 Ian X. McLeod
Ian X. McLeod Ruchi Saxena
Ruchi Saxena Zachary Carico
Zachary Carico You-Wen He
You-Wen He- Department of Immunology, Duke University Medical Center, Durham, NC, United States
Autophagy, a highly conserved intracellular process, has been identified as a novel mechanism regulating T lymphocyte homeostasis. Herein, we demonstrate that both starvation- and T cell receptor-mediated autophagy induction requires class I phosphatidylinositol-3 kinases to produce PI(3)P. In contrast, common gamma chain cytokines are suppressors of autophagy despite their ability to activate the PI3K pathway. T cells lacking the PI3KI regulatory subunits, p85 and p55, were almost completely unable to activate TCR-mediated autophagy and had concurrent defects in PI(3)P production. Additionally, T lymphocytes upregulate polyinositol phosphatases in response to autophagic stimuli, and the activity of the inositol phosphatases Inpp4 and SHIP are required for TCR-mediated autophagy induction. Addition of exogenous PI(3,4)P2 can supplement cellular PI(3)P and accelerate the outcome of activation-induced autophagy. TCR-mediated autophagy also requires internalization of the TCR complex, suggesting that this kinase/phosphatase activity is localized in internalized vesicles. Finally, HIV-induced bystander CD4+ T cell autophagy is dependent upon PI3KI. Overall, our data elucidate an important pathway linking TCR activation to autophagy, via induction of PI3KI activity and inositol phosphatase upregulation to produce PI(3)P.
Introduction
Autophagy is a major pro-survival mechanism, involved in intracellular remodeling and delivery of membrane-bound structures, termed autophagosomes, to the lysosome for turnover of the encompassed components into constituent building blocks (Bento et al., 2016). The autophagosome goes through a maturation process, reminiscent of late endosomal maturation, whereby the compartment becomes acidified when it fuses with lysosomes, during which further substrates can be imported for degradation, through the activity of adaptor molecules and autophagosomal receptors (Deter, 1975). Autophagy plays an important role in orchestrating innate and adaptive immune responses (Schmid and Munz, 2007; Cui et al., 2019). It plays specialized functions in antigen processing and presentation (Münz, 2016; Øynebråten, 2020), and thymic education of CD4+CD8+ double positive thymocytes (Nedjic et al., 2008a,b). In lymphoid-lineage cells, autophagy plays homeostatic roles (Bronietzki et al., 2015), such as control of mitochondria levels and related gene functions (Miller et al., 2008; Pua et al., 2009) and ER membrane trimming (Jia et al., 2011) and regulation of cytokine production (Galle-Treger et al., 2020; Singh et al., 2020).
In most systems studied, the autophagy induction machinery utilizes the coordinated activity of the class III PI3K complex, containing Vps34, to produce PI(3)P (Petiot et al., 2000). PI(3)P is a signal for the recruitment of effectors with FYVE- or GLUE-domains, in order to flex and fuse membranes (Nascimbeni et al., 2017), as well as an important signal in inward vesiculation and multi-vesicular body (MVB) formation (Zeng et al., 2006). In yeast, it has been shown that PI(3)P is imported into the autophagosome during autophagy (Axe et al., 2008). Vps34 has been shown to directly produce PI(3)P from phosphatidylinsotiol (PI), and has been demonstrated in several systems to be the major PI3K responsible for the production of cellular PI(3)P for autophagy (Yang and Van Kaer, 2020). T cells in which Vps34 was deleted entirely had slightly defective autophagy (Willinger and Flavell, 2012). However, Vps34 null kinase domain knockout T cells have intact autophagy (Zhou et al., 2010; McLeod et al., 2011). Importantly, autophagy is regulated by general PI3K inhibitors, wortmannin, and 3-methyladenine (3MA) (Li et al., 2006; Pua et al., 2007; McLeod et al., 2011) and other classes of PI3K are also involved in the production of PI(3)P and the progression of autophagy in T lymphocytes (Bilanges et al., 2019). Additionally, since other classes of PI3K, especially class Ia isoforms, are activated during TCR engagement with peptide-MHC complexes (von Willebrand et al., 1994), we hypothesized these kinases are responsible for linking TCR activation to autophagy. Additionally, it has also been shown that Class I PI3K regulates autophagy by modulating protein synthesis and the Beclin 1 signaling pathway in malignant blood cells (Wang et al., 2017). The class I PI3K, P110β, functions as a positive regulator of autophagy by serving as a scaffolding protein (Dou et al., 2010, 2013). In contrast, knockdown of the class I PI3K, P110δ, in myeloma cells results in the potent activation of autophagy (Ikeda et al., 2010). Clearly, different isoforms of the class I PI3K have varying functions (Yu et al., 2015). This could be due in part to varying subcellular localizations or differing upstream receptors and adaptors.
Various inositol phosphatases are present in metazoan cells to modify the products of inositol kinases. The 3′ inositol phosphatase, PTEN, is responsible for the silencing of PI3K signals during TCR activation (Aquila et al., 2020), and enforces the need for costimulation through CD28 (Buckler et al., 2006). Knockout of another 3′ phosphatase, Jumpy, led to decreased autophagic flux (Vergne et al., 2009). Therefore, 3′ inositol phosphatases are important regulators of autophagy (Vergne and Deretic, 2010). However, the production of PI(3)P is likely influenced by other inositol phosphatases. 4′ phosphatases, including Inpp4a and Inpp4b can modify PI(3,4,5)P3, and are directly linked to PI3K activity by virtue of being in the same protein complex in human platelets (Munday et al., 1999). This complex is necessary for platelet aggregation through the coordinated activity of PI3KI and Inpp4 (Munday et al., 1999). Additionally, 5′ inositol polyphosphatases, including SHIP1 and SHIP2, are important in the modification of TCR induced signals, by dephosphorylating PI(3,4,5)P3 to PI(3,4)P2, and can form a complex with p85, the regulatory subunit of class I PI3K, and directly modify its activity (Jackson et al., 1995). The increased expression of SHIP was also shown to correlate with increased autophagy (Ngoh et al., 2015). It is heretofore unknown whether 4′ and 5′ inositol phosphatases affect autophagy induction and progression. In this study, we show that class I PI3K catalytic activity is essential for the induction of autophagy in T lymphocytes. We demonstrate that 4′ and 5′ inositol phosphatase activity is also required for autophagy induction by TCR stimulation, and that addition of PI(3,4)P2 can modify autophagy induction and progression. Additionally, we show that IL-7 inhibits autophagy in T cells, despite activating the PI3K pathway, while downregulating the transcription of Inpp4 and SHIP mRNAs. Finally, we demonstrate that HIV glycoprotein induced autophagy in human lymphocytes is dependent upon class I PI3K activity.
Materials and Methods
Mice
P85 conditional knockout mice (Luo et al., 2005) were purchased from the Jackson Laboratory, and crossed to ER-cre mice (Hayashi and McMahon, 2002) (The Jackson Laboratory). Genomic deletion of p85 was assessed by PCR primers detecting the floxed (1,275 bp) and deleted (298 bp) alleles (forward, GGT TTC TTA CTT TAG ACG GAG CTG; reverse, CCA GTT ACT TTC AAA TCA GCA CAG). Constitutively active P110α, mice (Srinivasan et al., 2009) were purchased from the Jackson Laboratory. Vps34-deficient T lymphocytes were generated by crossing Vps34-floxed mice (Zhou et al., 2010) to Lck-Cre transgenic mice (The Jackson Laboratory). All mice were bred and housed in Duke’s specific pathogen-free facilities in accordance with IACUC regulations.
Human Samples
Human CD4+ T cells were isolated from the blood of healthy volunteers according to protocol approved by Duke’s Institutional Review Board. PBMC’s were isolated from whole blood using Ficoll-Paque Plus (GE Healthcare Bio-Sciences AB) according to the manufacturer’s instructions.
Antibodies and Reagents
FITC, PE, PE-cy5, APC, APC-cy7, or Pacific Blue conjugated anti-CD3, -CD4, -CD8, -CD44, and -CD62L, were purchased from BioLegend, eBioscience, and BD Pharmingen. Anti-LC3 (PD015) and -p62 were purchased from MBL. Anti-PI(3)P, -PI(3,4)P2, -PI(3,4,5)P3, and PI(3,4)P2 lipid were purchased from Echelon Biosciences. IL-7, IL-4, and IL-15 were purchased from PeproTech. Acridine orange was purchased from Sigma. CytoID was purchased from Enzo Life Sciences. PIK75 was purchased from Cayman Chemical. iSHIP (AS1949490) and Dynasore were purchased from Tocris.
Flow Cytometry
Single cell suspensions with RBC’s lysed were incubated with FcR blocker (2.4G2; eBioscience) and were immunostained for all surface markers. For intracellular staining, cells were fixed in 4% PFA for 20 min, washed, and permeabilized in 0.1% saponin for 20 min. All stains were performed in 0.1% saponin thereafter. Acridine Orange and CytoID were used at 0.1 μg/mL for 20 min at RT just prior to FACS analysis. All FACS utilized a BD Facscanto II (BD).
