 Logan Meurer
Logan Meurer Leonard Ferdman1
Leonard Ferdman1 Troy Camarata
Troy Camarata
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 19 August 2021
Sec. Molecular and Cellular Oncology
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.707854
The sine oculis (SIX) family of transcription factors are key regulators of developmental processes during embryogenesis. Members of this family control gene expression to promote self-renewal of progenitor cell populations and govern mechanisms of cell differentiation. When the function of SIX genes becomes disrupted, distinct congenital defects develops both in animal models and humans. In addition to the embryonic setting, members of the SIX family have been found to be critical regulators of tumorigenesis, promoting cell proliferation, epithelial-to-mesenchymal transition, and metastasis. Research in both the fields of developmental biology and cancer research have provided an extensive understanding of SIX family transcription factor functions. Here we review recent progress in elucidating the role of SIX family genes in congenital disease as well as in the promotion of cancer. Common themes arise when comparing SIX transcription factor function during embryonic and cancer development. We highlight the complementary nature of these two fields and how knowledge in one area can open new aspects of experimentation in the other.
The sine oculis (SIX) homeobox family of transcription factors play important developmental roles in a wide range of species from fruit flies to humans. The founding member, sine oculis (so), was first identified in Drosophila melanogaster where it was discovered to be required for compound eye formation (Cheyette et al., 1994; Serikaku and O’Tousa, 1994). Subsequent research in fruit flies identified two additional SIX genes, optix, and DSix4 (reviewed in Kawakami et al., 2000). All three transcription factors were found to share a conserved N-terminal SIX domain adjacent to a homeodomain (HD), which function as protein-protein and DNA binding domains, respectively (Figure 1; Kawakami et al., 2000). Gene duplication during evolution expanded the SIX family of genes and created three subfamilies in vertebrates, which are composed of the so subfamily (Six1 and Six2), the optix subfamily (Six3 and Six6), and the DSix4 subfamily (Six4 and Six5). The vertebrate orthologs contain the same SIX and HD domains as the ancestral Drosophila proteins with significant amino acid sequence identity between the functional domains of family members. For example, the mouse SIX domain amino acid identity ranges from 63 to 93% while sequence identity in the HD ranges from 59 to 98% between family members, with Six3, Six4, and Six5 showing the most divergence. For a more complete review of SIX family protein structure and sequence comparisons see Kawakami et al. (2000) and Kumar (2009).
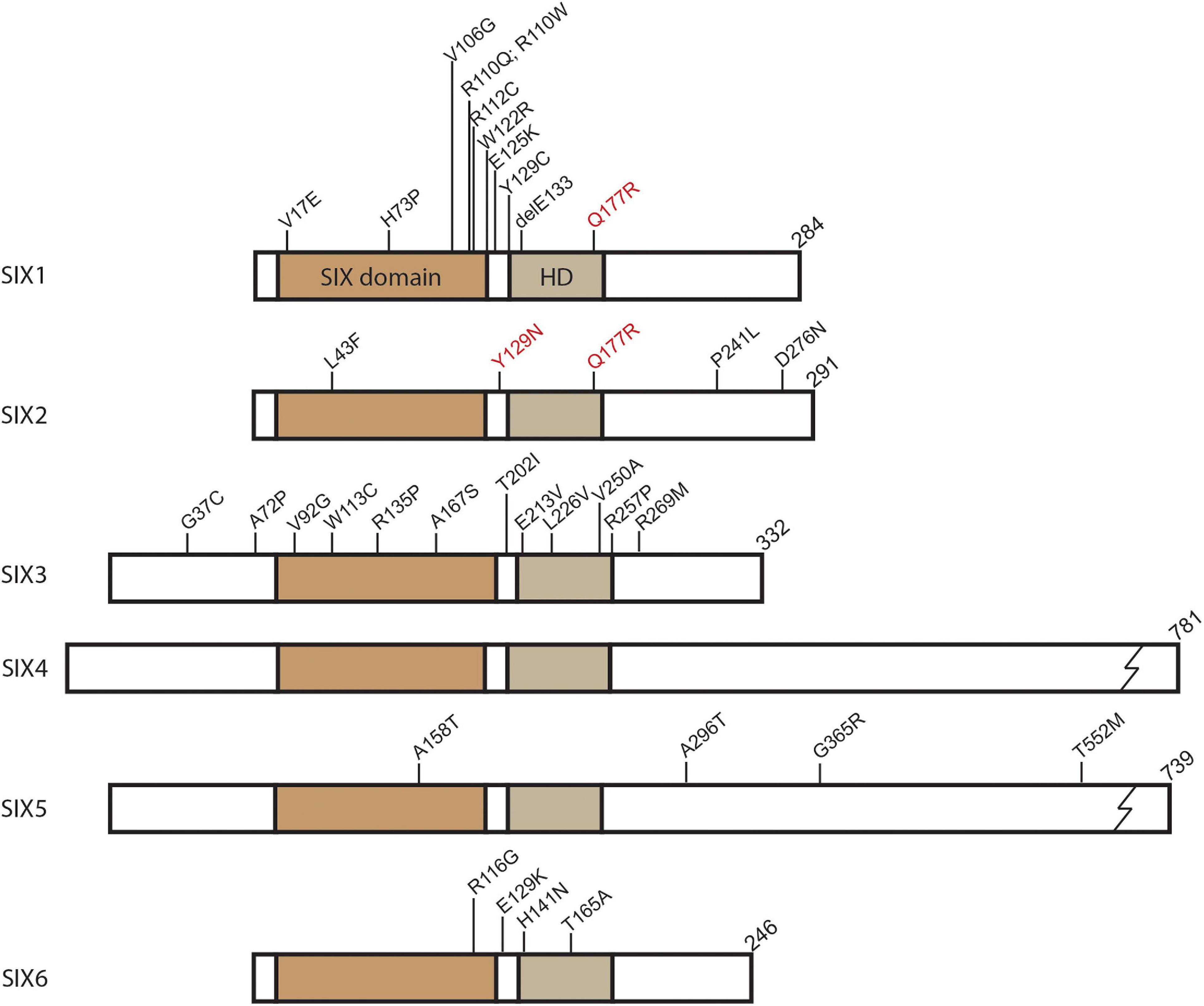
Figure 1. SIX protein domain schematic and identified human mutations. Position of conserved domains shown for SIX domain and Homeodomain (HD). Mutations identified in SIX genes related to congenital conditions are labeled at their relative amino acid position. In addition to the mutations shown, single allele deletions have been identified for SIX2, SIX3, and SIX6. SIX1 and SIX2 hyperactive mutations associated with Wilms tumor denoted in red. Common identified SIX3 mutations shown. For a complete list of SIX3 mutations identified in holoprosencephaly see Lacbawan et al. (2009).
In vertebrates, SIX genes play critical roles in tissue formation and organogenesis, such as for the head, ear, retina, nose, brain, skeletal muscle, and kidney (Oliver et al., 1995; Ohto et al., 1998; Jean et al., 1999; Kobayashi et al., 2001; Laclef et al., 2003; Lagutin et al., 2003; Li et al., 2003; Xu et al., 2003; Self et al., 2006). In these tissues, the SIX family of transcription factors function as regulators of progenitor cell maintenance and differentiation. They can act as transcriptional activators or repressors depending on interactions with other highly conserved regulators including Paired-box (Pax), Eyes absent (Eya), Dachshund (Dach), and Groucho (Grg) proteins (see reviews Kawakami et al., 2000; Kumar, 2009). Correlating with their important functions during embryogenesis, several congenital defects in humans are associated with mutations in SIX genes. In cancer, SIX genes have been found to be ectopically- or over-expressed and experimental interrogation suggests SIX proteins can drive disease pathogenesis. This review will focus on recent developments on SIX gene function and regulation in order to provide insight into congenital diseases along with how SIX genes become exploited in the context of cancer formation. Common themes emerge when comparing roles of SIX genes in developmental and cancer biology which relate to progenitor cell maintenance, cell behavior, and regulation; generating new questions and opportunities for research in each field.
Members of the SIX family of transcription factors are widely expressed in the developing vertebrate embryo where they play important regulatory roles in tissue and organ formation. Unsurprisingly, several human congenital conditions have been associated with haploinsufficiency or hypomorphic mutations in SIX genes (Figure 1 and Table 1). Many human SIX gene mutations were located within the SIX domain or homeodomain, which were suggestive of impaired protein-protein interactions or DNA binding. However, mutations have been detected outside of these domains and their functional significance remains unclear (Figure 1). Mutations in human SIX1 and SIX2 have been associated with multiple congenital disorders such as branchio-oto-renal syndrome (BOR), renal dysplasia, hearing loss, and frontonasal dysplasia syndrome (Ruf et al., 2004; Kochhar et al., 2008; Weber et al., 2008; Mosrati et al., 2011; Guan et al., 2016; Hufnagel et al., 2016). BOR is an autosomal dominant disorder that is characterized by a triad of clinical symptoms including branchial arch defects, hearing loss, and renal abnormalities (Melnick et al., 1976). The most commonly identified mutations in BOR are in the SIX1 binding partner EYA1 (Abdelhak et al., 1997), however, mutations have been identified in SIX1 that disrupt DNA binding or impact the ability to form a functional transcriptional complex with EYA1 (Ruf et al., 2004; Patrick et al., 2009). Independent of BOR, dominantly inherited hearing loss has been associated with mutations in both SIX1 and SIX2 (Mosrati et al., 2011; Guan et al., 2016). Additionally, SIX2 mutations were detected in patients with renal hypodysplasia (Weber et al., 2008) and more recently in frontonasal dysplasia syndrome (Hufnagel et al., 2016).

Table 1. Associated congenital conditions and related mouse model data for SIX family members.
The involvement of SIX1 and SIX2 in human syndromes is supported by functional studies in animal models. Aspects of BOR are evident in mouse knock-out models as Six1–/– null mice fail to develop a thymus and kidney, in addition to having significant defects in structures of the inner ear and nose (Zheng et al., 2003; Xu et al., 2003; Ruf et al., 2004; Ozaki et al., 2004; Zou et al., 2006). Further analysis has shown Six1 to be required for otic vesicle and cochlea development as well as differentiation of hair cells in the ear (Ozaki et al., 2004; Zhang et al., 2017). Identified human BOR SIX1 mutations expressed in Xenopus embryos disrupted otic vesicle formation and ear morphology, further connecting SIX1 function to BOR phenotypes (Shah et al., 2020). Six2 manipulation in animals models has also been informative in its role during development. For example, Six2 knock-out mice develop renal hypoplasia, where the metaneprhic kidney forms but is significantly smaller than normal (Self et al., 2006; Kobayashi et al., 2008). Mouse Six2 mutants also display features associated with frontonasal dysplasia. Two independent Six2 animal models, a genetic knockout and a mutant from an X-ray irradiation screen (Brachyrrhine), exhibited defects of the cranial base and cleft palate (Self et al., 2006; Fogelgren et al., 2008; He et al., 2010; Sweat et al., 2020). These phenotypes were similar to a dominantly inherited form of frontonasal dysplasia syndrome linked to a chromosomal deletion of SIX2 at 2p21 (Hufnagel et al., 2016). Six1 has also been linked to craniofacial development as disruption of Six1 function leads to micrognathia and defects of the nose, mandible, and midface structures (Ozaki et al., 2004; Guo et al., 2011; Tavares et al., 2017). Furthermore, both so homologs have been shown to participate together in craniofacial development as Six1/Six2 double knockout animals have significantly more severe phenotypes than single mutants, including agenesis of the frontal and parietal bones of the skull (Liu et al., 2019a).
SIX3 and SIX6 of the optix subfamily have been associated with specific congenital defects of the developing forebrain in humans. Mutations in SIX3 result in holoprosencephaly (HPE), a heterogeneous collection of forebrain malformations, with incomplete penetrance due to SIX3 haploinsufficiency or hypomorphic function (Table 1; Wallis et al., 1999; Lacbawan et al., 2009; Hehr et al., 2010; Stokes et al., 2018). Mutations in additional genes are also known to cause HPE including Sonic Hedgehog (SHH) and other members of this signaling pathway (Dubourg et al., 2007). Mutations in SIX3 and SHH have both been implicated in schizencephaly, a syndrome which clinically overlaps with HPE (Hehr et al., 2010). SIX6 has been connected with various eye malformations including anophthalmia and microphthalmia (Gallardo et al., 1999, 2004), primary open angle glaucoma (POAG) (Iglesias et al., 2014; Carnes et al., 2014; Mohanty et al., 2018), and optic disk anomalies and macular atrophy (Yariz et al., 2015). Similar to other family members, mutations such as point mutations and allele deletions in SIX6 were thought to result in haploinsufficiency or hypomorphic function (Figure 1 and Table 1).
In the mammalian embryo, Six3 and Six6 are expressed in derivatives of the anterior neural plate including the hypothalamus, pituitary gland, olfactory placodes, and regions of the developing eye (Oliver et al., 1995; Jean et al., 1999). Functional studies have shown a requirement for Six3 in forebrain development, for example, Six3 knock-out mice do not form telencephalic or optic vesicles (Lagutin et al., 2003). Shh, which has been shown to be involved in forebrain formation and HPE presentations (Shimamura and Rubenstein, 1997; Fuccillo et al., 2004), is directly regulated by Six3 (Jeong et al., 2008; Geng et al., 2008). The Six3-Shh interaction in the anterior neural plate is required to repress Wnt signaling, allowing for proper dorsoventral patterning of the telencephalon (Geng et al., 2008; Jeong et al., 2008; Liu et al., 2010; Carlin et al., 2012). Manipulation of Six3 expression in animal models has also provided insight into the incomplete penetrance observed in familial HPE. Knock-in of human mutations or the creation of variable expressing hypomorphs in mice has modeled both semilobar and alobar HPE with variable penetrance (Geng et al., 2008, 2016). The ultimate result of haploinsufficiency or hypomorphic alleles of SIX3 is reduced SHH signaling and defective forebrain formation. Compared to Six3, Six6 was found to be more restricted during forebrain development with expression in the forming hypothalamus, pituitary, and retina (Jean et al., 1999). Disruption of Six6 function in animal models has led to defects in the visual system ranging from small eye phenotypes in zebrafish to hypoplasia of the neural retina in mice (Li et al., 2002; Iglesias et al., 2014; Carnes et al., 2014; Teotia et al., 2017; Diacou et al., 2018). Conditional knockout of Six3 or Six6 have provided additional insights into the roles of the transcription factors in eye development. Neural retina maintenance and differentiation is dependent upon the function of both Six3 and Six6 (Zhu et al., 2002; Liu et al., 2006, 2010; Manavathi et al., 2007; Samuel et al., 2016; Takata et al., 2017; Liu and Cvekl, 2017) and both genes are required to repress Wnt signaling during eye development (Diacou et al., 2018). Coordinated and overlapping roles for the two transcription factors has also been implicated in the hypothalamus and pituitary gland where they may regulate the neurons that express gonadotropin-releasing hormone and differentiation of the receptive pituitary gonadotropes (Larder et al., 2011; Xie et al., 2015).
