 Yu-Chen S. H. Yang1
Yu-Chen S. H. Yang1 Zi-Lin Li2,3Tung-Yung Huang2,3Kuan-Wei Su4Chi-Yu Lin5Chi-Hung Huang6Han-Yu Chen2,3Mei-Chin Lu7,8
Zi-Lin Li2,3Tung-Yung Huang2,3Kuan-Wei Su4Chi-Yu Lin5Chi-Hung Huang6Han-Yu Chen2,3Mei-Chin Lu7,8 Haw-Ming Huang5Sheng-Yang Lee5,9,10Jaqueline Whang-Peng3,11
Haw-Ming Huang5Sheng-Yang Lee5,9,10Jaqueline Whang-Peng3,11 Hung-Yun Lin3,11,12,13,14*
Hung-Yun Lin3,11,12,13,14* Paul J. Davis14,15Kuan Wang2
Paul J. Davis14,15Kuan Wang2- 1Joint Biobank, Office of Human Research, Taipei Medical University, Taipei, Taiwan
- 2Graduate Institute of Nanomedicine and Medical Engineering, College of Medical Engineering, Taipei Medical University, Taipei, Taiwan
- 3Graduate Institute of Cancer Biology and Drug Discovery, College of Medical Science and Technology, Taipei Medical University, Taipei, Taiwan
- 4Department of Dentistry, Hsinchu MacKay Memorial Hospital, Hsinchu, Taiwan
- 5School of Dentistry, College of Oral Medicine, Taipei Medical University, Taipei, Taiwan
- 6Division of Cardiology, Department of Internal Medicine, Cathay General Hospital, Taipei, Taiwan
- 7National Museum of Marine Biology and Aquarium, Pingtung, Taiwan
- 8Graduate Institute of Marine Biology, National Dong Hwa University, Pingtung, Taiwan
- 9Center for Tooth Bank and Dental Stem Cell Technology, Taipei Medical University, Taipei, Taiwan
- 10Department of Dentistry, Wan-Fang Medical Center, Taipei Medical University, Taipei, Taiwan
- 11Cancer Center, Wan Fang Hospital, Taipei Medical University, Taipei, Taiwan
- 12TMU Research Center of Cancer Translational Medicine, Taipei Medical University, Taipei, Taiwan
- 13Traditional Herbal Medicine Research Center of Taipei Medical University Hospital, Taipei Medical University, Taipei, Taiwan
- 14Pharmaceutical Research Institute, Albany College of Pharmacy and Health Sciences, Rensselaer, NY, United States
- 15Department of Medicine, Albany Medical College, Albany, NY, United States
Estrogen (E2) has multiple functions in breast cancers including stimulating cancer growth and interfering with chemotherapeutic efficacy. Heteronemin, a marine sesterterpenoid-type natural product, has cytotoxicity on cancer cells. Breast cancer cell lines, MCF-7 and MDA-MB-231, were used for investigating mechanisms involved in inhibitory effect of E2 on heteronemin-induced anti-proliferation in breast cancer cells with different estrogen receptor (ER) status. Cytotoxicity was detected by cell proliferation assay and flow cytometry, gene expressions were determined by qPCR, mechanisms were investigated by Western blot and Mitochondrial ROS assay. Heteronemin exhibited potent cytotoxic effects against both ER-positive and ER-negative breast cancer cells. E2 stimulated cell growth in ER-positive breast cancer cells. Heteronemin induced anti-proliferation via suppressing activation of ERK1/2 and STAT3. Heteronemin suppressed E2-induced proliferation in both breast cancer cells although some gene expressions and anti-proliferative effects were inhibited in the presence of E2 in MCF-7 and MDA-MB-231 cells with a higher concentration of heteronemin. Heteromenin decreased the Bcl-2/Bax ratio to inhibit proliferation in MDA-MB-231 but not in MCF-7 cells. Both heteronemin and E2 increased mitochondrial reactive oxygen species but combined treatment reversed superoxide dismutase (SOD)s accumulation in MCF-7 cells. Heteronemin caused G0/G1 phase arrest and reduced the percentage of cells in the S phase to suppress cancer cell growth. In conclusion, Heteronemin suppressed both ER-positive and ER-negative breast cancer cell proliferation. Interactions between E2 and heteronemin in signal transduction, gene expressions, and biological activities provide insights into the complex pathways by which anti-proliferation is induced by heteronemin in E2-replete environments.
Introduction
Although early detection methods and effective treatments have been developed for breast cancer, it is still the most common diagnosed cancer among women (Bray et al., 2018). Despite advancements in diagnostics and systemic treatments, up to one-third of patients with breast cancer undergo a mastectomy as initial surgical treatment to achieve local control (Anampa et al., 2015). Adjuvant systemic treatment, including chemotherapy, reduces the risks of distant recurrence and breast cancer mortality.
Generally, 17β-estradiol (E2) binds with estrogen receptor-α (ER-α) as a transcription factor complex to regulate expression of target genes and proteins that are important for biological functions (Hofseth et al., 1999). However, estrogen can initiate breast cancer development, and promotes breast cancer cell growth. Recently, we showed that estrogen is able to bind αvβ3, a cell surface integrin, to activate signal transduction and cancer growth (Cody et al., 2007); however, the mechanisms are not fully revealed. Although the ER-regulated signaling transduction pathway plays a vital role in breast cancer growth, it does not involve in breast cancer initiation (Cavalieri and Rogan, 2014). On the other hand, strong evidence indicates that estrogen oxidative metabolism may initiate breast cancer development majorly (Cavalieri et al., 1997; Devanesan et al., 2001).
Reactive oxygen species (ROS) include hydroxyl radicals (OH⋅), hydrogen peroxide H2O2, and superoxide (O2−) (Poillet-Perez et al., 2015). Both aerobic glycolysis and mitochondrial oxidative phosphorylation are cellular sources of ROS. During oxidation, electrons leak from the electron transport chain, transfer to oxygen, and convert approximately 1∼5% of the oxygen into superoxide (Bhat et al., 2015). However, additional electrons that prematurely leak from the respiratory chain under stressful conditions exacerbate superoxide production, thus causing detrimental effects (Poillet-Perez et al., 2015). At low concentrations, they are essential signaling molecules. On the other hand, high ROS quantities can cause damage to DNA and other macromolecules to trigger senescence (Cobbaut and Van Lint, 2018). High concentrations of ROS also permeabilize mitochondria leading to the release of cytochrome c which induces apoptosis (Cobbaut and Van Lint, 2018). Breast cancer cells modify metabolic pathways to facilitate increased proliferation and cell survival resulting in glucose and glutamine (Gwangwa et al., 2019). Cancer cells increase ROS production during cancer cell proliferation. Estrogen-mediated high ROS accumulation plays a key role in driving carcinogenesis (Tian et al., 2016). Excessive ROS serve as important effectors to increase genomic instability and activate redox-associated signaling pathways. Physiologically available concentrations of estrogens or estrogen metabolites that directly act on the mitochondria of mammary epithelial cells produce ROS, which subsequently enhances the phosphorylation of kinases to activate redox-sensitive transcription factors (Okoh et al., 2013). Therefore, ROS are important mediators of estrogen-induced cancer. Two metabolites of estrogen, 2-OHE1E2, and 4-OHE1E2, are highly redox-active and generate ROS in breast epithelial cells (Fussell et al., 2011). Long-term exposure to estrogen induces ROS overproduction and increases mitochondrial (mt)DNA mutations and mitochondrial protein damage. A recent study indicated that 4-OHE1E2 induces ROS and causes malignant transformation of MCF-10A cells. However, biological or chemical ROS scavengers prevent 4-OHE1E2-induced carcinogenesis in MCF-10A cells (Okoh et al., 2013). Excess ROS generated by repeated exposure to 4-OHE1(E2) caused malignancy of human mammary epithelial cells in nude mice (Okoh et al., 2013).
Heteronemin, the most abundant secondary metabolite in the sponge Hippospongia sp., exhibited potent cytotoxic activities against several cancer cell lines. It induced apoptosis in different types of cancer cells (Kollmann et al., 2015; Lee Y. S. et al., 2018; Lin et al., 2018; Cheng et al., 2019; Huang et al., 2020). It inhibited activation of extracellular signal-regulated kinase 1/2 (ERK1/2) and signal transducer and activator of transcription 3 (STAT3) (Huang et al., 2020). The role of nuclear factor (NF)-κB in heteronemin-induced anti-proliferation is controversial (Schumacher et al., 2010; Chen et al., 2018). It upregulated talin expression and talin phosphorylation in leukemia Molt4 cells (Chen et al., 2018). Heteronemin was shown to modulate mitochondrial (mt)ROS and oxidative phosphorylation (OXPHOS) (Cheng et al., 2019). Some studies indicated that heteronemin induces apoptosis via inhibition of the transforming growth factor (TGF)-β signal transduction pathway in cholangiocarcinomas (Lin et al., 2018). In addition, it inhibits p53 expression but does not affect apoptosis (Lin et al., 2018). Altogether, results have shown that this compound has potential as an anti-inflammatory and anti-cancer agent (Schumacher et al., 2010). Mitochondria are major sites for apoptosis, and they are highly regulated by the Bcl-2 family of proteins comprising both anti-apoptotic (Bcl-2 and Bcl-xL) and proapoptotic (Bax and Bak) members (el-Deiry et al., 1994; Chipuk et al., 2004; Papi et al., 2008; Weng et al., 2009; Xu et al., 2009; Gwangwa et al., 2019). Therefore, targeting mitochondria is a novel strategy for cancer therapy. Heteronemin was shown to target mitochondrial-mediated apoptosis (Wu et al., 2015; Chen et al., 2018; Cheng et al., 2019); however, evidence indicates there are other death pathways involved. Heteronemin induced a novel type of programmed cell death, “Ferroptosids” (Chang et al., 2021).
In the present study, we investigated the inhibitory effect of E2 on heteronemin-induced cytotoxic effects via suppressing activation of ERK1/2 and STAT3 in breast cancer cells. Heteronemin and E2 showed different effects on expressions of proliferation, angiogenesis, and growth factor receptor genes in breast cancer cells. E2 showed inhibitory effects on heteronemin-induced signal transduction, gene expression, and anti-proliferation in breast cancer cells. However, heteronemin induced anti-proliferation via different patterns in ER-positive and negative breast cancer cells. In the presence of E2, heteronemin induced anti-proliferation via modulating ROS in MCF-7 cells. On the other hand, it decreased Bcl-2/Bax ratio to inhibit cancer growth in MDA-MB-231 cells.