Fluorescence Microscopy
All images were captured with a custom-built Zeiss Observer D1 using a Zeiss 100x objective lens and a 1.4 NA. Images were captured using a Photometrics CoolSNAP HQ2 and analyzed using Metamorph software for punctae number, size, and intensity. Images were deconvoluted and thresholded using Autoquant X2 software. Deconvolution was done blind at 40 iterations. LC3 punctate structures were defined as at least 10 pixels in size with fluorescence intensities of at least twice that of average background intensities. Flurochromes used included Pacific Blue, cy5, cy3, and FITC.
siRNA Knockdown
Clones were purchased from Dharmacon. Pools of siRNA clones for Inpp4A were GCCGAGAGGUUUGGCGAUA, CAUC AUAGGUUGCAUUUAA, GAUCGAAAGCCAAAUAGUU, and GAUGAGAGUUCAAGACGAU. Pools for Inpp4B were ACG AGAACAUUACGUGGUA, GAAGGAUUGUUAAGUACAU, A AUGAUGUAUUGCCAGUUA, and CGAUGAAAUUGGAAU GUUA. Pools for SHIP1 were CGACAGGGAUGAAGUACAA, GAAUUGCGUUUACAUUAC, GCAUUGCCCUUCGGUUAG A, and UGACAGCGACGAAUCCUAU. Pools for SHIP2 were UCAAGGAGCUUACGGAUCU, GUCAGUACGUCCAGUGU GA, CCAAGAAAGGGCUCUCAAA, and GCACACGUAUCGC AUUCUG. siRNA clones were electroporated into freshly isolated human peripheral blood mononuclear cells (hPBMC’s) and allowed to recover for 3 days before treatments for AVO formation. Primers used to detect knockdown efficiency included TAAGCTGAGGAACTGCCTGCATGA and TGGA AGTGGCCTGAGTGACTTTGA for Inpp4A, GTGGCGGCAA CAATGATGGAGAAA and TACGCAAGTTCCTGAAGGAGCA CA for Inpp4B, TCGGGACAAATACGCCTACACCAA and TGGGAAGTGACTCCTGCCTCAAAT for SHIP1, and TAA GTCCCAGCGTGTCCAGAACAA and TTCCCATGTTCCAG GTGCCTATGA for SHIP2.
Statistical Analysis
Graph Pad Prism was used for statistical analysis and the differences between groups was calculated using Student’s t-test. p-values < 0.05 were considered statistically significant.
Results
Class I PI3K Activity Is Required for T Cell Starvation-Induced Autophagy
Since kinase activity of the class III PI3K, Vps34, is dispensable for autophagy induction in T cells (McLeod et al., 2011), if not the entire protein itself (Willinger and Flavell, 2012), we investigated what other classes of PI3K were required for starvation-induced autophagy. We measured acidic vesicular organelle (AVO) formation in naïve T cells from WT mice with acridine orange staining to screen for compounds of interest. Starved mouse CD4+ T cells had increased AVOs, while the addition of interleukin 7 (IL-7), 3-methyladenine (3MA), and the class I PI3K inhibitor, PIK75, all inhibited autophagy (Figure 1A). The use of PIK75 at 20 nM reflected an IC50 for PI3KIα, while 100 nM reflects an IC50 of PI3KIγ and δ. A 2 μM PIK75 inhibits all four class I isoforms. Atg3f/fLck-cre CD4+ T cells were used as a negative control, as they have severely impaired autophagy (Jia and He, 2011). The pan-PI3K inhibitor, 3MA, and 100 nM PIK75 had the greatest impact upon AVO formation (Figures 1A,B), suggesting that PI3Kγ and/or δ play important roles in starvation-induced autophagy in T cells.
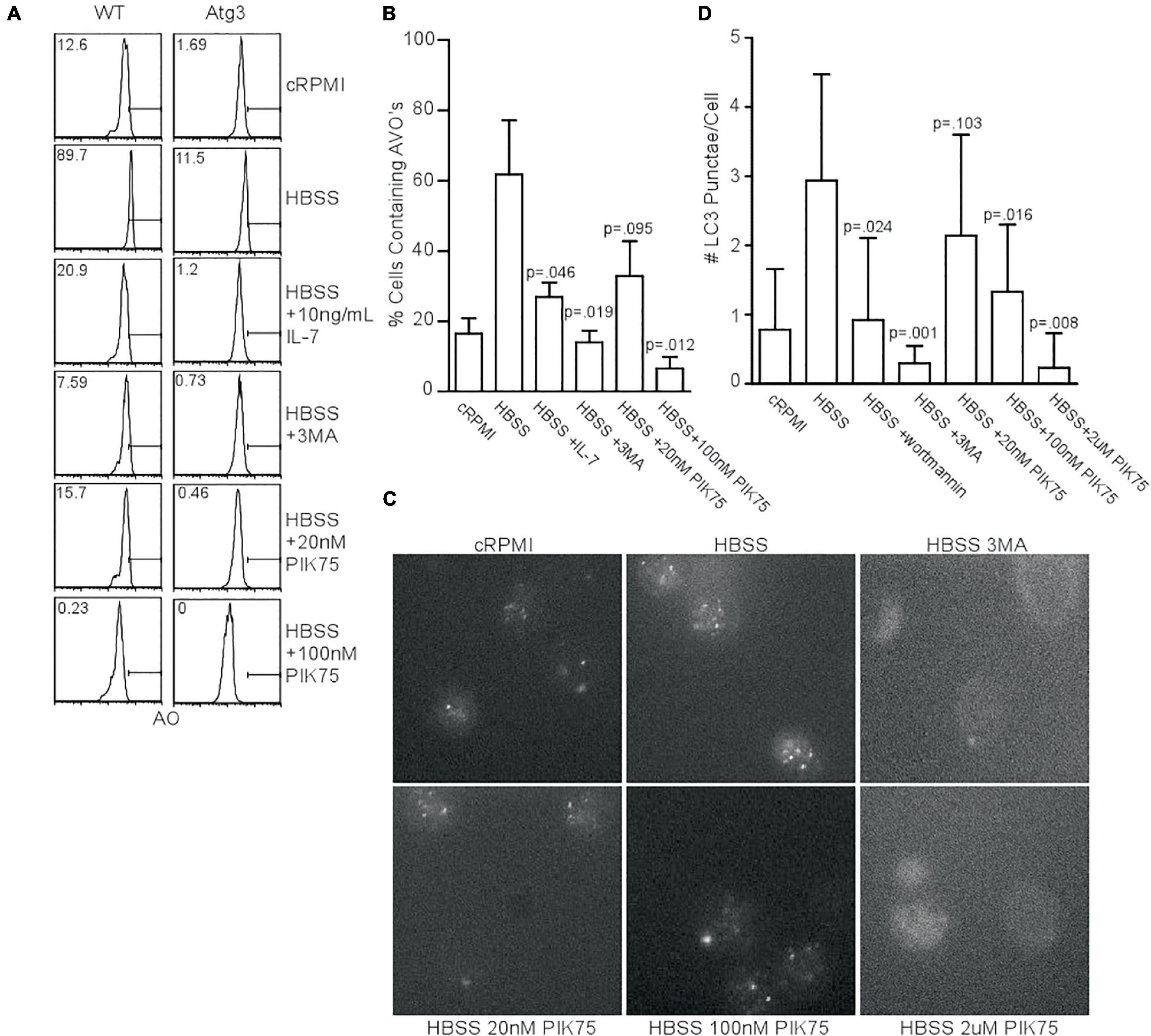
Figure 1. Class 1 PI3K activity is necessary for starvation-induced autophagy. (A) AVO profiles gated on live CD4 T lymphocytes starved for 48 h in HBSS and treated with the indicated activators inhibitors of PI3K. A 5mM 3MA and higher doses of PIK75 drastically inhibited AVO formation, while 20 nM PIK75 and 10 ng/mL IL-7 had slightly less potent effects. Atg3f/fLck-cre T cells were used as a negative control to assess background lysosomal contribution. Gates and numbers indicate the percentage of cells that have upregulated autophagy above basal levels. (B) Quantitation of (A), representative of at least 4 independent experiments per condition. (C) Fluorescence microscopy of endogenous LC3 punctate structures 48 h post starvation with the indicated inhibitors. (D) Quantitation of starvation-induced LC3 aggregates 48 h post starvation. Data are representative of at least 2 independent experiments per condition with at least 30 cells counted in each replicate.
We next sought to validate the AVO’s observed in Figure 1A were de facto autophagosomes using endogenous LC3II punctate formation as a measure of autophagy induction (McLeod et al., 2011). Starvation increased the number of LC3 structures, while 3MA, wortmannin, 100 nM PIK75, and 2 μM PIK75 reduced the number of LC3 structures (Figures 1C,D). A 20 nM PIK75 had a much smaller inhibitory effect on autophagy induction, suggesting that P110α is only minimally involved in starvation-induced autophagy in primary T cells (Figure 1D). To further investigate the role of P110α in autophagy induction, mouse T cells were induced to express a constitutively active form of P110α (P110caf/fER-cre). Forty eight hours h post-induction, CD4+ T cells had increased Akt activity, demonstrating a constitutively active PI3K pathway (Supplementary Figure 1A). Although P110ca expressing naïve T cells cultured in complete media had slightly increased levels of LC3+ punctae (Supplementary Figure 1B), no difference in AVO formation could be detected 48 h post-starvation (Supplementary Figures 1C,D), further suggesting that P110α is not the major isoform responsible for autophagy induction.
TCR-Mediated Autophagy Requires Class I PI3K
T cell receptor (TCR) signaling is a potent activator of autophagy in T cells (Pua et al., 2007; Botbol et al., 2016). TCR engagement is known to activate the class I PI3K pathway, suggesting a link between the processes. To investigate this, we activated splenocytes with anti-CD3 and anti-CD28 for 24 or 48 h in the presence of various PI3K inhibitors, and examined AVO induction using CytoID, a cationic amphiphilic tracer dye specific to autophagosomes (Kauntz et al., 2011). TCR stimulation induced AVO formation, which was inhibited by 100 nM and 2 μM PIK75, suggesting p110β, γ, or δ isoforms are potentially involved in TCR-induced autophagy (Figures 2A,B). However, p110 kinase activity results in the production of PI(3,4,5)P3, with very little PI(3)P (Auger et al., 1989). However, PI(3)P is required for autophagy (Dall’Armi et al., 2013). This suggests the involvement of other enzymes related to phosphatidylinositol phosphate production. Two different inositol phosphatase families, with linked activity, would be necessary to derive PI(3)P from PI(3,4,5)P3. A 5′ inositol phosphatase, such as SHIP, known to be involved in TCR signal quenching, could generate PI(3,4)P2 for PI(3)P production.
