cGAMP) synthase (cGAS) is one such PRR and binds cytosolic double-stranded DNA (dsDNA) derived from micronuclei, mitochondria, or invading pathogens. dsDNA binding to cGAS leads to its enzymatic activation and the generation of the second messenger cGAMP. cGAMP activates stimulator of interferon genes (STING) at the endoplasmic reticulum (ER), resulting in the recruitment of the tank binding kinase 1 (TBK1) and activation of the IFN signalling pathway (Rongvaux et al., 2014; White et al., 2014; West et al., 2015). While the release of mtDNA can support the antiviral response orchestrated by PRRs, it can also cause chronic inflammation in disease. Notably, exposed mtDNA is also recognised by endosomal TLR9 (Zhang et al., 2010; Shepard, 2020) or, if oxidised, can stimulate the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome (Nakahira et al., 2011; Shimada et al., 2012). It is often unclear what determines the recognition of mtDNA by different PRRs leading to different immune responses. The same mitochondrial DAMPs can even trigger different PRRs in a tissue and cell-type specific manner (Rai et al., 2021).
Recent comprehensive reviews dissect the plethora of scenarios, in which mtDNA release has been observed and linked to mechanisms of immune activation (West and Shadel, 2017; Riley and Tait, 2020). Here, we explore how mitochondria provide a metabolic platform during innate immune signalling and how mtDNA release and innate immunity are coupled to metabolism. Firstly, we look at how mitochondria integrate metabolic regulation within effective innate immune responses to viral infection. We then go on to summarise the machinery and mechanisms that govern the release of mtDNA and discuss the emerging evidence that mtDNA localisation is controlled by cellular metabolism. Finally, we explore the metabolic outcomes of mtDNA release and touch upon the plethora of metabolic consequences of innate immune signalling.
During viral infection, metabolic signals converge on the innate immune regulatory protein MAVS at the mitochondrial surface (Jacobs and Coyne, 2013). The multi-layered crosstalk between metabolism and MAVS at the OM drives innate immune responses to viral infection. The metabolic control of MAVS is dictated by direct metabolite and metabolic enzyme interactions and coupled to the dynamic behaviour of mitochondria.
The metabolic control of MAVS was described by Zhang et al. (2019), who identified lactate as a regulator of MAVS and RIG-I signalling. MAVS associates with the glycolytic enzyme hexokinase 2 (HK2) at the mitochondrial surface, where HK2 activity is regulated (John et al., 2011; Roberts and Miyamoto, 2015). Lactate binds to MAVS and prevents its aggregation, which is required for the activation of TBK1 and IFN production (Zhang et al., 2019; Figure 1). Limiting lactate production, for instance at low glucose levels or upon inhibition of lactate dehydrogenase A (LDHA), increases the IFN response to viral infection. Furthermore, MAVS activation leads to the dissociation and inactivation of HK2, which further decreases lactate levels (Zhang et al., 2019). Interestingly, similar to many cancer cells, virally infected cells shift their metabolism to aerobic glycolysis (DeBerardinis and Chandel, 2020; Zhou et al., 2021) and increase the production of lactate, which may be considered as an evolving mechanism employed by viruses to evade immune surveillance.
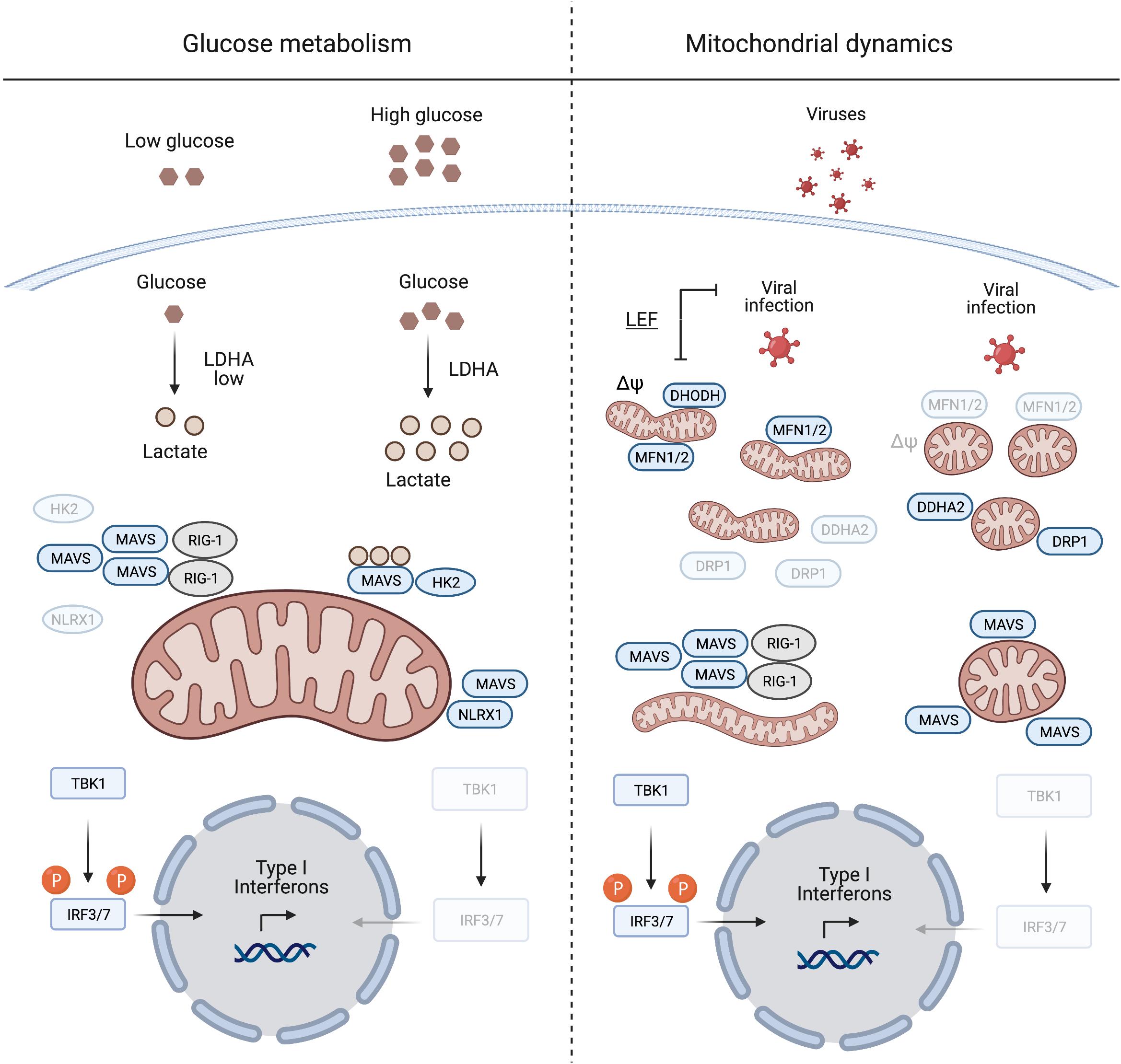
Figure 1. Mitochondrial antiviral signalling protein regulation induced by glucose metabolism and mitochondrial dynamics. During viral infection MAVS aggregation is essential for mounting an IFN response to suppress viral replication. Glucose and lactate levels (together with NLRX1) are negative regulators of MAVS aggregation (left panel). The right panel illustrates the pivotal role of mitochondrial dynamics in mounting an IFN response during viral infection. Inducing mitochondrial tubulation by expressing MFN2 or preventing DRP1 mitochondrial localisation mediated by DDHA2, as well as treating cells with the DHODH inhibitor leflunomide (LEF), supports MAVS aggregation and subsequent IFN response. Δψ, mitochondrial membrane potential. Created with BioRender.com.
Another innate immune regulatory protein that localises to mitochondria and is regulated by metabolic cues, is the nucleotide binding oligomerisation domain (NOD)-like receptor 1 (NLRX1). It negatively regulates the IFN response after viral infection via several mechanisms (Nagai-Singer et al., 2019; Pickering and Booty, 2021). During infection, NLRX1 interacts with MAVS to promote its ubiquitination and subsequent degradation (Qin et al., 2017), while the depletion of NLRX1 stabilises the MAVS-RIG-1 interaction and constitutively activates MAVS (Allen et al., 2011). Glucose depletion leads to decreased levels of NLRX1, which under these conditions will further unleash MAVS activation (Soares et al., 2014).
