
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 19 August 2021
Sec. Molecular and Cellular Pathology
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.689533
This article is part of the Research Topic Skeletal Muscle – From Developmental Concepts to Therapy View all 7 articles
 Shanshan Yao1
Shanshan Yao1 Zihao Chen1
Zihao Chen1 Yuanyuan Yu2
Yuanyuan Yu2 Ning Zhang1
Ning Zhang1 Hewen Jiang1
Hewen Jiang1 Ge Zhang2*
Ge Zhang2* Zongkang Zhang1*
Zongkang Zhang1* Baoting Zhang1*
Baoting Zhang1*Duchenne muscular dystrophy (DMD) is a lethal, X-linked neuromuscular disorder caused by the absence of dystrophin protein, which is essential for muscle fiber integrity. Loss of dystrophin protein leads to recurrent myofiber damage, chronic inflammation, progressive fibrosis, and dysfunction of muscle stem cells. There is still no cure for DMD so far and the standard of care is principally limited to symptom relief through glucocorticoids treatments. Current therapeutic strategies could be divided into two lines. Dystrophin-targeted therapeutic strategies that aim at restoring the expression and/or function of dystrophin, including gene-based, cell-based and protein replacement therapies. The other line of therapeutic strategies aims to improve muscle function and quality by targeting the downstream pathological changes, including inflammation, fibrosis, and muscle atrophy. This review introduces the important developments in these two lines of strategies, especially those that have entered the clinical phase and/or have great potential for clinical translation. The rationale and efficacy of each agent in pre-clinical or clinical studies are presented. Furthermore, a meta-analysis of gene profiling in DMD patients has been performed to understand the molecular mechanisms of DMD.
As an X-linked neuromuscular disorder, Duchenne muscular dystrophy (DMD), is one of the most severe inherited muscle diseases and there is still no cure for DMD (Verhaart and Aartsma-Rus, 2019). The incidence of DMD is 1 per 5,136 male births (Crisafulli et al., 2020). The symptoms of DMD in infants, such as the slow motor development, will not be noticed until 3 years of age. Between 8 and 10 years of age, it is hard for patients to stand and walk, which may need the assistance of braces or wheelchairs. Cardiorespiratory failure is the leading cause of death in DMD patients (Meyers and Townsend, 2019; Silva et al., 2019). The life span of DMD patients is around 20 years, but the condition is fatal in 100% of cases (Chemello et al., 2020). DMD not only makes patients physically and mentally suffer tremendously but also imposes a heavy economic burden on their families and society (Gocheva et al., 2019).
Duchenne muscular dystrophy is caused by the mutations in the DMD gene (Monaco et al., 1986; Hoffman et al., 1987; Haidet et al., 2008; Okubo et al., 2020). The severity of DMD depends on the mutation type. ‘Out of frame’ mutations disrupt the reading frame and further produce the dysfunctional dystrophin, which result in the severe DMD phenotype (Aravind et al., 2019). On the other hand, ‘In-frame’ mutations perverse the frame of DMD gene and generate partial functional dystrophin protein, which lead to less severe Becker muscular dystrophy (BMD) phenotype (Haidet et al., 2008). In DMD, the absence of functional dystrophin and consequential loss of the dystrophin-associated protein complex (DAPC) increase the fragility of muscle fibers and lead to adverse cycles of muscle degeneration and regeneration (Ribeiro et al., 2019). In this condition, a continuous pathological environment including deteriorations of calcium homeostasis, activation of Ca2+-dependent proteases, inflammatory cytokines and mitochondrial dysfunction will further promote fibrosis and fatty tissue replacement (Klingler et al., 2012). To study the pathobiology of DMD, different animal models have been generated and studied (McGreevy et al., 2015). The most widely used animal model is mdx mice (Willmann et al., 2009), which carry a point mutation in exon 23 of the Dmd gene that results in the absence of full-length dystrophin (Sicinski et al., 1989). Mdx mice exhibit similar pathological features to DMD patients, including intense muscle fiber inflammation, muscle fibrosis and loss of muscle fibers (Stedman et al., 1991; Kupatt et al., 2021). However, two other proteins, utrophin and α7-integrin, fulfill the function of dystrophin in a compensatory way and minimize the dystrophic phenotype in mdx mice. Therefore, utrophin/dystrophin and integrin/dystrophin double-knockout mice have been generated, which have shown more similar pathological features with DMD patients (Guo et al., 2006; Rooney et al., 2006). Dystrophin-deficient dogs are also regarded as an excellent DMD model (Duan, 2011), which share remarkably similar clinical symptoms and histological lesions with DMD boys (Duan et al., 2015).
There are two lines of therapeutic strategies for DMD so far. One is dystrophin-targeted therapies that aim to restore the function of dystrophin. And the other one is to target the downstream pathological changes (Guiraud and Davies, 2017). Dystrophin-targeted therapies include gene therapies, cell therapies and protein replacement therapies. Despite massive pre-clinical and clinical studies have been carried on, there are challenges exist for the dystrophin-targeted therapies (Chamberlain and Chamberlain, 2017). The first concern is that the dystrophin-targeted therapies can just slow down the progression of DMD, but not restore the function of abnormal muscle tissues due to the degenerative nature of the DMD. Secondly, it is difficult for dystrophin-targeted therapies to target all muscle tissues, because muscle tissue is highly abundant and widely distributed throughout the whole body (Verhaart and Aartsma-Rus, 2019). Therefore, in addition to investigating strategies to cure dystrophin deficiency, increasing efforts are being placed on improving muscle function by targeting the downstream pathological changes of DMD. However, the curative effects of downstream specific therapies are not fully unity and the accurate mechanisms are obscure. It is desirable to have an in-depth understanding of the secondary pathological changes and develop more effective therapies.
This review introduces the current therapeutic strategies for DMD that have entered the clinical phase and/or have great potential for clinical translation. Furthermore, a meta-analysis of gene profiling in DMD patients is performed to understand the molecular mechanisms of DMD. The hub genes achieved from a meta-analysis may provide the potential targets for the treatment of DMD.
Gene-based therapeutic strategies have the potential to provide long-lasting and one-time treatment for DMD. In DMD, the deleted exons concentrate in the region from exon 43 to 55 (Asher et al., 2020), which make up roughly 60–65% of the events (Zimowski et al., 2014). Several gene-based therapeutic strategies including exon skipping, stop codon readthrough, gene-addition and gene-editing therapy have been designed.
Exon-skipping therapies target mutant codons with predesigned antisense oligonucleotides (AONs) to generate the proteins that can display partial function of dystrophin (Echevarría et al., 2018). AONs are used to control pre-mRNA splicing in specific sites and further obtain an in-frame message by removing one proximal exon to the deletion breakpoint (Kinali et al., 2009). In 2016, the US Food and Drug Administration (FDA) have approved the Eteplirsen (Exondys 51) that can specifically skip exon 51 in defective gene variants for DMD (Lim et al., 2017). Eteplirsen shows promising results for 6-min walk test (6MWT) that measures the distance a patient can independently walk within 6 min (Cirak et al., 2011). In 2019 and 2020, Golodirsen and Viltolarsen have been approved by the FDA, respectively (Heo, 2020; Al Musaimi et al., 2021). Both drugs are designed for patients with mutation that is amenable to exon 53 skipping (Dzierlega and Yokota, 2020). Forty to eighty week Golodirsen treatment significantly increases dystrophin protein expression in muscle of DMD patients (Frank et al., 2020). Similar beneficial result in dystrophin level is also observed in patients who received Viltolarsen treatment (Iftikhar et al., 2020).
However, the significant obstacle of exon-skipping therapies is that they can only provide treatment for patients with specific mutations. For example, skipping exon 51 therapy and skipping exon 53 therapy are applicable to around 14 and 8% of DMD patients, respectively (Echevarría et al., 2018). Besides, the long-term safety and efficacy of exon-skipping therapies are still needed to be tested. For example, Eteplirsen, as the first drug approved by FDA, its long-term efficacy has not been confirmed yet (Dzierlega and Yokota, 2020). Moreover, hypersensitivity reactions including upper respiratory tract infection, cough, and pyrexia have been reported in the latest clinical data of Viltolarsen and Golodirsen (Clemens et al., 2020; Heo, 2020).
Except for single exon skipping therapies, double exon skipping therapies have also been undertaken in clinical trials (which target two different exons). However, unlike in vitro studies for double exon skipping, the therapy transferring two AONs into myofibers shows reduced efficacy in vivo (Mitrpant et al., 2009; Niks and Aartsma-Rus, 2017). Therefore, more extensive studies are required.
Some nonsense mutations can result in premature stop codons (PSCs) and produce dysfunctional dystrophin (Ng et al., 2021). Around 13% of DMD patients are caused by these nonsense mutations (Aartsma-Rus et al., 2016). Unlike frameshift mutations, the PSCs do not disrupt the reading frame but prematurely terminate the translation during protein synthesis. Some small molecules have been designed to combine with these PSCs and stimulate stop codon readthrough (Malik et al., 2010; Aartsma-Rus et al., 2016). One representative is Gentamicin (Baradaran-Heravi et al., 2017). However, continual use of Gentamicin is prohibited due to the side effects including renal toxicity and ototoxicity (Vitiello et al., 2019). Two derivatives of Gentamicin, NB74 and NB84, show reduced cell toxicity and improved readthrough efficiency in vitro (Nudelman et al., 2010). Another aminoglycoside antibiotic Ataluren (NPC-14) has shown effective readthrough activities in pre-clinical studies (Guiraud and Davies, 2017) and could potentially provide treatment for around 11% of all DMD patients with a nonsense mutation (Namgoong and Bertoni, 2016). In 2016, Ataluren has been approved by the European Medicines Agency (EMA) for the treatment of DMD (Finkel et al., 2013; McDonald et al., 2017). However, a Phase 3 clinical trial reported that patients aged 7–16 after receiving Ataluren for 48 weeks did not show significant changes in 6MWD compared to placebo-treated patients (McDonald et al., 2017). Based on this, the drug failed to receive approval from the FDA (McDonald et al., 2017).
Because DMD is caused by the mutations in the DMD gene. A highly appealing gene therapy is to replace the dysfunctional DMD gene with normal gene (Ricci et al., 2019). Adeno-associated virus (AAV) is a viral vector that can infect skeletal muscle with high efficiency (Zhang et al., 2020), which could be used as a delivery vector. However, the transgene capacity of AAV is limited and full-length dystrophin mRNA transcripts of the DMD gene are exceptionally large for AAV (Duan, 2018). To address this obstacle, the whole dystrophin gene is abbreviated to partially functional micro-dystrophin gene and packaged into AAV (Banks et al., 2010; Ramos et al., 2019). Numerous AAV serotypes have been explored in animal models (Wasala et al., 2019). Among several serotypes of AAV, serotypes 1, 6, 8, and 9 are outstanding candidates for DMD therapy as they exhibit a potent tropism for striated muscles (Inagaki et al., 2006; Leborgne et al., 2019). Recently, there are several micro-dystrophins AAV-based clinical trials ongoing (NCT03362502, NCT03375164, and NCT03368742; Table 1). The data from the 19 patients who received 1-year treatment in a Phase 1b trial (NCT03362502) showed that PF-06939926 treatment could induce continuous production of the partial functional dystrophin protein in muscle tissues and improvements in motor function (Belluscio et al., 2021). PF-06939926 has received orphan drug designation by both FDA and EMA and has entered Phase 3 trial (NCT04281485). Another micro-dystrophin transgene (rAAVrh74.MHCK7.micro-dystrophin) has been designed and possesses higher delivery efficacy than PF-06939926 (Potter et al., 2021). In mdx mice, after 3-month rAAVrh74.MHCK7.micro-dystrophin treatment, the enhancement in the specific force of diaphragm and tibialis anterior muscle has been observed (Potter et al., 2021).
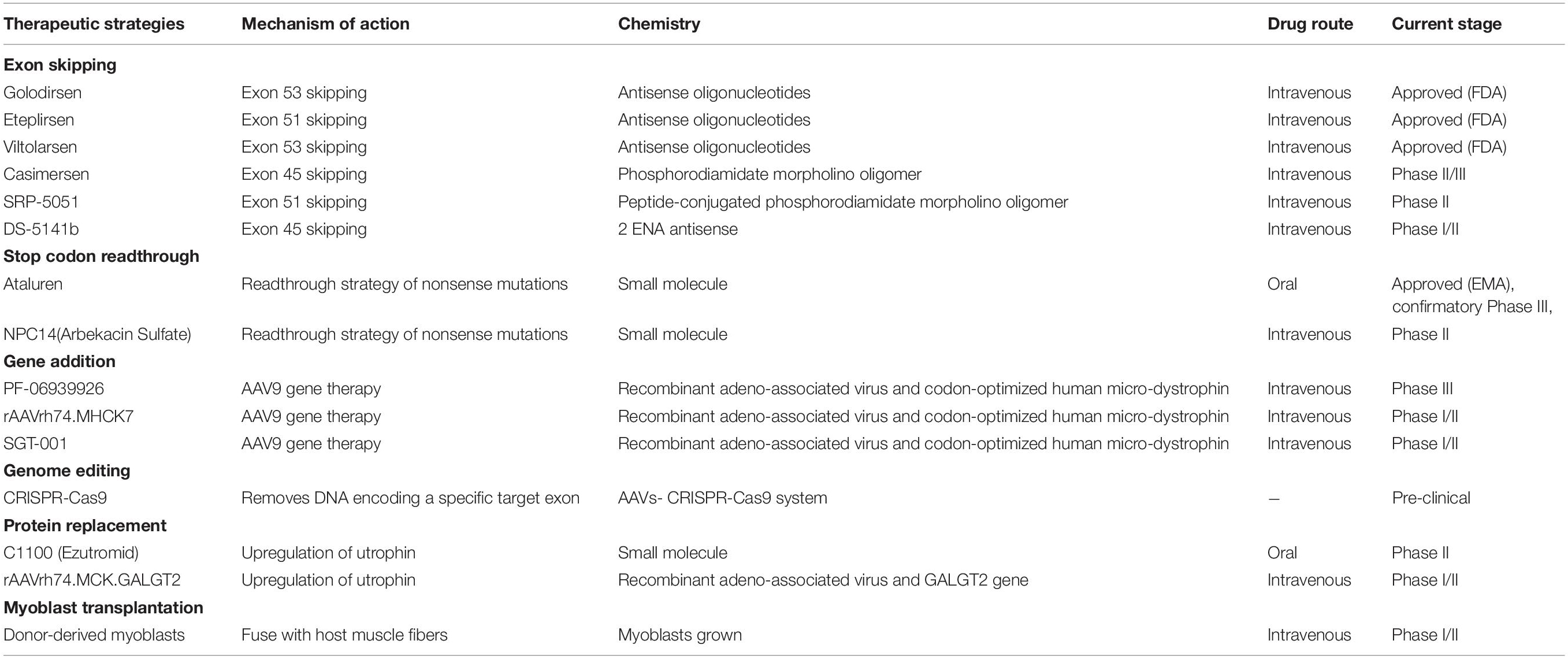
Table 1. Status of the current therapeutic strategies targeting the primary defect of Duchenne muscular dystrophy (https://www.clinicaltrials.gov).
CRISPR (clustered regularly interspaced short palindromic repeats) Cas 9 (CRISPR associated protein) gene editing as a powerful tool to model or even to rectify genetic abnormalities has been used for correcting mutations in the DMD gene (Wang et al., 2017; Lek et al., 2020). CRISPR/Cas9 is an adaptive immune system in bacteria against invading bacteriophages and later developed as a tool for DNA editing (Jinek et al., 2012). The specific double-stranded DNA breaks can be induced by CRISPR/Cas9 technology in the genome and further activate DNA repair systems (Karimian et al., 2019). And the DNA repair systems will correct missense mutations by homology-directed repair-based germline editing or non-homologous end joining (NHEJ)-based postnatal editing (Long et al., 2014, 2016; Bengtsson et al., 2017; Zhang et al., 2017). The AAVs have been wildly used for the delivery of CRISPR/Cas9 in vivo (Samulski and Muzyczka, 2014). In mdx mice, the correction of exon 23 deletion by AAV8-CRISPR/Cas9 therapy can restore the expression of functional dystrophin protein in skeletal myofibers and significantly enhance the muscle force (Nelson et al., 2016). Besides, another study has shown that AAV6-CRISPR/Cas9 can correct both exon 52 and 53 deletions and significantly increase the force generation in mdx mice (Bengtsson et al., 2017). CRISPR/Cas9 gene editing delivered by AAV vectors can potentially bring clinical benefits to about 60% of DMD patients (Echigoya et al., 2015; Ousterout et al., 2015a, b; Taglia et al., 2015). However, the efficiency of CRISPR/Cas9-mediated genome editing is highly dependent on the dose of AAV (Min et al., 2019b). Inevitably, the excessive immune responses caused by exogenous AAVs and Cas9 might pose a challenge to their effective clinical application (Zhang et al., 2020). Further studies are still needed to understand the mechanism and frequency of off-target effects (Echigoya et al., 2015; Feng et al., 2015; Hollinger and Chamberlain, 2015). The related research is ongoing to generate more-specific Cas9 enzymes (Zheng et al., 2018).
Cell-based therapies aim to insert normal genomes into dystrophic muscles by transplanting normal muscle precursor cells (Sun et al., 2020). The ideal cell source can deliver a copy of the intact DMD gene and affect both limb muscles and heart (Ramos and Chamberlain, 2015). Different types of stem cells have been applied in DMD animal studies and clinical trials (Sun et al., 2020). The transplantation of satellite cells from muscle biopsies leads to the increased expression of dystrophin in mdx mice (Xu X. et al., 2015). Besides, CD133+ cells, the multipotent stem cells that are derived from normal human skeletal muscles, also have myogenic potential (Meng et al., 2018). In mdx mice, the transplantation of CD133+ cells can generate satellite cells and dystrophin-positive myofibers (Meng et al., 2014). Unfortunately, both satellite cells and CD133+ cells show no obvious functional improvement in DMD patients (Torrente et al., 2007; Xu X. et al., 2015). The engraftment of human-induced pluripotent stem cells (hiPSCs) holds great promise to correct mutations in DMD patients (Uchimura et al., 2017; Mondragon-Gonzalez and Perlingeiro, 2018). The hiPSCs are obtained from DMD patients and can be genetically corrected to restore normal dystrophin expression by CRISPR/Cas9 mediated gene editing (Hochheiser et al., 2018). Transplantation of genetically corrected hiPSCs can successfully restore the expression of dystrophin in mdx mice (Young et al., 2016). Although these cell-based therapies have shown apparent engraftment in mdx mice, their limited abilities for cell survival and migration after transplantation negatively influenced their clinical application (Sun et al., 2020). Therefore, more attempts are needed to improve the engraftment rates of stem cells (Hicks et al., 2018; Wu et al., 2018).