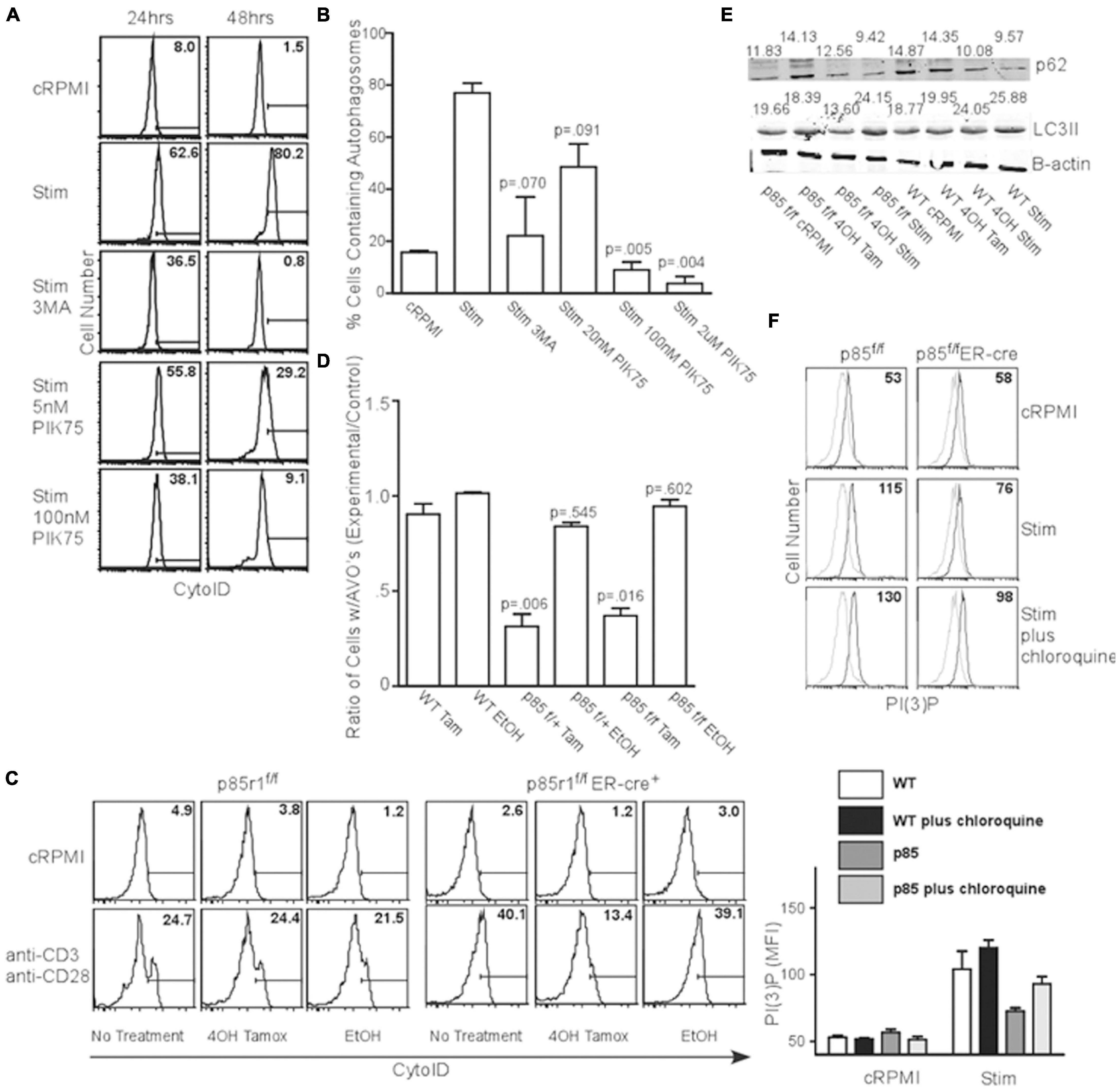
Figure 2. TCR-induced autophagy is dependent upon class I PI3K and 5′ phosphatase activity. (A) AVO formation histograms gated on live CD4 T lymphocytes stimulated with 1ug/mL soluble anti-CD3 and anti-CD28 for 24 and 48 h with the indicated reagents. A 5 mM 3MA, 100 nM PIK75, and 2 μM PIK75 potently reduced AVO formation, especially at 48 h. Histograms are representative of at least 3 independent experiments under each condition. (B) Quantitation of AVO formation from (A) summarizing at least 3 independent experiments per condition. (C) AVO formation in p85f/f and p85f/fER-cre CD4 T cells demonstrates the requirement for class I PI3K in TCR-mediated autophagy induction. Splenocyte and lymphocyte mixtures were cultured for 4 days in 1 ng/mL IL-7 and either 500 nM 4OH Tamoxifen or EtOH, and stimulated for 2 days with 1 μg/mL soluble anti-CD3 and anti-CD28 for 48 h. (D) Quantification of (C). Ratios of AVO formation of CD4 T cells pre-treated with 4OH Tamoxifen or EtOH to those kept in complete media for 96 h. Data are compiled from 3 independent experiments. (E) p62 degradation and LC3 lipidation are impaired in p85-deficient CD4 T cells. Cells were treated as in (C). Numbers indicate band densities compared to those of β-actin. (F) PI(3)P levels measured 24 h after activation through soluble anti-CD3 and anti-CD28 as in (C).
In order to fully inhibit class IA PI3K, we crossed mice with loxP sites flanking the p85 regulatory subunit of PI3KI (which also contains the p55 and p50 splice variants that act as regulatory subunits for most class IA PI3K) to an estrogen inducible cre recombinase to create p85f/fER-cre mice (Vooijs et al., 2001; Luo et al., 2005). Ninety six hours after deletion with 4-OH Tamoxifen, all the floxed allele was absent, leaving only the deleted allele (Supplementary Figure 1E). These mice are deficient in all three p85a isoforms including full length p85a and the truncated proteins p55a and p50a (Luo et al., 2005; Vanhaesebroeck et al., 2005). When the TCR of p85f/fER-cre or p85f/+ER-cre CD4+ T lymphocytes were stimulated, they showed a 60-80% impairment in AVO formation (Figures 2C,D). Additionally, stimulated p85f/fER-cre T cells had a 50% reduction in LC3II processing from undeleted controls (Figure 2E lower panel, quantified in Supplementary Figure 2D). p85 and p55 deletion almost completely inhibited autophagic flux, as assessed by the degradation of p62, after TCR stimulation, whereas WT T cells had a 40% degradative efficiency (Figure 2E upper panel, quantified in Supplementary Figure 2C). Furthermore, p85-deficient T cells had a production of PI(3)P that was only half of WT controls 24 h post TCR stimulation (Figure 2F), though the lysosomal turnover of PI(3)P was not impaired, as chloroquine enhanced PI(3)P levels in both WT and p85-deficient T cells (Figure 2F).
However, T cells were still able to be fully activated, both after treatment with various concentrations of PIK75, and in the absence of p85 as assessed by the upregulation of activation markers CD44 and CD69, as well as growth of T cells into blasts (Supplementary Figures 2A,B). Only treatment with 3MA and the highest concentration of PIK75 has any discernible effect on T cell activation (Supplementary Figure 2A). These results show PI3K kinase activity is required for TCR-induced autophagy.
Common γc Cytokines Inhibit Autophagy Induction
Since the addition of IL-7 was able to prevent AVO formation (Figure 1A), despite the ability of the γc chain to activate the PI3K pathway, we sought to determine the capacity of other homeostatic T cell cytokines to inhibit autophagy. IL-4 and IL-7 both had an inhibitory effect on AVO formation in CD4+ T cells, while IL-15 had a reduced potency (Figure 3A). The low inhibitory potential of IL15 compared to other cytokines could be due to low expression of its receptor CD122 on CD4+ T cells which is critical for binding and signaling of IL15 (Zhang et al., 1998; Keller et al., 2020). The capacity of IL-4, IL-7, and IL-15 to regulate T cell autophagy was confirmed using LC3 punctate formation (Figures 3B,C). Each cytokine was able to significantly reduce autophagy in naïve T lymphocytes. IL-4 and other Th2 cytokines have been previously shown to reduce autophagosomal delivery of M. tuberculosis to lysosomal compartments in macrophages (Harris et al., 2007). However, this paradigm can be extended to other homeostatic T cell cytokines. Though PI(3,4,5)P3 is generated during γc signaling and during TCR activation, the progression of autophagy from these two signals is remarkably different. This would suggest that additional factors are involved in mediating an autophagic response downstream of PI3K.
