 Chong Zhang
Chong Zhang Rui Huang
Rui Huang Jiehui Chen
Jiehui Chen Li Li
Li Li Honghui Huang
Honghui Huang- Key Laboratory of Freshwater Fish Reproduction and Development, Ministry of Education, State Key Laboratory Breeding Base of Eco-Environments and Bio-Resources of the Three Gorges Reservoir Region, School of Life Sciences, Southwest University, Chongqing, China
Ribosome biogenesis is a fundamental activity in cells. Ribosomal dysfunction underlies a category of diseases called ribosomopathies in humans. The symptomatic characteristics of ribosomopathies often include abnormalities in craniofacial skeletons, digestive organs, and hematopoiesis. Consistently, disruptions of ribosome biogenesis in animals are deleterious to embryonic development with hypoplasia of digestive organs and/or impaired hematopoiesis. In this study, ltv1, a gene involved in the small ribosomal subunit assembly, was knocked out in zebrafish by clustered regularly interspaced short palindromic repeats (CRISPRs)/CRISPR associated protein 9 (Cas9) technology. The recessive lethal mutation resulted in disrupted ribosome biogenesis, and ltv1Δ14/Δ14 embryos displayed hypoplastic craniofacial cartilage, digestive organs, and hematopoiesis. In addition, we showed that the impaired cell proliferation, instead of apoptosis, led to the defects in exocrine pancreas and hematopoietic stem and progenitor cells (HSPCs) in ltv1Δ14/Δ14 embryos. It was reported that loss of function of genes associated with ribosome biogenesis often caused phenotypes in a P53-dependent manner. In ltv1Δ14/Δ14 embryos, both P53 protein level and the expression of p53 target genes, Δ113p53 and p21, were upregulated. However, knockdown of p53 failed to rescue the phenotypes in ltv1Δ14/Δ14 larvae. Taken together, our data demonstrate that LTV1 ribosome biogenesis factor (Ltv1) plays an essential role in digestive organs and hematopoiesis development in zebrafish in a P53-independent manner.
Introduction
The ribosome is a fundamental macromolecular machine, found within all living cells, that synthesizes proteins according to mRNA sequences. Ribosome biogenesis is a very intricate process in cells (Warner, 2001). Eukaryotic ribosome consists of the large 60S and small 40S subunits, which are assembled to form the functional 80S ribosome. In addition to the four ribosomal RNAs (rRNAs) and 82 core ribosomal proteins, which are the components of the 80S ribosome, over 200 non-ribosomal proteins are involved in ribosome biogenesis. This precisely controlled process is inextricably associated with many fundamental cellular activities, such as growth and division (Panse and Johnson, 2010). Disruption of ribosome biogenesis leads to a class of human genetic diseases, collectively termed as ribosomopathies (Narla and Ebert, 2010). Although these diseases are all related to the ribosome dysfunction, ribosomopathies display different clinical manifestations and mechanisms. The symptomatic features of ribosomopathies often include craniofacial defects, digestive organs dysplasia, hematological abnormalities, and the increased risk of some blood cancers (Narla and Ebert, 2010). Ribosomopathies with defects in digestive organs and/or hematological abnormalities include Shwachman-Diamond syndrome (SDS), 5q-syndrome, Diamond-Blackfan anemia (DBA), X-linked dyskeratosis congenita (DC), Treacher Collins syndrome (TCS), and North American Indian childhood cirrhosis (NAIC) (Armistead and Triggs-Raine, 2014).
Numerous genetic models have been established for the investigation of the mechanisms underlying ribosomopathies. In mice, conditional deletion of the syntenic region, including Rps14, absent in 5q-syndrome leads to macrocytic anemia, which is the key clinical feature of the disease (Barlow et al., 2010). In zebrafish, knockdown of rps19 expression causes hematopoietic and developmental abnormalities that is similar to the symptoms of DBA (Danilova et al., 2008). Besides DBA, zebrafish models of SDS (Provost et al., 2012; Carapito et al., 2017; Oyarbide et al., 2020), 5q-syndrome (Ear et al., 2016), NAIC (Wilkins et al., 2013), and DC (Zhang et al., 2012; Anchelin et al., 2013) were generated and characterized. These models are valuable resources to develop potential therapies of ribosomopathies according to the underlying mechanisms. In addition to causative genes in ribosomopathies, some other genes involved in ribosome biogenesis were either mutated or knocked down in zebrafish, such as bms1l (Wang et al., 2012, 2016), kri1l (Jia et al., 2015), nol9 (Bielczyk-Maczynska et al., 2015), nom1 (Qin et al., 2014), and pwp2h (Boglev et al., 2013). Depletion of these genes causes disrupted rRNA processing and leads to defects in digestive organs and/or hematological abnormalities, which suggest common roles for ribosome biogenesis factors in organogenesis.
Evidences from a number of animal models of ribosomopathies suggest that P53 is often activated in ribosome dysfunction (Danilova et al., 2008, 2011; Jones et al., 2008; Fumagalli et al., 2009; Barlow et al., 2010; Pereboom et al., 2011; Taylor et al., 2012; Zhang et al., 2012; Boglev et al., 2013; Wilkins et al., 2013; Qin et al., 2014; Bielczyk-Maczynska et al., 2015; Ear et al., 2016). In some cases, inhibition of p53 is able to rescue the phenotypes (Danilova et al., 2008, 2011; Jones et al., 2008; Barlow et al., 2010; Pereboom et al., 2011; Taylor et al., 2012; Zhang et al., 2012; Bielczyk-Maczynska et al., 2015; Ear et al., 2016), but not in others (Provost et al., 2012; Boglev et al., 2013; Jia et al., 2015). These studies suggest that targeting the P53 pathway could be a therapeutic strategy. However, it should be noted that being a tumor suppression gene, p53 inhibition may increase risk of cancer development.
LTV1 ribosome biogenesis factor (Ltv1) is a non-ribosomal factor required for the processing of 40S ribosomal subunit (Ameismeier et al., 2018; Collins et al., 2018). Alterations of LTV1 can cause aberrant processing of 18S rRNA in yeast, fruit fly and human cells (Seiser et al., 2006; Tafforeau et al., 2011; Ghalei et al., 2015; Kressler et al., 2015). In ΔLTV1 yeast cells, the accumulation of 18S rRNA precursors (20S, 21S, and 23S rRNA) is evident, accounted by the decreased pre-rRNA cleavage at sites A0, A1, and A2 (Seiser et al., 2006). Similarly, in fruit fly and human cells, LTV1 deficiency leads to increased level of 21S rRNA, hence a reduced production of the final 18S rRNA and eventually a higher than expected ratio of 28S/18S rRNA (Tafforeau et al., 2011; Kressler et al., 2015). Cell growth is inhibited in LTV1 loss-of-function yeast strains (Seiser et al., 2006). The fruit fly LTV1 mutant larvae exhibit development delay and lethality at the second larvae stage (Kressler et al., 2015). These studies suggest the conserved role of LTV1 in ribosome biogenesis and cell growth from yeast to multicellular animals. However, the function of LTV1 in vertebrate development remains poorly understood.
Here, we reported that knockout of ltv1 in zebrafish embryo disrupted ribosome biogenesis. The zebrafish ltv1Δ14/Δ14 larvae displayed aberrant cartilage structure, defects in digestive organs, characterized by smaller size of liver, intestine and exocrine pancreas, and impaired definitive hematopoiesis. Further characterization of ltv1Δ14/Δ14 larvae showed that the decreased proliferation gave rise to the dysplastic features of exocrine pancreas and hematopoietic stem and progenitor cells (HSPCs). Although P53 and its target genesΔ113p53 and p21 were upregulated, knockdown of p53 failed to rescue the developmental abnormalities in ltv1Δ14/Δ14 mutant.
Results
Craniofacial Cartilage Was Defective in ltv1Δ14/Δ14 Zebrafish Mutant Embryo
Ltv1 is highly conserved by amino acid sequence homology between human and zebrafish, with approximately 60.9% identity and 76.2% similarity (Supplementary Figure 1). To determine the function of ltv1, zebrafish ltv1–/– mutants were generated using the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR associated protein 9 (Cas9)-mediated approach, and a guide RNA (gRNA) was designed to target the exon 7 of ltv1. Two F1 mutant alleles were identified with 14 and 7 bp nucleotides deletion, respectively in the coding region (Figure 1A). Both mutations were predicted to result in frame shifts and premature stop codons in mutant transcripts, encoding two truncated Ltv1 proteins with 284 and 283 N-terminal and 32 and 31 missense amino acids, respectively (Figure 1B). These two mutant alleles could not genetically complement each other, and the ltv1Δ14/Δ14 mutant allele was used for the following experiments. RNA whole mount in situ hybridization (WISH) showed that ltv1 transcripts were almost absent in ltv1Δ14/Δ14 mutant at 3 days post fertilization (dpf), indicating that the knockout of ltv1 was successful (Figure 1C). The mutant mRNA probably underwent a nonsense-mediated decay.
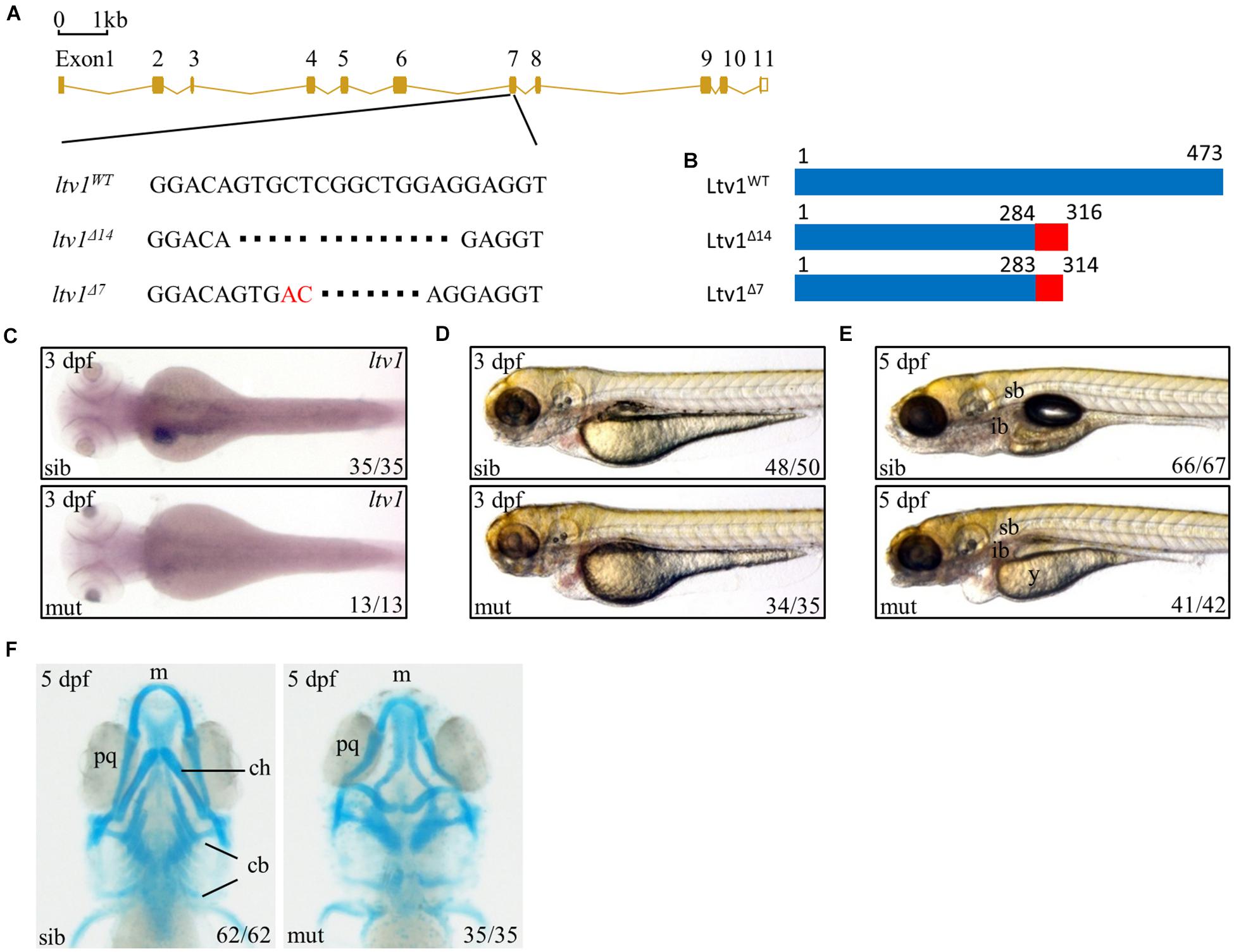
Figure 1. Knockout of ltv1 and craniofacial defects in ltv1Δ14/Δ14 mutant. (A) The sequences flanking the deletion site in ltv1 mutants (ltv1Δ7,14) are shown. The deleted nucleotides are denoted by dashes. In ltv1Δ7/Δ7 mutant, 9 bp nucleotides are deleted and 2 bp nucleotides (red font) are inserted. (B) Two ltv1 mutant alleles (ltv1Δ14 and ltv1Δ7) harbor frameshift mutations that are predicted to give rise to truncated Ltv1 proteins as shown. (C) WISH using ltv1 antisense RNA probe at 3 dpf. (D,E) Representative images of ltv1Δ14/Δ14 mutants and siblings at 3 and 5 dpf. (F) Alcian blue-labeled craniofacial cartilage in ventral view at 5 dpf. Abbreviations used: sb, swimming bladder; ib, intestinal bulb; y, yolk; m, Meckel’s; pq, palatoquadrate; ch, ceratohyal; cb, ceratobranchial.