Utrophin is a structural and functional autosomal paralog of dystrophin (Loro et al., 2020). Due to the similar structure and function, utrophin has been proposed as an alternate for dystrophin (Guiraud et al., 2019). In mdx mice, the upregulated expression of full-length utrophin can prevent the development of muscular dystrophy (Péladeau et al., 2018). Therefore, it is a promising strategy to upregulate utrophin levels in DMD patients. C1100 (Ezutromid) is a small molecule utrophin modulator to treat DMD (Tinsley et al., 2011). In mdx mice, the treatment of C1100 leads to better muscle physiology and increases overall muscle strength (Tinsley et al., 2011). However, a Phase 2 clinical trial (NCT02383511) indicated that C1100 did not provide any significant benefit for DMD patients and the development of C1100 therapy was discontinued (Ricotti et al., 2016). The GALGT2 is an enzyme that can generate the cytotoxic T-cell (CT) glycan to protect dystrophic skeletal myofibers from injury (Xu R. et al., 2015). In mdx mice, the overexpression of GALGT2 increases the expression of utrophin and can greatly strengthen the muscle membrane’s resistance to injury (Xu et al., 2009). rAAVrh74.MCK.GALGT2 is the surrogate gene therapy that using rhesus serotype 74 (rAAVrh74) to deliver human GALGT2 in the heart and skeletal muscle (Chicoine et al., 2014; Zygmunt et al., 2019). The phase1/2 trial of rAAVrh74.MCK.GALGT2 is ongoing (NCT03333590). Although utrophin exerts favorable effects as a compensate for dystrophin, it still differs from dystrophin in some functions. One of the most significant difference is that utrophin cannot prevent functional ischemia during muscle contraction (Lai et al., 2009; Li et al., 2010). Besides, dystrophin binds microtubules to form a rectilinear lattice beneath the sarcolemma, whereas utrophin cannot (Belanto et al., 2014). Therefore, the combination of utrophin-based therapies with other dystrophin-targeted treatments for DMD is well worth considering (Guiraud et al., 2019).
The loss of dystrophin triggers multiple pathological pathways, including fibrosis, inflammation, loss of calcium homeostasis, oxidative stress, ischemia, and muscle atrophy (Birnkrant et al., 2018). In addition to the therapeutic strategies that aim to restore the functions of dystrophin, the agents targeting the downstream pathological changes caused by dystrophin deficiency attract more and more attentions (Table 2).
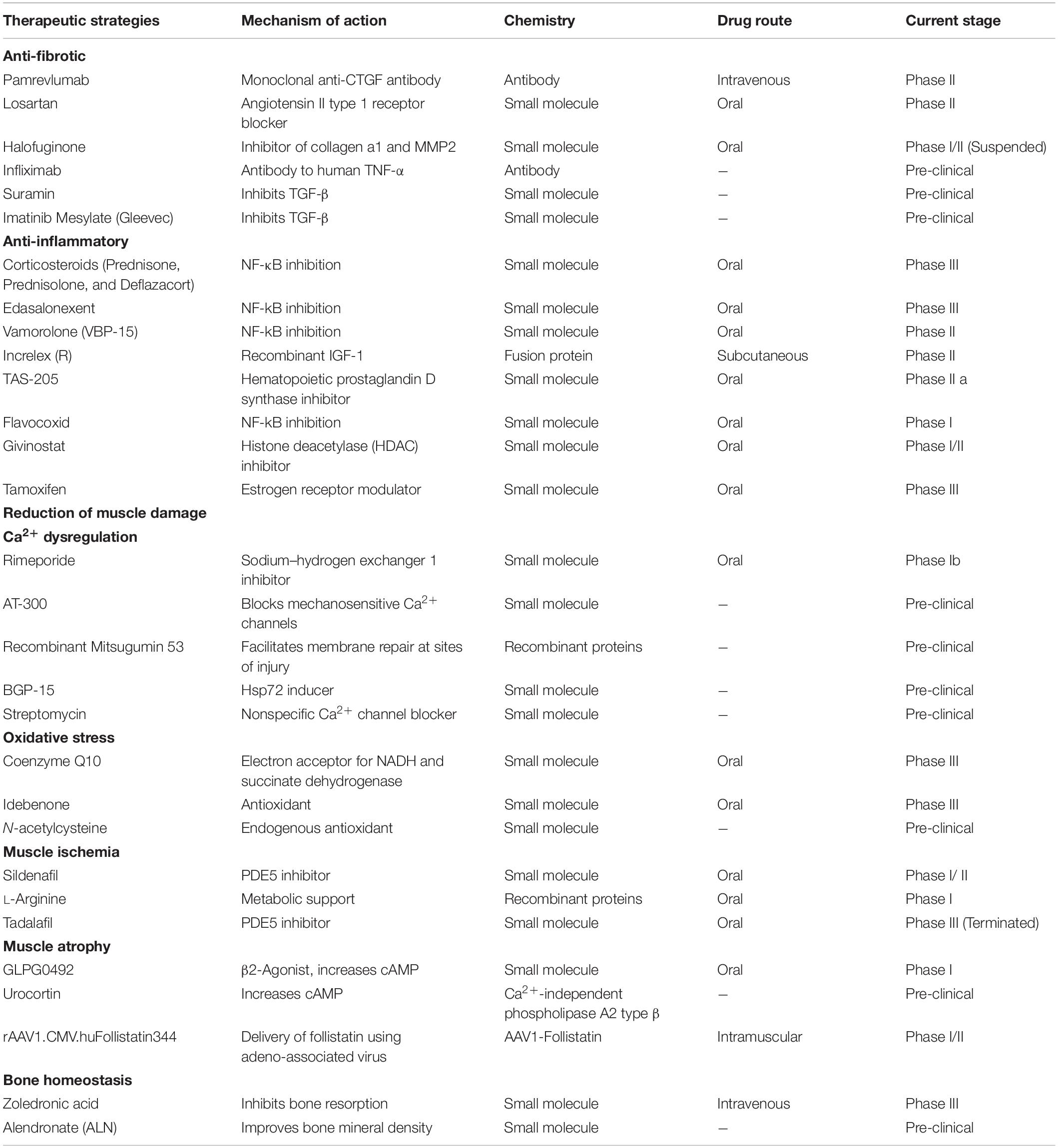
Table 2. Status of the current therapeutic strategies acting on downstream pathological mechanisms of Duchenne muscular dystrophy (https://www.clinicaltrials.gov).
Fibrosis is a prominent pathological change in DMD (Zhou and Lu, 2010). As a response to chronic tissue injury and long-term over-immunosuppression, fibrosis is a harmful and uncontrolled wound-healing process characterized by excessive accumulation of extracellular matrix (ECM) proteins (Wynn, 2007). Fibrosis can affect all tissues/organs and is the major cause of mortality in DMD patients (Wynn, 2008). A longitudinal study showed that endomysium fibrosis on the initial muscle biopsies correlated with poor muscle strength and age at loss of ambulation in DMD patients (Desguerre et al., 2009). Fibrogenic cytokines can activate tissue fibroblasts to produce ECM proteins (Suntar et al., 2020). Transforming growth factor-β (TGF-β), connective tissue growth factor (CTGF) and tumor necrosis factor-α (TNF-α) play significant roles in this process (Walker et al., 2019). TGF-β is a major mediator of the fibrotic response which can promote the build-up of ground substance as well as regulate the expression of catabolic enzymes and other mediators (Mázala et al., 2020). CTGF is one of the important downstream mediators of the effects of TGF-β on fibroblasts (Gonzalez and Brandan, 2019), which correlates with fibrotic activities (Sakai et al., 2017).
It has been proved that the expression patterns of TGF-β, as well as its receptors, are associated with dystrophin deficiency in DMD (Mázala et al., 2020). Type I (TβRI) and type II (TβRII) are two receptors of TGF-β. TGF-β can directly bind to the TβRII and then activate TβRI. TβRI subsequently phosphorylates downstream Smad proteins and activated Smad proteins further regulate the transcription of fibrotic genes (Kemaladewi et al., 2014). The TGF-β pathway could be an important therapeutic target to reduce excessive fibrotic tissue deposition in DMD (Mázala et al., 2020). It is known that angiotensin II independently or cooperatively works with TGF-β to induce fibrosis via the angiotensin type 1 receptor (AT1) in a multitude of tissues outside of the cardiovascular and renal systems (Acuña et al., 2014). Losartan is an AT1 receptor blocker that can attenuate TGF-β signaling (Lee et al., 2015). Losartan has been reported to significantly decrease fibrosis of the diaphragm and restore muscle strength of extensor digitorum longus muscle in mdx mice (Cohn et al., 2007). However, subsequent studies showed a minimal functional benefit of Losartan in mdx mice (Bish et al., 2011). A limited human clinical study showed that 1-year Losartan treatment significantly improved the cardiac function of DMD patients, but there was no impact on the fibrosis in skeletal muscle (Allen et al., 2013). Halofuginone (Halo) is another small molecule that can inhibit phosphorylation of TGF-β dependent Smad3 (Turgeman et al., 2008). Halo could decrease the established fibrosis (diaphragm and quadriceps), collagen I and III expression and smooth muscle actin content after 10-week treatment in mdx mice (Huebner et al., 2008). However, in a Phase 1/2 clinical trial, the patient experienced serious adverse reactions and died after receiving a high dose of Halo for 2 weeks (NCT02525302). The corresponding clinical trials were terminated. Suramin is a TGF-β inhibitor that has shown antifibrotic function in mdx mice (De Oliveira Moreira et al., 2013). The treatment of Suramin reduces the level of creatine kinase and attenuates fibrosis in skeletal muscles in mdx mice (De Oliveira Moreira et al., 2013, 2017). Besides, Suramin can protect muscles against exercise-induced damage in mdx mice (Taniguti et al., 2011). Imatinib mesylate (Gleevec®), an antineoplastic drug, also shows the ability to inhibit the pro-fibrogenic activation of TGF-β. In mdx mice, Imatinib mesylate significantly reduces fibrosis and improves hindlimb grip strength (Huang et al., 2009). However, in contrast with the results of B10-mdx, imatinib has not improved the fibrosis in the diaphragm of D2-mdx mice, and an unanticipated side effect of imatinib in heart has been reported (Ito et al., 2013). Therefore, more-specific inhibitors with less side effects are needed.
Connective tissue growth factor is a central mediator in the pathogenesis of fibrosis and plays important roles in the proliferation of fibroblasts, ECM production, vascular regeneration or other biological activities (Song et al., 2017). CTGF is widespread in endothelial, smooth muscle, fibroblast, and cartilage. TGF-β1 can significantly increase the expression level of CTGF in human fibroblasts (Ramazani et al., 2018). Moreover, CTGF promotes the activation of TGF-β signaling (Ramazani et al., 2018). In mdx mice, CTGF correlates with the number of necrotic-regenerative foci in dystrophic muscle (Morales et al., 2018). the mdx mice with hemizygous CTGF deletion (mdx–Ctgf+/–) shows better muscle strength and attenuated skeletal muscle impairment and fibrosis (Morales et al., 2013). It has been reported that raised expression levels of CTGF and TGF-β1 in the skeletal muscle are positively associated with the clinical severity of DMD (Song et al., 2017). These studies demonstrate that CTGF plays a central role in regulating fibrosis of DMD, which implied that CTGF could be a promising target for DMD treatment. CTGF is composed of four functionally conserved domains: insulin-like growth factor binding protein (IGFBP), von Willebrand factor type C repeat (VWC), thrombospondin type-1 repeat (TSP1 or TSR), and cysteine knot-containing domain (CT) (Chen et al., 2020). CTGF can activate TGF-β signaling by directly binding to TGF-β1 through the VWC domain (O’Leary et al., 2004). Besides, integrin αvβ3 can be activated by binding to the CT domain (Chen et al., 2020). The activated integrin signaling pathway can trigger the expression of CTGF and further promote the accumulation of ECM constituents (Riquelme-Guzmán et al., 2018). FG-3019 is a fully human monoclonal antibody that interferes with the action of CTGF by specifically targeting the VWC domain of CTGF. Pre-clinical studies suggest that FG-3019 can penetrate into tissues to reduce effective tissue levels of CTGF (Morales et al., 2013). A Phase 2 trial of FG-3019 in non-ambulatory subjects with DMD is ongoing (NCT01890265).
In DMD, an increased level of Ca2+ leads to fiber necrosis and subsequently causes a severe inflammatory response (Allen et al., 2016). This process is mainly mediated by NF-kB pro-inflammatory pathway (Altamirano et al., 2012). The NF-κB pathway is regulated by TNF-α and interleukin 6 (IL-6), which are responsible for assembling the I-κB kinase (IKK) catalytic complex subunits (De Pasquale et al., 2012). Activation of NF-κB can induce the translocation of p50/p65 into the nucleus, and subsequent transcription of genes encoding cytokines and response factors involving in immune response (Baldwin, 2001).
Currently, the anti-inflammatory treatment for DMD is long-term dosing with glucocorticoids, including prednisone, prednisolone, deflazacort, and vamorolone, via inhibiting the NF-κB pathway (St-Pierre et al., 2004; Biggar et al., 2006). Glucocorticoids have been shown to prolong independent ambulation, improve pulmonary function, and delay the onset of cardiomyopathy in DMD patients (Hammers et al., 2020). However, there are also severe side effects for long-term glucocorticoid treatments, such as weight gain and hypertension (Matthews et al., 2016). Even though glucocorticoids share the same functional principles, there are still differences in the effectiveness and severity of side effects. For example, prednisone is superior to prednisolone and deflazacort in improving strength and pulmonary function (Gloss et al., 2016). And in terms of side effects, deflazacort can cause less weight gain than prednisone after 1-year treatment (Matthews et al., 2016). The glucocorticoid alternatives with fewer side effects are needed (Gloss et al., 2016). Vamorolone is an optimized glucocorticoid drug that shows consistent anti-inflammatory efficacy and reduced side effects compared to prednisolone (Sreetama et al., 2018). A Phase 1/2 study of Vamorolone in DMD patients showed an improved profile with reduced typical glucocorticoid-induced safety concerns after 2-week treatment (Hoffman et al., 2019).
Flavocoxid is another anti-inflammatory small molecule that can inhibit activities of COX and 5-LOX enzymes and further inhibit the NF-κB, TNF-α, and MAPKs pathways (Norwood et al., 2010; Vita et al., 2021). A Phase 1/2 study of flavocoxid indicated that the treatment was well tolerated and there was a significant reduction in serum IL-1β and TNF-α in the group of DMD boys (NCT01335295). However, the exploratory outcome measures failed to show significant effects for short-term treatment (Vita et al., 2021).
TNF-α is one of the pro-inflammatory cytokines that can regulate inflammatory response (Grounds and Torrisi, 2004). In mdx mice, proinflammatory cytokine TNF-α strongly contributes to necrosis in muscle fibers (de Senzi Moraes Pinto et al., 2013). Therefore, the anti-TNF-α therapies may be an effective treatment for DMD. Remicade (Infliximab) treatment can significantly increase skeletal muscle strength and decrease both cardiac and skeletal fibrosis by blocking the TNF-α signaling pathway in mdx mice (Grounds and Torrisi, 2004; Ermolova et al., 2014). However, the long-term TNF-α blockade treatment leads to deleterious changes in heart function (Ermolova et al., 2014).
Tamoxifen (TAM) is a selective estrogen receptor modulator that can either mimic or antagonize estrogens in a tissue-dependent manner (Dorchies et al., 2013). TAM can elevate the level of pro-inflammatory cytokines and growth factors involved in muscle regeneration and fibrosis, such as TGF-β, insulin-like growth factor 1 (IGF1) and osteopontin (Nagy et al., 2019). And then leads to an increased capacity of muscle-purified mitochondria to buffer cytosolic calcium (Nagy et al., 2019). It has been reported that TAM treatment can improve the body force and leg muscle force in mdx mice (Dorchies et al., 2013). Recently, a Phase 3 safety and efficacy trial of TAM in DMD is ongoing (NCT03354039). However, there is no result posted so far. The exact mode of action of TAM on dystrophic muscle is not clearly understood and some concerns about possible oncogenic side-effects in eventual life-long use have been raised (Vitiello et al., 2019).
Inhibition of histone deacetylase (HDAC) could lead to decreased expression of inflammatory genes in mdx mice (Nguyen-Tran et al., 2014). Givinostat (ITF2357) is an HDAC inhibitor that is currently being developed for the treatment of DMD (Bettica et al., 2016). Givinostat has shown several functional and morphological beneficial effects in mdx mice, such as increased cross-sectional area of myofibers, restored muscle force, decreased inflammatory infiltrate and prevention from fibrotic scars, which contribute to counteracting the muscle loss and the functional decline that are typically observed in mdx mice (Colussi et al., 2008). A Phase 2 clinical trial of Givinostat (NCT03373968) for DMD has been completed (Bettica et al., 2016). The trial showed that Givinostat treatment could increase the number of muscle fibers and suppress fibrosis. However, the study was not able to show improvement in functional tests and authors attributed this to the small sample size (Bettica et al., 2016). Recently, a Phase 3 trial of Givinostat is ongoing (NCT02851797).
Edasalonexent is a small molecule drug that can significantly inhibit p65-dependent inflammatory responses (Hammers et al., 2016). Long-term administration of Edasalonexent, or the related analog CAT-1041 with equivalent pharmacology, has been demonstrated several disease-modifying characteristics, including attenuated fatigue in skeletal muscle and increased muscle mass in mdx mice (Hammers et al., 2016). In addition, suppressed inflammation and fibrosis result in increased exercise endurance and improved diaphragm function in DMD animal models (Donovan et al., 2017). A Phase 3 study of Edasalonexent (NCT03703882) reported a significant down-regulation of NF-κB pathway-related gene expression in peripheral mononuclear cells after 2-week treatment. However, mild diarrhea and headache were the most common adverse events of Edasalonexent (Donovan et al., 2017).
Increlex is a recombinant human IGF-1 (rhIGF-1) that also offers potential treatments for DMD patients (Rutter et al., 2020). rhIGF-1 can reduce the inflammatory response and macrophage infiltration by down-regulating the expression of the NF-κB signal pathway in mdx mice (Ji et al., 2020). Systemic treatment with rhIGF-1 can improve muscle function and regeneration in mdx mice (Barton et al., 2002; Gregorevic et al., 2002; Shavlakadze et al., 2004; Schertzer et al., 2007). The data of a 6-month trial showed that rhIGF-1 improved linear growth but did not change motor function (Rutter et al., 2020).
Prostaglandin D2 (PGD2) is an inflammatory mediator and Hematopoietic prostaglandin D synthase (HPGDS) can promote the production of PGD2 (Ricciotti and FitzGerald, 2011). The overproduction of PGD2 by HPGDS can aggravate inflammation and exacerbate muscle tissue damage (Nakagawa et al., 2013). PGD2-mediated inflammation is involved in the pathology of DMD but the mechanism is not clarified yet (Nakagawa et al., 2013). The inhibition of PGD2 production via HPGDS inhibition may be a potential therapy for DMD. A Phase 1/2 study showed a favorable safety profile of TAS-205, HPGDS inhibitor, in DMD patients (Takeshita et al., 2018). However, a larger trial is required to ensure the effectiveness of TAS-205 (Komaki et al., 2020).