These studies thus further support the notion that the suppression of glucose metabolism activates MAVS. It should be noted, however, that NLRX1 inhibition of MAVS activity at the OM remains controversial. Studies performed in primary murine fibroblasts and mice lacking NLRX1 did not show any deficiency in elucidating an IFN response after viral infection (Rebsamen et al., 2011; Soares et al., 2013). Moreover, NLRX1 harbours a mitochondrial targetting sequence and was shown to translocate to the mitochondrial matrix (Arnoult et al., 2009).
Mitochondrial antiviral signalling protein signalling is linked to mitochondrial fission and fusion, which drive the dynamic adaptation of the mitochondrial network to metabolic cues (Mishra and Chan, 2016; Wai and Langer, 2016). Mitochondrial fusion is controlled at the OM by the mitochondrial fusion proteins mitofusins 1 and 2 (MFN1/2), which regulate MAVS activity (Figure 1). Inhibition of fusion in response to viral infection decreases MAVS activity and IFN production (Koshiba et al., 2011; Pourcelot and Arnoult, 2014). On the other hand, increased mitochondrial fusion upon chemical inhibition of dihydroorotate dehydrogenase (DHODH), a mitochondrial inner membrane (IM) enzyme involved in de novo pyrimidine synthesis, prevents viral replication (Miret-Casals et al., 2018; Coelho and Oliveira, 2020; Xiong et al., 2020). Thus, mitochondrial fusion appears to be required for innate immune signalling. In line with this concept, the nitric oxide (NO) producer dimethylarginine dimethylaminohydrolase 2 (DDAH2) inhibits MAVS activity during viral infection by promoting mitochondrial fission (Huang et al., 2021). NO produced by DDAH2 in response to viral infection stimulates the phosphorylation of the dynamin-related protein-1 (DRP-1) and promotes mitochondrial fission. Cells lacking DDAH2 maintained an elongated mitochondrial network and MAVS-dependent IFN production after infection. Moreover, viral infection induces the expression of numerous miRNAs, amongst them miR-302b and miR-372, which target MAVS and limit the IFN response by promoting DRP-1 phosphorylation and mitochondrial fission (Yasukawa et al., 2020).
Together, these studies highlight the importance of a tubular mitochondrial network for mounting a MAVS dependent IFN response during infection. This agrees with the metabolic regulation of MAVS, since increased mitochondrial tubulation is usually associated with OXPHOS dependent cell growth, while glycolytic cells harbour a more fragmented mitochondrial network (Chen and Chan, 2017). Although it remains to be determined how mitochondrial dynamics and the shape of mitochondria affect immune signalling, an intricate interplay between antiviral signalling, mitochondrial dynamics and metabolism appears to exist. Further work will undoubtedly reveal other mechanisms by which MAVS translate metabolic changes initiated during viral infection into an effective IFN response. For the remainder of this review, we turn our attention to another metabolically sensitive regulator of innate immunity that normally resides in mitochondria: mtDNA.
Mitochondria exhibit a pivotal role in mediating the IFN response upon infection by releasing mtDNA (or other DAMPs) to the cytosol, further boosting IFN production and immunosurveillance toward pathogens (Rongvaux et al., 2014; White et al., 2014; West et al., 2015; Sun et al., 2017). However, the presence of cytosolic or circulating mtDNA is also associated with chronic inflammation in a plethora of metabolic diseases and with ageing independent of bacterial or viral infection.
While the release of mtDNA from mitochondria is well established, the mechanisms allowing transfer to the cytosol are less clear. First insight was obtained by analysing the escape of mtDNA into the cytosol of apoptotic cells upon mitochondrial damage or various insults to tissue integrity and cellular homoeostasis. During apoptosis, mtDNA-protein assemblies termed nucleoids are released by herniation of the IM through large assemblies of BAX and BAK in the OM and subsequent IM permeabilisation (McArthur et al., 2018; Riley et al., 2018; Figure 2). mtDNA molecules are released under these conditions in association with the mitochondrial transcription factor A (TFAM), which serves as an mtDNA packaging factor. While this form of mtDNA release is predominantly immunologically silent owing to the activation of apoptotic caspases, a pro-inflammatory role for mtDNA release likely exists during the sub-lethal engagement of the mitochondrial apoptosis pathway, termed minority mitochondrial outer membrane permeabilisation (MOMP) (Ichim et al., 2015). Indeed, various pathogens were found to trigger minority MOMP and cytokine secretion from infected cells (Brokatzky et al., 2019).
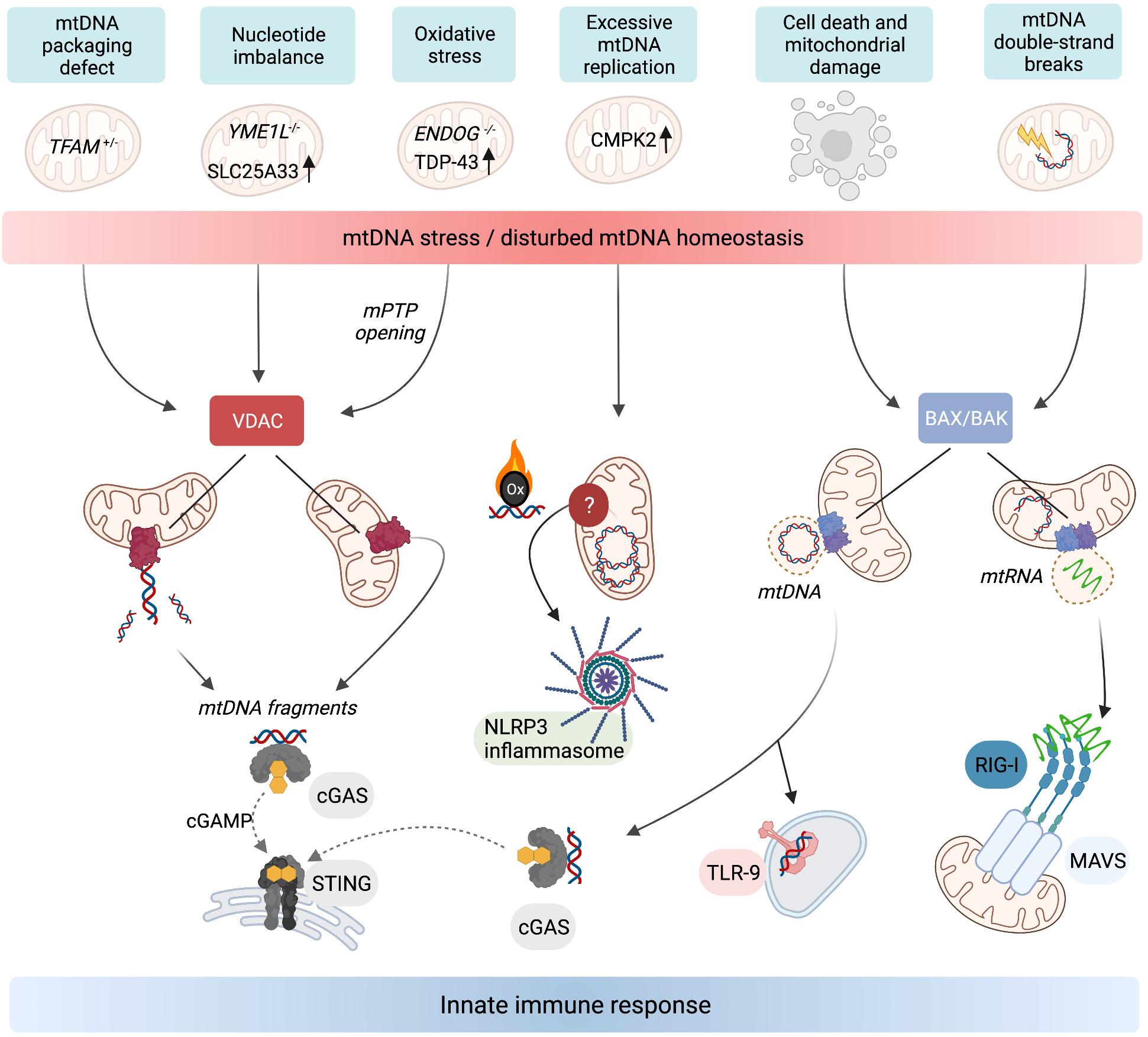
Figure 2. Release of mtDNA/RNA and the induction of innate immunity. Stressors, genetic, and metabolic perturbations triggering mtDNA/RNA-dependent innate immune responses. Fragments of mtDNA and mitochondrial nucleoids can be released along different pathways. VDAC oligomerisation at the OM allows the release of mtDNA fragments, which also requires concurrent opening of the mPTP in the mitochondria of ENDOG–/– cells and neurons expressing mutant mitochondrial localised TDP-43. Oxidised mtDNA fragments are also released in macrophages undergoing excessive mtDNA replication and trigger the NLRP3 inflammasome. TFAM-bound mtDNA nucleoids are released from mitochondria in response to cell death signalling upon herniation of the IM through BAX/BAK pores in the OM. If the cell does not undergo apoptosis, the cytosolic mtDNA triggers innate immune signalling upon recognition by DNA-binding receptors including cGAS and TLR-9. BAX/BAK pores also facilitate the release of mtRNA from mitochondria that have been exposed to stressors which cause double-stranded breaks in mtDNA. Cytosolic mtRNA is recognised by RIG-I and triggers a RIG-I-MAVS-dependent innate immune response. Created with BioRender.com.