Materials and Methods
Cell Lines
Human ER-positive MCF-7 breast cancer cells (ATCC® HTB-22TM), ER-negative MDA-MB-231 cells (ATCC® HTB-26TM) and normal epithelial cell line Vero (ATCC® CCL-81TM) was established from kidney of normal adult African green monkey were obtained from American Type Culture Collection (ATCC, Manassas, VA, United States). These cell lines were tested and authenticated by BCRC (isoenzyme analysis, Mycoplasma, cytogenetics, tumorigenesis, and receptor expression testing). Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Life Technologies, Carlsbad, CA, United States), supplemented with 10% fetal bovine serum (FBS). Incubation conditions were 5% CO2 at 37°C. Before the study, cells were placed in a 0.25% hormone-depleted serum-supplemented medium for 2 days.
Cell Viability Assay
MCF-7, MDA-MB-231, and Vero cells were plated at a density of 4 × 103 cells/well in 96-well plates. Cell viability was determined by using the Alamar Blue® Assay Kit (Thermo Fisher Scientific, Watertown, MA, United States) at 72 h after treatment. Medium containing different drugs was replaced daily. At the time of detection, medium was removed, and cells were incubated with Alamar Blue® reagent for 2 h at 37°C according to the manufacturer’s instructions. Plates were then analyzed using a VersaMax Microplate reader (Molecular Devices, San Jose, CA, United States) at a wavelength of 570 nm, with 600 nm as a reference.
Real-Time Quantitative Polymerase Chain Reaction (qPCR)
Total RNA was extracted with genomic DNA removed with an Illustra RNAspin Mini RNA Isolation Kit (GE Healthcare Life Sciences, Buckinghamshire, United Kingdom). DNase I-treated total RNA (1 μg) was reverse-transcribed using a RevertAid H Minus First Strand cDNA Synthesis Kit (Life Technologies) into complementary (c)DNA. cDNAs were used as the template for the real-time PCR and analysis. Real-time PCRs were conducted using a QuantiNovaTM SYBR® Green PCR Kit (Qiagen, Hilden, Germany) on a CFX ConnectTM Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA, United States). The reaction procedure involved initial denaturation at 95°C for 5 min, followed by 40 cycles of denaturing at 95°C for 5 s and combined annealing/extension at 60°C for 10 s, as shown in detail in the manufacturer’s instructions. Primer sequences are listed in Table 1. The relative gene expression (normalized to 18s reference gene) was calculated according to the ΔΔCT method. The fidelity of the PCR was determined by a melting temperature analysis.
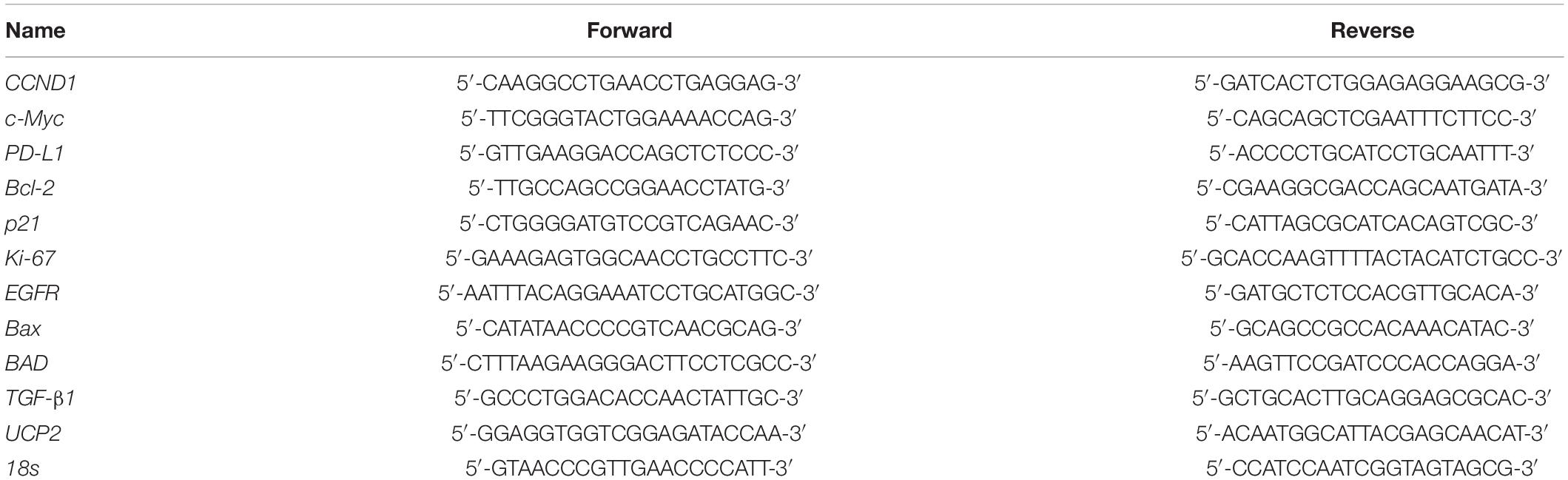
Table 1. Primer sequences for the qPCR.
Western Blot Analysis
To examine the effects of E2 and heteronemin on signaling pathways, Western blot analyses were conducted to quantify protein expression levels of phosphorylated (p)STAT3-S727, p-protein kinase Cα (PKCα)-T497, and pERK1/2 in MCF-7 and MDA-MB-231 cells. For the Western blot analyses, cells were lysed, and extracted protein samples were separated by 10% sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE). A 30-μg quantity of protein was loaded into each well with 5 × sample buffer and samples were resolved by electrophoresis at 100 V for 2 h. Resolved proteins were transferred from the polyacrylamide gel to Millipore Immobilon-PSQ Transfer polyvinylidene difluoride membranes (Millipore, Billerica, MA, United States) with Mini Trans-Blot® Cells (Bio-Rad, Hercules, CA, United States). Membranes were blocked with a solution of 2% bovine serum albumin (BSA) in Tris-buffered saline. Membranes were treated with primary antibodies from Cell Signaling Technology: pSTAT3-S727 (catalog no. 9136), pERK1/2 (catalog no. 4377), and GeneTex: pPKCα (catalog no. 130433) and GAPDH (catalog no. 100118). All antibodies were incubated at 4 °C overnight. Proteins were detected with horseradish peroxidase (HRP)-conjugated secondary antibodies and ImmobilonTM Western HRP Substrate Luminol Reagent (Millipore, St. Charles, MO, United States). Western blots were visualized and recorded with an Amersham Imager 600 (GE Healthcare Life Sciences, Pittsburgh, PA, United States). Intensities of the protein bands representing expression levels were quantitated using Image J 1.47 software (NIH, Bethesda, MD, United States) according to the software instructions.
Mitochondrial ROS Assay
Changes in mitochondrial ROS that occurred during apoptosis were detected with a fluorescence-based assay. Mitochondrial ROS was detected with a Mitochondrial Superoxide Detection Kit (ab219943, Abcam, Cambridge, United Kingdom). MCF-7 cells were cultured as previously described. After being starved with 0.25% stripped FBS-containing medium for 2 days, cells were re-fed using 5% stripped FBS-containing medium and treated with E2 and heteronemin. Antimycin A at 50 μM was used as a positive control. After 24 h, cells were processed with the Mitochondrial Superoxide Detection Kit according to the manufacturer’s instructions. Excitation at 540 nm and emission at 590 nm was read with a spectral scanning multimode reader (Thermo Fisher Scientific Varioskan Flash, Waltham, MA, United States).
Cell Cycle Assay
MCF-7 and MDA-MB-231 cells were seeded at a density of 1.5 × 105 cells/well in six-well plates. After starvation with 0.25% stripped FBS-containing medium for 2 days, cells were re-fed using 5% stripped FBS-containing medium and treated with agents for 24 h. Cells were trypsinized and fixed with 70% ethanol and stored at −20°C for 2 weeks prior to propidium iodide (PI) staining and a flow cytometric analysis. Cells were incubated with 1 ml of phosphate-buffered saline (PBS) containing 0.5% Triton X-100 and 0.05% RNase A for 1 h, then stained with PI/RNase Staining Buffer (BD, San Jose, CA, United States) in the dark at room temperature for 30 min. Flow cytometry was carried out on an Invitrogen AttuneTM NxT Acoustic Focusing Cytometer (Thermo Fisher Scientific, MA, United States). Percentages of DNA contents were analyzed using Attune NxT Flow Cytometer software (ver. 4.2) to determine the fractions of each phase of the cell cycle (sub G0/G1, G0/G1, S, and G2/M).
Statistical Analysis
All collected data for immunoblots, nucleotide densities, and cell densities were analyzed by IBMS®PSS® Statistics software vers. 19.0 (SPSS, Chicago, IL, United States). Student’s t-test was conducted, and p-values of < 0.05 (*,#, or $), 0.01 **, ##, or $$), and 0.001 (***, ###, or $$$) as thresholds of significance, were used to evaluate the significance of effects of estrogen, heteronemin, and their combined treatment.
Results
Heteronemin Induces Anti-proliferation and Reverses E2 –Induced Proliferation in ER-Positive and ER-Negative Breast Cancer Cells
Heteronemin inhibited cell proliferation in different kinds of cancer cells (Lee Y. S. et al., 2018; Lin et al., 2018; Huang et al., 2020). We examined the inhibitory effect of heteronemin on human ER-positive and ER-negative breast cancer cells. ER-positive MCF-7 cells and ER-negative breast cancer MDA-MB-231 cells were treated with different concentrations of heteronemin that were refreshed daily for 3 days, and a cell viability assay was conducted.
Heteronemin significantly inhibited the viability of MCF-7 cells at concentrations was higher than 0.625 μM and IC50 = 0.8779 μM (Figure 1A). In MDA-MB-231 cells, there was significant inhibition at concentrations higher than 0.3125 μM and IC50 = 0.8672 μM (Figure 1B). However, with less toxicity in Vero cells that are the normal kidney epithelial cells extracted from an African green monkey and IC50 = 3.5676 μM (Figure 1C). These results suggest that both ER-positive and ER-negative breast cancer cells were sensitive to heteronemin treatment. Moreover, E2 induced cell proliferation and reversed anti-proliferation partially induced by heteronemin in MCF-7 cells (Figure 1D). On the other hand, E2 did not promote proliferation in MDA-MB-231 cells. However, it slightly reversed heteronemin-induced anti-proliferation at 1.25 μM in MBA-MD-231 cells (Figure 1E). Although E2 affect cell proliferation in breast cancer cells, the cell viability were suppressed in the co-treatments. Additionally, combined E2 and heteronemin also do not affect normal cell proliferation (Figure 1E).