In DMD, the deficiency of functional dystrophin leads to membrane tears and Ca2+ leakage (Whitehead et al., 2006). High resting and active Ca2+ levels are observed in myofibers of DMD patients (Law et al., 2020). The excessive cytosolic Ca2+ can further aggravate dystrophic pathology by promoting Ca2+-dependent proteinase cleaving intracellular proteins (Townsend et al., 2007). Streptomycin can be used as a non-specific blocker of Ca2+ channels (Teichmann et al., 2008). Long-term Streptomycin treatment can alleviate limb muscle pathology, including reduced fibrosis, increased sarcolemmal stability and promoted muscle regeneration in mdx mice (Whitehead et al., 2006). However, there is no positive effect of streptomycin treatment on the diaphragm and heart muscle (Jørgensen et al., 2011). Another Ca2+ channel blocker, AT-300 (GsMTx4), which can specifically block mechanosensitive Ca2+ channels and improve muscle force production and decreases muscle degeneration in mdx model (Yeung et al., 2005; Bush et al., 2017). AT-300 has been granted orphan drug designation by the FDA. However, it still takes time to conduct pre-clinical testing before clinical application (Spinazzola and Kunkel, 2016).
Another approach is to repair the plasma membrane (Weisleder et al., 2012). Mitsugumin 53 (MG53) is an essential and muscle-specific member in cell membrane repairing, which plays a vital role in protecting skeletal and cardiac muscle cells from numerous types of acute injuries or chronic physiological stresses (Cai et al., 2009). Injection of recombinant human MG53 protein leads to reduced tissue damage in skeletal muscles of mdx mice (Weisleder et al., 2012).
The Na+-H+ exchanger 1 (NHE1/Rimeporide) is a transmembrane protein not only regulates the cellular volume of Na+ and H+ but also participates in the regulation of intracellular Ca2+ (Chen et al., 2004). There is an increase of intracellular Na+ concentration in the skeletal muscle of mdx mice and DMD patients. The intracellular Na+ overload in muscles is accompanied with muscle edema, which plays a role in muscle degeneration (Weber et al., 2012; Mijares et al., 2014). Rimeporide is an inhibitor of NHE-1 and shows anti-inflammatory and anti-fibrotic effects in mdx mice (Ghaleh et al., 2020). The positive effects on several pharmacodynamic biomarkers, including IGFBP1 and IGFBP6, have been reported in the Phase 1b study of Rimeporide (Gidaro et al., 2017; Previtali et al., 2020).
Oxidative stress is one of the hallmarks of dystrophic muscles. Overload of Ca2+ enhances the production of reactive oxygen species (ROS) and results in the elevated oxidative stress (Allen et al., 2016). ROS further exacerbates the Ca2+ induced mitochondrial dysfunction and necrosis (Allen et al., 2016). Coenzyme Q10 (CoQ10) is one potential additional therapy targeting oxidative stress in DMD (Spurney et al., 2011). CoQ10 is an antioxidant that functions as an electron acceptor for respiratory complexes I (NADH) and II (SDH) in the electron transport pathway. Exogenous supplement of CoQ10 into mitochondria can increase the oxidative activity of NADH, and further provide metabolic support to muscles (Mizobuti et al., 2019). Besides, CoQ10 also helps to reduce the excessive oxidation radicals and intracellular Ca2+ concentration in DMD muscles (Mizobuti et al., 2019). Pre-clinical studies have demonstrated that CoQ10 treatment can preserve muscle strength by 42% compared to controls in mdx mice (Mizobuti et al., 2019). A Phase 3 clinical study reported that CoQ10 could prevent the loss of respiratory function in DMD patients. The favorable safety and tolerability profiles of CoQ10 make it a potential option to ameliorate the ROS in DMD (Salehi et al., 2017).
Another drug that targets ROS is Idebenone. As a synthetic short-chain benzoquinone, Idebenone can reduce ROS and improve mitochondrial function, therefore alleviate the muscle damage induced by dystrophin deficiency (Servais et al., 2020). Idebenone has cardioprotective effect and improves exercise performance of mdx mice (Buyse et al., 2009). A Phase 3 clinical trial (NCT01027884) indicated the potential disease-modifying effect of Idebenone on the progression of early functional cardiac and respiratory parameters in DMD. However, there has no significant improvement been observed in upper limb muscle strength (Buyse et al., 2015).
N-acetylcysteine (NAC) is an endogenous antioxidant (McKenna et al., 2006). Several pre-clinical studies have shown that NAC administration can reduce the dystrophic pathology in both skeletal and cardiac muscle in mdx mice (Whitehead et al., 2008; Fauconnier et al., 2010; Terrill et al., 2012; de Senzi Moraes Pinto et al., 2013), which implies that NAC could be a potential therapy for DMD boys. However, the treatment of NAC shows significant suppression on body weight gain and muscle weight in mdx mice (Pinniger et al., 2017). Therefore, these potential adverse effects should be given more considerations in the future clinical application of NAC.
Dystrophin deficiency leads to down-regulated expression and mislocalization of the source of nitric oxide (NO) in skeletal muscle (Brescia et al., 2020). NO is used to stimulate the production of cyclic guanosine monophosphate (cGMP), and the NO-cGMP signaling pathway is required to balance muscle oxygenation and prevent excessive vasoconstriction caused by exercising (Percival et al., 2011). In mdx mice, the suppression of the NO-cGMP signaling pathway promotes skeletal muscle weakness, vascular dysfunction and defective exercise performance (Asai et al., 2007; Garbincius and Michele, 2015; Hörster et al., 2015; Brescia et al., 2020). The inhibitors of phosphodiesterase 5 (PDE5) can suppress cGMP breakdown and further amplify the signal of NO-cGMP pathways (Dombernowsky et al., 2018). Two PDE5 inhibitors Tadalafil (Cialis®) and Sildenafil (Viagra®) can significantly reduce dystrophic pathology in mdx mice (Khairallah et al., 2008; Kobayashi et al., 2008; Percival et al., 2011, 2012). These studies indicate that PDE5 inhibitors can be potential therapeutic strategies for DMD, even though the molecular mechanisms remain unclear. However, a Phase 3 clinical trial demonstrated that Tadalafil failed to attenuate the decline in ambulatory ability in DMD patients (NCT01865084). And the related study for tadalafil is being terminated for lack of efficacy (Victor et al., 2017). The other inhibitor, Sildenafil, showed no significant improvement in cardiac function in a clinical trial (Leung et al., 2014). Besides, due to the higher number of side-effects, the study of Sildenafil has been terminated (Leung et al., 2014).
In DMD, loss of dystrophin also reduces the activity of Nitric oxide synthases (NO) (Dombernowsky et al., 2018). In normal muscle tissue, intramuscular L-arginine can convert to NO. However, DMD patients have elevated synthesis of asymmetric dimethylarginine (ADMA) and diminished synthesis of Homoarginine (hArg), which results in reduced bioavailability of NO (Hafner et al., 2016). Therefore, to elevate L-arginine level seems to be a promising strategy to stimulate mitochondrial function and reduce the oxidative stress of DMD. It has been proved that L-arginine can decrease inflammation and enhance muscle regeneration in mdx mice (Hnia et al., 2008). Furthermore, a Phase 3 study indicated that treatment with a combination of L-arginine and metformin slowed muscle function decline in DMD patients (Hafner et al., 2019).
The maintenance of skeletal muscle mass mainly relies on the regulation of muscle protein synthesis and degradation (Selathurai et al., 2019). β2-agonists have been found to initiate satellite cell proliferation and promote muscle protein production, as well as to increase lean body mass in muscle injury models (Lynch et al., 2000; Zeman et al., 2000; Harcourt et al., 2007). DT-200 is a β2-agonists originally investigated to treat sarcopenia and cachexia (Jones et al., 2010; Cozzoli et al., 2013). In mdx mice, DT-200 significantly reduces the muscle atrophy in the hindlimb and improves the running performance (Cozzoli et al., 2013). However, trials in DMD patients have not yet begun.
Urocortins (Ucns) are neuropeptides related to the hypothalamic corticotropin-releasing factor (CRF) (Manning et al., 2017). CRF1R and CRF2R are two receptors of Ucns and positively couple to G-proteins, which can activate adenylyl cyclase and promote cAMP formation (Kishimoto et al., 1995; Samuelsson et al., 2004). The activation of the CRF2R and downstream cAMP upregulation can prevent the degeneration of the diaphragm and attenuate the loss of its force (Reutenauer-Patte et al., 2012; Silveira et al., 2020). Further studies in mdx mice demonstrated that Ucns improved dystrophic muscle structure and function (Reutenauer-Patte et al., 2012; Burns et al., 2018), implying that Ucns might have great potential as the therapeutic target for DMD.
As a member of the TGF-β superfamily, myostatin is a muscle-specific secretory protein that negatively regulates muscle growth (Han and Mitch, 2011; Tang et al., 2020). In myogenic cells, myostatin inhibits the differentiation of myoblast and decreases the expression of transcriptional regulators for cell proliferation (Langley et al., 2002). Therefore, myostatin inhibition is a promising approach for treating DMD (Al-Zaidy et al., 2015). In mdx, the inactivation of myostatin resulted in a significant increase in muscle mass (Ran et al., 2020). Follistatin (FST) is a myostatin-binding protein that can enhance muscle formation by neutralizing inflammation induced by myostatin (Tang et al., 2020). The AAV-mediated gene delivery of FST in mdx animals showed reversed muscle pathology and improved strength (Mendell et al., 2015). Follistatin-344 is an isoform of FST, has the beneficial effects on muscle size and function (Haidet et al., 2008). Recently, a Phase 1/2 trial of rAAV1.CMV.huFollistatin344 in DMD patients is ongoing (NCT02354781).
In DMD, the combination of different causes, including long-term glucocorticoid treatment, decrease of physical activity, release of dystrophin-deficient induced cytokines, and insufficiency of vitamin D, will lead to osteoporosis (Buckner et al., 2015; Mah, 2016). In mdx mice, low bone mineral density and bone strength have been reported (Rufo et al., 2011). Furthermore, DMD patients show a trend of increased bone loss and bone fragility (Bianchi et al., 2003; Perera et al., 2016; Tian et al., 2016). Therefore, bone health management is an important part of lifelong care for patients with DMD. Pamidronate and Zoledronic acid are two bone metastases treatment drugs (Yang and Du, 2015). A small sample study indicated that Pamidronate or Zoledronic acid could stabilize or even increase the height of compressed vertebrae and alleviate back pain in prepubertal DMD patients (Hawker et al., 2005; Sbrocchi et al., 2012). Besides, in an uncontrolled study of 16 DMD boys, alendronate maintained the BMD Z-scores of the total body and lumbar spine (Hawker et al., 2005).
In addition to dystrophin, numerous pathophysiological consequences of dystrophin deficiency also offer various downstream targets for DMD (Ziemba et al., 2021). However, not all the therapeutic strategies based on these targets are promising, and the concerns of efficacy and safety limit the clinical application of most strategies. To eliminate the unpromising targets, further understanding of the molecular mechanisms of these pathological changes is needed. Recently, computational approaches and network analysis have been extensively and successfully applied to find new gene–disease associations and identify novel therapeutic targets of diseases (Barabási et al., 2011; Nabirotchkin et al., 2020). A meta-analysis based on the publicly available differential gene expression profiles and literature-extracted expression regulation network of patients with DMD has been conducted in this review to identify the key regulators of these pathological changes.
A search for gene expression datasets publicly available on the National Center for Biotechnology Information (NCBI), Gene Expression Omnibus (GEO) and the European Bioinformatics Institute (EBI) Array Express (AE) was retrieved. Student’s t-tests and fold-change (FC) filtering were conducted to screen for the differentially expressed genes (DEGs) between DMD tissues and paired healthy tissues using the R software ‘limma’ package (Ritchie et al., 2015). The expression profiling information of each DEG was log2-transformed and then used to calculate the mean value (M) and standard deviation (SD) for control and DMD groups. In addition, the ‘meta’ package was used for performing a comprehensive meta-analysis of data aggregated from multiple sources (Balduzzi et al., 2019). The DEGs which satisfied the criterion of P-value < 0.05 and standardized mean difference (SMD) > 0.5 in the meta-analysis were identified as robust DEGs (Wang et al., 2019). Genes (STRING) database1 (version: 11.0) and Cytoscape software (version: 3.7.0) were utilized to construct protein–protein Interactions (PPI) networks on the robust DEGs (Olsen et al., 2014). A novel Cytoscape plugin cytoHubba (Chin et al., 2014) which could explore important nodes in biological networks was used for selecting hub genes by the cut-off value of Maximal Clique Centrality (MCC) score > 6 (Yu et al., 2020). Gene Ontology (GO) enrichment analysis of the robust DEGs was implemented with R package ‘cluster profiler’ and the Kyoto Encyclopedia of Genes and the Genomes (KEGG) was utilized on the robust DEGs (Yu et al., 2020).
As shown in Figure 1, GO enrichment analysis results of DEGs indicated that changes in the cellular component of DEGs were mainly enriched in immune activation, ECM organization and regulation of fibrosis (Table 3). The KEGG signaling pathway analysis of DEGs showed that hub genes were mostly involved in the PI3K-Akt signaling pathway, human papillomavirus infection pathway and focal adhesion pathway. Moreover, 10 hub genes from the network were identified with the cut-off value of MCC score > 5, using the cytoHubba plugin of Cytoscape software. As shown in Table 4, Fibronectin 1 (FN1), CTGF, Secreted Phosphoprotein 1(SPP1), Periostin (POSTN), THBS2 (Thrombospondin 2), COL4A2, and TGF-β1 were involved in ECM formation. FN1 is an important component of ECM and serum FN1 level is a promising biomarker to monitor disease progression in DMD (Cynthia Martin et al., 2014). Both TGF-β1 and CTGF are the key regulators of fibrosis. Furthermore, as the upstream of TGF-β1, SPP1 can active TGF-β1 to induce collagen expression in skeletal muscle fibroblasts (Kramerova et al., 2019). COL4A2 and THBS2 also are two genes involved in the fibrotic process such as liver fibrosis and renal fibrosis (Ishikawa et al., 2015; Chen et al., 2019). However, further studies are needed to identify their roles in DMD. POSTN may participate in the decline of muscle necrosis in mdx mice (Ohlendieck and Swandulla, 2017). Except for the ECM-related genes, Galectin 3 (LGALS3), Platelet and Endothelial Cell Adhesion Molecule1 (PECAM1) and C-C Motif Chemokine Ligand 2 (CCL2) are mainly involved in the immune response. In the STRING database and Cytoscape software, robust DEGs were mapped into the PPI network and 89 interactions were selected by median confidence (more than 0.5) (Figure 2). The PPI networks are the networks of protein complexes formed as the results of biochemical or electrostatic forces. PPI network is crucial for molecular processes and abnormal PPI is the basis of many diseases. In this study, PPI network indicated that FN1, CTGF, PECAM1, POSTN, and CCL2 may play important roles in DMD (Figure 3).
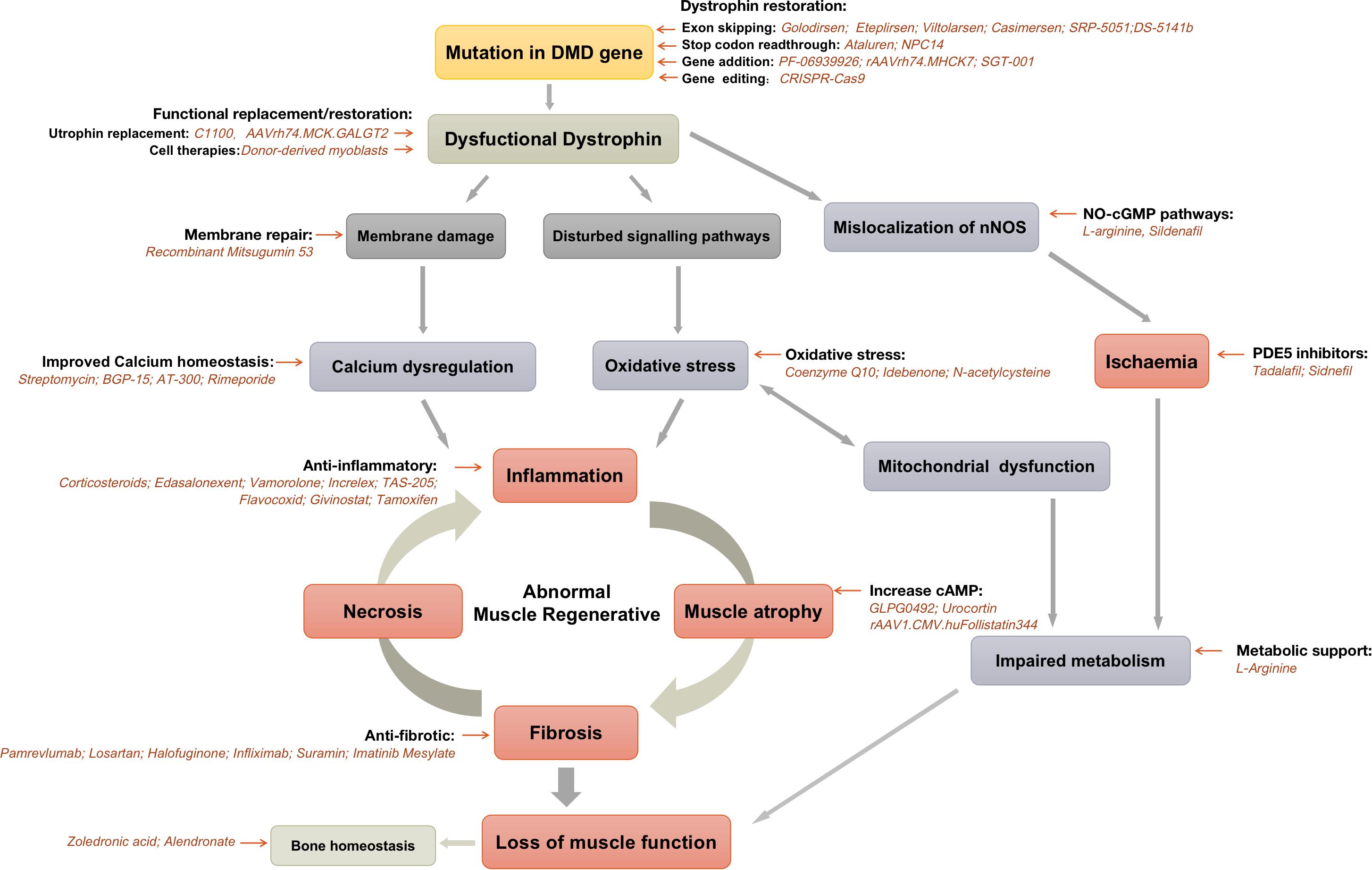
Figure 1. Current pharmacological strategies for Duchenne muscular dystrophy.

Table 3. GO and KEGG pathways.
