
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 26 August 2021
Sec. Molecular and Cellular Pathology
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.712931
This article is part of the Research Topic Omics Data Integration towards Mining of Phenotype Specific Biomarkers in Cancer, Volume II View all 65 articles
 Lei Chen1,2†
Lei Chen1,2† Xianchao Zhou3,4†
Xianchao Zhou3,4† Tao Zeng5
Tao Zeng5 Xiaoyong Pan6
Xiaoyong Pan6 Yu-Hang Zhang7
Yu-Hang Zhang7 Tao Huang5,8*
Tao Huang5,8* Zhaoyuan Fang9*
Zhaoyuan Fang9* Yu-Dong Cai1*
Yu-Dong Cai1*Cancer has been generally defined as a cluster of systematic malignant pathogenesis involving abnormal cell growth. Genetic mutations derived from environmental factors and inherited genetics trigger the initiation and progression of cancers. Although several well-known factors affect cancer, mutation features and rules that affect cancers are relatively unknown due to limited related studies. In this study, a computational investigation on mutation profiles of cancer samples in 27 types was given. These profiles were first analyzed by the Monte Carlo Feature Selection (MCFS) method. A feature list was thus obtained. Then, the incremental feature selection (IFS) method adopted such list to extract essential mutation features related to 27 cancer types, find out 207 mutation rules and construct efficient classifiers. The top 37 mutation features corresponding to different cancer types were discussed. All the qualitatively analyzed gene mutation features contribute to the distinction of different types of cancers, and most of such mutation rules are supported by recent literature. Therefore, our computational investigation could identify potential biomarkers and prediction rules for cancers in the mutation signature level.
Cancer is generally defined as a systematic disease with abnormal cell proliferation and invasion potentials. The general symptoms of cancer include cough, weight loss, lump, and abnormal bleeding, depending on the pathological regions of cancer. Such symptoms are common but not specific in cancers, which may also be shared in other diseases. Genetic factors from either noxious environmental factors or inherited variations trigger the initiation and progression of cancers (Anand et al., 2008; Tang et al., 2018). The regulation and functioning of genetic alterations vary at different omics levels with alternative pathological potentials. However, one of the shared and most common pathological alterations at the phenotypic level is abnormal cell proliferation, which is consistently regulated at the transcriptomics and proteomics level, apart from genomic regulations (Feitelson et al., 2015; Li Z. et al., 2020). Generated from and regulated by tumor microenvironment, cancer is consistently shaped by surrounding cells and tissues, making tumor microenvironment a crucial factor for tumorigenesis in most kinds of cancers, including lung, colorectal, cervical, and breast cancers.
Various biological or pathological behavior affect cancer during initiation, progression, and metastasis. For instance, a bidirectional functioning has been identified on autophagy, one of the most common biological behavior during tumorigenesis. Autophagy suppresses malignant transformation during cancer initiation (Kondo et al., 2005); however, with the invasion and progression of cancer, autophagy may promote the malignant proliferation of cancer cells (de Visser et al., 2006; Nadia and Ramana, 2020). Apart from biological processes, such as autophagy, cancer immune responses are another key factor affecting cancer initiation, progression, and metastasis. Complicated relationships between immune cells and general tumor biological processes, such as initiation, progression, and metastasis, have been recognized in multiple cancer subtypes. Similarly, oxygen-mediated cell damage (Srinivas et al., 2019), growth factor abnormality (Normanno et al., 2017; Umino et al., 2018), and various similar malignant alterations have been recognized during tumorigenesis, describing a complicated profiling of tumor biology at different omics levels.
Clustering cancer related SNPs have been one of the most important research fields in cancer biology for a long time. Multiple previous clustering methods have been presented to summarize the mutational patterns of different cancers. According to one of the most famous cancer mutation databases, COSMIC (Catalogue of Somatic Mutations In Cancer) (Tate et al., 2019), three groups of mutational signatures, including Single Base Substitution (SBS), Doublet Base Substitution (DBS), and Small insertions and deletions (ID), have been summarized. However, such clustering only involves in the molecular biological features of cancer mutations without functional interpretation. Furthermore, in 2019, investigators from University Medical Centre Utrecht summarized current contribution of cancer mutations on clinical diagnosis and identified functional interpretations of existed identified cancer mutation biomarkers (Van Hoeck et al., 2019). However, such results only summarized the effects of mutations with validated functional interpretation. As for computational contribution of cancer mutation clustering, few methods have been presented and these algorithms can only recognize single nucleotide variants (SNVs) and copy number variants (CNVs) (Maura et al., 2019), but not mutation patterns involving multiple base pairs. Therefore, as a summary, the method we presented here have two major innovations: (1) we are targeting essential malignant signatures at the level of mutation patterns not just SNVs or just CNVs; (2) we established quantitative rules to evaluate the contribution of mutation patterns to tumorigenesis.
Among such biological characteristics, cancer-associated genetic mutations are one of the key and essential malignant signatures related to the initiation, invasion, and metastasis of cancers. Specific mutation patterns (ACA to AAA, ACC to AAC, ACG to AAG, etc.) on target regions/genes of the genome have been widely reported to participate in tumorigenesis. For instance, mutant KAI1, regulating a batch of downstream tumor-associated genes, has been observed during metastasis in human prostate cancer (Dong et al., 1996; Yang L. et al., 2020). hMLH1 functioning in DNA mismatch repair is one of the most important molecular biomarkers for hereditary non-polyposis colon cancer at the genomics level (Bronner et al., 1994; Ran et al., 2020). The frequency of Smad4 gene mutation in human colorectal cancer is higher than in any type of cancer (Miyaki et al., 1999; Zhang et al., 2020). Other genes, such as P53 (Fujimoto et al., 1992) and VHL (Kim and Kaelin, 2004), influence cancer development to some extent. However, studies on the mutation features and rules that affect cancers are limited. In the present study, we gave a computational investigation on mutation profiles of cancer samples in 27 types. The powerful feature selection method, Monte Carlo Feature Selection (MCFS) (Dramiński et al., 2007), was adopted to analyze such profiles. A feature list was generated. Then, the incremental feature selection (IFS) (Liu and Setiono, 1998) was applied to this list to extract essential mutation features, find out interesting rules and construct efficient classifiers. As a result, 207 rules and many mutation features related to the 27 types of cancers were obtained. We discussed the top 37 mutation features corresponding to different types of cancers. Our study may serve as a reference in establishing a novel qualitative and quantitative standard in identifying tumor type-specific mutation patterns for tumor classification, and thus provide a new tool for the study of tumorigenesis mechanism based on mutation signatures.
We downloaded the relative mutation frequency of 96 mutation types in 2,892 patients from 27 cancer types (Lawrence et al., 2013). The sample sizes of each cancer type are listed in Table 1. The relative mutation frequency of each mutation type in each cancer patient was calculated by Lawrence et al. (2013) and defined as , where s is the sample, c is the mutation type, ncs is the number of observed mutations, and Ncs is the number of bases with enough coverage (≥14 reads in tumor cases and ≥8 reads in normal cases) to observe mutation. The mutation types were summarized according to their base pair changes identified in a data set of 3,083 tumor–normal pairs across 27 tumor types (Lawrence et al., 2013). Mutations were specified in the middle of three base pair patterns with all possible mutational directions. The detailed mutation types are provided in Supplementary Table 1. Several studies have suggested that the mutation signatures of different cancers vary and involve combinations of the above mutation types (Alexandrov et al., 2013; Lawrence et al., 2013; Huang et al., 2018; Wojtowicz et al., 2019). We investigated the cancer mutation signatures quantitatively through advanced machine learning methods and identified the mutation rules for explaining and understanding each cancer type.

Table 1. Sample sizes of 27 cancer types.
We applied feature selection to discriminate influential mutation types from the unrelated ones in the dataset. First, MCFS (Dramiński et al., 2007) was used to evaluate the importance of each mutation type. A feature list was generated. Then, a set of optimal mutation types with strong distinctions between different cancer types was obtained by applying IFS (Liu and Setiono, 1998; Ma et al., 2020) with a supervised classifier on such list.