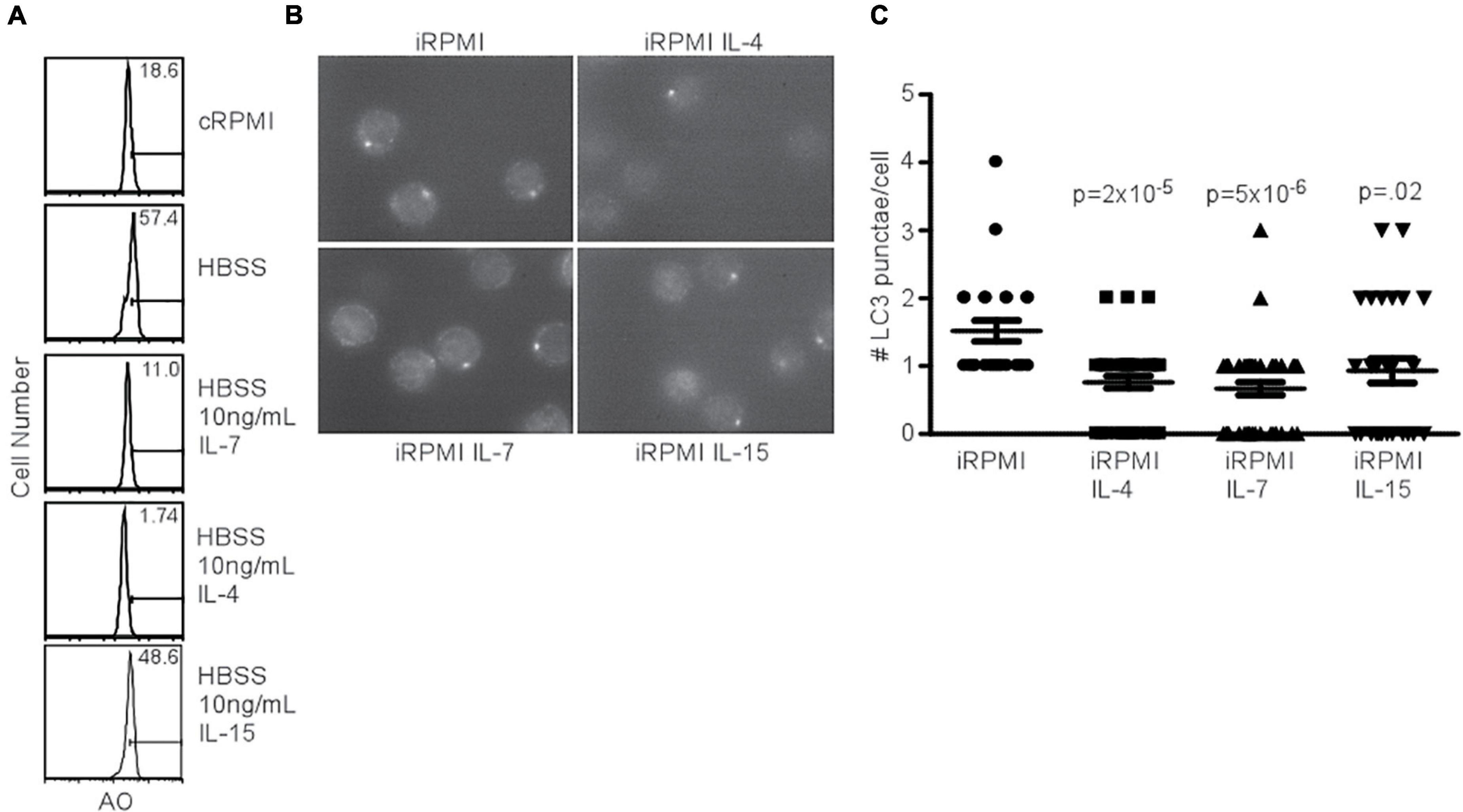
Figure 3. Common gamma chain cytokines inhibit starvation-induced autophagy. (A) AVO profiles of naïve CD4 T lymphocytes starved in HBSS for 48 h with the indicated cytokines. Histograms are representative of 3 independent experiments. IL-4 and IL-7 had potent effects on starvation-induced AVO formation, while IL-15 had a less drastic effect. (B) Fluorescence microscopy of LC3 aggregates in naïve, starved CD4 T lymphocytes treated with the indicated γc cytokines for 48 h, permeabilized, and stained with anti-LC3. Data are representative of fields observed. (C) Quantitation of (B), with at least 30 cells counted for each condition in two independent experiments. The indicated cytokines significantly reduced LC3 punctate formation. P-values are from unpaired, two-tailed Student’s t-tests.
Role of TCR Endocytosis in TCR-Induced Autophagy
Since p85 is required for the induction of autophagy in T lymphocytes, we hypothesized that the production of PI(3)P occurs on endomembranes after the internalization of the TCR complex. Subsequently, early endosomes are highly enriched for PI(3)P, and Vps34 is localized to early endosomes in T cells as well (McLeod et al., 2011). Utilizing dynasore to inhibit dynamin function, including the internalization of TCR complexes after TCR stimulation (Chaturvedi et al., 2011), AVO formation was examined. T cells treated with dynamin failed to upregulate AVO’s and had an almost complete block in TCR downregulation from the cell surface (Figures 4A,B), whereas rapamycin treated cells efficiently upregulated AVOs with no consequent TCR internalization, and 3MA treated cells had a block in AVO formation, but complete TCR internalization (Figures 4A,B). Hence, the signaling events required for TCR-induced autophagy induction occur post-internalization of the TCR complex on signaling endomembranes.
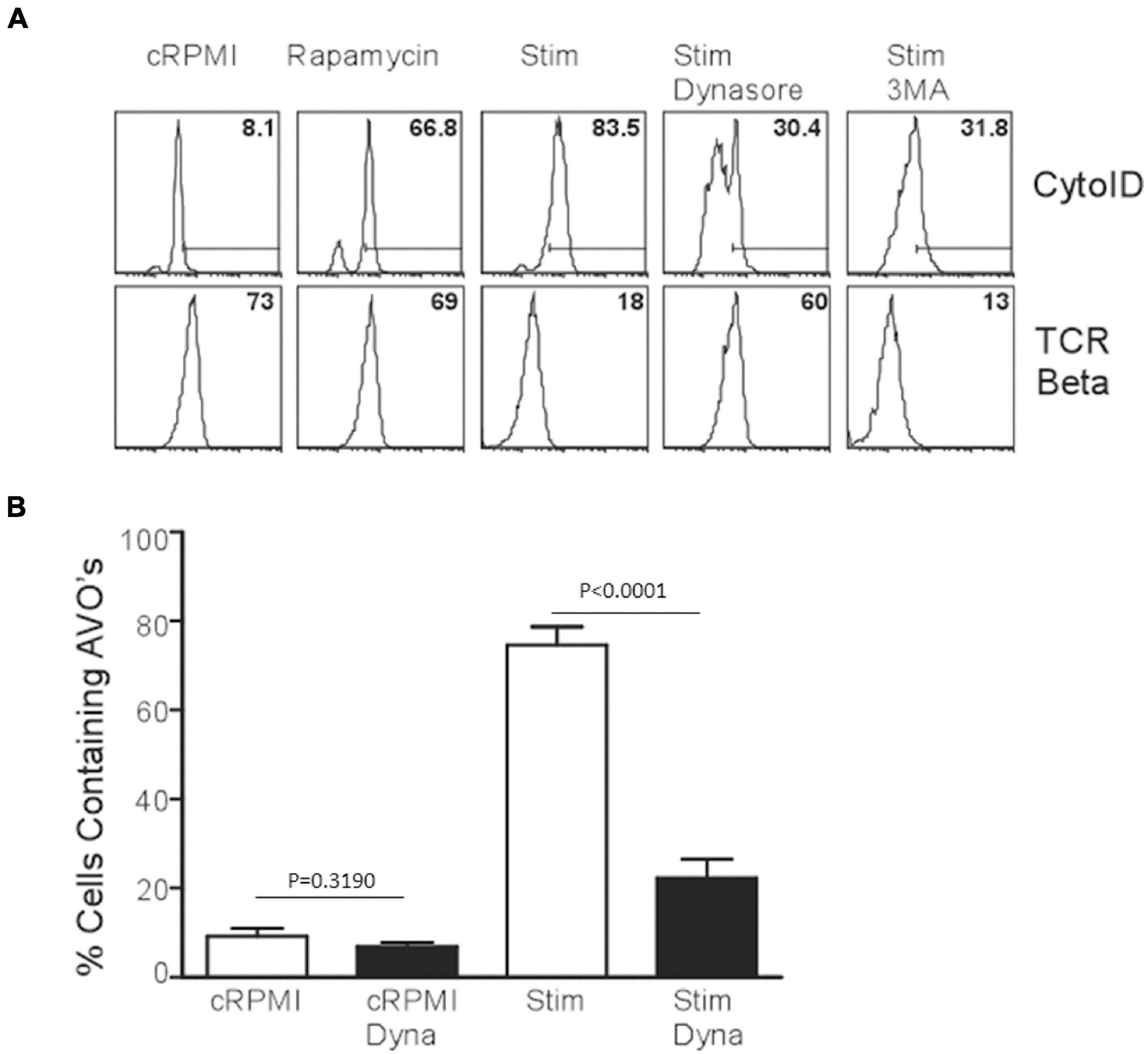
Figure 4. TCR internalization is required for TCR-mediated autophagy induction. (A) AVO formation in CD4 T cells pre-treated with 100 μM Dynasore or 5 mM 3MA for 1 h and stimulated for 24 h with 1 μg/mL soluble anti-CD3 and anti-CD28. Lower panel indicates the relative levels of TCR-β on the cell surface to demonstrate the efficacy of Dynasore versus other treatments. (B) Quantification of AVO formation in (A), data represents 3 independent experiments.
SHIP 5′ Phosphatase Activity Is Required for TCR-Induced Autophagy
As PI(3)P could be derived from PI(3,4,5)P3 catalyzed by SHIP in T cells, we tested the effect of pharmacological inhibition of SHIP on autophagy induction in T cells. The compound AS1949490 (Suwa et al., 2009), abbreviated iSHIP here, displayed inhibition of TCR-induced autophagy. We further sought to dissect the SHIP pathway by investigating the activity of both SHIP1 and SHIP2. Using 500 nM iSHIP, selectively inhibiting SHIP2 activity, and 15 μM iSHIP, inhibiting both SHIP2 and SHIP1, TCR-mediated AVO formation was measured. Although 500 nM iSHIP had only a minor impact on AVO formation, 15 μM almost completely inhibited any AVO upregulation (Figures 5A,B).