Another form of mtDNA release occurs in non-apoptotic cells and does not require the formation of BAX/BAK pores but oligomerisation of VDAC in the OM instead (Figure 2). This mechanism, first described in cells lacking mitochondrial endonuclease G (ENDOG), involves the release of short mtDNA fragments (100–200 bp) rather than nucleoids and is also immunologically active, efficiently eliciting IFN response via cGAS-STING signalling (Kim et al., 2019). Chronic leakage of mtDNA through VDAC pores is associated with the induction of autoimmune diseases including Lupus (Kim et al., 2019) and amyotrophic lateral sclerosis (ALS) (Yu et al., 2020). The release of mtDNA fragments through VDAC pores in the OM is indeed emerging as a common pathway for the release of mtDNA to the cytosol, since treatment of cells with the VDAC oligomerisation inhibitor, VBIT4, is sufficient to blunt mtDNA-dependent innate immune responses in a number of contexts (Kim et al., 2019; Torres-Odio et al., 2020; Yu et al., 2020; Sprenger et al., 2021).
The mechanism(s) by which the IM is permeabilised to permit mtDNA export remain largely enigmatic. Opening the mitochondrial permeability transition pore (mPTP) can lead to mtDNA release (Patrushev et al., 2004; Garcia and Chavez, 2007). The pharmacological or genetic inactivation of mPTP is sufficient to block VDAC-dependent mtDNA release in EndoG–/– cells and cells expressing a mutant form of TDP-43 associated with ALS (Kim et al., 2019; Yu et al., 2020). It remains to be seen whether mPTP opening is a pre-requisite for mtDNA release through VDAC pores in all cases and to what degree mPTP opening and VDAC oligomerisation are co-ordinated.
We are only beginning to understand what triggers the escape of mtDNA from mitochondria. mtDNA release during chronic inflammation in ageing and disease is mainly considered as the unwanted consequence of a decline in mitochondrial integrity. Mitophagy ensures the removal of damaged mitochondria and was found to counteract the release of mtDNA and the inflammatory response (Sliter et al., 2018). Mice lacking the pro-mitophagy protein Parkin, an E3 ubiquitin ligase mutated in familial cases of Parkinson’s disease, accumulate dysfunctional mitochondria and contain higher levels of circulating mtDNA following exhaustive exercise or genetic induction of mtDNA mutations (Sliter et al., 2018). Loss of Parkin drives inflammatory phenotypes under these conditions, which depend on STING. These data indicate that released mtDNA stimulates inflammation in these mice by activating the cGAS-STING pathway and highlights the immunosuppressive role of Parkin relevant to Parkinson’s disease (Matheoud et al., 2016; Sliter et al., 2018). Similar observations were made in tissues and cells derived from mice lacking the autophagy regulator protein immunity-related GTPase family M protein 1 (IRGM1) (Rai et al., 2021). In Irgm1–/– fibroblasts, the release of mtDNA from dysfunctional mitochondria triggered an IFN response via cytosolic cGAS-STING, whereas the IFN response in Irgm1–/– macrophages required the endosomal RNA sensor TLR7. This indicates an immunostimulatory role of extra-mitochondrial mtRNA in Irgm1–/– macrophages, which may be another consequence of inefficient mitophagy and a decline in mitochondrial integrity (Rai et al., 2021).
Many forms of metabolic perturbations compromise mitochondrial function and integrity, which lead to the cytosolic release of mtDNA (Figure 2). First evidence for the release of mtDNA independent of apparent gross mitochondrial dysfunction came from studies in cells heterozygous for the mitochondrial transcription factor and mtDNA packaging protein TFAM. TFAM-depleted cells exhibit mitochondrial stress linked to perturbed DNA packaging and constitutively release mtDNA from mitochondria to trigger cGAS-STING dependent ISG expression (West et al., 2015). It is unclear what stimulates the release of mis-packaged mtDNA and whether it is under metabolic control. Evidence of mtDNA oxidative stress upon TFAM depletion exists in vivo (Woo et al., 2012), which may signal its release or exacerbate the immunogenic potential of mtDNA (Caielli et al., 2016; Lai et al., 2018). The translation of TFAM mRNA is under control of the master metabolic regulator mTORC1 (Liu et al., 2017) but it remains to be seen whether TFAM levels or other factors affecting mtDNA stability or expression, such as the endonuclease ENDOG (Kim et al., 2019) or caseinolytic mitochondrial matrix peptidase subunit CLPP (Torres-Odio et al., 2020), are directly regulated by metabolic pathways to promote or repress mtDNA release.
An inflammatory response is also mounted against the accumulation of errors during mtDNA replication. Mitochondrial polymerase gamma (POLG) mutator mice harbour excessive mtDNA mutations due to insufficient proof-reading and exhibit pathologies common in human mitochondrial diseases and premature ageing (Trifunovic et al., 2004; Kujoth et al., 2005). New results indicate that mutator mice exhibit enhanced activation of the cGAS-STING pathway associated with mtDNA release after LPS challenge and innate immune stimuli (Lei et al., 2021) or in the absence of mitophagy (Sliter et al., 2018). Blocking the hyperactive type I IFN response alleviates the pro-inflammatory metabolic phenotypes of mutator mice, including oxidative stress and aerobic glycolysis, and extends lifespan (Lei et al., 2021). Tigano et al. (2021) recently discovered that excessive mtDNA breaks can cause BAX/BAK-dependent release of mtRNA to activate a RIG-I-MAVS-dependent immune response. The cytosolic release of mtRNA has also been observed upon an excessive accumulation of double-stranded RNA in mitochondria (Dhir et al., 2018). Further work is necessary to understand how mtDNA integrity or replication rates are sensed and translated into immune responses downstream of different mitochondrial DAMPs.
Mitochondrial genome maintenance is coupled to cellular nucleotide metabolism and homoeostasis (El-Hattab et al., 2017). Recent work revealed that nucleotide synthesis and balance impacts mtDNA stability and regulates its release into the cytosol (Zhong et al., 2018; Sprenger et al., 2021). Macrophage priming with LPS causes the cytosolic exposure of mtDNA and subsequent activation of the NLRP3 inflammasome (Nakahira et al., 2011; Zhong et al., 2018). LPS-primed macrophages undergo a surge in mtDNA synthesis upon transcriptional upregulation of the mitochondrial deoxyribonucleotide kinase CMPK2 (Zhong et al., 2018). CMPK2 phosphorylates dCMP to dCDP in the mitochondrial deoxynucleoside triphosphate (dNTP) salvage pathway and is rate limiting for the accumulation of newly synthesised oxidised mtDNA in primed macrophages, which stimulates the inflammasome upon exposure to the cytosol (Zhong et al., 2018; Figure 2). These findings demonstrate that mitochondrial nucleotide metabolism is coupled to the replication and release of mtDNA.
