 Qiuzhen Li
Qiuzhen Li Kata Dudás1†
Kata Dudás1† Lajos Haracska
Lajos Haracska- 1HCEMM-BRC Mutagenesis and Carcinogenesis Research Group, Institute of Genetics, Biological Research Centre, Szeged, Hungary
- 2Mutagenesis and Carcinogenesis Research Group, Institute of Genetics, Biological Research Centre, Szeged, Hungary
DNA interstrand crosslinks (ICLs) are covalently bound DNA lesions, which are commonly induced by chemotherapeutic drugs, such as cisplatin and mitomycin C or endogenous byproducts of metabolic processes. This type of DNA lesion can block ongoing RNA transcription and DNA replication and thus cause genome instability and cancer. Several cellular defense mechanism, such as the Fanconi anemia pathway have developed to ensure accurate repair and DNA replication when ICLs are present. Various structure-specific nucleases and translesion synthesis (TLS) polymerases have come into focus in relation to ICL bypass. Current models propose that a structure-specific nuclease incision is needed to unhook the ICL from the replication fork, followed by the activity of a low-fidelity TLS polymerase enabling replication through the unhooked ICL adduct. This review focuses on how, in parallel with the Fanconi anemia pathway, PCNA interactions and ICL-induced PCNA ubiquitylation regulate the recruitment, substrate specificity, activity, and coordinated action of certain nucleases and TLS polymerases in the execution of stalled replication fork rescue via ICL bypass.
Introduction
Our genome is constantly exposed to different exogenous and endogenous DNA damaging factors. Chemotherapeutic drugs, such as cisplatin or mitomycin C and metabolites like those from lipid peroxidation can cause interstrand crosslinks (ICLs), covalent links between the opposite strands of the DNA (reviewed in Stone et al., 2008). ICLs prevent strand separation, physically blocking replication and transcription. Stalled replication forks may collapse, causing DNA double-strand breaks, which can lead to chromosomal rearrangements, carcinogenesis, or cell death (reviewed in Negrini et al., 2010; Lenart and Krejci, 2016). ICLs have significant clinical relevance; inactivation of the Fanconi Anemia (FA) ICL repair pathway leads to FA. Patients diagnosed with FA suffer from progressive bone marrow failure and have a higher risk of developing cancer (reviewed in Shimamura and Alter, 2010). Due to their high cytotoxicity, ICL-inducing agents are the earliest and most commonly applied chemotherapeutic drugs (Rosenberg et al., 1969).
Since ICLs pose a high risk to cell survival and genome integrity, cells have developed multiple pathways to repair this type of DNA lesion. Nucleotide excision repair (NER) is able to recognize and remove ICL lesions in non-S-phase cells as well, while when ICLs block ongoing replication forks, in higher eukaryotic cells, the Fanconi anemia complementation group (FANC) DNA repair proteins that belong to the Fanconi Anemia (FA) pathway are believed to be the main operating defense system (reviewed in Wood, 2010). Activation of the FA pathway leads to the recruitment of structure-specific nucleases and translesion DNA synthesis (TLS) polymerases to enable the bypass of the lesion as well as facilitate the recombination-dependent rescue system (Howlett et al., 2002; Sarkar et al., 2006; Hicks et al., 2010; Kim and D’Andrea, 2012). However, a general defense system, the so-called Rad6-18 postreplication repair system, also comes into play when replication encounters unrepaired DNA damage, such as ICLs (Shen et al., 2006). Rad6-Rad18-dependent monoubiquitination of proliferating cell nuclear antigen (PCNA) initiates a number of subsequent replication fork rescue processes and is believed to serve as a key regulatory step in stalled replication fork rescue (reviewed in Chang and Cimprich, 2009).
Although the FA as well as the Rad6-Rad18 postreplication repair pathway become activated when replication stalls at ICLs, their interplay has been less characterized. In this review, we are placing the focus on sensors of ICLs, nucleases for ICL unhooking, and TLS polymerases for ICL adduct bypass with particular emphasis on parallels and possible interplays between their regulation by ubiquitylation of the FANCI and FANCD2 heterodimer (ID2) and ubiquitylation of PCNA during the rescue of replication forks stalled at ICLs.
Sensors and Transducers of ICL Repair Pathways
ICL repair is mostly activated during the S phase, but there are secondary mechanisms that are active in quiescent cells as well (Williams et al., 2012). Although these pathways have distinct mechanisms depending on the cell cycle, they all have common key steps. First, the lesion is recognized by sensor proteins that recruit other downstream regulators. During the G0/G1 phase, mainly nucleotide excision repair (NER) pathways monitor the genome, searching for ICL-caused distortion in the DNA. The XPA (Xeroderma pigmentosum A) and RPA (replicative protein A) protein, after having been recruited to the damaged area, load the structure-specific nuclease ERCC1-XPF onto the DNA (Volker et al., 2001; Tsodikov et al., 2007).
In the S phase of the cell cycle, ICLs cause replication fork stalling followed by activation of Ataxia telangiectasia (ATR)-dependent damage signaling, which will prevent dormant replication fork firing, while stabilizing the stalled replication fork (Luke-Glaser et al., 2010; Schwab et al., 2010). As shown in Figure 1A, the binding of FANCM at the site of the ICL has an essential role since it provides a platform for anchoring other FA proteins (Collis et al., 2008; Deans and West, 2009). Although FANCM seems to be an upstream regulator of ATR, its activity is also induced by ATR-dependent phosphorylation (Ciccia et al., 2007; Collis et al., 2008; Singh et al., 2013). When the phosphorylated FANCM recognizes ICLs, it recruits the FA core complex, which has a ubiquitin ligase activity transferring the ubiquitin with the help of FANCL, a RING-domain containing E3, from the UBE2T/FANCT, an E2 enzyme, to the FANCI and FANCD2 heterodimer (ID2) at lysines 523 and 561, respectively (Smogorzewska et al., 2007; Alpi et al., 2008; Huang et al., 2014). Prior to monoubiquitination, ID2 seems to be recruited by the FANC core complex to the damage where its DNA-binding generates the needed conformational change in FANCD2 for its ubiquitylation. Ubiquitylation closes the ID2 complex into a clamp conformation (Liang et al., 2015; Alcón et al., 2020; Tan et al., 2020). The activated ID2 complex then serves as a central hub for subsequent molecular events by enabling the recruitment of proteins that provide finally the rescue of the replication fork stalled at the ICL. These proteins include nucleases for unhooking ICLs, TLS polymerases for bypass of the unhooked adduct, repair factors for ICL elimination, and several factors of homologous recombination (Figure 1A).