Of the DSix4 subfamily members, Six4 and Six5, only SIX5 has thus far been linked to congenital disease in humans (Table 1). Along with SIX1, mutations in SIX5 have been identified in patients with BOR that may impact DNA binding (Figure 1 and Table 1; Hoskins et al., 2007). However, more recent investigations have questioned the causative impact of SIX5 mutations in BOR where either no mutations were detected or mutations in other genes had been identified in addition to mutations in SIX5 (Krug et al., 2011; Wang et al., 2012; Song et al., 2013). Renal malformations are part of the complex BOR phenotype and a screen of 749 patients with congenital anomalies of the kidney and urinary tract (CAKUT) did detect one family with a mutation in SIX5 (Hwang et al., 2014). Further research is needed to more concretely connect SIX5 mutations as a causative factor in BOR and associated renal malformations. Another disease connected to SIX5 expression is Myotonic dystrophy (DM1). DM1 is dominantly inherited and presents with myotonia, muscle wasting, cardiac conduction defects, fertility defects, and cataracts (Harper, 1975). The underlying genetic cause of DM1 is a CTG trinucleotide repeat expansion in the 3′ UTR of the DMPK gene on chromosome 19 (Brook et al., 1992; Fu et al., 1992; Mahadevan et al., 1992). The repeat expansion appears to disrupt the expression of neighboring genes, including SIX5, which has reduced expression in DM1 (Klesert et al., 1997; Thornton et al., 1997).
Investigation into Six5 function in animal models has begun to tease out its role in multigenic DM1. Knock-out mice for Six5, both heterozygous and homozygous animals, develop cataracts with variable penetrance replicating observed DM1 phenotypes (Klesert et al., 2000; Sarkar et al., 2000). Additional studies have shown a requirement for Six5 in spermatogonia viability and spermatozoa differentiation in male mice (Sarkar et al., 2004). Furthermore, cardiac conduction defects have been observed in heterozygous mutant mice (Wakimoto et al., 2002). One of the hallmark features of DM1 is progressive muscle wasting and hypotonia. Thorough interrogation of Six5 function in mouse models does not support a direct role for the transcription factor in muscle phenotypes of DM1 despite expression in the developing myotome and skeletal muscle (Murakami et al., 1998; Klesert et al., 2000; Personius et al., 2005; Matynia et al., 2010). Although, triple and quadruple gene knockout of loci suspected in DM1, including Six5, does result in the array of multisystem defects present in myotonic dystrophy (Yin et al., 2020). Therefore, it appears the independent role of SIX5 in DM1 is limited to the eye, spermatogonia, and cardiac conduction system.
The lack of SIX4 mutations identified in human congenital disease is not surprising based upon studies in animal models. For example, Six4 knock-out mice were found to be viable with no obvious developmental or progressive defects (Ozaki et al., 2001). However, Six4 has been found to cooperate with other SIX family members in several developmental processes. Both Six5 and Six4 were identified to function together during vertebrate body wall development where loss of both genes resulted in omphalocele (Takahashi et al., 2018). Further mouse compound knock-out studies have uncovered Six4 functional cooperation with Six1 during myogenesis (Grifone et al., 2005; Relaix et al., 2013; Wurmser et al., 2020), gonadogenesis (Fujimoto et al., 2013); thymus development (Zou et al., 2006), neurogenesis (Konishi et al., 2006; Chen et al., 2009), and kidney development (Kobayashi et al., 2007; Xu and Xu, 2015). Based upon these studies, it appears Six4 function is compensated by other family members but still plays an important supportive role during embryonic development.
The embryonic phenotypes associated with SIX gene manipulation in animal models or from human mutation greatly correlate with the developmental expression profile of each family member (Table 1; reviewed in Kawakami et al., 2000). However, associations with adult gene expression and disease have not been as clear. Adult expression has been detected in skeletal muscle and satellite progenitor cells for Six1, Six2, Six4, and Six5 (reviewed in Maire et al., 2020). Six1 has been detected in the adult thymus as well as the salivary gland, trachea, and at low levels in the mouse mammary gland (Ford et al., 1998; Coletta et al., 2004; Guo et al., 2011). Six1 expression has also been demonstrated to be induced in differentiated mouse immune cells following infection (Liu et al., 2019b). SIX2 and SIX3 gene expression has been detected in adult pancreatic β-cells (Arda et al., 2016; Bevacqua et al., 2021) while SIX3 and SIX6 were found to be expressed in the adult pituitary (Aijaz et al., 2005; Xie et al., 2015). SIX5 was detected in the epithelium of the Fallopian tube and cervix but not in the ovary or glandular epithelium (Winchester et al., 2000). Most other adult tissues appear to be negative or express very low levels of SIX genes including the lymph nodes, lung, and kidney (Ford et al., 1998; Kobayashi et al., 2008; Guo et al., 2011). The lack of significant expression of SIX transcription factors in adult tissue is of greater consequence in the context of cancer, where SIX genes become ectopically or re-expressed to drive tumorigenesis.
The features of the SIX family of transcription factors that make them critical in the development of specific tissues and organ systems also makes them potentially deleterious when ectopically expressed in adult tissues. Promotion of cell proliferation or migration by SIX proteins ectopically expressed in adult tissues can and often contribute to the formation, survival, and metastasis of a variety of tumor types (Table 2).

Table 2. SIX transcription factors in cancer biology.
The so subfamily (SIX1 and SIX2), particularly SIX1, have been frequently implicated in the promotion, invasion, and survival of a variety of cancers (Blevins et al., 2015). SIX1 alone has been shown to be overexpressed in many forms of cancer such as breast (Ford et al., 1998; Coletta et al., 2004; Iwanaga et al., 2012), ovarian (Behbakht et al., 2007), cervical (Sun et al., 2016), Wilms tumor (Wegert et al., 2015), osteosarcoma (Hua et al., 2014; Chao et al., 2017), rhabdomyosarcoma (Yu et al., 2004), and several others (Table 2). Increased levels of SIX1 gene or protein expression was often found to be strongly correlated with poor prognosis regardless of tumor type (Blevins et al., 2015). SIX2, like SIX1, overexpression was detected in breast cancer (Wang et al., 2014; Oliphant et al., 2019) and appeared to promote increased survival, self-renewal, and metastasis of tumor cells (Table 2). All of these characteristics contribute to poor prognosis and decreased patient survival. SIX2 has been detected in other cancers such as hepatocellular carcinoma (HCC) (Zhu et al., 2016; Li et al., 2018; Wan et al., 2019), non-small cell lung cancer (Hou et al., 2019) and colorectal cancer (Wu et al., 2017). As one might predict, increased levels of SIX2 are highly correlated with cancers involving the kidney. SIX2 overexpression has been identified in the pediatric cancer Wilms tumor (Murphy et al., 2012; Pierce et al., 2014; Walz et al., 2015; Wegert et al., 2015) as well as in renal cell carcinoma (Senanayake et al., 2013; Cheng et al., 2019) and nephroblastoma (Senanayake et al., 2013). Specific point mutations in SIX1 and SIX2 have been detected in Wilms tumor cells where they potentially increase transcriptional activity (mutations denoted in red of Figure 1; Wegert et al., 2015).
Recently, SIX6 has been found to be associated with T-cell acute lymphoblastic leukemia (T-ALL) (Laukkanen et al., 2020), though the researchers of the study concluded that SIX6 most likely belonged to a larger regulatory gene network and increased levels of SIX6 alone were not sufficient to induce development of T-ALL. The study did conclude, however, that higher levels of SIX6 was associated with inferior treatment response and poor prognosis (Laukkanen et al., 2020). Increased SIX6 levels have also been associated with poor overall survival in non-small cell lung carcinoma and breast cancer (Liu et al., 2016; Xu H. X. et al., 2016). In stark contrast to other SIX transcription factors, increased levels of SIX3 appear to play a tumor suppressive role rather than an oncogenic one. Higher levels of SIX3 were associated with decreased tumor proliferation and metastasis, leading to better survival outcomes and/or prognosis in breast cancer (Zheng et al., 2018), astrocytoma (Yu et al., 2017), glioblastoma (Zhang B. et al., 2017), and lung adenocarcinoma (Mo et al., 2013).
SIX4 expression has been detected in non-small cell lung (Tang et al., 2019), breast (Sun et al., 2020), colorectal (Li et al., 2017; Sun et al., 2019), and hepatocellular cancers (He et al., 2020). Over- or ectopic expression of SIX4 promoted metastasis by inducing epithelial-to-mesenchymal transition and angiogenesis (Sun et al., 2019). Furthermore, significant correlations have been identified between expression levels of SIX4, tumor cell metastasis, and poor patient prognosis (Li et al., 2017; Tang et al., 2019; Sun et al., 2019, 2020; He et al., 2020). To date, few studies have analyzed SIX5 function in cancer (Table 2). In one report, SIX5 was detected in normal ovarian epithelium, as well as in malignant ovarian and borderline tumors suggesting that SIX5 could be used as a marker for epithelial differentiation in ovarian tissue rather than a specific marker for cancer (Winchester et al., 2000). A recent meta-analysis showed that high SIX5 expression levels correlated with poor overall survival in lung squamous cell carcinoma (Liu et al., 2016).
Several common functions and modes of regulation have been identified for SIX genes, not just amongst family members, but also between roles during embryonic and cancer development (Table 3).

Table 3. Common functions and pathways of SIX family genes in development and cancer biology.
Cell survival and proliferation are key functions of the SIX family of transcription factors. Therefore, it is not surprising that many of the gross morphological phenotypes detected from SIX gene mutations, either in animal studies or human syndromes, are attributed to improper maintenance of progenitor cell populations (Xu et al., 2003; Ozaki et al., 2004; Sarkar et al., 2004; Self et al., 2006; Gaston-Massuet et al., 2008; Jeong et al., 2008; Kobayashi et al., 2008; Guo et al., 2011; Wang et al., 2011; Fujimoto et al., 2013; Lu et al., 2013; Riddiford and Schlosser, 2017; Liu and Cvekl, 2017). Disruption or knock-out of SIX gene function in experimental studies has resulted in increased progenitor cell apoptosis concomitant with reduced proliferation in several developing tissues (Table 1). In the context of cancer, ectopic or overexpression of SIX genes has resulted in increased tumor cell proliferation as well as maintenance of cancer stem cells (Table 2; McCoy et al., 2009; Farabaugh et al., 2012; Cheng et al., 2019; Oliphant et al., 2019). The one exception may be Six3 where its role in proliferation is not as clear. Overexpression of Six3 has been shown to promote progenitor cell proliferation in the developing forebrain of zebrafish, medaka, and Xenopus (Kobayashi et al., 1998; Carl et al., 2002). However, other developmental studies utilizing Six3 loss-of-function approaches have not detected expected proliferation defects (Lagutin et al., 2003; Geng et al., 2008; Liu et al., 2010; Carlin et al., 2012). It is unclear whether the observed differences in proliferation is due to experimental approach or compensation by other family members. In cancer studies, SIX3, appeared to function as a tumor suppressor, where overexpression in cancer cells was associated with decreased proliferation (Mo et al., 2013; Yu et al., 2017; Zheng et al., 2018). One possible explanation for the difference in cell cycle regulation between SIX3 and other family members may be attributed to different core DNA binding sequences. Six1, Six2, Six4, Six5, and Six6 have been shown to bind a TCAGGTTC core sequence identified in the Myogenin MEF3 promoter (Spitz et al., 1998; Harris et al., 2000; Hu et al., 2008). However, both Six3 and Six6 were found to bind to a core ATTA sequence utilized by other homeodomain containing proteins (Zhu et al., 2002; Hu et al., 2008). Six3 seems to be unique among the SIX family in transcriptional targets based upon DNA binding sequences.
The ability to regulate progenitor cell populations, both during embryonic development and in cancer, stems from the ability of SIX proteins to directly regulate the cell cycle (Table 3). Six1 has been found to transcriptionally regulate genes encoding cyclin A1 and cyclin D1 in developmental and cancer contexts (Coletta et al., 2004; Yu et al., 2006; Li et al., 2013). For the related Six2, direct interaction with cyclin promoters has not been demonstrated. However, cyclin D1 expression has been shown to be dependent upon Six2 in the developing palate (Okello et al., 2017) while studies in kidney progenitor cells have detected Six2 binding sites in the ccnd1 (cyclin D1) promoter region (O’Brien et al., 2018). Six6, along with Dachous (Dach) proteins, promoted cell proliferation by directly repressing the expression of cyclin-dependent kinase inhibitors (Li et al., 2002; Iglesias et al., 2014) while Six4 regulated the expression of Yap1 and c-Met to promote cell proliferation in HCC (He et al., 2020). Cell cycle control by SIX proteins is also accomplished via protein interactions. For example, Geminin (a cell cycle inhibitor) binding with either Six3 or Six6 inhibited cell cycle progression (Del Bene et al., 2004; Turcu et al., 2019).
Enhancing cell proliferation by SIX proteins may also occur via crosstalk with other progenitor cell markers such as Sox2. The transcriptional regulator Sox2 has been shown to be directly regulated in developmental and cancer contexts by Six1 (Zhang T. et al., 2017; De Lope et al., 2019), Six2 (Cheng et al., 2019; Oliphant et al., 2019), Six3 (Liu et al., 2006), and Six6 (Diacou et al., 2018) to further promote stem/progenitor cell phenotypes. SIX family transcription factors seem to promote progenitor cell self-renewal through both direct cell cycle regulation and via indirect mechanisms by activating additional pro-stem cell identity genes.