The ltv1Δ14/Δ14 mutant embryos were morphologically indistinguishable from siblings before 2 dpf with normal blood flow and heart beating. However, at 3 dpf, ltv1Δ14/Δ14 mutants displayed pericardial edema and aplasia in the head (Figure 1D). At 5 dpf, mutants exhibited underdeveloped intestine, smaller liver, uninflated swim bladder, and impaired yolk absorption (Figure 1E). These phenotypes were completely penetrant and the mutant larvae died from 8 to 11 dpf.
Disruption of ribosome biogenesis can cause abnormal craniofacial skeletons in zebrafish (Mayer and Fishman, 2003; Provost et al., 2012; Qin et al., 2014). Thus, Alcian blue staining was performed to check the craniofacial cartilage structure of ltv1Δ14/Δ14 mutant. At 5 dpf, mutants displayed severe abnormalities in craniofacial cartilage, including smaller Meckel’s cartilage, curly palatoquadrate and lack of ceratohyal, and five ceratobranchial cartilage (Figure 1F).
Digestive Organs Were Hypoplastic in ltv1Δ14/Δ14 Mutant Embryo
To further characterize the digestive organ phenotype observed in bright field, WISH was performed to analyze the specific organ formation. Both the liver (marked by fabp10) and exocrine pancreas (marked by trypsin) of ltv1Δ14/Δ14 displayed a smaller size compared with sibling at 3 dpf (Figures 2A–C). However, no visible defect was found in the endocrine pancreas (marked by insulin) (Figure 2D). In zebrafish, differentiated intestinal cells include three types: enterocytes, goblet cells, and enteroendocrine cells (Chen et al., 2009). In ltv1Δ14/Δ14 mutant, enterocytes (marked by fabp2) at 3 dpf (Figure 2E) and goblet cells (Alcian blue-stained) at 5 dpf (Figures 2F,H) were substantially decreased in number. In zebrafish, both the enteroendocrine and goblet cells of the intestine could be labeled by 2F11 monoclonal antibody (Roach et al., 2013). In zebrafish, goblet cells are only distributed in the posterior part (Roach et al., 2013), not in the intestine bulb, so the 2F11 antibody marked cells in the intestine bulb are enteroendocrine cells. The number of enteroendocrine cells in the intestine bulb was reduced significantly in ltv1Δ14/Δ14 mutant at 4 dpf (Figures 2G,H). To examine the gut morphology, DCFH-DA, a dye that could label zebrafish gut lumen, was used to visualize the intestine. At 5 dpf, although the overall shape of the mutant intestine resembled that of the sibling, the lumen was narrower than that of the sibling (Figure 2I).
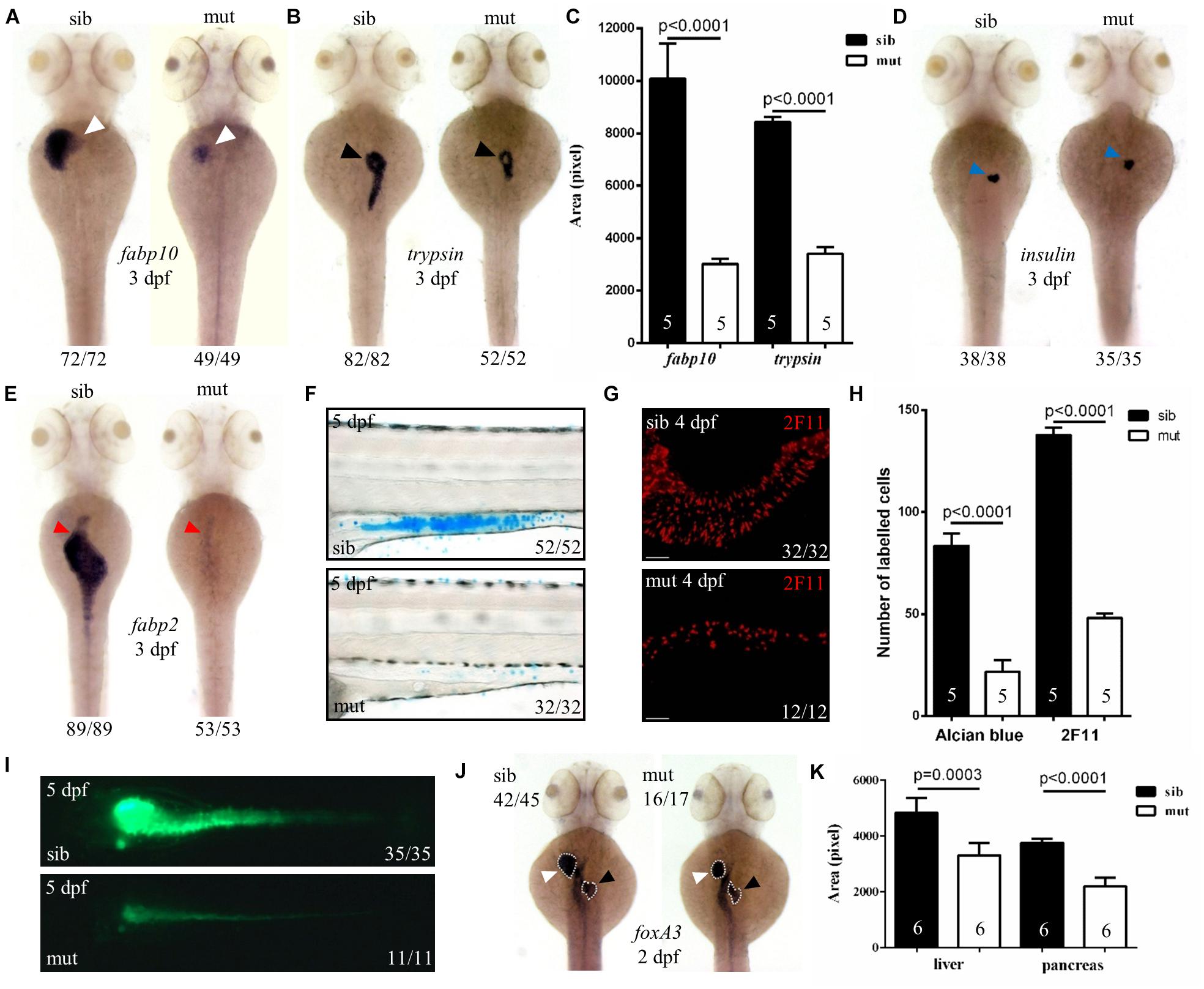
Figure 2. Defects in the development of digestive organs in ltv1Δ14/Δ14 mutant. (A,B,D,E) WISH using the hepatocyte marker fabp10, acinar cell marker trypsin, beta cell marker insulin, and enterocyte marker fabp2 at 3 dpf. (C) Quantification of fabp10 (siblings, N = 5; mutants, N = 5) or trypsin (siblings, N = 5; mutants, N = 5) positive area in panel (A) or (B), respectively. Bars represent means with SD. (F) Alcian blue-stained goblet cells at 5 dpf. (G) Enteroendocrine cells (2F11+) in the intestine bulb at 4 dpf. (H) Quantification of Alcian blue-stained (siblings, N = 5; mutants, N = 5) or 2F11-positive (siblings, N = 5; mutants, N = 5) cells in panel (F) or (G), respectively. Bars represent means with SD. (I) Gut lumen labeled by DCFH-DA at 5 dpf. (J) WISH using the pan-endodermal marker foxA3 at 2 dpf. (K) Quantification of the liver and pancreas area labeled by foxA3 (siblings, N = 6; mutants, N = 6) in panel (J). Bars represent means with SD. White arrowhead: liver; black arrowhead: exocrine pancreas; blue arrowhead: endocrine pancreas; red arrowhead: intestine.
Developmental defects of digestive organs could be due to impaired differentiation of endodermal cells. The genes foxA1, foxA3, and gata6 are early endodermal markers that can also label digestive organ primordia in zebrafish (Tao and Peng, 2009). These three genes expressed normally in the ltv1Δ14/Δ14 mutant endoderm at 1 dpf (data not shown). Both liver and pancreatic buds were found to be smaller in the mutant than that in the sibling, while the intestine seemed normal at 2 dpf (Figures 2J,K and Supplementary Figures 2A,B). These data suggested that the process from the endoderm to bud initiation was intact whereas bud expansion, taking place at a later stage, was affected in the mutant. To test whether liver specification was impaired in the ltv1Δ14/Δ14 mutant, two of the earliest markers of hepatoblasts, prox1 and hhex, were analyzed (Ober et al., 2006). Consistent with foxA1, foxA3, and gata6, the expression of prox1 and hhex revealed a slightly smaller liver bud in the mutant compared with the sibling at 2 dpf (Supplementary Figures 2C,D). A noticeable hypoplastic liver phenotype in the ltv1Δ14/Δ14 mutants could be observed at 34 hours post fertilization (hpf) by tracing the prox1 expression at earlier developmental time points (Supplementary Figures 2E,F). There are two types of glandular tissue in the zebrafish pancreas: exocrine pancreas and endocrine pancreas (Field et al., 2003). By checking pdx1 (precursor cell of endocrine pancreas), gcga (alpha cell), insulin (beta cell), and sst2 (delta cell) expression at 2 dpf, no obvious defect was observed in ltv1Δ14/Δ14 mutants (Supplementary Figures 2G–J). These data suggested that cell differentiation of endocrine pancreas was not affected in the mutant. However, the number of ptf1a+ cells (exocrine pancreas progenitor cells) decreased significantly at 2 dpf in the ltv1Δ14/Δ14 mutant on the ptf1a:gfp background (Supplementary Figures 2K,L). Thus, hypoplasia of digestive organs in ltv1Δ14/Δ14 mutant embryos could be a consequence of impaired organ progenitor cell expansion.