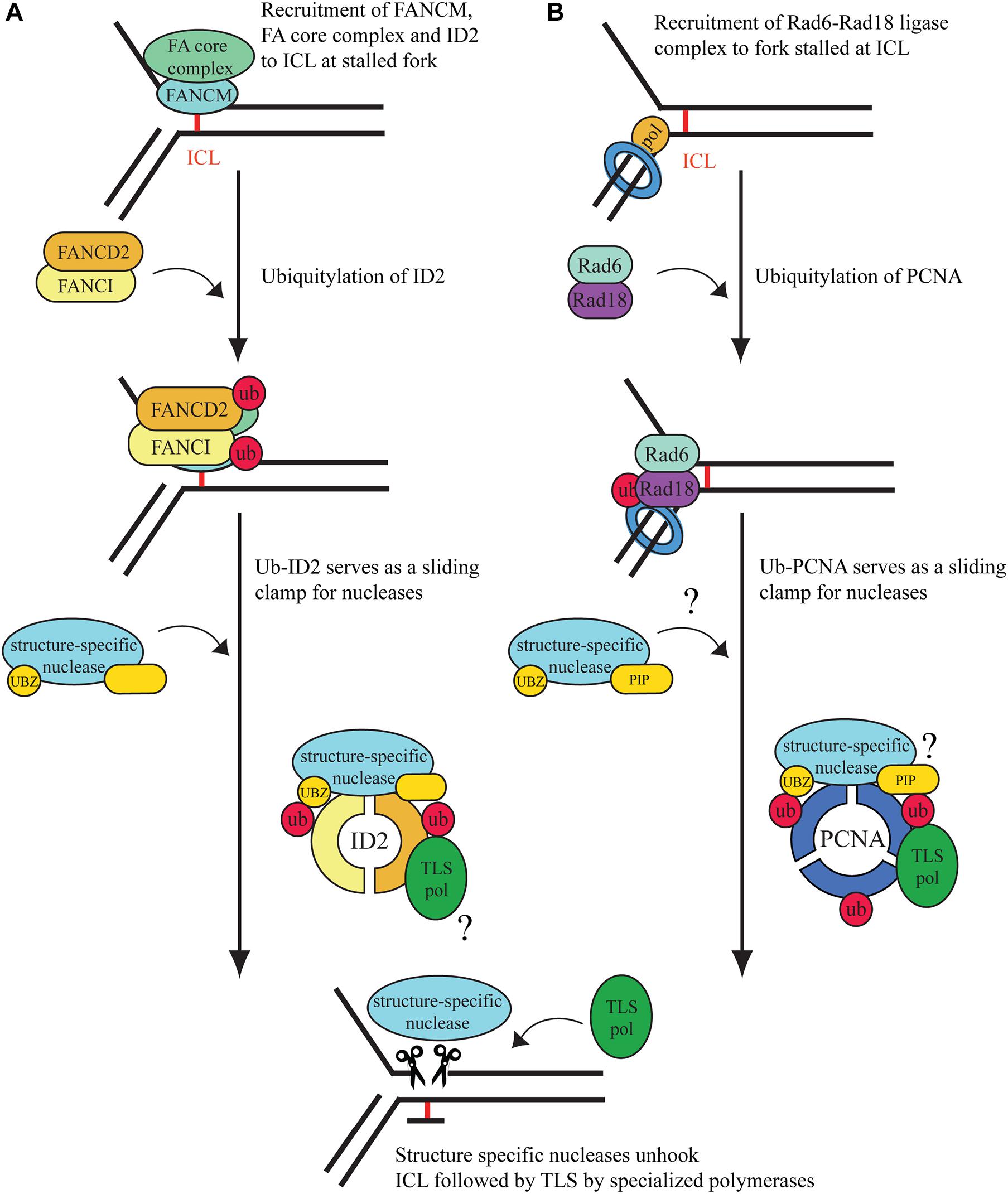
Figure 1. Interstrand crosslink repair pathways at the stalled replication fork. (A) Replication-dependent ICL repair is considered to be carried out mainly by the Fanconi anemia pathway. After damage recognition, the ID2 complex is ubiquitylated by the activated FA core complex. The monoubiquitinated ID2 complex can further recruit structure-specific nucleases for unhooking. Gap filling is carried out by TLS polymerases, but the mechanism behind the recruitment of the polymerases is yet unknown. We hypothesize that Ub-ID2 serves as a DNA- encircling sliding clamp for TLS polymerases and nucleases. (B) When the ongoing DNA replication fork is blocked by DNA lesions, such as ICLs, upstream factors recognize it, and PCNA is monoubiquitinated by RAD6/RAD18. We hypothesize that monoubiquitinated PCNA acts as a hub for the recruitment of certain PIP box- and UBZ domain-containing structure-specific nucleases to unhook the ICL lesion. Translesion synthesis polymerases are also recruited to the site of damage by Ub-PCNA, which complete the bypass of the DNA damage. The covalent bond of the monoadduct is removed by nucleotide excision repair.
Interestingly, in addition to the FANCM-dependent mechanism, other ICL-sensors, such as the Ubiquitin-Like PHD And RING Finger Domain-Containing Protein 1 (UHRF1) was also identified (Liang et al., 2015). UHRF1 and its paralog, UHRF2, can recruit FANCD2 to the site of the ICL and stimulate monoubiquitination of the ID2 complex (Motnenko et al., 2018).