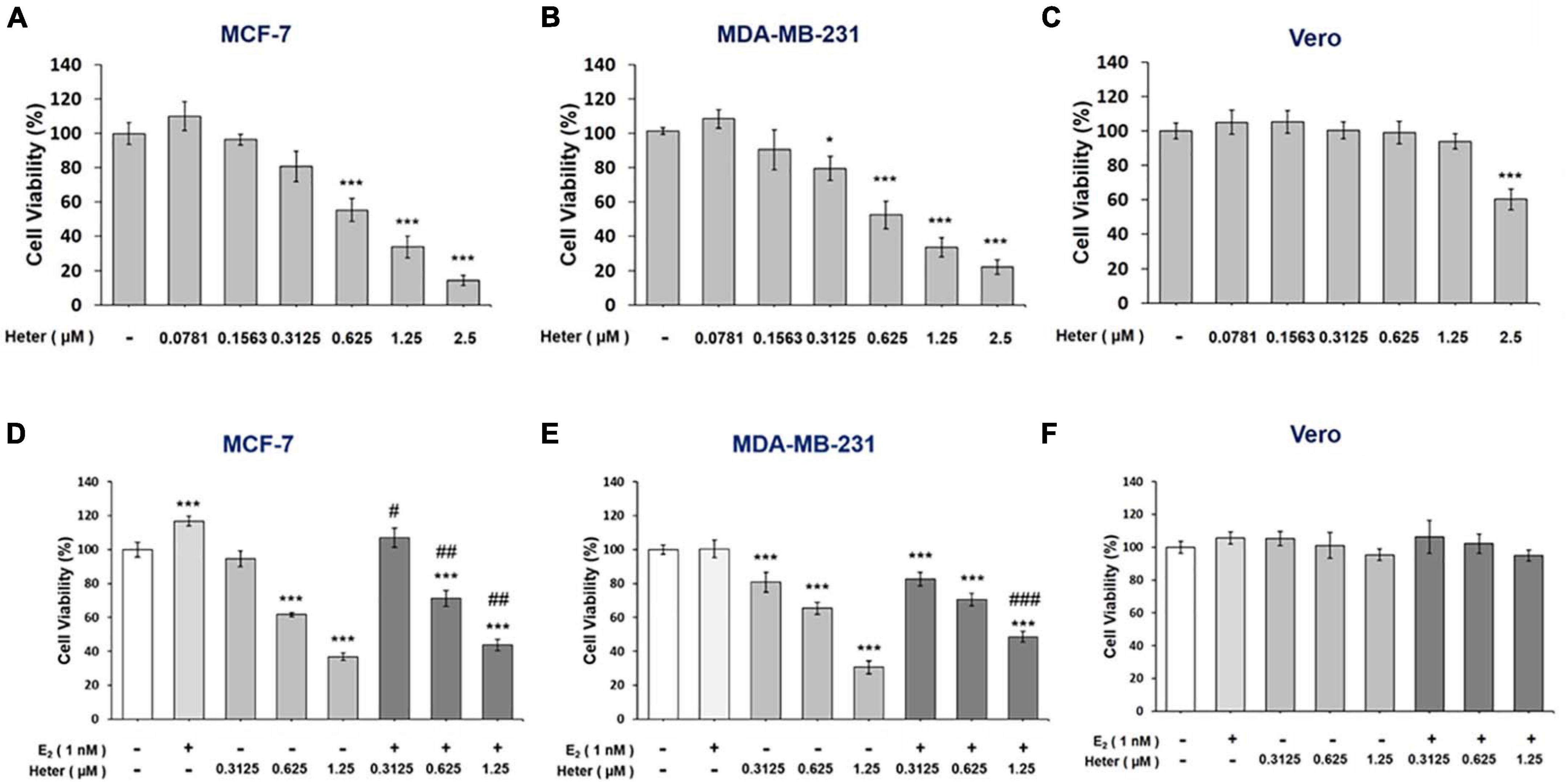
Figure 1. Estrogen reverses heteronemin induces anti-proliferation in breast cancer cells. (A) MFC-7, (B) MDA-MB-231, and (C) Vero cells were treated with various concentrations of heteronemin with medium containing reagents refreshed daily for 72 h. (D) MCF-7, (E) MDA-MB-231, and (F) Vero cells were co-treated with various concentrations of heteronemin in the presence and absence of 1 nM E2 for 72 h. Cell viability was detected with an Alamar Blue® Assay Kit. The number of independent studies (n) = 4. Results were expressed as the mean ± SD. *p < 0.05, ***p < 0.001 compared to the untreated control. #p < 0.05, ##p < 0.01, ###p < 0.001, compared to the same dosage of heteronemin treatment only. Heter: Heteronemin.
Heteronemin Regulates Gene Expressions Differently in ER-Positive and ER-Negative Breast Cancer Cells
We further studied the effects of heteronemin on gene expressions. Both MCF-7 and MDA-MB-231 cells were treated with different concentrations of heteronemin for 24 h. Cells were harvested and RNA was extracted. qPCR studies were conducted for Ki-67, CCND1, c-Myc, Bcl-2, PD-L1, and p21. Heteronemin significantly inhibited expressions of the proliferation genes, Ki-67 and CCND-1, in concentration-dependent manners in both MCF-7 and MDA-MB-231 cells. It decreased c-Myc expression only in MDA-MB-231 cells, while in MCF-7 cells, c-Myc expression significantly increased with heteronemin treatment (Figure 2).
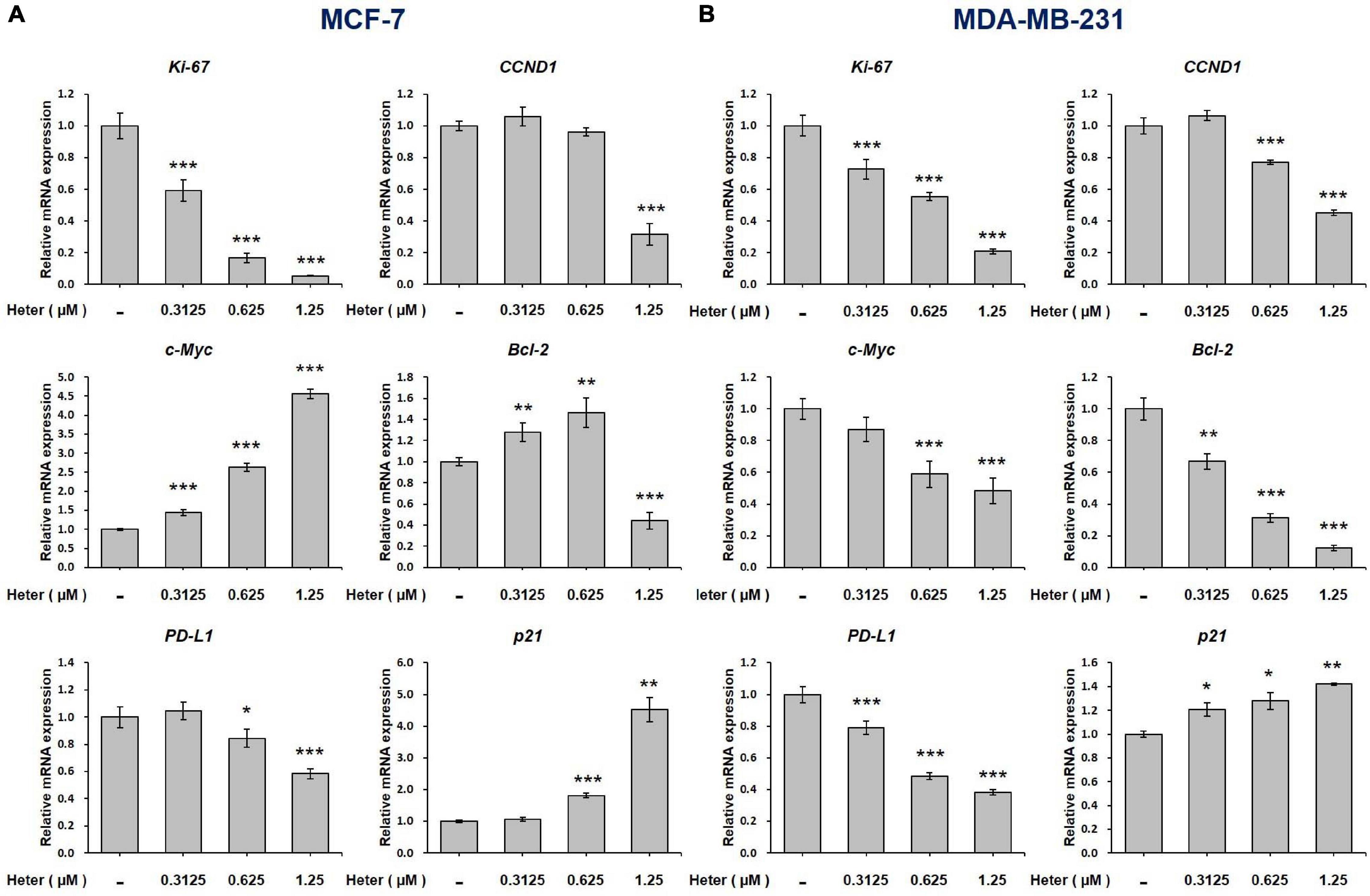
Figure 2. Heteronemin regulates expression of ki-67, CCND1, c-Myc, Bcl-2, PD-L1, and p21 in breast cancer cells. (A) MCF-7 and (B) MDA-MB-231 cells were treated with various concentrations of heteronemin for 24 h. Total RNA was extracted. qPCR analyses were conducted for Ki-67, CCND1, c-Myc, Bcl-2, PD-L1, and p21. The number of independent studies (n) = 3. *p < 0.05, **p < 0.01, ***p < 0.001, compared to the untreated control.
