 Baoyu Chen1†
Baoyu Chen1† Yifei Feng
Yifei Feng
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 22 September 2021
Sec. Molecular and Cellular Pathology
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.697614
Intestinal fibrosis is one of the common pathophysiological processes in inflammatory bowel diseases (IBDs). Previously it has been demonstrated that epithelial-mesenchymal transition (EMT) can contribute to the development of intestinal fibrosis. Here we report that conditional ablation of SIRT1, a class III lysine deacetylase, in intestinal epithelial cells exacerbated 2, 4, 6-trinitro-benzene sulfonic acid (TNBS) induced intestinal fibrosis in mice. SIRT1 activity, but not SIRT1 expression, was down-regulated during EMT likely due to up-regulation of its inhibitor deleted in breast cancer 1 (DBC1). TGF-β augmented the recruitment of KDM4A, a histone H3K9 demethylase, to the DBC1 promoter in cultured intestinal epithelial cells (IEC-6) leading to DBC1 trans-activation. KDM4A depletion or inhibition abrogated DBC1 induction by TGF-β and normalized SIRT1 activity. In addition, KDM4A deficiency attenuated TGF-β induced EMT in IEC-6 cells. In conclusion, our data identify a KDM4-DBC1-SIRT1 pathway that regulates EMT to contribute to intestinal fibrosis.
Inflammatory bowl diseases (IBDs) refer to a group of relapsing and heterogeneous intestinal disorders exemplified by Crohn’s disease and ulcerative colitis (Ananthakrishnan, 2015). Documented as early as the nineteenth century, IBDs are more pervasive in the industrialized nations although recent epidemiology studies have indicated that there is a growing trend in the prevalence and incidence of IBDs in the developing countries. For instance, the population affected by IBDs in China alone is projected to reach 1.5 million by 2025 creating considerable socioeconomic burdens (Kaplan, 2015). Patients diagnosed with IBDs typically present chronic abdominal pain, fever, abscess, and fistula; these patients, in most severe cases and in the long term, can develop intestinal cancer (D’Haens, 2010). IBDs are equally influenced by the genetics and the environment. Thus far, over 160 loci have been identified to confer altered susceptibility to the development of IBDs in humans (Jostins et al., 2012). On the other hand, smoking, diets, life style, pollution, and even personal hygiene have been reported as potential environmental risk factors for IBDs (Leong, 2010). Although IBDs are clinically considered as a gastroenterological disorder, extraintestinal systems, the immune system in particular, play pivotal roles (Digby-Bell et al., 2020).
Intestinal fibrosis is one of the most common complications observed in IBD patients that are characterized by excessive scarring in the region of inflammation (Latella et al., 2015). Limited or mild intestinal fibrosis in IBD patients may evade detection with no overt clinical manifestation. Severe cases of intestinal fibrosis, however, often require surgical resections (Rieder and Fiocchi, 2009). Myofibroblasts are believed to be the major mediator of intestinal fibrosis although their origins are often debated and remain controversial (Valatas et al., 2017). Resident fibroblasts, stellate cells, bone marrow stem cells, fibrocytes, pericytes, epithelial cells, and endothelial cells have been reported to contribute to the pool of mature myofibroblasts in the course of intestinal fibrosis (Lawrance et al., 2017). Kalluri and colleagues have shown, aided by genetic lineage tracing technique, that Villin+ intestinal epithelial cells could trans-differentiate into myofibroblasts, in a process known as epithelial-mesenchymal transition (EMT), thereby contributing to intestinal fibrosis in mice (Flier et al., 2010). EMT and the closely related EndMT (endothelial-mesenchymal transition) are conserved pathobiological processes playing essential roles in organogenesis; EMT taking place post-developmentally is usually associated with such devastating diseases as malignant cancer and tissue fibrosis (Kalluri and Weinberg, 2009). During EMT, the cells shed the expression of epithelial signature genes (e.g., E-Cadherin encoded by CDH1 and ZO-1 encoded by TJP1) in favor of mesenchymal marker genes (e.g., α-SMA encoded by ACTA2 and collagen type I encoded by COL1A1).
Silent information regulator 1 (SIRT1) is the founding member of the mammalian sirtuin family of lysine deacetylases. SIRT1 regulates a wide range of pathobiological processes by differentially modulating the acetylation status of target proteins (Zschoernig and Mahlknecht, 2008). Mounting evidence suggests that SIRT1 generally plays a protective role in tissue fibrosis in multiple organs (Huang et al., 2014; Zerr et al., 2016; Bugyei-Twum et al., 2018; Li M. et al., 2018). Here we report that epithelial conditional ablation of SIRT1 exacerbates intestinal fibrosis in a TNBS-induced mouse model of colitis. SIRT1 activity, but not SIRT1 expression, is down-regulated during EMT owing to KDM4A mediated induction of DBC1 transcription.
All the animal protocols were reviewed and approved by the intramural Ethics Committee on Humane Treatment of Experimental Animals. The Sirt1flox/flox strain (Li et al., 2017) was crossbred with the Villin-Cre strain (Sun et al., 2017) to generate intestinal epithelium-specific SIRT1 knockout (CKO) mice. Colitis was induced in the mice by intrarectal injection of TNBS (2.5% wt/vol) as previously described (Wirtz et al., 2017). The mice were euthanized 42 days after the onset of injection.
The rat intestinal epithelial cell IEC-6 (ATCC) was maintained in DMEM supplemented with 10% FBS. TGF-β was purchased from R&D. EX-527 and CP2 were purchased from Selleck. Small interfering RNAs were purchased from Dharmacon. Transient transfections were performed with Lipofectamine 2000 as previously described (Chen et al., 2021; Kong et al., 2021b; Liu et al., 2021a,b; Zhang et al., 2021).
Whole cell lysates were obtained by re-suspending cell pellets in RIPA buffer (50 mM Tris pH7.4, 150 mM NaCl, 1% Triton X-100) with freshly added protease inhibitor (Roche) as previously described (Hong et al., 2020; Dong et al., 2021; Kong et al., 2021a; Li et al., 2021). Nuclear proteins were extracted using the NE-PER Kit (Pierce) following manufacturer’s recommendation. Specific antibodies or pre-immune IgGs were added to and incubated with cell lysates overnight before being absorbed by Protein A/G-plus Agarose beads (Santa Cruz). Precipitated immune complex was released by boiling with 1X SDS electrophoresis sample buffer. Western blot analyses were performed with anti-SIRT1 (Santa Cruz, sc-74465), anti-α-SMA (Sigma, A2547), anti-E-Cadherin (Abcam, ab1416), anti-β-actin (Sigma, A2228), anti-DBC1 (Abcam ab215852), and anti-KDM4A (Abcam, ab191433) antibodies. For densitometrical quantification, densities of target proteins were normalized to those of β-actin. Data are expressed as relative protein levels compared to the control group which is arbitrarily set as 1.
RNA was extracted with the RNeasy RNA isolation kit (Qiagen). Reverse transcriptase reactions were performed using a SuperScript First-strand Synthesis System (Invitrogen) as previously described (Sun L. et al., 2020; Wu T. et al., 2020; Wu X. et al., 2020; Yang et al., 2020a,b). Real-time PCR reactions were performed on an ABI Prism 7500 system with the following primers: mouse Cdh1, 5′-AAGTGACCGATGATGATGCC-3′ and 5′-CTTCTCTGTCCATCTCAGCG-3′; mouse Tjp1, 5′-GATCCCTGTAAGTCACCCAGA-3′ and 5′-CTCCCTGCTT GCACTCCTATC-3′; mouse Col1a1, 5′-GACGCCATCAAGG TCTACTG-3′ and 5′-ACGGGAATCCATCGGTCA-3′; mouse Acta2, 5′-CTGAGCGTGGCTATTCCTTC-3′ and 5′-CTTCTG CATCCTGTCAGCAA-3′; rat Cdh1, 5′-GGAGAAGAAGAC CAGGACTTTG-3′ and 5′-GATGAAGTTCCCGATTTCATCAG-3′; rat Acta2, 5′-AGGATGCAGAAGGAGATCACAG-3′; and 5′-CTGGAAGGTAGATAGAGAAGCC-3′; rat Tjp1, 5′-GCAGTGTGAACATGGATTGAA-3′ and 5′-AGCCAATGCCT GACAGTTCT-3′; rat Col1a1, 5′-ATCTCCTGGTGCTGATG GAC-3′ and 5′-ACCTTGTTTGCCAGGTTCAC-3′; rat Dbc1, 5′-TCTCCAAGTCTCGCCTGTG-3′ and 5′-CTCTGTTGCC TCCAACCAGT-3′; mouse Dbc1. Ct values of target genes were normalized to the Ct values of housekeeping control gene (18s, 5′-CGCGGTTCTATTTTGTTGGT-3′ and 5′-TCGTCTTCGAAACTCCGACT-3′ for both human and mouse genes) using the ΔΔCt method and expressed as relative mRNA expression levels compared to the control group which is arbitrarily set as 1.
Chromatin Immunoprecipitation (ChIP) assays were performed essentially as described before (Coarfa et al., 2020; Hu et al., 2020; Jehanno et al., 2020; Maity et al., 2020; Mallik et al., 2020; Moon et al., 2020; Shen et al., 2020; Wang J. N. et al., 2020; Wang S. et al., 2020; Zhang et al., 2020; Zhao et al., 2020; Marti et al., 2021; Peng et al., 2021; Rashid et al., 2021). In brief, chromatin in control and treated cells were cross-linked with 1% formaldehyde. Cells were incubated in lysis buffer (150 mM NaCl, 25 mM Tris pH 7.5, 1% Triton X-100, 0.1% SDS, 0.5% deoxycholate) supplemented with protease inhibitor tablet and PMSF. DNA was fragmented into ∼200 bp pieces using a Branson 250 sonicator. Aliquots of lysates containing 200 μg of protein were used for each immunoprecipitation reaction with anti-acetyl H3 (Millipore, 06-599), anti-trimethyl H3K4 (Millipore, 07-473), anti-trimethyl H3K9 (Diagenode, C15410193), anti-trimethyl H3K27 (Millipore, 04-449), anti-trimethyl H4K20 (Millipore, 07-463), anti-KDM4A (Abcam, ab191433), anti-KDM4B (Bethyl Laboratories, A301-477A), anti-KDM4C (Bethyl Laboratories, A300-885A), or anti-KDM4D (Proteintech, 22591-1-AP) antibodies.