In parallel with regulating the cell cycle, SIX proteins also appear to influence apoptotic pathways. Loss of SIX function in several animal models resulted in increased progenitor cell apoptosis. Furthermore, silencing of SIX genes overexpressed in cancer cells resulted in increased cell death. The mechanisms of SIX regulation of apoptosis is not clear and most investigations have focused on SIX1 in cancer cell lines. SIX1 has been shown to post-translationally regulate p53, where the levels of the two proteins are inversely related in cancer (Towers et al., 2015). Protein–protein interactions between DACH1 and SIX1, which normally behaves as a transcriptional repressor complex, can stabilize p53 levels in HCC (Cheng et al., 2018). However, DACH1 is commonly downregulated in HCC, especially in cases with high levels of SIX1, allowing for the reduction of p53 and cell survival. Inverse relationships have also been detected between SIX1 and caspases. In osteosarcoma cells, overexpression of SIX1 led to decreased caspase-3 and caspase-7 with reduced apoptosis (Yu et al., 2018). The opposite result was detected following SIX1 knock-down where increased cell apoptosis and caspase levels were observed. Similar observations have been found in mouse trigeminal ganglia where double knock-out of Six1 and Six4 resulted in increased caspase-3 dependent apoptosis (Konishi et al., 2006). In addition, SIX1 and SIX4 have been shown to upregulate PI3K/AKT signaling in osteosarcoma and colorectal cancer, respectively, possibly through the downregulation of PTEN to further suppress apoptosis (Li et al., 2017; Yu et al., 2018; Na et al., 2019; Ji et al., 2020). In other studies, SIX1 overexpression reduced TRAIL-mediated apoptosis (Behbakht et al., 2007). Taken together, SIX genes play critical roles in promoting progenitor cell self-renewal by directly regulating the cell cycle as well as inhibiting apoptotic pathways. However, questions remain about the mechanisms of apoptosis inhibition by SIX transcription factors such as the post-translational stabilization of p53 by a SIX1/DACH1 complex.
One of the more devastating aspects of SIX gene overexpression in cancer appears to be from driving metastasis in part by inducing epithelial-to-mesenchymal transition (EMT). Studies in several cancer types have shown a relationship between SIX gene expression and increased EMT, cell migration, and tumor invasion (Table 2). A common mechanism for SIX proteins to induce EMT is through indirect regulation of Cadherin-1 (CDH1), which encodes for the epithelial marker, E-cadherin. Increased expression of either SIX1 or SIX2 in several cancer types reduced the level of CDH1 through either activating known repressors of CDH1, such as Zeb proteins, or by CDH1 promoter methylation (McCoy et al., 2009; Wang et al., 2014; Li J. W. et al., 2018; Hou et al., 2019). Similar EMT-promoting mechanisms may also be present during development. Six1 appears to regulate both N-cadherin and E-cadherin in auditory epithelium (Zhang T. et al., 2017) while forced expression of genetic factors, including Six1 and Six2, in kidney epithelial cells induces EMT and reduces E-cadherin expression (Hendry et al., 2013). Additionally, Six2 expression was required to suppress epithelialization of renal progenitor cells and Six2 null embryonic mouse kidney explants showed expanded E-cadherin expression, suggesting regulation by the transcription factor (Self et al., 2006; McMahon, 2016). Conversely, continual expression of Six2 in renal progenitor cells in vivo inhibited Cdh1 expression and mesenchymal-to-epithelial transition (Chung et al., 2016). SIX genes also appear to activate pathways known to promote EMT and cell migration such as the c-Met/HGF pathway. One of the downstream targets of c-Met/HGF is Snail, a known repressor of E-cadherin (Wang et al., 2020). It has been demonstrated that SIX4 could directly activate c-Met expression in HCC providing a mechanism for promoting EMT, cell migration, and metastasis (He et al., 2020). In support of the finding in HCC, both Six1 and Six4 have been shown to activate met expression in both embryonic mouse and zebrafish and this activation was required for skeletal muscle precursor cell migration (Grifone et al., 2005; Talbot et al., 2019).
SIX protein function has been connected to a number of regulatory and signal transduction pathways, however, interactions with Wnt, Notch, and TGF-β pathways appear to be shared most amongst family members in both development and cancer (Table 3). Wnt signaling regulation has been connected to Six1, Six2, Six3, Six4, and Six6. Six1 overexpression upregulated Wnt pathway genes and promoted β-catenin nuclear localization in mammary gland tumors and colorectal cancer cell lines (McCoy et al., 2009; Song et al., 2019). In developing auditory sensory epithelium, Six1 binding sites indicative of gene activation have been detected upstream of Wnt5a and other Wnt targets (Li et al., 2020). During kidney development, Wnt/beta-catenin and Six2 have opposing functions of cell differentiation and self-renewal, respectively (Park et al., 2007). Wnt/beta-catenin repressed Six2 expression in renal progenitor cells to help control mesenchymal-to-epithelial transition (Park et al., 2012). A similar opposing interaction has been observed in the pediatric kidney tumor, Wilms tumor. Six2 overexpression in Wilms tumor cells resulted in downregulation of Wnt pathway genes (Pierce et al., 2014). However, Six2 has been shown to be activated by Wnt in limb tendon precursor cells suggesting context dependent regulation (Yamamoto-Shiraishi and Kuroiwa, 2013). In the developing forebrain, Six3 directly repressed the expression of Wnt1 as well as Wnt8b (Lagutin et al., 2003; Liu et al., 2010). Repression of Wnt1 by Six3 has been detected in mammary glands as well as breast cancer cells (Kumar et al., 2010). Both Six3 and Six6 suppressed Wnt signaling during retinal development to maintain retinal progenitor cells (Diacou et al., 2018). In breast cancer, Six3 was transcriptionally targeted for repression by metastatic tumor antigen 1 (MTA1) which in turn upregulated Wnt1 (Kumar et al., 2010). Wnt signaling has been shown to play a significant role in cancer stem cell maintenance and metastasis (Zhan et al., 2017) and repression of Wnt by Six3 supports its role as a tumor suppressor. Finally, repression of Six4 by Wnt signaling has been implicated in neuronal placode development in the chick model (Litsiou et al., 2005).
Notch signaling and SIX transcription factor function have been associated in different developmental and cancer contexts. Six1, for example, has been shown to regulate Notch pathway targets hes8 and neurog1 during Xenopus neurogenesis (Riddiford and Schlosser, 2017) as well as jagged1 in mouse mandibular arch formation (Tavares et al., 2017). In breast cancer cells, Notch signaling was found to be upregulated with Six1 overexpression (Smith et al., 2012). Six1 also appeared to be a downstream effector of Notch2 in the developing olfactory epithelium and in lung adenocarcinoma cells (Rodriguez et al., 2008; Mimae et al., 2012). Overexpression of both SIX1 and NOTCH2 in lung cancer was associated with poor overall survival (Mimae et al., 2012). Further supporting a SIX/Notch pathway, Six2 has been shown to be regulated by Notch in kidney progenitor cells during renal organogenesis (Chung et al., 2016). Additionally, Notch1 expression is dependent upon both Six3 and Six6 in retinal development (Diacou et al., 2018) and expression of human SIX6 glaucoma risk alleles in Xenopus embryos downregulated the Notch pathway (Teotia et al., 2017).
Compelling evidence has emerged connecting the TGF-β pathway with SIX gene overexpression in various cancers. For example, SIX1 overexpression in breast cancer cell lines activated TGF-β signaling and activity of both factors correlated with poor prognosis in breast cancer (Micalizzi et al., 2009). Further investigation showed that SIX1 could bind to the promoter of TBRI and regulate its transcription (Micalizzi et al., 2010). The Six1/TGF-β pathway appears to switch cells toward a pro-EMT fate, an important step toward tumor metastasis (Micalizzi et al., 2009; Farabaugh et al., 2012; Smith et al., 2012). Similar interactions have been detected in models of cervical cancer and esophageal squamous cell carcinoma (Liu et al., 2014a; Nishimura et al., 2017). To date, SIX/TGF-β networks have not been thoroughly studied during embryonic development. However, components of the TGF-β pathway were found to be downregulated in Six1/Six4 double knockout mouse Pax7 + muscle precursor cells (Wurmser et al., 2020) while Six2 expression in metanephric mesenchyme progenitor cells may be controlled by TBRII/Smad3 (Mao et al., 2017).
SIX family transcriptions factors can behave as transcriptional activators or repressors. For example, mouse Six1 has been shown to activate gene expression in skeletal muscle cells (Li et al., 2003) and human SIX1 functioned in HCC as either an activator or repressor of gene expression (Cheng et al., 2018). The use of ChIP-seq has demonstrated that Six2 in mouse kidney progenitor cells functioned as a gene activator to promote progenitor cell self-renewal or as a repressor to inhibit cell differentiation (O’Brien et al., 2018). Six3 was found to repress AURKA and AURKB genes in astrocytoma cells (Yu et al., 2017) while the transcription factor was found to activate the expression of rhodopsin in the mouse retina (Manavathi et al., 2007). What determines the activation or repression function of the SIX transcription factors appears to the presence of interacting proteins such as Eya, Dach, and Grg. Eya proteins have been shown to bind to Six1, Six2, Six4, Six5, and Six6 to promote gene activation (Ohto et al., 1999; Ikeda et al., 2002; Li et al., 2003; Hu et al., 2008; Xu J. et al., 2016). Interestingly, Six3 has not been demonstrated to interact with Eya proteins (Zhu et al., 2002). In contrast to activation SIX/Eya complexes, interactions with Dach function to repress gene transcription. For example, Six6/Dach interactions have been demonstrated to act as a repressor complex in mouse retina and pituitary gland (Li et al., 2002). Although, the presence of Eya proteins can convert the repressive function of SIX/Dach complex toward gene activation (Li et al., 2003). Similarly, interactions demonstrated between Six3 or Six6 with Grg acted as repressor complexes (Kobayashi et al., 2001; Lopez-Rios et al., 2003). Zebrafish Six2 and Six4 were also found to bind to Grg proteins suggesting conservation of the repressor complex (Kobayashi et al., 2001). In cell culture experiments, Six3 was shown to activate promoter sites when binding alone, however, in the presence of Geminin, the two proteins complexed and repressed promoters (Del Bene et al., 2004). Eya, Grg, and Dach are commonly co-expressed with SIX transcription factors during development. The determination of whether the transcriptional complexes promote or repress gene expression remains unclear. Of clinical significance, repressive factors such as DACH are commonly downregulated in cancer while EYA genes are overexpressed (reviewed in Blevins et al., 2015; Kingsbury et al., 2019) which may provide a permissive environment for SIX dependent tumor growth and metastasis.
Appropriate control of SIX gene expression is required for normal tissue development and homeostasis. However, how members of the SIX family are regulated in development and reactivated in cancer remains unclear. Several studies have interrogated the upstream promoters of SIX genes to gain insight into their regulation. Binding sites for several transcription factors have been identified in the Six1 promoter which included, Sox, Pax, Fox, Tcf/Lef, Smad, E-box binding basic helix-loop-helix, and nuclear hormone receptor proteins (Sato et al., 2012; Sato et al., 2015). Additional promoter characterization for Six1 detected consensus sites for MyoD, Creb, and Pax7 (Wei et al., 2017). Six2 was shown to be regulated by Hox proteins, Hoxa2 (Kutejova et al., 2008; Yallowitz et al., 2009) and Hox11 (Yallowitz et al., 2009; Park et al., 2012; O’Brien et al., 2018) as well as β-catenin, Wilms tumor 1 and Odd-skipped related 1 (Park et al., 2012; O’Brien et al., 2018). Both Six1 and Six2 are capable of autoregulation and Six2 binding was detected on the Six1 promoter (Brodbeck et al., 2004; O’Brien et al., 2016). The presence of Tcf/Lef, β-catenin, and smad binding sites in the Six1 and Six2 promoters supports experimental evidence of Wnt and TGF-β regulation of SIX function (Table 3). Pax6 has been demonstrated to activate both Six3 and Six6 expression (Goudreau et al., 2002) along with other activators such as Prox1, Sox2, Sox3, and Lhx2 (Lengler and Graw, 2001; Tetreault et al., 2009; Lee et al., 2012). Six3 has been shown to autorepress its expression along with other repressor proteins including Msx2 and MTA1 (Lengler and Graw, 2001; Manavathi et al., 2007). Six6 promoter repression has been demonstrated for FoxD1 and Onecut (Ledford et al., 2017). Despite these findings, a great deal remains to be learned about the regulation of SIX genes during organ development and especially how these genes become reactivated in cancer cells.
Experimental evidence has suggested a significant role for epigenetic regulation in controlling SIX gene expression. Two emerging epigenetic mechanisms appear to be DNA methylation and targeting with microRNA (miRNA). Differential methylation patterns have been identified during embryonic development and in cancerous tissues for Six2, Six3, Six5, and Six6 (Table 4). For pro-oncogenic functions of Six2 and Six6, hypomethylation of promoter and genic regions correlated with increased expression (Marcinkiewicz and Gudas, 2014; Song et al., 2015; Berdasco et al., 2017; Sun et al., 2018). Two reports have associated hypermethylation of SIX6 with cancer types, however, expression levels of SIX6 were not addressed and it remains unclear the significance of gene methylation in these instances (Zhao et al., 2013; Lindqvist et al., 2014). Reduced SIX5 expression was associated with increased DNA and repressive-associated histone methylation linked to trinucleotide expansion in DM1, suggesting a similar epigenetic mechanism to control SIX gene expression (Filippova et al., 2001; Yanovsky-Dagan et al., 2015). For SIX3, the tumor suppressor appeared to be hypermethylated in lung cancer and glioblastoma accounting for decreased expression in these cancer types (Mo et al., 2013; Yu et al., 2020).

Table 4. Epigenetic regulation of SIX transcription factor genes.