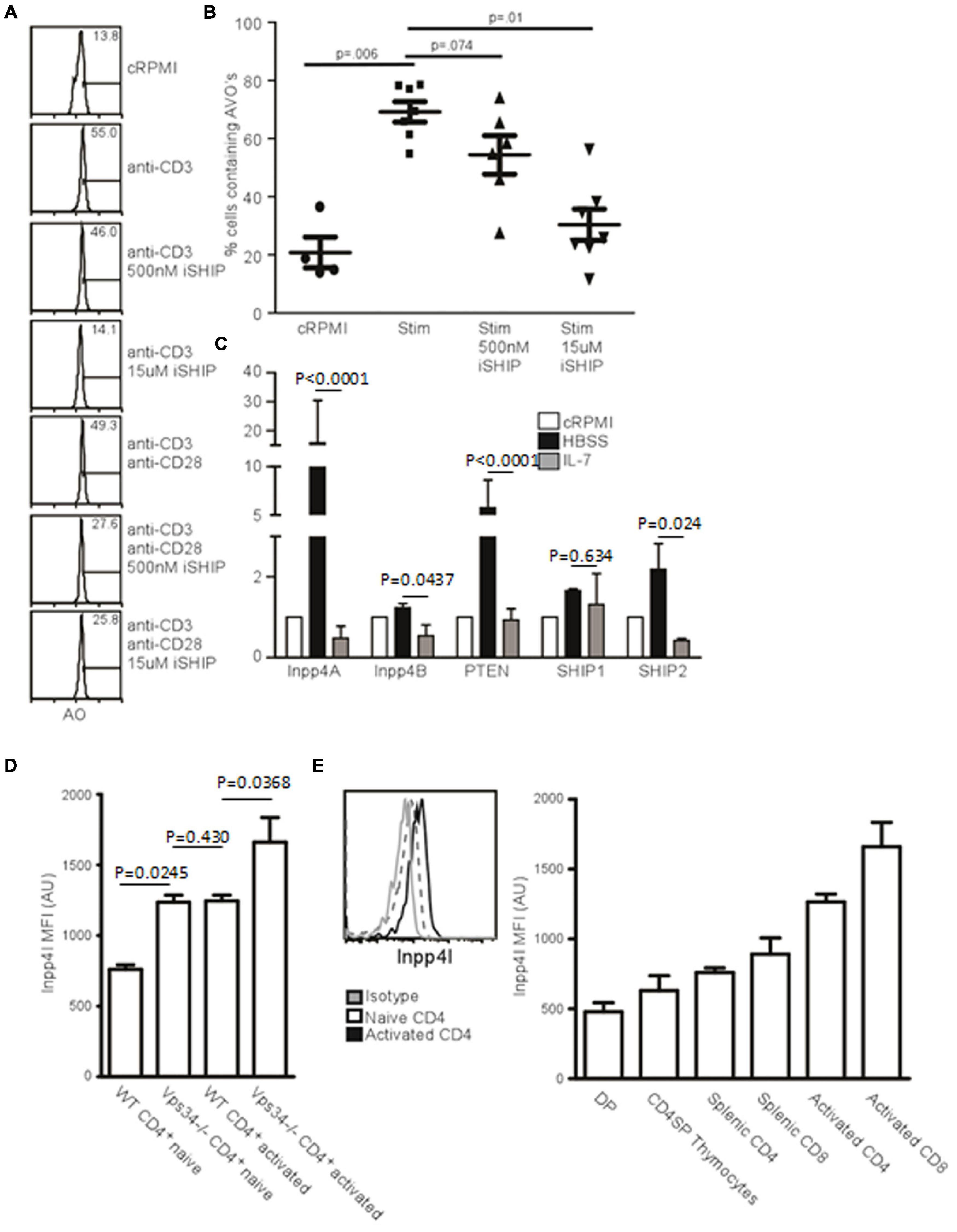
Figure 5. Inositol phosphatases respond to autophagic stimuli. (A) AVO formation in CD4 T lymphocytes pre-treated with 500 nM or 15 uM Anchor (AS1949490) for 1 h and stimulated with either 1 μg/mL soluble anti-CD3 alone or with anti-CD28 for 24 h. 500 nM Anchor inhibits SHIP2 activity and 15 μM inhibits both SHIP1 and SHIP2. (B) Quantification of AVO’s from (A). p-values are from paired, two tailed, Student’s t-tests from 6 replicates in 3 independent experiments. (C) Relative levels of inositol phosphatase mRNA in CD4 T cells 48 h after starvation in HBSS or with 10 ng/mL IL-7. QPCR results were normalized to that of GAPDH. Data are representative of 3 independent experiments with 3 replicates per experiment. (D) Histogram of Inpp4A protein level in naïve or 24 h stimulated CD4 T cells, quantified in the right panel as 4 replicates from 3 independent experiments. Gray histogram represents the isotype control, the dotted histogram is the naïve level, and black histogram is the level in TCR activated cells. (E) Intracellular stains for Inpp4A protein levels in WT or Vps34f/fLck-cre CD4 T cells (Vps34-/-). Data are from 3 replicates in 2 independent experiments.
Inpp4 Levels Are Regulated by Autophagic Stimuli
If SHIP-mediated hydrolysis of PI(3,4,5)P3 is required for the production of PI(3,4)P, a 4′ phosphatase activity would be required for the production of PI(3)P from PI(3,4)P2. Two 4′ phosphatases have been described, Inpp4A and Inpp4B (Dyson et al., 2012). We used quantitative PCR to measure how autophagic stimuli affect the levels of 4’ inositol phosphatases in T cells. IL-7 treatment decreased the levels of Inpp4A and Inpp4B, as well as the levels of SHIP1 and SHIP2 (Figure 5C). However, starvation had the opposite effect, with levels of Inpp4A increasing dramatically (Figure 5C). While Inpp4A is expressed at low levels by naïve T cells, TCR stimulation increases the level of Inpp4A twofold (Figure 5D). Additionally, naïve Vps34f/fLck-cre T cells have increased Inpp4A and that level further increases upon TCR stimulation (Figure 5E). This could be a compensatory mechanism that allows further processing of PI(3,4)P2 into PI(3)P in the absence of Vps34. Hence the levels of various polyinositol phosphatases are positively regulated by autophagic stimulators and are negatively regulated by IL-7. This could account for the difference seen in the autophagic response between IL-7 treatment and TCR stimulation of T lymphocytes.
PI(3,4)P2 Can Be Hydrolyzed to PI(3)P, Accelerating Autophagy
If PI(3,4)P2 is a relevant intermediate in TCR-mediated autophagy, the addition of exogenous phospholipids should impact the induction and progression of autophagy. To test this, we added PI(3,4)P2 to T cell cultures and measured both PI(3)P production and AVO formation. Naïve CD4+ T cells have very little PI(3)P and the addition of PI(3,4)P2 only slightly increased the amount of PI(3)P, despite the efficient loading of cells with PI(3,4)P2 (Figures 6A,B). However, TCR stimulation caused cells loaded with PI(3,4)P2 to produce twice the amount of PI(3)P and more efficiently hydrolyze PI(3,4)P2 (Figures 6A,B). This had a major impact on autophagic progression. Although by 24 h, AVO formation was only slightly enhanced (Figure 6A), 120 h later AVO’s had been completely turned over in stimulated T cells loaded with PI(3,4)P2, while TCR-stimulated cells in the absence of exogenous lipid were still undergoing some autophagy (Figure 6A right panels). Naïve T cells started to undergo autophagy after a few days, despite being kept in complete media culture conditions, possibly due to a stress response (Figure 6A). Consistent with our model, PI(3,4)P2 loaded naïve T cells did not upregulate autophagy over unloaded controls, since no upregulation of inositol phosphatases occurred (Figure 5C).
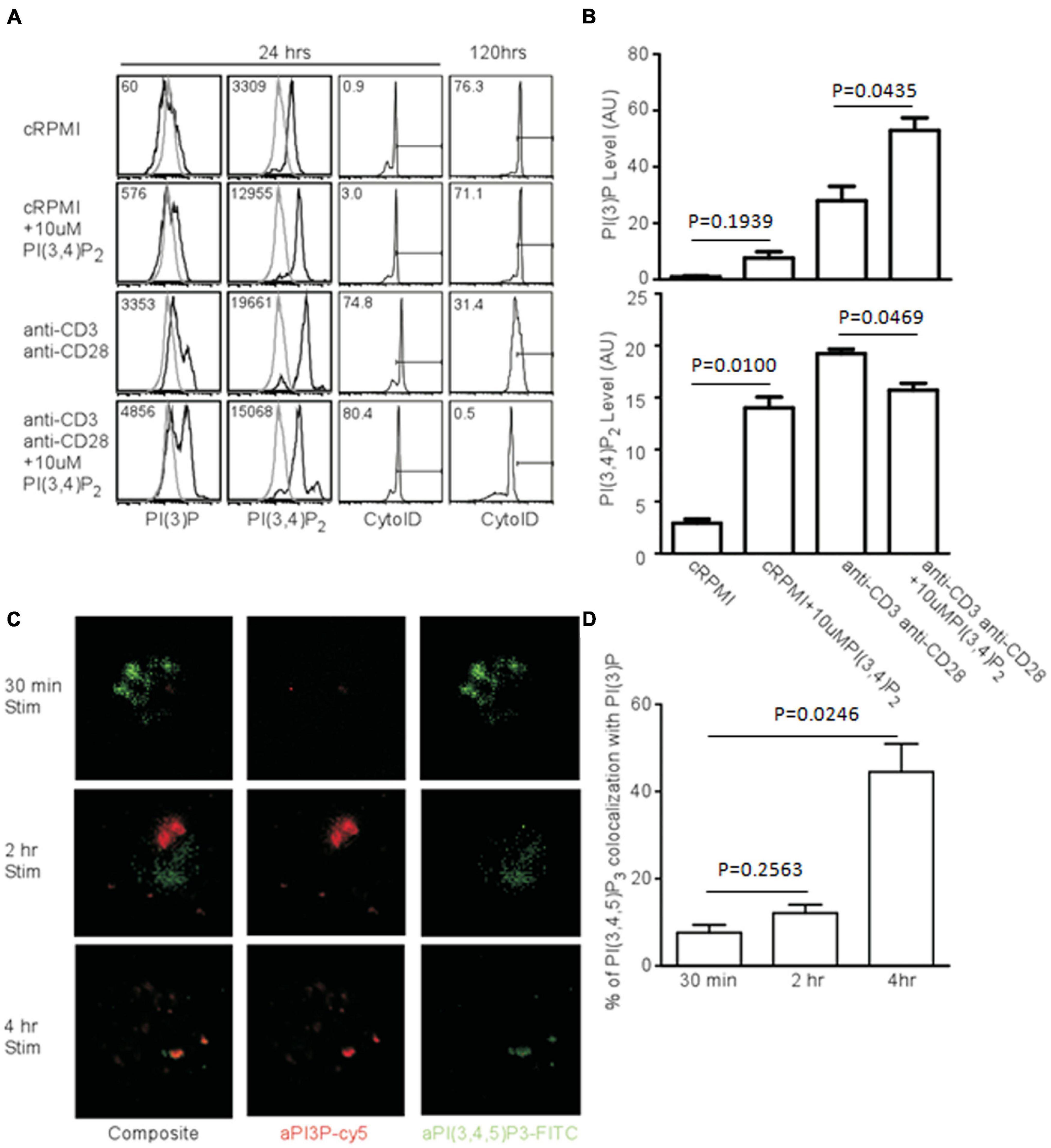
Figure 6. Exogenous PI(3,4)P2 alters the autophagic profile and is converted to PI(3)P. (A, left panel) Histograms of relative levels of PI(3)P and PI(3,4)P2 CD4 T cells were loaded with 10 μM PI(3,4)P2 and stimulated with 1 μg/mL soluble anti-CD3 and anti-CD28 for 24 and 120 h. PI(3,4)P2 is largely converted to PI(3)P only after TCR stimulation. (A, right panel) AVO formation of CD4 T cells loaded with PI(3,4)P2. Exogenous PI(3,4)P2 effects an accelerated autophagic profile that is resolved by 120 h compared to unloaded controls. (B) Quantification of phosphatidyl inositol levels from CD4 T cells left untreated or loaded with PI(3,4)P2 and stimulated for 24 h as in (A). Data represent 4 replicates from 3 independent experiments. (C) Fluorescence microscopy of phosphatidylinositol species in T cells activated with plate-bound anti-CD3 and anti-CD28 for the indicated time periods. (D) Quantitation of colocalization of phosphatidylinositol species from (C).