In this study, we used the MCFS method to assess the importance of mutation types. MCFS is a feature selection method based on the random features of the original features (Dramiński et al., 2007). Given a dataset with d features (in this study, 96 mutation types were deemed as features), MCFS first randomly constructed p feature subsets, each of which contains m features, where m is much smaller than d. Second, for each feature subset, t decision trees are built. Each tree is constructed based on 66% samples that are randomly selected from the original dataset, and the rest samples are used to test such tree. Thus, p × t trees can be constructed in total. Based on these trees, the importance of each feature, called relative importance (RI) score in MCFS, is evaluated by the following equation
where wAcc is the weighted accuracy of the decision tree τ and nf(τ) is a node of feature f in decision tree τ. The information gain of nf(τ) is expressed as IG(nf(τ)), and no.in nf(τ) is the number of training samples in nf(τ). u and v are two different weighting factors.
The MCFS program used in this study was retrieved from http://www.ipipan.eu/staff/m.draminski/mcfs.html. For convenience, default parameters were adopted. In detail, u = v = 1, p = 3,000, t = 5, m = 5. The 96 features (mutation types) were analyzed by the MCFS program. Each feature was assigned a RI score. Evidently, a feature with a high RI score was more important than that with a low RI score. Thus, we sorted all 96 features with the decreasing order of their RI scores. For formulation, this list was denoted by F.
Incremental Feature Selection (IFS) (Liu and Setiono, 1998) is a feature selection method that filters out a set of optimal features to accurately distinguish different sample classes. As mentioned in section “Monte Carlo Feature Selection,” a feature list F was generated by MCFS method. Clearly, the high-ranked features should have positive contributions to classification and can help the classification algorithm to produce good performance. To perform IFS, we first created a series of feature subsets with a step 1 from the feature list F. In detail, the first feature subset included the top feature in the list F, the second feature subset contained the top two features, and so forth. For each constructed feature subset, a random forest (RF) classifier was built based on samples represented by features in the subset and it was further evaluated by 10-fold cross-validation (Kohavi, 1995; Li J. et al., 2020; Zhou et al., 2020; Liu et al., 2021; Pan et al., 2021; Zhang et al., 2021a, b, c; Zhu et al., 2021). After testing all feature subsets, we obtained the optimal feature subset with the optimal performance. This feature subset was termed as the optimal feature subset and the classifier with such subset was called the optimal classifier.
Synthetic Minority Over-Sampling Technique (SMOTE) (Chawla et al., 2002; Chao et al., 2019; Yang X.F. et al., 2020) is a classic technology used to address the potential sample imbalance issue during classification learning. SMOTE can add new samples into the minority class as the same number of samples in the majority class, in an oversampling manner. SMOTE includes several computational steps: (1) it randomly selects a sample x in the minority class; (2) it finds k neighboring samples in such class with x; (3) it randomly select again a sample y from these neighboring samples to generate a new sample z by a linear combination of x and y; (4) it places each new sample z into the minority class; and (5) it repeats the above steps with predefined times. We directly adopted SMOTE in WEKA (Witten and Frank, 2005).
When evaluating the performance of classifiers in the IFS method, SMOTE was used to decrease the influence of imbalanced problem. In detail, in each round of 10-fold cross-validation, the training dataset was processed by SMOTE so that all classes had same number of samples. The classifier was built on such dataset and further used to predict testing samples.
A RF is a classifier that is a predictive model for establishing classification and regression problems. It determines the output class for one sample by aggregating votes from different decision trees (Breiman, 2001). As one of the common methods of machine learning, we built a RF by constructing a large number of decision trees. Averaging the predictions of all decision trees to reduce the variance will slightly increase the bias of the predictions, but at the same time, the performance of the model will be considerably improved while avoiding over-learning. As one of the common methods in the field of machine learning, it has wide applications in tackling different biological problems (Pugalenthi et al., 2011; Pan et al., 2014; Marques et al., 2016; Zhao X. et al., 2018; Ru et al., 2019; Zhang et al., 2019; Zhao R. et al., 2019; Zhao X. et al., 2019; Jia et al., 2020; Liang et al., 2020). In this study, we used the tool “RandomForest” in Weka (Witten and Frank, 2005), which implements the RF. Default parameters were used, where the number of decision tree was 10.
In this study, we also used the interpretable machine learning method repeated incremental pruning to produce error reduction (RIPPER) (Cohen, 1995) to learn the classification rules. In RIPPER, each rule is an IF-ELSE statement; for instance, if gene1 > 1.3 and gene2 < 5, then breast cancer occurs. The learned rules can be used to make predictions for new samples. We used the RIPPER implemented tool “JRip” in WEKA.
Matthew’s Correlation Coefficient (MCC) (Matthews, 1975) is a commonly used method for estimating performance measurements of classification models (Chen et al., 2017a, b, 2018; Cui and Chen, 2019). However, the original MCC was designed for binary classification problem. In this study, 27 cancer types were involved. Thus, we used the MCC in multi-class version, which was proposed by Gorodkin (2004). To calculate such MCC, two matrices X and Y must be constructed first, where X stands for the true labels of all samples and Y represents the predicted labels of all samples. Then, the MCC in multi-class can be computed by
where cov(⋅) indicates the correlation coefficient of two matrices. Similar to the original MCC, MCC in multi-class ranges between –1 and 1. A high MCC implies the good performance. For convenience, we still called MCC in multi-class as MCC in the following text.
Besides, we also reported the accuracy of each cancer type and overall accuracy to give a complete picture on the performance of each classifier.
In this study, several machine learning methods were adopted to investigate the mutation profiles of cancer samples in 27 types. The entire procedures are illustrated in Figure 1.
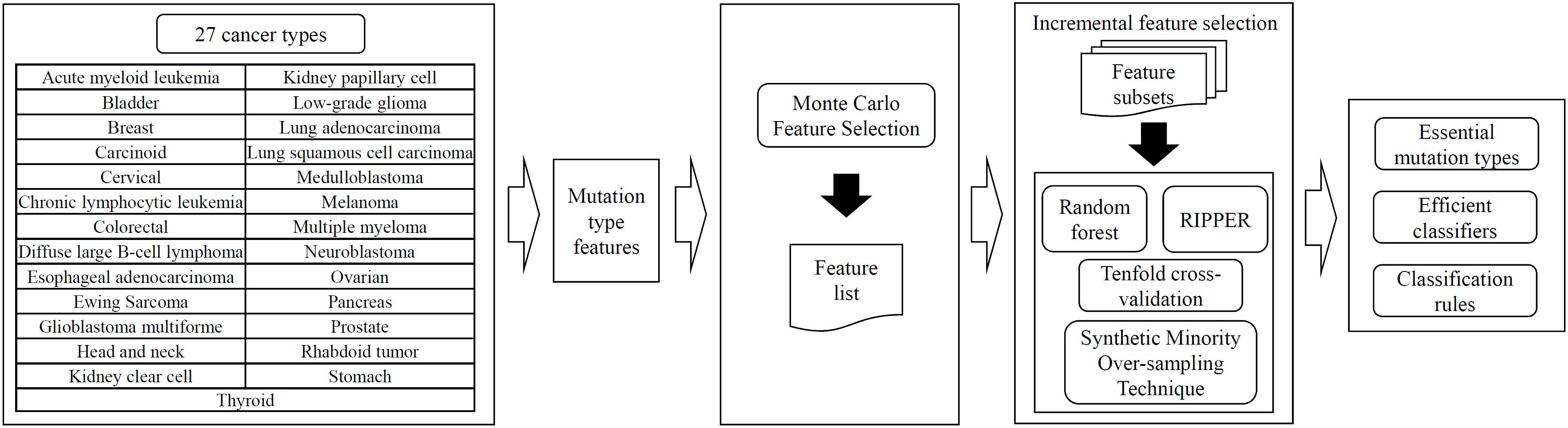
Figure 1. Entire procedures for the investigation on mutation profiles of cancer samples in 27 types. The profiles are analyzed by the MCFS method, resulting in a feature list. Such list is fed into the incremental feature selection, incorporating random forest or RIPPER as the classification algorithm, to extract essential mutation types, build efficient classifiers and construct classification rules.