Whole cell lysates or tissue homogenates were prepared using the NETN buffer (20 mM Tris pH8.0, 100 mM NaCl, 1 mM EDTA, 0.5% NP-40) with freshly added protease inhibitor tablet and SIRT1 activities were measured with a SIRT1 assay kit (Sigma) according to vender’s recommendations.
Histologic analyses were performed essentially as described before (Chen et al., 2020a,b,c; Dong et al., 2020; Fan et al., 2020; Li et al., 2020a,b,c; Lv et al., 2020). Briefly, paraffin-embedded sections were stained with picrosirius red (Sigma-Aldrich) or Masson’s trichrome (Sigma-Aldrich) according to standard procedures. Pictures were taken using an Olympus IX-70 microscope (Olympus, Tokyo, Japan).
One-way ANOVA with post-hoc Scheffe analyses were performed by SPSS software (IBM SPSS v18.0, Chicago, IL, United States). Unless otherwise specified, values of p < 0.05 were considered statistically significant.
To specifically delete SIRT1 in the intestinal epithelial cells, the Sirt1f/f mice were crossed with the Villin-Cre mice. We first evaluated the development of intestinal fibrosis in the WT mice and the CKO mice. Quantitative PCR showed that TNBS injection significantly induced the expression of smooth muscle cell actin (Acta2) and collagen type I (Col1a1), two prototypical markers of myofibroblasts, in the small intestines (Figure 1A) and colons (Figure 1B). Compared to the WT mice, CKO mice displayed significantly augmented expression of pro-fibrogenic genes indicative of accelerated intestinal fibrosis. Picrosirius red staining confirmed that deposition of collagenous tissues in the small intestines and the colons was up-regulated in the mice following TNBS injection whereas the CKO mice exhibited more prominent intestinal fibrosis than the WT mice (Figure 1C).
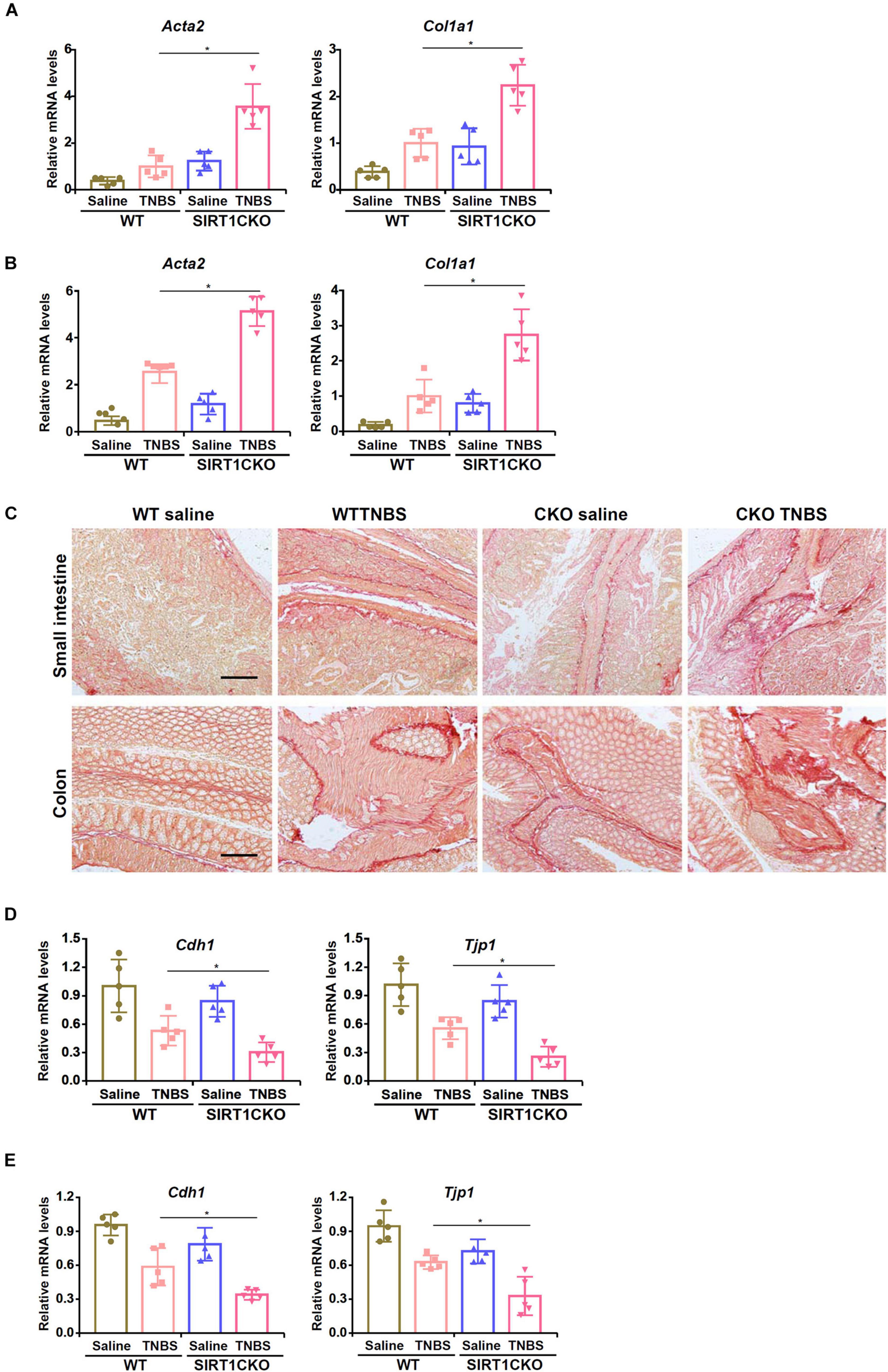
Figure 1. Intestinal epithelial deletion of SIRT1 exacerbates TNBS induced intestinal fibrosis. Intestine-conditional SIRT1 knockout (CKO) mice and wild type littermates were induced to develop colitis and intestinal fibrosis by TNBS injection. (A) Expression levels of pro-fibrogenic molecules in the small intestines were examined by qPCR. (B) Expression levels of pro-fibrogenic molecules in the colons were examined by qPCR. (C) Paraffin sections were stained with picrosirius red. Scale bar, 100 mm. (D) Expression levels of epithelial marker genes in the small intestines were examined by qPCR. (E) Expression levels of epithelial marker genes in the colons were examined by qPCR. N = 5 mice for each group.
Previously Kalluri and colleagues have demonstrated that epithelial-mesenchymal transition (EMT) plays a role in intestinal fibrosis in mice (Flier et al., 2010). Now that SIRT1 deficiency in epithelial cells resulted in up-regulation of mesenchymal marker genes (Acta2 and Col1a1), we hypothesized that SIRT1 deficiency might promote EMT leading to accelerated loss of epithelial markers. Indeed, TNBS injection down-regulated the expression of E-Cadherin (Cdh1) and ZO-1 (Tjp1) in the intestines and the loss of Cdh1 and Tjp1 expression was more severe in the CKO mice than in the WT mice (Figures 1D,E).
Transforming growth factor (TGF-β) is considered one of the most potent inducers of EMT. TGF-β expression has been observed to elevate in animal models of IBD (Wengrower et al., 2004; Fichtner-Feigl et al., 2007) and in IBD patients (Li et al., 2013). Consistent with prior observations, TGF-β (Tgfb1) expression was up-regulated in the TNBS treated mice compared to the saline treated mice although the induction of TGF-β expression by TNBS was comparable between the WT and the CKO mice (Supplementary Figure 1). In a cell model of EMT wherein the rat intestinal epithelial IEC-6 cells were treated with TGF-β, over-expression of a wild type (WT) SIRT1, but not an enzymatically inactive (HY) SIRT1, partially reversed the effect of TGF-b (Supplementary Figure 2). Similarly, pre-treatment with a specific SIRT1 agonist (SRT1720) dose-dependently attenuated TGF-β induced EMT in IEC-6 cells (Supplementary Figure 3). Collectively, these data suggest that intestinal epithelial deletion of SIRT1 exacerbates TNBS induced intestinal fibrosis likely by promoting the EMT process.
We then asked whether SIRT1 expression and/or activity would be altered during intestinal fibrosis. As shown in Figure 2A, mice exposed to TNBS displayed significant down-regulation of SIRT1 activity in the intestines compared to the control mice. Of note, neither SIRT1 mRNA expression (Figure 2B) nor protein expression (Figure 2C) was affected by TNBS exposure in the mouse intestines suggesting that dampening of SIRT1 activity by TNBS may be attributed to a post-transcriptional mechanism. It has been previously reported that deleted in breast cancer 1 (DBC1) allosterically inhibits SIRT1 activity without altering its expression (Kim et al., 2008). Indeed, DBC1 expression was significantly up-regulated in the intestines in the TNBS-treated mice compared to the control mice (Figures 2B,C). To cement the role of TGF-β in DBC1 induction, we pre-treated the cells with a selective TβRI/II inhibitor (LY2109761). Indeed, blocking TGF-β engagement to its receptor by LY2109761 largely abrogated DBC1 up-regulation and prevented EMT in IEC-6 cells (Supplementary Figure 4A). Concordantly, suppression of SIRT1 activity was alleviated (Supplementary Figure 4B).