Additionally, Heteronemin inhibited programmed death-ligand 1 (PD-L1) expression and stimulated expression of the proapoptotic gene, p21 in dose-dependent manners in both MCF-7 and MDA-MB-231 cells (Figure 2). Heteronemin inhibited Bcl-2 expression only with a high concentration treatment of 1.25 μM, while Bcl-2 expression increased at a low concentration in MCF-7 cells. In MDA-MB-231 cells, Bcl-2 expression was inhibited at all concentrations of heteronemin. It is interesting that heteronemin inhibited expressions of c-Myc and Bcl-2 in ER-negative breast cancer cells but stimulated their expressions in ER-positive breast cancer cells (Figure 2).
Heteronemin Reverses E2 –Induced Gene Expression in Breast Cancer Cells
We also studied the effect of E2 on genes regulated by heteronemin in ER-positive and ER-negative breast cancer cells. In co-treatment, we chose 0.625 and 0.3125 μM heteronemin, respectively, in MCF-7 and MDA-MB-231 cells which exhibited significant inhibition of cell viability (Figure 1). MCF-7 and MDA-MB-231 cells were treated with E2, heteronemin, and combined E2 and heteronemin for 24 h. E2 induced expression of Ki-67, CCND1, EGFR, and PD-L1 significantly in MCF-7 cells. Heteronemin suppressed expression of Ki-67, EGFR, and PD-L1 significantly and inhibited E2’s stimulatory effect. While 0.625 μM heteronemin treatment did not inhibit CCND1 expression, it reversed the effect of E2 in combined treatment (Figure 3A). In addition, E2 not only increased Bax expression and the Bcl-2/Bax ratio but also inhibited expression of BAD and p21, two important pro-apoptotic genes in MCF-7 cells (Figure 3A). Heteronemin increased expression of Bax, BAD, and p21. E2 reduced the effect and the Bcl-2/Bax ratio increased with combined treatment in MCF-7 cells (Figure 3A).
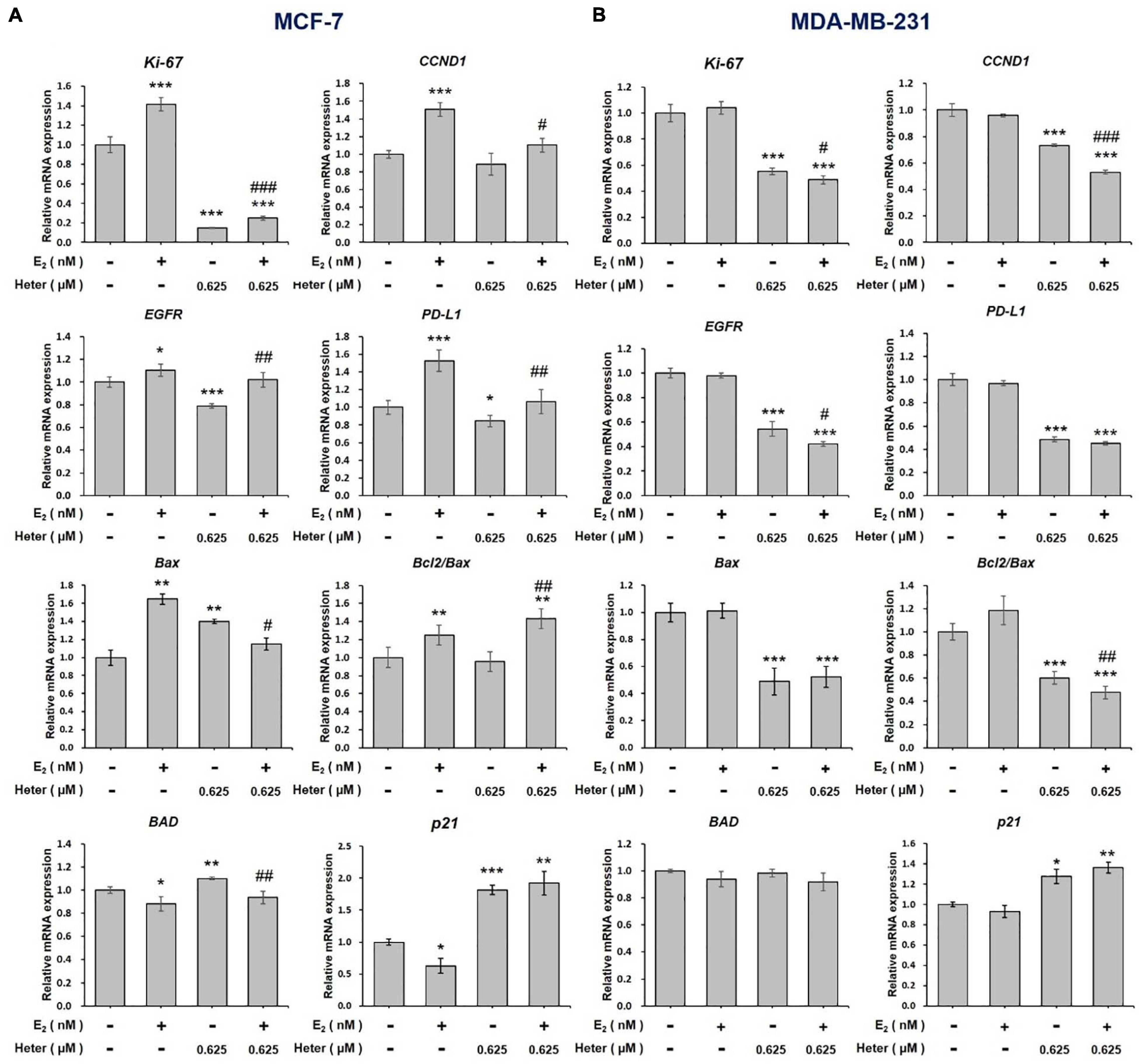
Figure 3. Estrogen and heteronemin regulate expressions of Ki-67, CCND1, EGFR, PD-L1, Bax, Bcl/Bax, BAD, and p21 in breast cancer cells. (A) MCF-7 and (B) MDA-MB-231 cells were treated with heteronemin (0.625 μM) in the presence and absence of 1 nM E2 for 24 h. RNA was extracted and qPCR analyses were conducted for Ki-67, CCND1, EGFR, PD-L1, Bax, Bcl-2/Bax, BAD, and p21. The number of independent studies (n) = 3. *p < 0.05, **p < 0.01, ***p < 0.001, compared to the untreated control. #p < 0.05, ##p < 0.01, ###p < 0.001, compared to the same dosage of heteronemin treatment only.
In ER-negative MDA-MB-231 cells, E2 did not affect gene expressions of Ki-67, CCND1, EGFR, and PD-L1 (Figure 3B). On the other hand, heteronemin alone or co-treatment with E2 suppressed expression of Ki-67, EGFR, and PD-L1 (Figure 3B). E2 did not affect Bax, BAD, and p21 expressions. Heteronemin suppressed expression of Bax but stimulated expression of BAD and p21 significantly (Figure 3B). It also reduced Bcl-2/Bax ratio. The combined treatment inhibited Bcl-2/Bax ratio significantly, but increased expression of BAD and p21 significantly (Figure 3B). In ER-positive MCF-7 cells, E2 stimulated expressions of proliferation-related genes and inhibited pro-apoptotic gene expression. Nevertheless, heteronemin could rescue the effects of E2. E2 did not affect those gene expressions in ER-negative MDA-MB-231 cells. Additionally, results also suggested that Bcl-2/Bax ratio may play a vital role in cell fate in MDA-MB-231 cells but not in MCF-7 cells.
Heteronemin Inhibits Signal Transduction Pathways in Breast Cancer Cells
We investigated the mechanisms involved in the heteronemin-induced anti-cancer ability in breast cancer cells. Previous studies showed that activation of STAT3, ERK1/2, and PKC plays an important role in proliferation in cancer cells (Lønne et al., 2009; Nana et al., 2018; Chin et al., 2019; Huang et al., 2020). Results shown E2 induced phosphorylation of ERK1/2, PKCα, and STAT3. Alternatively, heteronemin inhibited activation of ERK1/2 and STAT3, but it increased PKCα phosphorylation in MCF-7 cancer cells (Figure 4). The increased phosphorylated STAT3 and ERK1/2 induced by E2 was reversed by heteronemin. In the presence of a PKC inhibitor, sotrastaurin (SOT), all activities of ERK1/2, PKCα, and STAT3 were inhibited in MCF-7 cells. Heteronemin enhanced the inhibitory effect of SOT on activities of ERK1/2 and STAT3 (Figure 4).