The mutation profiles were first analyzed by the MCFS method to assess the importance of each mutation type. Each mutation type was assigned a RI score, which is listed in Supplementary Table 2. Then, all mutation types were sorted by the decreasing order of their RI scores, resulting in a feature list F. Such list is also provided in Supplementary Table 2.
A feature list F was generated according to the results of MCFS. Such list was fed into the IFS method. Two classification algorithms: RF and RIPPER, were integrated in the IFS method. For RF, its performance on each feature subset is provided in Supplementary Table 3. An IFS curve was plotted, as shown in Figure 2, for an easy observation. The number of features was set as X-axis and MCC was set as the Y-axis. It can be observed that the highest MCC was 0.772 when top 96 features were adopted. Surprisingly, all mutation features were used in this case, indicating that each mutation type gave less or more contributions to the distinction of different cancer types. Accordingly, all mutation features comprised the optimal feature subset for RF and the RF classifier with these features was called the optimal RF classifier. The overall accuracy of such classifier was 0.784, as listed in Table 2. Its detailed performance on 27 cancer types is illustrated in Figure 3. 14 cancer types received the accuracies higher than 0.900. All these suggested the good performance of such RF classifier. Furthermore, we also employed the rule learning algorithm, RIPPER, to do the same procedures. The performance of RIPPER on all feature subsets is also available in Supplementary Table 3. Likewise, an IFS curve was plotted, as illustrated in Figure 2. Evidently, the highest MCC was 0.408 when top 61 features were adopted. Subsequently, the optimal RIPPER classifier was constructed using these 61 features and these features constituted the optimal feature subset for RIPPER. The MCC (0.408) was much lower than that of the optimal RF classifier (0.772). The overall accuracy was 0.443, as listed in Table 2, also much lower than that of the optimal RF classifier (0.784). The detailed performance of the optimal RIPPER classifier on all cancer types is illustrated in Figure 3. It can be observed that almost all cancer types received lower accuracies than those of the optimal RF classifier. Thus, the optimal RF classifier was much superior to the optimal RIPPER classifier. However, the optimal RF classifier was an absolute black-box classifier, few insights can be extracted from such classifier. The RIPPER classifier was much better in this regard because it can learn some classification rules for explaining and understanding particular cancer differences. In detail, generally, several mutation features were involved in one classification rule that can be used to predict one cancer type. These mutation features together with their corresponding thresholds can comprise a mutation pattern on such cancer type. Further investigation on such pattern was helpful to understand the mechanism of this cancer type in the mutation signature level.
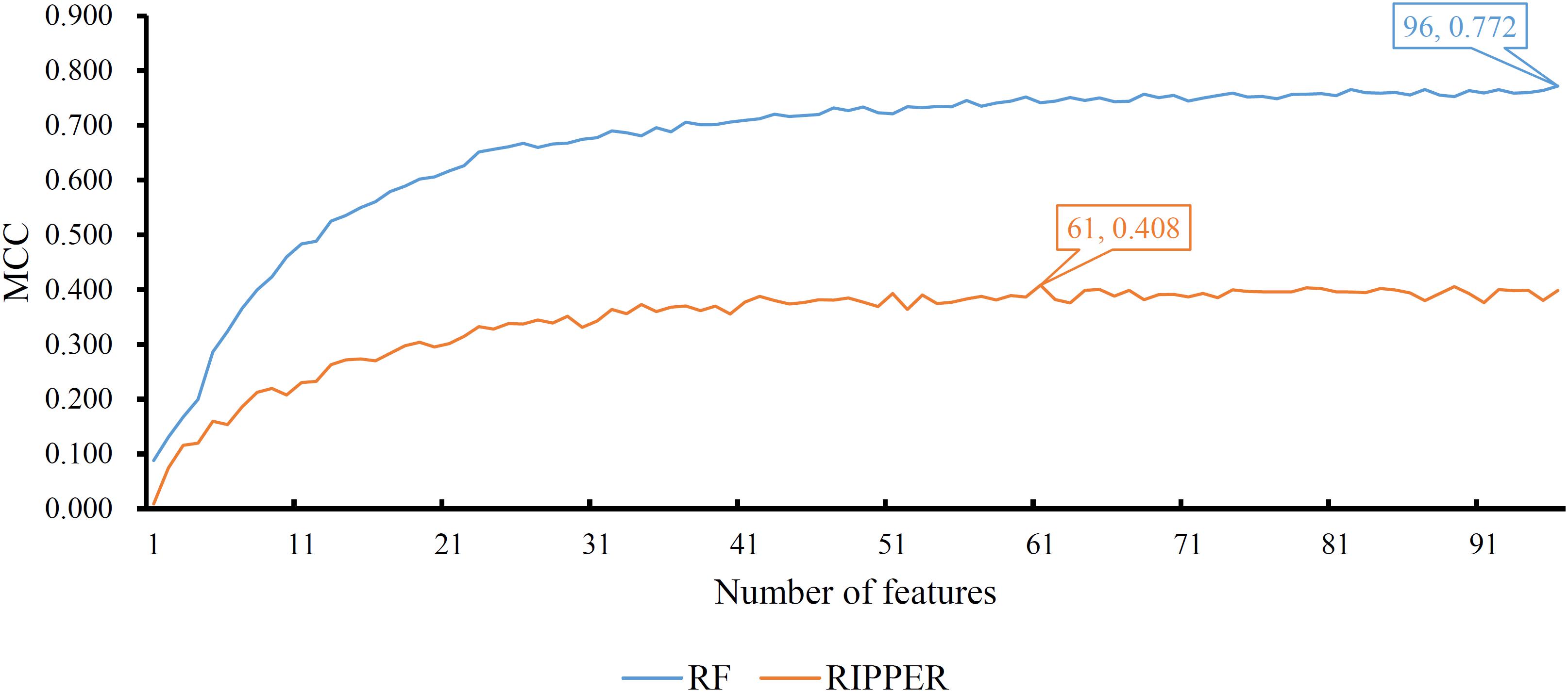
Figure 2. Performance of RF and RIPPER on different feature subsets. The RF yields the highest MCC of 0.772 when all features are used, whereas RIPPER generates the highest MCC of 0.408 when top 61 features are used.
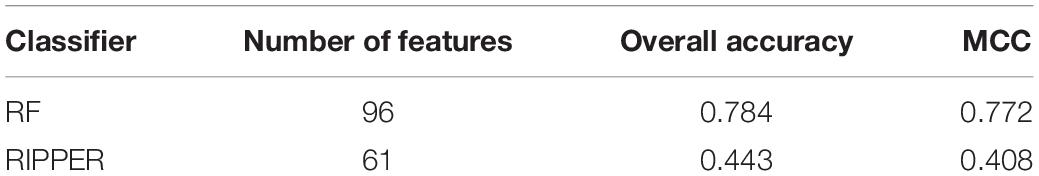
Table 2. Performance of IFS with RF and RIPPER for classifying samples from different cancers.