PCNA Ubiquitylation, a Sensor and Signal Transducer in the Replication of ICL Lesions
The homotrimer PCNA is the master regulator of the replication fork. PCNA forms a sliding clamp over the DNA double strand and serves as a processivity factor for replicative polymerases. Encountering an unrepaired DNA lesion, such as an ICL results in the stalling of the replicative polymerase, which leads to the recruitment of Rad6-Rad18, a ubiquitin conjugating-ligase protein complex, which facilitates PCNA monoubiquitination (Bailly et al., 1997; Yoon et al., 2012). Monoubiquitinated PCNA can recruit TLS polymerases, which synthesize DNA across the damaged region either in an error-free or an error-prone mode, depending on the actual lesion and the TLS polymerase accessed (reviewed in Prakash et al., 2005). Many TLS polymerases, such as Pol eta, Pol kappa, and Pol iota exhibit conserved PCNA-interacting (PIP) as well as ubiquitin-binding (UBD) domains, by which they can strongly associate with monoubiquitinated PCNA (Ub-PCNA), which provides their timely access to the primer ends at stalled forks when replication encounters a barrier (Haracska et al., 2001a; Plosky et al., 2006). The PIP and UBD domains are also exhibited in certain nucleases implicated in ICL repair, such as SNM1A and FAN1, which raises the possibility that, similarly to Ub-ID2, Ub-PCNA can also play a role in their targeting to replication-stalling ICLs (Yang et al., 2010; Porro et al., 2017). Monoubiquitinated PCNA can also undergo polyubiquitylation in an Mms2–Ubc13 (E2)- and HTLF-SHPRH (E3)-dependent manner (Unk et al., 2008). Polyubiquitylated PCNA can initiate template switching, an error-free pathway for stalled replication fork rescue, which can involve fork reversal. Fork reversal can place ICL back to a general double-stranded DNA region, making it accessible for excision repair pathways, such as NER. Interestingly, RAD18 also has a function in ICL repair, independent of ubiquitinated PCNA. RAD18 has been shown to indirectly regulate the ubiquitination and loading of FANCD2 and FANCI in S phase, which suggests a possible role for RAD18 as an E3 ligase of the FA core complex (Williams et al., 2011). Recently, the RING finger and WD repeat domain-containing protein 3 (RFWD3) has been identified as an activator of various rescue mechanisms at the stalled replication fork, such as translesion synthesis and homologous recombination (HR) (Elia et al., 2015; Inano et al., 2017). Importantly for ICL repair, RFWD3 was suggested to be a FA gene since its biallelic mutation was found in a child with FA, which was also supported by results in cellular systems and animal model (Knies et al., 2017). Indicating complex interplays, the interaction of RFWD3 and PCNA stabilizes RFWD3 to the replication fork, and PCNA also interacts with FANCM, pointing to its possible function in lesion recognition (Rohleder et al., 2016; Lin et al., 2018).
Structure-Specific Nucleases as Executors of ICL Unhooking at the Stalled Replication Fork
Following lesion recognition, the ICL is released from one of the DNA strands by a process called unhooking. During unhooking, structure-specific nucleases nick the DNA on both sides of the ICL, removing it from one of the parental strands. Several nucleases have been implicated in ICL unhooking, such as XPF-ERCC1, MUS81-EME1, SLX1, SNM1A, and FAN1 the deficiency of which renders cells sensitive to ICL-generating agents. These nucleases alone or in collaboration can cleave the DNA on both sites of the ICL, leaving a gap, which can be filled in subsequently by TLS polymerases (Figure 2). XPF and SLX1 nucleases are considered to incise at 3′- and 5′-sides of the ICL (Kuraoka et al., 2000; Fricke and Brill, 2003), respectively, while MUS81 cuts at 3′-sides in specific cases (Ciccia et al., 2003). Various interactions can modulate the cleavage specificity of these enzymes; the nuclease activity of SLX1 can be extremely enhanced by its interaction with SLX4 (Fricke and Brill, 2003). As a scaffold protein, SLX4 can interact with several other nucleases and via its UBZ-domain-mediated binding to Ub-ID2 it can recruit XPF-ERCC1-MUS81-EME1-SLX1-SLX4 to the ICL (Fekairi et al., 2009; Castor et al., 2013). Ub-ID2 might play a role in the recruitment of other nucleases, as well, as it was proposed for FAN1; however, here we put more focus on interaction of SNM1A and FAN1 with Ub-PCNA in the ICL repair process. The SNM1A nuclease contains a ubiquitin−binding zinc finger (UBZ) domain at the N-terminal and a PIP box in the middle region of the protein (Yang et al., 2010). SNM1A has an intrinsic 5′ to 3′ exonuclease activity and was shown to be epistatic with the XPF-ERCC1 endonuclease that can nick the DNA 5′ from the ICL (Wang et al., 2011). Their coordinated action in ICL unhooking was shown; the XPF-ERCC1-generated nick provided an entry point for SNM1A exonuclease activity (Wang et al., 2011). Recently, SNM1A has been shown to have single-strand-specific endonuclease activity as well (5′ and 3′overhangs, hairpins, flaps, and gapped substrates) (Buzon et al., 2018).