Nucleotides are synthesised de novo from amino acids and ribose precursors in the cytosol or assembled via the mitochondrial dNTP salvage pathway from free nucleosides and bases. Increasing evidence suggests that nucleotide uptake into mitochondria limits the accumulation of mtDNA in proliferating cells (Favre et al., 2010; Di Noia et al., 2014; Sprenger et al., 2021). We recently observed that disturbances in cellular nucleotide homoeostasis, including deregulated nucleotide uptake, triggers the release of mtDNA (Sprenger et al., 2021). Mouse retina and cultured cells lacking the mitochondrial protease YME1L exhibit a specific innate immune response that, in cultured cells, depends on mtDNA-dependent cGAS-STING activation (Sprenger et al., 2021). YME1L is a multifaceted metabolic regulator in the IM that programmes the mitochondrial proteome to maintain pyrimidine nucleotide synthesis via glutaminolysis and to modulate the transport of pyrimidines across the IM via proteolysis of the pyrimidine nucleotide carrier SLC25A33 (MacVicar et al., 2019; Sprenger et al., 2021). Loss of YME1L results in the accumulation of SLC25A33 and cellular depletion of pyrimidine nucleotides, which triggers the release of mtDNA into the cytosol (Figure 2). mtDNA release occurs in a BAX/BAK-independent manner but depends on VDAC oligomerisation in the OM, which also governs the release of mtDNA fragments in cells lacking ENDOG (Kim et al., 2019). Both accumulation of SLC25A33 or inhibition of pyrimidine synthesis independently are sufficient to induce mtDNA release from mitochondria. Depletion of cellular pyrimidines via genetic intervention or treatment with the pyrimidine nucleoside analogue 5-fluorouracil (5-FU) triggered mtDNA-dependent cGAS-STING activation, demonstrating that this mtDNA release pathway may be of relevance in a number of clinical contexts (Sprenger et al., 2021). For instance, cGAS-STING dependent type I IFN production is triggered by 5-FU in cancer cells and supports effective anti-tumour immunity in colorectal cancer (Tian et al., 2021). Intriguingly, YME1L was found to be frequently mutated in colorectal cancer tissue, which may drive the migration of mtDNA to the nucleus observed in these cancers (Srinivasainagendra et al., 2017).
It is exciting to consider that pyrimidine depletion is a common trigger of mtDNA release and innate immunity. Cytosolic exposure of mtDNA contributes to cellular antiviral responses since enhanced expression of the specific panel of ISGs downstream of mtDNA-dependent cGAS-STING signalling supports antiviral immunity (West et al., 2015). Of note, pyrimidine analogues are employed as antiviral agents and the fact that they induce type I IFN responses has been appreciated for some time (Lucas-Hourani et al., 2013; Shin et al., 2018). It will be important to ascertain whether mtDNA exposure supports innate immune responses to nucleoside analogues in antiviral and anti-tumour signalling. It will also be pertinent to establish the degree to which mtDNA release synergises with nuclear DNA damage responses to nucleotide depletion (Hastak et al., 2008; Hamalainen et al., 2019). Interestingly, some viral infections such as Dengue virus induce innate immune responses associated with mtDNA stress and release (Sun et al., 2017; Lai et al., 2018) and mtDNA has been detected in the blood of hospitalised COVID-19 patients (Scozzi et al., 2021). While the presence of circulating mtDNA in virus infected individuals likely results mainly from tissue damage and cell death, it remains to be seen whether infected cells also sense nucleotide imbalance during viral infection and respond by releasing mtDNA to drive innate immunity.
In addition to nucleotides, the metabolic homoeostasis of other macromolecules, especially lipids, has also been linked to mtDNA-dependent innate immunity. Disturbances in the synthesis of lipids in adipose tissue impair mitochondrial respiration and enhance ROS production (Bai et al., 2017). This is associated with the release of mtDNA and the induction of cGAS-STING-dependent inflammation and insulin resistance during obesity (Bai et al., 2017). Lipid metabolism also regulates mtDNA release in immune cells. Regulatory T cells (Tregs) suppress immune responses by secreting cytokines such as IL-10 to arrest effector T cell proliferation (Chaudhry et al., 2011). The suppressive capability of Tregs is enhanced upon acute inhibition of the fatty acid binding protein FABP5, which was associated with a decline in fatty acid oxidation (Field et al., 2020). Pharmacological inhibition of FABP5 triggered the release of mtDNA and stimulated a type I IFN response in a cGAS-STING dependent manner. A similar innate immune response was engaged in Tregs within solid tumours indicating that mtDNA-dependent signalling may occur upon exposure to a lipid-depleted microenvironment (Field et al., 2020). While the presence of cytosolic mtDNA in Tregs correlated with a decline in OXPHOS and disturbed mitochondrial cristae morphology, it is unclear how mtDNA is released from these mitochondria especially since limiting OXPHOS and disturbing cristae morphogenesis in these cells by other means does not trigger mtDNA release (Field et al., 2020). Finally, metabolic dysregulation in renal tubule cells that rely heavily on fatty acid oxidation is associated with transcriptional repression of the mtDNA binding protein TFAM and mtDNA-dependent cGAS-STING activation in kidney disease and fibrosis (Chung et al., 2019), further indicating a link between disturbed fatty acid metabolism and mtDNA release.
Compelling evidence demonstrates that metabolic signals direct mitochondria-dependent immune responses and ISG expression, which restrict viral replication. However, only a subset of ISGs limits viral replication directly and the antiviral function for the majority of ISGs is unknown. Many ISGs reshape cellular and mitochondrial metabolism to activate immune cells and suppress infection indirectly (Figure 3). For instance, reprogramming host cell nucleotide metabolism in response to infection can limit the pool of dNTPs available for viral DNA replication (Lahouassa et al., 2012) and IFN signalling rewires oxidative metabolism and alters citric acid cycle metabolite levels to facilitate the reprogramming of dendritic cells and macrophages (O’Neill and Pearce, 2016; Timblin et al., 2021). The reciprocal relationship between innate immune response and metabolism is also illustrated by the metabolic phenotypes associated with chronic immune activation in disease (Fritsch and Weichhart, 2016; Raniga and Liang, 2018). While many intriguing aspects of immunometabolism lie beyond the scope of this review, understanding more about the function(s) of individual ISGs specifically expressed during mtDNA-dependent innate immunity will help us to decipher the metabolic consequences of mtDNA release and its role in disease.