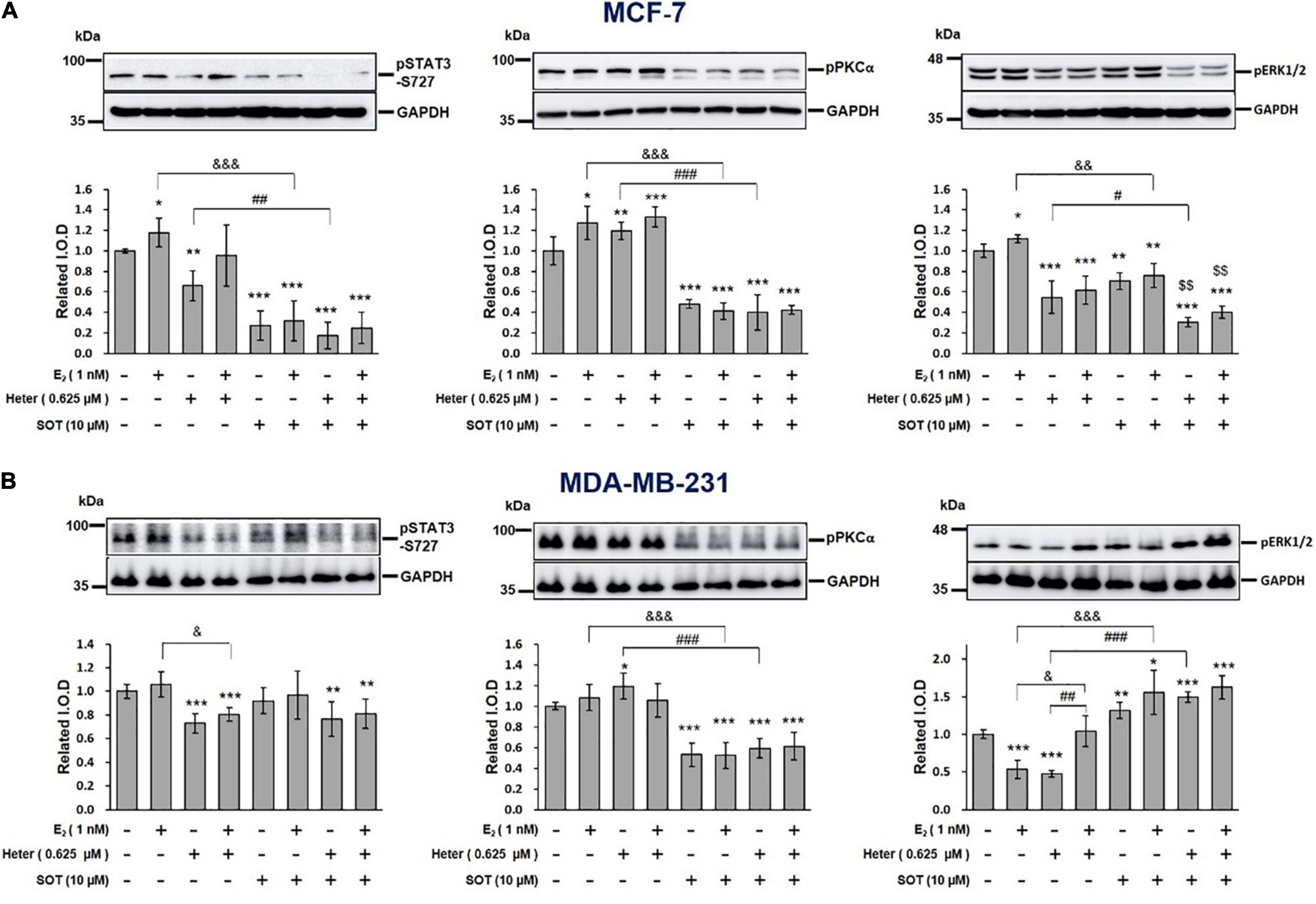
Figure 4. Estrogen, heteronemin, and sotrastaurin (SOT) affect the signal transduction pathway in breast cancer cells. (A) MCF-7 and (B) MDA-MB-231 cells were treated with 1 nM E2, 0.625 μM heteronemin, and their combination in the presence and absence of the PKC inhibitor, SOT for 24 h. Cells were harvested and total proteins were extracted. Western blot analyses were conducted for phosphorylated (p)STAT3, pPKCα, and pERK1/2. The number of independent studies (n) = 4. *p < 0.05, **p < 0.01, ***p < 0.001, compared to the untreated control. #p < 0.05, ##p < 0.01, ###p < 0.001 compared to heteronemin. &p < 0.05, &&p < 0.01, &&&p < 0.001, compared to E2.
In MDA-MB-231 cells, E2 did not affect STAT3 and PKCα phosphorylation. Heteronemin treatment at 0.625 μM suppressed the phosphorylation of STAT3 and ERK1/2 but slightly increased PKCα activation. Interestingly, E2 reduced the inhibition of pERK1/2 but did not reduce the inhibition of pSTAT3 induced by heteronemin (Figure 4). The result of PKCα was similar from MCF-7 cells, in which SOT just suppressed PKCα activation.
Both E2 and Heteronemin Induce Mitochondrial ROS Production in MCF-7 Cells
Activation of signal transduction is linked to ROS production. To investigate whether E2 or heteronemin plays a direct role in mitochondrial function, we performed a mitochondrial ROS assay. MCF-7 cells were treated with E2 and different concentrations of heteronemin for 24 h. E2 increased TGF-β1 expression which has been shown to increase ROS production (Joo et al., 2008) but downregulated UCP2 expression (Figures 5A, B). On the other hand, heteronemin stimulated TGF-β1 expression at 0.625 μM but inhibited its expression at 1.25 μM (Figure 5A). Expression of UCP2 was inhibited by heteronemin in a concentration-dependent manner (Figure 5B). The stimulatory effect of E2 on TGF-β1 was inhibited by heteronemin treatment (Figure 5A). The inhibitory effect of E2 on UCP2 was further enhanced by heteronemin treatment (Figure 5B). Not only did E2 significantly increase mitochondrial ROS production, but heteronemin also significantly increased mitochondrial ROS production in a dose-dependent manner (Figure 5C). In the presence of E2, 0.3125 μM heteronemin increased mitochondrial ROS production compared to heteronemin only (Figure 5C). Interestingly, both E2 and heteronemin increased accumulation of superoxide dismutase (SOD)s (Figure 5D). However, the combination of E2 and heteronemin reversed the accumulation of SOD1 and, SOD2 was reversed only at the higher concentration heteronemin (Figure 5D).
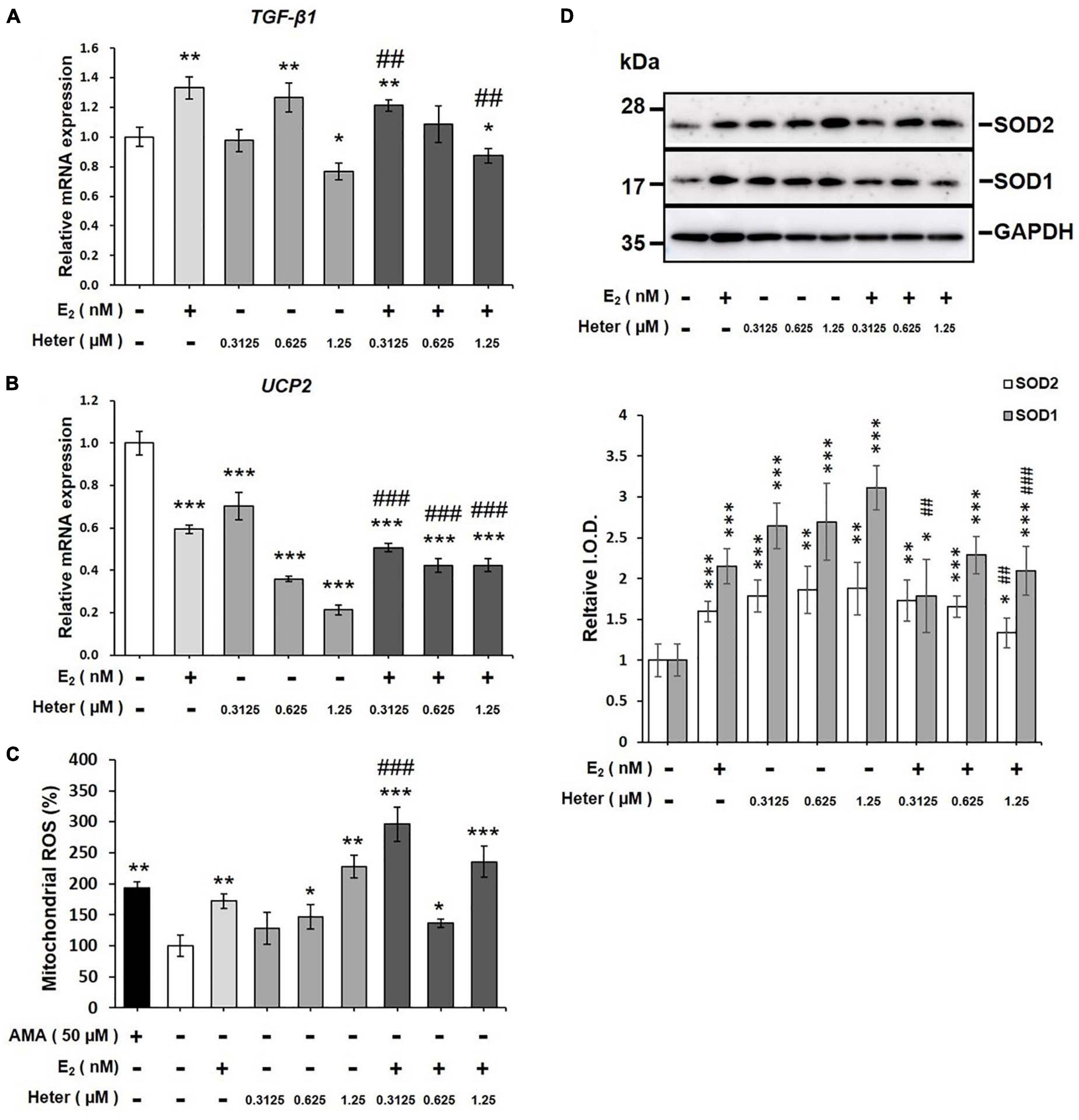
Figure 5. Estrogen and heteronemin regulate expressions of TGF-β1 and UCP2 and induce mitochondrial reactive oxygen species (ROS) production in MCF-7 cancer cells. Cells were treated with different concentrations of heteronemin in the presence and absence of 1 nM E2 for 24 h. Cells were harvested and analyzed for (A) TGF-β1 expression, (B) UCP2 expression, (C) mitochondrial ROS production, and (D) SODs accumulation. The number of independent studies (n) = 4. *p < 0.05, **p < 0.01, ***p < 0.001, compared to the untreated control. ##p < 0.01, ###p < 0.001, compared to the same dosage of heteronemin treatment only.
Heteronemin Induces Sub G0/G1 Increase and Arrests Cell Cycle at G0/G1 Phase to Block Cell Proliferation
To understand whether E2 or heteronemin is involved in regulating cell death in breast cancer cells, we performed propidium iodide (PI) staining for further exploration. Percentages of the various cell phases in MCF-7 cells are shown in Figure 6. Cells were treated with E2 and 0.625 μM heteronemin in the presence and absence of SOT for 24 h. The results indicated that G0/G1 phase was reduced and S phase and G2/M phase were increased with E2 treatment. Heteronemin treatment not only increased sub G0/G1 and G0/G1 phase population but also decreased S phase. Compared with heteronemin treatment, co-treatment of E2 and heteronemin decreased sub G0/G1 and G0/G1 phase but increased S phase. These data suggest that heteronemin caused cell apoptosis, G0/G1 arrest, and then reduced cell proliferation. In addition, heteronemin could reverse the effects induced by E2. SOT, an inhibitor of PKC, enhanced the G0/G1 phase increasing and S phase decreasing by heteronemin.