PI(3)P Is Produced in PI(3,4,5)P3 Compartments
Since PI(3)P is produced from PI(3,4,5)P3 after TCR stimulation, this process might occur at distinct sites. To visualize this process, we stimulated naïve T cells. After 30 min, there was a huge production of PI(3,4,5)P3, but very little PI(3)P, with little compartmental overlap between the two species at 30 min and 2 h (Figures 6C,D). However, by 4 h, much of the PI(3,4,5)P3 had disappeared, and a significant proportion of what remained was bound up in PI(3)P-bearing vesicles, which were quite prominent at this time point (Figures 6C,D). Thus, PI(3)P accumulates in activated T cells contemporaneously with the quenching of PI(3,4,5)P3 in vesicles containing both phospholipids at a time point prior to the onset of autophagy.
Human T Cells Undergo Autophagy Regulated by Class I PI3K and Inositol Phosphatases
HIV glycoprotein binding to CXCR4 and CD4 on human T lymphocytes has been proposed as a major mechanism by which uninfected, bystander cells die through autophagy, suggesting that CXCR4-mediated signaling induces autophagy in T cells (Espert et al., 2006; Espert et al., 2008). We first examined the effect of PI3K inhibitors on TCR-stimulation induced autophagy in human T cells. TCR stimulation-induced autophagy in human T cells was inhibited by 3MA, PIK75, and iSHIP (Figure 7A). To verify autophagy was initiated, we measured LC3 punctate formation in human T cells 48 h after TCR stimulation, and confirmed 100 nM PIK75 and iSHIP were able to ablate autophagy (Figures 7B,C). Furthermore, when either Inpp4A or Inpp4B were knocked down with siRNA cocktails, or when both SHIP1 and SHIP2 were knocked down (Supplementary Figures 3A,B), AVO formation via TCR stimulation was severely impaired (Figures 7D,E). SHIP1 and SHIP2 seemed to have some redundancy during this process, while Inpp4A and Inpp4B did not. However, the loss of both Inpp4 isoforms had no synergistic effect, suggesting they may function within the same complex. Thus, TCR-induced autophagy in human CD4 T cells is dependent on the coordinated activity of class I PI3K and inositol phosphatases.
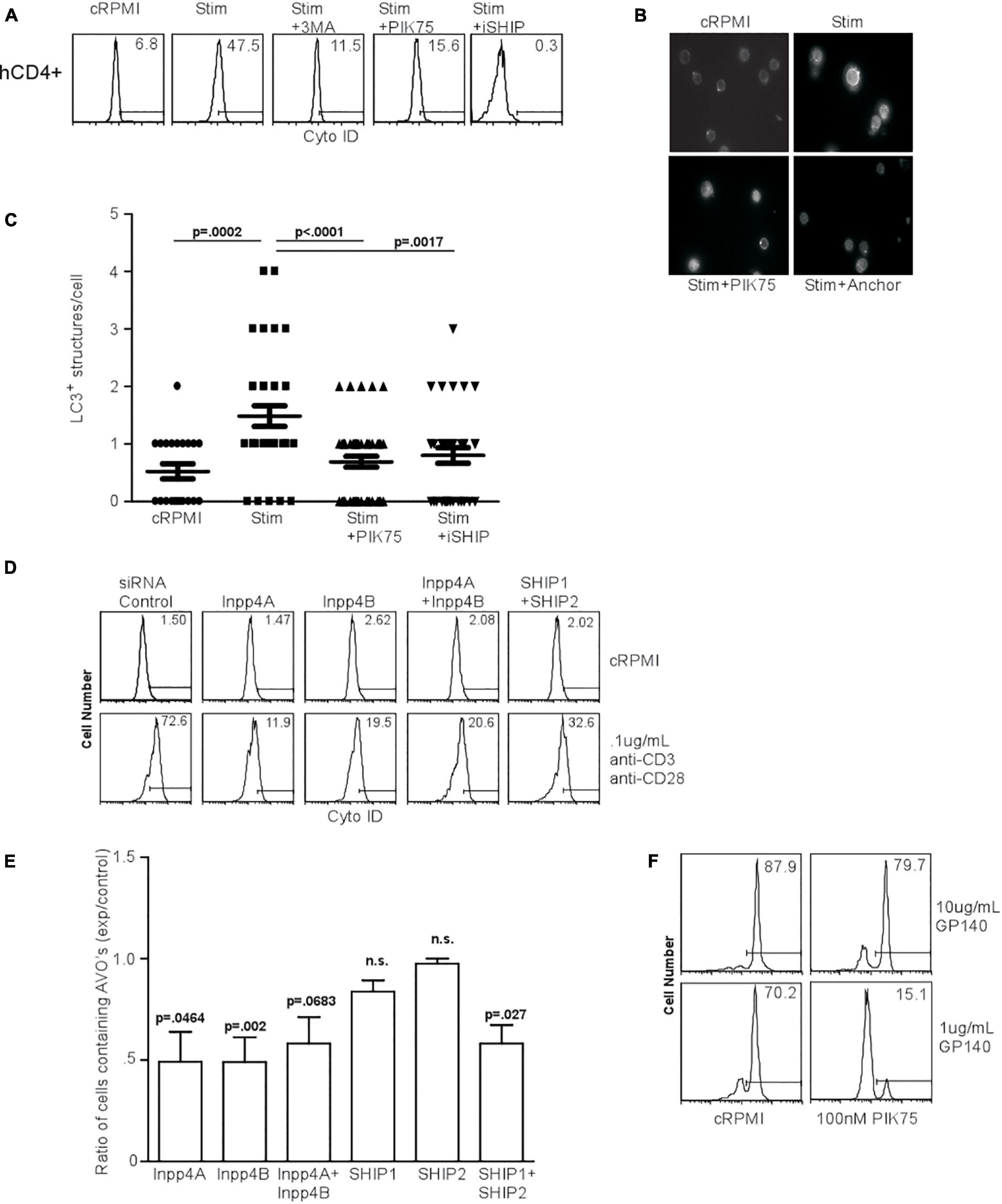
Figure 7. Human T cell autophagy is dependent upon class I PI3K activity and inositol phosphatase activity. (A) AVO formation in human lymphocytes. Human CD4+ T cells were isolated from healthy donors, purified by Ficol gradient centrifugation, and stimulated with 0.5 μg/mL anti-CD3 and anti-CD28 for 24 h. AVO formation was inhibited by 100 nM PIK75, 3MA, and 15 μM Anchor. Data are representative of at least 3 independent experiments per condition. (B) LC3 punctate formation in stimulated human CD4+ T cells for 48 h as in (A), permeabilized, and stained for endogenous LC3. (C) Quantification of (B), representative of two replicates with at least 30 CD4 cells counted per condition. (D) AVO formation in siRNA mediated knockdown of Inpp4A, Inpp4B, and both SHIP1 and SHIP2. Human CD4+ T cells were purified as in (A) and electroporated to allow uptake of 5 μg/5 × 106 cells siRNA cocktails, allowed to recover in complete media for 72 h and stimulated with 0.5 μg/mL anti-CD3 and anti-CD28 for 48 h. (E) Quantification of stimulated CD4 T cells in (D), comparing the percentage of cells upregulating AVO formation in the siRNA knockdown samples to a scrambled control siRNA. Data are representative of cells from 5 donors in 4 independent experiments. (F) AVO formation in human CD4+ T cells exposed to soluble HIV GP140 fusion protein at the indicated concentrations. Total PBMC’s were treated with the glycoprotein and 100 nM PIK75 for 48 h.
HIV Glycoprotein-Induced Autophagy Is Mediated by PI3K
The HIV glycoproteins GP40 and GP120 can bind to CXCR4 on uninfected T lymphocytes and induce autophagy (Espert et al., 2006). CXCR4 is a G-protein coupled receptor (GPCR), the downstream signaling of which often occurs through the class Ib PI3K, PI3Kγ (Suzuki et al., 2001). To test whether autophagy downstream of HIV glycoprotein binding was dependent on PI3K, we subjected human CD4 T cells to a soluble fusion protein consisting of the exodomains of GP41 and GP120, termed GP140. Co-culture of CD4 T cells with GP140 induced a high level of autophagy, which was sensitive to 100 nM PIK75 treatment, suggesting that HIV glycoprotein autophagy induction uses the same machinery as TCR-mediated autophagy induction (Figures 2, 7F).