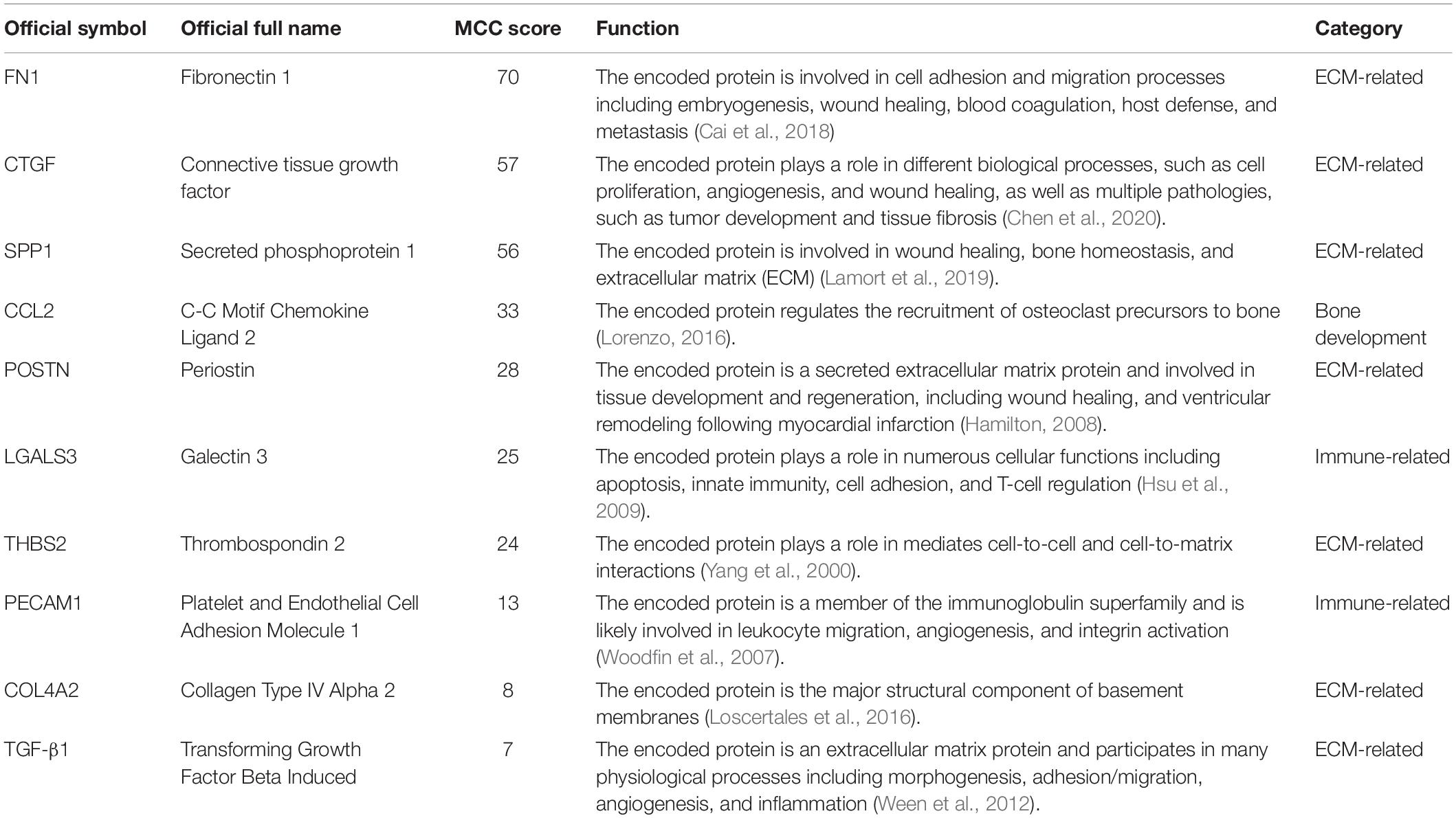
Table 4. Hub genes selected by Maximal Clique Centrality (MCC) score.
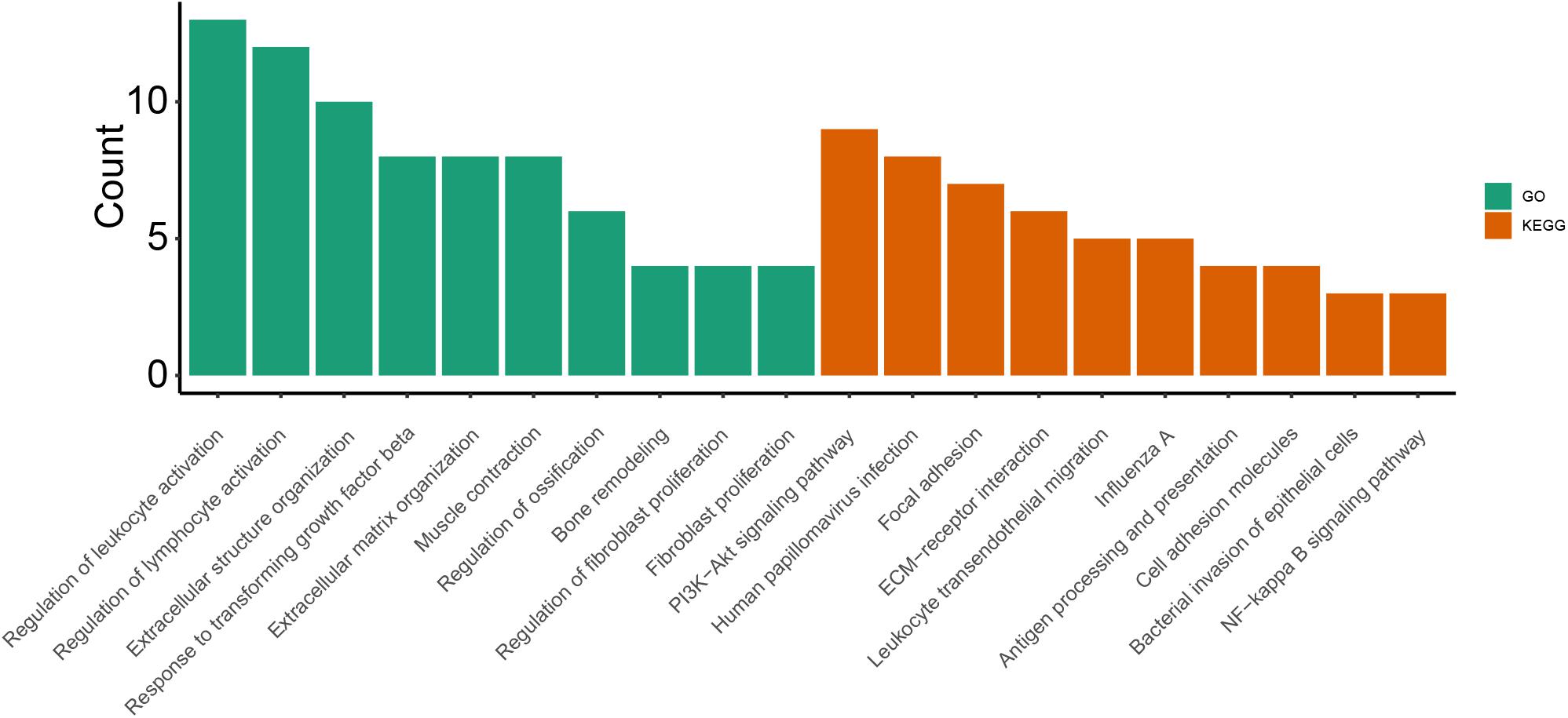
Figure 2. Enriched GO terms and KEGG pathways.
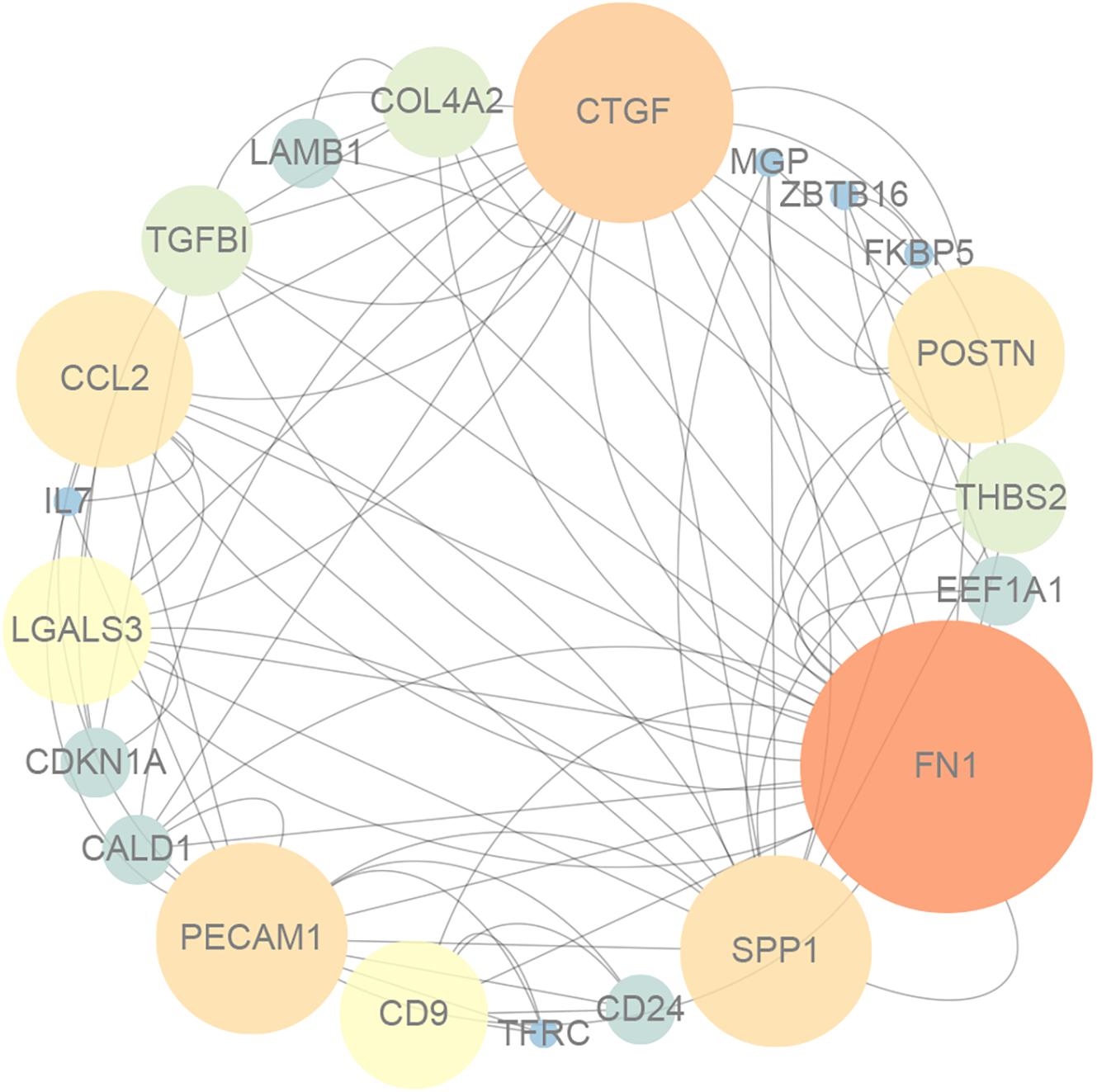
Figure 3. The protein–protein interaction network for DEGs, and the nodes with lighter color and bigger size represent the hub genes.
The apparent enrichment of ECM-related genes suggests that most of the samples might be obtained from patients at an advantaged stage of disease progression. Indeed, the subjects might at different stages of DMD when the samples were obtained. Unfortunately, there is no information about the disease stage of the patients in each data set. The disease stages are largely associated with the age of patients since DMD is a progressive disease. We reviewed the clinical data of total 12 data sets and 7 of them (GSE1004, GSE1007, GSE6011, GSE13608, GSE38417, GSE43698, and GSE109178) showed the information of subject age. The mean age of subjects were calculated as following: GSE1004: 36.3 (months); GSE1007: 36.2 (months); GSE6011: 14.3 (months); GSE13608: 11 (months); GSE38417: 44 (months); GSE43698: 48 (months); GSE109178: 40.1 (months); All samples: 31.65 (months). The mean age of data sets GSE13608 is the lowest. The 10 top enriched pathways in GSE13608 are: hsa05416: Viral myocarditis; hsa05330: Allograft rejection; hsa05332: Graft-versus-host disease; hsa04940: Type I diabetes mellitus; hsa05320: Autoimmune thyroid disease; hsa04612: Antigen processing and presentation; hsa04514: Cell adhesion molecules; hsa04610: Complement and coagulation cascades; hsa05150: Staphylococcus aureus infection; hsa04145: Phagosome. The immune-related pathway (Antigen processing and presentation) and ECM-related pathway (Cell adhesion molecules) are the top enriched pathways in early age of DMD patients, implying that the ECM- and immune-related pathways have been involved in the pathological process in early stage of DMD.
Different therapeutic strategies for DMD have been developed. Numerous researches and clinical trials have been performed as detailed above. Nevertheless, there is still no effective disease-modifying therapy and many questions remain unsolved.
Among the dystrophin-targeted therapeutic strategies, the therapies of exon skipping and readthrough blazed the new ways for DMD treatment (Ryan, 2014; Kole and Krieg, 2015). Three exon-skipping drugs, Golodirsen, Eteplirsen and Viltolarsen have received approvals from the FDA for DMD treatment and dozens more are being tested in clinical trials (Niks and Aartsma-Rus, 2017). Readthrough-based drug Ataluren has been marketed in some European countries for the treatment of DMD patients with nonsense mutations (Ryan, 2014). However, the most prominent problem is that exon skipping and readthrough therapies only available for DMD patients with specific mutation types. Therefore, more efforts are needed to improve the applicability of both therapies. Further innovations have been made in the field, such as multi-exons skipping therapies potentially benefit up to 45% of DMD patients. However, the efficacy of multi-exons skipping therapy is still controversial (Dzierlega and Yokota, 2020).
Gene-addition and gene-editing therapies are mainly restricted by the limitations of delivery with AAV vectors. Currently, AAV vectors are the most promising gene transfer tools due to their outstanding ability to target muscle (Wang et al., 2017). Unfortunately, AAV vectors have a limited carrying capacity and cannot deliver full-length DMD gene (Duan, 2018). One potential solution is to optimize AVV-based micro-dystrophin delivery therapies, which aim to restore the partial function of dystrophin. Another challenge for AAV-based therapies is an immunological risk, because high levels of AAV can lead to toxicity (Duan, 2018). The AAV vector with less immunotoxicity has been developed. AAVrh74 as a delivery vehicle has been found to be safe and tolerable in human studies, with minimal immune response (Mendell et al., 2009). Besides, the rAAVrh74.MHCK7.micro-dystrophin treatment has shown relatively low-risk adverse events in clinical studies (Mendell et al., 2020). However, some side effects, such as loss of appetite and vomiting, have still been observed in DMD patients (Nelson et al., 2017). And for gene-editing therapies, the effects of potential off-target events need to be investigated rigorously to ensure the long-term safety (Bengtsson et al., 2017; Min et al., 2019a).
Stem cells bring the promise for cell-based therapies due to their advantageous regeneration capability. Especially, the hiPSCs-based therapies, which can genetically correct and express functional dystrophin, hold the keys to treat DMD (Sun et al., 2020). However, many technical challenges and safety concerns still limit the clinical applications of cell-based therapies. Cultured satellite cells show limited transplantation efficiency and most satellite cells die after transplantation. Therefore, the delivery route needs to be optimized to obtain a higher muscle engraftment rate in vivo. Besides, the off-target effect of the gene-editing tools that used for hiPSCs construction, such as CRISPR/Cas9, is a big concern. Progress on overcoming these limitations is being made, including the identification of specific cell surface markers to separate the myogenic cells from the other types of cells, as well as the use of new generation CRISPR-based systems (Magli et al., 2017; Hicks et al., 2018).
To sum up, the restoration of dystrophin and the DAPC in DMD could prevent further muscle damage and slow disease progression theoretically. However, challenges such as tailoring to specific DMD mutations and achieving widespread delivery still need a long time to overcome.
Besides the dystrophin-targeted therapeutic strategies, great efforts have also been made to target the downstream pathogenic changes of dystrophin deficiency. The major advantage of these pharmacological strategies is that they are applicable to all DMD patients irrespective of their mutation type. Furthermore, most therapies introduced above have already been approved or under clinical studies for other diseases, which facilitates their clinical applications for DMD. Compared with gene-targeted and cell-based therapies, these molecular drugs with histories of clinical application show less unexplored safety problems. However, DMD is a multi-system disorder of obscure pathogenesis with some symptom complexes. It is desirable to have an insight into the molecular mechanisms of DMD in different organs/tissues and discover potential promising therapeutic targets.
Fibrosis has been increasingly accepted as the principal cause of muscle dysfunction in DMD (Desguerre et al., 2009), and it is an impediment for AVV-based gene delivery and stem cell delivery. Over the past few years, the molecular mechanisms underlying muscle fibrosis and the potential antifibrotic therapies have been studied. Most fibrosis-targeted therapeutic strategies exhibit promising results in pre-clinical experiments (Henderson et al., 2020). For example, the pro-fibrotic role of CTGF has been well-studied in several pathologies characterized by the development of fibrosis (Gonzalez and Brandan, 2019). Reduction of CTGF levels in mdx mice can significantly decrease fibrosis and improve skeletal muscle phenotype as well as functions. The CTGF-specific antibody, FG-3019, has shown promising anti-fibrosis effect in patients with idiopathic pulmonary fibrosis (IPF) and pancreatic cancer (Raghu et al., 2016). Furthermore, compared with the therapeutic strategies that target TGF-β, the therapeutic strategies targeting CTGF are considered safer since sustained systemic inhibition of TGF-β can result in side effects such as cardiac valve problems (Henderson et al., 2020). Besides, the TGF-β1 blockade can also induce carcinogenesis, owing to the role of TGF-β1 as an anti-proliferative mediator for most epithelial cell types (Henderson et al., 2020).
Most notably, even though some endpoints about muscle wasting (i.e., muscle mass) could be ameliorated with some of the tested drugs, most trials failed to show significant improvement in the defined clinical endpoints. More effective and safer potential targets for DMD are desired.
Many gene variants are contained in the human genome, and some special gene variants can interact with the disease-causing mutation and further modulate the phenotype of DMD (Bello and Pegoraro, 2019). The bioinformatic analysis could be an effective tool for the identification of potential genetic targets. In this study, 133 DMD samples and 116 normal control samples from multiple microarray datasets were selected for the identification of DEGs. However, there is no gene that could be consistently identified as DEG in all 12 datasets. Possible reasons for this include: (i) The patient sample might be collected at different stages of disease progression. (ii) The number of genes was low in some datasets such as GSE32720 which only contains 3266 genes. (iii) Stringent quality control criteria were implanted to detect and discard low-quality genes. A meta-analysis that could combine information from multiple datasets and increase the reliability and generalizability identified a robust meta-gene profile comprising 121 DEGs. Hub genes identification and enrichment analysis on the DEGs demonstrated that ECM formation, immune response, bone development exerted significant roles in DMD pathogenesis. Furthermore, there are also several limitations in this meta-analysis. Firstly, the result of this meta-analysis might be also affected by the relatively small sample size of the included studies. Studies with a larger sample size and cross-validation in different populations and ethnic groups are warranted. Secondly, the present study utilized the public database to process secondary data mining and conduct bioinformatic analysis, which lacked clinical verification.
Despite these limitations, our meta-analysis still provides an overview of the involved biomarkers and DEGs in the pathogenesis of DMD for further experimental validation or consideration. Among 10 hub genes identified from the meta-analysis, seven genes, including FN1, CTGF, SPP1, POSTN, THBS2, COL4A2, and TGF-β1, are involved in ECM formation, implying anti-fibrotic therapy might serve as a necessary and important way to treat DMD, in addition to gene and cell therapies, in the future.
SY, ZC, GZ, ZZ, BZ, and YY made contributions to the concept design of the article, acquisition, and analysis of the review. SY and ZC drafted the manuscript. ZC covered the meta-analysis. NZ and HJ interpreted the data for the meta-analysis. All the authors reviewed the manuscript and approved the final version of the manuscript.