The ltv1Δ14/Δ14 Mutation Impaired Definitive Hematopoiesis During Embryogenesis
Hematopoietic defects are usually related to the ribosome biogenesis gene deficiency in zebrafish (Oyarbide et al., 2019). Therefore, to figure out the role of ltv1 in hematopoiesis, different blood cell lineages were examined. HSPCs (marked by c-myb, ikaros, and runx1 transgene) in the mutant were pronouncedly reduced in the caudal hematopoietic tissue (CHT), thymus and kidney at 4 dpf (Figures 3A–D). In addition, blood cell lineage markers, such as gata1 (erythrocyte progenitors), αe1 (erythrocytes), mfap4 (macrophages), csf1ra (macrophages), lyz (neutrophils), rag1 (lymphocytes), and Sudan Black (neutrophils) staining, were all significantly reduced in the ltv1Δ14/Δ14 mutant at 4 dpf (Figures 3E–L), indicating the impaired development of definitive erythrocytes, myeloid cells, and lymphocytes.
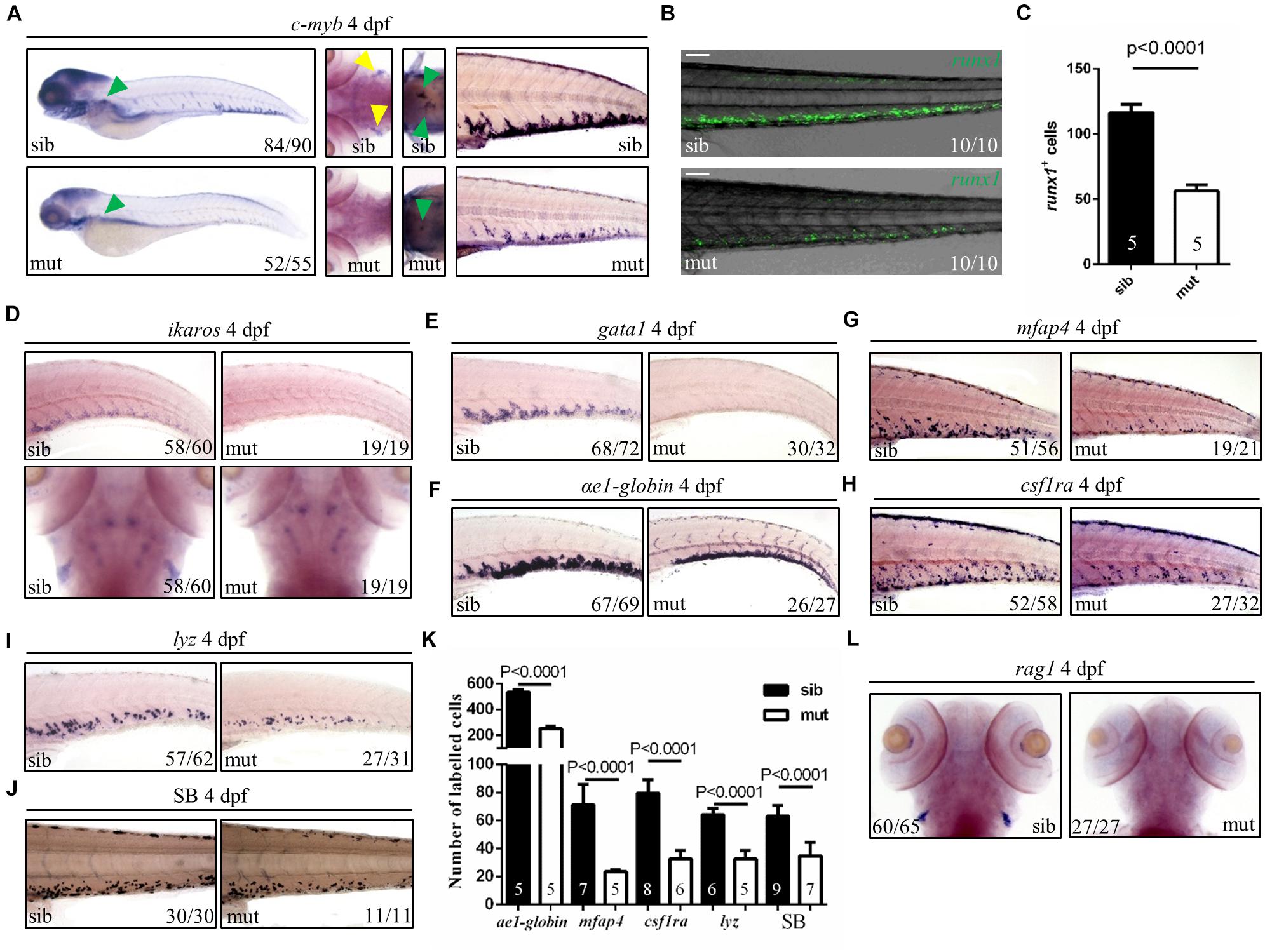
Figure 3. Defects of definitive hematopoiesis in ltv1Δ14/Δ14 mutant. (A) WISH of HSPC marker c-myb in the thymus, kidney and CHT at 4 dpf. (B) Representative confocal images of ltv1Δ14/Δ14; runx1:en-gfp mutants and siblings at 4 dpf. (C) Quantification of runx1+ cells of ltv1Δ14/Δ14; runx1:en-gfp mutants (N = 5) and siblings (N = 5) at 4 dpf. Bars represent means with SD. (D) WISH of HSPC marker ikaros in the CHT and thymus at 4 dpf. (E–I) WISH of erythrocyte progenitor marker gata1 (E), erythrocyte marker αe1-globin (F), macrophage markers mfap4 (G) and csf1ra (H), and neutrophil marker lyz (I) in the CHT at 4 dpf. (J) Sudan Black-stained neutrophils in the CHT at 4 dpf. (K) Quantification of αe1-globin, mfap4, csf1ra, lyz, and SB-positive (Sudan Black) cells in panels (F–J), respectively. Numbers of each group are shown in the respective columns. Bars represent means with SD. (L) WISH of lymphocyte marker rag1 at 4 dpf. Yellow arrowhead: thymus; green arrowhead: kidney. Scale bar: 100 μm.
Two waves of hematopoiesis are involved in zebrafish: the primitive wave and the definitive wave (Jagannathan-Bogdan and Zon, 2013). To assess the status of primitive hematopoiesis in the ltv1Δ14/Δ14 mutant, two genes regulating the primitive erythroid and myeloid fates, gata1 and pu.1, were examined using WISH at 20 and 22 hpf, respectively, and no visible defect was observed in the mutant (Supplementary Figures 3A,B). The results of c-myb expression from 2 to 4 dpf showed that a decreased c-myb expression was detectable starting from 3 dpf (Figure 3A and Supplementary Figures 4A–C). Taken together, the primitive hematopoiesis was unaffected while the definitive hematopoiesis was impaired in the ltv1Δ14/Δ14 mutant, probably due to the reduced HSPCs.
To confirm whether ltv1 mutation is indeed responsible for the mutant phenotypes observed, zebrafish wild type and mutant form ltv1 mRNAs were used for rescue experiments. At 3 dpf, both the smaller liver and reduced HSPC phenotypes in the ltv1Δ14/Δ14 mutant were rescued by zebrafish wild-type ltv1 mRNA efficiently but not by the mutant form (Supplementary Figures 5A–D).
Proliferation of Exocrine Pancreas Progenitor Cells and Hematopoietic Stem and Progenitor Cells in ltv1Δ14/Δ14 Mutant Embryo Was Significantly Reduced
Disrupted cell proliferation and/or enhanced apoptosis may account for the digestive organs and hematopoiesis defects. The terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay revealed no apoptotic cell in the pancreas region in sectioned ltv1Δ14/Δ14 mutant and sibling at 3 dpf, indicating that apoptosis is not the reason of the dysplastic exocrine pancreas in the mutant (Supplementary Figure 6A). In order to detect the level of cell proliferation, phospho-Histone H3 (pH3) immunostaining and bromodeoxyuridine (BrdU) labeling experiments were performed in mutants and siblings on ptf1a:gfp background at 2 dpf. In ltv1Δ14/Δ14 mutants, both pH3 and BrdU-labeled ptf1a+ cells were significantly reduced after normalizing for total pancreatic cells (Figures 4A,B,A’,B’). Thus, the impaired exocrine pancreas in ltv1Δ14/Δ14 mutants would be most likely due to the decreased cell proliferation of the exocrine pancreas progenitor cells.

Figure 4. Impaired proliferation of exocrine pancreas progenitors and HSPCs in ltv1Δ14/Δ14 mutant. (A,B) Representative confocal images of pH3 immunostaining (A) or BrdU labeling (B) in ltv1Δ14/Δ14; ptf1a:gfp mutants and siblings at 2 dpf. (A’,B’) The percentage of pH3+ (A’, siblings, N = 6; mutants, N = 9) or BrdU+ (B’, siblings, N = 6; mutants, N = 6) cells within the ptf1a+ population in ltv1Δ14/Δ14 mutants and siblings at 2 dpf. Bars represent means with SD. (C,D) Representative confocal images of pH3 immunostaining (C) and BrdU labeling (D) in ltv1Δ14/Δ14; runx1:en-gfp mutants and siblings at 2.5 dpf. (C’,D’) The percentage of pH3+ (C’, siblings, N = 6; mutants, N = 7) and BrdU+ (D’, siblings, N = 6; mutants, N = 11) cells within the runx1+ population in ltv1Δ14/Δ14 mutants and siblings at 2.5 dpf. Bars represent means with SD. White arrow: merged cell. Scale bar: 10 μm.
Consistent with the results observed in the exocrine pancreas, TUNEL assay revealed similar apoptotic level of HSPCs in the CHT between ltv1Δ14/Δ14 mutants and siblings at 2.5 dpf (Supplementary Figures 6B,C). The proliferation of HSPCs was also reduced in ltv1Δ14/Δ14 mutants as indicated by the decreased pH3 and BrdU signals of HSPCs in the CHT at 2.5 dpf (Figures 4C,D,C’,D’). Thus, defects in the definitive hematopoiesis were most likely attributed to decreased proliferation of HSPCs, instead of cell death.
ltv1 Expression Was Enriched in Digestive Organs During Embryogenesis
To investigate the reason behind tissue specificity of the mutant phenotypes observed, the expression pattern of ltv1 in zebrafish embryos was examined by WISH using the antisense ltv1 RNA probe. The sense probe was used as a negative control. At one-cell stage, ltv1 mRNA was easily detected (Figures 5A,E), which suggested that ltv1 was a maternal expression gene. From 50%-epiboly to 13 hpf, ltv1 transcripts were distributed ubiquitously (Figures 5B,C), while no positive staining was detected for sense probe (Figures 5F,G). At 24 hpf, ltv1 transcripts were found in the eyes and pharyngeal primordia (Figures 5D,H,I). Between 48 and 72 hpf, ltv1 transcripts were abundant in the eyes, liver, intestine, and pancreas (Figures 5J,K). At 96 and 120 hpf, ltv1 was highly expressed in the exocrine pancreas (Figures 5L,M). The digestive organ and pharyngeal primordia-specific expression pattern of ltv1 was consistent with the hypoplastic phenotypes of these tissues during embryogenesis in the mutant. HSPCs and differentiated hematopoietic lineages were also affected severely in ltv1Δ14/Δ14 mutants; however, no clear ltv1 mRNA signal was detected in the aorta-gonad-mesonephros (AGM) or CHT by WISH using ltv1 RNA probe from 24 to 120 hpf.