In addition to DNA methylation, several miRNAs have been identified as epigenetic regulators that down regulate SIX expression (Table 4). Much of the research focus has been on various cancer cell lines and tumor types where miRNAs that normally target and suppress SIX mRNA transcripts were downregulated, allowing for SIX transcription factor induced cell proliferation and EMT. Several miRNAs have been identified that target Six1, Six2, and Six4 in a diverse array of cancers (Table 4). In addition to the cancer studies, examples of miRNA regulated SIX gene expression have been detected during embryonic development. Six1 was found to be directly regulated by miR30a during zebrafish skeletal muscle development (O’Brien et al., 2014). The related Six2 has been shown to be regulated by miR-181b in cultured metanephric mesenchyme cells and the interaction may function to control cell differentiation (Lyu et al., 2013). Finally, Six3 may potentially be regulated by miRNAs during eye development, although these results need to be further investigated to confirm this mechanism (Qiu et al., 2009).
The fields of developmental biology and cancer research together have provided great insight into the important functions of the SIX gene family in vertebrates. However, many questions remain including how the gene family is regulated, the regulation of activation or repression complexes, and whether SIX proteins are viable therapeutic targets. Efforts have been made to identify transcriptional regulators and characterize the promoters of SIX genes. For example, complex ChIP-seq analysis identified regulatory regions of Six2 in kidney progenitor cells and how Six2 may function in complex transcriptional regulatory networks (Park et al., 2012; O’Brien et al., 2018). The functional importance of the putative Six2 regulatory sequences and how they function in the balance between kidney progenitor cell maintenance and differentiation remains to be determined. It is also unclear whether any of the identified transcription factor binding sites upstream of Six2 or other SIX family members become re-engaged in cancer. In addition to the specific proteins that regulate SIX family enhancers and promoters, it is of interest to further identify upstream signal transduction pathways that can induce SIX gene expression. Evidence has shown a role for Notch, Wnt, and TGF-β pathways to not only be targets of SIX proteins but also to feedback onto SIX genes (Table 3). Identification of upstream pathways may be helpful in the context of congenital disease where clinical manifestations are commonly the result of SIX gene haploinsufficiency and compensation by wild-type alleles could reduce disease severity. Upstream activation pathways of SIX expression would also be of interest in cancer where they could provide new opportunities to reduce SIX induced tumorogenesis. Another tumorogenic target to reduce SIX function would be miRNAs, which have been found to be reduced in several SIX family associated cancers (Table 4). MicroRNAs appear to be an intriguing class of therapeutic targets and reintroduction into cancer cells may allow for specific downregulation of SIX expression in cancer (Rupaimoole and Slack, 2017).
A second area to further investigate is the regulation of activator vs. repressor complexes involving SIX proteins and other conserved factors such as Eya and Dach proteins. During development, these three protein families are commonly co-expressed to maintain a balance between progenitor cell proliferation and differentiation. Does the presence of Eya proteins always promote transcriptional activation even in the presence of repressors such as Dach (Li et al., 2003) or is there more complex regulation of additional SIX binding factors involved. Efforts have begun to better elucidate how SIX factors coordinate with other transcription factors to regulate target genes (O’Brien et al., 2018; Ogawa et al., 2019; Xu et al., 2021). The mechanisms regulating SIX transcriptional activation or repression would provide great insights for both developmental and cancer biology.
Traditionally, transcription factors have been seen as poor drug targets despite the central role they can play in disease such as cancer (Bushweller, 2019). Past difficulties have included targeting protein-DNA or protein-protein interactions due to the charge and flat shape of binding surfaces (Arkin et al., 2014). However, great progress has been made in better understanding protein structure, identifying the residues required for protein interactions, and the regulation of protein function through post-translational modifications. All of these areas are potential targets to control SIX transcription factors in the context of cancer (Bushweller, 2019). One approach has already been demonstrated in a breast cancer model where a small molecule inhibited the interaction between SIX1 and EYA2, reducing downstream TGF-β signaling and EMT leading to reduced metastasis in mouse xenografts (Zhou et al., 2020). Further exploration should identify additional novel regulators of SIX protein function and in conjunction with other therapeutic modalities, such as epigenetic modifiers, could prove effective strategies to combat SIX induced tumorogensis. Such therapeutic insights will only arise from the continual integration of developmental and cancer biology research into the function of the SIX family of transcription factors.
LM, LF, and TC conceived and developed the manuscript outline, and wrote and edited the manuscript. LM, LF, BB, and TC researched the literature and developed the tables. LM and TC created the figure. All the authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
This work was supported by NYIT College of Osteopathic Medicine.
The authors would like to thank laboratory members for helpful discussions and Tony Slieman for critical comments on the manuscript.
Abdelhak, S., Kalatzis, V., Heilig, R., Compain, S., Samson, D., Vincent, C., et al. (1997). Clustering of mutations responsible for branchio-oto-renal (BOR) syndrome in the eyes absent homologous region (eyaHR) of EYA1. Hum. Mol. Genet. 6, 2247–2255. doi: 10.1093/hmg/6.13.2247
Aijaz, S., Allen, J., Tregidgo, R., Van Heyningen, V., Hanson, I., and Clark, B. J. (2005). Expression analysis of SIX3 and SIX6 in human tissues reveals differences in expression and a novel correlation between the expression of SIX3 and the genes encoding isocitrate dehyhrogenase and cadherin 18. Genomics 86, 86–99. doi: 10.1016/j.ygeno.2005.03.002
Arda, H. E., Li, L., Tsai, J., Torre, E. A., Rosli, Y., Peiris, H., et al. (2016). Age-dependent pancreatic gene regulation reveals mechanisms governing human beta cell function. Cell Metab. 23, 909–920. doi: 10.1016/j.cmet.2016.04.002
Arkin, M. R., Tang, Y., and Wells, J. A. (2014). Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem. Biol. 21, 1102–1114. doi: 10.1016/j.chembiol.2014.09.001
Behbakht, K., Qamar, L., Aldridge, C. S., Coletta, R. D., Davidson, S. A., Thorburn, A., et al. (2007). Six1 overexpression in ovarian carcinoma causes resistance to TRAIL-mediated apoptosis and is associated with poor survival. Cancer Res. 67, 3036–3042. doi: 10.1158/0008-5472.can-06-3755
Berdasco, M., Gomez, A., Rubio, M. J., Catala-Mora, J., Zanon-Moreno, V., Lopez, M., et al. (2017). DNA methylomes reveal biological networks involved in human eye development, functions and associated disorders. Sci. Rep. 7:11762.
Bevacqua, R. J., Lam, J. Y., Peiris, H., Whitener, R. L., Kim, S., Gu, X., et al. (2021). SIX2 and SIX3 coordinately regulate functional maturity and fate of human pancreatic beta cells. Genes Dev. 35, 234–249. doi: 10.1101/gad.342378.120
Blevins, M. A., Towers, C. G., Patrick, A. N., Zhao, R., and Ford, H. L. (2015). The SIX1-EYA transcriptional complex as a therapeutic target in cancer. Exp. Opin. Ther. Targets 19, 213–225. doi: 10.1517/14728222.2014.978860
Brodbeck, S., Besenbeck, B., and Englert, C. (2004). The transcription factor Six2 activates expression of the Gdnf gene as well as its own promoter. Mech. Dev. 121, 1211–1222. doi: 10.1016/j.mod.2004.05.019
Brook, J. D., Mccurrach, M. E., Harley, H. G., Buckler, A. J., Church, D., Aburatani, H., et al. (1992). Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 69:385.
Bushweller, J. H. (2019). Targeting transcription factors in cancer - from undruggable to reality. Nat. Rev. Cancer 19, 611–624. doi: 10.1038/s41568-019-0196-7
Carl, M., Loosli, F., and Wittbrodt, J. (2002). Six3 inactivation reveals its essential role for the formation and patterning of the vertebrate eye. Development 129, 4057–4063. doi: 10.1242/dev.129.17.4057
Carlin, D., Sepich, D., Grover, V. K., Cooper, M. K., Solnica-Krezel, L., and Inbal, A. (2012). Six3 cooperates with Hedgehog signaling to specify ventral telencephalon by promoting early expression of Foxg1a and repressing Wnt signaling. Development 139, 2614–2624. doi: 10.1242/dev.076018
Carnes, M. U., Liu, Y. P., Allingham, R. R., Whigham, B. T., Havens, S., Garrett, M. E., et al. (2014). Discovery and functional annotation of SIX6 variants in primary open-angle glaucoma. PLoS Genet. 10:e1004372. doi: 10.1371/journal.pgen.1004372
Chao, L., Liu, J., and Zhao, D. (2017). Increased Six1 expression is associated with poor prognosis in patients with osteosarcoma. Oncol. Lett. 13, 2891–2896. doi: 10.3892/ol.2017.5803
Chen, B., Kim, E. H., and Xu, P. X. (2009). Initiation of olfactory placode development and neurogenesis is blocked in mice lacking both Six1 and Six4. Dev. Biol. 326, 75–85. doi: 10.1016/j.ydbio.2008.10.039
Cheng, N., Li, H., Han, Y., and Sun, S. (2019). Transcription factor Six2 induces a stem cell-like phenotype in renal cell carcinoma cells. FEBS Open Bio 9, 1808–1816. doi: 10.1002/2211-5463.12721
Cheng, Q., Ning, D., Chen, J., Li, X., Chen, X. P., and Jiang, L. (2018). SIX1 and DACH1 influence the proliferation and apoptosis of hepatocellular carcinoma through regulating p53. Cancer Biol. Ther. 19, 381–390.
Cheyette, B. N., Green, P. J., Martin, K., Garren, H., Hartenstein, V., and Zipursky, S. L. (1994). The Drosophila sine oculis locus encodes a homeodomain-containing protein required for the development of the entire visual system. Neuron 12, 977–996. doi: 10.1016/0896-6273(94)90308-5
Chung, E., Deacon, P., Marable, S., Shin, J., and Park, J. S. (2016). Notch signaling promotes nephrogenesis by downregulating Six2. Development 143, 3907–3913. doi: 10.1242/dev.143503
Coletta, R. D., Christensen, K., Reichenberger, K. J., Lamb, J., Micomonaco, D., Huang, L., et al. (2004). The Six1 homeoprotein stimulates tumorigenesis by reactivation of cyclin A1. Proc. Natl. Acad. Sci. U.S.A. 101, 6478–6483. doi: 10.1073/pnas.0401139101
De Lope, C., Martin-Alonso, S., Auzmendi-Iriarte, J., Escudero, C., Mulet, I., Larrasa-Alonso, J., et al. (2019). SIX1 represses senescence and promotes SOX2-mediated cellular plasticity during tumorigenesis. Sci. Rep. 9:1412.
Del Bene, F., Tessmar-Raible, K., and Wittbrodt, J. (2004). Direct interaction of geminin and Six3 in eye development. Nature 427, 745–749. doi: 10.1038/nature02292
Diacou, R., Zhao, Y., Zheng, D., Cvekl, A., and Liu, W. (2018). Six3 and Six6 are jointly required for the maintenance of multipotent retinal progenitors through both positive and negative regulation. Cell Rep. 25, 2510.e4–2523.e4.
Dubourg, C., Bendavid, C., Pasquier, L., Henry, C., Odent, S., and David, V. (2007). Holoprosencephaly. Orphanet J. Rare Dis. 2:8.
Farabaugh, S. M., Micalizzi, D. S., Jedlicka, P., Zhao, R., and Ford, H. L. (2012). Eya2 is required to mediate the pro-metastatic functions of Six1 via the induction of TGF-beta signaling, epithelial-mesenchymal transition, and cancer stem cell properties. Oncogene 31, 552–562. doi: 10.1038/onc.2011.259
Filippova, G. N., Thienes, C. P., Penn, B. H., Cho, D. H., Hu, Y. J., Moore, J. M., et al. (2001). CTCF-binding sites flank CTG/CAG repeats and form a methylation-sensitive insulator at the DM1 locus. Nat. Genet. 28, 335–343. doi: 10.1038/ng570
Fogelgren, B., Kuroyama, M. C., Mcbratney-Owen, B., Spence, A. A., Malahn, L. E., Anawati, M. K., et al. (2008). Misexpression of Six2 is associated with heritable frontonasal dysplasia and renal hypoplasia in 3H1 Br mice. Dev. Dyn. 237, 1767–1779. doi: 10.1002/dvdy.21587
Ford, H. L., Kabingu, E. N., Bump, E. A., Mutter, G. L., and Pardee, A. B. (1998). Abrogation of the G2 cell cycle checkpoint associated with overexpression of HSIX1: a possible mechanism of breast carcinogenesis. Proc. Natl. Acad. Sci. U.S.A. 95, 12608–12613. doi: 10.1073/pnas.95.21.12608
Fu, Y. H., Pizzuti, A., Fenwick, R. G. Jr., King, J., Rajnarayan, S., Dunne, P. W., et al. (1992). An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 255, 1256–1258. doi: 10.1126/science.1546326
Fuccillo, M., Rallu, M., Mcmahon, A. P., and Fishell, G. (2004). Temporal requirement for hedgehog signaling in ventral telencephalic patterning. Development 131, 5031–5040. doi: 10.1242/dev.01349
Fujimoto, Y., Tanaka, S. S., Yamaguchi, Y. L., Kobayashi, H., Kuroki, S., Tachibana, M., et al. (2013). Homeoproteins Six1 and Six4 regulate male sex determination and mouse gonadal development. Dev. Cell 26, 416–430. doi: 10.1016/j.devcel.2013.06.018
Gallardo, M. E., Lopez-Rios, J., Fernaud-Espinosa, I., Granadino, B., Sanz, R., Ramos, C., et al. (1999). Genomic cloning and characterization of the human homeobox gene SIX6 reveals a cluster of SIX genes in chromosome 14 and associates SIX6 hemizygosity with bilateral anophthalmia and pituitary anomalies. Genomics 61, 82–91. doi: 10.1006/geno.1999.5916
Gallardo, M. E., Rodriguez De Cordoba, S., Schneider, A. S., Dwyer, M. A., Ayuso, C., and Bovolenta, P. (2004). Analysis of the developmental SIX6 homeobox gene in patients with anophthalmia/microphthalmia. Am. J. Med. Genet. A 129A, 92–94.
Gaston-Massuet, C., Andoniadou, C. L., Signore, M., Sajedi, E., Bird, S., Turner, J. M., et al. (2008). Genetic interaction between the homeobox transcription factors HESX1 and SIX3 is required for normal pituitary development. Dev. Biol. 324, 322–333. doi: 10.1016/j.ydbio.2008.08.008
Geng, X., Acosta, S., Lagutin, O., Gil, H. J., and Oliver, G. (2016). Six3 dosage mediates the pathogenesis of holoprosencephaly. Development 143, 4462–4473.