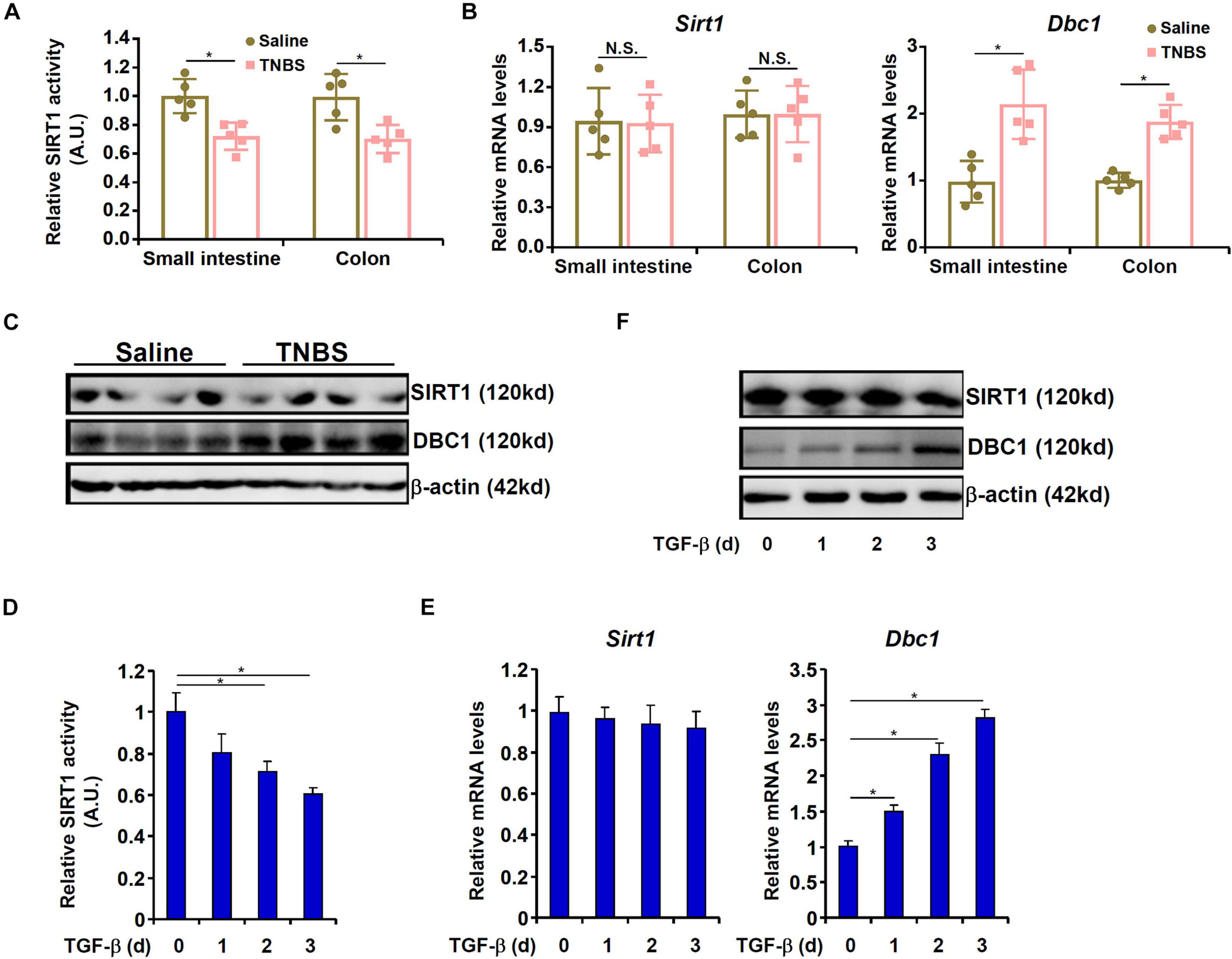
Figure 2. SIRT1 activity is down-regulated by TNBS in vivo and by TGF-β in epithelial cells in vitro. (A–C) Colitis was induced in C57B/6 mice by TNBS as described in section “Materials and Methods.” SIRT1 activity was determined by a colorimetric kit. Expression levels of SIRT1 and DBC1 were examined by qPCR and Western. N = 5 mice for each group. (D–F) IEC-6 cells were treated with TGF-β (2 ng/ml) and harvested at indicated time points. SIRT1 activity was determined by a colorimetric kit. Expression levels of SIRT1 and DBC1 were examined by qPCR and Western. All experiments were performed in triplicate wells and repeated three times. One representative experiment is shown.
TGF-β treatment suppressed SIRT1 activity in a time course dependent manner without influencing SIRT1 expression in IEC-6 cells (Figures 2D–F). On the other hand, TGF-β stimulated DBC1 expression mirroring the suppression of SIRT1 activity in IEC-6 cells (Figures 2D–F). Furthermore, stronger association between DBC1 and SIRT1 was detected by co-immunoprecipitation in the TNBS-treated intestines (Supplementary Figure 5A) and in TGF-treated IEC-6 cells (Supplementary Figure 5B). Together, these data suggest that loss of SIRT1 activity, but not SIRT1 expression, likely due to increased DBC1 expression and enhanced DBC1-SIRT1 interaction may contribute to EMT in intestinal epithelial cells.
In order to ascertain that DBC1 might mediate the suppressive effect of TGF-β on SIRT1 activity, siRNAs targeting DBC1 were transfected into IEC-6 cells. DBC1 depletion restored SIRT1 activity despite the presence of TGF-β (Figures 3A–C). Of note, DBC1 knockdown did not significantly alter SIRT1 expression consistent with its role as an allosteric inhibitor of SIRT1.
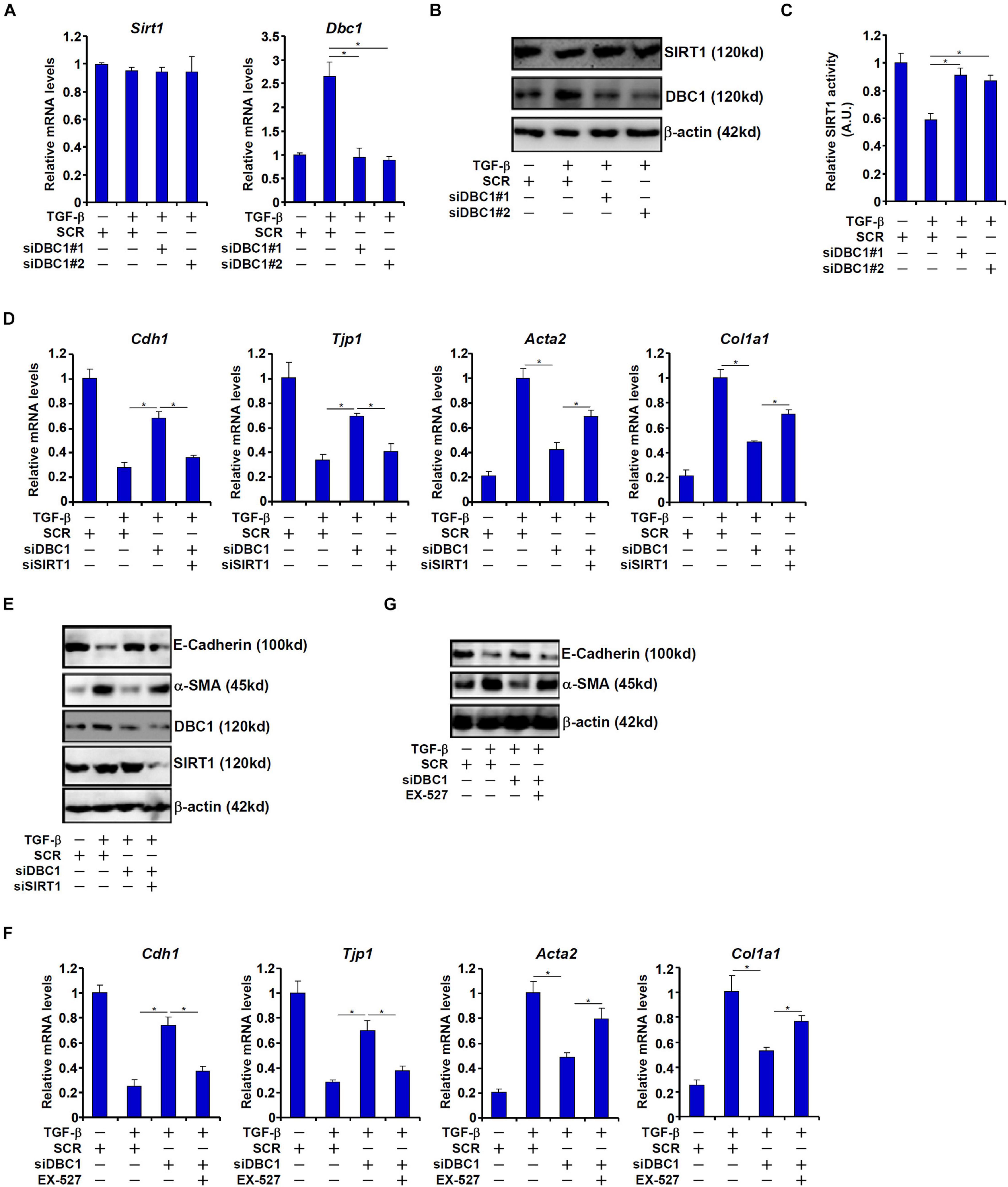
Figure 3. DBC1 contributes to TGF-β induced EMT by repressing SIRT1 activity. (A–C) IEC-6 cells were transfected with siRNAs targeting DBC1 or scrambled siRNAs followed by treatment with TGF-β (2 ng/ml). Expression levels of SIRT1 and DBC1 were examined by qPCR and Western. SIRT1 activity was determined by a colorimetric kit. (D,E) IEC-6 cells were transfected with indicated siRNAs followed by treatment with TGF-β (2 ng/ml) for 48 h. Gene expression levels were examined by qPCR and Western. (F,G) IEC-6 cells were transfected with indicated siRNAs followed by treatment with TGF-β (2 ng/ml) in the presence or absence of EX527 (10 mM) for 48 h. Gene expression levels were examined by qPCR and Western. All experiments were performed in triplicate wells and repeated three times. One representative experiment is shown.
Next, we asked whether DBC1 could contribute to TGF-β induced EMT. As shown in Figures 3D,E, DBC1 knockdown attenuated down-regulation of Cdh1 expression and Tjp1 expression and up-regulation of Acta2 expression and Col1a1 expression in TGF-β treated IEC-6 cells. Simultaneous depletion of SIRT1, however, blocked the anti-EMT effect of DBC1 knockdown, suggesting that DBC1 may rely on its function as a SIRT1 inhibitor to regulate EMT. Similarly, SIRT1 inhibition by EX-527 largely abrogated the normalization of epithelial/mesenchymal signature genes by DBC1 knockdown and enabled TGF-β to stimulate EMT in IEC-6 cells (Figures 3F,G).