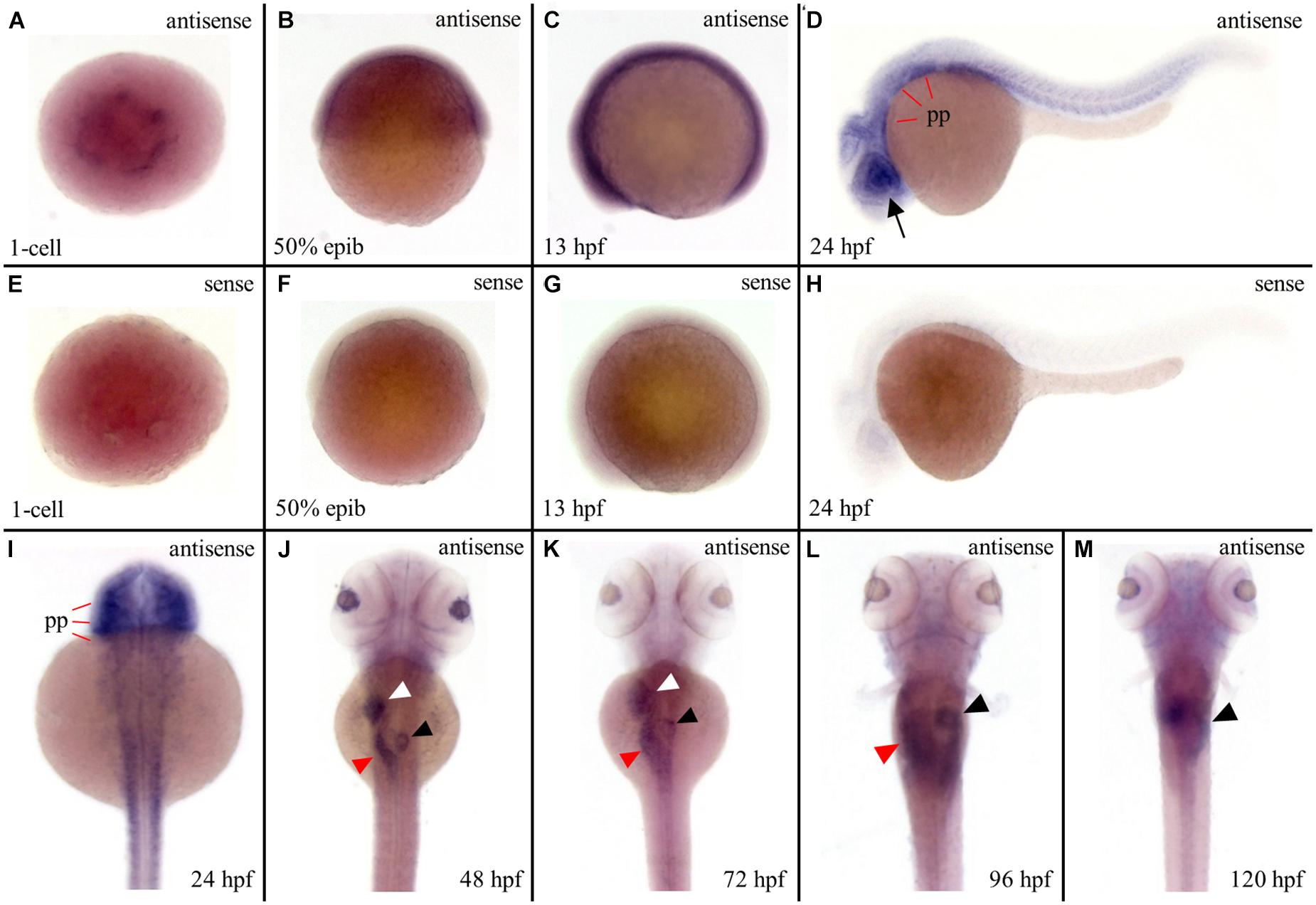
Figure 5. Strong expression of ltv1 in digestive organs. (A,I–M) Dorsal views. (B–D) Lateral views, anterior to the left. (A–D,I–M) WISH using ltv1 antisense probe in wild-type embryos at the stages as indicated in each panel. (E–H) As a negative control, ltv1 sense probe is used to perform the WISH. Black arrow: eye. White arrowhead: liver. Red arrowhead: intestine. Black arrowhead: exocrine pancreas. pp: pharyngeal primordia.
Ribosome Biogenesis Was Disrupted in ltv1Δ14/Δ14 Mutant Embryo
In eukaryotic cells, the 28S, 18S, and 5.8S rRNAs are cleaved by various nucleases from a single primary transcript, known as the pre-rRNA. It was reported that deletion of ltv1 could lead to aberrant processing of 18S rRNA in yeast, fruit fly, and human cells and accumulation of its precursor 20S (yeast) or 21S rRNA (fruit fly and human cells), implying a conserved role of ltv1 in 18S rRNA processing. To test if it were the case in zebrafish, Northern blot was used to analyze rRNA processing using the probes that could hybridize the ETS, ITS1, ITS2 (ETS/ITS: external/internal-transcribed spacer region), and 18S rRNA (Azuma et al., 2006). ETS, ITS1, and ITS2 probes could mark the rRNA precursor and the intermediate and some minor products (Figure 6A). The ETS and ITS1 probes revealed that the full-length precursor “a” accumulated significantly in ltv1Δ14/Δ14 mutants, indicating the disruption of rRNA processing (Figure 6B). The “d,” which might correspond to the 20S rRNA in yeast or 21S rRNA in human cells, accumulated while the “c” decreased, showing the impaired 18S rRNA processing in the mutants (Figure 6B). Consistently, the amount of 18S rRNA was declined slightly in the mutants (Figure 6C). Although the “e” increased slightly, the “b” showed no obvious difference in the mutants (Figure 6B), suggesting the intact 28S rRNA processing in ltv1Δ14/Δ14 mutants. To quantify the amount of 18S and 28S rRNA, E-bioanalyzer analysis was performed and the results showed that the 18S rRNA was reduced obviously in ltv1Δ14/Δ14 mutants at 5 dpf, while the amount of 28S rRNA remained comparable (Figure 6D). The altered quantity of 18S rRNA therefore caused the imbalance of the 28S/18S ratio in mutants, which is 3.1, compared with 2.0 in siblings (Figure 6E). Consistent with rRNA quantification data, the ribosome fractionation results showed that the amount of 40S subunits and 80S monosomes decreased, while that of the 60S subunits increased about twofold (Figures 6F,G).
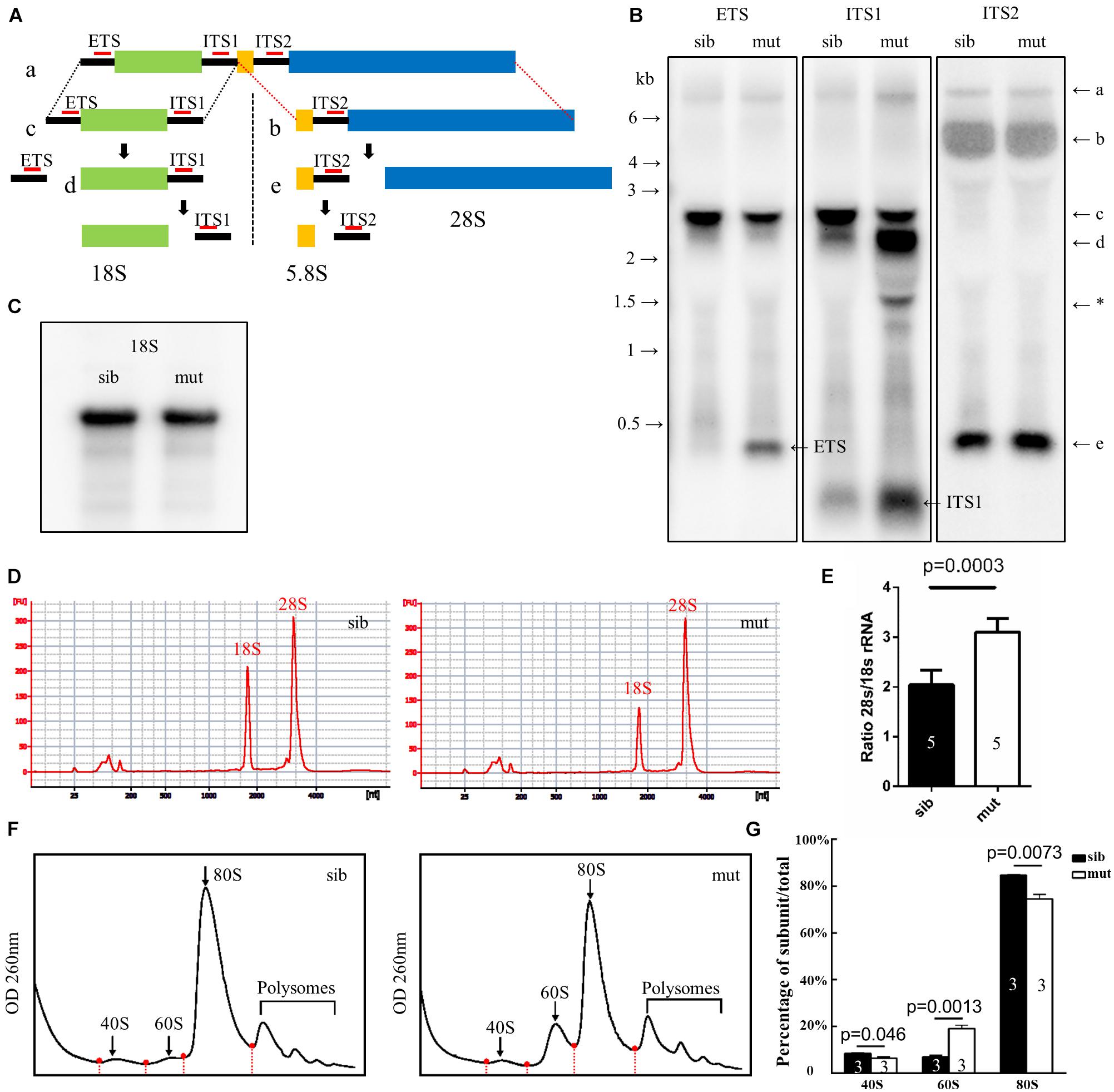
Figure 6. Defects in 18S rRNA processing in ltv1Δ14/Δ14 mutant. (A) Schematic diagram showing the processing pathway of 28S, 18S, and 5.8S rRNAs. The hybridization sites of each probe are indicated by red bars, respectively. (B,C) Northern blot analysis using the corresponding probes as indicated to detect 18S rRNA or the intermediate products of rRNA processing. Asterisk: unidentified rRNA intermediate product. (D) Representative analysis result by E-bioanalyzer. (E) The ratio of 28S/18S rRNA is increased in ltv1Δ14/Δ14 mutants (N = 5), as compared with siblings (N = 5). Bars represent means with SD. ETS, external transcribed spacer; ITS, internal transcribed spacer. (F) Representative ribosome fractionation results of siblings and ltv1Δ14/Δ14 mutants at 4 dpf. The peaks of 40S, 60S, and 80S are indicated by arrows. Red dots on the curves represent the lowest points flanking the peaks. The area under respective peaks of 40S, 60S, and 80S, circled by curves, dashed lines, and baselines are measured. (G) Percentages of 40S, 60S, and 80S in total lysate in siblings and ltv1Δ14/Δ14 mutants at 4 dpf. Bars represent means with SD. Quantifications of three independent experiments are analyzed.