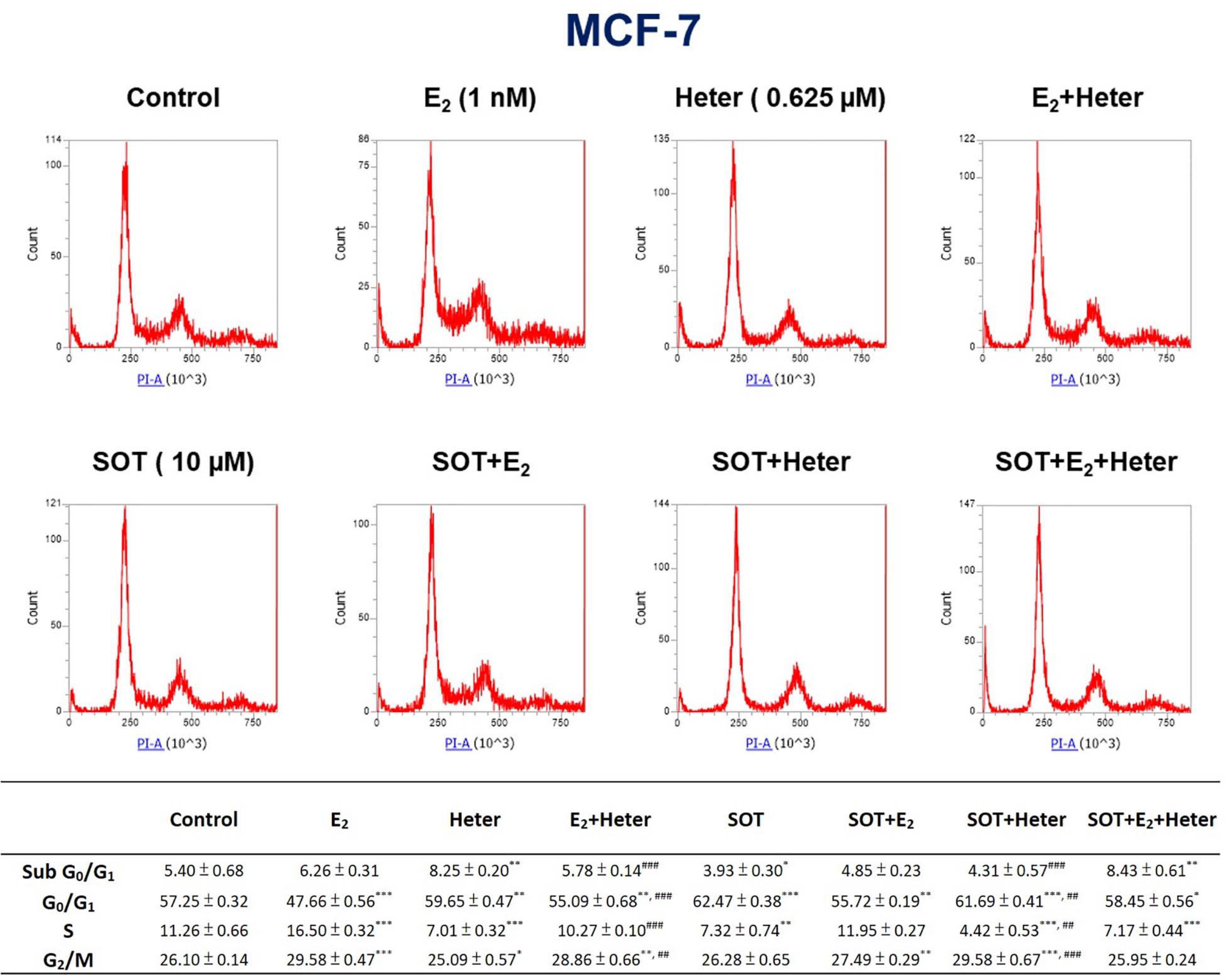
Figure 6. Estrogen, heteronemin, and sotrastaurin (SOT) affect proliferation that induces apoptosis and cell cycle arrest in breast cancer cells. MCF-7 cells were treated with 1 nM E2, 0.625 μM heteronemin in the presence and absence of 10 μM SOT for 24 h. Cells were harvested, and a flow cytometric assay was conducted as described in section “Materials and Methods.” n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, compared to the untreated control. ##p < 0.01, ###p < 0.001, compared to heteronemin treatment only.
In summary, heteronemin inhibited activation of ERK1/2 and STAT3 via different pathways in MCF-7 cells and MDA-MB-231 cells. The former was PKC-dependent. Heteronemin also inhibited proliferation-related gene expressions, increased proapoptotic gene expressions, and suppressed cell viability. Bcl-2/Bax ratio was downgraded in MDA-MB-231 cells but no change in MCF-7 cells after heteronemin treatment. Heteronemin was able to increase mitochondrial ROS production and SODs accumulation in MCF-7 cells. In the presence of E2, ROS production was increased but SODs were decreased to reverse heteronemin-induced anti-proliferative effects. Thus, E2 rescued heteronemin-induced anti-proliferative effects in both MCF-7 and MDA-MB-231 breast cancer cells via different mechanisms.
Discussion
Heteronemin caused anti-proliferation via cell apoptosis and cell cycle arrest in breast cancer cell lines are observed (Figures 1, 6). Heteronemin inhibited expressions of Ki-67, CCND-1, and PD-L1 in concentration-dependent manners and stimulated p21 expression in both MCF-7 and MDA-MB-231 cells (Figure 2). These results confirmed our recent observations in oral cancer cells (Huang et al., 2020) and another report (Kopf et al., 2013). On the other hand, heteronemin inhibited expressions of c-Myc and Bcl-2 in MDA-MB-231 cells but stimulated their expression in MCF-7 cells (Figure 2). In addition, E2 stimulated expression of those genes and reversed some inhibitory effects of heteronemin (Figure 3). Heteronemin reduced Bcl-2/Bax ratio in MDA-MB-231 cells but not in MCF-7 cells (Figure 3). Heteronemin also affected the protein expressions of the Bcl-2 family (Supplementary Figure 1), In MCF-7 cells, heteronemin increased Bcl-2, Bax, and Bid expression. However, Bcl-2 expression was suppressed by heteronemin in MDA-MB-231. Thus, Bcl-2/Bax ratio was downgraded in MDA-MB-231 cells but no change in MCF-7 cells after heteronemin treatment. E2 inhibited stimulatory effects of heteronemin and the Bcl-2/Bax ratio increased with combined treatment (Figure 3A). In MDA-MB-231 cells, E2 did not stimulate Bax expression although suppressed BAD expression slightly (Figure 3B). These results suggested that the ratio of Bcl-2/Bax should play a critical role in determining fate in MDA-MB-231 cells but not in MCF-7 cells. On the other hand, E2 activated ERK1/2, PKCα, and STAT3, and reversed effects of heteronemin on activation of Bcl-2 and reduction of BAD. Therefore, increased Bcl-2 expression in MCF-7 cells but reduced Bcl-2 expression in MDA-MB-231 cells by heteronemin may reflect different mechanisms in different types of breast cancer cells.
E2 treatment can stimulate PKC activation through ERα followed by the activation of PKCδ or via ERα directly upregulating PKC (Ronda et al., 2010). Therefore, activated PKC plays an important role in estrogen-stimulated breast cancer cell proliferation. In addition, the PKC inhibitor, AEB071 (sotrastaurin), was shown to be effective against triple-negative breast cancer cells (Byerly et al., 2016). Heteronemin repressed STAT3 (Wu et al., 2016). Heteronemin inhibited activation of STAT3 and ERK1/2 (Figure 4). Sotrastaurin inhibits Akt phosphorylation, NF-κB/STAT3 activation, and Mcl-1 upregulation. It also renders cells sensitive to arsenic trioxide (Amigo-Jiménez et al., 2015). Therefore, we used sotrastaurin to block PKC to reduce the NF-κB/STAT3 signaling pathway and investigated mechanisms in breast cancer cells.
Estrogen reversed the inhibitory effect of heteronemin against activation of ERK1/2 and STAT3 (Figure 4) in MCF-7 cancer cells and activation of PKC and STAT3 in MDA-MB-231 cancer cells (Figure 4) but diminished ERK1/2 in MDA-MB-231 cancer cells. These results suggested that E2 activates different signaling pathways in MCF-7 and MDA-MB-231 cancer cells. Activation of E2-induced PKC may mediate E2-dependent biological activities including cell proliferation. The PKC inhibitor, sotrastaurin, more completely inhibited STAT3 activation than did heteronemin in MCF-7 cells (Figure 4), however, but not in MDA-MB-231 cells (Figure 4). Estradiol binds to the αvβ3 integrin (Cody et al., 2007), and this possibly explains the result that cell viability of MDA-MB-231 triple-negative breast cancer cells was affected by E2.
Heteronemin is a farnesyl transferase inhibitor (FTI) that inhibits the cytarabine-induced, farnesyl transferase-mediated Ras activation and inhibits Ras downstream signal transduction pathways such as mitogen-activated protein kinases (MAPKs), activator protein (AP)-1, nuclear factor (NF)-κB, and c-Myc (Saikia et al., 2018). Overproduced ROS by conjugated estrogens during estrogen metabolism activates IκB kinase (IKK)-α and -β to increase the translocation of nuclear NF-κB (Kim et al., 2000). Alterations of mitochondrial metabolism may induce ROS overproduction to involve in estrogen-mediated carcinogenesis via induction of oxidative DNA damage (Tian et al., 2016). Additionally, blocking estrogen attenuates respiratory and metabolic responses and superoxide accumulation in estrogen-responsive breast cancer cells (Tan et al., 2002; Doan et al., 2004). Oxidative stress was postulated to be one of the mechanisms underlying E2’s carcinogenic effect in breast cancer. E2 increases mitochondrial-derived ROS by an unknown mechanism (Sastre-Serra et al., 2010). E2 significantly increased ROS production in MCF-7 cells (Figure 5C). UCPs regulate energy efficiency in mitochondria and production of ROS (Chouchani et al., 2016; Pierelli et al., 2017; Cadenas, 2018). They function as an adaptive anti-oxidant defense to protect against over-productive oxidation (Pierelli et al., 2017). E2 downregulated UCP expression and significantly increased mitochondrial ROS production in ER-positive MCF-7 cells (Figure 5). Heteronemin inhibited UCP2 expression in a dose-dependent manner (Figure 5B). Heteronemin also significantly increased ROS production at 0.625 and 1.25 μM (Figure 5C). Lu’s group showed that heteronemin treatment (2.56 μM) increased ROS levels in LNCaP cells (Lee Y. S. et al., 2018). These results suggest that heteronemin may suppress UCP2 accumulation to increase ROS production. UCP2 was shown to negatively modulate intracellular ROS production (Pierelli et al., 2017). In the presence of E2, 0.3125 μM heteronemin increased ROS production compared to heteronemin only (Figure 5C). Combined treatment with E2 and heteronemin consistently suppressed UCP2 expression (Figure 5B), suggesting that downregulation of UCP2 may play an important role in heteronemin-induced anti-proliferation in ER-positive breast cancer cells in the presence of estradiol. Overall, these results suggest that through an ER-dependent mechanism, E2 may increase mitochondrial ROS production by repressing UCPs, which offers a new perspective on the understanding of why E2 is a risk factor for breast cancer. Heteronemin stimulated TGF-β1 expression at 0.625 μM but inhibited its expression at 1.25 μM (Figure 5A). The stimulatory effect of E2 on TGF-β was inhibited by heteronemin treatment (Figure 5A). The inhibitory effect of E2 on UCP2 was further enhanced by heteronemin treatment (Figure 5B). Downregulation of UCP2 expression by TGF-β-SMAD4 signaling was shown to play a regulatory role in mitochondrial ROS formation (Kim and Lee, 2018). Thus, heteronemin stimulated TGF-β1 expression to downregulate UCP expression and further increase ROS production in MCF-7 cells at 0.625 μM.