We then performed ChIP assays to explore the epigenetic mechanism whereby TGF-β activates DBC1 expression. High levels of acetylated histone H3 (Figure 4A) and trimethylated H3K4 (Figure 4B), two typical markers for actively transcribed chromatin, were detected on the DBC1 promoter under basal conditions and were not noticeably up-regulated by TGF-β treatment. On the contrary, levels of trimethylated H3K9, a marker for silenced chromatins, were down-regulated by TGF-b treatment (Figure 4C). By comparison, trimethylated H3K27 (Figure 4D) and trimethylated H4K20 (Figure 4E), both reported to be associated with transcriptional repression, were not significantly altered by TGF-β treatment.
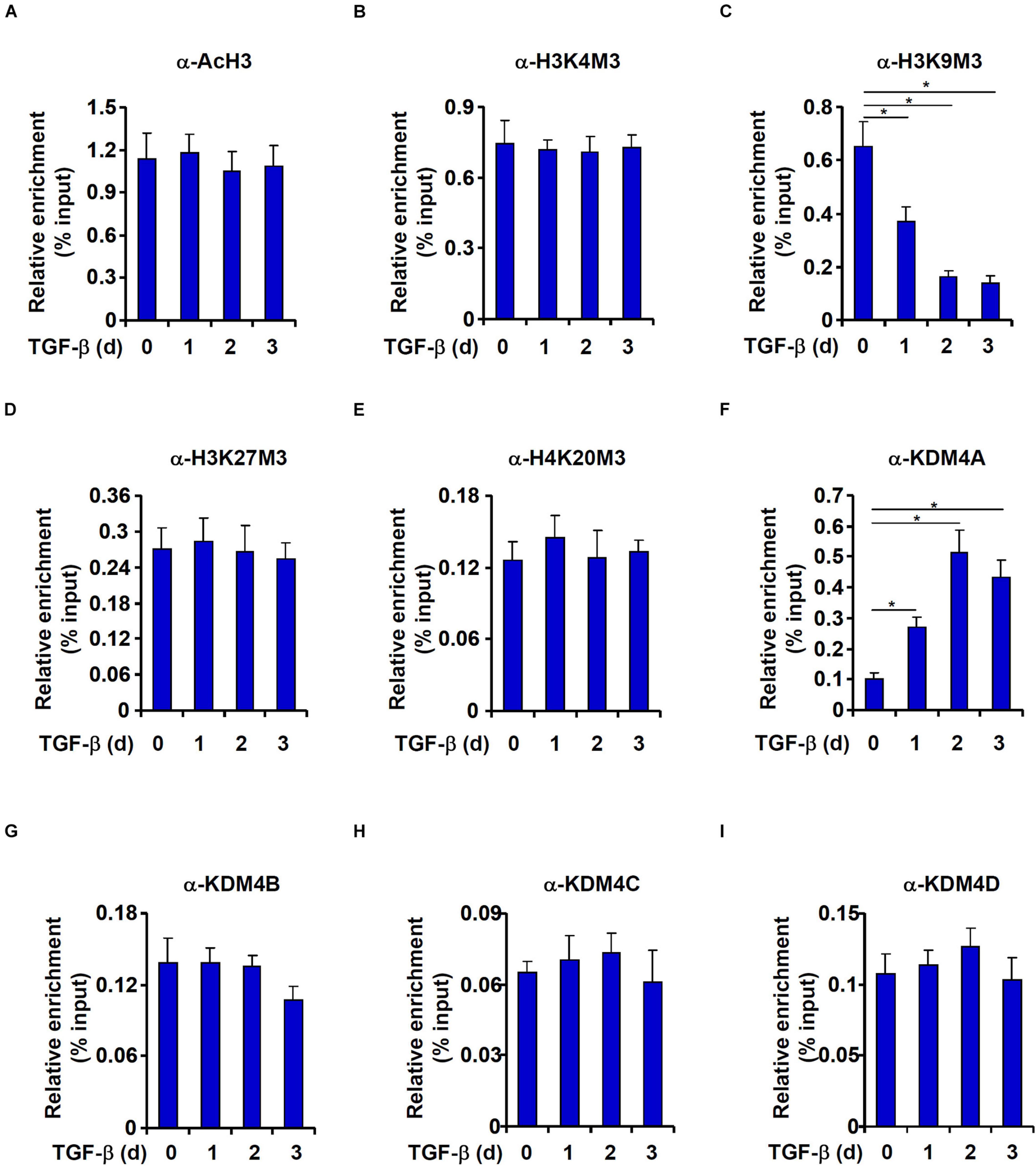
Figure 4. TGF-β promotes KDM4A recruitment to the DBC1 promoter. (A–I) IEC-6 cells were treated with TGF-β(2 ng/ml) and collected at indicated time points. ChIP assays were performed anti-acetyl H3, anti-trimethyl H3K4, anti-trimethyl H3K9, anti-trimethyl H3K27, anti-trimethyl H4K20, anti-KDM4A, anti-KDM4B, anti-KDM4C, or anti-KDM4D. All experiments were performed in triplicate wells and repeated three times. One representative experiment is shown.
Because removal of trimethyl H3K9 from the DBC1 promoter was observed in TGF-β induced EMT process, we hypothesized that TGF-β treatment might enhance the occupancies of specific histone H3K9 demethylases. Indeed, KDM4A association with the DBC1 promoter was enhanced by TGF-β treatment with a kinetics closely mirroring that of H3K9M3 removal (Figure 4F). In contrast, occupancies of KDM4B (Figure 4G), KDM4C (Figure 4H), or KDM4D (Figure 4I) on the DBC1 promoter were not impacted by TGF-β treatment.
To confirm that KDM4A was responsible for DBC1 induction by TGF-β and its relevance to EMT, the following experiments were performed. KDM4A knockdown by two separate pairs of siRNAs dampened the DBC1 induction in TGF-β treated IEC-6 cells at mRNA (Figure 5A) and protein (Figure 5B) levels. Consistent with the changes in DBC1 expression, a concomitant change in H3K9M3 levels was detected on the DBC1 promoter (Figure 5C). As a result, inhibition of SIRT1 activity was partially rescued (Figure 5D). In a second set of experiments, CP2 was used to inhibit KDM4A activity (Kawamura et al., 2017). KDM4A inhibition similarly antagonized DBC1 induction (Figures 5E,F) and removal of H3K9M3 (Figure 5G) by TGF-β. Consequently, CP2 treatment led to a partial normalization of SIRT1 activity (Figure 5H).
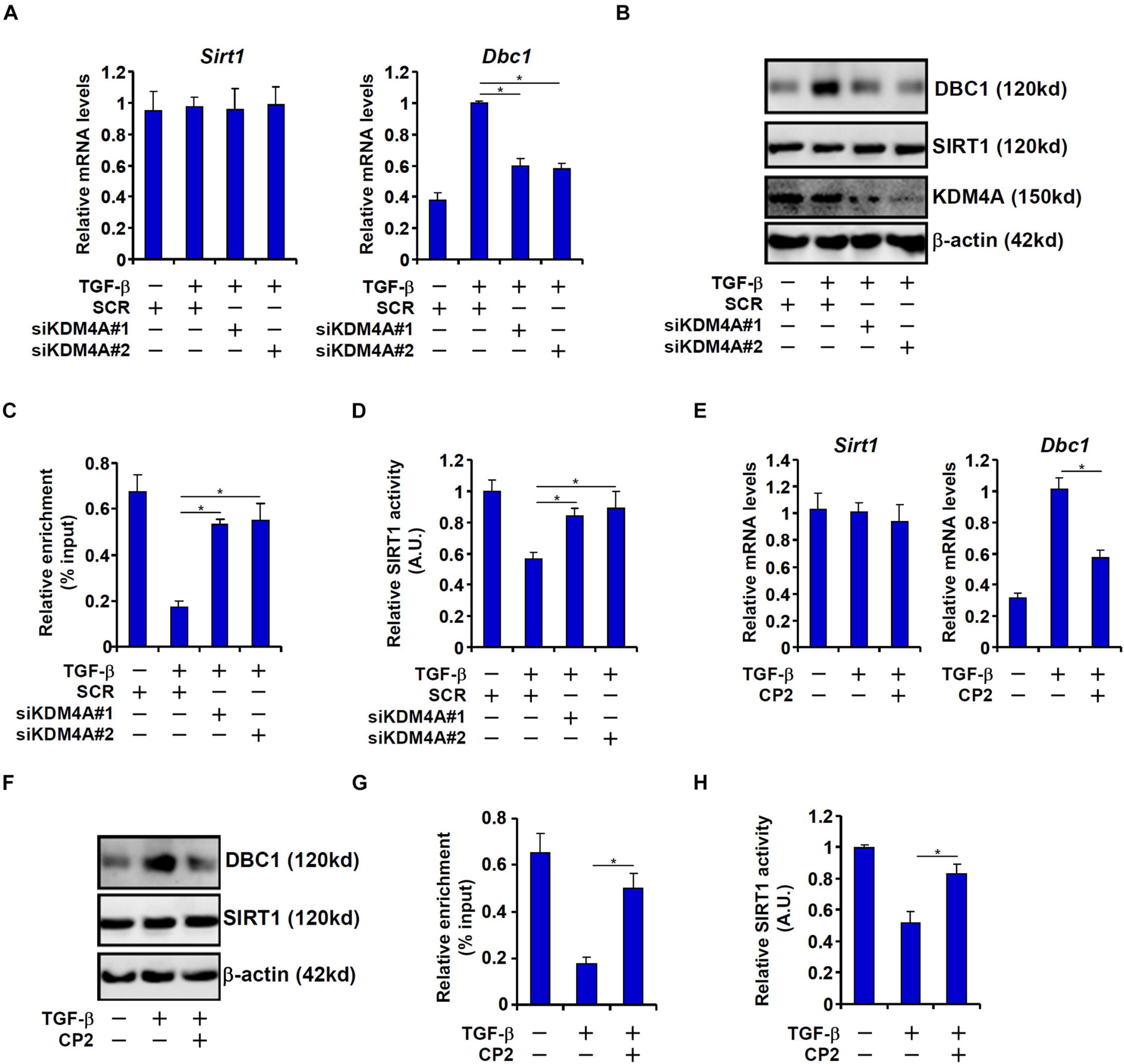
Figure 5. KDM4A mediates TGF-β induced DBC1 transcription. (A–D) IEC-6 cells were transfected with siRNAs targeting KDM4A or scrambled siRNAs followed by treatment with TGF-β (2 ng/ml). DBC1 expression levels were examined by qPCR and Western. ChIP assay was performed with anti-trimethyl H3K9. SIRT1 activity was determined by a colorimetric kit. (E–H) IEC-6 cells were treated with TGF-β (2 ng/ml) in the presence or absence of CP2 (1 μM). DBC1 expression levels were examined by qPCR and Western. ChIP assay was performed with anti-trimethyl H3K9. SIRT1 activity was determined by a colorimetric kit. All experiments were performed in triplicate wells and repeated three times. One representative experiment is shown.