Phenotypes in ltv1Δ14/Δ14 Mutant Were Independent of P53
A growing number of studies suggest that P53 may play a vital role in phenotypes relevant to ribosome dysfunction (Armistead and Triggs-Raine, 2014). In ltv1Δ14/Δ14 mutants, there was a clear increase in the expression level of p53 at 3 dpf, as indicated by WISH using a p53 probe which can detect both p53 andΔ113p53 (Figure 7A). In addition, the P53 protein level was upregulated obviously in mutants (Figure 7B). Then mRNA levels of Δ113p53 and p21, downstream genes of p53, were evaluated by quantitative polymerase chain reaction (PCR). Consistently, bothΔ113p53 and p21 mRNA levels were increased significantly in ltv1Δ14/Δ14 mutant, which further suggested the activation of p53 pathway (Figure 7C). To determine if the downregulation of p53 could rescue the mutant phenotypes, knockdown of p53 was achieved by the p53ATG morpholino injection. The increased P53 expression was attenuated in the mutant, which validated the efficacy of p53 knockdown (Figure 7B). However, neither the smaller liver nor the reduced HSPC phenotype in ltv1Δ14/Δ14 mutant could be alleviated by p53 knockdown (data not shown), suggesting that the mutant phenotypes were independent of P53.
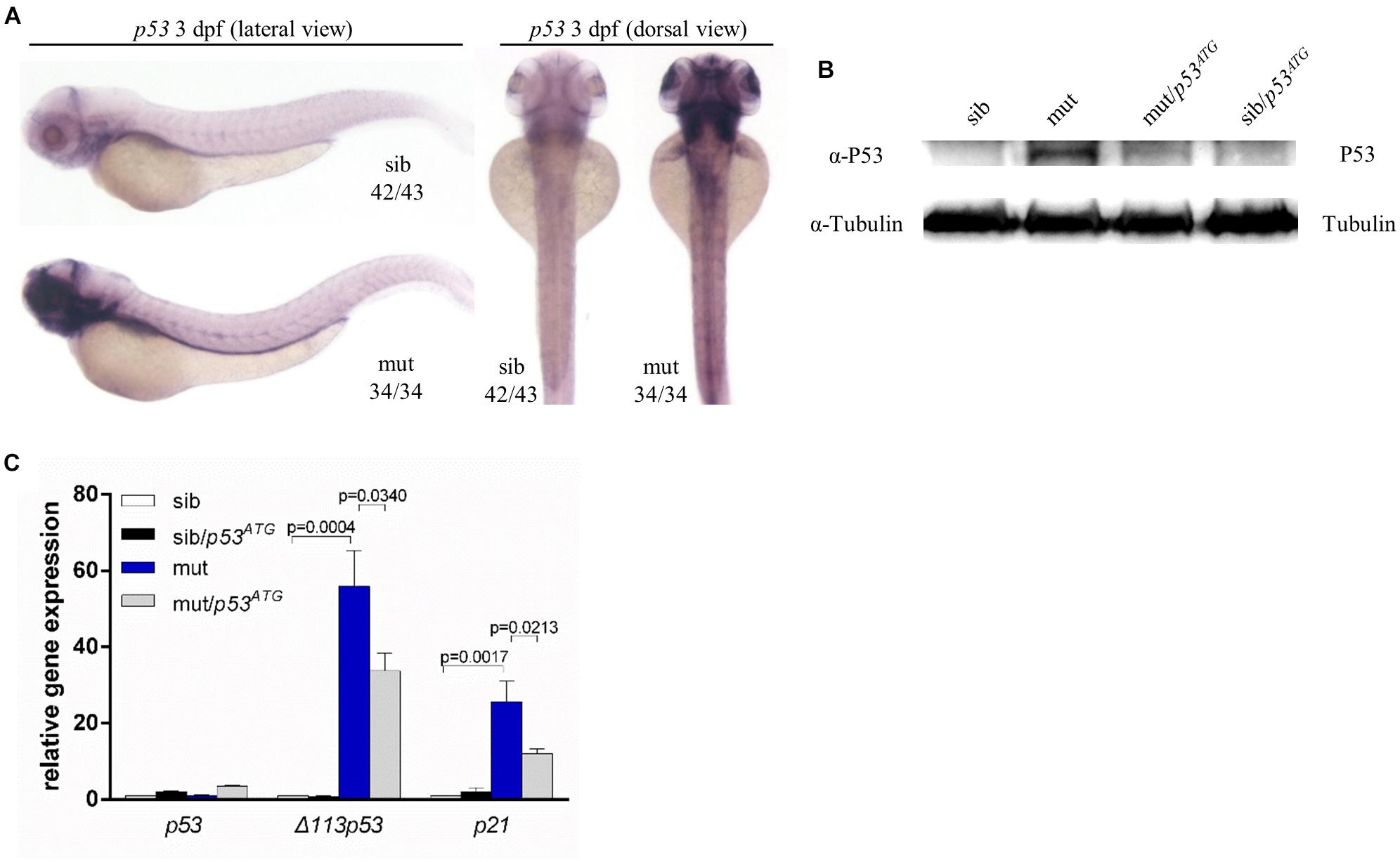
Figure 7. P53 Independence of phenotypes in ltv1Δ14/Δ14 mutant. (A) WISH of p53 probe for detecting both p53 and Δ113p53 at 3 dpf. (B) Western blot results of P53 protein of p53ATG MO-treated or MO-untreated mutants and siblings at 3 dpf. (C) Evaluation of expression of p53 and its transcriptional targets Δ113p53 and p21 by quantitative PCR.
Discussion
Ltv1 is a non-ribosomal protein essential for 18S rRNA processing in yeast, fruit fly, and human cells (Seiser et al., 2006; Tafforeau et al., 2011; Ghalei et al., 2015; Kressler et al., 2015). In this report, Ltv1 was demonstrated functionally conserved in zebrafish as illustrated by disrupted 18S rRNA processing in the ltv1 mutants. Deletion of zebrafish ltv1 resulted in defective growth of liver, exocrine pancreas, intestine, abnormal craniofacial structures and impaired development of HSPCs, definitive erythrocytes, myeloid cells, and lymphocytes. These phenotypic features resembled some specific ribosomopathy models in zebrafish studies (Provost et al., 2012; Carapito et al., 2017; Oyarbide et al., 2019, 2020).
Ltv1 is an assembly factor that can facilitate the incorporation of Rps3 and Rps10 into the small ribosomal subunit in yeast. Ltv1 deficiency led to mispositioned Rps3 in ribosomes (Collins et al., 2018). In zebrafish, knockdown of rps3 could result in morphological defects, including reduced head size, pericardial edema, and erythropoiesis failure (Yadav et al., 2014). These phenotypic features are consistent with those in ltv1Δ14/Δ14 zebrafish mutants. Hence, it is interesting to investigate whether ltv1 functions through rps3 in digestive system development and hematopoiesis. However, it should be noticed that the morphological defects of rps3 morphants could be rescued by knockdown of p53, while the erythroid failure could not be alleviated (Yadav et al., 2014). In ltv1Δ14/Δ14 mutants, none of the defects in morphology, digestive organogenesis, or hematopoiesis could be rescued by p53 knockdown. It was reported that Rps3 could directly interact with P53 and MDM2 (Yadavilli et al., 2009). This may be underlying the P53-dependent recovery of morphological deformities of the rps3 morphants. Further genetic investigation is required to validate the relationship between ltv1 and rps3. Ribosomes from Ltv1-deficient yeast harbored less Rps10 protein (Collins et al., 2018). Rps10 was found to be mutated in 6.4% of patients with DBA (Doherty et al., 2010). To the edge of our knowledge, no zebrafish mutant of rps10 has been constructed. It will be meaningful to analyze the phenotypes of rps10 mutants and investigate the genetic interaction among ltv1, rps3, and rps10 in zebrafish.
What is the justification for dysfunction in a macromolecule as ubiquitous and essential as the ribosome causing ribosomopathies with defects in selective tissues? Xue and Barna (2012) believed that the tissue specificity of gene expression in ribosomal biogenesis was the cause. Agreed with this point, ltv1 was found highly expressed in digestive organs during embryogenesis, which may partially explain the phenotypes of ltv1 mutants in these organs. However, despite of no detectable expression of ltv1 in the AGM and CHT, HSPCs and differentiated hematopoietic lineages were impaired severely. Some zebrafish models of ribosomopathy, such as sbds (Provost et al., 2012), rpl11 (Danilova et al., 2011), rpl24, rpl35a (Yadav et al., 2014), etc., all displayed hematopoietic defects at different levels. While all the respective genes were highly expressed in digestive organs, no description of gene expression in AGM or CHT was reported (Venkatasubramani and Mayer, 2008; Provost et al., 2013), similar to that observed in ltv1. One possible explanation is that these genes deficiencies may lead to impaired hematopoiesis indirectly, probably by impairment of the niche of HSPCs. Like ltv1, zebrafish nol9 encoded a non-ribosomal protein, and nol9 mutants displayed defects in both digestive organs and hematopoiesis. Transmission electron microscopy (TEM) analysis revealed great changes in the CHT niche in nol9 mutants, including extracellular matrix (ECM) and endothelial cells (Bielczyk-Maczynska et al., 2015).
It has been demonstrated here that Ltv1 is essential for ribosome biogenesis and organogenesis of digestive system and hematopoiesis. Among the existing zebrafish models with deficient ribosome biogenesis, most of them exhibited hypoplasia of liver, pancreas, and intestine, including nil per os (npo) (Mayer and Fishman, 2003), titania (tti) (Boglev et al., 2013), bms1-like (bms1l) (Wang et al., 2012), and nucleolar protein with MIF4G domain 1 (nom1) (Qin et al., 2014), while some displayed defects in definitive hematopoiesis, for example, kri1l (Jia et al., 2015). To the best of our knowledge, only one mutant nol9 (Bielczyk-Maczynska et al., 2015) described both phenotypes. Interestingly, in line with ltv1Δ14/Δ14 mutants, nol9 mutants showed arrested development of exocrine pancreas and HSPCs as a result of reduced proliferative rate. Although nol9 and ltv1 are involved in the 28S and 18S rRNA processing, respectively, the similar phenotypes in these two models suggest the conserved function of ribosome biogenesis genes during embryogenesis.
Several studies revealed that excess free ribosomal proteins, while ribosome biogenesis was impaired, could outcompete P53 in binding the E3 ubiquitin ligase MDM2, consequently protecting P53 from degradation (Zhang et al., 2003; Dai and Lu, 2004; Dai et al., 2004). In some ribosome biogenesis-deficient models, phenotypes could be rescued by inhibition of P53 (Zhang et al., 2012; Bielczyk-Maczynska et al., 2015; Ear et al., 2016). However, in some other cases, P53-independent cell apoptosis and cell proliferation arrest have also been described (Provost et al., 2012; Boglev et al., 2013; Qin et al., 2014; Yadav et al., 2014; Jia et al., 2015). Although P53 protein and target genesΔ113p53 and p21 were upregulated in ltv1Δ14/Δ14 mutants, knockdown of p53 could not rescue the defects of the liver or HSPCs, suggesting a p53-independent mechanism was involved, which agreed with the fact that the abnormal rRNA processing in LTV1-deficient human cells was P53 independent (Tafforeau et al., 2011). In addition to P53, some other pathways were reported to be involved in the ribosome-deficient zebrafish models. In zebrafish kri1l mutants, an increased level of autophagy was observed, and blocking autophagy could significantly restore the definitive hematopoiesis (Jia et al., 2015). In contrast, inhibition of autophagy reduced the lifespan of zebrafish mutants of pwp2h gene, which encoded a protein promoting the small ribosomal subunit processing. In pwp2h mutants, autophagy was considered a survival mechanism triggered by ribosomal efficiency (Boglev et al., 2013). The question that whether autophagy is involved in the ltv1 function in zebrafish is required further validation. Rpl35a was mutated in 3.3% DBA (Farrar et al., 2008). In zebrafish rpl35a knockdown embryos, upregulation of mammalian target of rapamycin (mTOR) could rescue the morphological defects and the erythroid failure (Yadav et al., 2014). This case suggested that mTOR functioned downstream of rpl35a. Urb1, a protein promoting the big ribosomal subunit assembly in zebrafish, was demonstrated to play a role downstream of mTOR in digestive organ formation (He et al., 2017). It is also possible that mTOR pathway is involved in ltv1-dependent digestive organ development and hematopoiesis.