This work was supported by the Hong Kong General Research Fund (14100218 and 14103420) and Hong Kong Theme-based Research Scheme (T12-201/20-R).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aartsma-Rus, A., Ginjaar, I. B., and Bushby, K. (2016). The importance of genetic diagnosis for Duchenne muscular dystrophy. J. Med. Genet. 53, 145–151. doi: 10.1136/jmedgenet-2015-103387
Acuña, M. J., Pessina, P., Olguin, H., Cabrera, D., Vio, C. P., Bader, M., et al. (2014). Restoration of muscle strength in dystrophic muscle by angiotensin-1-7 through inhibition of TGF-β signalling. Hum. Mol. Genet. 23, 1237–1249. doi: 10.1093/hmg/ddt514
Al Musaimi, O., Al Shaer, D., Albericio, F., and de la Torre, B. G. (2021). 2020 FDA TIDES (peptides and oligonucleotides) harvest. Pharmaceuticals (Basel) 14:145. doi: 10.3390/ph14020145
Allen, D. G., Whitehead, N. P., and Froehner, S. C. (2016). Absence of dystrophin disrupts skeletal muscle signaling: roles of ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol. Rev. 96, 253–305. doi: 10.1152/physrev.00007.2015
Allen, H. D., Flanigan, K. M., Thrush, P. T., Dvorchik, I., Yin, H., Canter, C., et al. (2013). A randomized, double-blind trial of lisinopril and losartan for the treatment of cardiomyopathy in duchenne muscular dystrophy. PLoS Curr. 5:6. doi: 10.1371/currents.md.2cc69a1dae4be7dfe2bcb420024ea865
Altamirano, F., López, J. R., Henríquez, C., Molinski, T., Allen, P. D., and Jaimovich, E. (2012). Increased resting intracellular calcium modulates NF-κB-dependent inducible nitric-oxide synthase gene expression in dystrophic mdx skeletal myotubes. J. Biol. Chem. 287, 20876–20887. doi: 10.1074/jbc.M112.344929
Al-Zaidy, S. A., Sahenk, Z., Rodino-Klapac, L. R., Kaspar, B., and Mendell, J. R. (2015). Follistatin gene therapy improves ambulation in becker muscular dystrophy. J. Neuromuscul. Dis. 2, 185–192. doi: 10.3233/JND-150083
Aravind, S., Ashley, B., Mannan, A., Ganapathy, A., Ramesh, K., Ramachandran, A., et al. (2019). Targeted sequencing of the DMD locus: a comprehensive diagnostic tool for all mutations. Indian J. Med. Res. 150, 282–289. doi: 10.4103/ijmr.IJMR_290_18
Asai, A., Sahani, N., Kaneki, M., Ouchi, Y., Martyn, J. A., and Yasuhara, S. E. (2007). Primary role of functional ischemia, quantitative evidence for the two-hit mechanism, and phosphodiesterase-5 inhibitor therapy in mouse muscular dystrophy. PLoS One 2:e806. doi: 10.1371/journal.pone.0000806
Asher, D. R., Thapa, K., Dharia, S. D., Khan, N., Potter, R. A., Rodino-Klapac, L. R., et al. (2020). Clinical development on the frontier: gene therapy for duchenne muscular dystrophy. Expert. Opin. Biol. Ther. 20, 263–274. doi: 10.1080/14712598.2020.1725469
Balduzzi, S., Rücker, G., and Schwarzer, G. (2019). How to perform a meta-analysis with R: a practical tutorial. Evid. Based Mental Health 22, 153–160.
Baldwin, A. S. Jr. (2001). Series introduction: the transcription factor NF-kappaB and human disease. J. Clin. Invest. 107, 3–6. doi: 10.1172/JCI11891
Banks, G. B., Judge, L. M., Allen, J. M., and Chamberlain, J. S. (2010). The polyproline site in hinge 2 influences the functional capacity of truncated dystrophins. PLoS Genet. 6:e1000958. doi: 10.1371/journal.pgen.1000958
Barabási, A. L., Gulbahce, N., and Loscalzo, J. (2011). Network medicine: a network-based approach to human disease. Nat. Rev. Genet. 12, 56–68. doi: 10.1038/nrg2918
Baradaran-Heravi, A., Niesser, J., Balgi, A. D., Choi, K., Zimmerman, C., South, A. P., et al. (2017). Gentamicin B1 is a minor gentamicin component with major nonsense mutation suppression activity. Proc. Natl. Acad. Sci. U.S.A. 114, 3479–3484. doi: 10.1073/pnas.1620982114
Barton, E. R., Morris, L., Musaro, A., Rosenthal, N., and Sweeney, H. L. (2002). Muscle-specific expression of insulin-like growth factor I counters muscle decline in mdx mice. J. Cell Biol. 157, 137–148. doi: 10.1083/jcb.200108071
Belanto, J. J., Mader, T. L., Eckhoff, M. D., Strandjord, D. M., Banks, G. B., Gardner, M. K., et al. (2014). Microtubule binding distinguishes dystrophin from utrophin. Proc. Natl. Acad. Sci. U.S.A. 111, 5723–5728. doi: 10.1073/pnas.1323842111
Bello, L., and Pegoraro, E. (2019). The “Usual Suspects”: genes for inflammation, fibrosis, regeneration, and muscle strength modify duchenne muscular dystrophy. J. Clin. Med. 8.
Belluscio, B., Beaverson, K., Garnier, N., Ryan, K., Moorehead, T., Yong, F., et al. (2021). “Safety and efficacy of PF-06939926 gene therapy in boys with Duchenne muscular dystrophy: update on data from the phase 1b study,” in Proceedings of the MDA Clinical and Scientific Conference 2021, (Rye, NY: Muscular Dystrophy Association’s (MDA)).
Bengtsson, N. E., Hall, J. K., Odom, G. L., Phelps, M. P., Andrus, C. R., Hawkins, R. D., et al. (2017). Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 8:14454. doi: 10.1038/ncomms16007
Bettica, P., Petrini, S., D’Oria, V., D’Amico, A., Catteruccia, M., Pane, M., et al. (2016). Histological effects of givinostat in boys with Duchenne muscular dystrophy. Neuromuscul. Disord. 26, 643–649. doi: 10.1016/j.nmd.2016.07.002
Bianchi, M. L., Mazzanti, A., Galbiati, E., Saraifoger, S., Dubini, A., Cornelio, F., et al. (2003). Bone mineral density and bone metabolism in Duchenne muscular dystrophy. Osteoporos. Int. 14, 761–767. doi: 10.1007/s00198-003-1443-y
Biggar, W. D., Harris, V. A., Eliasoph, L., and Alman, B. (2006). Long-term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscul. Disord. 16, 249–255. doi: 10.1016/j.nmd.2006.01.010
Birnkrant, D. J., Bushby, K., Bann, C. M., Alman, B. A., Apkon, S. D., Blackwell, A., et al. (2018). Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 17, 347–361. doi: 10.1016/S1474-4422(18)30025-5
Bish, L. T., Yarchoan, M., Sleeper, M. M., Gazzara, J. A., Morine, K. J., Acosta, P., et al. (2011). Chronic losartan administration reduces mortality and preserves cardiac but not skeletal muscle function in dystrophic mice. PLoS One 6:e20856. doi: 10.1371/journal.pone.0020856
Brescia, M., Chao, Y. C., Koschinski, A., Tomek, J., and Zaccolo, M. (2020). Multi-compartment, early disruption of cGMP and cAMP signalling in cardiac myocytes from the mdx model of duchenne muscular dystrophy. Int. J. Mol. Sci. 21:7056. doi: 10.3390/ijms21197056
Buckner, J. L., Bowden, S. A., and Mahan, J. D. (2015). Optimizing bone health in duchenne muscular dystrophy. Int. J. Endocrinol. 2015:928385. doi: 10.1155/2015/928385
Burns, D. P., Canavan, L., Rowland, J., O’Flaherty, R., Brannock, M., Drummond, S. E., et al. (2018). Recovery of respiratory function in mdx mice co-treated with neutralizing interleukin-6 receptor antibodies and urocortin-2. J. Physiol. 596, 5175–5197. doi: 10.1113/JP276954
Bush, E., Ward, C., Suchyna, T., Sacks, F., Blaustein, M., and Escolar, D. (2017). AT-300, a calcium modulator, improves muscle force production and decreases muscle degeneration in D2-mdx model of Duchenne muscular dystrophy. Neuromusc. Disord. 27:S192. doi: 10.1016/j.nmd.2017.06.358
Buyse, G. M., Van der Mieren, G., Erb, M., D’Hooge, J., Herijgers, P., Verbeken, E., et al. (2009). Long-term blinded placebo-controlled study of SNT-MC17/idebenone in the dystrophin deficient mdx mouse: cardiac protection and improved exercise performance. Eur. Heart J. 30, 116–124. doi: 10.1093/eurheartj/ehn406
Buyse, G. M., Voit, T., Schara, U., Straathof, C. S. M., D’Angelo, M. G., Bernert, G., et al. (2015). Efficacy of idebenone on respiratory function in patients with Duchenne muscular dystrophy not using glucocorticoids (DELOS): a double-blind randomised placebo-controlled phase 3 trial. Lancet 385, 1748–1757. doi: 10.1016/S0140-6736(15)60025-3
Cai, C., Masumiya, H., Weisleder, N., Matsuda, N., Nishi, M., Hwang, M., et al. (2009). MG53 nucleates assembly of cell membrane repair machinery. Nat. Cell Biol. 11, 56–64. doi: 10.1038/ncb1812
Cai, X., Liu, C., Zhang, T. N., Zhu, Y. W., Dong, X., and Xue, P. (2018). Down-regulation of FN1 inhibits colorectal carcinogenesis by suppressing proliferation, migration, and invasion. J. Cell. Biochem. 119, 4717–4728.
Chamberlain, J. R., and Chamberlain, J. S. (2017). Progress toward gene therapy for duchenne muscular dystrophy. Mol. Ther. 25, 1125–1131. doi: 10.1016/j.ymthe.2017.02.019
Chemello, F., Wang, Z., Li, H., McAnally, J. R., Liu, N., Bassel-Duby, R., et al. (2020). Degenerative and regenerative pathways underlying Duchenne muscular dystrophy revealed by single-nucleus RNA sequencing. Proc. Natl. Acad. Sci. U.S.A. 117, 29691–29701. doi: 10.1073/pnas.2018391117
Chen, L., Chen, C. X., Gan, X. T., Beier, N., Scholz, W., and Karmazyn, M. (2004). Inhibition and reversal of myocardial infarction-induced hypertrophy and heart failure by NHE-1 inhibition. Am. J. Physiol. Heart Circ. Physiol. 286, H381–H387. doi: 10.1152/ajpheart.00602.2003
Chen, W., Wu, X., Yan, X., Xu, A., Yang, A., and You, H. (2019). Multitranscriptome analyses reveal prioritized genes specifically associated with liver fibrosis progression independent of etiology. Am. J. Physiol. Gastrointest. Liver Physiol. 316, G744–G754. doi: 10.1152/ajpgi.00339.2018
Chen, Z., Zhang, N., Chu, H. Y., Yu, Y., Zhang, Z. K., Zhang, G., et al. (2020). Connective tissue growth factor: from molecular understandings to drug discovery. Front. Cell Dev. Biol. 8:593269.
Chicoine, L. G., Rodino-Klapac, L. R., Shao, G., Xu, R., Bremer, W. G., Camboni, M., et al. (2014). Vascular delivery of rAAVrh74.MCK.GALGT2 to the gastrocnemius muscle of the rhesus macaque stimulates the expression of dystrophin and laminin α2 surrogates. Mol. Ther. 22, 713–724. doi: 10.1038/mt.2013.246
Chin, C. H., Chen, S. H., Wu, H. H., Ho, C. W., Ko, M. T., and Lin, C. Y. (2014). cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 8 (Suppl. 4):S11. doi: 10.1186/1752-0509-8-S4-S11
Cirak, S., Arechavala-Gomeza, V., Guglieri, M., Feng, L., Torelli, S., Anthony, K., et al. (2011). Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet 378, 595–605. doi: 10.1016/S0140-6736(11)60756-3
Clemens, P. R., Rao, V. K., Connolly, A. M., Harper, A. D., Mah, J. K., Smith, E. C., et al. (2020). Safety, tolerability, and efficacy of viltolarsen in boys with duchenne muscular dystrophy amenable to exon 53 skipping: a phase 2 randomized clinical trial. JAMA Neurol. 77, 982–991. doi: 10.1001/jamaneurol.2020.1264
Cohn, R. D., van Erp, C., Habashi, J. P., Soleimani, A. A., Klein, E. C., Lisi, M. T., et al. (2007). Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat. Med. 13, 204–210. doi: 10.1038/nm1536
Colussi, C., Mozzetta, C., Gurtner, A., Illi, B., Rosati, J., Straino, S., et al. (2008). HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc. Natl. Acad. Sci. U.S.A. 105, 19183–19187. doi: 10.1073/pnas.0805514105
Cozzoli, A., Capogrosso, R. F., Sblendorio, V. T., Dinardo, M. M., Jagerschmidt, C., Namour, F., et al. (2013). GLPG0492, a novel selective androgen receptor modulator, improves muscle performance in the exercised-mdx mouse model of muscular dystrophy. Pharmacol. Res. 72, 9–24. doi: 10.1016/j.phrs.2013.03.003
Crisafulli, S., Sultana, J., Fontana, A., Salvo, F., Messina, S., and Trifirò, G. (2020). Global epidemiology of Duchenne muscular dystrophy: an updated systematic review and meta-analysis. Orphanet. J. Rare Dis. 15:141. doi: 10.1186/s13023-020-01430-8
Cynthia Martin, F., Hiller, M., Spitali, P., Oonk, S., Dalebout, H., Palmblad, M., et al. (2014). Fibronectin is a serum biomarker for Duchenne muscular dystrophy. Proteomics Clin. Appl. 8, 269–278. doi: 10.1002/prca.201300072
De Oliveira Moreira, D., Pereira, J. A., Taniguti, A. P., Matsumura, C. Y., Ramos, L. A., Areas, M. A., et al. (2013). Suramin attenuates dystrophin-deficient cardiomyopathy in the mdx mouse model of duchenne muscular dystrophy. Muscle Nerve 48, 911–919. doi: 10.1002/mus.23858
De Oliveira Moreira, D., Santo Neto, H., and Marques, M. J. (2017). P2Y(2) purinergic receptors are highly expressed in cardiac and diaphragm muscles of mdx mice, and their expression is decreased by suramin. Muscle Nerve 55, 116–121. doi: 10.1002/mus.25199
De Pasquale, L., D’Amico, A., Verardo, M., Petrini, S., Bertini, E., and De Benedetti, F. (2012). Increased muscle expression of interleukin-17 in Duchenne muscular dystrophy. Neurology 78, 1309–1314. doi: 10.1212/WNL.0b013e3182518302
de Senzi Moraes Pinto, R., Ferretti, R., Moraes, L. H., Neto, H. S., Marques, M. J., and Minatel, E. (2013). N-acetylcysteine treatment reduces TNF-α levels and myonecrosis in diaphragm muscle of mdx mice. Clin. Nutr. 32, 472–475. doi: 10.1016/j.clnu.2012.06.001
Desguerre, I., Mayer, M., Leturcq, F., Barbet, J. P., Gherardi, R. K., and Christov, C. (2009). Endomysial fibrosis in Duchenne muscular dystrophy: a marker of poor outcome associated with macrophage alternative activation. J. Neuropathol. Exp. Neurol. 68, 762–773. doi: 10.1097/NEN.0b013e3181aa31c2
Dombernowsky, N. W., Ölmestig, J. N. E., Witting, N., and Kruuse, C. (2018). Role of neuronal nitric oxide synthase (nNOS) in Duchenne and Becker muscular dystrophies – Still a possible treatment modality? Neuromuscul. Disord. 28, 914–926. doi: 10.1016/j.nmd.2018.09.001
Donovan, J. M., Zimmer, M., Offman, E., Grant, T., and Jirousek, M. (2017). A novel NF-κB Inhibitor, edasalonexent (CAT-1004), in development as a disease-modifying treatment for patients with duchenne muscular dystrophy: phase 1 safety, pharmacokinetics, and pharmacodynamics in adult subjects. J. Clin. Pharmacol. 57, 627–639. doi: 10.1002/jcph.842
Dorchies, O. M., Reutenauer-Patte, J., Dahmane, E., Ismail, H. M., Petermann, O., Patthey- Vuadens, O., et al. (2013). The anticancer drug tamoxifen counteracts the pathology in a mouse model of duchenne muscular dystrophy. Am. J. Pathol. 182, 485–504. doi: 10.1016/j.ajpath.2012.10.018
Duan, D. (2011). Duchenne muscular dystrophy gene therapy: lost in translation? Res. Rep. Biol. 2011, 31–42. doi: 10.2147/RRB.S13463
Duan, D. (2018). Systemic AAV micro-dystrophin gene therapy for duchenne muscular dystrophy. Mol. Ther. 26, 2337–2356. doi: 10.1016/j.ymthe.2018.07.011
Duan, D., Hakim, C. H., Ambrosio, C. E., Smith, B. F., and Sweeney, H. L. (2015). Early loss of ambulation is not a representative clinical feature in Duchenne muscular dystrophy dogs: remarks on the article of Barthélémy et al. Dis. Model Mech. 8, 193–194. doi: 10.1242/dmm.019216
Dzierlega, K., and Yokota, T. (2020). Optimization of antisense-mediated exon skipping for Duchenne muscular dystrophy. Gene Ther. 27, 407–416. doi: 10.1038/s41434-020-0156-6
Echevarría, L., Aupy, P., and Goyenvalle, A. (2018). Exon-skipping advances for Duchenne muscular dystrophy. Hum. Mol. Genet. 27, R163–R172. doi: 10.1093/hmg/ddy171
Echigoya, Y., Aoki, Y., Miskew, B., Panesar, D., Touznik, A., Nagata, T., et al. (2015). Long-term efficacy of systemic multiexon skipping targeting dystrophin exons 45-55 with a cocktail of vivo-morpholinos in mdx52 mice. Mol. Ther. Nucleic Acids 4:e225. doi: 10.1038/mtna.2014.76
Ermolova, N. V., Martinez, L., Vetrone, S. A., Jordan, M. C., Roos, K. P., Sweeney, H. L., et al. (2014). Long-term administration of the TNF blocking drug remicade (cV1q) to mdx mice reduces skeletal and cardiac muscle fibrosis, but negatively impacts cardiac function. Neuromuscul. Disord. 24, 583–595. doi: 10.1016/j.nmd.2014.04.006
Fauconnier, J., Thireau, J., Reiken, S., Cassan, C., Richard, S., Matecki, S., et al. (2010). Leaky RyR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. U.S.A. 107, 1559–1564. doi: 10.1073/pnas.0908540107
Feng, W., Dai, Y., Mou, L., Cooper, D. K., Shi, D., and Cai, Z. (2015). The potential of the combination of CRISPR/Cas9 and pluripotent stem cells to provide human organs from chimaeric pigs. Int. J. Mol. Sci. 16, 6545–6556. doi: 10.3390/ijms16036545
Finkel, R. S., Flanigan, K. M., Wong, B., Bönnemann, C., Sampson, J., Sweeney, H. L., et al. (2013). Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dystrophy. PLoS One 8:e81302. doi: 10.1371/journal.pone.0081302
Frank, D. E., Schnell, F. J., Akana, C., El-Husayni, S. H., Desjardins, C. A., Morgan, J., et al. (2020). Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology 94, e2270–e2282. doi: 10.1212/WNL.0000000000009233
Garbincius, J. F., and Michele, D. E. (2015). Dystrophin-glycoprotein complex regulates muscle nitric oxide production through mechanoregulation of AMPK signaling. Proc. Natl. Acad. Sci. U.S.A. 112, 13663–13668. doi: 10.1073/pnas.1512991112
Ghaleh, B., Barthélemy, I., Wojcik, J., Sambin, L., Bizé, A., Hittinger, L., et al. (2020). Protective effects of rimeporide on left ventricular function in golden retriever muscular dystrophy dogs. Int. J. Cardiol. 312, 89–95. doi: 10.1016/j.ijcard.2020.03.031
Gidaro, T., Servais, L., Previtali, S., Zambon, A., Pitchforth, J., Maresh, K., et al. (2017). P.398 - Rimeporide: safety, tolerability and pharmacokinetic results from a phase Ib study in DMD boys as well as exploratory biomarkers. Neuromuscul. Disord. 27, S215–S216. doi: 10.1016/j.nmd.2017.06.438
Gloss, D., Moxley, R. T. III, Ashwal, S., and Oskoui, M. (2016). Practice guideline update summary: corticosteroid treatment of duchenne muscular dystrophy: report of the guideline development subcommittee of the american academy of neurology. Neurology 86, 465–472. doi: 10.1212/WNL.0000000000002337
Gocheva, V., Schmidt, S., Orsini, A. L., Hafner, P., Schaedelin, S., Weber, P., et al. (2019). Psychosocial adjustment and parental stress in duchenne muscular dystrophy. Eur. J. Paediatr. Neurol. 23, 832–841. doi: 10.1016/j.ejpn.2019.09.008
Gonzalez, D., and Brandan, E. (2019). CTGF/CCN2 from skeletal muscle to nervous system: impact on neurodegenerative diseases. Mol. Neurobiol. 56, 5911–5916. doi: 10.1007/s12035-019-1490-9
Gregorevic, P., Plant, D. R., Leeding, K. S., Bach, L. A., and Lynch, G. S. (2002). Improved contractile function of the mdx dystrophic mouse diaphragm muscle after insulin-like growth factor-I administration. Am. J. Pathol. 161, 2263–2272. doi: 10.1016/S0002-9440(10)64502-6
Grounds, M. D., and Torrisi, J. (2004). Anti-TNFalpha (Remicade) therapy protects dystrophic skeletal muscle from necrosis. FASEB J. 18, 676–682. doi: 10.1096/fj.03-1024com
Guiraud, S., and Davies, K. E. (2017). Pharmacological advances for treatment in Duchenne muscular dystrophy. Curr. Opin. Pharmacol. 34, 36–48. doi: 10.1016/j.coph.2017.04.002
Guiraud, S., Edwards, B., Babbs, A., Squire, S. E., Berg, A., Moir, L., et al. (2019). The potential of utrophin and dystrophin combination therapies for Duchenne muscular dystrophy. Hum. Mol. Genet. 28, 2189–2200. doi: 10.1093/hmg/ddz049
Guo, C., Willem, M., Werner, A., Raivich, G., Emerson, M., Neyses, L., et al. (2006). Absence of alpha 7 integrin in dystrophin-deficient mice causes a myopathy similar to Duchenne muscular dystrophy. Hum. Mol. Genet. 15, 989–998. doi: 10.1093/hmg/ddl018
Hafner, P., Bonati, U., Erne, B., Schmid, M., Rubino, D., Pohlman, U., et al. (2016). Improved muscle function in duchenne muscular dystrophy through L-arginine and metformin: an investigator-initiated, open-label, single-center, proof-of-concept-study. PLoS One 11:e0147634. doi: 10.1371/journal.pone.0147634
Hafner, P., Bonati, U., Klein, A., Rubino, D., Gocheva, V., Schmidt, S., et al. (2019). Effect of combination l-citrulline and metformin treatment on motor function in patients with duchenne muscular dystrophy: a randomized clinical trial. JAMA Netw. Open 2:e1914171. doi: 10.1001/jamanetworkopen.2019.14171
Haidet, A. M., Rizo, L., Handy, C., Umapathi, P., Eagle, A., Shilling, C., et al. (2008). Long-term enhancement of skeletal muscle mass and strength by single gene administration of myostatin inhibitors. Proc. Natl. Acad. Sci. U.S.A. 105, 4318–4322. doi: 10.1073/pnas.0709144105
Hamilton, D. W. (2008). Functional role of periostin in development and wound repair: implications for connective tissue disease. J. Cell Commun. Signal. 2:9.