On the other hand, KDM4A knockdown, similar to DBC1 knockdown, resulted in up-regulation of Cdh1 expression and Tjp1 expression and down-regulation of Acta2 expression and Col1a1 expression, a trend reversed by simultaneous SIRT1 depletion (Figures 6A,B). SIRT1 inhibition by EX-527 similarly circumvented KDM4A deficiency to allow TGF-β to down-regulate Cdh1 expression and Tjp1 expression and up-regulate Acta2 expression and Col1a1 expression (Figures 6C,D).
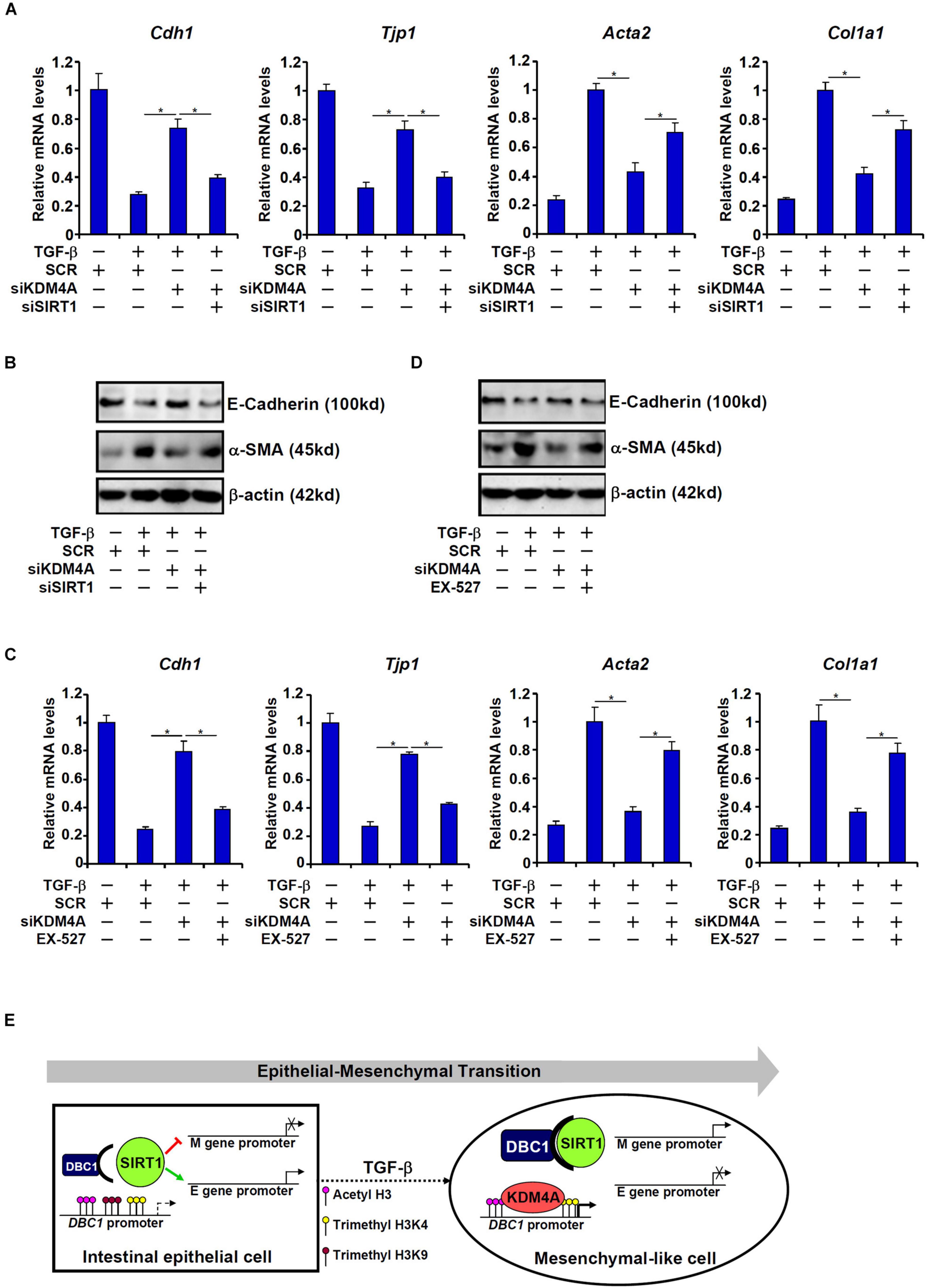
Figure 6. KDM4A silencing or inhibition attenuates TGF-β induced EMT in a SIRT1-dependent manner. (A,B) IEC-6 cells were transfected with indicated siRNAs followed by treatment with TGF-β (2 ng/ml) for 48 h. Gene expression levels were examined by qPCR and Western. (C,D) IEC-6 cells were transfected with indicated siRNAs followed by treatment with TGF-β (2 ng/ml) in the presence or absence of EX527 (10 μM) for 48 h. Gene expression levels were examined by qPCR and Western. All experiments were performed in triplicate wells and repeated three times. One representative experiment is shown. (E) A schematic model.
Intestinal fibrosis, one of the most common complications of IBDs, remains a daunting challenge due to the lack of effective therapeutic solutions. Previous investigations have suggested that the developmentally conserved EMT process plays a role in the pathogenesis of intestinal fibrosis (Lovisa et al., 2019). Here we provide evidence that supports the involvement of a KDM4A-DBC1-SIRT1 axis in the regulation of EMT potentially contributing to intestinal fibrosis.
We show here that down-regulation of SIRT1 activity, but not SIRT1 expression, is a determining factor in TNBS-induced intestinal fibrosis. Further, our data suggest that DBC1 is responsible for the suppression of SIRT1 activity in intestinal epithelial cells in the process of EMT, which alludes to the possibility that DBC1 might drive EMT and intestinal fibrosis. It is noteworthy that this hypothesis is buttressed by several indirect pieces of evidence. First, Gao et al. (2015) have reported that mice with systemic DBC1 deletion are resistant to colitis although the underlying mechanism is proposed to be mediated by inhibition of Foxp3, a key anti-inflammatory transcription factor, by DBC1 in regulatory T cells; it remains undetermined whether the mice with epithelial-specific deletion of DBC1 would phenocopy the systemic DBC1 KO mice in the pathogenesis of intestinal fibrosis. Second, DBC1 activates the expression of MMP7, a mesenchymal cell marker, to pivot breast cancer cells to a more malignant phenotype by interacting with the zinc finger transcription factor ZFN326 (Yu et al., 2018). Third, a recent investigation by Kim et al. (2018) has revealed a role for DBC1 in the regulation of colorectal cancer metastasis; DBC1 directly interacts with β-catenin and stabilizes the β-catenin/LEF1 complex to activate the transcription of metastasis-associated in colon cancer 1 (MACC1), an EMT-related molecule. It would be of great interest to, along this line of investigations, further delineate the role of DBC1 in intestinal fibrosis.
Our data suggest that the lysine demethylase KDM4A can contribute to TGF-β induced EMT by stimulating DBC1 transcription to inhibit SIRT1 activity. KDM4A knockdown or pharmaceutical inhibition normalizes DBC1 expression and SIRT1 activity leading to stagnation of EMT. This is consistent with a report by Ishiguro et al. (2017) showing that pharmaceutical inhibition of KDM4A alleviates colitis in mice likely through the IL-6 signaling. However, these observations do not entirely foreclose the possibility that KDM4A may contribute to intestinal fibrosis via alternative mechanisms. Pezone et al. (2020) have recently reported that KDM4A can be recruited to the SNAI1 promoter and activate SNAI1 transcription in response to TGF-β stimulation in mammalian epithelial cells. The E-box binding transcriptional repressor SNAIL, which is encoded by SNAI1, subsequently binds to the promoters of epithelial signature genes to repress transcription thus initiating the EMT process. Alternatively, Sun S. et al. (2020) have shown that KDM4A can directly bind to the promoter and activate the transcription of Myc, a master regulator of EMT, in lung cancer cells. It should be pointed out that our proposed model (Figure 6E) and any alternative scenarios alluded to by other investigations are not mutually exclusive. For instance, it has been observed that SIRT1 can directly interact with Myc and deacetylate Myc leading to Myc degradation by the ubiquitin-proteasome system (Yuan et al., 2009). Alternatively, SIRT1 has been shown to deacetylate SNAIL and modulate SNAIL activity in lung cancer cells (Yang et al., 2021). Further analysis is warranted to delineate the functional interplay between KDM4A and SIRT1.