In ltv1Δ14/Δ14 mutants, the proliferation was inhibited and p53 was activated. It was reported that activation of p53 could lead to cell cycle arrest via p21 upregulation (Georgakilas et al., 2017). However, it is possibly not the case in ltv1Δ14/Δ14 mutants because inhibition of p53 could not restore the growth of the liver and HSPCs. The P53-independent mechanism underlying the cell cycle arrest might be the key way through which ltv1 functions in zebrafish. Pescadillo was a protein that played an essential role in 28S rRNA processing and the zebrafish pescadillo-deficient embryos displayed underdeveloped liver, gut, and craniofacial cartilage (Allende et al., 1996; Lapik et al., 2004; Provost et al., 2012). Cyclin D1 was indispensable for cell proliferation in cells. As a cyclin-dependent kinase inhibitor, P27 could decrease catalytic activity of cyclin D1 through direct interaction, and so that led to cell cycle arrest (Razavipour et al., 2020). It was reported that the cell cycle arrest in pescadillo-deficient cells was due to cyclin D1 downregulation and activation of P27, which was independent of P53 (Li et al., 2009). In erythroid cell lines, ribosome synthesis defects could lead to decreased level of PIM1, a kinase implicated in cell proliferation. The reduction of PIM1 induced cell cycle arrest via accumulated P27 in a P53-independent way (Iadevaia et al., 2010). It is interesting to investigate that whether the ltv1Δ14/Δ14 mutants share the same P27-regulated mechanism, regardless of P53, underlying the cell proliferation inhibition with these two cases.
It should be highlighted that the phenotypes of ltv1Δ14/Δ14 and sbds-deficient embryos share some similar features such as the affected exocrine pancreas and hematopoiesis (Burroughs et al., 2009). Although the Sbds protein plays a role in 60S ribosomal subunit biogenesis (Menne et al., 2007), Ltv1 is required for the assembly of 40S ribosomal subunit (Ameismeier et al., 2018; Collins et al., 2018). The expression pattern of sbds is very similar to that of ltv1 in zebrafish (Venkatasubramani and Mayer, 2008). In line with ltv1Δ14/Δ14 larvae, morpholino knockdown of sbds in zebrafish causes defects in exocrine pancreas, of which could not be rescued by p53 downregulation, while the endocrine pancreas is normal (Provost et al., 2012). Together with other animal models with ribosome biogenesis dysfunction, ltv1 zebrafish mutant not only provides new candidate genes for the screening of ribosomopathies with unknown genetic deterioration but also serves as a tool to investigate molecular and cellular mechanisms underlying ribosomal genes deficiency phenotypes. As a powerful model for chemical screen, the corresponding zebrafish mutants may be used for the identification of potential compounds for treating specific ribosomopathies.
Materials and Methods
Zebrafish Strains and Embryos Collection
Wild-type Tübingen fish line, transgenic line ptf1a:gfp (Godinho et al., 2005), and runx1:en-gfp (He et al., 2015) were used and maintained under standard conditions.
Genomic DNA Extraction
Embryos or fish scales were lysed in the buffer (10 mM Tris–HCl, 50 mM KCl, 0.3% Tween-20, 0.3% NP40, and 1/10 volume proteinase K, Invitrogen, Waltham, MA, United States) at 55°C for 12 h and then the reaction was inactivated by increasing the temperature to 95°C for 20 min. The crude lysate could be used as the template for PCR directly.
Generation of ltv1 Mutants by Clustered Regularly Interspaced Short Palindromic Repeat/Cas9 System
The gRNA was designed to target a site in the exon 7 of ltv1 at the sequence GGACAGTGCTCGGCTGGAGG (PAM site in italics). Zebrafish Cas9 mRNA and the ltv1 gRNA were synthesized as described (Chang et al., 2013; Ear et al., 2016). At one-cell stage, Cas9 mRNA (300 pg) and gRNA (50 pg) were injected into wild-type embryos. At 36 hpf, about 10 embryos were pooled and lysed, and the primers (ltv1 fw: 5′-TGGTAAGGAGTCTGATTATC-3′ and ltv1 rv: 5′-CCAATCCATGTGATGCATAC-3′) were used to amplified DNA fragment harboring gRNA targeted site. PCR products were subjected to sequencing to identify potential indels in the region. Upon detecting mutation, the rest of the embryos were raised to adults (F0). Pooled F1 embryos obtained by crossing F0 with wild-type fish were examined for indels in ltv1 gene using the PCR method described above. The nature of indels could be obtained by sequencing and mutant allele specific primers were then designed according to specific indels.
Genotyping of ltv1Δ14/Δ14 Mutants
The common forward primer (ltv1 fw) was described above. The mutant and wild-type allele-specific reverse primers were listed as follow: wt rv: 5′-CTTTGATGACCTCCTC-3′ and ltv1Δ14 rv: 5′-CTTTGATGACCTCTGT-3′.
RNA Whole Amount in situ Hybridization
The following digoxigenin-labeled antisense RNA probes were used: fabp10, trypsin, insulin, fabp2, foxA1, foxA3, gata6, hhex, prox1, pdx1, gcga, sst2, c-myb, ikaros, gata1, αe1-globin, mfap4, csf1ra, lyz, rag1, apoe, and pu.1. WISH was performed as described previously (Huang et al., 2008; Li et al., 2011).
Mutant Rescue
Zebrafish wild-type ltv1 cDNAs were cloned into pCS2 + vector. Mutant form cDNA was obtained by site-directed mutagenesis. The microinjection was performed at one-cell stage, 0.5 ng in vitro transcribed either zebrafish wild-type ltv1 mRNA or ltv1Δ14 mRNA was used to try to rescue the mutant phenotypes. At 3 days post injection (dpi), embryos were fixed for WISH using the liver-specific fabp10 or c-myb probe.
Immunochemistry Staining
Immunohistochemistry staining was performed as described previously (Chen et al., 2005). The primary antibodies were goat anti-GFP (Abcam, Cambridge, MA, United States; 1:400), rabbit anti-pH3 (Santa Cruz Biotechnology, Santa Cruz, CA, United States; 1:200), and mouse 2F11 (Abcam, 1:1,000). GFP antibody was used to enhance the GFP signal in runx1:en-gfp.
Bromodeoxyuridine Labeling
For BrdU labeling, BrdU (Roche Diagnostics, Indianapolis, IN, United States; 1 nl, 30 mM) was injected into the pericardium of embryos. Consequently, the embryos were incubated for 1.5–2 h at 28.5°C. After three times of washing with PBST, the embryos were fixed using 4% PFA. After being treated with 2 N HCl for 1 h, the embryos were incubated with mouse anti-BrdU (Roche Diagnostics, United States; 1:50) and goat anti-GFP (Abcam, United States; 1:400) antibodies at 4°C overnight, and finally visualized by Alexa Fluor 555 donkey anti-mouse (Life Technology, Carlsbad, CA, United States; 1:400) and Alexa Fluor 488 donkey anti-goat (Life Technology, United States; 1:400) antibodies.
TUNEL Assay
Whole embryo and cryosectioned samples were prepared for TUNEL assay, and in situ cell death detection kit, TMR Red (Roche Diagnostics, United States) was used following the manuals provided.
Dyes Staining
Alcian blue, Neutral red, and Sudan black (Sigma-Aldrich, Burlington, MA, United States) staining was conducted as described previously (Herbomel et al., 2001; Le Guyader et al., 2008; Chen et al., 2009; Li et al., 2011). DCFH-DA (Wako, Japan) was used as described previously (Shi et al., 2014).
Northern Blot
Total RNA was extracted from mutant and sibling embryos at 120 hpf using TriPure Isolation Reagent (Roche Diagnostics, United States). The DIG-labeled DNA probes were PCR-amplified using previously described primers (Azuma et al., 2006). Equal amount of total RNAs were subjected to electrophoresis. The probe hybridization and detection process were carried out as previously described (Chen et al., 2005).
Quantification of 18S and 28S Ribosomal RNA
Total RNA was extracted from ltv1Δ14/Δ14 mutants and siblings at 5 dpf. Then, RNA were subjected to E-Bioanalyzer (Agilent 2100) analysis according to the manual.
Ribosome Fractionation
For each group, 300 embryos at 4 dpf were collected and rinsed by pre-chilled PBS (containing 100 μg/ml cycloheximide) for three times. After removing the yolk through 23 G needles, the embryos were resuspended in 500 μl pre-chilled lysis buffer (5 mM Tris–HCl pH 7.5, 2.5 mM MgCl2, 1.5 mM KCl, 100 μg/ml cycloheximide, 2 mM DTT, 0.32 U/μl RNase inhibitor, 0.5% Triton X-100, 0.5% sodium deoxycholate, 1 × EDTA-free Protease Inhibitor Cocktail, Abcam) and sheared on ice through 23, 25, and 27 G needles gradually. Then the lysate was centrifuged at 15,000× g for 10 min at 4°C to remove the nuclei and cellular debris pellet. The liquid supernatant was gently loaded on 5–50% gradient sucrose solution (containing 20 mM HEPES pH 7.6, 0.1 M KCl, 5 mM MgCl2, 10 μg/ml cycloheximide, 0.1× EDTA-free Protease Inhibitor Cocktail, 32 U/ml RNase inhibitor), which was made by Gradient Master (Biocomp, Gyeongju, South Korea) and centrifuged at 36,000 rpm for 4 h at 4°C in SW41 Ti rotor. The fractions were collected using the Piston Gradient Fractionator (Biocomp) and scanned continuously by Triax Flow Cells detector (Biocomp) to measure the absorbance at 260 nm.
p53 Knockdown With Morpholinos
To knock down p53, p53-morpholino (MO)ATG (5′-GCGCCATTGCTTTGCAAGAATTG-3′, Gene Tools, 1 nl, 0.7 mM) was injected to one-cell stage embryos (Chen et al., 2005). The morpholino against human β-globin was used as the negative control (Chen et al., 2005).
Quantitative PCR
Total RNA was extracted from whole embryos using TriPure Isolation Reagent (Roche Diagnostics, United States). cDNAs were generated using Oligo (dT) and SuperScript III reverse transcriptase (Life Technologies, United States). For each sample, three parallel repeated tests were performed. The measured expression of ef1α was the internal control for each gene. The primers used: ef1α fw: 5′-CTTCTCAGGCTGACTGTGC-3′, ef1α rv: 5′-CCGCTAGCATTACCCTCC-3′, p53 fw: 5′-TGGAGAGGAGGTCGGCAAAATCAA-3′, p53 rv: 5′-GAC TGCGGGAACCTGAGCCTAAAT-3′, Δ113p53 fw: 5′-ATAT CCTGGCGAACATTTGGAGGG-3′, Δ113p53 rv: 5′-CCTCCT GGTCTTGTAATGTCAC-3′, p21 fw: 5′-GAAGCGCAAA CAGACCAACAT-3′ and p21 rv: 5′-GCAGCTCAATTACGA TAAAGA-3′.