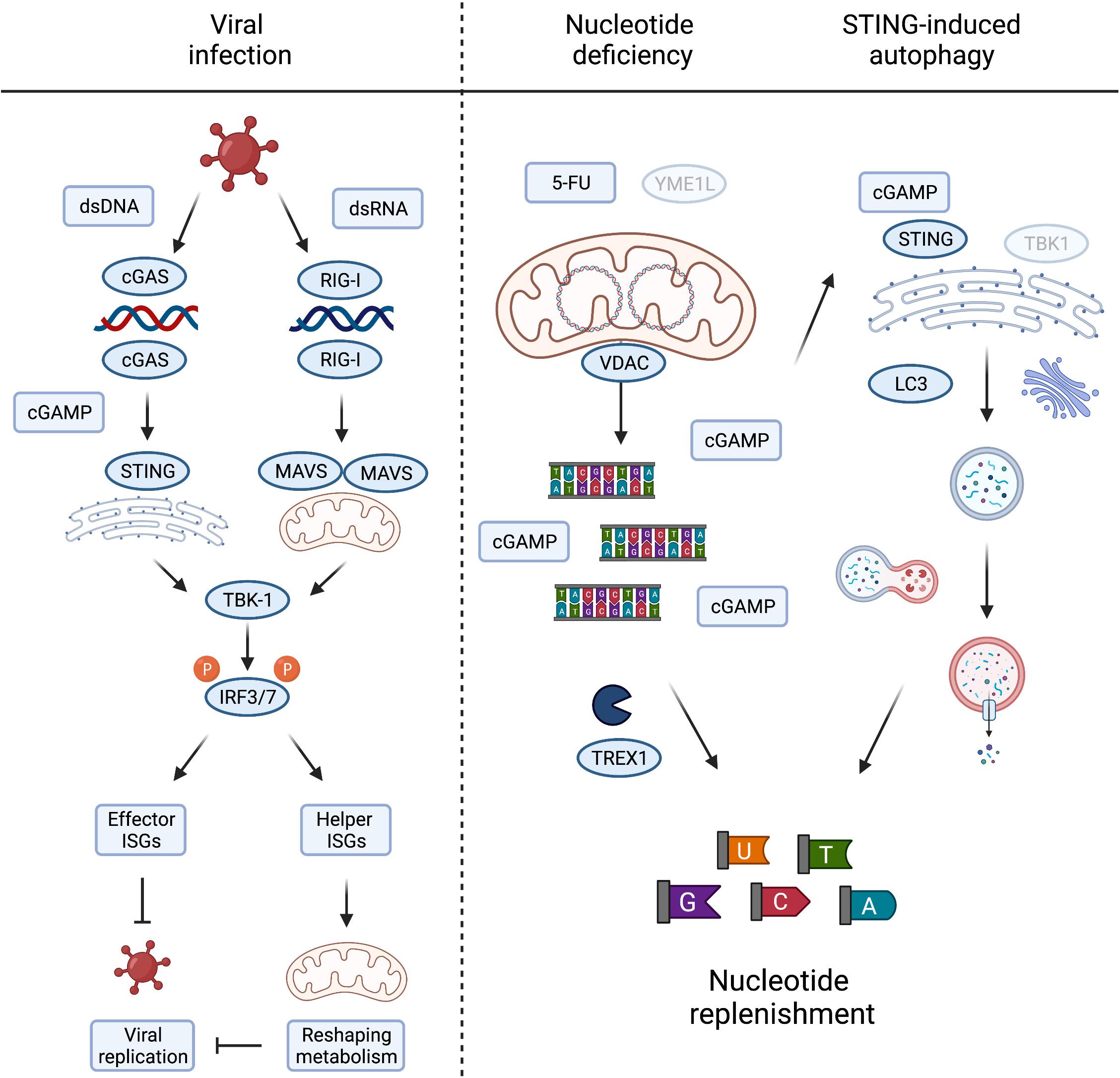
Figure 3. Metabolic consequences of mtDNA release and innate immune signaling. Viral infection, either by DNA or RNA viruses, leads to the activation of TBK1 and subsequent phosphorylation of IRF3/7 to induce their nuclear translocation to activate type I interferons. The interferon stimulated genes (ISGs) prevent viral growth, either by direct inhibition of viral replication, termed here “effector ISGs,” or indirectly by rewiring mitochondrial and cellular metabolism (“helper ISGs”). The latter could be achieved by inducing metabolic conditions supporting/favouring the release of mtDNA (left panel). Nucleotide deficiency induced by loss of YME1L or treatment with thymidylate synthase (TS) inhibitor 5-FU leads to the release of mtDNA. TREX1 competes with cGAS for the binding and degradation of mitochondrial dsDNA to single nucleotides, replenishing cellular nucleotide pools in the process. cGAS binding of mtDNA generates cGAMP to activate STING-induced autophagy, independent of TBK1 activation, and innate immunity, by LC3 lipidation and autophagosome formation. The autophagy/lysosome system further contribute to the replenishment of cytosolic nucleotide pools (right panel). Created with BioRender.com.
It is fascinating to consider that mtDNA release to the cytosol during sterile inflammation serves additional metabolic purposes besides engaging innate immune signalling pathways. The loss of the evolutionary conserved mitochondrial protease YME1L induces mtDNA release and cGAS-STING signalling in mouse and human cells. The stimulation of mtDNA release in YME1L deficient cells inherently devoid of inflammation signalling pathways, such as yme1 mutant yeast, argues that extra-mitochondrial mtDNA could have alternative conserved roles to play (Thorsness et al., 1993). This includes the migration of mitochondrial genetic material to the nucleus (Thorsness and Fox, 1990; Thorsness et al., 1993) but may also fulfil other signalling duties when triggered by nucleotide depletion. It is another intriguing possibility that mtDNA also serves as a storage for nucleotides that can be released into the cytosol under conditions when cytosolic nucleotide pools are limiting (Sprenger et al., 2021). Consistently, depletion of the cytosolic three prime repair exonuclease 1 (TREX1) boosted cGAS-dependent immune signalling in YME1L–/– cells suggesting competition between degradation of cytosolic mtDNA and cGAS binding (Sprenger et al., 2021).
Pools of nucleotides and their metabolic precursors can also be maintained via the autophagy pathway and lysosomal recycling (Guo et al., 2016; Liu et al., 2018). Pyrimidine and purine depletion can induce autophagy via different pathways (Hoxhaj et al., 2017; Mimura et al., 2021) that may synergise with the release of mtDNA to replenish nucleotide pools. Gui et al. (2019) recently described the induction of autophagy as a primordial function of dsDNA recognition by cGAS-STING, whereby cGAMP triggers STING translocation to the ER-Golgi intermediate compartment to trigger autophagosome formation independent of IFN production. Delineation and functional analysis of the autophagosome-inducing STING domain revealed that the autophagy activity exists in ancient STING homologues, which are otherwise incapable of activating IFNs via TBK1 (Kranzusch et al., 2015; Gui et al., 2019). While autophagy induction by STING does not appear to be sufficient to drive an effective STING-mediated antiviral response in mice, which rather depends on STING-TBK1 signalling (Yum et al., 2021), autophagy can help defend cells against pathogens by delivering them to the lysosome for degradation and supports the clearance of cytosolic DNA (Gui et al., 2019). It is therefore tempting to speculate that mtDNA release supports autophagy induction via cGAS-STING signalling in a further attempt to restore depleted nucleotide levels (Figure 3). In line with this possibility, yeast autophagy mutants under starvation conditions were shown to degrade mtDNA, perhaps further releasing it to replenish cytosolic nucleotides otherwise limiting under these conditions (Medeiros et al., 2018). Further work will be required to investigate the contribution of cytosolic mtDNA to cellular nucleotide metabolism, which will bear significance in scenarios where nucleotide balance is disturbed, including tumour cell responses to chemotherapy and host cell responses to pathogen infection.
Mitochondria play essential roles at the crossroads of metabolism and innate immunity. The dynamic mitochondrial network integrates MAVS signalling with glucose metabolism during viral infection and releases mtDNA to drive inflammatory responses to distinct metabolic signals. mtDNA release is frequently observed in pathological scenarios and evidence is accumulating that metabolic signals can induce it. On the other hand, a sentinel role played by cytosolic mtDNA in metabolism and innate immune signalling is emerging.