Geng, X., Speirs, C., Lagutin, O., Inbal, A., Liu, W., Solnica-Krezel, L., et al. (2008). Haploinsufficiency of Six3 fails to activate Sonic hedgehog expression in the ventral forebrain and causes holoprosencephaly. Dev. Cell 15, 236–247. doi: 10.1016/j.devcel.2008.07.003
Goudreau, G., Petrou, P., Reneker, L. W., Graw, J., Loster, J., and Gruss, P. (2002). Mutually regulated expression of Pax6 and Six3 and its implications for the Pax6 haploinsufficient lens phenotype. Proc. Natl. Acad. Sci. U.S.A. 99, 8719–8724. doi: 10.1073/pnas.132195699
Grifone, R., Demignon, J., Houbron, C., Souil, E., Niro, C., Seller, M. J., et al. (2005). Six1 and Six4 homeoproteins are required for Pax3 and Mrf expression during myogenesis in the mouse embryo. Development 132, 2235–2249. doi: 10.1242/dev.01773
Guan, J., Wang, D., Cao, W., Zhao, Y., Du, R., Yuan, H., et al. (2016). SIX2 haploinsufficiency causes conductive hearing loss with ptosis in humans. J. Hum. Genet. 61, 917–922. doi: 10.1038/jhg.2016.86
Guo, C., Sun, Y., Zhou, B., Adam, R. M., Li, X., Pu, W. T., et al. (2011). A Tbx1-Six1/Eya1-Fgf8 genetic pathway controls mammalian cardiovascular and craniofacial morphogenesis. J. Clin. Invest. 121, 1585–1595. doi: 10.1172/jci44630
Harper, P. S. (1975). Congenital myotonic dystrophy in Britain. I. Clinical aspects. Arch. Dis. Child 50, 505–513. doi: 10.1136/adc.50.7.505
Harris, S. E., Winchester, C. L., and Johnson, K. J. (2000). Functional analysis of the homeodomain protein SIX5. Nucleic Acids Res. 28, 1871–1878. doi: 10.1093/nar/28.9.1871
He, G., Tavella, S., Hanley, K. P., Self, M., Oliver, G., Grifone, R., et al. (2010). Inactivation of Six2 in mouse identifies a novel genetic mechanism controlling development and growth of the cranial base. Dev. Biol. 344, 720–730. doi: 10.1016/j.ydbio.2010.05.509
He, Q., Lin, Z., Wang, Z., Huang, W., Tian, D., Liu, M., et al. (2020). SIX4 promotes hepatocellular carcinoma metastasis through upregulating YAP1 and c-MET. Oncogene 39, 7279–7295. doi: 10.1038/s41388-020-01500-y
Hehr, U., Pineda-Alvarez, D. E., Uyanik, G., Hu, P., Zhou, N., Hehr, A., et al. (2010). Heterozygous mutations in SIX3 and SHH are associated with schizencephaly and further expand the clinical spectrum of holoprosencephaly. Hum. Genet. 127, 555–561. doi: 10.1007/s00439-010-0797-4
Hendry, C. E., Vanslambrouck, J. M., Ineson, J., Suhaimi, N., Takasato, M., Rae, F., et al. (2013). Direct transcriptional reprogramming of adult cells to embryonic nephron progenitors. J. Am. Soc. Nephrol. 24, 1424–1434. doi: 10.1681/asn.2012121143
Hoskins, B. E., Cramer, C. H., Silvius, D., Zou, D., Raymond, R. M., Orten, D. J., et al. (2007). Transcription factor SIX5 is mutated in patients with branchio-oto-renal syndrome. Am. J. Hum. Genet. 80, 800–804. doi: 10.1086/513322
Hou, H., Yu, X., Cong, P., Zhou, Y., Xu, Y., and Jiang, Y. (2019). Six2 promotes non-small cell lung cancer cell stemness via transcriptionally and epigenetically regulating E-cadherin. Cell Prolif 52:e12617.
Hu, S., Mamedova, A., and Hegde, R. S. (2008). DNA-binding and regulation mechanisms of the SIX family of retinal determination proteins. Biochemistry 47, 3586–3594. doi: 10.1021/bi702186s
Hua, L., Fan, L., Aichun, W., Yongjin, Z., Qingqing, C., and Xiaojian, W. (2014). Inhibition of Six1 promotes apoptosis, suppresses proliferation, and migration of osteosarcoma cells. Tumour Biol. 35, 1925–1931. doi: 10.1007/s13277-013-1258-1
Hufnagel, R. B., Zimmerman, S. L., Krueger, L. A., Bender, P. L., Ahmed, Z. M., and Saal, H. M. (2016). A new frontonasal dysplasia syndrome associated with deletion of the SIX2 gene. Am. J. Med. Genet. A 170A, 487–491. doi: 10.1002/ajmg.a.37441
Hwang, D. Y., Dworschak, G. C., Kohl, S., Saisawat, P., Vivante, A., Hilger, A. C., et al. (2014). Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney Int. 85, 1429–1433. doi: 10.1038/ki.2013.508
Iglesias, A. I., Springelkamp, H., Van Der Linde, H., Severijnen, L. A., Amin, N., Oostra, B., et al. (2014). Exome sequencing and functional analyses suggest that SIX6 is a gene involved in an altered proliferation-differentiation balance early in life and optic nerve degeneration at old age. Hum. Mol, Genet. 23, 1320–1332. doi: 10.1093/hmg/ddt522
Ikeda, K., Watanabe, Y., Ohto, H., and Kawakami, K. (2002). Molecular interaction and synergistic activation of a promoter by Six, Eya, and Dach proteins mediated through CREB binding protein. Mol. Cell Biol. 22, 6759–6766. doi: 10.1128/mcb.22.19.6759-6766.2002
Imam, J. S., Buddavarapu, K., Lee-Chang, J. S., Ganapathy, S., Camosy, C., Chen, Y., et al. (2010). MicroRNA-185 suppresses tumor growth and progression by targeting the Six1 oncogene in human cancers. Oncogene 29, 4971–4979. doi: 10.1038/onc.2010.233
Iwanaga, R., Wang, C. A., Micalizzi, D. S., Harrell, J. C., Jedlicka, P., Sartorius, C. A., et al. (2012). Expression of Six1 in luminal breast cancers predicts poor prognosis and promotes increases in tumor initiating cells by activation of extracellular signal-regulated kinase and transforming growth factor-beta signaling pathways. Breast Cancer Res. 14:R100.
Jean, D., Bernier, G., and Gruss, P. (1999). Six6 (Optx2) is a novel murine Six3-related homeobox gene that demarcates the presumptive pituitary/hypothalamic axis and the ventral optic stalk. Mech. Dev. 84, 31–40. doi: 10.1016/s0925-4773(99)00068-4
Jeong, Y., Leskow, F. C., El-Jaick, K., Roessler, E., Muenke, M., Yocum, A., et al. (2008). Regulation of a remote Shh forebrain enhancer by the Six3 homeoprotein. Nat. Genet. 40, 1348–1353. doi: 10.1038/ng.230
Ji, Q., Zhu, J., Fang, C. L., Jin, H., Zhan, D. P., and Huang, J. (2020). Down-regulation of MIAT suppresses osteosarcoma progression by acting as a ceRNA for miR-141-3p to regulate SIX1-mediated PI3K/AKT pathway. Eur. Rev. Med. Pharmacol. Sci. 24, 2218–2228.
Jia, Q., Ye, L., Xu, S., Xiao, H., Xu, S., Shi, Z., et al. (2020). Circular RNA 0007255 regulates the progression of breast cancer through miR-335-5p/SIX2 axis. Thorac. Cancer 11, 619–630. doi: 10.1111/1759-7714.13306
Jin, A., Xu, Y., Liu, S., Jin, T., Li, Z., Jin, H., et al. (2014). Sineoculis homeobox homolog 1 protein overexpression as an independent biomarker for pancreatic ductal adenocarcinoma. Exp. Mol. Pathol. 96, 54–60. doi: 10.1016/j.yexmp.2013.11.003
Kawakami, K., Sato, S., Ozaki, H., and Ikeda, K. (2000). Six family genes–structure and function as transcription factors and their roles in development. Bioessays 22, 616–626. doi: 10.1002/1521-1878(200007)22:7<616::aid-bies4>3.0.co;2-r
Kingsbury, T. J., Kim, M., and Civin, C. I. (2019). Regulation of cancer stem cell properties by SIX1, a member of the PAX-SIX-EYA-DACH network. Adv. Cancer Res. 141, 1–42. doi: 10.1016/bs.acr.2018.12.001
Klesert, T. R., Cho, D. H., Clark, J. I., Maylie, J., Adelman, J., Snider, L., et al. (2000). Mice deficient in Six5 develop cataracts: implications for myotonic dystrophy. Nat. Genet. 25, 105–109. doi: 10.1038/75490
Klesert, T. R., Otten, A. D., Bird, T. D., and Tapscott, S. J. (1997). Trinucleotide repeat expansion at the myotonic dystrophy locus reduces expression of DMAHP. Nat. Genet. 16, 402–406. doi: 10.1038/ng0897-402
Kobayashi, A., Valerius, M. T., Mugford, J. W., Carroll, T. J., Self, M., Oliver, G., et al. (2008). Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell 3, 169–181. doi: 10.1016/j.stem.2008.05.020
Kobayashi, H., Kawakami, K., Asashima, M., and Nishinakamura, R. (2007). Six1 and Six4 are essential for Gdnf expression in the metanephric mesenchyme and ureteric bud formation, while Six1 deficiency alone causes mesonephric-tubule defects. Mech. Dev. 124, 290–303. doi: 10.1016/j.mod.2007.01.002
Kobayashi, M., Nishikawa, K., Suzuki, T., and Yamamoto, M. (2001). The homeobox protein Six3 interacts with the Groucho corepressor and acts as a transcriptional repressor in eye and forebrain formation. Dev. Biol. 232, 315–326. doi: 10.1006/dbio.2001.0185
Kobayashi, M., Toyama, R., Takeda, H., Dawid, I. B., and Kawakami, K. (1998). Overexpression of the forebrain-specific homeobox gene six3 induces rostral forebrain enlargement in zebrafish. Development 125, 2973–2982. doi: 10.1242/dev.125.15.2973
Kochhar, A., Orten, D. J., Sorensen, J. L., Fischer, S. M., Cremers, C. W., Kimberling, W. J., et al. (2008). SIX1 mutation screening in 247 branchio-oto-renal syndrome families: a recurrent missense mutation associated with BOR. Hum. Mutat. 29:565. doi: 10.1002/humu.20714
Kong, D., Li, A., Liu, Y., Cui, Q., Wang, K., Zhang, D., et al. (2019). SIX1 activates STAT3 signaling to promote the proliferation of thyroid carcinoma via EYA1. Front. Oncol. 9:1450. doi: 10.3389/fonc.2019.01450
Konishi, Y., Ikeda, K., Iwakura, Y., and Kawakami, K. (2006). Six1 and Six4 promote survival of sensory neurons during early trigeminal gangliogenesis. Brain Res. 1116, 93–102. doi: 10.1016/j.brainres.2006.07.103
Krug, P., Moriniere, V., Marlin, S., Koubi, V., Gabriel, H. D., Colin, E., et al. (2011). Mutation screening of the EYA1, SIX1, and SIX5 genes in a large cohort of patients harboring branchio-oto-renal syndrome calls into question the pathogenic role of SIX5 mutations. Hum. Mutat. 32, 183–190. doi: 10.1002/humu.21402
Kumar, J. P. (2009). The sine oculis homeobox (SIX) family of transcription factors as regulators of development and disease. Cell Mol. Life Sci. 66, 565–583. doi: 10.1007/s00018-008-8335-4
Kumar, R., Balasenthil, S., Manavathi, B., Rayala, S. K., and Pakala, S. B. (2010). Metastasis-associated protein 1 and its short form variant stimulates Wnt1 transcription through promoting its derepression from Six3 corepressor. Cancer Res. 70, 6649–6658. doi: 10.1158/0008-5472.can-10-0909
Kutejova, E., Engist, B., Self, M., Oliver, G., Kirilenko, P., and Bobola, N. (2008). Six2 functions redundantly immediately downstream of Hoxa2. Development 135, 1463–1470. doi: 10.1242/dev.017624
Lacbawan, F., Solomon, B. D., Roessler, E., El-Jaick, K., Domene, S., Velez, J. I., et al. (2009). Clinical spectrum of SIX3-associated mutations in holoprosencephaly: correlation between genotype, phenotype and function. J. Med. Genet. 46, 389–398.
Laclef, C., Hamard, G., Demignon, J., Souil, E., Houbron, C., and Maire, P. (2003). Altered myogenesis in Six1-deficient mice. Development 130, 2239–2252. doi: 10.1242/dev.00440
Lagutin, O. V., Zhu, C. C., Kobayashi, D., Topczewski, J., Shimamura, K., Puelles, L., et al. (2003). Six3 repression of Wnt signaling in the anterior neuroectoderm is essential for vertebrate forebrain development. Genes Dev. 17, 368–379. doi: 10.1101/gad.1059403
Larder, R., Clark, D. D., Miller, N. L., and Mellon, P. L. (2011). Hypothalamic dysregulation and infertility in mice lacking the homeodomain protein Six6. J. Neurosci. 31, 426–438. doi: 10.1523/jneurosci.1688-10.2011
Laukkanen, S., Oksa, L., Nikkila, A., Lahnalampi, M., Parikka, M., Seki, M., et al. (2020). SIX6 is a TAL1-regulated transcription factor in T-ALL and associated with inferior outcome. Leuk Lymphoma 61, 3089–3100. doi: 10.1080/10428194.2020.1804560
Ledford, K. L., Martinez-De Luna, R. I., Theisen, M. A., Rawlins, K. D., Viczian, A. S., and Zuber, M. E. (2017). Distinct cis-acting regions control six6 expression during eye field and optic cup stages of eye formation. Dev. Biol. 426, 418–428. doi: 10.1016/j.ydbio.2017.04.003
Lee, B., Rizzoti, K., Kwon, D. S., Kim, S. Y., Oh, S., Epstein, D. J., et al. (2012). Direct transcriptional regulation of Six6 is controlled by SoxB1 binding to a remote forebrain enhancer. Dev. Biol. 366, 393–403. doi: 10.1016/j.ydbio.2012.04.023
Lengler, J., and Graw, J. (2001). Regulation of the human SIX3 gene promoter. Biochem. Biophys. Res. Commun. 287, 372–376. doi: 10.1006/bbrc.2001.5605
Lerbs, T., Bisht, S., Scholch, S., Pecqueux, M., Kristiansen, G., Schneider, M., et al. (2017). Inhibition of Six1 affects tumour invasion and the expression of cancer stem cell markers in pancreatic cancer. BMC Cancer 17:249. doi: 10.1186/s12885-017-3225-5
Li, G., Hu, F., Luo, X., Hu, J., and Feng, Y. (2017). SIX4 promotes metastasis via activation of the PI3K-AKT pathway in colorectal cancer. PeerJ 5:e3394. doi: 10.7717/peerj.3394
Li, H. L., Sun, J. J., Ma, H., Liu, S. J., Li, N., Guo, S. J., et al. (2019). MicroRNA-23a inhibits endometrial cancer cell development by targeting SIX1. Oncol. Lett. 18, 3792–3802.