Western Blot
p53ATG MO-injected or p53ATG MO-uninjected embryos were de-yolked at 3 dpf. Then the embryos were treated with pre-extraction buffer (1 mM EDTA, 0.1 mM Na3VO4, 20 mM NaF, 1 mM DTT, 100 mM PMSF, and a work concentration of Protease Inhibitor Cocktail in PBS) and lysed in SDS lysis buffer. Subsequently, the lysate was used in Western blot. The primary antibodies used were α-P53 antibody (Abcam, 1:1,000) and α-tubulin antibody (Pierce, 1:500).
Statistical Methods
The experimental data were analyzed in GraphPad Prism 6.0. The unpaired Student’s t-test was used for comparing the means of two groups.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The animal study was reviewed and approved by Institutional Animal Care and Use Committee in Southwest University, China.
Author Contributions
HH and HR designed the project. CZ, RH, XM, JC, and XH performed the experiments. CZ and HH wrote the manuscript. LLi and LLu commented on the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Key R&D Program of China 2018YFA0801000 and 2018YFA0800502, National Natural Science Foundation of China 31471365.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Li Jan Lo (Zhejiang University, China) for suggestion on the manuscript, LLi (Southwest University, China) for blood cell markers, and Bo Zhang and Jingwei Xiong (Peking University, China) for plasmids for CRISPR/Cas9 system.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.704730/full#supplementary-material
Supplementary Figure 1 | Ltv1 is conserved among human, mouse and zebrafish. Alignment of Ltv1 protein sequence from human, mouse, and zebrafish.
Supplementary Figure 2 | The liver bud growth and exocrine pancreas expansion are affected in ltv1Δ14/Δ14 mutant. (A–J) Embryos at 2 dpf, 34 hpf, and 30 hpf were subjected to WISH to analyze the liver and pancreas formation. The probes used include: pan-endodermal markers foxA1, gata6; hepatic markers hhex, prox1; pancreas marker pdx1; endocrine pancreas markers gcga, insulin, and sst2. (K) Representative confocal images of ltv1Δ14/Δ14/ptf1a:gfp mutants and siblings at 2 dpf. (L) Quantification of ptf1a+ cells of ltv1Δ14/Δ14/ptf1a:gfp mutants and siblings at 2 dpf. Bars represent means with SD. White arrowhead: liver. Blue arrowhead: endocrine pancreas. Scale bar: 10 μm.
Supplementary Figure 3 | The primitive hematopoiesis is normal in ltv1Δ14/Δ14 mutant. (A,B) WISH of primitive hematopoiesis regulators gata1 (A) and pu.1 (B) at 20 and 22 hpf, respectively.
Supplementary Figure 4 | The defects of HSPC is observable at 3 dpf. (A) WISH of c-myb in the CHT at 2 dpf. (B) Quantification of c-myb+ HSPCs in the CHT of ltv1Δ14/Δ14 mutants and siblings at 2 dpf. (C) ltv1Δ14/Δ14 mutants displayed significantly decreased c-myb expression in the CHT at 3 dpf compared with the siblings.
Supplementary Figure 5 | The mutant phenotypes can be rescued by zebrafish ltv1 mRNA injection. (A,C) Representative images of fabp10 or c-myb expression of unrescued and partially and fully rescued mutants after the injection of zebrafish wild-type ltv1 mRNA. (B,D) The rescue efficiency of the liver or HSPC phenotypes in mutants after the injection of zebrafish wild-type or Δ14 ltv1 mRNA (N ≥ 30 in every group). Bars represent means with SD.
Supplementary Figure 6 | Apoptotic level of the exocrine pancreas and HSPCs are not increased in ltv1Δ14/Δ14 mutant. (A) No apoptotic cells are found in the pancreas region in cryosectioned samples from both ltv1Δ14/Δ14 mutants and siblings at 3 dpf. (B) Representative confocal images of the ltv1Δ14/Δ14/runx1:en-gfp mutants and siblings after TUNEL assay at 2.5 dpf. (C) Ratio of apoptotic cells in runx1+ HSPCs in ltv1Δ14/Δ14 mutants (N = 6) and siblings (N = 6) at 2.5 dpf. Bars represent means with SD. White arrow: merged cell. Scale bar: 10 μm.
References
Allende, M. L., Amsterdam, A., Becker, T., Kawakami, K., Gaiano, N., and Hopkins, N. (1996). Insertional mutagenesis in zebrafish identifies two novel genes, pescadillo and dead eye, essential for embryonic development. Genes Dev. 10, 3141–3155. doi: 10.1101/gad.10.24.3141
Ameismeier, M., Cheng, J., Berninghausen, O., and Beckmann, R. (2018). Visualizing late states of human 40S ribosomal subunit maturation. J. Cell Biol. 558, 249–253.
Anchelin, M., Alcaraz-Pérez, F., Martínez, C. M., Bernabé-García, M., Mulero, V., and Cayuela, M. L. (2013). Premature aging in telomerase-deficient zebrafish. Dis. Model. Mech. 6, 1101–1112. doi: 10.1242/dmm.011635
Armistead, J., and Triggs-Raine, B. (2014). Diverse diseases from a ubiquitous process: the ribosomopathy paradox. FEBS Lett. 588, 1491–1500. doi: 10.1016/j.febslet.2014.03.024
Azuma, M., Toyama, R., Laver, E., and Dawid, I. B. (2006). Perturbation of rRNA synthesis in the bap28 mutation leads to apoptosis mediated by p53 in the zebrafish central nervous system. J. Biol. Chem. 281, 13309–13316. doi: 10.1074/jbc.M601892200
Barlow, J. L., Drynan, L. F., Hewett, D. R., Holmes, L. R., Lorenzo-Abalde, S., Lane, A. L., et al. (2010). A p53-dependent mechanism underlies macrocytic anemia in a mouse model of human 5q- syndrome. Nat. Med. 16, 59–66. doi: 10.1038/nm.2063
Bielczyk-Maczynska, E., Lam Hung, L., Ferreira, L., Fleischmann, T., Weis, F., Fernandez-Pevida, A., et al. (2015). The ribosome biogenesis protein nol9 is essential for definitive hematopoiesis and pancreas morphogenesis in zebrafish. PLoS Genet. 11:e1005677. doi: 10.1371/journal.pgen.1005677
Boglev, Y., Badrock, A. P., Trotter, A. J., Du, Q., Richardson, E. J., Parslow, A. C., et al. (2013). Autophagy induction is a Tor- and Tp53-independent cell survival response in a zebrafish model of disrupted ribosome biogenesis. PLoS Genet. 9:e1003279. doi: 10.1371/journal.pgen.1003279
Burroughs, L., Woolfrey, A., and Shimamura, A. (2009). Shwachman-diamond syndrome: a review of the clinical presentation, molecular pathogenesis, diagnosis, and treatment. Hematol. Oncol. Clin. North Am. 23, 233–248. doi: 10.1016/j.hoc.2009.01.007
Carapito, R., Konantz, M., Paillard, C., Miao, Z., Pichot, A., Leduc, M. S., et al. (2017). Mutations in signal recognition particle SRP54 cause syndromic neutropenia with Shwachman-diamond-like features. J. Clin. Invest. 127, 4090–4103. doi: 10.1172/jci92876
Chang, N., Sun, C., Gao, L., Zhu, D., Xu, X., Zhu, X., et al. (2013). Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res. 23, 465–472. doi: 10.1038/cr.2013.45
Chen, J., Ruan, H., Ng, S. M., Gao, C., Soo, H. M., Wu, W., et al. (2005). Loss of function of def selectively up-regulates Delta113p53 expression to arrest expansion growth of digestive organs in zebrafish. Genes Dev. 19, 2900–2911. doi: 10.1101/gad.1366405
Chen, Y. H., Lu, Y. F., Ko, T. Y., Tsai, M. Y., Lin, C. Y., Lin, C. C., et al. (2009). Zebrafish cdx1b regulates differentiation of various intestinal cell lineages. Dev. Dyn. 238, 1021–1032. doi: 10.1002/dvdy.21908
Collins, J. C., Ghalei, H., Doherty, J. R., Huang, H., and Culver, R. N. (2018). Ribosome biogenesis factor Ltv1 chaperones the assembly of the small subunit head. J. Cell Biol. 217, 4141–4154. doi: 10.1083/jcb.201804163
Dai, M. S., and Lu, H. (2004). Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J. Biol. Chem. 279, 44475–44482. doi: 10.1074/jbc.M403722200
Dai, M. S., Zeng, S. X., Jin, Y., Sun, X. X., David, L., and Lu, H. (2004). Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol. Cell Biol. 24, 7654–7668. doi: 10.1128/mcb.24.17.7654-7668.2004
Danilova, N., Sakamoto, K. M., and Lin, S. (2008). Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood 112, 5228–5237. doi: 10.1182/blood-2008-01-132290
Danilova, N., Sakamoto, K. M., and Lin, S. (2011). Ribosomal protein L11 mutation in zebrafish leads to haematopoietic and metabolic defects. Br. J. Haematol. 152, 217–228. doi: 10.1111/j.1365-2141.2010.08396.x
Doherty, L., Sheen, M. R., Vlachos, A., Choesmel, V., O’Donohue, M. F., Clinton, C., et al. (2010). Ribosomal protein genes RPS10 and RPS26 are commonly mutated in Diamond-Blackfan anemia. Am. J. Hum. Genet. 86, 222–228. doi: 10.1016/j.ajhg.2009.12.015
Ear, J., Hsueh, J., Nguyen, M., Zhang, Q., Sung, V., Chopra, R., et al. (2016). A zebrafish model of 5q-syndrome using CRISPR/Cas9 targeting RPS14 reveals a p53-independent and p53-dependent mechanism of erythroid failure. J. Genet. Genomics 43, 307–318. doi: 10.1016/j.jgg.2016.03.007
Farrar, J. E., Nater, M., Caywood, E., McDevitt, M. A., Kowalski, J., Takemoto, C. M., et al. (2008). Abnormalities of the large ribosomal subunit protein, Rpl35a, in Diamond-Blackfan anemia. Blood 112, 1582–1592. doi: 10.1182/blood-2008-02-140012
Field, H. A., Dong, P. D. S., Beis, D., and Stainier, D. Y. R. (2003). Formation of the digestive system in zebrafish. II. Pancreas morphogenesis⋆. Dev. Biol. 261, 197–208. doi: 10.1016/s0012-1606(03)00308-7
Fumagalli, S., Di Cara, A., Neb-Gulati, A., Natt, F., Schwemberger, S., Hall, J., et al. (2009). Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an rpL11-translation-dependent mechanism of p53 induction. Nat. Cell Biol. 11, 501–508. doi: 10.1038/ncb1858
Georgakilas, A. G., Martin, O. A., and Bonner, W. M. (2017). p21: a two-faced genome guardian. Trends Mol. Med. 23, 310–319. doi: 10.1016/j.molmed.2017.02.001
Ghalei, H., Schaub, F. X., Doherty, J. R., Noguchi, Y., Roush, W. R., Cleveland, J. L., et al. (2015). Hrr25/CK1delta-directed release of Ltv1 from pre-40S ribosomes is necessary for ribosome assembly and cell growth. J. Cell Biol. 208, 745–759.