Many important questions await to be answered, including how these metabolic signals are relayed to the mitochondria in order to expel mtDNA and what determines whether mtDNA is released in fragments via VDAC or as whole nucleoids via BAX/BAK pores. It will be fascinating to address whether metabolic signalling programmes control the selective packaging and extracellular extrusion of mtDNA via exosomes. Recently, Todkar et al. (2021) reported that mitochondrial derived vesicles (Sugiura et al., 2014) control the incorporation of mitochondrial proteins and mtDNA into exosomes in cultured cells. Finally, what is the physiological role of regulated mtDNA release and can it be manipulated, for example in order to improve chemotherapy or treat autoimmune diseases? It has become clear that cytosolic mtDNA can have profound effects on cellular metabolism and it will be exciting to see if targetting these metabolic pathways prove beneficial in therapy.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This review was supported by grants from the Max-Planck-Society and the Deutsche Forschungsgemeinschaft (SFB1403, project number 414786233).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Allen, I. C., Moore, C. B., Schneider, M., Lei, Y., Davis, B. K., Scull, M. A., et al. (2011). NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-kappaB signaling pathways. Immunity 34, 854–865. doi: 10.1016/j.immuni.2011.03.026
Arnoult, D., Carneiro, L., Tattoli, I., and Girardin, S. E. (2009). The role of mitochondria in cellular defense against microbial infection. Semin. Immunol. 21, 223–232. doi: 10.1016/j.smim.2009.05.009
Bahat, A., and Gross, A. (2019). Mitochondrial plasticity in cell fate regulation. J. Biol. Chem. 294, 13852–13863. doi: 10.1074/jbc.REV118.000828
Bai, J., Cervantes, C., Liu, J., He, S., Zhou, H., Zhang, B., et al. (2017). DsbA-L prevents obesity-induced inflammation and insulin resistance by suppressing the mtDNA release-activated cGAS-cGAMP-STING pathway. Proc. Natl. Acad. Sci. U S A 114, 12196–12201. doi: 10.1073/pnas.1708744114
Brokatzky, D., Dorflinger, B., Haimovici, A., Weber, A., Kirschnek, S., Vier, J., et al. (2019). A non-death function of the mitochondrial apoptosis apparatus in immunity. EMBO J. 38:100907. doi: 10.15252/embj.2018100907
Brubaker, S. W., Bonham, K. S., Zanoni, I., and Kagan, J. C. (2015). Innate immune pattern recognition: a cell biological perspective. Annu. Rev. Immunol. 33, 257–290. doi: 10.1146/annurev-immunol-032414-112240
Caielli, S., Athale, S., Domic, B., Murat, E., Chandra, M., Banchereau, R., et al. (2016). Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med 213, 697–713. doi: 10.1084/jem.20151876
Chakrabarty, R. P., and Chandel, N. S. (2021). Mitochondria as Signaling Organelles Control Mammalian Stem Cell Fate. Cell Stem Cell 28, 394–408. doi: 10.1016/j.stem.2021.02.011
Chaudhry, A., Samstein, R. M., Treuting, P., Liang, Y., Pils, M. C., Heinrich, J. M., et al. (2011). Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 34, 566–578. doi: 10.1016/j.immuni.2011.03.018
Chen, H. C., and Chan, D. C. (2017). Mitochondrial Dynamics in Regulating the Unique Phenotypes of Cancer and Stem Cells. Cell Metab. 26, 39–48. doi: 10.1016/j.cmet.2017.05.016
Chung, K. W., Dhillon, P., Huang, S., Sheng, X., Shrestha, R., Qiu, C., et al. (2019). Mitochondrial Damage and Activation of the STING Pathway Lead to Renal Inflammation and Fibrosis. Cell Metab. 30, 784–799e785. doi: 10.1016/j.cmet.2019.08.003
Coelho, A. R., and Oliveira, P. J. (2020). Dihydroorotate dehydrogenase inhibitors in SARS-CoV-2 infection. Eur. J. Clin. Invest. 50:e13366. doi: 10.1111/eci.13366
Collins, L. V., Hajizadeh, S., Holme, E., Jonsson, I. M., and Tarkowski, A. (2004). Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J. Leukoc Biol. 75, 995–1000. doi: 10.1189/jlb.0703328
DeBerardinis, R. J., and Chandel, N. S. (2020). We need to talk about the Warburg effect. Nat Metab. 2, 127–129. doi: 10.1038/s42255-020-0172-2)
Dhir, A., Dhir, S., Borowski, L. S., Jimenez, L., Teitell, M., Rotig, A., et al. (2018). Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 560, 238–242. doi: 10.1038/s41586-018-0363-0)
Di Noia, M. A., Todisco, S., Cirigliano, A., Rinaldi, T., Agrimi, G., Iacobazzi, V., et al. (2014). The human SLC25A33 and SLC25A36 genes of solute carrier family 25 encode two mitochondrial pyrimidine nucleotide transporters. J. Biol. Chem. 289, 33137–33148. doi: 10.1074/jbc.M114.610808
Dixit, E., Boulant, S., Zhang, Y., Lee, A. S., Odendall, C., Shum, B., et al. (2010). Peroxisomes are signaling platforms for antiviral innate immunity. Cell 141, 668–681. doi: 10.1016/j.cell.2010.04.018
El-Hattab, A. W., Craigen, W. J., and Scaglia, F. (2017). Mitochondrial DNA maintenance defects. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 1539–1555. doi: 10.1016/j.bbadis.2017.02.017
Favre, C., Zhdanov, A., Leahy, M., Papkovsky, D., and O’Connor, R. (2010). Mitochondrial pyrimidine nucleotide carrier (PNC1) regulates mitochondrial biogenesis and the invasive phenotype of cancer cells. Oncogene 29, 3964–3976. doi: 10.1038/onc.2010.146
Field, C. S., Baixauli, F., Kyle, R. L., Puleston, D. J., Cameron, A. M., Sanin, D. E., et al. (2020). Mitochondrial Integrity Regulated by Lipid Metabolism Is a Cell-Intrinsic Checkpoint for Treg Suppressive Function. Cell Metab. 31, 422–437e425. doi: 10.1016/j.cmet.2019.11.021)
Flores-Romero, H., Ros, U., and Garcia-Saez, A. J. (2020). Pore formation in regulated cell death. EMBO J. 39:e105753. doi: 10.15252/embj.2020105753
Fritsch, S. D., and Weichhart, T. (2016). Effects of Interferons and Viruses on Metabolism. Front. Immunol. 7:630. doi: 10.3389/fimmu.2016.00630
Garcia, N., and Chavez, E. (2007). Mitochondrial DNA fragments released through the permeability transition pore correspond to specific gene size. Life Sci. 81, 1160–1166. doi: 10.1016/j.lfs.2007.08.019
Gong, T., Liu, L., Jiang, W., and Zhou, R. (2020). DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 20, 95–112. doi: 10.1038/s41577-019-0215-7
Gui, X., Yang, H., Li, T., Tan, X., Shi, P., Li, M., et al. (2019). Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 567, 262–266. doi: 10.1038/s41586-019-1006-9
Guo, J. Y., Teng, X., Laddha, S. V., Ma, S., Van Nostrand, S. C., Yang, Y., et al. (2016). Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 30, 1704–1717. doi: 10.1101/gad.283416.116
Hamalainen, R.H., Landoni, J.C., Ahlqvist, K.J., Goffart, S., Ryytty, S., Rahman, M.O., et al. (2019). Defects in mtDNA replication challenge nuclear genome stability through nucleotide depletion and provide a unifying mechanism for mouse progerias. Nat. Metab. 1:958. doi: 10.1038/s42255-019-0120-1
Hastak, K., Paul, R. K., Agarwal, M. K., Thakur, V. S., Amin, A. R., Agrawal, S., et al. (2008). DNA synthesis from unbalanced nucleotide pools causes limited DNA damage that triggers ATR-CHK1-dependent p53 activation. Proc. Natl. Acad. Sci. U S A 105, 6314–6319. doi: 10.1073/pnas.0802080105
Horner, S. M., Liu, H. M., Park, H. S., Briley, J., and Gale, M. Jr. (2011). Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. U S A 108, 14590–14595. doi: 10.1073/pnas.1110133108
Hou, F., Sun, L., Zheng, H., Skaug, B., Jiang, Q. X., and Chen, Z. J. (2011). MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 146, 448–461. doi: 10.1016/j.cell.2011.06.041
Hoxhaj, G., Hughes-Hallett, J., Timson, R. C., Ilagan, E., Yuan, M., Asara, J. M., et al. (2017). The mTORC1 Signaling Network Senses Changes in Cellular Purine Nucleotide Levels. Cell Rep. 21, 1331–1346. doi: 10.1016/j.celrep.2017.10.029
Huang, S., Li, Z., Wu, Z., Liu, C., Yu, M., Wen, M., et al. (2021). DDAH2 suppresses RLR-MAVS-mediated innate antiviral immunity by stimulating nitric oxide-activated, Drp1-induced mitochondrial fission. Sci. Signal 14:7931. doi: 10.1126/scisignal.abc7931
Ichim, G., Lopez, J., Ahmed, S. U., Muthalagu, N., Giampazolias, E., Delgado, M. E., et al. (2015). Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol. Cell 57, 860–872. doi: 10.1016/j.molcel.2015.01.018
Imre, G. (2020). Cell death signalling in virus infection. Cell Signal 76:109772. doi: 10.1016/j.cellsig.2020.109772
Jacobs, J. L., and Coyne, C. B. (2013). Mechanisms of MAVS Regulation at the Mitochondrial Membrane. J. Mol. Biol. 425, 5009–5019. doi: 10.1016/j.jmb.2013.10.007
John, S., Weiss, J. N., and Ribalet, B. (2011). Subcellular localization of hexokinases I and II directs the metabolic fate of glucose. PLoS One 6:e17674. doi: 10.1371/journal.pone.0017674
Kawai, T., Takahashi, K., Sato, S., Coban, C., Kumar, H., Kato, H., et al. (2005). IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6, 981–988. doi: 10.1038/ni1243
Kim, J., Gupta, R., Blanco, L. P., Yang, S. T., Shteinfer-Kuzmine, A., Wang, K. N., et al. (2019). VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 366:1531. doi: 10.1126/science.aav4011
Koshiba, T., Yasukawa, K., Yanagi, Y., and Kawabata, S. (2011). Mitochondrial Membrane Potential Is Required for MAVS-Mediated Antiviral Signaling. Sci. Signal 4:ra7. doi: 10.1126/scisignal.2001147
Kranzusch, P. J., Wilson, S. C., Lee, A. S., Berger, J. M., Doudna, J. A., and Vance, R. E. (2015). Ancient Origin of cGAS-STING Reveals Mechanism of Universal 2′,3′ cGAMP Signaling. Mol. Cell 59, 891–903.