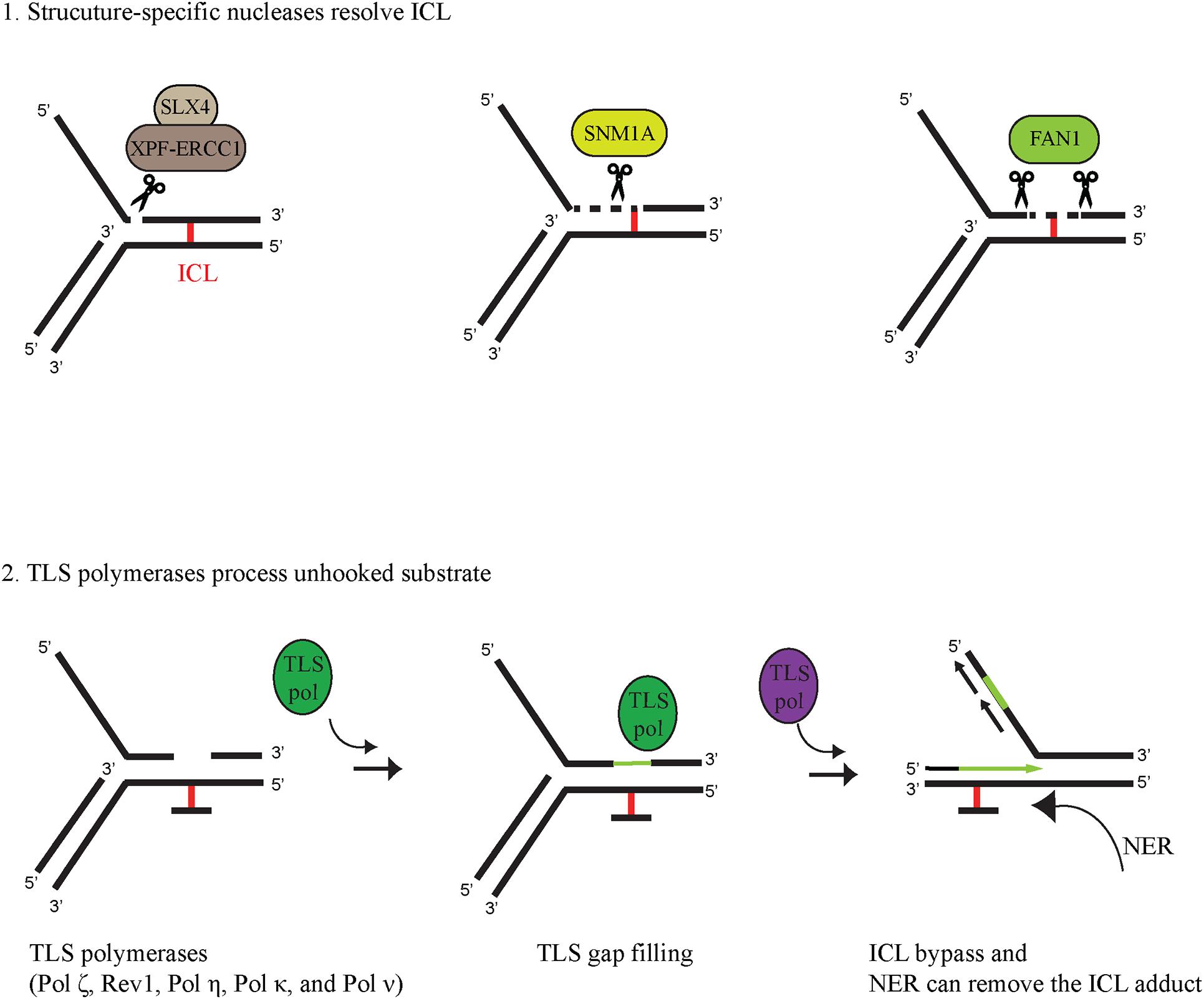
Figure 2. Coordinated activity of structure-specific nucleases and TLS polymerases in ICL bypass (1) Structure-specific nucleases unhook ICL-containing DNA. SLX4 serves as a scaffold protein for XPF-ERCC1 to digest the dsDNA phosphodiester bond at the 5′ primer of the junction between dsDNA and ssDNA. The digested substrate can be further processed by the 5′-3′ exonuclease activity of SNM1A. FAN1 can completely unhook the ICL-containing substrate due to its 5′-3′ endo/exonuclease activity. (2) TLS polymerases can process the unhooked ICL-containing DNA by filling in the gap generated by ICL-unhooking nucleases and by extending the primer through the unhooked ICL adduct. Finally, the ICL monoadduct can be removed by another DNA repair pathway, such as nucleotide excision repair (NER).
The FAN1 nuclease also possesses a UBZ domain and a PIP box at the N-terminal end (Smogorzewska et al., 2007; Pennell et al., 2014). Originally, several reports described FAN1 as a member of the FA pathway since its action on ICL-containing DNA was described to be dependent on monoubiquitinated FANCD2 (Kratz et al., 2010; Liu et al., 2010; MacKay et al., 2010). However, later on, it turned out that patients with biallelic FAN1 mutations do not develop FA, and FAN1 does not show epistasis with other FA genes, which indicates some other role for FAN1 in ICL repair (Zhou et al., 2012; Lachaud et al., 2016b). FAN1 has an endonuclease activity and is able to incise 5′ to the ICL at the 4th nucleotide after the replication fork junction on the 5′ flap model substrate (Figure 2). FAN1 has a 5′→ 3′ exonuclease activity as well, initiating cleavage 4 nt from the 5′ end on single- and double-stranded DNA (MacKay et al., 2010; Lachaud et al., 2016a). FAN1 is able to unhook nitrogen mustard-induced interstrand crosslinks in vitro due to its nuclease activity (Pizzolato et al., 2015). FAN1 also interacts with ubiquitin-PCNA and enhances PCNA ubiquitylation after mitomycin C treatment (Porro et al., 2017).
TLS Polymerases Process Substrates Unhooked by Nucleases
Replicative polymerases, such as the human Pol δ and Pol ε have high fidelity and possess 3′→ 5′ exonuclease activity as a proofreading function to ensure precise DNA replication (Johnson et al., 2015). However, there is a cost of high fidelity since DNA contains many lesions, such as base adducts, photoproducts, intrastrand and interstrand crosslinks, which cannot be accommodated by the tight active sites of the replicative polymerases, leading to blocked replication fork machinery (Bezalel-Buch et al., 2020). Stalling of replication can lead to strand breaks, chromosomal rearrangement, and other genome-destabilizing events; to avoid this, the so-called DNA damage tolerance (DDT) pathways, such as the Rad6-Rad18-dependent PCNA-monoubiquitination-mediated one come into play upon fork stalling (Davies et al., 2008). One sub-branch of these DDT pathways is translesion synthesis, in which, at the damage, low-fidelity polymerases take over the 3′ primer end from the replicative polymerase and insert either the correct or uncorrect nucleotide opposite the lesion, leading to error-free or error-prone bypass. As shown in Figure 2, bypass of ICL may involve TLS polymerase action at two points; in the gap filling after the nuclease has unhooked the ICL and in the replication through the adduct. Based on genetic assays with TLS polymerase-deficient cells as well as biochemical findings, many TLS polymerases have been implicated in the bypass of the unhooked ICL (Ho et al., 2011; Roy et al., 2016; Bezalel-Buch et al., 2020). Sensitivity assays performed by treating cells with various crosslinking agents revealed ICL-repair functions for Pol ζ, Rev1, Pol η, Pol κ, and Pol ν (reviewed in Ho and Schärer (2010). Although various crosslinking agents can produce a wide variety of ICLs requiring different TLS polymerases for bypass, based on cisplatin and mitomycin C exposure, REV3-encoded Pol ζ and REV1 are believed to be among the main players of ICL bypass (Hicks et al., 2010). Genetic evidence also supports the involvement of Pol η in a more general role in ICL bypass, while Pol κ seems to be more restricted to minor groove DNA adducts and Pol ν to major groove ICL bypass (Zhang et al., 2000; Acharya et al., 2008; Yamanaka et al., 2010; Roy et al., 2016). TLS polymerases can bypass DNA lesions in a two-step fashion, first inserting a nucleotide opposite the lesion and then extending opposite from the lesion (Johnson et al., 2000; Haracska et al., 2001b). Certain TLS polymerases can carry out both steps, but often it requires the collaboration of two polymerases: an inserter and an extender (reviewed in Ho and Schärer, 2010). Even though the deficiency of a certain TLS polymerase does not cause strong hypersensitivity to crosslinking agents, its involvement in ICL bypass cannot be ruled out because cells can use multiple TLS polymerases as inserters as well as extenders. Although purified REV1 together with Pol ζ show complete bypass synthesis, experiments using Xenopus egg extracts indicate that Pol ζ and REV1 are required only for the extension step past a cisplatin-induced ICL (Bezalel-Buch et al., 2020). Pol η alone is able to carry out both the insertion and the extension steps across various major ICL lesions (Roy et al., 2016). Pol η and many other TLS polymerases, such as Pol κ and Pol ι contain PIP and ubiquitin-binding motif (UBM)/UBZ domains allowing them to interact with Ub-PCNA, which can target them to the site of the stalled fork at the ICL as well as stimulate their synthetic activity (Haracska et al., 2001a; Plosky et al., 2006).