There are still major limitations associated with the present study. First, although we focused on SIRT1 as a regulator of TGF-induced EMT and intestinal fibrosis, it is highly likely that a group of factors, rather than a single factor alone (e.g., SIRT1), collectively mediate the TGF-β effect. For instance, the E-box recognizing family of transcriptional repressors, including Snail, Slug, Twist, and Zeb, are well characterized mediators of EMT in various systems (Lamouille et al., 2014). Recently, Scharl et al. (2013) have observed that Snail1 appears to play a critical role in TGF induced EMT in monolayer intestinal epithelial cells and IEC spheroids. However, the potential contribution by these proteins and the model as supported by our data are not necessarily mutually exclusive because SIRT1 has been reported to influence the activities of these transcription factors via protein-protein interactions or altering expression levels (Choupani et al., 2018). Additional investigation is warrant to sort out the entanglement of these EMT factors. Second, our proposal that a KDM4A-DBC1 axis mediates TGF-β induced SIRT1 repression remains to be validated in vivo. This is partly due to the lack of appropriate model animals (for instance, mice harboring epithelial-specific KDM4A/DBC1/TβRI/II deletion). Further compounding the issue is the fact that despite the development of a plethora of TGF-β targeting compounds (Teixeira et al., 2020), none have been demonstrated to confer protective effects on intestinal fibrosis. Therefore, a large disconnect exists between the in vitro observations and the in vivo pathology. Third, the precise mechanism whereby SIRT1 regulates EMT and intestinal fibrosis awaits further investigation. It is generally agreed that SIRT1 relies on its activity as a lysine deacetylase to participate in pathophysiological processes, a notion supported by our observation (Supplementary Figure 3). However, SIRT1 can use both histones and non-histone factors as substrates to orchestrate downstream events. For instance, pro-EMT/fibrogenic transcription factors, including NF-κB (Li X. et al., 2018), β-catenin (Simic et al., 2013), c-Myc (Yuan et al., 2009), and SMAD (Chen et al., 2014), can all be modulated by SIRT1-mediated lysine deacetylation. In addition, SIRT1 is able to bind to and directly deacetylate histones H3/H4 surrounding the pro-fibrogenic gene promoters (Guo et al., 2017). It would be helpful to exploit transcriptomic and proteomic tools to comprehensively profile the influence of SIRT1 on the acetylation status of both histones and non-histone factors in the context of EMT and intestinal fibrosis. Finally, although we propose here that DBC1 might contribute to TGF induced EMT and intestinal fibrosis via SIRT1 suppression, the role of other DBC1-interacting factors cannot be fully excluded. For instance, Chini et al. (2010) have identified HDAC3, a class I lysine deacetylase, as a binding partner for DBC1; binding of DBC1 to HDAC3 inhibits HDAC3 activity. A string of recent reports have implicated HDAC3 as an important regulator of EMT and/or tissue fibrosis (Ning et al., 2021). Alternatively, interaction with DBC1 stabilizes the nuclear receptor Rev-erbα (Chini et al., 2013). Recent studies have shown that Rev-erbaα is an emerging target for tissue fibrosis (Li et al., 2014; Welch et al., 2017). It is plausible that a panel of DBC1-interacting proteins, rather than SIRT1 alone, collectively determine its function as a modulator of EMT and/or intestinal fibrosis, which certainly deserves to be thoroughly investigated in future studies.
In summary, evidence has been mounting that targeting SIRT1 may be considered as a viable approach to treat tissue fibrosis. Indeed, small-molecule compounds that boost SIRT1 activity has been exploited to mitigate tissue fibrosis with success at least in model animals (Ponnusamy et al., 2015; Liu et al., 2017; Yu et al., 2017; Chu et al., 2018). Our data as reported here reinforce the notion that suppression of SIRT1 activity represents a key pathophysiological event in the pathogenesis of intestinal fibrosis and argue for the screening of small-molecule SIRT1 agonists in the intervention of intestinal fibrosis.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Nanjing Medical University Ethics Committee on Humane Treatment of Laboratory Animals.
YF conceived the project. BC, WD, TS, XM, YG, and XL designed and performed the experiments, and collected data. All authors contributed to the drafting and editing of the manuscript.
This work was supported by the Postgraduate Fellowship Grants from the Jiangsu Province Education Commission.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.697614/full#supplementary-material
Ananthakrishnan, A. N. (2015). Epidemiology and risk factors for IBD. Nat. Rev. Gastroenterol. Hepatol. 12, 205–217. doi: 10.1038/nrgastro.2015.34
Bugyei-Twum, A., Ford, C., Civitarese, R., Seegobin, J., Advani, S. L., Desjardins, J. F., et al. (2018). Sirtuin 1 activation attenuates cardiac fibrosis in a rodent pressure overload model by modifying Smad2/3 transactivation. Cardiovasc. Res. 114, 1629–1641. doi: 10.1093/cvr/cvy131
Chen, B., Fan, Z., Sun, L., Chen, J., Feng, Y., Fan, X., et al. (2020a). Epigenetic activation of the small GTPase TCL contributes to colorectal cancer cell migration and invasion. Oncogenesis 9:86.
Chen, B., Yuan, Y., Sun, L., Chen, J., Yang, M., Yin, Y., et al. (2020b). MKL1 mediates TGF-β induced RhoJ transcription to promote breast cancer cell migration and invasion. Front. Cell Dev. Biol. 8:832. doi: 10.3389/fcell.2020.00832
Chen, B., Zhao, Q., Xu, T., Yu, L., Zhuo, L., Yang, Y., et al. (2020c). BRG1 activates PR65A transcription to regulate NO bioavailability in vascular endothelial cell. Front. Cell Dev. Biol. 8:774. doi: 10.3389/fcell.2020.00774
Chen, B., Zhu, Y., Chen, J., Feng, Y., and Xu, Y. (2021). Activation of TCL transcription by lysine demethylase KDM4B in colorectal cancer cells. Front. Cell Dev. Biol. 9:617549. doi: 10.3389/fcell.2021.617549
Chen, I. C., Chiang, W. F., Huang, H. H., Chen, P. F., Shen, Y. Y., and Chiang, H. C. (2014). Role of SIRT1 in regulation of epithelial-to-mesenchymal transition in oral squamous cell carcinoma metastasis. Mol. Cancer 13:254.
Chini, C. C., Escande, C., Nin, V., and Chini, E. N. (2010). HDAC3 is negatively regulated by the nuclear protein DBC1. J. Biol. Chem. 285, 40830–40837. doi: 10.1074/jbc.m110.153270
Chini, C. C., Escande, C., Nin, V., and Chini, E. N. (2013). DBC1 (Deleted in Breast Cancer 1) modulates the stability and function of the nuclear receptor Rev-erbalpha. Biochem. J. 451, 453–461. doi: 10.1042/bj20121085
Choupani, J., Mansoori Derakhshan, S., Bayat, S., Alivand, M. R., and Shekari Khaniani, M. (2018). Narrower insight to SIRT1 role in cancer: a potential therapeutic target to control epithelial-mesenchymal transition in cancer cells. J. Cell. Physiol. 233, 4443–4457. doi: 10.1002/jcp.26302
Chu, H., Jiang, S., Liu, Q., Ma, Y., Zhu, X., Liang, M., et al. (2018). Sirtuin1 protects against systemic sclerosis-related pulmonary fibrosis by decreasing proinflammatory and profibrotic processes. Am. J. Respir. Cell Mol. Biol. 58, 28–39. doi: 10.1165/rcmb.2016-0192oc
Coarfa, C., Grimm, S. L., Katz, T., Zhang, Y., Jangid, R. K., Walker, C. L., et al. (2020). Epigenetic response to hyperoxia in the neonatal lung is sexually dimorphic. Redox Biol. 37:101718. doi: 10.1016/j.redox.2020.101718
D’Haens, G. R. (2010). Top-down therapy for IBD: rationale and requisite evidence. Nat. Rev. Gastroenterol. Hepatol. 7, 86–92. doi: 10.1038/nrgastro.2009.222
Digby-Bell, J. L., Atreya, R., Monteleone, G., and Powell, N. (2020). Interrogating host immunity to predict treatment response in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 17, 9–20. doi: 10.1038/s41575-019-0228-5
Dong, W., Kong, M., Zhu, Y., Shao, Y., Wu, D., Lu, J., et al. (2020). Activation of TWIST transcription by chromatin remodeling protein BRG1 contributes to liver fibrosis in mice. Front. Cell Dev. Biol. 8:340. doi: 10.3389/fcell.2020.00340
Dong, W., Zhu, Y., Zhang, Y., Fan, Z., Zhang, Z., Fan, X., et al. (2021). BRG1 links TLR4 trans-activation to LPS-induced SREBP1a expression and liver injury. Front. Cell Dev. Biol. 9:617073. doi: 10.3389/fcell.2021.617073
Fan, Z., Kong, M., Li, M., Hong, W., Fan, X., and Xu, Y. (2020). Brahma related gene 1 (Brg1) regulates cellular cholesterol synthesis by acting as a Co-factor for SREBP2. Front. Cell Dev. Biol. 8:259. doi: 10.3389/fcell.2020.00259
Fichtner-Feigl, S., Fuss, I. J., Young, C. A., Watanabe, T., Geissler, E. K., Schlitt, H. J., et al. (2007). Induction of IL-13 triggers TGF-beta1-dependent tissue fibrosis in chronic 2,4,6-trinitrobenzene sulfonic acid colitis. J. Immunol. 178, 5859–5870. doi: 10.4049/jimmunol.178.9.5859
Flier, S. N., Tanjore, H., Kokkotou, E. G., Sugimoto, H., Zeisberg, M., and Kalluri, R. (2010). Identification of epithelial to mesenchymal transition as a novel source of fibroblasts in intestinal fibrosis. J. Biol. Chem. 285, 20202–20212. doi: 10.1074/jbc.m110.102012
Gao, Y., Tang, J., Chen, W., Li, Q., Nie, J., Lin, F., et al. (2015). Inflammation negatively regulates FOXP3 and regulatory T-cell function via DBC1. Proc. Natl. Acad. Sci. U.S.A. 112, E3246–E3254.