It is not surprising to observe that E2 increased SODs accumulation (Figure 5D) as reported (Rao et al., 2008). On the other hand, anti-oxidant also stimulates ROS production in previous studies (Heo et al., 2018; Kim et al., 2019). Both ceramide and resveratrol stimulate mitochondrial potential. Thus, at certain concentrations, heteronemin may increase SOD to activate mitochondrial potential (Figure 5). However, the combination of heteronemin and E2 decreased SOD accumulation to reach the balance between oxidation and anti-oxidation.
The ROS scavenger, N-acetyl cysteine (NAC), inhibited heteronemin-induced mitochondrial ROS production and cell apoptosis (Lin et al., 2018). Heteronemin significantly increased both cellular ROS and mtROS. It also induced the loss of the mitochondrial membrane potential (MMP) in lung cancer cells (Cheng et al., 2019). It increased the percentage of apoptotic cells and ROS in Molt4 cells (Chen et al., 2018). Heteronemin -treated lung cancer cells showed a significant increase in both cellular ROS and mtROS, which in turn caused the loss of the MMP. Heteronemin decreased expressions of the anti-oxidant enzymes Cu/ZnSOD, MnSOD, and catalase (Cheng et al., 2019). Pretreatment with the mitochondrion-targeted anti-oxidant, MitoTEMPO, reduced heteronemin-induced apoptosis through a mitochondrion-dependent apoptotic pathway, which was accompanied by increased cell viability, decreased mtROS, enhanced MMP, and suppressed expressions of cleaved caspase-3 and caspase-9 proteins (Cheng et al., 2019). Oxidative phosphorylation performed in mitochondria and glycolysis in the cytoplasm were inhibited, which subsequently reduced downstream ATP production (Cheng et al., 2019). Additionally, Chang et al., 2021 reported heteronemin may with high toxicity and causing animal death (Chang et al., 2021). However, Lee M. G. et al. (2018) also shown heteronemin can using safely with a lower dosage.
These results suggest heteronemin inhibited activation of ERK1/2 and STAT3 in both ER-positive and ER-negative breast cancer cells. In addition, heteronemin downregulated the expression of Ki-67, CCND1, EGFR, and PD-L1, but upregulated p21 and BAD expression. However, Bcl-2/Bax ratio was downgraded in MDA-MB-231 cells but no change in MCF-7 cells after heteronemin treatment. It restrained UCP2 expression, extended ROS production, increased SODs accumulation, induced G0/G1 arrest, and caused anti-proliferation in breast cancer MCF-7 cells. On the other hand, E2 activated ERK1/2, PKC, and STAT3, increased ROS production, and recued heteronemin-induced biological activities.
Conclusion
In conclusion, heteronemin inhibited ER-positive and ER-negative breast cancer cell proliferation via different mechanisms (Figure 7), but less effect on normal cells. Additionally, heteronemin also could overcome E2 stimulated proliferation. Those results suggest Heteronemin had a strong capacity to inhibit proliferation in both MCF-7 and MDA-MB-231 breast cancer cells. Thus, heteronemin has a potential as an anti-cancer drug.
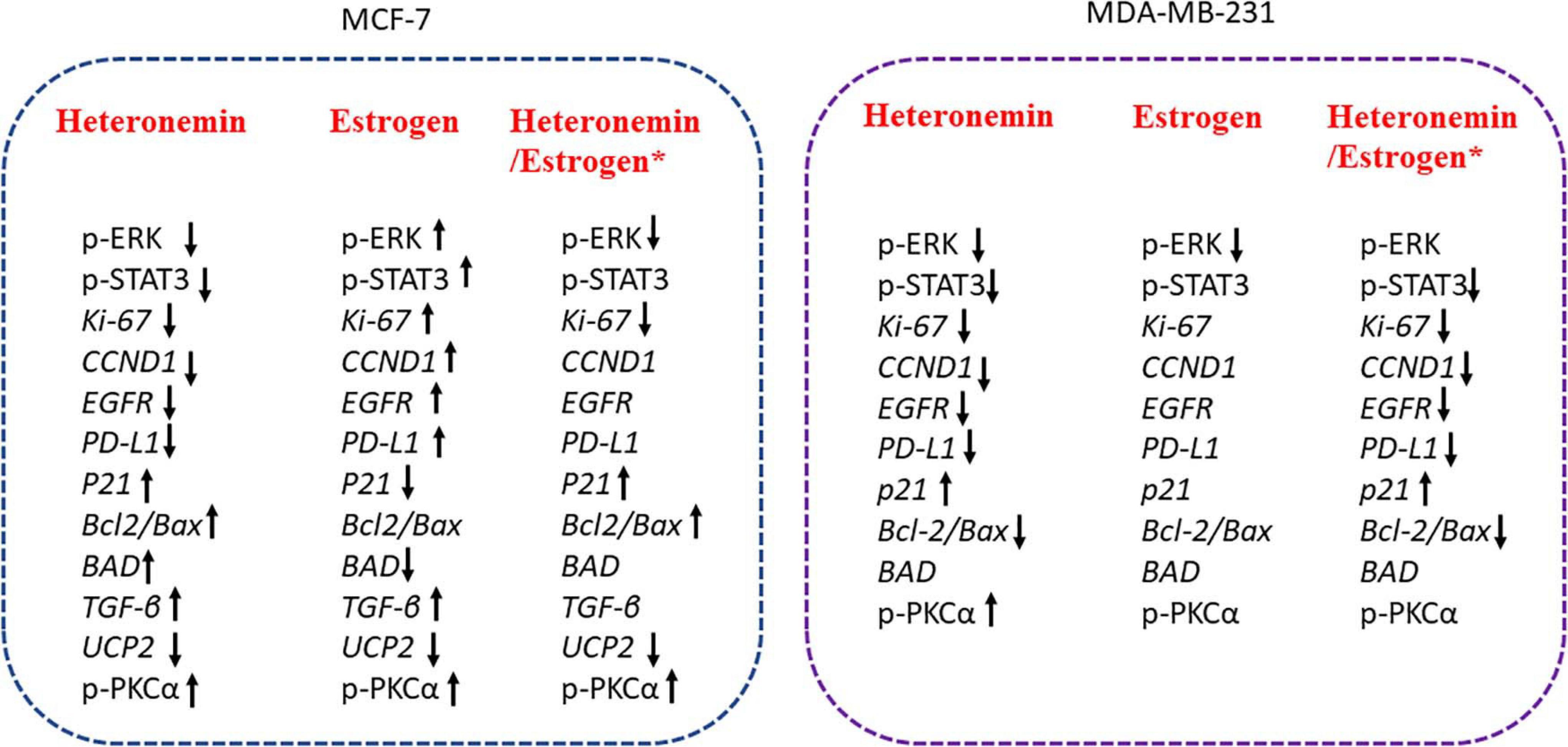
Figure 7. The schematic of heteronemin reverses E2–induced mechanism in breast cancer cells. *Significantly different compare with the untreated control group. Heteronemin: 0.625 μM, E2: 1nM.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
Y-CY, M-CL, and H-YL contributed the study concept and methodology design of this study. Z-LL, T-YH, and H-YC conducted the experiments. Z-LL, K-WS, and H-YC analyzed and interpreted the data. Y-CY and H-YL wrote the manuscript. Z-LL, H-YC, T-YH, C-YL, H-MH, S-YL, JW-P, PD, and KW helped in revising the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported in part by the Chair Professor Research Fund to KW and JW-P, the TMU Research Center of Cancer Translational Medicine from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project, by the Ministry of Education (MOE) in Taiwan (DP2-107-20000), and by grants from the Ministry of Science and Technology, Taiwan (MOST109-2314-B-038-038 to Y-CY; MOST109-2314-B-038-125 to H-YL; and MOST109-2124-M-038-001 to JW-P).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.688607/full#supplementary-material
References
Amigo-Jiménez, I., Bailón, E., Aguilera-Montilla, N., Terol, M. J., García-Marco, J. A., and García-Pardo, A. (2015). Bone marrow stroma-induced resistance of chronic lymphocytic leukemia cells to arsenic trioxide involves Mcl-1 upregulation and is overcome by inhibiting the PI3Kδ or PKCβ signaling pathways. Oncotarget 6, 44832–44848.
Anampa, J., Makower, D., and Sparano, J. A. (2015). Progress in adjuvant chemotherapy for breast cancer: an overview. BMC Med. 13:195. doi: 10.1186/s12916-015-0439-8
Bhat, A. H., Dar, K. B., Anees, S., Zargar, M. A., Masood, A., Sofi, M. A., et al. (2015). Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 74, 101–110.
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., and Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424. doi: 10.3322/caac.21492
Byerly, J., Halstead-Nussloch, G., Ito, K., Katsyv, I., and Irie, H. Y. (2016). PRKCQ promotes oncogenic growth and anoikis resistance of a subset of triple-negative breast cancer cells. Breast Cancer Res. 18:95.