Discussion
Cytotoxic ICL lesions pose a considerable threat to cells regardless of cell phase. Cells can remove ICLs by NER during the G0/G1 phase, but some may escape repair and cause stalling of the replication machinery. In higher eukaryotic cells, the FA pathway is considered the main defense system to repair ICLs during the S phase of the cell cycle. In this review, we compare the FA pathway to a more general defense system, the Rad6-RAD18-dependent PCNA monoubiquitination, which is activated when replication stalls at various lesions, including ICLs. We also point out the similarities between the FA and Rad6-Rad18 pathways in dealing with the ICL at the stalled replication fork (Figure 1) and summarize our current knowledge on ICL-unhooking nucleases and ICL bypass polymerases (Figure 2), and reach the following conclusions. First, when replication encounters an ICL, monoubiquitation of the central hub proteins, ID2 and PCNA, is a critical step for the operation of the FA and Rad6-Rad18 pathways, respectively. Second, several proteins implicated in ICL repair can interact with Ub-ID2 as well as Ub-PCNA, such as the FAN1 nuclease, which exhibits PIP and UBZ domains for timely binding to the stalled fork (Buzon et al., 2018). Interestingly, deficiency of FAN1 can be partially complemented by the SNM1A nuclease, which also exhibits PIP and UBZ domains (Yang et al., 2010; Buzon et al., 2018). Third, ubiquitylation can provide access to unhooked ICL adduct bypass of various TLS polymerases exhibiting PIP and, in most cases, UBZ domains as well, which indicates similarities between the recruitment of nucleases and polymerases. Fourth, after completion of the repair process, both FA and Rad6-Rad18 DDT pathways are terminated by USP1-dependent deubiquitylation of their central hub proteins (Nijman et al., 2005; Huang et al., 2006). Importantly, PCNA encircles DNA as a homotrimeric sliding clamp, and each of its subunit can be ubiquitylated, providing three binding surfaces for PIP- and UBZ-domain-containing proteins. Thus, it is possible that proteins exhibiting these two domains, such as an ICL-unhooking nuclease like FAN1 and a TLS polymerase like Pol η, can bind to a Ub-PCNA ring at the same time, which would provide a high degree of coordination between ICL unhooking and bypass. Interestingly, recent structural studies revealed that ubiquitylation of the ID2 complex results in its conformational change which converts the ID2 to a clamp encircling the DNA (Alcón et al., 2020; Wang et al., 2020). The structural analogy between Ub-PCNA and Ub-ID2 is tempting and forces one to speculate whether Ub-ID2 can serve as a sliding clamp for TLS polymerases in ICL bypass. Also, it would be interesting to explore whether the Ub-PCNA and Ub-ID2 sliding clamps can bind ICL nucleases and TLS polymerases at the same time for efficient bypass. Finally, it remains to be explored whether Ub-PCNA and Ub-ID2 rings can interact and provide a joint sliding clamp for protein exchange and higher coordination between the FA and Ub-PCNA ICL damage bypass pathways.
Author Contributions
QL and KD reviewed the literature and wrote the first draft of the manuscript. GT and LH revised and edited the manuscript. All authors approved the final version of the manuscript for submission.
Funding
This project received funding from the European Union’s Horizon 2020 Research and Innovation Programme under Grant Agreement No. 739593. This work was also supported by the National Research, Development and Innovation Office GINOP-2.3.2–15-2016–00024 and GINOP-2.3.2–15-2016–00026.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Acharya, N., Yoon, J.-H., Gali, H., Unk, I., Haracska, L., Johnson, R. E., et al. (2008). Roles of PCNA-binding and ubiquitin-binding domains in human DNA polymerase η in translesion DNA synthesis. Proc. Natl. Acad. Sci. 105, 17724–17729. doi: 10.1073/pnas.0809844105
Alcón, P., Shakeel, S., Chen, Z. A., Rappsilber, J., Patel, K. J., and Passmore, L. A. (2020). FANCD2–FANCI is a clamp stabilized on DNA by monoubiquitination of FANCD2 during DNA repair. Nat. Struct. Mol. Biol. 27, 240–248. doi: 10.1038/s41594-020-0380-1
Alpi, A. F., Pace, P. E., Babu, M. M., and Patel, K. J. (2008). Mechanistic insight into site-restricted monoubiquitination of FANCD2 by Ube2t, FANCL, and FANCI. Mol. Cell 32, 767–777. doi: 10.1016/j.molcel.2008.12.003
Bailly, V., Lauder, S., Prakash, S., and Prakash, L. (1997). Yeast DNA repair proteins Rad6 and Rad18 form a heterodimer that has ubiquitin conjugating, DNA binding, and ATP hydrolytic activities. J. Biol. Chem. 272, 23360–23365. doi: 10.1074/jbc.272.37.23360