Guo, J., Zhao, W., Cao, X., Yang, H., Ding, J., Tan, Z., et al. (2017). SIRT1 promotes tumor-like invasion of fibroblast-like synoviocytes in rheumatoid arthritis via targeting TIMP1. Oncotarget 8, 88965–88973. doi: 10.18632/oncotarget.21628
Hong, W., Kong, M., Qi, M., Bai, H., Fan, Z., Zhang, Z., et al. (2020). BRG1 mediates nephronectin activation in hepatocytes to promote T lymphocyte infiltration in ConA-induced hepatitis. Front. Cell Dev. Biol. 8:587502. doi: 10.3389/fcell.2020.587502
Hu, K., Li, Y., Wu, W., Xie, L., Yan, H., Cai, Y., et al. (2020). ATM-dependent recruitment of BRD7 is required for transcriptional repression and DNA repair at DNA breaks flanking transcriptional active regions. Adv. Sci. (Weinh) 7:2000157. doi: 10.1002/advs.202000157
Huang, X. Z., Wen, D., Zhang, M., Xie, Q., Ma, L., Guan, Y., et al. (2014). Sirt1 activation ameliorates renal fibrosis by inhibiting the TGF-beta/Smad3 pathway. J. Cell. Biochem. 115, 996–1005. doi: 10.1002/jcb.24748
Ishiguro, K., Watanabe, O., Nakamura, M., Yamamura, T., Matsushita, M., Goto, H., et al. (2017). Inhibition of KDM4A activity as a strategy to suppress interleukin-6 production and attenuate colitis induction. Clin. Immunol. 180, 120–127. doi: 10.1016/j.clim.2017.05.014
Jehanno, C., Fernandez-Calero, T., Habauzit, D., Avner, S., Percevault, F., Jullion, E., et al. (2020). Nuclear accumulation of MKL1 in luminal breast cancer cells impairs genomic activity of ERalpha and is associated with endocrine resistance. Biochim. Biophys. Acta Gene Regul. Mech. 1863:194507. doi: 10.1016/j.bbagrm.2020.194507
Jostins, L., Ripke, S., Weersma, R. K., Duerr, R. H., Mcgovern, D. P., Hui, K. Y., et al. (2012). Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124.
Kalluri, R., and Weinberg, R. A. (2009). The basics of epithelial-mesenchymal transition. J. Clin. Invest. 119, 1420–1428. doi: 10.1172/jci39104
Kaplan, G. G. (2015). The global burden of IBD: from 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 12, 720–727. doi: 10.1038/nrgastro.2015.150
Kawamura, A., Munzel, M., Kojima, T., Yapp, C., Bhushan, B., Goto, Y., et al. (2017). Highly selective inhibition of histone demethylases by de novo macrocyclic peptides. Nat. Commun. 8:14773.
Kim, H. J., Moon, S. J., Kim, S. H., Heo, K., and Kim, J. H. (2018). DBC1 regulates Wnt/beta-catenin-mediated expression of MACC1, a key regulator of cancer progression, in colon cancer. Cell Death Dis. 9:831.
Kim, J. E., Chen, J., and Lou, Z. (2008). DBC1 is a negative regulator of SIRT1. Nature 451, 583–586. doi: 10.1038/nature06500
Kong, M., Dong, W., Xu, H., Fan, Z., Miao, X., Guo, Y., et al. (2021a). Choline kinase alpha is a novel transcriptional target of the Brg1 in hepatocyte: implication in liver regeneration. Front. Cell Dev. Biol. 9:705302. doi: 10.3389/fcell.2021.705302
Kong, M., Zhu, Y., Shao, J., Fan, Z., and Xu, Y. (2021b). The chromatin remodeling protein BRG1 regulates SREBP maturation by activating SCAP transcription in hepatocytes. Front. Cell Dev. Biol. 9:622866. doi: 10.3389/fcell.2021.622866
Lamouille, S., Xu, J., and Derynck, R. (2014). Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15, 178–196.
Latella, G., Di Gregorio, J., Flati, V., Rieder, F., and Lawrance, I. C. (2015). Mechanisms of initiation and progression of intestinal fibrosis in IBD. Scand. J. Gastroenterol. 50, 53–65. doi: 10.3109/00365521.2014.968863
Lawrance, I. C., Rogler, G., Bamias, G., Breynaert, C., Florholmen, J., Pellino, G., et al. (2017). Cellular and molecular mediators of intestinal fibrosis. J. Crohns Colitis 11, 1491–1503.
Leong, R. W. (2010). Environmental risk factors in inflammatory bowel disease. J. Gastroenterol. Hepatol. 25, 227–228. doi: 10.1111/j.1440-1746.2010.06257.x
Li, C., Flynn, R. S., Grider, J. R., Murthy, K. S., Kellum, J. M., Akbari, H., et al. (2013). Increased activation of latent TGF-beta1 by alphaVbeta3 in human Crohn’s disease and fibrosis in TNBS colitis can be prevented by cilengitide. Inflamm. Bowel Dis. 19, 2829–2839. doi: 10.1097/mib.0b013e3182a8452e
Li, M., Hong, W., Hao, C., Li, L., Wu, D., Shen, A., et al. (2018). SIRT1 antagonizes liver fibrosis by blocking hepatic stellate cell activation in mice. FASEB J. 32, 500–511. doi: 10.1096/fj.201700612r
Li, M., Hong, W., Hao, C., Li, L., Xu, H., Li, P., et al. (2017). Hepatic stellate cell-specific deletion of SIRT1 exacerbates liver fibrosis in mice. Biochim. Biophys. Acta 1863, 3202–3211. doi: 10.1016/j.bbadis.2017.09.008
Li, N., Liu, S., Zhang, Y., Yu, L., Hu, Y., Wu, T., et al. (2020a). Transcriptional activation of matricellular protein Spondin2 (SPON2) by BRG1 in vascular endothelial cells promotes macrophage chemotaxis. Front. Cell Dev. Biol. 8:794. doi: 10.3389/fcell.2020.00794
Li, T., Eheim, A. L., Klein, S., Uschner, F. E., Smith, A. C., Brandon-Warner, E., et al. (2014). Novel role of nuclear receptor Rev-erbalpha in hepatic stellate cell activation: potential therapeutic target for liver injury. Hepatology 59, 2383–2396. doi: 10.1002/hep.27049
Li, X., Jiang, Z., and Zhang, X. (2018). SIRT1 overexpression protects non-small cell lung cancer cells against osteopontin-induced epithelial-mesenchymal transition by suppressing NF-kappaB signaling. Onco Targets Ther. 11, 1157–1171. doi: 10.2147/ott.s137146
Li, Z., Kong, X., Zhang, Y., Yu, L., Guo, J., and Xu, Y. (2020b). Dual roles of chromatin remodeling protein BRG1 in angiotensin II-induced endothelial-mesenchymal transition. Cell Death Dis. 11:549.
Li, Z., Zhang, Y., Yu, L., Xiao, B., Li, T., Kong, X., et al. (2020c). BRG1 stimulates endothelial derived alarmin MRP8 to promote macrophage infiltration in an animal model of cardiac hypertrophy. Front. Cell Dev. Biol. 8:569. doi: 10.3389/fcell.2020.00569
Li, Z., Zhao, Q., Lu, Y., Zhang, Y., Li, L., Li, M., et al. (2021). DDIT4 S-Nitrosylation Aids p38-MAPK signaling complex assembly to promote hepatic reactive oxygen species production. Adv. Sci. (Weinh) 15:e2101957.
Liu, L., Zhao, Q., Kong, M., Mao, L., Yang, Y., and Xu, Y. (2021a). Myocardin-related transcription factor A (MRTF-A) regulates integrin beta 2 transcription to promote macrophage infiltration and cardiac hypertrophy in mice. Cardiovasc. Res. cvab110.
Liu, L., Zhao, Q., Lin, L., Yang, G., Yu, L., Zhuo, L., et al. (2021b). Myeloid MKL1 disseminates cues to promote cardiac hypertrophy in mice. Front. Cell Dev. Biol. 9:583492. doi: 10.3389/fcell.2021.583492
Liu, X., Hu, D., Zeng, Z., Zhu, W., Zhang, N., Yu, H., et al. (2017). SRT1720 promotes survival of aged human mesenchymal stem cells via FAIM: a pharmacological strategy to improve stem cell-based therapy for rat myocardial infarction. Cell Death Dis. 8:e2731. doi: 10.1038/cddis.2017.107
Lovisa, S., Genovese, G., and Danese, S. (2019). Role of epithelial-to-mesenchymal transition in inflammatory bowel disease. J. Crohns Colitis 13, 659–668. doi: 10.1093/ecco-jcc/jjy201
Lv, F., Li, N., Kong, M., Wu, J., Fan, Z., Miao, D., et al. (2020). CDKN2a/p16 antagonizes hepatic stellate cell activation and liver fibrosis by modulating ROS levels. Front. Cell Dev. Biol. 8:176. doi: 10.3389/fcell.2020.00176
Maity, J., Deb, M., Greene, C., and Das, H. (2020). KLF2 regulates dental pulp-derived stem cell differentiation through the induction of mitophagy and altering mitochondrial metabolism. Redox Biol. 36:101622. doi: 10.1016/j.redox.2020.101622
Mallik, R., Prasad, P., Kundu, A., Sachdev, S., Biswas, R., Dutta, A., et al. (2020). Identification of genome-wide targets and DNA recognition sequence of the Arabidopsis HMG-box protein AtHMGB15 during cold stress response. Biochim. Biophys. Acta Gene Regul. Mech. 1863:194644. doi: 10.1016/j.bbagrm.2020.194644
Marti, J. M., Garcia-Diaz, A., Delgado-Bellido, D., O’valle, F., Gonzalez-Flores, A., Carlevaris, O., et al. (2021). Selective modulation by PARP-1 of HIF-1alpha-recruitment to chromatin during hypoxia is required for tumor adaptation to hypoxic conditions. Redox Biol. 41:101885. doi: 10.1016/j.redox.2021.101885
Moon, Y., Kim, I., Chang, S., Park, B., Lee, S., Yoo, S., et al. (2020). Hypoxia regulates allele-specific histone modification of the imprinted H19 gene. Biochim. Biophys. Acta Gene Regul. Mech. 1863:194643. doi: 10.1016/j.bbagrm.2020.194643
Ning, L., Rui, X., Bo, W., and Qing, G. (2021). The critical roles of histone deacetylase 3 in the pathogenesis of solid organ injury. Cell Death Dis. 12:734.