Discussion
The role of phosphatidyl-3 kinases has long been appreciated in pro-survival signaling (Shanware et al., 2013). Even as autophagy has vaulted to the forefront as a pro-survival mechanism, many aspects of its regulation remain unknown. Through yeast studies implicating Vps34 as the main proponent of PI(3)P production, and the use of various PI3K inhibitors, which we now know are non-specific, Vps34 has been assumed to be the main producer of cellular PI(3)P. However, studies from our lab showing Vps34 kinase activity is dispensable for autophagy induction in T lymphocytes, suggest that other PI3K classes important. The reduction in autophagy observed using specific PI3K inhibitors, and a knockout of p85/p55 in T cells, demonstrate that class I PI3K is the major player in autophagic activity. This was confirmed by observing autophagic flux in p85-deficient T cells. The specific isoform of PI3KI utilized by the cell is most likely dependent upon the nature of the autophagic stimulus. This could help explain the different degrees of inhibition observed when class Iα was pharmacologically deactivated with 20 nM PIK75 between starvation- and TCR-induced autophagy. Furthermore, we have not ruled out contributions of class II PI3K or class 1b PI3K on autophagy induction, as even the p85/p55/p50 knockout T cells still had a nominal level of autophagy.
Our study also investigated the requirement of inositol phosphatases to translate TCR signaling into autophagy. Using iSHIP, or by knocking down Inpp4 or SHIP, we demonstrated the necessity of these proteins to sustain PI(3)P production. Interestingly, both SHIP1 and SHIP2 had to be inhibited or removed before a significant reduction in autophagy could be observed, suggesting these proteins can compensate for one another. However, Inpp4A and Inpp4B could not compensate for one another, and knockdown of either one was sufficient to reduce autophagy. However, knockdown of both had no synergistic effect on AVO formation. This suggests that they operate within a single complex or pathway. The significance of the Inpp4 complex to participate in TCR-mediated autophagy was further confirmed by the ability of T cells to convert exogenous PI(3,4)P2 into PI(3)P, only after TCR stimulation, leading to a more rapidly resolved autophagy. This correlates very well with the upregulation of Inpp4A after TCR stimulation.
Indeed, the levels of all the polyinositol phosphatases correlated very well with autophagy. Starvation leads to the upregulation of Inpp4A, Inpp4B, SHIP1, and SHIP2, while IL-7 treatment leads to repression of all these phosphatases. The regulation of Inpp4A and SHIP2 are especially dynamic, suggesting that they might be the limiting factors in substrate conversion. It is the levels of these inositol phosphatases that we believe to account for the difference in autophagy observed in TCR-stimulated versus IL-7 treated T cells, despite the production in PI(3,4,5)P3 in both cases. This is important because pro-survival cues such as TCR stimulation and IL-7 treatment need to be quenched to avoid pathogenic T cell activation and proliferation. The activities of inositol phosphatases ensure that higher ordered species of inositol phosphates are not only removed, but also converted into intermediates for alternative pro-survival pathways such as autophagy. This paradigm goes beyond PI(3)P production for PI(3,4,5)P3, and can be applied to any membranes that need to be converted into another type of organelle membrane with an alternative phosphatidyl inositol coat (Botelho, 2009). We wish to stress that PI(3)P is a complex signal governing many functions. Though this signal is very low in naïve T cells (Figure 6A), TCR signaling produces much PI(3)P, of which only a fraction is ever bound for an autophagosome (Figures 6C,B). Much of this signal is undoubtedly used to degrade signaling molecules, or to assist with the immense intracellular organization that occurs during the T cell response.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by the Duke University IACUC.
Author Contributions
IM designed experiments, wrote the manuscript, crossed the mice, and analyzed the data. ZC performed experiments and analyzed the data. RS performed the experiments and wrote the manuscript. Y-WH designed the experiments, analyzed the data, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the NIH grants AI074944 and AI125552 to Y-WH.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.709398/full#supplementary-material
Abbreviations
AVO, Acidic vesicular organelle; Inpp4, inositol polyphosphate 4-phosphatase; LC3, Microtubule associated protein light chain 3; PI3K, Phosphatidylinositol 3 Kinase; PI(3)P, Phosphatidylinositol 3-phosphate; SHIP, SH2 domain-containing inositol 5′-phosphatase; TCR, T cell receptor.
References
Aquila, S., Santoro, M., Caputo, A., Panno, M. L., Pezzi, V., and De Amicis, F. (2020). The tumor suppressor pten as molecular switch node regulating cell metabolism and autophagy: implications in immune system and tumor microenvironment. Cells 9:1725. doi: 10.3390/cells9071725
Auger, K. R., Serunian, L. A., Soltoff, S. P., Libby, P., and Cantley, L. C. (1989). PDGF-dependent tyrosine phosphorylation stimulates production of novel polyphosphoinositides in intact cells. Cell 57, 167–175. doi: 10.1016/0092-8674(89)90182-7
Axe, E. L., Walker, S. A., Manifava, M., Chandra, P., Roderick, H. L., Habermann, A., et al. (2008). Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 182, 685–701. doi: 10.1083/jcb.200803137
Bento, C. F., Renna, M., Ghislat, G., Puri, C., Ashkenazi, A., Vicinanza, M., et al. (2016). Mammalian autophagy: how does it work? Annu. Rev. Biochem. 85, 685–713. doi: 10.1146/annurev-biochem-060815-014556
Bilanges, B., Posor, Y., and Vanhaesebroeck, B. (2019). PI3K isoforms in cell signalling and vesicle trafficking. Nat. Rev. Mol. Cell Biol. 20, 515–534. doi: 10.1038/s41580-019-0129-z
Botbol, Y., Guerrero-Ros, I., and Macian, F. (2016). Key roles of autophagy in regulating T-cell function. Eur. J. Immunol. 46, 1326–1334. doi: 10.1002/eji.201545955
Botelho, R. J. (2009). Changing phosphoinositides “on the fly”: how trafficking vesicles avoid an identity crisis. Bioessays 31, 1127–1136. doi: 10.1002/bies.200900060
Bronietzki, A. W., Schuster, M., and Schmitz, I. (2015). Autophagy in T-cell development, activation and differentiation. Immunol. Cell Biol. 93, 25–34. doi: 10.1038/icb.2014.81
Buckler, J. L., Walsh, P. T., Porrett, P. M., Choi, Y., and Turka, L. A. (2006). Cutting edge: T cell requirement for CD28 costimulation is due to negative regulation of TCR signals by PTEN. J. Immunol. 177, 4262–4266. doi: 10.4049/jimmunol.177.7.4262
Chaturvedi, A., Martz, R., Dorward, D., Waisberg, M., and Pierce, S. K. (2011). Endocytosed BCRs sequentially regulate MAPK and Akt signaling pathways from intracellular compartments. Nat. Immunol. 12, 1119–1126. doi: 10.1038/ni.2116ni.2116
Cui, B., Lin, H., Yu, J., Yu, J., and Hu, Z. (2019). Autophagy and the immune response. Adv. Exp. Med. Biol. 1206, 595–634. doi: 10.1007/978-981-15-0602-4_27
Dall’Armi, C., Devereaux, K. A., and Di Paolo, G. (2013). The role of lipids in the control of autophagy. Curr. Biol. 23, R33–R45. doi: 10.1016/j.cub.2012.10.041
Deter, R. L. (1975). Analog modeling of glucagon-induced autophagy in rat liver. II. Evaluation of iron labeling as a means for identifying telolysosome, autophagosome and autolysosome populations. Exp. Cell Res. 94, 127–139. doi: 10.1016/0014-4827(75)90539-x
Dou, Z., Chattopadhyay, M., Pan, J. A., Guerriero, J. L., Jiang, Y. P., Ballou, L. M., et al. (2010). The class IA phosphatidylinositol 3-kinase p110-beta subunit is a positive regulator of autophagy. J. Cell Biol. 191, 827–843. doi: 10.1083/jcb.201006056
Dou, Z., Pan, J. A., Dbouk, H. A., Ballou, L. M., DeLeon, J. L., Fan, Y., et al. (2013). Class IA PI3K p110β subunit promotes autophagy through Rab5 small GTPase in response to growth factor limitation. Mol. Cell 50, 29–42. doi: 10.1016/j.molcel.2013.01.022
Dyson, J. M., Fedele, C. G., Davies, E. M., Becanovic, J., and Mitchell, C. A. (2012). Phosphoinositide phosphatases: just as important as the kinases. Subcell. Biochem. 58, 215–279. doi: 10.1007/978-94-007-3012-0_7
Espert, L., Codogno, P., and Biard-Piechaczyk, M. (2008). What is the role of autophagy in HIV-1 infection? Autophagy 4, 273–275. doi: 10.4161/auto.5211
Espert, L., Denizot, M., Grimaldi, M., Robert-Hebmann, V., Gay, B., Varbanov, M., et al. (2006). Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J. Clin. Invest. 116, 2161–2172. doi: 10.1172/JCI26185
Galle-Treger, L., Hurrell, B. P., Lewis, G., Howard, E., Jahani, P. S., Banie, H., et al. (2020). Autophagy is critical for group 2 innate lymphoid cell metabolic homeostasis and effector function. J. Allergy Clin. Immunol. 145, 502–517. doi: 10.1016/j.jaci.2019.10.035
Harris, J., De Haro, S. A., Master, S. S., Keane, J., Roberts, E. A., Delgado, M., et al. (2007). T helper 2 cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity 27, 505–517. doi: 10.1016/j.immuni.2007.07.022
Hayashi, S., and McMahon, A. P. (2002). Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev. Biol. 244, 305–318. doi: 10.1006/dbio.2002.0597
Ikeda, H., Hideshima, T., Fulciniti, M., Perrone, G., Miura, N., Yasui, H., et al. (2010). PI3K/p110{delta} is a novel therapeutic target in multiple myeloma. Blood 116, 1460–1468.