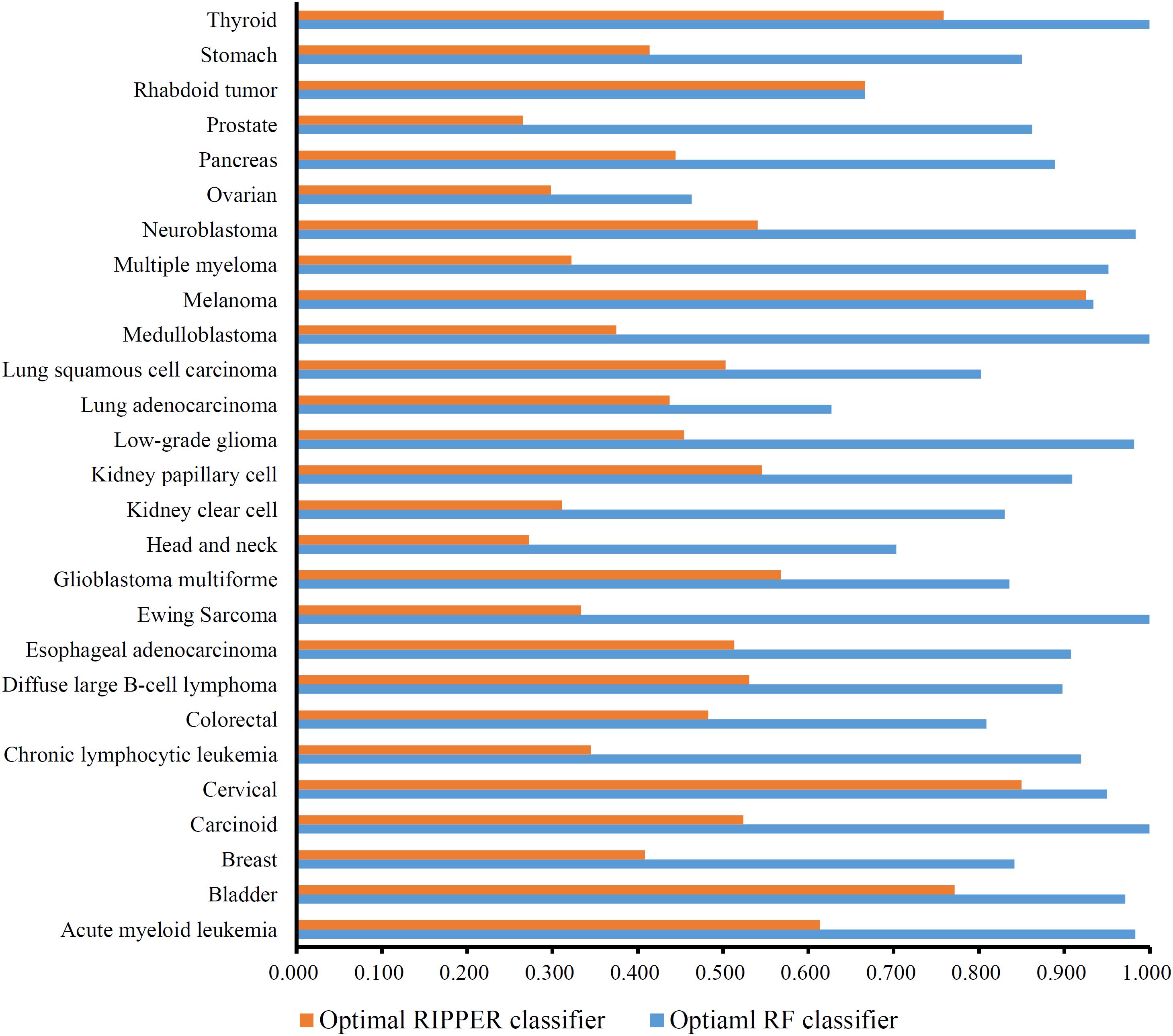
Figure 3. Accuracy on each cancer type yielded by the optimal RF and RIPPER classifiers. The optimal RF classifier gives high performance (accuracy > 0.900) on 14 cancer types, indicating its good performance. The optimal RIPPER classifier provides much lower performance.
From Table 1, we can see that some cancer types contained much more samples than other types, that is, the type sizes were of great differences. Here, we investigated the performance of two optimal classifiers on cancer types with different sizes. To this end, we classified 27 cancer types into three categories. The first category contained types with less than 10 samples, the second category included the types with 10–100 samples, and the third category contained types with more than 100 samples. For convenience, these three categories were called small, middle and large cancer types. The performance of the optimal RF and RIPPER classifiers on three categories is illustrated in Figure 4. It can be observed that the middle cancer type received relative higher accuracies, whereas the small cancer type received slightly higher accuracies than the large cancer type. The reason may be the application of SMOTE. It can increase the performance on minor classes (small cancer type) and decrease the performance on major classes (large cancer type).
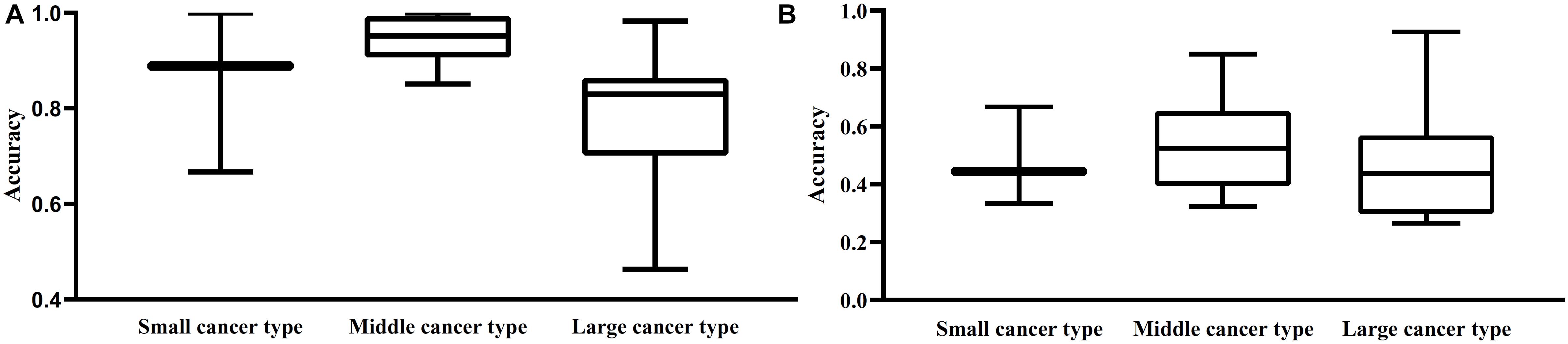
Figure 4. Box plot to show the performance of the optimal RF and RIPPER classifier on three different cancer types. Small cancer type includes the types with less than 10 samples; middle cancer type contains types containing 10–100 samples; larger cancer type contains types with more than 100 samples. (A) Box plot for the optimal RF classifier; (B) Box plot for the optimal RIPPER classifier. The performance on middle cancer type of two optimal classifiers is best.
The optimal RIPPER classifier used the top 61 mutation features. Accordingly, the RIPPER was applied on all cancer samples that were represented by these 61 features. As a result, 207 rules were obtained, which are provided in Supplementary Table 4. Each cancer type had at least one rules. The number of rules on each cancer type is shown in Figure 5. The cancer types “Esophageal adenocarcinoma” and “Neuroblastoma” had most rules, whereas “Kidney clear cell” and “Rhabdoid tumor” had only one rule. In section “Mutation Pattern Rules Associated With Cancer Subgrouping,” some representative rules would be discussed.