Bezalel-Buch, R., Cheun, Y. K., Roy, U., Schärer, O. D., and Burgers, P. M. (2020). Bypass of DNA interstrand crosslinks by a Rev1–DNA polymerase ζ complex. Nucleic Acids Res. 48, 8461–8473. doi: 10.1093/nar/gkaa580
Buzon, B., Grainger, R., Huang, S., Rzadki, C., and Junop, M. S. (2018). Structure-specific endonuclease activity of SNM1A enables processing of a DNA interstrand crosslink. Nucleic Acids Res. 46, 9057–9066. doi: 10.1093/nar/gky759
Castor, D., Nair, N., Déclais, A. C., Lachaud, C., Toth, R., Macartney, T. J., et al. (2013). Cooperative control of holliday junction resolution and DNA repair by the SLX1 and MUS81-EME1 nucleases. Mol. Cell 52, 221–233. doi: 10.1016/j.molcel.2013.08.036
Chang, D. J., and Cimprich, K. A. (2009). DNA damage tolerance: when it’s ok to make mistakes. Nat. Chem. Biol. 5, 82–90. doi: 10.1038/nchembio.139
Ciccia, A., Constantinou, A., and West, S. C. (2003). Identification and characterization of the human Mus81-Emel endonuclease. J. Biol. Chem. 278, 25172–25178. doi: 10.1074/jbc.m302882200
Ciccia, A., Ling, C., Coulthard, R., Yan, Z., Xue, Y., Meetei, A. R., et al. (2007). Identification of FAAP24, a fanconi anemia core complex protein that interacts with FANCM. Mol. Cell 25, 331–343. doi: 10.1016/j.molcel.2007.01.003
Collis, S. J., Ciccia, A., Deans, A. J., Horejsí, Z., Martin, J. S., Maslen, S. L., et al. (2008). FANCM and FAAP24 function in ATR-mediated checkpoint signaling independently of the Fanconi anemia core complex. Mol. Cell 32, 313–324. doi: 10.1016/j.molcel.2008.10.014
Davies, A. A., Huttner, D., Daigaku, Y., Chen, S., and Ulrich, H. D. (2008). Activation of ubiquitin-dependent DNA damage bypass is mediated by replication protein a. Mol. Cell 29, 625–636. doi: 10.1016/j.molcel.2007.12.016
Deans, A. J., and West, S. C. (2009). FANCM connects the genome instability disorders bloom’s syndrome and fanconi anemia. Mol. Cell 36, 943–953. doi: 10.1016/j.molcel.2009.12.006
Elia, A. E. H., Wang, D. C., Willis, N. A., Boardman, A. P., Hajdu, I., Adeyemi, R. O., et al. (2015). RFWD3-dependent ubiquitination of RPA regulates repair at stalled replication forks. Mol. Cell 60, 280–293. doi: 10.1016/j.molcel.2015.09.011
Fekairi, S., Scaglione, S., Chahwan, C., Taylor, E. R., Tissier, A., Coulon, S., et al. (2009). Human SLX4 Is a holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell 138, 78–89. doi: 10.1016/s9999-9994(09)20375-8
Fricke, W. M., and Brill, S. J. (2003). Slx1 - Slx4 is a second structure-specific endonuclease functionally redundant with Sgs1 - Top3. Genes Dev. 17, 1768–1778. doi: 10.1101/gad.1105203
Haracska, L., Kondratick, C. M., Unk, I., Prakash, S., and Prakash, L. (2001a). Interaction with PCNA is essential for yeast DNA polymerase eta function. Mol. Cell 8, 407–415. doi: 10.1016/s1097-2765(01)00319-7
Haracska, L., Unk, I., Johnson, R. E., Johansson, E., Burgers, P. M., Prakash, S., et al. (2001b). Roles of yeast DNA polymerases delta and zeta and of Rev1 in the bypass of abasic sites. Genes Dev. 15, 945–954. doi: 10.1101/gad.882301
Hicks, J. K., Chute, C. L., Paulsen, M. T., Ragland, R. L., Howlett, N. G., Guéranger, Q., et al. (2010). Differential roles for DNA polymerases Eta, Zeta, and REV1 in lesion bypass of intrastrand versus interstrand DNA cross-links. Mol. Cell. Biol. 30, 1217–1230. doi: 10.1128/mcb.00993-09
Ho, T. V., and Schärer, O. D. (2010). Translesion DNA synthesis polymerases in DNA interstrand crosslink repair. Environ. Mol. Mutagen. 51, 552–566.
Ho, T. V., Guainazzi, A., Derkunt, S. B., Enoiu, M., and Schärer, O. D. (2011). Structure-dependent bypass of DNA interstrand crosslinks by translesion synthesis polymerases. Nucleic Acids Res. 39, 7455–7464. doi: 10.1093/nar/gkr448
Howlett, N. G., Taniguchi, T., Olson, S., Cox, B., Waisfisz, Q., De Die-Smulders, C., et al. (2002). Biallelic inactivation of BRCA2 in Fanconi anemia. Science 297, 606–609. doi: 10.1126/science.1073834
Huang, T. T., Nijman, S. M. B., Mirchandani, K. D., Galardy, P. J., Cohn, M. A., Haas, W., et al. (2006). Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat. Cell Biol. 8, 341–347. doi: 10.1038/ncb1378
Huang, Y., Leung, J. W. C., Lowery, M., Matsushita, N., Wang, Y., Shen, X., et al. (2014). Modularized functions of the Fanconi anemia core complex. Cell Rep. 7, 1849–1857. doi: 10.1016/j.celrep.2014.04.029
Inano, S., Sato, K., Katsuki, Y., Kobayashi, W., Tanaka, H., Nakajima, K., et al. (2017). RFWD3-mediated ubiquitination promotes timely removal of both RPA and RAD51 from DNA damage sites to facilitate homologous recombination. Mol. Cell 66, 622–634.e8.