Jackson, S. P., Schoenwaelder, S. M., Matzaris, M., Brown, S., and Mitchell, C. A. (1995). Phosphatidylinositol 3,4,5-trisphosphate is a substrate for the 75 kDa inositol polyphosphate 5-phosphatase and a novel 5-phosphatase which forms a complex with the p85/p110 form of phosphoinositide 3-kinase. EMBO J. 14, 4490–4500. doi: 10.1002/j.1460-2075.1995.tb00128.x
Jia, W., and He, Y. W. (2011). Temporal regulation of intracellular organelle homeostasis in T lymphocytes by autophagy. J. Immunol. 186, 5313–5322. doi: 10.4049/jimmunol.1002404
Jia, W., Pua, H. H., Li, Q. J., and He, Y. W. (2011). Autophagy regulates endoplasmic reticulum homeostasis and calcium mobilization in T lymphocytes. J. Immunol. 186, 1564–1574. doi: 10.4049/jimmunol.1001822
Kauntz, H., Bousserouel, S., Gosse, F., and Raul, F. (2011). Silibinin triggers apoptotic signaling pathways and autophagic survival response in human colon adenocarcinoma cells and their derived metastatic cells. Apoptosis 16, 1042–1053. doi: 10.1007/s10495-011-0631-z
Keller, H. R., Kim, H. K., Jo, Y., Gress, R. E., Hong, C., and Park, J. H. (2020). The abundance and availability of cytokine receptor IL-2Rbeta (CD122) constrain the lymphopenia-induced homeostatic proliferation of naive CD4 T Cells. J. Immunol. 204, 3227–3235. doi: 10.4049/jimmunol.1901276
Li, C., Capan, E., Zhao, Y., Zhao, J., Stolz, D., Watkins, S. C., et al. (2006). Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J. Immunol. 177, 5163–5168. doi: 10.4049/jimmunol.177.8.5163
Luo, J., McMullen, J. R., Sobkiw, C. L., Zhang, L., Dorfman, A. L., Sherwood, M. C., et al. (2005). Class IA phosphoinositide 3-kinase regulates heart size and physiological cardiac hypertrophy. Mol. Cell Biol. 25, 9491–9502. doi: 10.1128/mcb.25.21.9491-9502.2005
McLeod, I. X., Zhou, X., Li, Q. J., Wang, F., and He, Y. W. (2011). The class III kinase Vps34 promotes T lymphocyte survival through regulating IL-7Ralpha surface expression. J. Immunol. 187, 5051–5061. doi: 10.4049/jimmunol.1100710
Miller, B. C., Zhao, Z., Stephenson, L. M., Cadwell, K., Pua, H. H., Lee, H. K., et al. (2008). The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy 4, 309–314.
Munday, A. D., Norris, F. A., Caldwell, K. K., Brown, S., Majerus, P. W., and Mitchell, C. A. (1999). The inositol polyphosphate 4-phosphatase forms a complex with phosphatidylinositol 3-kinase in human platelet cytosol. Proc. Natl. Acad. Sci. USA 96, 3640–3645. doi: 10.1073/pnas.96.7.3640
Münz, C. (2016). Autophagy proteins in antigen processing for presentation on MHC molecules. Immunol. Rev. 272, 17–27. doi: 10.1111/imr.12422
Nascimbeni, A. C., Codogno, P., and Morel, E. (2017). Phosphatidylinositol-3-phosphate in the regulation of autophagy membrane dynamics. Febs. J. 284, 1267–1278. doi: 10.1111/febs.13987
Nedjic, J., Aichinger, M., Emmerich, J., Mizushima, N., and Klein, L. (2008a). Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature 455, 396–400. doi: 10.1038/nature07208
Nedjic, J., Aichinger, M., and Klein, L. (2008b). A novel role for autophagy in T cell education. Autophagy 4, 1090–1092. doi: 10.4161/auto.7139
Ngoh, E. N., Brugger, H. K., Monajemi, M., Menzies, S. C., Hirschfeld, A. F., Del Bel, K. L., et al. (2015). The Crohn’s disease-associated polymorphism in ATG16L1 (rs2241880) reduces SHIP gene expression and activity in human subjects. Genes Immun. 16, 452–461. doi: 10.1038/gene.2015.30
Øynebråten, I. (2020). Involvement of autophagy in MHC class I antigen presentation. Scand. J. Immunol. 92:e12978. doi: 10.1111/sji.12978
Petiot, A., Ogier-Denis, E., Blommaart, E. F., Meijer, A. J., and Codogno, P. (2000). Distinct classes of phosphatidylinositol 3’-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. 275, 992–998. doi: 10.1074/jbc.275.2.992
Pua, H. H., Dzhagalov, I., Chuck, M., Mizushima, N., and He, Y. W. (2007). A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J. Exp. Med. 204, 25–31. doi: 10.1084/jem.20061303
Pua, H. H., Guo, J., Komatsu, M., and He, Y. W. (2009). Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J. Immunol. 182, 4046–4055. doi: 10.4049/jimmunol.0801143
Schmid, D., and Munz, C. (2007). Innate and adaptive immunity through autophagy. Immunity 27, 11–21. doi: 10.1016/j.immuni.2007.07.004
Shanware, N. P., Bray, K., and Abraham, R. T. (2013). The PI3K, metabolic, and autophagy networks: interactive partners in cellular health and disease. Annu. Rev. Pharmacol. Toxicol. 53, 89–106. doi: 10.1146/annurev-pharmtox-010611-134717
Singh, A., Anang, V., Kumar Rana, A., Verma, C., Surender Kumar, Saraswati, S., et al. (2020). Deciphering the role of calcium homeostasis in T cells functions during mycobacterial infection. Cell Immunol. 357:104198. doi: 10.1016/j.cellimm.2020.104198
Srinivasan, L., Sasaki, Y., Calado, D. P., Zhang, B., Paik, J. H., DePinho, R. A., et al. (2009). PI3 kinase signals BCR-dependent mature B cell survival. Cell 139, 573–586. doi: 10.1016/j.cell.2009.08.041
Suwa, A., Yamamoto, T., Sawada, A., Minoura, K., Hosogai, N., Tahara, A., et al. (2009). Discovery and functional characterization of a novel small molecule inhibitor of the intracellular phosphatase. SHIP2. Br. J. Pharmacol. 158, 879–887. doi: 10.1111/j.1476-5381.2009.00358.x
Suzuki, Y., Rahman, M., and Mitsuya, H. (2001). Diverse transcriptional response of CD4(+) T cells to stromal cell-derived factor (SDF)-1: cell survival promotion and priming effects of SDF-1 on CD4(+) T cells. J. Immunol. 167, 3064–3073. doi: 10.4049/jimmunol.167.6.3064
Vanhaesebroeck, B., Ali, K., Bilancio, A., Geering, B., and Foukas, L. C. (2005). Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biochem. Sci. 30, 194–204. doi: 10.1016/j.tibs.2005.02.008
Vergne, I., and Deretic, V. (2010). The role of PI3P phosphatases in the regulation of autophagy. FEBS Lett. 584, 1313–1318. doi: 10.1016/j.febslet.2010.02.054
Vergne, I., Roberts, E., Elmaoued, R. A., Tosch, V., Delgado, M. A., Proikas-Cezanne, T., et al. (2009). Control of autophagy initiation by phosphoinositide 3-phosphatase Jumpy. EMBO J. 28, 2244–2258. doi: 10.1038/emboj.2009.159
von Willebrand, M., Baier, G., Couture, C., Burn, P., and Mustelin, T. (1994). Activation of phosphatidylinositol-3-kinase in Jurkat T cells depends on the presence of the p56lck tyrosine kinase. Eur. J. Immunol. 24, 234–238. doi: 10.1002/eji.1830240137
Vooijs, M., Jonkers, J., and Berns, A. (2001). A highly efficient ligand-regulated Cre recombinase mouse line shows that LoxP recombination is position dependent. EMBO Rep. 2, 292–297. doi: 10.1093/embo-reports/kve064
Wang, S., Li, J., Du, Y., Xu, Y., Wang, Y., Zhang, Z., et al. (2017). The Class I PI3K inhibitor S14161 induces autophagy in malignant blood cells by modulating the Beclin 1/Vps34 complex. J. Pharmacol. Sci. 134, 197–202. doi: 10.1016/j.jphs.2017.07.001
Willinger, T., and Flavell, R. A. (2012). Canonical autophagy dependent on the class III phosphoinositide-3 kinase Vps34 is required for naive T-cell homeostasis. Proc. Natl. Acad. Sci. USA 109, 8670–8675. doi: 10.1073/pnas.1205305109
Yang, G., and Van Kaer, L. (2020). PIK3C3/VPS34 links T-cell autophagy to autoimmunity. Cell Death Dis. 11:334. doi: 10.1038/s41419-020-2568-z
Yu, X., Long, Y. C., and Shen, H. M. (2015). Differential regulatory functions of three classes of phosphatidylinositol and phosphoinositide 3-kinases in autophagy. Autophagy 11, 1711–1728. doi: 10.1080/15548627.2015.1043076
Zeng, X., Overmeyer, J. H., and Maltese, W. A. (2006). Functional specificity of the mammalian Beclin-Vps34 PI 3-kinase complex in macroautophagy versus endocytosis and lysosomal enzyme trafficking. J. Cell Sci. 119, 259–270. doi: 10.1242/jcs.02735
Zhang, X., Sun, S., Hwang, I., Tough, D. F., and Sprent, J. (1998). Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity 8, 591–599. doi: 10.1016/s1074-7613(00)80564-6
Keywords: autophagy, PI3K I, T cells, lipid kinase, lipid phosphatase, cytokines, HIV
Citation: McLeod IX, Saxena R, Carico Z and He Y-W (2021) Class I PI3K Provide Lipid Substrate in T Cell Autophagy Through Linked Activity of Inositol Phosphatases. Front. Cell Dev. Biol. 9:709398. doi: 10.3389/fcell.2021.709398
Received: 13 May 2021; Accepted: 21 July 2021;
Published: 12 August 2021.
Edited by:
Jianke Zhang, Thomas Jefferson University, United StatesReviewed by:
Binfeng Lu, University of Pittsburgh, United StatesDeng Liufu, Shanghai Jiao Tong University, China
Copyright © 2021 McLeod, Saxena, Carico and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: You-Wen He, eW91d2VuLmhlQGR1a2UuZWR1