Godinho, L., Mumm, J. S., Williams, P. R., Schroeter, E. H., Koerber, A., Park, S. W., et al. (2005). Targeting of amacrine cell neurites to appropriate synaptic laminae in the developing zebrafish retina. Development 132, 5069–5079. doi: 10.1242/dev.02075
He, J., Yang, Y., Zhang, J., Chen, J., Wei, X., He, J., et al. (2017). Ribosome biogenesis protein Urb1 acts downstream of mTOR complex 1 to modulate digestive organ development in zebrafish. J. Genet. Genomics 44, 567–576. doi: 10.1016/j.jgg.2017.09.013
He, Q., Zhang, C., Wang, L., Zhang, P., Ma, D., Lv, J., et al. (2015). Inflammatory signaling regulates hematopoietic stem and progenitor cell emergence in vertebrates. Blood 125, 1098–1106. doi: 10.1182/blood-2014-09-601542
Herbomel, P., Thisse, B., and Thisse, C. (2001). Zebrafish early macrophages colonize cephalic mesenchyme and developing brain, retina, and epidermis through a M-CSF receptor-dependent invasive process. Dev. Biol. 238, 274–288. doi: 10.1006/dbio.2001.0393
Huang, H., Ruan, H., Aw, M. Y., Hussain, A., Guo, L., Gao, C., et al. (2008). Mypt1-mediated spatial positioning of Bmp2-producing cells is essential for liver organogenesis. Development 135, 3209–3218. doi: 10.1242/dev.024406
Iadevaia, V., Caldarola, S., Biondini, L., Gismondi, A., Karlsson, S., Dianzani, I., et al. (2010). PIM1 kinase is destabilized by ribosomal stress causing inhibition of cell cycle progression. Oncogene 29, 5490–5499. doi: 10.1038/onc.2010.279
Jagannathan-Bogdan, M., and Zon, L. I. (2013). Hematopoiesis. Development 140, 2463–2467. doi: 10.1242/dev.083147
Jia, X. E., Ma, K., Xu, T., Gao, L., Wu, S., Fu, C., et al. (2015). Mutation of kri1l causes definitive hematopoiesis failure via PERK-dependent excessive autophagy induction. Cell Res. 25, 946–962. doi: 10.1038/cr.2015.81
Jones, N. C., Lynn, M. L., Gaudenz, K., Sakai, D., Aoto, K., Rey, J. P., et al. (2008). Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat. Med. 14, 125–133. doi: 10.1038/nm1725
Kressler, D., Pertschy, B., Kim, W., Kim, H. D., Jung, Y., Kim, J., et al. (2015). Drosophila low temperature viability protein 1 (LTV1) is required for ribosome biogenesis and cell growth downstream of Drosophila Myc (dMyc). J. Biol. Chem. 290, 13591–13604.
Lapik, Y. R., Fernandes, C. J., Lau, L. F., and Pestov, D. G. (2004). Physical and functional interaction between Pes1 and Bop1 in mammalian ribosome biogenesis. Mol. Cell 15, 17–29. doi: 10.1016/j.molcel.2004.05.020
Le Guyader, D., Redd, M. J., Colucci-Guyon, E., Murayama, E., Kissa, K., Briolat, V., et al. (2008). Origins and unconventional behavior of neutrophils in developing zebrafish. Blood 111, 132–141. doi: 10.1182/blood-2007-06-095398
Li, J., Yu, L., Zhang, H., Wu, J., Yuan, J., Li, X., et al. (2009). Down-regulation of pescadillo inhibits proliferation and tumorigenicity of breast cancer cells. Cancer Sci. 100, 2255–2260. doi: 10.1111/j.1349-7006.2009.01325.x
Li, L., Jin, H., Xu, J., Shi, Y., and Wen, Z. (2011). Irf8 regulates macrophage versus neutrophil fate during zebrafish primitive myelopoiesis. Blood 117, 1359–1369. doi: 10.1182/blood-2010-06-290700
Mayer, A. N., and Fishman, M. C. (2003). Nil per os encodes a conserved RNA recognition motif protein required for morphogenesis and cytodifferentiation of digestive organs in zebrafish. Development 130, 3917–3928.
Menne, T. F., Goyenechea, B., Sanchez-Puig, N., Wong, C. C., Tonkin, L. M., Ancliff, P. J., et al. (2007). The Shwachman-Bodian-Diamond syndrome protein mediates translational activation of ribosomes in yeast. Nat. Genet. 39, 486–495. doi: 10.1038/ng1994
Narla, A., and Ebert, B. L. (2010). Ribosomopathies: human disorders of ribosome dysfunction. Blood 115, 3196–3205. doi: 10.1182/blood-2009-10-178129
Ober, E. A., Verkade, H., Field, H. A., and Stainier, D. Y. (2006). Mesodermal Wnt2b signalling positively regulates liver specification. Nature 442, 688–691. doi: 10.1038/nature04888
Oyarbide, U., Shah, A. N., Amaya-Mejia, W., Snyderman, M., Kell, M. J., Allende, D. S., et al. (2020). Loss of Sbds in zebrafish leads to neutropenia and pancreas and liver atrophy. JCI Insight 5:e134309. doi: 10.1172/jci.insight.134309
Oyarbide, U., Topczewski, J., and Corey, S. J. (2019). Peering through zebrafish to understand inherited bone marrow failure syndromes. Haematologica 104, 13–24. doi: 10.3324/haematol.2018.196105
Panse, V. G., and Johnson, A. W. (2010). Maturation of eukaryotic ribosomes: acquisition of functionality. Trends Biochem. Sci. 35, 260–266. doi: 10.1016/j.tibs.2010.01.001
Pereboom, T. C., van Weele, L. J., Bondt, A., and MacInnes, A. W. (2011). A zebrafish model of dyskeratosis congenita reveals hematopoietic stem cell formation failure resulting from ribosomal protein-mediated p53 stabilization. Blood 118, 5458–5465. doi: 10.1182/blood-2011-04-351460
Provost, E., Wehner, K. A., Zhong, X., Ashar, F., Nguyen, E., Green, R., et al. (2012). Ribosomal biogenesis genes play an essential and p53-independent role in zebrafish pancreas development. Development 139, 3232–3241. doi: 10.1242/dev.077107
Provost, E., Weier, C. A., and Leach, S. D. (2013). Multiple ribosomal proteins are expressed at high levels in developing zebrafish endoderm and are required for normal exocrine pancreas development. Zebrafish 10, 161–169. doi: 10.1089/zeb.2013.0884
Qin, W., Chen, Z., Zhang, Y., Yan, R., Yan, G., Li, S., et al. (2014). Nom1 mediates pancreas development by regulating ribosome biogenesis in zebrafish. PLoS One 9:e100796. doi: 10.1371/journal.pone.0100796
Razavipour, S. F., Harikumar, K. B., and Slingerland, J. M. (2020). p27 as a transcriptional regulator: new roles in development and cancer. Cancer Res. 80, 3451–3458. doi: 10.1158/0008-5472.can-19-3663
Roach, G., Heath Wallace, R., Cameron, A., Emrah Ozel, R., Hongay, C. F., Baral, R., et al. (2013). Loss of ascl1a prevents secretory cell differentiation within the zebrafish intestinal epithelium resulting in a loss of distal intestinal motility. Dev. Biol. 376, 171–186. doi: 10.1016/j.ydbio.2013.01.013
Seiser, R. M., Sundberg, A. E., Wollam, B. J., Zobel-Thropp, P., Baldwin, K., Spector, M. D., et al. (2006). Ltv1 is required for efficient nuclear export of the ribosomal small subunit in Saccharomyces cerevisiae. Genetics 174, 679–691. doi: 10.1074/jbc.M109.040774
Shi, Y., Zhang, Y., Zhao, F., Ruan, H., Huang, H., Luo, L., et al. (2014). Acetylcholine serves as a derepressor in loperamide-induced opioid-induced bowel dysfunction (OIBD) in zebrafish. Sci. Rep. 4:5602. doi: 10.1038/srep05602
Tafforeau, L., Zorbas, C., Langhendries, J.-L., Mullineux, S.-T., Stamatopoulou, V., Mullier, R., et al. (2011). The complexity of human ribosome biogenesis revealed by systematic nucleolar screening of pre-rRNA processing factors. Mol. Cell 51, 539–551. doi: 10.1016/j.molcel.2013.08.011
Tao, T., and Peng, J. (2009). Liver development in zebrafish (Danio rerio). J. Genet. Genomics 36, 325–334. doi: 10.1016/s1673-8527(08)60121-6
Taylor, A. M., Humphries, J. M., White, R. M., Murphey, R. D., Burns, C. E., and Zon, L. I. (2012). Hematopoietic defects in rps29 mutant zebrafish depend upon p53 activation. Exp. Hematol. 40, 228–237e225.
Venkatasubramani, N., and Mayer, A. N. (2008). A zebrafish model for the Shwachman-Diamond syndrome (SDS). Pediatr. Res. 63, 348–352. doi: 10.1203/PDR.0b013e3181659736
Wang, Y., Luo, Y., Hong, Y., Peng, J., and Lo, L. (2012). Ribosome biogenesis factor Bms1-like is essential for liver development in zebrafish. J. Genet. Genomics 39, 451–462. doi: 10.1016/j.jgg.2012.07.007
Wang, Y., Zhu, Q., Huang, L., Zhu, Y., Chen, J., Peng, J., et al. (2016). Interaction between Bms1 and Rcl1, two ribosome biogenesis factors, is evolutionally conserved in zebrafish and human. J. Genet. Genomics 43, 467–469. doi: 10.1016/j.jgg.2016.05.001
Wilkins, B. J., Lorent, K., Matthews, R. P., and Pack, M. (2013). p53-mediated biliary defects caused by knockdown of cirh1a, the zebrafish homolog of the gene responsible for North American Indian Childhood Cirrhosis. PLoS One 8:e77670. doi: 10.1371/journal.pone.0077670
Xue, S., and Barna, M. (2012). Specialized ribosomes: a new frontier in gene regulation and organismal biology. Nat. Rev. Mol. Cell Biol. 13, 355–369. doi: 10.1038/nrm3359
Yadav, G. V., Chakraborty, A., Uechi, T., and Kenmochi, N. (2014). Ribosomal protein deficiency causes Tp53-independent erythropoiesis failure in zebrafish. Int. J. Biochem. Cell Biol. 49, 1–7. doi: 10.1016/j.biocel.2014.01.006
Yadavilli, S., Mayo, L. D., Higgins, M., Lain, S., Hegde, V., and Deutsch, W. A. (2009). Ribosomal protein S3: a multi-functional protein that interacts with both p53 and MDM2 through its KH domain. DNA Repair 8, 1215–1224. doi: 10.1016/j.dnarep.2009.07.003
Zhang, Y., Morimoto, K., Danilova, N., Zhang, B., and Lin, S. (2012). Zebrafish models for dyskeratosis congenita reveal critical roles of p53 activation contributing to hematopoietic defects through RNA processing. PLoS One 7:e30188. doi: 10.1371/journal.pone.0030188
Keywords: Ltv1, ribosome biogenesis, digestive organs, hematopoiesis, P53
Citation: Zhang C, Huang R, Ma X, Chen J, Han X, Li L, Luo L, Ruan H and Huang H (2021) The Ribosome Biogenesis Factor Ltv1 Is Essential for Digestive Organ Development and Definitive Hematopoiesis in Zebrafish. Front. Cell Dev. Biol. 9:704730. doi: 10.3389/fcell.2021.704730
Received: 03 May 2021; Accepted: 13 September 2021;
Published: 07 October 2021.
Edited by:
Wolfgang Knabe, University of Münster, GermanyReviewed by:
Nadine Hein, Australian National University, AustraliaAnthony Henras, Université de Toulouse, France
Seth Corey, Cleveland Clinic, United States
Copyright © 2021 Zhang, Huang, Ma, Chen, Han, Li, Luo, Ruan and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hua Ruan, cnVhbmh1YTIzQDEyNi5jb20=; Honghui Huang, aG9uZ2h1aWhAMTI2LmNvbQ==