Hammers, D. W., Hart, C. C., Patsalos, A., Matheny, M. K., Wright, L. A., Nagy, L., et al. (2020). Glucocorticoids counteract hypertrophic effects of myostatin inhibition in dystrophic muscle. JCI Insight 5, e133276. doi: 10.1172/jci.insight.133276
Hammers, D. W., Sleeper, M. M., Forbes, S. C., Coker, C. C., Jirousek, M. R., Zimmer, M., et al. (2016). Disease-modifying effects of orally bioavailable NF-κB inhibitors in dystrophin-deficient muscle. JCI Insight 1:e90341. doi: 10.1172/jci.insight.90341
Han, H. Q., and Mitch, W. E. (2011). Targeting the myostatin signaling pathway to treat muscle wasting diseases. Curr. Opin. Support Palliat Care 5, 334–341. doi: 10.1097/SPC.0b013e32834bddf9
Harcourt, L. J., Schertzer, J. D., Ryall, J. G., and Lynch, G. S. (2007). Low dose formoterol administration improves muscle function in dystrophic mdx mice without increasing fatigue. Neuromuscul. Disord. 17, 47–55. doi: 10.1016/j.nmd.2006.08.012
Hawker, G. A., Ridout, R., Harris, V. A., Chase, C. C., Fielding, L. J., and Biggar, W. D. (2005). Alendronate in the treatment of low bone mass in steroid-treated boys with Duchennes muscular dystrophy. Arch. Phys. Med. Rehabil. 86, 284–288. doi: 10.1016/j.apmr.2004.04.021
Henderson, N. C., Rieder, F., and Wynn, T. A. (2020). Fibrosis: from mechanisms to medicines. Nature 587, 555–566. doi: 10.1038/s41586-020-2938-9
Hicks, M. R., Hiserodt, J., Paras, K., Fujiwara, W., Eskin, A., Jan, M., et al. (2018). ERBB3 and NGFR mark a distinct skeletal muscle progenitor cell in human development and hPSCs. Nat. Cell Biol. 20, 46–57. doi: 10.1038/s41556-017-0010-2
Hnia, K., Gayraud, J., Hugon, G., Ramonatxo, M., De La Porte, S., Matecki, S., et al. (2008). L-arginine decreases inflammation and modulates the nuclear factor-kappaB/matrix metalloproteinase cascade in mdx muscle fibers. Am. J. Pathol. 172, 1509–1519. doi: 10.2353/ajpath.2008.071009
Hochheiser, K., Kueh, A. J., Gebhardt, T., and Herold, M. J. (2018). CRISPR/Cas9: a tool for immunological research. Eur. J. Immunol. 48, 576–583. doi: 10.1002/eji.201747131
Hoffman, E., Monaco, A., Feener, C., and Kunkel, L. (1987). Conservation of the Duchenne muscular dystrophy gene in mice and humans. Science 238, 347–350. doi: 10.1126/science.3659917
Hoffman, E. P., Schwartz, B. D., Mengle-Gaw, L. J., Smith, E. C., Castro, D., Mah, J. K., et al. (2019). Vamorolone trial in Duchenne muscular dystrophy shows dose-related improvement of muscle function. Neurology 93, e1312–e1323. doi: 10.1212/WNL.0000000000008168
Hollinger, K., and Chamberlain, J. S. (2015). Viral vector-mediated gene therapies. Curr. Opin. Neurol. 28, 522–527. doi: 10.1097/WCO.0000000000000241
Hörster, I., Weigt-Usinger, K., Carmann, C., Chobanyan-Jürgens, K., Köhler, C., Schara, U., et al. (2015). The L-arginine/NO pathway and homoarginine are altered in Duchenne muscular dystrophy and improved by glucocorticoids. Amino Acids 47, 1853–1863. doi: 10.1007/s00726-015-2018-x
Huang, P., Zhao, X. S., Fields, M., Ransohoff, R. M., and Zhou, L. (2009). Imatinib attenuates skeletal muscle dystrophy in mdx mice. FASEB J. 23, 2539–2548. doi: 10.1096/fj.09-129833
Huebner, K. D., Jassal, D. S., Halevy, O., Pines, M., and Anderson, J. E. (2008). Functional resolution of fibrosis in mdx mouse dystrophic heart and skeletal muscle by halofuginone. Am. J. Physiol. Heart Circ. Physiol. 294, H1550–H1561. doi: 10.1152/ajpheart.01253.2007
Hsu, D. K., Chen, H. Y., and Liu, F. T. (2009). Galectin-3 regulates T-cell functions. Immunol. Rev. 230, 114–127.
Iftikhar, M., Frey, J., Shohan, M. J., Malek, S., and Mousa, S. A. (2020). Current and emerging therapies for Duchenne muscular dystrophy and spinal muscular atrophy. Pharmacol. Ther. 220:107719. doi: 10.1016/j.pharmthera.2020.107719
Inagaki, K., Fuess, S., Storm, T. A., Gibson, G. A., McTiernan, C. F., Kay, M. A., et al. (2006). Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol. Ther. 14, 45–53. doi: 10.1016/j.ymthe.2006.03.014
Ishikawa, K., Yoshida, S., Kobayashi, Y., Zhou, Y., Nakama, T., Nakao, S., et al. (2015). Microarray analysis of gene expression in fibrovascular membranes excised from patients with proliferative diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 56, 932–946. doi: 10.1167/iovs.14-15589
Ito, T., Ogawa, R., Uezumi, A., Ohtani, T., Watanabe, Y., Tsujikawa, K., et al. (2013). Imatinib attenuates severe mouse dystrophy and inhibits proliferation and fibrosis-marker expression in muscle mesenchymal progenitors. Neuromuscul. Disord. 23, 349–356. doi: 10.1016/j.nmd.2012.10.025
Ji, G., Sun, S., Wu, H., Chen, X., Cui, H., Tang, J., et al. (2020). Effect of AAV9-hIGF-1 on inflammatory reaction in mdx mice and its mechanism. Am. J. Transl. Res. 12, 4488–4497. doi: 10.21037/tcr-19-2588
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. doi: 10.1126/science.1225829
Jones, A., Hwang, D. J., Narayanan, R., Miller, D. D., and Dalton, J. T. (2010). Effects of a novel selective androgen receptor modulator on dexamethasone-induced and hypogonadism-induced muscle atrophy. Endocrinology 151, 3706–3719. doi: 10.1210/en.2010-0150
Jørgensen, L. H., Blain, A., Greally, E., Laval, S. H., Blamire, A. M., Davison, B. J., et al. (2011). Long-term blocking of calcium channels in mdx mice results in differential effects on heart and skeletal muscle. Am. J. Pathol. 178, 273–283. doi: 10.1016/j.ajpath.2010.11.027
Karimian, A., Azizian, K., Parsian, H., Rafieian, S., Shafiei-Irannejad, V., Kheyrollah, M., et al. (2019). CRISPR/Cas9 technology as a potent molecular tool for gene therapy. J. Cell Physiol. 234, 12267–12277. doi: 10.1002/jcp.27972
Kemaladewi, D. U., Pasteuning, S., van der Meulen, J. W., van Heiningen, S. H., van Ommen, G. J., Ten Dijke, P., et al. (2014). Targeting TGF-β signaling by antisense oligonucleotide-mediated knockdown of TGF-β Type I receptor. Mol. Ther. Nucleic Acids 3:e156. doi: 10.1038/mtna.2014.7
Khairallah, M., Khairallah, R. J., Young, M. E., Allen, B. G., Gillis, M. A., Danialou, G., et al. (2008). Sildenafil and cardiomyocyte-specific cGMP signaling prevent cardiomyopathic changes associated with dystrophin deficiency. Proc. Natl. Acad. Sci. U.S.A. 105, 7028–7033. doi: 10.1073/pnas.0710595105
Kinali, M., Arechavala-Gomeza, V., Feng, L., Cirak, S., Hunt, D., Adkin, C., et al. (2009). Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 8, 918–928. doi: 10.1016/S1474-4422(09)70211-X
Kishimoto, T., Pearse, R. V. II, Lin, C. R., and Rosenfeld, M. G. (1995). A sauvagine/corticotropin-releasing factor receptor expressed in heart and skeletal muscle. Proc. Natl. Acad. Sci. U.S.A. 92, 1108–1112. doi: 10.1073/pnas.92.4.1108
Klingler, W., Jurkat-Rott, K., Lehmann-Horn, F., and Schleip, R. (2012). The role of fibrosis in Duchenne muscular dystrophy. Acta Myol. 31, 184–195.
Kobayashi, Y. M., Rader, E. P., Crawford, R. W., Iyengar, N. K., Thedens, D. R., Faulkner, J. A., et al. (2008). Sarcolemma-localized nNOS is required to maintain activity after mild exercise. Nature 456, 511–515. doi: 10.1038/nature07414
Kole, R., and Krieg, A. M. (2015). Exon skipping therapy for Duchenne muscular dystrophy. Adv. Drug Deliv. Rev. 87, 104–107. doi: 10.1016/j.addr.2015.05.008
Komaki, H., Maegaki, Y., Matsumura, T., Shiraishi, K., Awano, H., Nakamura, A., et al. (2020). Early phase 2 trial of TAS-205 in patients with Duchenne muscular dystrophy. Ann. Clin. Transl. Neurol. 7, 181–190. doi: 10.1002/acn3.50978
Kramerova, I., Kumagai-Cresse, C., Ermolova, N., Mokhonova, E., Marinov, M., Capote, J., et al. (2019). Spp1 (osteopontin) promotes TGFβ processing in fibroblasts of dystrophin-deficient muscles through matrix metalloproteinases. Hum. Mol. Genet. 28, 3431–3442. doi: 10.1093/hmg/ddz181
Kupatt, C., Windisch, A., Moretti, A., Wolf, E., Wurst, W., and Walter, M. C. (2021). Genome editing for Duchenne muscular dystrophy: a glimpse of the future? Gene Therapy 28, 1–7.
Lai, Y., Thomas, G. D., Yue, Y., Yang, H. T., Li, D., Long, C., et al. (2009). Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J. Clin. Invest. 119, 624–635. doi: 10.1172/JCI36612
Lamort, A. S., Giopanou, I., Psallidas, I., and Stathopoulos, G. T. (2019). Osteopontin as a link between inflammation and cancer: the thorax in the spotlight. Cells 8.
Langley, B., Thomas, M., Bishop, A., Sharma, M., Gilmour, S., and Kambadur, R. (2002). Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J. Biol. Chem. 277, 49831–49840. doi: 10.1074/jbc.M204291200
Law, M. L., Cohen, H., Martin, A. A., Angulski, A. B. B., and Metzger, J. M. (2020). Dysregulation of calcium handling in duchenne muscular dystrophy-associated dilated cardiomyopathy: mechanisms and experimental therapeutic strategies. J. Clin. Med. 9:520. doi: 10.3390/jcm9020520
Leborgne, C., Latournerie, V., Boutin, S., Desgue, D., Quéré, A., Pignot, E., et al. (2019). Prevalence and long-term monitoring of humoral immunity against adeno-associated virus in Duchenne Muscular Dystrophy patients. Cell Immunol. 342:103780. doi: 10.1016/j.cellimm.2018.03.004
Lee, E. M., Kim, D. Y., Kim, A. Y., Lee, E. J., Kim, S. H., Lee, M. M., et al. (2015). Chronic effects of losartan on the muscles and the serologic profiles of mdx mice. Life Sci. 143, 35–42. doi: 10.1016/j.lfs.2015.10.023
Lek, A., Zhang, Y., Woodman, K. G., Huang, S., DeSimone, A. M., Cohen, J., et al. (2020). Applying genome-wide CRISPR-Cas9 screens for therapeutic discovery in facioscapulohumeral muscular dystrophy. Sci. Transl. Med. 12:eaay0271. doi: 10.1126/scitranslmed.aay0271
Leung, D. G., Herzka, D. A., Thompson, W. R., He, B., Bibat, G., Tennekoon, G., et al. (2014). Sildenafil does not improve cardiomyopathy in Duchenne/Becker muscular dystrophy. Ann. Neurol. 76, 541–549. doi: 10.1002/ana.24214
Li, D., Bareja, A., Judge, L., Yue, Y., Lai, Y., Fairclough, R., et al. (2010). Sarcolemmal nNOS anchoring reveals a qualitative difference between dystrophin and utrophin. J. Cell Sci. 123, 2008–2013. doi: 10.1242/jcs.064808
Lim, K. R., Maruyama, R., and Yokota, T. (2017). Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Devel. Ther. 11, 533–545. doi: 10.2147/DDDT.S97635
Long, C., Amoasii, L., Mireault, A. A., McAnally, J. R., Li, H., Sanchez-Ortiz, E., et al. (2016). Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 351, 400–403. doi: 10.1126/science.aad5725
Long, C., McAnally, J. R., Shelton, J. M., Mireault, A. A., Bassel-Duby, R., and Olson, E. N. (2014). Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science 345, 1184–1188. doi: 10.1126/science.1254445
Lorenzo, J. (2016). “Chapter 9 – The effects of immune cell products (cytokines and hematopoietic cell growth factors) on bone cells,” in Osteoimmunology, 2nd Edn, eds J. Lorenzo, M. C. Horowitz, Y. Choi, H. Takayanagi, and G. Schett (San Diego, CA: Academic Press), 143–167.
Loro, E., Sengupta, K., Bogdanovich, S., Whig, K., Schultz, D. C., Huryn, D. M., et al. (2020). High-throughput identification of post-transcriptional utrophin up-regulators for Duchenne muscle dystrophy (DMD) therapy. Sci. Rep. 10:2132. doi: 10.1038/s41598-020-60885-8
Loscertales, M., Nicolaou, F., Jeanne, M., Longoni, M., Gould, D. B., Sun, Y., et al. (2016). Type IV collagen drives alveolar epithelial-endothelial association and the morphogenetic movements of septation. BMC Biol. 14:59.