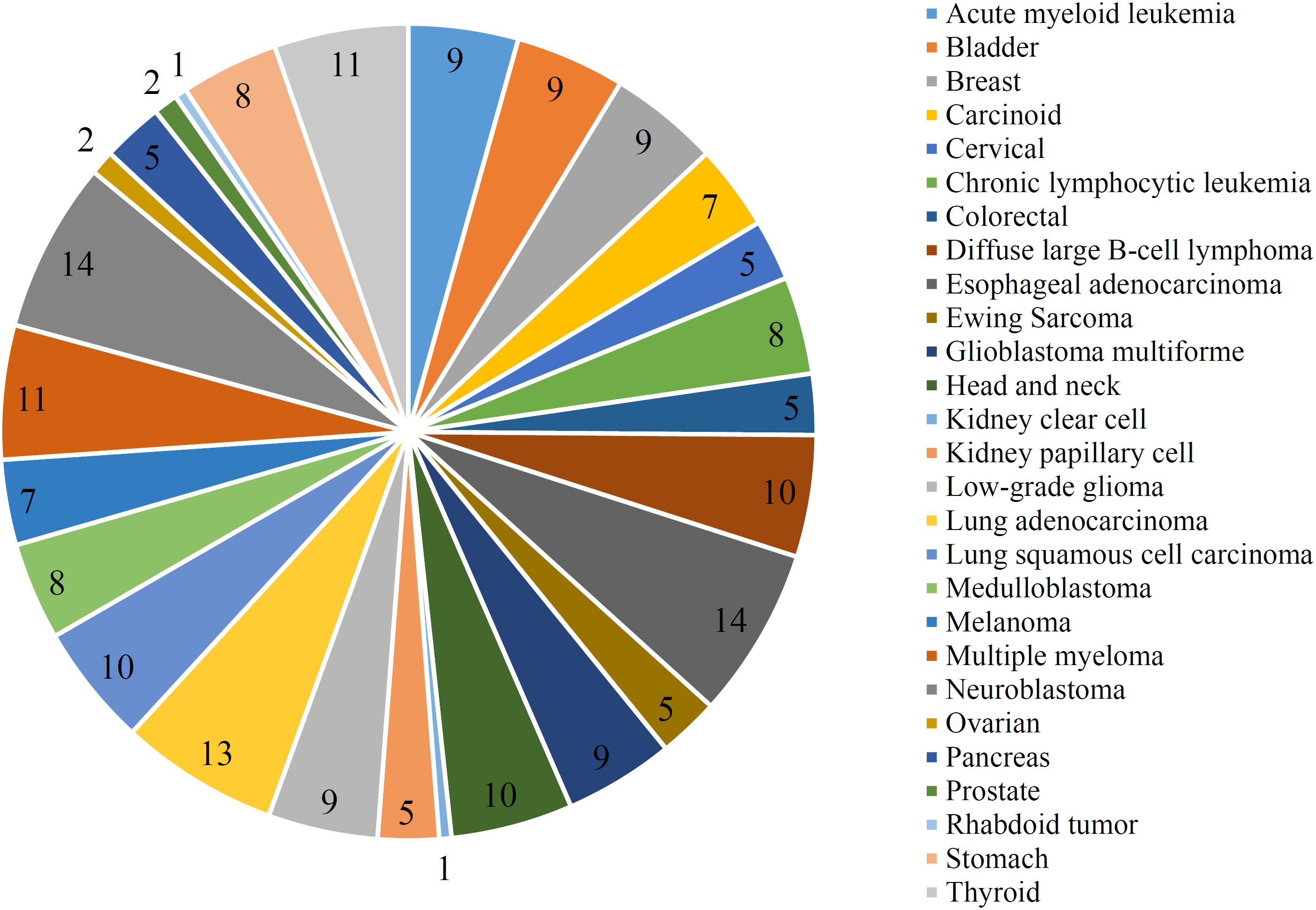
Figure 5. Pie chart to show the number of rules on each cancer type.
We summarized optimal features describing the distinct mutation types to generate an applicable classifier for all the 27 cancer subtypes. The contribution of typical mutation patterns was functionally connected to certain cancer biological behavior, such as the initiation and progression of tumorigenesis. The top 37 features were supported by previous studies as biomarker candidates. In addition to these potential cancer biomarkers, we identified 27 rules of mutation types associated with these 27 cancers. Such group of quantitative rules may contribute to the detailed classification of cancers and serve as guidance in future research.
First, we screened a group of functional mutation patterns with different distributions in various cancer types. Recent publications confirmed such mutation patterns consistent with our prediction. Here, we selected the top five mutation patterns for following detailed discussions.
The top mutation pattern ACG to ATG refers to amino acid substitution from Thr to Met. Recent publications indicated that such mutation pattern exists in multiple cancer-associated genes, including TP53, PRSS1, and XRCC3. For TP53, such mutation pattern has been recognized in multiple tumor types, especially for head and neck cancer (Boyle et al., 1993) and lung cancer (Veldore et al., 2015), rather than other malignant proliferative diseases, such as esophageal adenocarcinoma and melanoma; this finding reflects the distinctive potentials of such mutation pattern. For PRSS1, such mutation pattern has only been found in pancreatic cancer, validating our prediction (Yi et al., 2016). Similarly, XRCC3 has such a mutation pattern in multiple cancer types, including thyroid cancer (Wang et al., 2015) and breast cancer (Lee et al., 2007). Therefore, as we have mentioned above, this mutation pattern has been discovered in limited tumor types but not in all tumor categories, validating our prediction of mutation pattern distinctive with certain tumor types.
CCG to CTG, indicating the transition from Pro to Leu, is another mutation pattern we identified. According to previous studies, such mutation pattern in protein IL-10 contributes to the anti-tumor immune responses in prostate cancer but not in other tumor types, corresponding with our prediction (Bandil et al., 2017). Such mutation pattern has also been identified in the gene p16 in multiple tumor types but not in all of our candidate tumor types (Zhang and Peng, 2002), further validating our analysis.
The mutation pattern TCG to TTG describes the amino acid transition from serine to leucine. According to recent reports, such mutation pattern may also distinguish certain tumor types from the other ones, which has been validated to be pathogenic in a specific oncogene named RET for thyroid carcinoma (Colombo-Benkmann et al., 2008), confirming our prediction. Further, SMAD2 and SMAD4 as the two essential components of the TGF beta-Smad signaling pathway also have such variant pattern contributing to the tumorigenesis of head and neck squamous cell carcinoma (Qiu et al., 2007) in contrast with other tumor types.
Apart from the optimal mutation patterns analyzed above, the mutation patterns from GCG to GTG (i.e., from alanine to valine) and from TCA to TTA (i.e., from serine to leucine) (Zhang and Peng, 2002; Bandil et al., 2017) have been distinctively identified in different tumor types, which is consistent with to our prediction.
A total of 207 mutation rules are related to 27 cancers. This work focused on a few representative cancer types and several top mutation rules for each cancer.
The first rule is the mutation of codon CCC to CTC (Leu to Pro). In 1998, it was reported that the case of melanoma is particularly peculiar, showing Leu-to-Pro mutations in codons 31 and 35, both of which are located in the highly conserved regions (Kumar et al., 1998). P53 mutations in melanoma cell lines, metastases, and primary tumors include the Leu-to-Pro mutations (Zerp et al., 1999). The second rule is the mutations of codons from CCC to CTC, TCC to TTC, TAA to TTA, and the third rule includes the mutations from CCC to CTC, and CCG to CTG, which are some refinements of the first rule.
With typical symptoms as a lump or sore, head and neck cancer is a general description of all cancers related to mouth, nose, and other accessory organs around the head. The first rule for head and neck cancer involves mutations of codons TCA to TTA (Ser to Leu), CAG to CGG (Glu to Arg), and TAT to TGT (Cys to Tyr). In 2007, researchers identified an effective variation on SMAD2. Such variation located in exon 8 at codon 276 (Ser to Leu) contributes to the progression of human head and neck squamous cell carcinoma. The mutations of SMAD2 disrupts a famous tumor associated pathway named transforming growth factor β-Smad signaling pathway (Qiu et al., 2007). The second rule in head and neck cancer involves the mutation of codons TCA to TGA (Ser to a stop codon), ACG to ATG (Thr to Met), AAG to AGG (Lys to Arg), GAG to GGG (Glu to Gly), and TCC to TTC (Ser to Phe). TP53 mutation rate increases following the development and progression of head and neck cancer, and Thr to Met is a well-known p53 mutation (Boyle et al., 1993). The third rule in head and neck cancer includes mutations from codons CCG to CAG, GAC to GGC, and GCT to GTT; these mutations are especially related to CYP1 gene, and the genetic polymorphisms of CYP are associated with head and neck cancer (Gattás et al., 2010).