Johnson, R. E., Klassen, R., Prakash, L., and Prakash, S. (2015). A major role of DNA Polymerase δ in replication of both the leading and lagging DNA strands. Mol. Cell 59, 163–175. doi: 10.1016/j.molcel.2015.05.038
Johnson, R. E., Washington, M. T., Haracska, L., Prakash, S., and Prakash, L. (2000). Eukaryotic polymerases ι and ζ act sequentially to bypass DNA lesions. Nature 406, 1015–1019. doi: 10.1038/35023030
Kim, H., and D’Andrea, A. D. (2012). Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 26, 1393–1408. doi: 10.1101/gad.195248.112
Knies, K., Inano, S., Ramírez, M. J., Ishiai, M., Surrallés, J., Takata, M., et al. (2017). Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. J. Clin. Invest. 127, 3013–3027. doi: 10.1172/jci92069
Kratz, K., Schöpf, B., Kaden, S., Sendoel, A., Eberhard, R., Lademann, C., et al. (2010). Deficiency of FANCD2-associated nuclease KIAA1018/FAN1 sensitizes cells to interstrand crosslinking agents. Cell 142, 77–88. doi: 10.1016/j.cell.2010.06.022
Kuraoka, I., Kobertz, W. R., Ariza, R. R., Biggerstaff, M., Essigmann, J. M., and Wood, R. D. (2000). Repair of an interstrand DNA cross-link initiated by ERCC1-XPF repair/recombination nuclease. J. Biol. Chem. 275, 26632–26636. doi: 10.1074/jbc.c000337200
Lachaud, C., Moreno, A., Marchesi, F., Toth, R., Blow, J. J., and Rouse, J. (2016a). Ubiquitinated Fancd2 recruits Fan1 to stalled replication forks to prevent genome instability. Science 351, 846–849. doi: 10.1126/science.aad5634
Lachaud, C., Slean, M., Marchesi, F., Lock, C., Odell, E., Castor, D., et al. (2016b). Karyomegalic interstitial nephritis and DNA damage-induced polyploidy in Fan1 nuclease-defective knock-in mice. Genes Dev. 30, 639–644. doi: 10.1101/gad.276287.115
Lenart, P., and Krejci, L. (2016). DNA, the central molecule of aging. Mutat. Res. 786, 1–7. doi: 10.1016/j.mrfmmm.2016.01.007
Liang, C. C., Zhan, B., Yoshikawa, Y., Haas, W., Gygi, S. P., and Cohn, M. A. (2015). UHRF1 Is a sensor for DNA interstrand crosslinks and recruits FANCD2 to initiate the Fanconi anemia pathway. Cell Rep. 10, 1947–1956. doi: 10.1016/j.celrep.2015.02.053
Lin, Y. C., Wang, Y., Hsu, R., Giri, S., Wopat, S., Arif, M. K., et al. (2018). PCNA-mediated stabilization of E3 ligase RFWD3 at the replication fork is essential for DNA replication. Proc. Natl. Acad. Sci. U. S. A. 115, 13282–13287. doi: 10.1073/pnas.1814521115
Liu, T., Ghosal, G., Yuan, J., Chen, J., and Huang, J. (2010). FAN1 acts with FANCI-FANCD2 to promote DNA interstrand cross-link repair. Science 329, 693–696. doi: 10.1126/science.1192656
Luke-Glaser, S., Luke, B., Grossi, S., and Constantinou, A. (2010). FANCM regulates DNA chain elongation and is stabilized by S-phase checkpoint signalling. EMBO J. 29, 795–805. doi: 10.1038/emboj.2009.371
MacKay, C., Déclais, A.-C., Lundin, C., Agostinho, A., Deans, A. J., MacArtney, T. J., et al. (2010). Identification of KIAA1018/FAN1, a DNA repair nuclease recruited to DNA damage by monoubiquitinated FANCD2. Cell 142, 65–76. doi: 10.1016/j.cell.2010.06.021
Motnenko, A., Liang, C. C., Yang, D., Lopez-Martinez, D., Yoshikawa, Y., Zhan, B., et al. (2018). Identification of UHRF2 as a novel DNA interstrand crosslink sensor protein. PLoS Genet. 14:e1007643. doi: 10.1371/journal.pgen.1007643
Negrini, S., Gorgoulis, V. G., and Halazonetis, T. D. (2010). Genomic instability an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 11, 220–228. doi: 10.1038/nrm2858
Nijman, S. M. B., Huang, T. T., Dirac, A. M. G., Brummelkamp, T. R., Kerkhoven, R. M., D’Andrea, A. D., et al. (2005). The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol. Cell 17, 331–339. doi: 10.1016/j.molcel.2005.01.008
Pennell, S., Déclais, A.-C., Li, J., Haire, L. F., Berg, W., Saldanha, J. W., et al. (2014). FAN1 activity on asymmetric repair intermediates is mediated by an atypical monomeric virus-type replication-repair nuclease domain. Cell Rep. 8, 84–93. doi: 10.1016/j.celrep.2014.06.001
Pizzolato, J., Mukherjee, S., Schärer, O. D., and Jiricny, J. (2015). FANCD2-associated nuclease 1, but not exonuclease 1 or flap endonuclease 1, is able to unhook DNA interstrand cross-links in vitro. J. Biol. Chem. 290, 22602–22611. doi: 10.1074/jbc.m115.663666
Plosky, B. S., Vidal, A. E., Fernández, de Henestrosa, A. R., McLenigan, M. P., McDonald, J. P., et al. (2006). Controlling the subcellular localization of DNA polymerases iota and eta via interactions with ubiquitin. EMBO J. 25, 2847–2855. doi: 10.1038/sj.emboj.7601178
Porro, A., Berti, M., Pizzolato, J., Bologna, S., Kaden, S., Saxer, A., et al. (2017). FAN1 interaction with ubiquitylated PCNA alleviates replication stress and preserves genomic integrity independently of BRCA2. Nat. Commun. 8:1073.
Prakash, S., Johnson, R. E., and Prakash, L. (2005). Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu. Rev. Biochem. 74, 317–353. doi: 10.1146/annurev.biochem.74.082803.133250
Rohleder, F., Huang, J., Xue, Y., Kuper, J., Round, A., Seidman, M., et al. (2016). FANCM interacts with PCNA to promote replication traverse of DNA interstrand crosslinks. Nucleic Acids Res. 44, 3219–3232. doi: 10.1093/nar/gkw037
Rosenberg, B., Vancamp, L., Trosko, J. E., and Mansour, V. H. (1969). Platinum compounds: a new class of potent antitumour agents. Nature 222, 385–386. doi: 10.1038/222385a0
Roy, U., Mukherjee, S., Sharma, A., Frank, E. G., and Schärer, O. D. (2016). The structure and duplex context of DNA interstrand crosslinks affects the activity of DNA polymerase η. Nucleic Acids Res. 44, 7281–7291.