Li, J., Zhang, T., Ramakrishnan, A., Fritzsch, B., Xu, J., Wong, E. Y. M., et al. (2020). Dynamic changes in cis-regulatory occupancy by Six1 and its cooperative interactions with distinct cofactors drive lineage-specific gene expression programs during progressive differentiation of the auditory sensory epithelium. Nucleic Acids Res. 48, 2880–2896. doi: 10.1093/nar/gkaa012
Li, J. W., Huang, C. Z., Li, J. H., Yuan, J. H., Chen, Q. H., Zhang, W. F., et al. (2018). Six2 is negatively correlated with good prognosis and decreases 5-FU sensitivity via suppressing E-cadherin expression in hepatocellular carcinoma cells. Biomed. Pharmacother. 104, 204–210. doi: 10.1016/j.biopha.2018.05.032
Li, L., Liang, Y., Kang, L., Liu, Y., Gao, S., Chen, S., et al. (2018). Transcriptional regulation of the warburg effect in cancer by SIX1. Cancer Cell 33, 368.e7–385.e7.
Li, X., Oghi, K. A., Zhang, J., Krones, A., Bush, K. T., Glass, C. K., et al. (2003). Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature 426, 247–254. doi: 10.1038/nature02083
Li, X., Perissi, V., Liu, F., Rose, D. W., and Rosenfeld, M. G. (2002). Tissue-specific regulation of retinal and pituitary precursor cell proliferation. Science 297, 1180–1183.
Li, Z., Tian, T., Lv, F., Chang, Y., Wang, X., Zhang, L., et al. (2013). Six1 promotes proliferation of pancreatic cancer cells via upregulation of cyclin D1 expression. PLoS One 8:e59203. doi: 10.1371/journal.pone.0059203
Lindqvist, B. M., Wingren, S., Motlagh, P. B., and Nilsson, T. K. (2014). Whole genome DNA methylation signature of HER2-positive breast cancer. Epigenetics 9, 1149–1162. doi: 10.4161/epi.29632
Litsiou, A., Hanson, S., and Streit, A. (2005). A balance of FGF, BMP and WNT signalling positions the future placode territory in the head. Development 132, 4051–4062. doi: 10.1242/dev.01964
Liu, D., Li, L., Zhang, X. X., Wan, D. Y., Xi, B. X., Hu, Z., et al. (2014a). SIX1 promotes tumor lymphangiogenesis by coordinating TGFbeta signals that increase expression of VEGF-C. Cancer Res. 74, 5597–5607. doi: 10.1158/0008-5472.can-13-3598
Liu, D., Zhang, X. X., Xi, B. X., Wan, D. Y., Li, L., Zhou, J., et al. (2014b). Sine oculis homeobox homolog 1 promotes DNA replication and cell proliferation in cervical cancer. Int. J. Oncol. 45, 1232–1240. doi: 10.3892/ijo.2014.2510
Liu, P., Cai, S., and Li, N. (2020). Circular RNA-hsa-circ-0000670 promotes gastric cancer progression through the microRNA-384/SIX4 axis. Exp. Cell Res. 394:112141. doi: 10.1016/j.yexcr.2020.112141
Liu, Q., Li, A., Tian, Y., Liu, Y., Li, T., Zhang, C., et al. (2016). The expression profile and clinic significance of the SIX family in non-small cell lung cancer. J. Hematol. Oncol. 9:119.
Liu, W., and Cvekl, A. (2017). Six3 in a small population of progenitors at E8.5 is required for neuroretinal specification via regulating cell signaling and survival in mice. Dev. Biol. 428, 164–175. doi: 10.1016/j.ydbio.2017.05.026
Liu, W., Lagutin, O., Swindell, E., Jamrich, M., and Oliver, G. (2010). Neuroretina specification in mouse embryos requires Six3-mediated suppression of Wnt8b in the anterior neural plate. J. Clin. Invest. 120, 3568–3577. doi: 10.1172/jci43219
Liu, W., Lagutin, O. V., Mende, M., Streit, A., and Oliver, G. (2006). Six3 activation of Pax6 expression is essential for mammalian lens induction and specification. EMBO J. 25, 5383–5395. doi: 10.1038/sj.emboj.7601398
Liu, Z., Li, C., Xu, J., Lan, Y., Liu, H., Li, X., et al. (2019a). Crucial and overlapping roles of six1 and six2 in craniofacial development. J. Dent. Res. 98, 572–579. doi: 10.1177/0022034519835204
Liu, Z., Mar, K. B., Hanners, N. W., Perelman, S. S., Kanchwala, M., Xing, C., et al. (2019b). A NIK-SIX signalling axis controls inflammation by targeted silencing of non-canonical NF-kappaB. Nature 568, 249–253. doi: 10.1038/s41586-019-1041-6
Lopez-Rios, J., Tessmar, K., Loosli, F., Wittbrodt, J., and Bovolenta, P. (2003). Six3 and Six6 activity is modulated by members of the groucho family. Development 130, 185–195. doi: 10.1242/dev.00185
Lu, K., Reddy, R., Berika, M., Warburton, D., and El-Hashash, A. H. (2013). Abrogation of Eya1/Six1 disrupts the saccular phase of lung morphogenesis and causes remodeling. Dev. Biol. 382, 110–123. doi: 10.1016/j.ydbio.2013.07.019
Lyu, Z., Mao, Z., Wang, H., Fang, Y., Chen, T., Wan, Q., et al. (2013). MiR-181b targets Six2 and inhibits the proliferation of metanephric mesenchymal cells in vitro. Biochem. Biophys. Res. Commun. 440, 495–501. doi: 10.1016/j.bbrc.2013.09.059
Mahadevan, M., Tsilfidis, C., Sabourin, L., Shutler, G., Amemiya, C., Jansen, G., et al. (1992). Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science 255, 1253–1255. doi: 10.1126/science.1546325
Maire, P., Dos Santos, M., Madani, R., Sakakibara, I., Viaut, C., and Wurmser, M. (2020). Myogenesis control by SIX transcriptional complexes. Semin. Cell. Dev. Biol. 104, 51–64. doi: 10.1016/j.semcdb.2020.03.003
Manavathi, B., Peng, S., Rayala, S. K., Talukder, A. H., Wang, M. H., Wang, R. A., et al. (2007). Repression of Six3 by a corepressor regulates rhodopsin expression. Proc. Natl. Acad. Sci. U.S.A. 104, 13128–13133. doi: 10.1073/pnas.0705878104
Mao, Z., Lyu, Z., Huang, L., Zhou, Q., and Weng, Y. (2017). TbetaRII regulates the proliferation of metanephric mesenchyme cells through Six2 in vitro. Int. J. Mol. Sci. 18:853. doi: 10.3390/ijms18040853
Marcinkiewicz, K. M., and Gudas, L. J. (2014). Altered histone mark deposition and DNA methylation at homeobox genes in human oral squamous cell carcinoma. J. Cell Physiol. 229, 1405–1416. doi: 10.1002/jcp.24577
Matynia, A., Ng, C. H., Dansithong, W., Chiang, A., Silva, A. J., and Reddy, S. (2010). Muscleblind1, but not Dmpk or Six5, contributes to a complex phenotype of muscular and motivational deficits in mouse models of myotonic dystrophy. PLoS One 5:e9857. doi: 10.1371/journal.pone.0009857
McCoy, E. L., Iwanaga, R., Jedlicka, P., Abbey, N. S., Chodosh, L. A., Heichman, K. A., et al. (2009). Six1 expands the mouse mammary epithelial stem/progenitor cell pool and induces mammary tumors that undergo epithelial-mesenchymal transition. J. Clin. Invest. 119, 2663–2677. doi: 10.1172/jci37691
Melnick, M., Bixler, D., Nance, W. E., Silk, K., and Yune, H. (1976). Familial branchio-oto-renal dysplasia: a new addition to the branchial arch syndromes. Clin. Genet. 9, 25–34. doi: 10.1111/j.1399-0004.1976.tb01546.x
Micalizzi, D. S., Christensen, K. L., Jedlicka, P., Coletta, R. D., Baron, A. E., Harrell, J. C., et al. (2009). The Six1 homeoprotein induces human mammary carcinoma cells to undergo epithelial-mesenchymal transition and metastasis in mice through increasing TGF-beta signaling. J. Clin. Invest. 119, 2678–2690. doi: 10.1172/jci37815
Micalizzi, D. S., Wang, C. A., Farabaugh, S. M., Schiemann, W. P., and Ford, H. L. (2010). Homeoprotein Six1 increases TGF-beta type I receptor and converts TGF-beta signaling from suppressive to supportive for tumor growth. Cancer Res. 70, 10371–10380. doi: 10.1158/0008-5472.can-10-1354
Mimae, T., Okada, M., Hagiyama, M., Miyata, Y., Tsutani, Y., Inoue, T., et al. (2012). Upregulation of notch2 and six1 is associated with progression of early-stage lung adenocarcinoma and a more aggressive phenotype at advanced stages. Clin. Cancer Res. 18, 945–955. doi: 10.1158/1078-0432.ccr-11-1946
Mo, M. L., Okamoto, J., Chen, Z., Hirata, T., Mikami, I., Bosco-Clement, G., et al. (2013). Down-regulation of SIX3 is associated with clinical outcome in lung adenocarcinoma. PLoS One 8:e71816. doi: 10.1371/journal.pone.0071816
Mohanty, K., Dada, R., and Dada, T. (2018). Identification and genotype phenotype correlation of novel mutations in SIX6 gene in primary open angle glaucoma. Ophthalmic Genet. 39, 366–372. doi: 10.1080/13816810.2018.1432062
Mosrati, M. A., Hammami, B., Rebeh, I. B., Ayadi, L., Dhouib, L., Ben Mahfoudh, K., et al. (2011). A novel dominant mutation in SIX1, affecting a highly conserved residue, result in only auditory defects in humans. Eur. J. Med. Genet. 54, e484–e488.
Murphy, A. J., Pierce, J., De Caestecker, C., Taylor, C., Anderson, J. R., Perantoni, A. O., et al. (2012). SIX2 and CITED1, markers of nephronic progenitor self-renewal, remain active in primitive elements of Wilms’ tumor. J. Pediatr. Surg. 47, 1239–1249. doi: 10.1016/j.jpedsurg.2012.03.034
Murakami, Y., Ohto, H., Ikeda, U., Shimada, K., Momoi, T., and Kawakami, K. (1998). Promoter of mDMAHP/Six5: differential utilization of multiple transcription initiation sites and positive/negative regulatory elements. Hum. Mol. Genet. 7, 2103–2112. doi: 10.1093/hmg/7.13.2103
Na, X. Y., Shang, X. S., Zhao, Y., Ren, P. P., and Hu, X. Q. (2019). MiR-203a functions as a tumor suppressor in bladder cancer by targeting SIX4. Neoplasma 66, 211–221. doi: 10.4149/neo_2018_180512n312
Ng, K. T., Man, K., Sun, C. K., Lee, T. K., Poon, R. T., Lo, C. M., et al. (2006). Clinicopathological significance of homeoprotein Six1 in hepatocellular carcinoma. Br. J. Cancer 95, 1050–1055. doi: 10.1038/sj.bjc.6603399
Nishimura, T., Tamaoki, M., Komatsuzaki, R., Oue, N., Taniguchi, H., Komatsu, M., et al. (2017). SIX1 maintains tumor basal cells via transforming growth factor-beta pathway and associates with poor prognosis in esophageal cancer. Cancer Sci. 108, 216–225. doi: 10.1111/cas.13135
O’Brien, J. H., Hernandez-Lagunas, L., Artinger, K. B., and Ford, H. L. (2014). MicroRNA-30a regulates zebrafish myogenesis through targeting the transcription factor Six1. J. Cell Sci. 127, 2291–2301.
O’Brien, L. L., Guo, Q., Bahrami-Samani, E., Park, J. S., and Hasso, S. M. (2018). Transcriptional regulatory control of mammalian nephron progenitors revealed by multi-factor cistromic analysis and genetic studies. PLoS Genet. 14:e1007181. doi: 10.1371/journal.pgen.1007181
O’Brien, L. L., Guo, Q., Lee, Y., Tran, T., Benazet, J. D., Whitney, P. H., et al. (2016). Differential regulation of mouse and human nephron progenitors by the Six family of transcriptional regulators. Development 143, 595–608. doi: 10.1242/dev.127175
Ogawa, Y., Shiraki, T., Asano, Y., Muto, A., Kawakami, K., Suzuki, Y., et al. (2019). Six6 and Six7 coordinately regulate expression of middle-wavelength opsins in zebrafish. Proc. Natl. Acad. Sci. U.S.A. 116, 4651–4660. doi: 10.1073/pnas.1812884116
Ohto, H., Kamada, S., Tago, K., Tominaga, S. I., Ozaki, H., Sato, S., et al. (1999). Cooperation of six and eya in activation of their target genes through nuclear translocation of Eya. Mol. Cell Biol. 19, 6815–6824. doi: 10.1128/mcb.19.10.6815
Ohto, H., Takizawa, T., Saito, T., Kobayashi, M., Ikeda, K., and Kawakami, K. (1998). Tissue and developmental distribution of Six family gene products. Int. J. Dev. Biol. 42, 141–148.