Lynch, G. S., Hinkle, R. T., and Faulkner, J. A. (2000). Power output of fast and slow skeletal muscles of mdx (dystrophic) and control mice after clenbuterol treatment. Exp. Physiol. 85, 295–299. doi: 10.1111/j.1469-445X.2000.02018.x
Magli, A., Incitti, T., Kiley, J., Swanson, S. A., Darabi, R., Rinaldi, F., et al. (2017). PAX7 Targets, CD54, Integrin α9β1, and SDC2, Allow Isolation of Human ESC/iPSC-Derived Myogenic Progenitors. Cell Rep. 19, 2867–2877. doi: 10.1016/j.celrep.2017.06.005
Mah, J. K. (2016). Current and emerging treatment strategies for Duchenne muscular dystrophy. Neuropsychiatr. Dis. Treat. 12, 1795–1807. doi: 10.2147/NDT.S93873
Malik, V., Rodino-Klapac, L. R., Viollet, L., Wall, C., King, W., Al-Dahhak, R., et al. (2010). Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann. Neurol. 67, 771–780. doi: 10.1002/ana.22024
Manning, J., Buckley, M. M., O’Halloran, K. D., and O’Malley, D. (2017). Combined XIL-6R and urocortin-2 treatment restores MDX diaphragm muscle force. Muscle Nerve 56, E134–E140. doi: 10.1002/mus.25644
Matthews, E., Brassington, R., Kuntzer, T., Jichi, F., and Manzur, A. Y. (2016). Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database Syst. Rev. 5:Cd003725. doi: 10.1002/14651858.CD003725.pub4
Mázala, D. A., Novak, J. S., Hogarth, M. W., Nearing, M., Adusumalli, P., Tully, C. B., et al. (2020). TGF-β-driven muscle degeneration and failed regeneration underlie disease onset in a DMD mouse model. JCI Insight 5:e135703. doi: 10.1172/jci.insight.135703
McDonald, C. M., Campbell, C., Torricelli, R. E., Finkel, R. S., Flanigan, K. M., Goemans, N., et al. (2017). Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390, 1489–1498. doi: 10.1016/S0140-6736(17)31611-2
McGreevy, J. W., Hakim, C. H., McIntosh, M. A., and Duan, D. (2015). Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis. Model Mech. 8, 195–213. doi: 10.1242/dmm.018424
McKenna, M. J., Medved, I., Goodman, C. A., Brown, M. J., Bjorksten, A. R., Murphy, K. T., et al. (2006). N-acetylcysteine attenuates the decline in muscle Na+,K+-pump activity and delays fatigue during prolonged exercise in humans. J. Physiol. 576, 279–288. doi: 10.1113/jphysiol.2006.115352
Mendell, J. R., Rodino-Klapac, L. R., Rosales-Quintero, X., Kota, J., Coley, B. D., Galloway, G., et al. (2009). Limb-girdle muscular dystrophy type 2D gene therapy restores alpha-sarcoglycan and associated proteins. Ann. Neurol. 66, 290–297. doi: 10.1002/ana.21732
Mendell, J. R., Sahenk, Z., Lehman, K., Nease, C., Lowes, L. P., Miller, N. F., et al. (2020). Assessment of systemic delivery of rAAVrh74.MHCK7.micro-dystrophin in children with duchenne muscular dystrophy: a nonrandomized controlled trial. JAMA Neurol. 77, 1122–1131. doi: 10.1001/jamaneurol.2020.1484
Mendell, J. R., Sahenk, Z., Malik, V., Gomez, A. M., Flanigan, K. M., Lowes, L. P., et al. (2015). A phase 1/2a follistatin gene therapy trial for becker muscular dystrophy. Mol. Ther. 23, 192–201. doi: 10.1038/mt.2014.200
Meng, J., Chun, S., Asfahani, R., Lochmüller, H., Muntoni, F., and Morgan, J. (2014). Human skeletal muscle-derived CD133(+) cells form functional satellite cells after intramuscular transplantation in immunodeficient host mice. Mol. Ther. 22, 1008–1017. doi: 10.1038/mt.2014.26
Meng, J., Muntoni, F., and Morgan, J. (2018). CD133+ cells derived from skeletal muscles of Duchenne muscular dystrophy patients have a compromised myogenic and muscle regenerative capability. Stem Cell Res. 30, 43–52. doi: 10.1016/j.scr.2018.05.004
Meyers, T. A., and Townsend, D. (2019). Cardiac pathophysiology and the future of cardiac therapies in duchenne muscular dystrophy. Int. J. Mol. Sci. 20:4098. doi: 10.3390/ijms20174098
Mijares, A., Altamirano, F., Kolster, J., Adams, J. A., and López, J. R. (2014). Age-dependent changes in diastolic Ca(2+) and Na(+) concentrations in dystrophic cardiomyopathy: role of Ca(2+) entry and IP3. Biochem. Biophys. Res. Commun. 452, 1054–1059. doi: 10.1016/j.bbrc.2014.09.045
Min, Y. L., Bassel-Duby, R., and Olson, E. N. (2019a). CRISPR correction of duchenne muscular dystrophy. Annu. Rev. Med. 70, 239–255. doi: 10.1146/annurev-med-081117-010451
Min, Y. L., Li, H., Rodriguez-Caycedo, C., Mireault, A. A., Huang, J., Shelton, J. M., et al. (2019b). CRISPR-Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci. Adv. 5:eaav4324. doi: 10.1126/sciadv.aav4324
Mitrpant, C., Fletcher, S., Iversen, P. L., and Wilton, S. D. (2009). By-passing the nonsense mutation in the 4 CV mouse model of muscular dystrophy by induced exon skipping. J. Gene Med. 11, 46–56. doi: 10.1002/jgm.1265
Mizobuti, D. S., Fogaça, A. R., Moraes, F., Moraes, L. H. R., Mâncio, R. D., Hermes, T. A., et al. (2019). Coenzyme Q10 supplementation acts as antioxidant on dystrophic muscle cells. Cell Stress Chaperones 24, 1175–1185. doi: 10.1007/s12192-019-01039-2
Monaco, A. P., Neve, R. L., Colletti-Feener, C., Bertelson, C. J., Kurnit, D. M., and Kunkel, L. M. (1986). Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature 323, 646–650. doi: 10.1038/323646a0
Mondragon-Gonzalez, R., and Perlingeiro, R. C. R. (2018). Recapitulating muscle disease phenotypes with myotonic dystrophy 1 induced pluripotent stem cells: a tool for disease modeling and drug discovery. Dis. Model Mech. 11:dmm034728. doi: 10.1242/dmm.034728
Morales, M. G., Acuña, M. J., Cabrera, D., Goldschmeding, R., and Brandan, E. (2018). The pro-fibrotic connective tissue growth factor (CTGF/CCN2) correlates with the number of necrotic-regenerative foci in dystrophic muscle. J. Cell Commun. Signal. 12, 413–421. doi: 10.1007/s12079-017-0409-3
Morales, M. G., Gutierrez, J., Cabello-Verrugio, C., Cabrera, D., Lipson, K. E., Goldschmeding, R., et al. (2013). Reducing CTGF/CCN2 slows down mdx muscle dystrophy and improves cell therapy. Hum. Mol. Genet. 22, 4938–4951. doi: 10.1093/hmg/ddt352
Nabirotchkin, S., Peluffo, A. E., Rinaudo, P., Yu, J., Hajj, R., and Cohen, D. (2020). Next-generation drug repurposing using human genetics and network biology. Curr. Opin. Pharmacol. 51, 78–92. doi: 10.1016/j.coph.2019.12.004
Nagy, S., Hafner, P., Schmidt, S., Rubino-Nacht, D., Schädelin, S., Bieri, O., et al. (2019). Tamoxifen in Duchenne muscular dystrophy (TAMDMD): study protocol for a multicenter, randomized, placebo-controlled, double-blind phase 3 trial. Trials 20:637. doi: 10.1186/s13063-019-3740-6
Nakagawa, T., Takeuchi, A., Kakiuchi, R., Lee, T., Yagi, M., Awano, H., et al. (2013). A prostaglandin D2 metabolite is elevated in the urine of Duchenne muscular dystrophy patients and increases further from 8 years old. Clin. Chim. Acta 423, 10–14. doi: 10.1016/j.cca.2013.03.031
Namgoong, J. H., and Bertoni, C. (2016). Clinical potential of ataluren in the treatment of Duchenne muscular dystrophy. Degener. Neurol. Neuromuscul. Dis. 6, 37–48. doi: 10.2147/DNND.S71808
Nelson, C. E., Hakim, C. H., Ousterout, D. G., Thakore, P. I., Moreb, E. A., Castellanos Rivera, R. M., et al. (2016). In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 351, 403–407. doi: 10.1126/science.aad5143
Nelson, C. E., Robinson-Hamm, J. N., and Gersbach, C. A. (2017). Genome engineering: a new approach to gene therapy for neuromuscular disorders. Nat. Rev. Neurol. 13, 647–661. doi: 10.1038/nrneurol.2017.126
Ng, M. Y., Li, H., Ghelfi, M. D., Goldman, Y. E., and Cooperman, B. S. (2021). Ataluren and aminoglycosides stimulate read-through of nonsense codons by orthogonal mechanisms. Proc. Natl. Acad. Sci. U.S.A. 118:e2020599118. doi: 10.1073/pnas.2020599118
Nguyen-Tran, D. H., Hait, N. C., Sperber, H., Qi, J., Fischer, K., Ieronimakis, N., et al. (2014). Molecular mechanism of sphingosine-1-phosphate action in Duchenne muscular dystrophy. Dis. Model. Mech. 7, 41–54. doi: 10.1242/dmm.013631
Niks, E. H., and Aartsma-Rus, A. (2017). Exon skipping: a first in class strategy for Duchenne muscular dystrophy. Expert. Opin. Biol. Ther. 17, 225–236. doi: 10.1080/14712598.2017.1271872
Norwood, S., Liao, J., Hammock, B. D., and Yang, G. Y. (2010). Epoxyeicosatrienoic acids and soluble epoxide hydrolase: potential therapeutic targets for inflammation and its induced carcinogenesis. Am. J. Transl. Res. 2, 447–457.
Nudelman, I., Glikin, D., Smolkin, B., Hainrichson, M., Belakhov, V., and Baasov, T. (2010). Repairing faulty genes by aminoglycosides: development of new derivatives of geneticin (G418) with enhanced suppression of diseases-causing nonsense mutations. Bioorg. Med. Chem. 18, 3735–3746. doi: 10.1016/j.bmc.2010.03.060
Ohlendieck, K., and Swandulla, D. (2017). [Molecular pathogenesis of Duchenne muscular dystrophy-related fibrosis]. Pathologe 38, 21–29. doi: 10.1007/s00292-017-0265-1
Okubo, M., Noguchi, S., Hayashi, S., Nakamura, H., Komaki, H., Matsuo, M., et al. (2020). Exon skipping induced by nonsense/frameshift mutations in DMD gene results in Becker muscular dystrophy. Hum. Genet. 139, 247–255. doi: 10.1007/s00439-019-02107-4
O’Leary, J. M., Hamilton, J. M., Deane, C. M., Valeyev, N. V., Sandell, L. J., and Downing, A. K. (2004). Solution structure and dynamics of a prototypical chordin-like cysteine-rich repeat (von Willebrand Factor type C module) from collagen IIA. J. Biol. Chem. 279, 53857–53866. doi: 10.1074/jbc.M409225200
Olsen, L., Campos, B., Winther, O., Sgroi, D. C., Karger, B. L., and Brusic, V. (2014). Tumor antigens as proteogenomic biomarkers in invasive ductal carcinomas. BMC Med. Genomics 7 (Suppl. 3):S2. doi: 10.1186/1755-8794-7-S3-S2
Ousterout, D. G., Kabadi, A. M., Thakore, P. I., Majoros, W. H., Reddy, T. E., and Gersbach, C. A. (2015a). Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun. 6:6244. doi: 10.1038/ncomms7244
Ousterout, D. G., Kabadi, A. M., Thakore, P. I., Perez-Pinera, P., Brown, M. T., Majoros, W. H., et al. (2015b). Correction of dystrophin expression in cells from Duchenne muscular dystrophy patients through genomic excision of exon 51 by zinc finger nucleases. Mol. Ther. 23, 523–532. doi: 10.1038/mt.2014.234
Péladeau, C., Adam, N. J., and Jasmin, B. J. (2018). Celecoxib treatment improves muscle function in mdx mice and increases utrophin A expression. FASEB J. 32, 5090–5103. doi: 10.1096/fj.201800081R
Percival, J. M., Adamo, C. M., Beavo, J. A., and Froehner, S. C. (2011). Evaluation of the therapeutic utility of phosphodiesterase 5A inhibition in the mdx mouse model of duchenne muscular dystrophy. Handb. Exp. Pharmacol. 204, 323–344. doi: 10.1007/978-3-642-17969-3_14
Percival, J. M., Whitehead, N. P., Adams, M. E., Adamo, C. M., Beavo, J. A., and Froehner, S. C. (2012). Sildenafil reduces respiratory muscle weakness and fibrosis in the mdx mouse model of Duchenne muscular dystrophy. J. Pathol. 228, 77–87. doi: 10.1002/path.4054
Perera, N., Sampaio, H., Woodhead, H., and Farrar, M. (2016). Fracture in duchenne muscular dystrophy: natural history and vitamin D deficiency. J. Child. Neurol. 31, 1181–1187. doi: 10.1177/0883073816650034
Pinniger, G. J., Terrill, J. R., Assan, E. B., Grounds, M. D., and Arthur, P. G. (2017). Pre-clinical evaluation of N-acetylcysteine reveals side effects in the mdx mouse model of Duchenne muscular dystrophy. J. Physiol. 595, 7093–7107. doi: 10.1113/JP274229
Potter, R. A., Griffin, D. A., Heller, K. N., Peterson, E. L., Clark, E. K., Mendell, J. R., et al. (2021). Dose-escalation study of systemically delivered rAAVrh74.MHCK7.micro-dystrophin in the mdx mouse model of duchenne muscular dystrophy. Hum. Gene Ther. 32, 375–389. doi: 10.1089/hum.2019.255
Previtali, S. C., Gidaro, T., Díaz-Manera, J., Zambon, A., Carnesecchi, S., Roux-Lombard, P., et al. (2020). Rimeporide as a first- in-class NHE-1 inhibitor: results of a phase Ib trial in young patients with duchenne muscular dystrophy. Pharmacol. Res. 159:104999. doi: 10.1016/j.phrs.2020.104999
Raghu, G., Scholand, M. B., de Andrade, J., Lancaster, L., Mageto, Y., Goldin, J., et al. (2016). FG-3019 anti-connective tissue growth factor monoclonal antibody: results of an open-label clinical trial in idiopathic pulmonary fibrosis. Eur. Respir. J. 47, 1481–1491. doi: 10.1183/13993003.01030-2015
Ramazani, Y., Knops, N., Elmonem, M. A., Nguyen, T. Q., Arcolino, F. O., van den Heuvel, L., et al. (2018). Connective tissue growth factor (CTGF) from basics to clinics. Matrix Biol. 68-69, 44–66. doi: 10.1016/j.matbio.2018.03.007
Ramos, J., and Chamberlain, J. S. (2015). Gene therapy for duchenne muscular dystrophy. Expert. Opin. Orphan Drugs 3, 1255–1266. doi: 10.1517/21678707.2015.1088780
Ramos, J. N., Hollinger, K., Bengtsson, N. E., Allen, J. M., Hauschka, S. D., and Chamberlain, J. S. (2019). Development of novel micro-dystrophins with enhanced functionality. Mol. Ther. 27, 623–635. doi: 10.1016/j.ymthe.2019.01.002
Ran, N., Gao, X., Dong, X., Li, J., Lin, C., Geng, M., et al. (2020). Effects of exosome-mediated delivery of myostatin propeptide on functional recovery of mdx mice. Biomaterials 236:119826. doi: 10.1016/j.biomaterials.2020.119826
Reutenauer-Patte, J., Boittin, F. X., Patthey-Vuadens, O., Ruegg, U. T., and Dorchies, O. M. (2012). Urocortins improve dystrophic skeletal muscle structure and function through both PKA- and Epac-dependent pathways. Am. J. Pathol. 180, 749–762. doi: 10.1016/j.ajpath.2011.10.038
Ribeiro, A. F. Jr., Souza, L. S., Almeida, C. F., Ishiba, R., Fernandes, S. A., Guerrieri, D. A., et al. (2019). Muscle satellite cells and impaired late stage regeneration in different murine models for muscular dystrophies. Sci. Rep. 9:11842. doi: 10.1038/s41598-019-48156-7
Ricci, F., Vacchetti, M., Brusa, C., Vercelli, L., Davico, C., Vitiello, B., et al. (2019). New pharmacotherapies for genetic neuromuscular disorders: opportunities and challenges. Expert. Rev. Clin. Pharmacol. 12, 757–770. doi: 10.1080/17512433.2019.1634543
Ricciotti, E., and FitzGerald, G. A. (2011). Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 31, 986–1000. doi: 10.1161/ATVBAHA.110.207449
Ricotti, V., Spinty, S., Roper, H., Hughes, I., Tejura, B., Robinson, N., et al. (2016). Safety, tolerability, and pharmacokinetics of SMT C1100, a 2-Arylbenzoxazole utrophin modulator, following single- and multiple-dose administration to pediatric patients with duchenne muscular dystrophy. PLoS One 11:e0152840. doi: 10.1371/journal.pone.0152840
Riquelme-Guzmán, C., Contreras, O., and Brandan, E. (2018). Expression of CTGF/CCN2 in response to LPA is stimulated by fibrotic extracellular matrix via the integrin/FAK axis. Am. J. Physiol. Cell Physiol. 314, C415–C427. doi: 10.1152/ajpcell.00013.2017
Ritchie, M. E., Phipson, B., Wu, D., Hu, Y., Law, C. W., Shi, W., et al. (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43:e47. doi: 10.1093/nar/gkv007
Rooney, J. E., Welser, J. V., Dechert, M. A., Flintoff-Dye, N. L., Kaufman, S. J., and Burkin, D. J. (2006). Severe muscular dystrophy in mice that lack dystrophin and α7 integrin. J. Cell Sci. 119, 2185–2195. doi: 10.1242/jcs.02952
Rufo, A., Del Fattore, A., Capulli, M., Carvello, F., De Pasquale, L., Ferrari, S., et al. (2011). Mechanisms inducing low bone density in Duchenne muscular dystrophy in mice and humans. J. Bone Miner. Res. 26, 1891–1903. doi: 10.1002/jbmr.410
Rutter, M. M., Wong, B. L., Collins, J. J., Sawnani, H., Taylor, M. D., Horn, P. S., et al. (2020). Recombinant human insulin-like growth factor-1 therapy for 6 months improves growth but not motor function in boys with Duchenne muscular dystrophy. Muscle Nerve 61, 623–631. doi: 10.1002/mus.26846
Ryan, N. J. (2014). Ataluren: first global approval. Drugs 74, 1709–1714. doi: 10.1007/s40265-014-0287-4
Sakai, N., Nakamura, M., Lipson, K. E., Miyake, T., Kamikawa, Y., Sagara, A., et al. (2017). Inhibition of CTGF ameliorates peritoneal fibrosis through suppression of fibroblast and myofibroblast accumulation and angiogenesis. Sci. Rep. 7:5392. doi: 10.1038/s41598-017-05624-2
Salehi, F., Zeinaloo, A., Riasi, H. R., and Shamloo, A. S. (2017). Effectiveness of Coenzyme Q10 on echocardiographic parameters of patients with Duchenne muscular dystrophy. Electron. Phys. 9, 3896–3904. doi: 10.19082/3896
Samuelsson, S., Lange, J. S., Hinkle, R. T., Tarnopolsky, M., and Isfort, R. J. (2004). Corticotropin-releasing factor 2 receptor localization in skeletal muscle. J. Histochem. Cytochem. 52, 967–977. doi: 10.1369/jhc.4A6279.2004
Samulski, R. J., and Muzyczka, N. (2014). AAV-mediated gene therapy for research and therapeutic purposes. Annu. Rev. Virol. 1, 427–451. doi: 10.1146/annurev-virology-031413-085355
Sbrocchi, A. M., Rauch, F., Jacob, P., McCormick, A., McMillan, H. J., Matzinger, M. A., et al. (2012). The use of intravenous bisphosphonate therapy to treat vertebral fractures due to osteoporosis among boys with Duchenne muscular dystrophy. Osteoporos. Int. 23, 2703–2711. doi: 10.1007/s00198-012-1911-3
Schertzer, J. D., Gehrig, S. M., Ryall, J. G., and Lynch, G. S. (2007). Modulation of insulin-like growth factor (IGF)-I and IGF-binding protein interactions enhances skeletal muscle regeneration and ameliorates the dystrophic pathology in mdx mice. Am. J. Pathol. 171, 1180–1188. doi: 10.2353/ajpath.2007.070292
Selathurai, A., Kowalski, G. M., Mason, S. A., Callahan, D. L., Foletta, V. C., Della Gatta, P. A., et al. (2019). Phosphatidylserine decarboxylase is critical for the maintenance of skeletal muscle mitochondrial integrity and muscle mass. Mol. Metab. 27, 33–46. doi: 10.1016/j.molmet.2019.06.020
Servais, L., Straathof, C. S. M., Schara, U., Klein, A., Leinonen, M., Hasham, S., et al. (2020). Long-term data with idebenone on respiratory function outcomes in patients with Duchenne muscular dystrophy. Neuromuscul. Disord. 30, 5–16. doi: 10.1016/j.nmd.2019.10.008
Shavlakadze, T., White, J., Hoh, J. F., Rosenthal, N., and Grounds, M. D. (2004). Targeted expression of insulin-like growth factor-I reduces early myofiber necrosis in dystrophic mdx mice. Mol. Ther. 10, 829–843. doi: 10.1016/j.ymthe.2004.07.026
Sicinski, P., Geng, Y., Ryder-Cook, A. S., Barnard, E. A., Darlison, M. G., and Barnard, P. J. (1989). The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science 244, 1578–1580. doi: 10.1126/science.2662404
Silva, I. S., Pedrosa, R., Azevedo, I. G., Forbes, A. M., Fregonezi, G. A., Dourado Junior, M. E., et al. (2019). Respiratory muscle training in children and adults with neuromuscular disease. Cochrane Database Syst. Rev. 9:Cd011711. doi: 10.1002/14651858.CD011711.pub2
Silveira, W. A., Gonçalves, D. A., Machado, J., Lautherbach, N., Lustrino, D., Paula-Gomes, S., et al. (2020). cAMP-dependent protein kinase inhibits FoxO activity and regulates skeletal muscle plasticity in mice. FASEB J. 34, 12946–12962. doi: 10.1096/fj.201902102RR
Song, Y., Yao, S., Liu, Y., Long, L., Yang, H., Li, Q., et al. (2017). Expression levels of TGF-β1 and CTGF are associated with the severity of Duchenne muscular dystrophy. Exp. Ther. Med. 13, 1209–1214. doi: 10.3892/etm.2017.4105
Spinazzola, J. M., and Kunkel, L. M. (2016). Pharmacological therapeutics targeting the secondary defects and downstream pathology of Duchenne muscular dystrophy. Expert. Opin. Orphan Drugs 4, 1179–1194. doi: 10.1080/21678707.2016.1240613
Spurney, C. F., Rocha, C. T., Henricson, E., Florence, J., Mayhew, J., Gorni, K., et al. (2011). CINRG pilot trial of coenzyme Q10 in steroid-treated Duchenne muscular dystrophy. Muscle Nerve 44, 174–178. doi: 10.1002/mus.22047
Sreetama, S. C., Chandra, G., Van der Meulen, J. H., Ahmad, M. M., Suzuki, P., Bhuvanendran, S., et al. (2018). Membrane stabilization by modified steroid offers a potential therapy for muscular dystrophy due to dysferlin deficit. Mol. Ther. 26, 2231–2242. doi: 10.1016/j.ymthe.2018.07.021
Stedman, H. H., Sweeney, H. L., Shrager, J. B., Maguire, H. C., Panettieri, R. A., Petrof, B., et al. (1991). The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 352, 536–539. doi: 10.1038/352536a0
St-Pierre, S. J., Chakkalakal, J. V., Kolodziejczyk, S. M., Knudson, J. C., Jasmin, B. J., and Megeney, L. A. (2004). Glucocorticoid treatment alleviates dystrophic myofiber pathology by activation of the calcineurin/NF-AT pathway. FASEB J. 18, 1937–1939. doi: 10.1096/fj.04-1859fje
Sun, C., Serra, C., Lee, G., and Wagner, K. R. (2020). Stem cell-based therapies for Duchenne muscular dystrophy. Exp. Neurol. 323:113086. doi: 10.1016/j.expneurol.2019.113086
Suntar, I., Sureda, A., Belwal, T., Sanches Silva, A., Vacca, R. A., Tewari, D., et al. (2020). Natural products, PGC-1 α, and Duchenne muscular dystrophy. Acta Pharm. Sin B 10, 734–745. doi: 10.1016/j.apsb.2020.01.001
Taglia, A., Petillo, R., D’Ambrosio, P., Picillo, E., Torella, A., Orsini, C., et al. (2015). Clinical features of patients with dystrophinopathy sharing the 45-55 exon deletion of DMD gene. Acta Myol. 34, 9–13.