Arising from the esophagus, esophageal adenocarcinoma is one of the most common subtypes of malignancies affecting the digestive tract. The clinical symptoms of such cancer include difficulty in swallowing and weight loss. The first rule in esophageal cancer involves six mutations, such as AAG to ACG, CAT to CGT, GCC to GGC, CCG to CAG, GCA to GAA, and ACA to ATA. A highly significant association exists between P53 mutations in the molecular pathogenesis of esophageal adenocarcinoma and esophageal malignancy, and AAG to ACG is a remarkable type of TP53 mutation (Vaninetti et al., 2008). The second rule in esophageal adenocarcinoma comprises mutations, such as TAA to TTA, TCT to TGT, GCT to GTT, and CCT to CAT. The third rule involves mutations, such as AAG to AGG, CAT to CGT, CAG to CTG, TCG to TAG, and ACC to AAC. The missense polymorphism of human AGT gene (at codon 276, Ser to Leu) is pathogenic for such disease, revealing the specific characteristics of such diseases at the mutational pattern level. In addition, we confirmed the existence of a codon 84 genetic polymorphism previously, which converts leucine to phenylalanine (Deng et al., 1999).
With cancer-associated bone pain, neck, and chest lump as typical clinical symptoms, neuroblastoma derives from nerve tissues with high cellular diversity. The first rule in neuroblastoma is the involvement of five mutations, including ACG to ATG, GCA to GAA, CAC to CCC, CCA to CTA, and TCC to TTC. The second rule comprises the mutations, such as CAC to CCC, TCC to TAC, ACA to ATA, and CCA to CAA. The third comprises the mutations ACA to AAA, TCA to TTA, TCT to TAT, GCC to GTC, and ACC to ATC.
Here, we further compared our results with previously reported effective mutation patterns in COSMIC database with solid publication supports. Although in COSMIC, the mutations are summarized based on single base not a combination of three constitutive bases, we did find the consistency of COSMIC validated mutation patterns and our results.
For instance, in melanoma, we identified CCC to CTC, TCC to TTC, and CCG to CTG as three typical mutation patterns, which all involved specific cosmic mutation type as C > T. In COSMIC database, more than 23% of patients have such mutation pattern in melanoma associated genomic regions (Tate et al., 2019). Apart from that, such mutation patterns have been identified in melanoma associated genes, like OR4F5 and SAMD11, validating the specific role of such mutational patterns in melanoma.
Furthermore, as for esophageal adenocarcinoma, mutation patterns like GCA to GAA, CCT to CAT, and TCG to TAG are significant mutations identified in this study with the same single base pair alterations as C > A. According to COSMIC database, C > A pattern has been shown to be identified in more than 54% of all patients associated with esophageal adenocarcinoma (Tate et al., 2019), indicating the specific biological functions of such mutation patterns on such cancer subtypes.
Apart from mutation patterns associated with specific cancer subtypes, we also identified a group of effective mutation patterns associated with APOBEC (Chen et al., 2019). As an apolipoprotein B mRNA editing enzyme, APOBEC family has been shown to be associated with multiple cancer mutations and contribute to the variety of cancer mutation burdens. In our study, we also identified some specific APOBEC associated mutation patterns like TCG > TTG and TCA > TTA, validating that identified mutation patterns are associated with the initiation and progression of different cancer subtypes.
Therefore, the mutation patterns identified in this study to be associated with different cancer subtypes have been validated by COSMIC database, implying reliability of our results.
All of the qualitatively analyzed mutation signatures contribute to the distinction of different types of cancers. Most of the quantitative analyzed mutation rules are supported by recent literature. Our computational approach could efficiently identify mutation signatures and rules for cancers.
Publicly available datasets were analyzed in this study. This data can be found here: https://www.nature.com/articles/nature12213.
TH, ZF, and Y-DC designed the study. LC, XZ, and XP performed the experiments. TZ and Y-HZ analyzed the results. LC and XZ wrote the manuscript. All authors contributed to the research and reviewed the manuscript.
This research was funded by the Strategic Priority Research Program of Chinese Academy of Sciences (XDB38050200), the National Key R&D Program of China (2017YFC1201200), the Shanghai Municipal Science and Technology Major Project (2017SHZDZX01), the National Key R&D Program of China (2018YFC0910403), the National Natural Science Foundation of China (31701151), the Shanghai Sailing Program (16YF1413800), the Youth Innovation Promotion Association of Chinese Academy of Sciences (CAS) (2016245), and the Fund of the Key Laboratory of Tissue Microenvironment and Tumor of Chinese Academy of Sciences (202002).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.712931/full#supplementary-material
Supplementary Table 1 | Information of 96 mutation types.
Supplementary Table 2 | RI scores of mutation types.
Supplementary Table 3 | IFS corresponding to different numbers of features used.
Supplementary Table 4 | Classification rules generated by RIPPER.
Alexandrov, L. B., Nik-Zainal, S., Wedge, D. C., Aparicio, S. A., Behjati, S., Biankin, A. V., et al. (2013). Signatures of mutational processes in human cancer. Nature 500, 415–421.
Anand, P., Kunnumakara, A. B., Sundaram, C., Harikumar, K. B., Tharakan, S. T., Lai, O. S., et al. (2008). Cancer is a preventable disease that requires major lifestyle changes. Pharm. Res. 25, 2097–2116. doi: 10.1007/s11095-008-9661-9
Bandil, K., Singhal, P., Dogra, A., Rawal, S. K., Doval, D. C., Varshney, A. K., et al. (2017). Association of SNPs/haplotypes in promoter of TNF A and IL-10 gene together with life style factors in prostate cancer progression in Indian population. Inflamm. Res. 66, 1085–1097. doi: 10.1007/s00011-017-1088-5
Boyle, J. O., Hakim, J., Koch, W., Van Der Riet, P., Hruban, R. H., Roa, R. A., et al. (1993). The incidence of p53 mutations increases with progression of head and neck cancer. Cancer Res. 53:4477.
Bronner, C. E., Baker, S. M., Morrison, P. T., Warren, G., Smith, L. G., Lescoe, M. K., et al. (1994). Mutation in the DNA mismatch repair gene homologue hMLH 1 is associated with hereditary non-polyposis colon cancer. Nature 368:258. doi: 10.1038/368258a0
Chao, L., Wei, L., and Zou, Q. (2019). SecProMTB: a SVM-based Classifier for Secretory Proteins of Mycobacterium tuberculosis with Imbalanced Data Set. Proteomics 19:e1900007.
Chawla, N. V., Bowyer, K. W., Hall, L. O., and Kegelmeyer, W. P. (2002). SMOTE: synthetic minority over-sampling technique. J. Artif. Intell. Res. 16, 321–357. doi: 10.1613/jair.953
Chen, L., Chu, C., Zhang, Y.-H., Zheng, M.-Y., Zhu, L., Kong, X., et al. (2017a). Identification of drug-drug interactions using chemical interactions. Curr. Bioinform. 12, 526–534.
Chen, L., Pan, X., Hu, X., Zhang, Y. H., Wang, S., Huang, T., et al. (2018). Gene expression differences among different MSI statuses in colorectal cancer. Int. J. Cancer 143, 1731–1740. doi: 10.1002/ijc.31554
Chen, L., Wang, S., Zhang, Y.-H., Li, J., Xing, Z.-H., Yang, J., et al. (2017b). Identify key sequence features to improve CRISPR sgRNA efficacy. IEEE Access 5, 26582–26590. doi: 10.1109/access.2017.2775703
Chen, Z., Wen, W., Bao, J., Kuhs, K. L., Cai, Q., Long, J., et al. (2019). Integrative genomic analyses of APOBEC-mutational signature, expression and germline deletion of APOBEC3 genes, and immunogenicity in multiple cancer types. BMC Med. Genomics 12:131. doi: 10.1186/s12920-019-0579-3
Cohen, W. W. (1995). “Fast effective rule induction,” in Proceedings the Twelfth International Conference on Machine Learning, Tahoe City, CA, 115–123. doi: 10.1016/b978-1-55860-377-6.50023-2
Colombo-Benkmann, M., Li, Z., Riemann, B., Hengst, K., Herbst, H., Keuser, R., et al. (2008). Characterization of the RET protooncogene transmembrane domain mutation S649L associated with nonaggressive medullary thyroid carcinoma. Eur. J. Endocrinol. 158, 811–816. doi: 10.1530/eje-07-0817
Cui, H., and Chen, L. (2019). A binary classifier for the prediction of EC numbers of enzymes. Curr. Proteom. 16, 381–389.