Kujoth, G. C., Hiona, A., Pugh, T. D., Someya, S., Panzer, K., Wohlgemuth, S. E., et al. (2005). Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309, 481–484. doi: 10.1126/science.1112125
Lahouassa, H., Daddacha, W., Hofmann, H., Ayinde, D., Logue, E. C., Dragin, L., et al. (2012). SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 13, 223–228. doi: 10.1038/ni.2236
Lai, J. H., Wang, M. Y., Huang, C. Y., Wu, C. H., Hung, L. F., Yang, C. Y., et al. (2018). Infection with the dengue RNA virus activates TLR9 signaling in human dendritic cells. EMBO Rep. 19:46182. doi: 10.15252/embr.201846182
Lei, Y., Guerra Martinez, C., Torres-Odio, S., Bell, S. L., Birdwell, C. E., Bryant, J. D., et al. (2021). Elevated type I interferon responses potentiate metabolic dysfunction, inflammation, and accelerated aging in mtDNA mutator mice. Sci. Adv. 7:7548. doi: 10.1126/sciadv.abe7548
Liu, X., Zhang, Y., Ni, M., Cao, H., Signer, R. A. J., Li, D., et al. (2017). Regulation of mitochondrial biogenesis in erythropoiesis by mTORC1-mediated protein translation. Nat. Cell Biol. 19, 626–638. doi: 10.1038/ncb3527)
Liu, Y. B., Zou, W., Yang, P. G., Wang, L., Ma, Y., Zhang, H., et al. (2018). Autophagy-dependent ribosomal RNA degradation is essential for maintaining nucleotide homeostasis during C. elegans development. Elife 7:e36588.
Lucas-Hourani, M., Dauzonne, D., Jorda, P., Cousin, G., Lupan, A., Helynck, O., et al. (2013). Inhibition of pyrimidine biosynthesis pathway suppresses viral growth through innate immunity. PLoS Pathog. 9:e1003678. doi: 10.1371/journal.ppat.1003678
MacVicar, T., Ohba, Y., Nolte, H., Mayer, F. C., Tatsuta, T., Sprenger, H. G., et al. (2019). Lipid signalling drives proteolytic rewiring of mitochondria by YME1L. Nature 575, 361–365. doi: 10.1038/s41586-019-1738-6
Matheoud, D., Sugiura, A., Bellemare-Pelletier, A., Laplante, A., Rondeau, C., Chemali, M., et al. (2016). Parkinson’s Disease-Related Proteins PINK1 and Parkin Repress Mitochondrial Antigen Presentation. Cell 166, 314–327. doi: 10.1016/j.cell.2016.05.039)
McArthur, K., Whitehead, L. W., Heddleston, J. M., Li, L., Padman, B. S., Oorschot, V., et al. (2018). BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 359:6047. doi: 10.1126/science.aao6047
Medeiros, T. C., Thomas, R. L., Ghillebert, R., and Graef, M. (2018). Autophagy balances mtDNA synthesis and degradation by DNA polymerase POLG during starvation. J. Cell Biol. 217, 1601–1611. doi: 10.1083/jcb.201801168)
Meylan, E., Curran, J., Hofmann, K., Moradpour, D., Binder, M., Bartenschlager, R., et al. (2005). Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437, 1167–1172. doi: 10.1038/nature04193
Mimura, K., Sakamaki, J. I., Morishita, H., Kawazu, M., Mano, H., and Mizushima, N. (2021). Genome-wide CRISPR screening reveals nucleotide synthesis negatively regulates autophagy. J. Biol. Chem. 2021:100780.
Miret-Casals, L., Sebastian, D., Brea, J., Rico-Leo, E. M., Palacin, M., Fernandez-Salguero, P. M., et al. (2018). Identification of New Activators of Mitochondrial Fusion Reveals a Link between Mitochondrial Morphology and Pyrimidine Metabolism. Cell Chem. Biol. 25:268. doi: 10.1016/j.chembiol.2017.12.001
Mishra, P., and Chan, D. C. (2016). Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 212, 379–387. doi: 10.1083/jcb.201511036
Nagai-Singer, M. A., Morrison, H. A., and Allen, I. C. (2019). NLRX1 Is a Multifaceted and Enigmatic Regulator of Immune System Function. Front. Immunol. 10:2419. doi: 10.3389/fimmu.2019.02419
Nakahira, K., Haspel, J. A., Rathinam, V. A., Lee, S. J., Dolinay, T., Lam, H. C., et al. (2011). Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230. doi: 10.1038/ni.1980
O’Neill, L. A., and Pearce, E. J. (2016). Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 213, 15–23. doi: 10.1084/jem.20151570
Patrushev, M., Kasymov, V., Patrusheva, V., Ushakova, T., Gogvadze, V., and Gaziev, A. (2004). Mitochondrial permeability transition triggers the release of mtDNA fragments. Cell Mol Life Sci 61, 3100–3103. doi: 10.1007/s00018-004-4424-1
Pickering, R. J., and Booty, L. M. (2021). NLR in eXile: Emerging roles of NLRX1 in immunity and human disease. Immunology 162, 268–280.
Pourcelot, M., and Arnoult, D. (2014). Mitochondrial dynamics and the innate antiviral immune response. Febs J. 281, 3791–3802. doi: 10.1111/febs.12940
Qin, Y. W., Xue, B. B., Liu, C. Y., Wang, X. H., Tian, R. Y., Xie, Q. Y., et al. (2017). NLRX1 Mediates MAVS Degradation To Attenuate the Hepatitis C Virus-Induced Innate Immune Response through PCBP2. J. Virol. 91, e1264–e1217.
Rai, P., Janardhan, K. S., Meacham, J., Madenspacher, J. H., Lin, W. C., Karmaus, P. W. F., et al. (2021). IRGM1 links mitochondrial quality control to autoimmunity. Nat. Immunol. 22, 312–321. doi: 10.1038/s41590-020-00859-0
Raniga, K., and Liang, C. (2018). Interferons: Reprogramming the Metabolic Network against Viral Infection. Viruses 10:10010036. doi: 10.3390/v10010036
Rebsamen, M., Vazquez, J., Tardivel, A., Guarda, G., Curran, J., and Tschopp, J. (2011). NLRX1/NOD5 deficiency does not affect MAVS signalling. Cell Death Differ. 18:1387. doi: 10.1038/cdd.2011.64
Rehwinkel, J., and Gack, M. U. (2020). RIG-I-like receptors: their regulation and roles in RNA sensing. Nat. Rev. Immunol. 20, 537–551. doi: 10.1038/s41577-020-0288-3
Riley, J. S., and Tait, S. W. (2020). Mitochondrial DNA in inflammation and immunity. EMBO Rep. 21:e49799. doi: 10.15252/embr.201949799
Riley, J. S., Quarato, G., Cloix, C., Lopez, J., O’Prey, J., Pearson, M., et al. (2018). Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 37:99238. doi: 10.15252/embj.201899238
Roberts, D. J., and Miyamoto, S. (2015). Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 22, 248–257. doi: 10.1038/cdd.2014.173
Rongvaux, A., Jackson, R., Harman, C. C., Li, T., West, A. P., de Zoete, M. R., et al. (2014). Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159, 1563–1577. doi: 10.1016/j.cell.2014.11.037
Scozzi, D., Cano, M., Ma, L., Zhou, D., Zhu, J. H., O’Halloran, J. A., et al. (2021). Circulating mitochondrial DNA is an early indicator of severe illness and mortality from COVID-19. JCI Insight 6:143299. doi: 10.1172/jci.insight.143299
Seth, R. B., Sun, L., Ea, C. K., and Chen, Z. J. (2005). Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122, 669–682. doi: 10.1016/j.cell.2005.08.012
Shepard, C. R. (2020). TLR9 in MAFLD and NASH: At the Intersection of Inflammation and Metabolism. Front. Endocrinol. 11:613639. doi: 10.3389/fendo.2020.613639
Shimada, K., Crother, T. R., Karlin, J., Dagvadorj, J., Chiba, N., Chen, S., et al. (2012). Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414. doi: 10.1016/j.immuni.2012.01.009