Peng, H., Zhang, S., Peng, Y., Zhu, S., Zhao, X., Yang, S., et al. (2021). Yeast bromodomain factor 1 and its human homolog TAF1 play conserved roles in promoting homologous recombination. Adv. Sci. (Weinh) 8:e2100753.
Pezone, A., Taddei, M. L., Tramontano, A., Dolcini, J., Boffo, F. L., De Rosa, M., et al. (2020). Targeted DNA oxidation by LSD1-SMAD2/3 primes TGF-beta1/EMT genes for activation or repression. Nucleic Acids Res. 48, 8943–8958. doi: 10.1093/nar/gkaa599
Ponnusamy, M., Zhuang, M. A., Zhou, X., Tolbert, E., Bayliss, G., Zhao, T. C., et al. (2015). Activation of sirtuin-1 promotes renal fibroblast activation and aggravates renal fibrogenesis. J. Pharmacol. Exp. Ther. 354, 142–151. doi: 10.1124/jpet.115.224386
Rashid, M., Shah, S. G., Verma, T., Chaudhary, N., Rauniyar, S., Patel, V. B., et al. (2021). Tumor-specific overexpression of histone gene, H3C14 in gastric cancer is mediated through EGFR-FOXC1 axis. Biochim. Biophys. Acta Gene Regul. Mech. 1864:194703. doi: 10.1016/j.bbagrm.2021.194703
Rieder, F., and Fiocchi, C. (2009). Intestinal fibrosis in IBD–a dynamic, multifactorial process. Nat. Rev. Gastroenterol. Hepatol. 6, 228–235. doi: 10.1038/nrgastro.2009.31
Scharl, M., Frei, S., Pesch, T., Kellermeier, S., Arikkat, J., Frei, P., et al. (2013). Interleukin-13 and transforming growth factor beta synergise in the pathogenesis of human intestinal fistulae. Gut 62, 63–72. doi: 10.1136/gutjnl-2011-300498
Shen, T., Li, Y., Chen, Z., Liang, S., Qiu, Y., Zhu, L., et al. (2020). Activating transcription factor 6 (ATF6) negatively regulates Polo-like kinase 4 expression via recruiting C/EBPbeta to the upstream-promoter during ER stress. Biochim. Biophys. Acta Gene Regul. Mech. 1863:194488. doi: 10.1016/j.bbagrm.2020.194488
Simic, P., Williams, E. O., Bell, E. L., Gong, J. J., Bonkowski, M., and Guarente, L. (2013). SIRT1 suppresses the epithelial-to-mesenchymal transition in cancer metastasis and organ fibrosis. Cell Rep. 3, 1175–1186. doi: 10.1016/j.celrep.2013.03.019
Sun, L. N., Zhi, Z., Chen, L. Y., Zhou, Q., Li, X. M., Gan, W. J., et al. (2017). SIRT1 suppresses colorectal cancer metastasis by transcriptional repression of miR-15b-5p. Cancer Lett. 409, 104–115. doi: 10.1016/j.canlet.2017.09.001
Sun, L., Chen, B., Wu, J., Jiang, C., Fan, Z., Feng, Y., et al. (2020). Epigenetic regulation of a disintegrin and metalloproteinase (ADAM) promotes colorectal cancer cell migration and invasion. Front. Cell Dev. Biol. 8:581692. doi: 10.3389/fcell.2020.581692
Sun, S., Yang, F., Zhu, Y., and Zhang, S. (2020). KDM4A promotes the growth of non-small cell lung cancer by mediating the expression of Myc via DLX5 through the Wnt/beta-catenin signaling pathway. Life Sci. 262:118508. doi: 10.1016/j.lfs.2020.118508
Teixeira, A. F., Ten Dijke, P., and Zhu, H. J. (2020). On-Target anti-TGF-beta therapies are not succeeding in clinical cancer treatments: what are remaining challenges? Front Cell Dev Biol 8:605. doi: 10.3389/fcell.2020.00605
Valatas, V., Filidou, E., Drygiannakis, I., and Kolios, G. (2017). Stromal and immune cells in gut fibrosis: the myofibroblast and the scarface. Ann. Gastroenterol. 30, 393–404.
Wang, J. N., Yang, Q., Yang, C., Cai, Y. T., Xing, T., Gao, L., et al. (2020). Smad3 promotes AKI sensitivity in diabetic mice via interaction with p53 and induction of NOX4-dependent ROS production. Redox Biol. 32:101479. doi: 10.1016/j.redox.2020.101479
Wang, S., Chen, Z., Zhu, S., Lu, H., Peng, D., Soutto, M., et al. (2020). PRDX2 protects against oxidative stress induced by H. pylori and promotes resistance to cisplatin in gastric cancer. Redox Biol. 28:101319.
Welch, R. D., Billon, C., Valfort, A. C., Burris, T. P., and Flaveny, C. A. (2017). Pharmacological inhibition of REV-ERB stimulates differentiation, inhibits turnover and reduces fibrosis in dystrophic muscle. Sci. Rep. 7:17142.
Wengrower, D., Zanninelli, G., Pappo, O., Latella, G., Sestieri, M., Villanova, A., et al. (2004). Prevention of fibrosis in experimental colitis by captopril: the role of tgf-beta1. Inflamm. Bowel Dis. 10, 536–545. doi: 10.1097/00054725-200409000-00007
Wirtz, S., Popp, V., Kindermann, M., Gerlach, K., Weigmann, B., Fichtner-Feigl, S., et al. (2017). Chemically induced mouse models of acute and chronic intestinal inflammation. Nat. Protoc. 12, 1295–1309. doi: 10.1038/nprot.2017.044
Wu, T., Wang, H., Xin, X., Yang, J., Hou, Y., Fang, M., et al. (2020). An MRTF-A-Sp1-PDE5 axis mediates angiotensin-II-induced cardiomyocyte hypertrophy. Front. Cell Dev. Biol. 8:839. doi: 10.3389/fcell.2020.00839
Wu, X., Dong, W., Zhang, T., Ren, H., Wang, J., Shang, L., et al. (2020). Epiregulin (EREG) and myocardin related transcription factor A (MRTF-A) form a feedforward loop to drive hepatic stellate cell activation. Front. Cell Dev. Biol. 8:591246. doi: 10.3389/fcell.2020.591246
Yang, M., Li, Z., Tao, J., Hu, H., Zhang, Z., Cheng, F., et al. (2021). Resveratrol induces PD-L1 expression through snail-driven activation of Wnt pathway in lung cancer cells. J. Cancer Res. Clin. Oncol. 147, 1101–1113. doi: 10.1007/s00432-021-03510-z
Yang, Y., Li, Z., Guo, J., and Xu, Y. (2020a). Deacetylation of MRTF-A by SIRT1 defies senescence induced down-regulation of collagen type I in fibroblast cells. Biochim. Biophys. Acta Mol. Basis Dis. 1866:165723. doi: 10.1016/j.bbadis.2020.165723
Yang, Y., Yang, G., Yu, L., Lin, L., Liu, L., Fang, M., et al. (2020b). An interplay between MRTF-A and the histone acetyltransferase TIP60 mediates hypoxia-reoxygenation induced iNOS transcription in macrophages. Front. Cell Dev. Biol. 8:484. doi: 10.3389/fcell.2020.00484
Yu, L., Liu, X., Yuan, Z., Li, X., Yang, H., Sun, L., et al. (2017). SRT1720 alleviates ANIT-induced cholestasis in a mouse model. Front. Pharmacol. 8:256. doi: 10.3389/fphar.2017.00256
Yu, X., Wang, M., Han, Q., Zhang, X., Mao, X., Wang, X., et al. (2018). ZNF326 promotes a malignant phenotype of breast cancer by interacting with DBC1. Mol. Carcinog. 57, 1803–1815. doi: 10.1002/mc.22898
Yuan, J., Minter-Dykhouse, K., and Lou, Z. (2009). A c-Myc-SIRT1 feedback loop regulates cell growth and transformation. J. Cell Biol. 185, 203–211. doi: 10.1083/jcb.200809167
Zerr, P., Palumbo-Zerr, K., Huang, J., Tomcik, M., Sumova, B., Distler, O., et al. (2016). Sirt1 regulates canonical TGF-beta signalling to control fibroblast activation and tissue fibrosis. Ann. Rheum. Dis. 75, 226–233. doi: 10.1136/annrheumdis-2014-205740
Zhang, H., Lu, J., and Wu, S. (2020). Sp4 controls constitutive expression of neuronal serine racemase and NF-E2-related factor-2 mediates its induction by valproic acid. Biochim. Biophys. Acta Gene Regul. Mech. 1863:194597. doi: 10.1016/j.bbagrm.2020.194597
Zhang, Z., Chen, B., Zhu, Y., Zhang, T., Zhang, X., Yuan, Y., et al. (2021). The Jumonji domain-containing histone demethylase homolog 1D/lysine demethylase 7A (JHDM1D/KDM7A) is an epigenetic activator of RHOJ transcription in breast cancer cells. Front. Cell Dev. Biol. 9:664375. doi: 10.3389/fcell.2021.664375
Zhao, Z., Su, Z., Liang, P., Liu, D., Yang, S., Wu, Y., et al. (2020). USP38 couples histone ubiquitination and methylation via KDM5B to resolve inflammation. Adv. Sci. (Weinh) 7:2002680. doi: 10.1002/advs.202002680
Keywords: intestinal fibrosis, epithelial-mesenchymal transition, SIRT1, transcriptional regulation, epigenetics, histone demethylase
Citation: Chen B, Dong W, Shao T, Miao X, Guo Y, Liu X and Feng Y (2021) A KDM4-DBC1-SIRT1 Axis Contributes to TGF-b Induced Mesenchymal Transition of Intestinal Epithelial Cells. Front. Cell Dev. Biol. 9:697614. doi: 10.3389/fcell.2021.697614
Received: 20 April 2021; Accepted: 31 August 2021;
Published: 22 September 2021.
Edited by:
Rebecca Ann Wingert, University of Notre Dame, United StatesCopyright © 2021 Chen, Dong, Shao, Miao, Guo, Liu and Feng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yifei Feng, ZmVuZ3lpZmVpQG5qbXUuZWR1LmNu
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.