de Visser, K. E., Eichten, A., and Coussens, L. M. (2006). Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 6, 24–37. doi: 10.1038/nrc1782
Deng, C., Xie, D., Capasso, H., Zhao, Y., Wang, L. D., and Hong, J. Y. (1999). Genetic polymorphism of human O6-alkylguanine-DNA alkyltransferase: identification of a missense variation in the active site region. Pharmacogenetics 9, 81–87. doi: 10.1097/00008571-199902000-00011
Dong, J.-T., Suzuki, H., Pin, S. S., Bova, G. S., Schalken, J. A., Isaacs, W. B., et al. (1996). Down-Regulation of the <em>KAI1</em> metastasis suppressor gene during the progression of human prostatic cancer infrequently involves gene mutation or allelic loss. Cancer Res. 56, 4387–4390
Dramiński, M., Rada-Iglesias, A., Enroth, S., Wadelius, C., Koronacki, J., and Komorowski, J. (2007). Monte Carlo feature selection for supervised classification. Bioinformatics 24, 110–117. doi: 10.1093/bioinformatics/btm486
Feitelson, M. A., Arzumanyan, A., Kulathinal, R. J., Blain, S. W., Holcombe, R. F., Mahajna, J., et al. (2015). Sustained proliferation in cancer: mechanisms and novel therapeutic targets. Semin. Cancer Biol. 35(Suppl.), S25–S54.
Fujimoto, K., Yamada, Y., Okajima, E., Kakizoe, T., Sasaki, H., Sugimura, T., et al. (1992). Frequent association of p53 gene mutation in invasive bladder cancer. Cancer Res. 52, 1393–1398.
Gattás, G. J. F., Marcos Brasilino, D. C., Maria Salete, S., Curioni, O. A., Priscila, K., Jose, E. N., et al. (2010). Genetic polymorphisms of CYP1A1, CYP2E1, GSTM1, and GSTT1 associated with head and neck cancer. Head Neck 28, 819–826. doi: 10.1002/hed.20410
Gorodkin, J. (2004). Comparing two K-category assignments by a K-category correlation coefficient. Comput. Biol. Chem. 28, 367–374. doi: 10.1016/j.compbiolchem.2004.09.006
Huang, P. J., Chiu, L. Y., Lee, C. C., Yeh, Y. M., Huang, K. Y., Chiu, C. H., et al. (2018). mSignatureDB: a database for deciphering mutational signatures in human cancers. Nucleic Acids Res. 46, D964–D970.
Jia, Y., Zhao, R., and Chen, L. (2020). Similarity-Based machine learning model for predicting the metabolic pathways of compounds. IEEE Access 8, 130687–130696. doi: 10.1109/access.2020.3009439
Kim, W. Y., and Kaelin, W. G. (2004). Role of VHL gene mutation in human cancer. J. Clin. Oncol. 22, 4991–5004. doi: 10.1200/jco.2004.05.061
Kohavi, R. (1995). “A study of cross-validation and bootstrap for accuracy estimation and model selection,” in Proceedings of the International Joint Conference on Artificial Intelligence, (Mahwah, NJ: Lawrence Erlbaum Associates Ltd), 1137–1145.
Kondo, Y., Kanzawa, T., Sawaya, R., and Kondo, S. (2005). The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 5, 726–734. doi: 10.1038/nrc1692
Kumar, R., Rozell, B. L., Louhelainen, J., and Hemminki, K. (1998). Mutations in the CDKN2A (p16INK4a) gene in microdissected sporadic primary melanomas. Int. J. Cancer 75, 193–198. doi: 10.1002/(sici)1097-0215(19980119)75:2<193::aid-ijc5>3.0.co;2-p
Lawrence, M. S., Stojanov, P., Polak, P., Kryukov, G. V., Cibulskis, K., Sivachenko, A., et al. (2013). Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218.
Lee, S. A., Lee, K. M., Park, S. K., Choi, J. Y., Kim, B., Nam, J., et al. (2007). Genetic polymorphism of XRCC3 Thr241Met and breast cancer risk: case-control study in Korean women and meta-analysis of 12 studies. Breast Cancer Res. Treat. 103, 71–76. doi: 10.1007/s10549-006-9348-z
Li, J., Chang, M., Gao, Q., Song, X., and Gao, Z. (2020). Lung cancer classification and gene selection by combining affinity propagation clustering and sparse group lasso. Curr. Bioinform. 15, 703–712. doi: 10.2174/1574893614666191017103557
Li, Z., Zhang, T., Lei, H., Wei, L., Liu, Y., Shi, Y., et al. (2020). Research on gastric Cancer’s drug-resistant gene regulatory network model. Curr. Bioinform. 15, 225–234. doi: 10.2174/1574893614666190722102557
Liang, H., Chen, L., Zhao, X., and Zhang, X. (2020). Prediction of drug side effects with a refined negative sample selection strategy. Comput. Math. Methods Med. 2020:1573543.
Liu, H., Hu, B., Chen, L., and Lu, L. (2021). Identifying protein subcellular location with embedding features learned from networks. Curr. Proteom. 17.
Ma, X., Xi, B., Zhang, Y., Zhu, L., Sui, X., Tian, G., et al. (2020). A machine learning-based diagnosis of thyroid cancer using thyroid nodules ultrasound images. Curr. Bioinform. 15, 349–358. doi: 10.2174/1574893614666191017091959
Marques, Y. B., De Paiva Oliveira, A., Ribeiro Vasconcelos, A. T., and Cerqueira, F. R. (2016). Mirnacle: machine learning with SMOTE and random forest for improving selectivity in pre-miRNA ab initio prediction. BMC Bioinformatics 17:474. doi: 10.1186/s12859-016-1343-8
Matthews, B. (1975). Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta BBA Protein Struct. 405, 442–451. doi: 10.1016/0005-2795(75)90109-9
Maura, F., Degasperi, A., Nadeu, F., Leongamornlert, D., Davies, H., Moore, L., et al. (2019). A practical guide for mutational signature analysis in hematological malignancies. Nat. Commun. 10:2969.
Miyaki, M., Iijima, T., Konishi, M., Sakai, K., Ishii, A., Yasuno, M., et al. (1999). Higher frequency of Smad4 gene mutation in human colorectal cancer with distant metastasis. Oncogene 18, 3098–3103. doi: 10.1038/sj.onc.1202642
Nadia, and Ramana, J. (2020). The human OncoBiome database: a database of cancer microbiome datasets. Curr. Bioinform. 15, 472–477. doi: 10.2174/1574893614666190902152727
Normanno, N., Denis, M. G., Thress, K. S., Ratcliffe, M., and Reck, M. (2017). Guide to detecting epidermal growth factor receptor (EGFR) mutations in ctDNA of patients with advanced non-small-cell lung cancer. Oncotarget 8, 12501–12516. doi: 10.18632/oncotarget.13915
Pan, X. Y., Zhu, L., Fan, Y. X., and Yan, J. C. (2014). Predicting protein-RNA interaction amino acids using random forest based on submodularity subset selection. Comput. Biol. Chem. 53, 324–330. doi: 10.1016/j.compbiolchem.2014.11.002
Pan, X., Li, H., Zeng, T., Li, Z., Chen, L., Huang, T., et al. (2021). Identification of protein subcellular localization with network and functional embeddings. Front. Genet. 11:626500. doi: 10.3389/fgene.2020.626500
Pugalenthi, G., Kandaswamy, K., Chou, K.-C., Vivekanandan, S., and Kolatkar, P. (2011). RSARF: prediction of residue solvent accessibility from protein sequence using random forest method. Protein Peptide Lett. 19, 50–56. doi: 10.2174/092986612798472875
Qiu, W., Schonleben, F., Li, X., and Su, G. H. (2007). Disruption of transforming growth factor beta-Smad signaling pathway in head and neck squamous cell carcinoma as evidenced by mutations of SMAD2 and SMAD4. Cancer Lett. 245, 163–170. doi: 10.1016/j.canlet.2006.01.003
Ran, W., Chen, X., Wang, B., Yang, P., Li, Y., Xiao, Y., et al. (2020). Whole-exome sequencing of tumor-only samples reveals the association between somatic alterations and clinical features in pancreatic cancer. Curr. Bioinform. 15, 1160–1167. doi: 10.2174/1574893615999200626190346
Ru, X. Q., Li, L. H., and Zou, Q. (2019). Incorporating distance-based top-n-gram and random forest to identify electron transport proteins. J. Proteome Res. 18, 2931–2939. doi: 10.1021/acs.jproteome.9b00250
Srinivas, U. S., Tan, B. W., Vellayappan, B. A., and Jeyasekharan, A. D. (2019). ROS and the DNA damage response in cancer. Redox Biol. 25:101084. doi: 10.1016/j.redox.2018.101084
Tang, W., Wan, S., Yang, Z., Teschendorff, A. E., and Zou, Q. (2018). Tumor origin detection with tissue-specific miRNA and DNA methylation markers. Bioinformatics 34, 398–406. doi: 10.1093/bioinformatics/btx622
Tate, J. G., Bamford, S., Jubb, H. C., Sondka, Z., Beare, D. M., Bindal, N., et al. (2019). COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 47, D941–D947.