Okello, D. O., Iyyanar, P. P. R., Kulyk, W. M., Smith, T. M., Lozanoff, S., Ji, S., et al. (2017). Six2 Plays an intrinsic role in regulating proliferation of mesenchymal cells in the developing palate. Front. Physiol. 8:955. doi: 10.3389/fphys.2017.00955
Oliphant, M. U. J., Vincent, M. Y., Galbraith, M. D., Pandey, A., Zaberezhnyy, V., Rudra, P., et al. (2019). SIX2 mediates late-stage metastasis via direct regulation of SOX2 and induction of a cancer stem cell program. Cancer Res. 79, 720–734. doi: 10.1158/0008-5472.can-18-1791
Oliver, G., Mailhos, A., Wehr, R., Copeland, N. G., Jenkins, N. A., and Gruss, P. (1995). Six3, a murine homologue of the sine oculis gene, demarcates the most anterior border of the developing neural plate and is expressed during eye development. Development 121, 4045–4055. doi: 10.1242/dev.121.12.4045
Omer, D., Harari-Steinberg, O., Buzhor, E., Metsuyanim, S., Pleniceanu, O., Zundelevich, A., et al. (2013). Chromatin-modifying agents reactivate embryonic renal stem/progenitor genes in human adult kidney epithelial cells but abrogate dedifferentiation and stemness. Cell Reprogram 15, 281–292. doi: 10.1089/cell.2012.0087
Ono, H., Imoto, I., Kozaki, K., Tsuda, H., Matsui, T., Kurasawa, Y., et al. (2012). SIX1 promotes epithelial-mesenchymal transition in colorectal cancer through ZEB1 activation. Oncogene 31, 4923–4934. doi: 10.1038/onc.2011.646
Ozaki, H., Nakamura, K., Funahashi, J., Ikeda, K., Yamada, G., Tokano, H., et al. (2004). Six1 controls patterning of the mouse otic vesicle. Development 131, 551–562.
Ozaki, H., Watanabe, Y., Takahashi, K., Kitamura, K., Tanaka, A., Urase, K., et al. (2001). Six4, a putative myogenin gene regulator, is not essential for mouse embryonal development. Mol. Cell Biol. 21, 3343–3350. doi: 10.1128/mcb.21.10.3343-3350.2001
Park, J. S., Ma, W., O’brien, L. L., Chung, E., Guo, J. J., Cheng, J. G., et al. (2012). Six2 and Wnt regulate self-renewal and commitment of nephron progenitors through shared gene regulatory networks. Dev. Cell 23, 637–651. doi: 10.1016/j.devcel.2012.07.008
Park, J. S., Valerius, M. T., and Mcmahon, A. P. (2007). Wnt/beta-catenin signaling regulates nephron induction during mouse kidney development. Development 134, 2533–2539. doi: 10.1242/dev.006155
Patrick, A. N., Schiemann, B. J., Yang, K., Zhao, R., and Ford, H. L. (2009). Biochemical and functional characterization of six SIX1 Branchio-oto-renal syndrome mutations. J. Biol. Chem. 284, 20781–20790. doi: 10.1074/jbc.m109.016832
Personius, K. E., Nautiyal, J., and Reddy, S. (2005). Myotonia and muscle contractile properties in mice with SIX5 deficiency. Muscle Nerve 31, 503–505. doi: 10.1002/mus.20239
Pierce, J., Murphy, A. J., Panzer, A., De Caestecker, C., Ayers, G. D., Neblett, D., et al. (2014). SIX2 effects on wilms tumor biology. Transl. Oncol. 7, 800–811. doi: 10.1016/j.tranon.2014.09.005
Qiu, R., Liu, Y., Wu, J. Y., Liu, K., Mo, W., and He, R. (2009). Misexpression of miR-196a induces eye anomaly in Xenopus laevis. Brain Res. Bull. 79, 26–31. doi: 10.1016/j.brainresbull.2008.12.009
Relaix, F., Demignon, J., Laclef, C., Pujol, J., Santolini, M., Niro, C., et al. (2013). Six homeoproteins directly activate Myod expression in the gene regulatory networks that control early myogenesis. PLoS Genet. 9:e1003425. doi: 10.1371/journal.pgen.1003425
Reichenberger, K. J., Coletta, R. D., Schulte, A. P., Varella-Garcia, M., and Ford, H. L. (2005). Gene amplification is a mechanism of Six1 overexpression in breast cancer. Cancer Res. 65, 2668–2675. doi: 10.1158/0008-5472.CAN-04-4286
Riddiford, N., and Schlosser, G. (2017). Six1 and Eya1 both promote and arrest neuronal differentiation by activating multiple Notch pathway genes. Dev. Biol. 431, 152–167. doi: 10.1016/j.ydbio.2017.09.027
Rodriguez, S., Sickles, H. M., Deleonardis, C., Alcaraz, A., Gridley, T., and Lin, D. M. (2008). Notch2 is required for maintaining sustentacular cell function in the adult mouse main olfactory epithelium. Dev. Biol. 314, 40–58. doi: 10.1016/j.ydbio.2007.10.056
Ruf, R. G., Xu, P. X., Silvius, D., Otto, E. A., Beekmann, F., Muerb, U. T., et al. (2004). SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes. Proc. Natl. Acad. Sci. U.S.A. 101, 8090–8095. doi: 10.1073/pnas.0308475101
Rupaimoole, R., and Slack, F. J. (2017). MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat. Rev. Drug. Discov. 16, 203–222. doi: 10.1038/nrd.2016.246
Samuel, A., Rubinstein, A. M., Azar, T. T., Ben-Moshe Livne, Z., Kim, S. H., and Inbal, A. (2016). Six3 regulates optic nerve development via multiple mechanisms. Sci. Rep. 6:20267.
Sarkar, P. S., Appukuttan, B., Han, J., Ito, Y., Ai, C., Tsai, W., et al. (2000). Heterozygous loss of Six5 in mice is sufficient to cause ocular cataracts. Nat. Genet. 25, 110–114. doi: 10.1038/75500
Sarkar, P. S., Paul, S., Han, J., and Reddy, S. (2004). Six5 is required for spermatogenic cell survival and spermiogenesis. Hum. Mol. Genet. 13, 1421–1431. doi: 10.1093/hmg/ddh161
Sato, S., Ikeda, K., Shioi, G., Nakao, K., Yajima, H., and Kawakami, K. (2012). Regulation of Six1 expression by evolutionarily conserved enhancers in tetrapods. Dev. Biol. 368, 95–108. doi: 10.1016/j.ydbio.2012.05.023
Sato, S., Yajima, H., Furuta, Y., Ikeda, K., and Kawakami, K. (2015). Activation of Six1 expression in vertebrate sensory neurons. PLoS One 10:e0136666. doi: 10.1371/journal.pone.0136666
Self, M., Lagutin, O. V., Bowling, B., Hendrix, J., Cai, Y., Dressler, G. R., et al. (2006). Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J. 25, 5214–5228. doi: 10.1038/sj.emboj.7601381
Senanayake, U., Koller, K., Pichler, M., Leuschner, I., Strohmaier, H., Hadler, U., et al. (2013). The pluripotent renal stem cell regulator SIX2 is activated in renal neoplasms and influences cellular proliferation and migration. Hum. Pathol. 44, 336–345. doi: 10.1016/j.humpath.2012.05.021
Serikaku, M. A., and O’Tousa, J. E. (1994). sine oculis is a homeobox gene required for Drosophila visual system development. Genetics 138, 1137–1150. doi: 10.1093/genetics/138.4.1137
Shah, A. M., Krohn, P., Baxi, A. B., Tavares, A. L. P., Sullivan, C. H., Chillakuru, Y. R., et al. (2020). Six1 proteins with human branchio-oto-renal mutations differentially affect cranial gene expression and otic development. Dis. Model Mech. 13:dmm043489.
Shi, C., and Zhang, Z. (2017). MicroRNA-362 is downregulated in cervical cancer and inhibits cell proliferation, migration and invasion by directly targeting SIX1. Oncol. Rep. 37, 501–509. doi: 10.3892/or.2016.5242
Shimamura, K., and Rubenstein, J. L. (1997). Inductive interactions direct early regionalization of the mouse forebrain. Development 124, 2709–2718. doi: 10.1242/dev.124.14.2709
Smith, A. L., Iwanaga, R., Drasin, D. J., Micalizzi, D. S., Vartuli, R. L., Tan, A. C., et al. (2012). The miR-106b-25 cluster targets Smad7, activates TGF-beta signaling, and induces EMT and tumor initiating cell characteristics downstream of Six1 in human breast cancer. Oncogene 31, 5162–5171. doi: 10.1038/onc.2012.11
Song, D., Yue, L., Wu, G., Ma, S., Guo, L., Yang, H., et al. (2015). Assessment of promoter methylation and expression of SIX2 as a diagnostic and prognostic biomarker in Wilms’ tumor. Tumour. Biol. 36, 7591–7598. doi: 10.1007/s13277-015-3456-5
Song, M. H., Kwon, T. J., Kim, H. R., Jeon, J. H., Baek, J. I., Lee, W. S., et al. (2013). Mutational analysis of EYA1, SIX1 and SIX5 genes and strategies for management of hearing loss in patients with BOR/BO syndrome. PLoS One 8:e67236. doi: 10.1371/journal.pone.0067236
Song, W., Ma, J., Lei, B., Yuan, X., Cheng, B., Yang, H., et al. (2019). Sine oculis homeobox 1 promotes proliferation and migration of human colorectal cancer cells through activation of Wnt/beta-catenin signaling. Cancer Sci. 110, 608–616. doi: 10.1111/cas.13905
Spitz, F., Demignon, J., Porteu, A., Kahn, A., Concordet, J. P., Daegelen, D., et al. (1998). Expression of myogenin during embryogenesis is controlled by Six/sine oculis homeoproteins through a conserved MEF3 binding site. Proc. Natl. Acad. Sci. U.S.A. 95, 14220–14225. doi: 10.1073/pnas.95.24.14220
Stokes, B., Berger, S. I., Hall, B. A., Weiss, K., Martinez, A. F., Hadley, D. W., et al. (2018). SIX3 deletions and incomplete penetrance in families affected by holoprosencephaly. Congenit Anom (Kyoto) 58, 29–32. doi: 10.1111/cga.12234
Sun, S. H., Liu, D., Deng, Y. T., Zhang, X. X., Wan, D. Y., Xi, B. X., et al. (2016). SIX1 coordinates with TGFbeta signals to induce epithelial-mesenchymal transition in cervical cancer. Oncol. Lett. 12, 1271–1278. doi: 10.3892/ol.2016.4797
Sun, X., Hu, F., Hou, Z., Chen, Q., Lan, J., Luo, X., et al. (2019). SIX4 activates Akt and promotes tumor angiogenesis. Exp. Cell Res. 383:111495. doi: 10.1016/j.yexcr.2019.111495
Sun, X., Ma, J., Chen, Q., Hou, Z., Luo, X., Wang, G., et al. (2020). SIX4 promotes metastasis through STAT3 activation in breast cancer. Am. J. Cancer Res. 10, 224–236.
Sun, X. J., Wang, M. C., Zhang, F. H., and Kong, X. (2018). An integrated analysis of genome-wide DNA methylation and gene expression data in hepatocellular carcinoma. FEBS Open Bio 8, 1093–1103. doi: 10.1002/2211-5463.12433
Sweat, Y. Y., Sweat, M., Mansaray, M., Cao, H., Eliason, S., Adeyemo, W. L., et al. (2020). Six2 regulates Pax9 expression, palatogenesis and craniofacial bone formation. Dev. Biol. 458, 246–256. doi: 10.1016/j.ydbio.2019.11.010
Takahashi, M., Tamura, M., Sato, S., and Kawakami, K. (2018). Mice doubly deficient in Six4 and Six5 show ventral body wall defects reproducing human omphalocele. Dis. Model Mech. 11:dmm034611.
Takata, N., Abbey, D., Fiore, L., Acosta, S., Feng, R., Gil, H. J., et al. (2017). An eye organoid approach identifies Six3 suppression of R-spondin 2 as a critical step in mouse neuroretina differentiation. Cell Rep. 21, 1534–1549. doi: 10.1016/j.celrep.2017.10.041
Talbot, J. C., Teets, E. M., Ratnayake, D., Duy, P. Q., Currie, P. D., and Amacher, S. L. (2019). Muscle precursor cell movements in zebrafish are dynamic and require Six family genes. Development 146:dev171421.
Tang, X., Yang, Y., Song, X., Liu, X., Wang, X., Huang, F., et al. (2019). SIX4 acts as a master regulator of oncogenes that promotes tumorigenesis in non-small-cell lung cancer cells. Biochem. Biophys. Res. Commun. 516, 851–857. doi: 10.1016/j.bbrc.2019.06.114
Tavares, A. L. P., Cox, T. C., Maxson, R. M., Ford, H. L., and Clouthier, D. E. (2017). Negative regulation of endothelin signaling by SIX1 is required for proper maxillary development. Development 144, 2021–2031.
Teotia, P., Van Hook, M. J., Wichman, C. S., Allingham, R. R., Hauser, M. A., and Ahmad, I. (2017). Modeling glaucoma: retinal ganglion cells generated from induced pluripotent stem cells of patients with SIX6 risk allele show developmental abnormalities. Stem Cells 35, 2239–2252. doi: 10.1002/stem.2675
Tetreault, N., Champagne, M. P., and Bernier, G. (2009). The LIM homeobox transcription factor Lhx2 is required to specify the retina field and synergistically cooperates with Pax6 for Six6 trans-activation. Dev. Biol. 327, 541–550. doi: 10.1016/j.ydbio.2008.12.022
Thornton, C. A., Wymer, J. P., Simmons, Z., Mcclain, C., and Moxley, R. T. 3rd (1997). Expansion of the myotonic dystrophy CTG repeat reduces expression of the flanking DMAHP gene. Nat. Genet. 16, 407–409. doi: 10.1038/ng0897-407
Towers, C. G., Guarnieri, A. L., Micalizzi, D. S., Harrell, J. C., Gillen, A. E., Kim, J., et al. (2015). The Six1 oncoprotein downregulates p53 via concomitant regulation of RPL26 and microRNA-27a-3p. Nat. Commun. 6:10077.