Takeshita, E., Komaki, H., Shimizu-Motohashi, Y., Ishiyama, A., Sasaki, M., and Takeda, S. (2018). A phase I study of TAS-205 in patients with Duchenne muscular dystrophy. Ann. Clin. Transl. Neurol. 5, 1338–1349. doi: 10.1002/acn3.651
Tang, R., Harasymowicz, N. S., Wu, C. L., Collins, K. H., Choi, Y. R., Oswald, S. J., et al. (2020). Gene therapy for follistatin mitigates systemic metabolic inflammation and post-traumatic arthritis in high-fat diet-induced obesity. Sci. Adv. 6:eaaz7492. doi: 10.1126/sciadv.aaz7492
Taniguti, A. P., Pertille, A., Matsumura, C. Y., Santo Neto, H., and Marques, M. J. (2011). Prevention of muscle fibrosis and myonecrosis in mdx mice by suramin, a TGF-β1 blocker. Muscle Nerve 43, 82–87. doi: 10.1002/mus.21869
Teichmann, M. D., Wegner, F. V., Fink, R. H., Chamberlain, J. S., Launikonis, B. S., Martinac, B., et al. (2008). Inhibitory control over Ca(2+) sparks via mechanosensitive channels is disrupted in dystrophin deficient muscle but restored by mini-dystrophin expression. PLoS One 3:e3644. doi: 10.1371/journal.pone.0003644
Terrill, J. R., Radley-Crabb, H. G., Grounds, M. D., and Arthur, P. G. (2012). N-Acetylcysteine treatment of dystrophic mdx mice results in protein thiol modifications and inhibition of exercise induced myofibre necrosis. Neuromuscul. Disord. 22, 427–434. doi: 10.1016/j.nmd.2011.11.007
Tian, C., Wong, B. L., Hornung, L., Khoury, J. C., Miller, L., Bange, J., et al. (2016). Bone health measures in glucocorticoid-treated ambulatory boys with Duchenne muscular dystrophy. Neuromuscul. Disord. 26, 760–767. doi: 10.1016/j.nmd.2016.08.011
Tinsley, J. M., Fairclough, R. J., Storer, R., Wilkes, F. J., Potter, A. C., Squire, S. E., et al. (2011). Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PLoS One 6:e19189. doi: 10.1371/journal.pone.0019189
Torrente, Y., Belicchi, M., Marchesi, C., D’Antona, G., Cogiamanian, F., Pisati, F., et al. (2007). Autologous transplantation of muscle-derived CD133+ stem cells in Duchenne muscle patients. Cell Transplant. 16, 563–577. doi: 10.3727/000000007783465064
Townsend, D., Yasuda, S., and Metzger, J. (2007). Cardiomyopathy of Duchenne muscular dystrophy: pathogenesis and prospect of membrane sealants as a new therapeutic approach. Expert. Rev. Cardiovasc. Ther. 5, 99–109. doi: 10.1586/14779072.5.1.99
Turgeman, T., Hagai, Y., Huebner, K., Jassal, D. S., Anderson, J. E., Genin, O., et al. (2008). Prevention of muscle fibrosis and improvement in muscle performance in the mdx mouse by halofuginone. Neuromuscul. Disord. 18, 857–868. doi: 10.1016/j.nmd.2008.06.386
Uchimura, T., Otomo, J., Sato, M., and Sakurai, H. (2017). A human iPS cell myogenic differentiation system permitting high-throughput drug screening. Stem Cell Res. 25, 98–106. doi: 10.1016/j.scr.2017.10.023
Verhaart, I. E. C., and Aartsma-Rus, A. (2019). Therapeutic developments for Duchenne muscular dystrophy. Nat. Rev. Neurol. 15, 373–386. doi: 10.1038/s41582-019-0203-3
Victor, R. G., Sweeney, H. L., Finkel, R., McDonald, C. M., Byrne, B., Eagle, M., et al. (2017). A phase 3 randomized placebo-controlled trial of tadalafil for Duchenne muscular dystrophy. Neurology 89, 1811–1820. doi: 10.1212/WNL.0000000000004570
Vita, G. L., Sframeli, M., Licata, N., Bitto, A., Romeo, S., Frisone, F., et al. (2021). A phase 1/2 study of flavocoxid, an oral nf-κb inhibitor, in duchenne muscular dystrophy. Brain Sci. 11:115. doi: 10.3390/brainsci11010115
Vitiello, L., Tibaudo, L., Pegoraro, E., Bello, L., and Canton, M. (2019). Teaching an old molecule new tricks: drug repositioning for duchenne muscular dystrophy. Int. J. Mol. Sci. 20:6053. doi: 10.3390/ijms20236053
Walker, E. J., Heydet, D., Veldre, T., and Ghildyal, R. (2019). Transcriptomic changes during TGF-β-mediated differentiation of airway fibroblasts to myofibroblasts. Sci. Rep. 9:20377. doi: 10.1038/s41598-019-56955-1
Wang, J. Z., Wu, P., Shi, Z. M., Xu, Y. L., and Liu, Z. J. (2017). The AAV-mediated and RNA-guided CRISPR/Cas9 system for gene therapy of DMD and BMD. Brain Dev. 39, 547–556. doi: 10.1016/j.braindev.2017.03.024
Wang, Q., Zhang, Y., Wang, M., Song, W. M., Shen, Q., McKenzie, A., et al. (2019). The landscape of multiscale transcriptomic networks and key regulators in Parkinson’s disease. Nat. Commun. 10:5234. doi: 10.1038/s41467-019-13144-y
Wasala, L. P., Hakim, C. H., Yue, Y., Yang, N. N., and Duan, D. (2019). Systemic delivery of adeno-associated viral vectors in mice and dogs. Methods Mol. Biol. 1937, 281–294. doi: 10.1007/978-1-4939-9065-8_18
Weber, M. A., Nagel, A. M., Wolf, M. B., Jurkat-Rott, K., Kauczor, H. U., Semmler, W., et al. (2012). Permanent muscular sodium overload and persistent muscle edema in Duchenne muscular dystrophy: a possible contributor of progressive muscle degeneration. J. Neurol. 259, 2385–2392. doi: 10.1007/s00415-012-6512-8
Ween, M. P., Oehler, M. K., and Ricciardelli, C. (2012). Transforming growth factor-beta-induced protein (TGFBI)/(ßig-H3): a matrix protein with dual functions in ovarian cancer. Int. J. Mol. Sci. 13, 10461–10477.
Weisleder, N., Takizawa, N., Lin, P., Wang, X., Cao, C., Zhang, Y., et al. (2012). Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci. Transl. Med. 4:139ra185. doi: 10.1126/scitranslmed.3003921
Whitehead, N. P., Pham, C., Gervasio, O. L., and Allen, D. G. (2008). N-Acetylcysteine ameliorates skeletal muscle pathophysiology in mdx mice. J. Physiol. 586, 2003–2014. doi: 10.1113/jphysiol.2007.148338
Whitehead, N. P., Streamer, M., Lusambili, L. I., Sachs, F., and Allen, D. G. (2006). Streptomycin reduces stretch-induced membrane permeability in muscles from mdx mice. Neuromuscul. Disord. 16, 845–854. doi: 10.1016/j.nmd.2006.07.024
Willmann, R., Possekel, S., Dubach-Powell, J., Meier, T., and Ruegg, M. A. (2009). Mammalian animal models for Duchenne muscular dystrophy. Neuromuscul. Disord. 19, 241–249. doi: 10.1016/j.nmd.2008.11.015
Woodfin, A., Voisin, M. B., and Nourshargh, S. (2007). PECAM-1: a multi-functional molecule in inflammation and vascular biology. Arterioscler. Thromb. Vasc. Biol. 27, 2514–2523.
Wu, J., Matthias, N., Lo, J., Ortiz-Vitali, J. L., Shieh, A. W., Wang, S. H., et al. (2018). A Myogenic Double-Reporter Human Pluripotent Stem Cell Line Allows Prospective Isolation of Skeletal Muscle Progenitors. Cell Rep. 25, 1966–1981.e1964. doi: 10.1016/j.celrep.2018.10.067
Wynn, T. A. (2007). Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Invest. 117, 524–529. doi: 10.1172/JCI31487
Wynn, T. A. (2008). Cellular and molecular mechanisms of fibrosis. J. Pathol. 214, 199–210. doi: 10.1002/path.2277
Xu, R., DeVries, S., Camboni, M., and Martin, P. T. (2009). Overexpression of Galgt2 reduces dystrophic pathology in the skeletal muscles of alpha sarcoglycan-deficient mice. Am. J. Pathol. 175, 235–247. doi: 10.2353/ajpath.2009.080967
Xu, R., Singhal, N., Serinagaoglu, Y., Chandrasekharan, K., Joshi, M., Bauer, J. A., et al. (2015). Deletion of Galgt2 (B4Galnt2) reduces muscle growth in response to acute injury and increases muscle inflammation and pathology in dystrophin-deficient mice. Am. J. Pathol. 185, 2668–2684. doi: 10.1016/j.ajpath.2015.06.008
Xu, X., Wilschut, K. J., Kouklis, G., Tian, H., Hesse, R., Garland, C., et al. (2015). Human satellite cell transplantation and regeneration from diverse skeletal muscles. Stem Cell Rep. 5, 419–434. doi: 10.1016/j.stemcr.2015.07.016
Yang, Z., Kyriakides, T. R., and Bornstein, P. (2000). Matricellular proteins as modulators of cell-matrix interactions: adhesive defect in thrombospondin 2-null fibroblasts is a consequence of increased levels of matrix metalloproteinase-2. Mol. Biol. Cell 11, 3353–3364.
Yang, L., and Du, S. (2015). Efficacy and safety of zoledronic acid and pamidronate disodium in the treatment of malignant skeletal metastasis: a meta-analysis. Medicine (Baltimore) 94:e1822. doi: 10.1097/MD.0000000000001822
Yeung, E. W., Whitehead, N. P., Suchyna, T. M., Gottlieb, P. A., Sachs, F., and Allen, D. G. (2005). Effects of stretch-activated channel blockers on [Ca2+]i and muscle damage in the mdx mouse. J. Physiol. 562, 367–380. doi: 10.1113/jphysiol.2004.075275
Young, C. S., Hicks, M. R., Ermolova, N. V., Nakano, H., Jan, M., Younesi, S., et al. (2016). A single CRISPR-Cas9 deletion strategy that targets the majority of DMD patients restores dystrophin function in hiPSC-derived muscle cells. Cell Stem Cell 18, 533–540. doi: 10.1016/j.stem.2016.01.021
Yu, H., Zhao, F., Li, J., Zhu, K., Lin, H., Pan, Z., et al. (2020). TBX2 identified as a potential predictor of bone metastasis in lung adenocarcinoma via integrated bioinformatics analyses and verification of functional assay. J. Cancer 11, 388–402. doi: 10.7150/jca.31636
Zeman, R. J., Peng, H., Danon, M. J., and Etlinger, J. D. (2000). Clenbuterol reduces degeneration of exercised or aged dystrophic (mdx) muscle. Muscle Nerve 23, 521–528. doi: 10.1002/(SICI)1097-4598(200004)23:4<521::AID-MUS10>3.0.CO;2-8
Zhang, Y., Li, H., Min, Y. L., Sanchez-Ortiz, E., Huang, J., Mireault, A. A., et al. (2020). Enhanced CRISPR-Cas9 correction of Duchenne muscular dystrophy in mice by a self-complementary AAV delivery system. Sci. Adv. 6:eaay6812. doi: 10.1126/sciadv.aay6812
Zhang, Y., Long, C., Li, H., McAnally, J. R., Baskin, K. K., Shelton, J. M., et al. (2017). CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci. Adv. 3:e1602814. doi: 10.1126/sciadv.1602814
Zheng, L., Shi, J., and Mu, Y. (2018). Dynamics changes of CRISPR-Cas9 systems induced by high fidelity mutations. Phys. Chem. Chem. Phys. 20, 27439–27448. doi: 10.1039/C8CP04226H
Zhou, L., and Lu, H. (2010). Targeting fibrosis in Duchenne muscular dystrophy. J. Neuropathol. Exp. Neurol. 69, 771–776. doi: 10.1097/NEN.0b013e3181e9a34b
Ziemba, M., Barkhouse, M., Uaesoontrachoon, K., Giri, M., Hathout, Y., Dang, U. J., et al. (2021). Biomarker-focused multi-drug combination therapy and repurposing trial in mdx mice. PLoS One 16:e0246507. doi: 10.1371/journal.pone.0246507
Zimowski, J. G., Massalska, D., Holding, M., Jadczak, S., Fidziañska, E., Lusakowska, A., et al. (2014). MLPA based detection of mutations in the dystrophin gene of 180 Polish families with Duchenne/Becker muscular dystrophy. Neurol. Neurochir. Pol. 48, 416–422. doi: 10.1016/j.pjnns.2014.10.004
Keywords: Duchenne muscular dystrophy, pharmacological therapeutics, skeletal muscle, fibrosis, inflammation, regeneration, meta-analysis
Citation: Yao S, Chen Z, Yu Y, Zhang N, Jiang H, Zhang G, Zhang Z and Zhang B (2021) Current Pharmacological Strategies for Duchenne Muscular Dystrophy. Front. Cell Dev. Biol. 9:689533. doi: 10.3389/fcell.2021.689533
Received: 01 April 2021; Accepted: 23 July 2021;
Published: 19 August 2021.
Edited by:
Chrissa Kioussi, Oregon State University, United StatesReviewed by:
Angela Lek, Yale University, United StatesCopyright © 2021 Yao, Chen, Yu, Zhang, Jiang, Zhang, Zhang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ge Zhang, emhhbmdnZUBoa2J1LmVkdS5oaw==; Zongkang Zhang, bWF4emhhbmd6a0BjdWhrLmVkdS5oaw==; Baoting Zhang, emhhbmdiYW90aW5nQGN1aGsuZWR1Lmhr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.