Sarkar, S., Davies, A. A., Ulrich, H. D., and McHugh, P. J. (2006). DNA interstrand crosslink repair during G1 involves nucleotide excision repair and DNA polymerase zeta. EMBO J. 25, 1285–1294. doi: 10.1038/sj.emboj.7600993
Schwab, R. A., Blackford, A. N., and Niedzwiedz, W. (2010). ATR activation and replication fork restart are defective in FANCM-deficient cells. EMBO J. 29, 806–818. doi: 10.1038/emboj.2009.385
Shen, X., Jun, S., O’Neal, L. E., Sonoda, E., Bemark, M., Sale, J. E., et al. (2006). REV3 and REV1 play major roles in recombination-independent repair of DNA interstrand cross-links mediated by monoubiquitinated proliferating cell nuclear antigen (PCNA). J. Biol. Chem. 281, 13869–13872. doi: 10.1074/jbc.c600071200
Shimamura, A., and Alter, B. P. (2010). Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 24, 101–122. doi: 10.1016/j.blre.2010.03.002
Singh, T. R., Ali, A. M., Paramasivam, M., Pradhan, A., Wahengbam, K., Seidman, M. M., et al. (2013). ATR-dependent phosphorylation of FANCM at serine 1045 is essential for FANCM functions. Cancer Res. 73, 4300–4310. doi: 10.1158/0008-5472.can-12-3976
Smogorzewska, A., Matsuoka, S., Vinciguerra, P., McDonald, E. R., Hurov, K. E., Luo, J., et al. (2007). Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 129, 289–301. doi: 10.1016/j.cell.2007.03.009
Stone, M. P., Cho, Y., Huang, Kim, H.-Y., Kozekov, I. V., Kozekov, A., et al. (2008). Interstrand DNA cross-links induced by α,β-Unsaturated aldehydes derived from lipid peroxidation and environmental sources. Acc. Chem. Res. 41, 793–804. doi: 10.1021/ar700246x
Tan, W., van Twest, S., Leis, A., Bythell-Douglas, R., Murphy, V. J., Sharp, M., et al. (2020). Monoubiquitination by the human Fanconi anemia core complex clamps FANCI:FANCD2 on DNA in filamentous arrays. Elife 9:e54128.
Tsodikov, O. V., Ivanov, D., Orelli, B., Staresincic, L., Shoshani, I., and Oberman, R. (2007). Structural basis for the recruitment of ERCC1-XPF to nucleotide excision repair complexes by XPA. EMBO J. 26, 4768–4776. doi: 10.1038/sj.emboj.7601894
Unk, I., Hajdú, I., Fátyol, K., Hurwitz, J., Yoon, J. H., Prakash, L., et al. (2008). Human HLTF functions as a ubiquitin ligase for proliferating cell nuclear antigen polyubiquitination. Proc. Natl. Acad. Sci. U. S. A. 105, 3768–3773. doi: 10.1073/pnas.0800563105
Volker, M., Moné, M. J., Karmakar, P., van Hoffen, A., Schul, W., Vermeulen, W., et al. (2001). Sequential assembly of the nucleotide excision repair factors in vivo. Mol. Cell 8, 213–224. doi: 10.1016/s1097-2765(01)00281-7
Wang, A. T., Sengerová, B., Cattell, E., Inagawa, T., Hartley, J. M., Kiakos, K., et al. (2011). Human SNM1A and XPF-ERCC1 collaborate to initiate DNA interstrand cross-link repair. Genes Dev. 25, 1859–1870. doi: 10.1101/gad.15699211
Wang, R., Wang, S., Dhar, A., Peralta, C., and Pavletich, N. P. (2020). DNA clamp function of the monoubiquitinated Fanconi anaemia ID complex. Nature 580, 278–282. doi: 10.1038/s41586-020-2110-6
Williams, H. L., Gottesman, M. E., and Gautier, J. (2012). Replication-independent repair of DNA interstrand crosslinks. Mol. Cell 47, 140–147. doi: 10.1016/j.molcel.2012.05.001
Williams, S. A., Longerich, S., Sung, P., Vaziri, C., and Kupfer, G. M. (2011). The E3 ubiquitin ligase RAD18 regulates ubiquitylation and chromatin loading of FANCD2 and FANCI. Blood 117, 5078–5087. doi: 10.1182/blood-2010-10-311761
Wood, R. D. (2010). Mammalian nucleotide excision repair proteins and interstrand crosslink repair. Environ. Mol. Mutagen. 51, 520–526.
Yamanaka, K., Minko, I. G., Takata, K., Kolbanovskiy, A., Kozekov, I. D., Wood, R. D., et al. (2010). Novel enzymatic function of DNA Polymerase ν in translesion DNA synthesis past major groove DNA-Peptide and DNA-DNA cross-links. Chem. Res. Toxicol. 23, 689–695. doi: 10.1021/tx900449u
Yang, K., Moldovan, G. L., and D’Andrea, A. D. (2010). RAD18-dependent recruitment of SNM1A to DNA repair complexes by a ubiquitin-binding zinc finger. J. Biol. Chem. 285, 19085–19091. doi: 10.1074/jbc.m109.100032
Yoon, J.-H., Prakash, S., and Prakash, L. (2012). Requirement of Rad18 protein for replication through DNA lesions in mouse and human cells. Proc. Natl. Acad. Sci. 109, 7799–7804. doi: 10.1073/pnas.1204105109
Zhang, Y., Yuan, F., Wu, X., Wang, M., Rechkoblit, O., Taylor, J.-S., et al. (2000). Error-free and error-prone lesion bypass by human DNA polymerase κ in vitro. Nucleic Acids Res. 28, 4138–4146. doi: 10.1093/nar/28.21.4138
Keywords: interstrand crosslink, DNA repair, translesion synthesis polymerases, PCNA ubiquitylation, structure-specific nuclease
Citation: Li Q, Dudás K, Tick G and Haracska L (2021) Coordinated Cut and Bypass: Replication of Interstrand Crosslink-Containing DNA. Front. Cell Dev. Biol. 9:699966. doi: 10.3389/fcell.2021.699966
Received: 24 April 2021; Accepted: 07 June 2021;
Published: 28 June 2021.
Edited by:
Lin Deng, Shenzhen Bay Laboratory, ChinaReviewed by:
Jun Huang, Zhejiang University, ChinaIndrajit Chaudhury, University of Minnesota Morris, United States
Jing Zhang, Tongji University, China
Copyright © 2021 Li, Dudás, Tick and Haracska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lajos Haracska, aGFyYWNza2EubGFqb3NAYnJjLmh1
†These authors have contributed equally to this work