Umino, K., Fujiwara, S.-I., Ikeda, T., Toda, Y., Ito, S., Mashima, K., et al. (2018). Clinical outcomes of myeloid/lymphoid neoplasms with fibroblast growth factor receptor-1 (FGFR1) rearrangement. Hematology 23, 470–477. doi: 10.1080/10245332.2018.1446279
Van Hoeck, A., Tjoonk, N. H., Van Boxtel, R., and Cuppen, E. (2019). Portrait of a cancer: mutational signature analyses for cancer diagnostics. BMC Cancer 19:457. doi: 10.1186/s12885-019-5677-2
Vaninetti, N. M., Geldenhuys, L., Porter, G. A., Risch, H., Hainaut, P., Guernsey, D. L., et al. (2008). Inducible nitric oxide synthase, nitrotyrosine and p53 mutations in the molecular pathogenesis of Barrett’s esophagus and esophageal adenocarcinoma. Mol. Carcinog. 47, 275–285. doi: 10.1002/mc.20382
Veldore, V. H., Patil, S., Satheesh, C. T., Shashidhara, H. P., Tejaswi, R., Prabhudesai, S. A., et al. (2015). Genomic profiling in a homogeneous molecular subtype of non-small cell lung cancer: an effort to explore new drug targets. Indian J. Cancer 52, 243–248. doi: 10.4103/0019-509x.175843
Wang, X., Zhang, K., Liu, X., Liu, B., and Wang, Z. (2015). Association between XRCC1 and XRCC3 gene polymorphisms and risk of thyroid cancer. Int. J. Clin. Exp. Pathol. 8, 3160–3167.
Witten, I. H., and Frank, E. (2005). Data Mining: Practical Machine Learning Tools and Techniques. San Francisco, CA: Morgan Kaufmann Pub.
Wojtowicz, D., Sason, I., Huang, X., Kim, Y.-A., Leiserson, M. D. M., Przytycka, T. M., et al. (2019). Hidden Markov models lead to higher resolution maps of mutation signature activity in cancer. Genome Med. 11:49.
Yang, L., Gao, H., Wu, K., Zhang, H., Li, C., and Tang, L. (2020). Identification of cancerlectins by using cascade linear discriminant analysis and optimal g-gap tripeptide composition. Curr. Bioinform. 15, 528–537. doi: 10.2174/1574893614666190730103156
Yang, X.-F., Zhou, Y.-K., Zhang, L., Gao, Y., and Du, P.-F. (2020). Predicting LncRNA subcellular localization using unbalanced pseudo-k nucleotide compositions. Curr. Bioinform. 15, 554–562. doi: 10.2174/1574893614666190902151038
Yi, Q., Dong, F., Lin, L., Liu, Q., Chen, S., Gao, F., et al. (2016). PRSS1 mutations and the proteinase/antiproteinase imbalance in the pathogenesis of pancreatic cancer. Tumour Biol. 37, 5805–5810. doi: 10.1007/s13277-015-3982-1
Zerp, S. F., Elsas, A. V., Peltenburg, L. T. C., and Schrier, P. I. (1999). p53 mutations in human cutaneous melanoma correlate with sun exposure but are not always involved in melanomagenesis. Br. J. Cancer 79, 921–926. doi: 10.1038/sj.bjc.6690147
Zhang, B., and Peng, Z. Y. (2002). Structural consequences of tumor-derived mutations in p16INK4a probed by limited proteolysis. Biochemistry 41, 6293–6302. doi: 10.1021/bi0117100
Zhang, L., He, Y., Song, H., Wang, X., Lu, N., Sun, L., et al. (2020). Elastic net regularized softmax regression methods for multi-subtype classification in cancer. Curr. Bioinform. 15, 212–224. doi: 10.2174/1574893613666181112141724
Zhang, X., Chen, L., Guo, Z.-H., and Liang, H. (2019). Identification of human membrane protein types by incorporating network embedding methods. IEEE Access 7, 140794–140805. doi: 10.1109/access.2019.2944177
Zhang, Y.-H., Li, H., Zeng, T., Chen, L., Li, Z., Huang, T., et al. (2021a). Identifying transcriptomic signatures and rules for SARS-CoV-2 infection. Front. Cell Dev. Biol. 8:627302. doi: 10.3389/fcell.2020.627302
Zhang, Y.-H., Zeng, T., Chen, L., Huang, T., and Cai, Y.-D. (2021b). Detecting the multiomics signatures of factor-specific inflammatory effects on airway smooth muscles. Front. Genet. 11:599970. doi: 10.3389/fgene.2020.599970
Zhang, Y.-H., Zeng, T., Chen, L., Huang, T., and Cai, Y.-D. (2021c). Determining protein–protein functional associations by functional rules based on gene ontology and KEGG pathway. Biochim. Biophys. Acta BBA Proteins Proteom. 1869:140621. doi: 10.1016/j.bbapap.2021.140621
Zhao, R., Chen, L., Zhou, B., Guo, Z.-H., Wang, S., and Aorigele. (2019). Recognizing novel tumor suppressor genes using a network machine learning strategy. IEEE Access 7, 155002–155013. doi: 10.1109/access.2019.2949415
Zhao, X., Chen, L., and Lu, J. (2018). A similarity-based method for prediction of drug side effects with heterogeneous information. Math. Biosci. 306, 136–144. doi: 10.1016/j.mbs.2018.09.010
Zhao, X., Chen, L., Guo, Z.-H., and Liu, T. (2019). Predicting drug side effects with compact integration of heterogeneous networks. Curr. Bioinform. 14, 709–720. doi: 10.2174/1574893614666190220114644
Zhou, J.-P., Chen, L., and Guo, Z.-H. (2020). iATC-NRAKEL: an efficient multi-label classifier for recognizing anatomical therapeutic chemical classes of drugs. Bioinformatics 36, 1391–1396.
Keywords: cancer, subtype, mutation signature, pattern, rule, classification
Citation: Chen L, Zhou X, Zeng T, Pan X, Zhang Y-H, Huang T, Fang Z and Cai Y-D (2021) Recognizing Pattern and Rule of Mutation Signatures Corresponding to Cancer Types. Front. Cell Dev. Biol. 9:712931. doi: 10.3389/fcell.2021.712931
Received: 21 May 2021; Accepted: 02 July 2021;
Published: 26 August 2021.
Edited by:
Liang Cheng, Harbin Medical University, ChinaCopyright © 2021 Chen, Zhou, Zeng, Pan, Zhang, Huang, Fang and Cai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Huang, dG9odWFuZ3Rhb0AxMjYuY29t; Zhaoyuan Fang, ZmFuZ3poYW95dWFuQHNpYnMuYWMuY24=; Yu-Dong Cai, Y2FpX3l1ZEAxMjYuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.