Turcu, D. C., Lillehaug, J. R., and Seo, H. C. (2019). SIX3 and SIX6 interact with GEMININ via C-terminal regions. Biochem. Biophys. Rep. 20:100695. doi: 10.1016/j.bbrep.2019.100695
Wakimoto, H., Maguire, C. T., Sherwood, M. C., Vargas, M. M., Sarkar, P. S., Han, J., et al. (2002). Characterization of cardiac conduction system abnormalities in mice with targeted disruption of Six5 gene. J. Interv. Card. Electrophysiol. 7, 127–135.
Wallis, D. E., Roessler, E., Hehr, U., Nanni, L., Wiltshire, T., Richieri-Costa, A., et al. (1999). Mutations in the homeodomain of the human SIX3 gene cause holoprosencephaly. Nat. Genet. 22, 196–198. doi: 10.1038/9718
Walz, A. L., Ooms, A., Gadd, S., Gerhard, D. S., Smith, M. A., Guidry Auvil, J. M., et al. (2015). Recurrent DGCR8, DROSHA, and SIX homeodomain mutations in favorable histology Wilms tumors. Cancer Cell 27, 286–297. doi: 10.1016/j.ccell.2015.01.003
Wan, Z. H., Ma, Y. H., Jiang, T. Y., Lin, Y. K., Shi, Y. Y., Tan, Y. X., et al. (2019). Six2 is negatively correlated with prognosis and facilitates epithelial-mesenchymal transition via TGF-beta/Smad signal pathway in hepatocellular carcinoma. Hepatobiliary Pancreat Dis. Int. 18, 525–531. doi: 10.1016/j.hbpd.2019.09.005
Wang, C., Gargollo, P., Guo, C., Tang, T., Mingin, G., Sun, Y., et al. (2011). Six1 and Eya1 are critical regulators of peri-cloacal mesenchymal progenitors during genitourinary tract development. Dev. Biol. 360, 186–194. doi: 10.1016/j.ydbio.2011.09.020
Wang, C. A., Drasin, D., Pham, C., Jedlicka, P., Zaberezhnyy, V., Guney, M., et al. (2014). Homeoprotein Six2 promotes breast cancer metastasis via transcriptional and epigenetic control of E-cadherin expression. Cancer Res. 74, 7357–7370. doi: 10.1158/0008-5472.can-14-0666
Wang, H., Rao, B., Lou, J., Li, J., Liu, Z., Li, A., et al. (2020). The function of the HGF/c-Met axis in hepatocellular carcinoma. Front. Cell Dev. Biol. 8:55. doi: 10.3389/fcell.2020.00055
Wang, S. H., Wu, C. C., Lu, Y. C., Lin, Y. H., Su, Y. N., Hwu, W. L., et al. (2012). Mutation screening of the EYA1, SIX1, and SIX5 genes in an East Asian cohort with branchio-oto-renal syndrome. Laryngoscope 122, 1130–1136. doi: 10.1002/lary.23217
Weber, S., Taylor, J. C., Winyard, P., Baker, K. F., Sullivan-Brown, J., Schild, R., et al. (2008). SIX2 and BMP4 mutations associate with anomalous kidney development. J. Am. Soc. Nephrol. 19, 891–903. doi: 10.1681/asn.2006111282
Wegert, J., Ishaque, N., Vardapour, R., Georg, C., Gu, Z., Bieg, M., et al. (2015). Mutations in the SIX1/2 pathway and the DROSHA/DGCR8 miRNA microprocessor complex underlie high-risk blastemal type Wilms tumors. Cancer Cell 27, 298–311. doi: 10.1016/j.ccell.2015.01.002
Wei, D. W., Ma, X. Y., Zhang, S., Hong, J. Y., Gui, L. S., Mei, C. G., et al. (2017). Characterization of the promoter region of the bovine SIX1 gene: roles of MyoD, PAX7, CREB and MyoG. Sci. Rep. 7:12599.
Winchester, C., Robertson, S., Macleod, T., Johnson, K., and Thomas, M. (2000). Expression of a homeobox gene (SIX5) in borderline ovarian tumours. J. Clin. Pathol. 53, 212–217. doi: 10.1136/jcp.53.3.212
Wu, D. W., Lin, P. L., Wang, L., Huang, C. C., and Lee, H. (2017). The YAP1/SIX2 axis is required for DDX3-mediated tumor aggressiveness and cetuximab resistance in KRAS-wild-type colorectal cancer. Theranostics 7, 1114–1132. doi: 10.7150/thno.18175
Wurmser, M., Chaverot, N., Madani, R., Sakai, H., Negroni, E., Demignon, J., et al. (2020). SIX1 and SIX4 homeoproteins regulate PAX7+ progenitor cell properties during fetal epaxial myogenesis. Development 147: dev185975.
Xie, H., Hoffmann, H. M., Meadows, J. D., Mayo, S. L., Trang, C., Leming, S. S., et al. (2015). Homeodomain proteins SIX3 and SIX6 regulate gonadotrope-specific genes during pituitary development. Mol. Endocrinol. 29, 842–855. doi: 10.1210/me.2014-1279
Xu, H. X., Wu, K. J., Tian, Y. J., Liu, Q., Han, N., He, X. L., et al. (2016). Expression profile of SIX family members correlates with clinic-pathological features and prognosis of breast cancer: a systematic review and meta-analysis. Medicine (Baltimore) 95:e4085. doi: 10.1097/md.0000000000004085
Xu, J., Li, J., Zhang, T., Jiang, H., Ramakrishnan, A., Fritzsch, B., et al. (2021). Chromatin remodelers and lineage-specific factors interact to target enhancers to establish proneurosensory fate within otic ectoderm. Proc. Natl. Acad. Sci. U.S.A. 118:e2025196118. doi: 10.1073/pnas.2025196118
Xu, J., Wong, E. Y., Cheng, C., Li, J., Sharkar, M. T., Xu, C. Y., et al. (2014). Eya1 interacts with Six2 and Myc to regulate expansion of the nephron progenitor pool during nephrogenesis. Dev. Cell 31, 434–447. doi: 10.1016/j.devcel.2014.10.015
Xu, J., and Xu, P. X. (2015). Eya-six are necessary for survival of nephrogenic cord progenitors and inducing nephric duct development before ureteric bud formation. Dev. Dyn. 244, 866–873. doi: 10.1002/dvdy.24282
Xu, P. X., Zheng, W., Huang, L., Maire, P., Laclef, C., and Silvius, D. (2003). Six1 is required for the early organogenesis of mammalian kidney. Development 130, 3085–3094. doi: 10.1242/dev.00536
Yallowitz, A. R., Gong, K. Q., Swinehart, I. T., Nelson, L. T., and Wellik, D. M. (2009). Non-homeodomain regions of Hox proteins mediate activation versus repression of Six2 via a single enhancer site in vivo. Dev. Biol. 335, 156–165. doi: 10.1016/j.ydbio.2009.08.020
Yamamoto-Shiraishi, Y., and Kuroiwa, A. (2013). Wnt and BMP signaling cooperate with Hox in the control of Six2 expression in limb tendon precursor. Dev. Biol. 377, 363–374. doi: 10.1016/j.ydbio.2013.02.023
Yang, Z., Feng, Z., Gu, J., Li, X., Dong, Q., Liu, K., et al. (2017). microRNA-488 inhibits chemoresistance of ovarian cancer cells by targeting Six1 and mitochondrial function. Oncotarget 8, 80981–80993. doi: 10.18632/oncotarget.20941
Yang, Z. C., Qu, Z. H., Yi, M. J., Shan, Y. C., Ran, N., Xu, L., et al. (2019). MiR-448-5p inhibits TGF-beta1-induced epithelial-mesenchymal transition and pulmonary fibrosis by targeting Six1 in asthma. J. Cell. Physiol. 234, 8804–8814. doi: 10.1002/jcp.27540
Yanovsky-Dagan, S., Avitzour, M., Altarescu, G., Renbaum, P., Eldar-Geva, T., Schonberger, O., et al. (2015). Uncovering the role of hypermethylation by CTG expansion in myotonic dystrophy Type 1 using mutant human embryonic stem cells. Stem Cell Rep. 5, 221–231. doi: 10.1016/j.stemcr.2015.06.003
Yariz, K. O., Sakalar, Y. B., Jin, X., Hertz, J., Sener, E. F., Akay, H., et al. (2015). A homozygous SIX6 mutation is associated with optic disc anomalies and macular atrophy and reduces retinal ganglion cell differentiation. Clin. Genet. 87, 192–195. doi: 10.1111/cge.12374
Yin, Q., Wang, H., Li, N., Ding, Y., Xie, Z., Jin, L., et al. (2020). Dosage effect of multiple genes accounts for multisystem disorder of myotonic dystrophy type 1. Cell Res. 30, 133–145. doi: 10.1038/s41422-019-0264-2
Yu, C., Zhang, B., Li, Y. L., and Yu, X. R. (2018). SIX1 reduces the expression of PTEN via activating PI3K/AKT signal to promote cell proliferation and tumorigenesis in osteosarcoma. Biomed. Pharmacother. 105, 10–17. doi: 10.1016/j.biopha.2018.04.028
Yu, Y., Davicioni, E., Triche, T. J., and Merlino, G. (2006). The homeoprotein six1 transcriptionally activates multiple protumorigenic genes but requires ezrin to promote metastasis. Cancer Res. 66, 1982–1989. doi: 10.1158/0008-5472.can-05-2360
Yu, Y., Khan, J., Khanna, C., Helman, L., Meltzer, P. S., and Merlino, G. (2004). Expression profiling identifies the cytoskeletal organizer ezrin and the developmental homeoprotein Six-1 as key metastatic regulators. Nat. Med. 10, 175–181. doi: 10.1038/nm966
Yu, Z., Feng, J., Wang, W., Deng, Z., Zhang, Y., Xiao, L., et al. (2020). The EGFR-ZNF263 signaling axis silences SIX3 in glioblastoma epigenetically. Oncogene 39, 3163–3178. doi: 10.1038/s41388-020-1206-7
Yu, Z., Sun, Y., She, X., Wang, Z., Chen, S., Deng, Z., et al. (2017). SIX3, a tumor suppressor, inhibits astrocytoma tumorigenesis by transcriptional repression of AURKA/B. J. Hematol. Oncol. 10:115.
Zeng, J., Shi, R., Cai, C. X., Liu, X. R., Song, Y. B., Wei, M., et al. (2015). Increased expression of Six1 correlates with progression and prognosis of prostate cancer. Cancer Cell Int. 15:63.
Zhang, B., Shen, C., Ge, F., Ma, T., and Zhang, Z. (2017). Epigenetically controlled Six3 expression regulates glioblastoma cell proliferation and invasion alongside modulating the activation levels of WNT pathway members. J. Neurooncol. 133, 509–518. doi: 10.1007/s11060-017-2476-y
Zhang, M., Shi, H., Zhang, C., and Zhang, S. Q. (2019). MiRNA-621 inhibits the malignant progression of non-small cell lung cancer via targeting SIX4. Eur. Rev. Med. Pharmacol. Sci. 23, 4807–4814.
Zhang, T., Xu, J., Maire, P., and Xu, P. X. (2017). Six1 is essential for differentiation and patterning of the mammalian auditory sensory epithelium. PLoS Genet. 13:e1006967. doi: 10.1371/journal.pgen.1006967
Zhao, Y., Zhou, H., Ma, K., Sun, J., Feng, X., Geng, J., et al. (2013). Abnormal methylation of seven genes and their associations with clinical characteristics in early stage non-small cell lung cancer. Oncol. Lett. 5, 1211–1218. doi: 10.3892/ol.2013.1161
Zheng, W., Huang, L., Wei, Z. B., Silvius, D., Tang, B., and Xu, P. X. (2003). The role of Six1 in mammalian auditory system development. Development 130, 3989–4000. doi: 10.1242/dev.00628
Zheng, Y., Zeng, Y., Qiu, R., Liu, R., Huang, W., Hou, Y., et al. (2018). The homeotic protein SIX3 suppresses carcinogenesis and metastasis through recruiting the LSD1/NuRD(MTA3) complex. Theranostics 8, 972–989. doi: 10.7150/thno.22328
Zhou, H., Blevins, M. A., Hsu, J. Y., Kong, D., Galbraith, M. D., Goodspeed, A., et al. (2020). Identification of a small-molecule inhibitor that disrupts the SIX1/EYA2 complex, EMT, and metastasis. Cancer Res. 80, 2689–2702. doi: 10.1158/0008-5472.can-20-0435
Zhu, C. C., Dyer, M. A., Uchikawa, M., Kondoh, H., Lagutin, O. V., and Oliver, G. (2002). Six3-mediated auto repression and eye development requires its interaction with members of the Groucho-related family of co-repressors. Development 129, 2835–2849. doi: 10.1242/dev.129.12.2835
Zhu, S. M., Chen, C. M., Jiang, Z. Y., Yuan, B., Ji, M., Wu, F. H., et al. (2016). MicroRNA-185 inhibits cell proliferation and epithelial-mesenchymal transition in hepatocellular carcinoma by targeting Six2. Eur. Rev. Med. Pharmacol. Sci. 20, 1712–1719.
Keywords: transcription factor, SIX genes, developmental biology, cancer, congenital disease
Citation: Meurer L, Ferdman L, Belcher B and Camarata T (2021) The SIX Family of Transcription Factors: Common Themes Integrating Developmental and Cancer Biology. Front. Cell Dev. Biol. 9:707854. doi: 10.3389/fcell.2021.707854
Received: 10 May 2021; Accepted: 28 June 2021;
Published: 19 August 2021.
Edited by:
Monica Fedele, Istituto per l’Endocrinologia e l’Oncologia Gaetano Salvatore (CNR), ItalyReviewed by:
Giovanna Lucia Liguori, Institute of Genetics and Biophysics (CNR), ItalyCopyright © 2021 Meurer, Ferdman, Belcher and Camarata. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Troy Camarata, dGNhbWFyYXRAbnlpdC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.