Shin, H. J., Kim, C., and Cho, S. (2018). Gemcitabine and Nucleos(t)ide Synthesis Inhibitors Are Broad-Spectrum Antiviral Drugs that Activate Innate Immunity. Viruses 10:40211. doi: 10.3390/v10040211
Sliter, D. A., Martinez, J., Hao, L., Chen, X., Sun, N., Fischer, T. D., et al. (2018). Parkin and PINK1 mitigate STING-induced inflammation. Nature 561, 258–262. doi: 10.1038/s41586-018-0448-9
Soares, F., Tattoli, I., Rahman, M. A., Robertson, S. J., Belcheva, A., Liu, D., et al. (2014). The Mitochondrial Protein NLRX1 Controls the Balance between Extrinsic and Intrinsic Apoptosis. J. Biolog. Chem. 289, 19317–19330. doi: 10.1074/jbc.M114.550111
Soares, F., Tattoli, I., Wortzman, M. E., Arnoult, D., Philpott, D. J., and Girardin, S. E. (2013). NLRX1 does not inhibit MAVS-dependent antiviral signalling. Innate Immun. 19, 438–448. doi: 10.1177/1753425912467383
Sprenger, H., MacVicar, T., Bahat, A., Fiedler, K., Hermans, S., Ehrentraut, D., et al. (2021). Cellular pyrimidine imbalance triggers mitochondrial DNA-dependent innate immunity. Nat. Metab. 3, 636–650. doi: 10.1038/s42255-021-00385-9
Srinivasainagendra, V., Sandel, M. W., Singh, B., Sundaresan, A., Mooga, V. P., Bajpai, P., et al. (2017). Migration of mitochondrial DNA in the nuclear genome of colorectal adenocarcinoma. Genome Med. 9:31. doi: 10.1186/s13073-017-0420-6
Sugiura, A., McLelland, G. L., Fon, E. A., and McBride, H. M. (2014). A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J. 33, 2142–2156. doi: 10.15252/embj.201488104
Sun, B., Sundstrom, K. B., Chew, J. J., Bist, P., Gan, E. S., Tan, H. C., et al. (2017). Dengue virus activates cGAS through the release of mitochondrial DNA. Sci. Rep. 7:3594. doi: 10.1038/s41598-017-03932-1
Thorsness, P. E., and Fox, T. D. (1990). Escape of DNA from mitochondria to the nucleus in Saccharomyces cerevisiae. Nature 346, 376–379. doi: 10.1038/346376a0
Thorsness, P. E., White, K. H., and Fox, T. D. (1993). Inactivation of YME1, a member of the ftsH-SEC18-PAS1-CDC48 family of putative ATPase-encoding genes, causes increased escape of DNA from mitochondria in Saccharomyces cerevisiae. Mol. Cell Biol. 13, 5418–5426. doi: 10.1128/mcb.13.9.5418
Tian, J., Zhang, D., Kurbatov, V., Wang, Q., Wang, Y., Fang, D., et al. (2021). 5-Fluorouracil efficacy requires anti-tumor immunity triggered by cancer-cell-intrinsic STING. EMBO J 2021, e106065. doi: 10.15252/embj.2020106065
Tigano, M., Vargas, D. C., Tremblay-Belzile, S., Fu, Y., and Sfeir, A. (2021). Nuclear sensing of breaks in mitochondrial DNA enhances immune surveillance. Nature 591, 477–481. doi: 10.1038/s41586-021-03269-w)
Timblin, G. A., Tharp, K. M., Ford, B., Winchester, J. M., Wang, J., Zhu, S., et al. (2021). Mitohormesis reprogrammes macrophage metabolism to enforce tolerance. Nat. Metab. 3, 618–635. doi: 10.1038/s42255-021-00392-w
Todkar, K., Chikhi, L., Desjardins, V., El-Mortada, F., Pepin, G., and Germain, M. (2021). Selective packaging of mitochondrial proteins into extracellular vesicles prevents the release of mitochondrial DAMPs. Nat. Commun. 12:1971.
Torres-Odio, S., Lei, Y., Gispert, S., Maletzko, A., Key, J., Menissy, S., et al. (2020). Loss of mitochondrial protease CLPP activates type I interferon responses through the mtDNA-cGAS-STING signaling axis. bioRxiv 2020:274712. doi: 10.1101/2020.08.30.274712
Trifunovic, A., Wredenberg, A., Falkenberg, M., Spelbrink, J. N., Rovio, A. T., Bruder, C. E., et al. (2004). Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423. doi: 10.1038/nature02517
Wai, T., and Langer, T. (2016). Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 27, 105–117. doi: 10.1016/j.tem.2015.12.001
West, A. P., and Shadel, G. S. (2017). Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 17, 363–375. doi: 10.1038/nri.2017.21
West, A. P., Khoury-Hanold, W., Staron, M., Tal, M. C., Pineda, C. M., Lang, S. M., et al. (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557. doi: 10.1038/nature14156
White, M. J., McArthur, K., Metcalf, D., Lane, R. M., Cambier, J. C., Herold, M. J., et al. (2014). Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 159, 1549–1562. doi: 10.1016/j.cell.2014.11.036
Woo, D. K., Green, P. D., Santos, J. H., D’Souza, A. D., Walther, Z., Martin, W. D., et al. (2012). Mitochondrial genome instability and ROS enhance intestinal tumorigenesis in APC(Min/+) mice. Am. J. Pathol. 180, 24–31. doi: 10.1016/j.ajpath.2011.10.003)
Xiong, R., Zhang, L., Li, S., Sun, Y., Ding, M., Wang, Y., et al. (2020). Novel and potent inhibitors targeting DHODH are broad-spectrum antivirals against RNA viruses including newly-emerged coronavirus SARS-CoV-2. Protein Cell 11, 723–739. doi: 10.1007/s13238-020-00768-w
Xu, L. G., Wang, Y. Y., Han, K. J., Li, L. Y., Zhai, Z., and Shu, H. B. (2005). VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 19, 727–740. doi: 10.1016/j.molcel.2005.08.014
Yasukawa, K., Kinoshita, D., Yaku, K., Nakagawa, T., and Koshiba, T. (2020). The microRNAs miR-302b and miR-372 regulate mitochondrial metabolism via the SLC25A12 transporter, which controls MAVS-mediated antiviral innate immunity. J. Biol. Chem. 295, 444–457. doi: 10.1074/jbc.RA119.010511
Yu, C. H., Davidson, S., Harapas, C. R., Hilton, J. B., Mlodzianoski, M. J., Laohamonthonkul, P., et al. (2020). TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 183, 636–649.e618. doi: 10.1016/j.cell.2020.09.020
Yum, S., Li, M., Fang, Y., and Chen, Z. J. (2021). TBK1 recruitment to STING activates both IRF3 and NF-kappaB that mediate immune defense against tumors and viral infections. Proc. Natl. Acad. Sci. U S A 118:2021. doi: 10.1073/pnas.2100225118
Zhang, Q., Raoof, M., Chen, Y., Sumi, Y., Sursal, T., Junger, W., et al. (2010). Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107. doi: 10.1038/nature08780
Zhang, W. N., Wang, G. H., Xu, Z. G., Tu, H. Q., Hu, F. Q., Dai, J., et al. (2019). Lactate Is a Natural Suppressor of RLR Signaling by Targeting MAVS. Cell 178, 176. doi: 10.1016/j.cell.2019.05.003
Zhong, Z., Liang, S., Sanchez-Lopez, E., He, F., Shalapour, S., Lin, X. J., et al. (2018). New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560, 198–203. doi: 10.1038/s41586-018-0372-z
Keywords: mitochondria, metabolism, innate immunity, mitochondrial DNA, MAVS, CGAS, STING
Citation: Bahat A, MacVicar T and Langer T (2021) Metabolism and Innate Immunity Meet at the Mitochondria. Front. Cell Dev. Biol. 9:720490. doi: 10.3389/fcell.2021.720490
Received: 04 June 2021; Accepted: 05 July 2021;
Published: 27 July 2021.
Edited by:
Anna Maria Porcelli, University of Bologna, ItalyReviewed by:
Damien Arnoult, INSERM U1197 Unité Mixte de Recherche Interactions Cellules Souches-Niches, FranceCopyright © 2021 Bahat, MacVicar and Langer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Langer, VExhbmdlckBhZ2UubXBnLmRl
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.