Cadenas, S. (2018). Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta Bioenerg. 1859, 940–950. doi: 10.1016/j.bbabio.2018.05.019
Cavalieri, E., and Rogan, E. (2014). The molecular etiology and prevention of estrogen-initiated cancers: Ockham’s Razor: pluralitas non est ponenda sine necessitate. Plurality should not be posited without necessity. Mol. Aspects Med. 36, 1–55. doi: 10.1016/j.mam.2013.08.002
Cavalieri, E. L., Stack, D. E., Devanesan, P. D., Todorovic, R., Dwivedy, I., Higginbotham, S., et al. (1997). Molecular origin of cancer: catechol estrogen-3,4-quinones as endogenous tumor initiators. Proc. Natl. Acad. Sci. U.S.A. 94, 10937–10942. doi: 10.1073/pnas.94.20.10937
Chang, W. T., Bow, Y. D., Fu, P. J., Li, C. Y., Wu, C. Y., Chang, Y. H., et al. (2021). A marine terpenoid, heteronemin, induces both the apoptosis and ferroptosis of hepatocellular carcinoma cells and involves the ROS and MAPK pathways. Oxid. Med. Cell. Longev. 2021:7689045.
Chen, Y. C., Lu, M. C., El-Shazly, M., Lai, K. H., Wu, T. Y., Hsu, Y. M., et al. (2018). Breaking down leukemia walls: heteronemin, a sesterterpene derivative, induces apoptosis in leukemia Molt4 cells through Oxidative stress, mitochondrial dysfunction and induction of talin expression. Mar, Drugs 16:212. doi: 10.3390/md16060212
Cheng, M. H., Huang, H. L., Lin, Y. Y., Tsui, K. H., Chen, P. C., Cheng, S. Y., et al. (2019). BA6 induces apoptosis via stimulation of reactive oxygen species and inhibition of oxidative phosphorylation in human lung cancer cells. Oxid. Med. Cell. Longev. 2019:6342104.
Chin, Y. T., He, Z. R., Chen, C. L., Chu, H. C., Ho, Y., Su, P. Y., et al. (2019). Tetrac and NDAT induce anti-proliferation via integrin αvβ3 in colorectal cancers with different K-RAS status. Front. Endocrinol. 10:130.
Chipuk, J. E., Kuwana, T., Bouchier-Hayes, L., Droin, N. M., Newmeyer, D. D., Schuler, M., et al. (2004). Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303, 1010–1014. doi: 10.1126/science.1092734
Chouchani, E. T., Kazak, L., Jedrychowski, M. P., Lu, G. Z., Erickson, B. K., Szpyt, J., et al. (2016). Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature 532, 112–116. doi: 10.1038/nature17399
Cobbaut, M., and Van Lint, J. (2018). Function and regulation of protein kinase d in oxidative stress: a tale of isoforms. Oxid. Med. Cell. Longev. 2018:2138502.
Cody, V., Davis, P. J., and Davis, F. B. (2007). Molecular modeling of the thyroid hormone interactions with alpha v beta 3 integrin. Steroids 72, 165–170. doi: 10.1016/j.steroids.2006.11.008
Devanesan, P., Santen, R. J., Bocchinfuso, W. P., Korach, K. S., Rogan, E. G., and Cavalieri, E. (2001). Catechol estrogen metabolites and conjugates in mammary tumors and hyperplastic tissue from estrogen receptor-alpha knock-out (ERKO)/Wnt-1 mice: implications for initiation of mammary tumors. Carcinogenesis 22, 1573–1576. doi: 10.1093/carcin/22.9.1573
Doan, V. D., Gagnon, S., and Joseph, V. (2004). Prenatal blockade of estradiol synthesis impairs respiratory and metabolic responses to hypoxia in newborn and adult rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 287, R612–R618.
el-Deiry, W. S., Harper, J. W., O’connor, P. M., Velculescu, V. E., Canman, C. E., Jackman, J., et al. (1994). WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 54, 1169–1174.
Fussell, K. C., Udasin, R. G., Smith, P. J., Gallo, M. A., and Laskin, J. D. (2011). Catechol metabolites of endogenous estrogens induce redox cycling and generate reactive oxygen species in breast epithelial cells. Carcinogenesis 32, 1285–1293. doi: 10.1093/carcin/bgr109
Gwangwa, M. V., Joubert, A. M., and Visagie, M. H. (2019). Effects of glutamine deprivation on oxidative stress and cell survival in breast cell lines. Biol. Res. 52:15.
Heo, J. R., Kim, S. M., Hwang, K. A., Kang, J. H., and Choi, K. C. (2018). Resveratrol induced reactive oxygen species and endoplasmic reticulum stress-mediated apoptosis, and cell cycle arrest in the A375SM malignant melanoma cell line. Int. J. Mol. Med. 42, 1427–1435.
Hofseth, L. J., Raafat, A. M., Osuch, J. R., Pathak, D. R., Slomski, C. A., and Haslam, S. Z. (1999). Hormone replacement therapy with estrogen or estrogen plus medroxyprogesterone acetate is associated with increased epithelial proliferation in the normal postmenopausal breast. J. Clin. Endocrinol. Metab. 84, 4559–4565. doi: 10.1210/jc.84.12.4559
Huang, C. H., Huang, T. Y., Chang, W. J., Pan, Y. S., Chu, H. R., Li, Z. L., et al. (2020). Combined treatment of heteronemin and tetrac induces antiproliferation in oral cancer cells. Mar. Drugs 18:348. doi: 10.3390/md18070348
Joo, C. K., Kim, H. S., Park, J. Y., Seomun, Y., Son, M. J., and Kim, J. T. (2008). Ligand release-independent transactivation of epidermal growth factor receptor by transforming growth factor-beta involves multiple signaling pathways. Oncogene 27, 614–628. doi: 10.1038/sj.onc.1210649
Kim, D. W., Sovak, M. A., Zanieski, G., Nonet, G., Romieu-Mourez, R., Lau, A. W., et al. (2000). Activation of NF-kappaB/Rel occurs early during neoplastic transformation of mammary cells. Carcinogenesis 21, 871–879. doi: 10.1093/carcin/21.5.871
Kim, T. H., Park, J. H., and Woo, J. S. (2019). Resveratrol induces cell death through ROS-dependent downregulation of Notch1/PTEN/Akt signaling in ovarian cancer cells. Mol. Med. Rep. 19, 3353–3360.
Kim, Y. H., and Lee, S. H. (2018). TGF-β/SMAD4 mediated UCP2 downregulation contributes to Aspergillus protease-induced inflammation in primary bronchial epithelial cells. Redox Biol. 18, 104–113. doi: 10.1016/j.redox.2018.06.011
Kollmann, Z., Bersinger, N. A., Mckinnon, B. D., Schneider, S., Mueller, M. D., and Von Wolff, M. (2015). Anti-Müllerian hormone and progesterone levels produced by granulosa cells are higher when derived from natural cycle IVF than from conventional gonadotropin-stimulated IVF. Reprod. Biol. Endocrinol. 13:21.
Kopf, S., Viola, K., Atanasov, A. G., Jarukamjorn, K., Rarova, L., Kretschy, N., et al. (2013). In vitro characterisation of the anti-intravasative properties of the marine product heteronemin. Arch. Toxicol. 87, 1851–1861. doi: 10.1007/s00204-013-1045-1
Lee, M. G., Liu, Y. C., Lee, Y. L., El-Shazly, M., Lai, K. H., Shih, S. P., et al. (2018). Heteronemin, a marine sesterterpenoid-type metabolite, induces apoptosis in prostate LNcap cells via oxidative and ER stress combined with the inhibition of topoisomerase II and Hsp90. Mar. Drugs 16:204. doi: 10.3390/md16060204
Lee, Y. S., Chin, Y. T., Shih, Y. J., Nana, A. W., Chen, Y. R., Wu, H. C., et al. (2018). Thyroid hormone promotes beta-catenin activation and cell proliferation in colorectal cancer. Horm. Cancer 9, 156–165. doi: 10.1007/s12672-018-0324-y
Lin, H. Y., Tey, S. L., Ho, Y., Chin, Y. T., Wang, K., Whang-Peng, J., et al. (2018). Heteronemin induces anti-proliferation in cholangiocarcinoma cells via inhibiting TGF-β pathway. Mar. Drugs 16:489. doi: 10.3390/md16120489
Lønne, G. K., Masoumi, K. C., Lennartsson, J., and Larsson, C. (2009). Protein kinase Cdelta supports survival of MDA-MB-231 breast cancer cells by suppressing the ERK1/2 pathway. J. Biol. Chem. 284, 33456–33465. doi: 10.1074/jbc.m109.036186
Nana, A. W., Wu, S. Y., Yang, Y. S., Chin, Y. T., Cheng, T. M., Ho, Y., et al. (2018). Nano-diamino-tetrac (NDAT) enhances resveratrol-induced antiproliferation by action on the RRM2 pathway in colorectal cancers. Horm. Cancer 9, 349–360. doi: 10.1007/s12672-018-0334-9
Okoh, V. O., Felty, Q., Parkash, J., Poppiti, R., and Roy, D. (2013). Reactive oxygen species via redox signaling to PI3K/AKT pathway contribute to the malignant growth of 4-hydroxy estradiol-transformed mammary epithelial cells. PLoS One 8:e54206. doi: 10.1371/journal.pone.0054206
Papi, A., Orlandi, M., Bartolini, G., Barillari, J., Iori, R., Paolini, M., et al. (2008). Cytotoxic and antioxidant activity of 4-methylthio-3-butenyl isothiocyanate from Raphanus sativus L. (Kaiware Daikon) sprouts. J Agric Food Chem 56, 875–883. doi: 10.1021/jf073123c
Pierelli, G., Stanzione, R., Forte, M., Migliarino, S., Perelli, M., Volpe, M., et al. (2017). Uncoupling protein 2: a key player and a potential therapeutic target in vascular diseases. Oxid. Med. Cell. Longev. 2017:7348372.
Poillet-Perez, L., Despouy, G., Delage-Mourroux, R., and Boyer-Guittaut, M. (2015). Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 4, 184–192. doi: 10.1016/j.redox.2014.12.003
Rao, A. K., Ziegler, Y. S., Mcleod, I. X., Yates, J. R., and Nardulli, A. M. (2008). Effects of Cu/Zn superoxide dismutase on estrogen responsiveness and oxidative stress in human breast cancer cells. Mol. Endocrinol. 22, 11 13–1124.
Ronda, A. C., Buitrago, C., and Boland, R. (2010). Role of estrogen receptors, PKC and Src in ERK2 and p38 MAPK signaling triggered by 17β-estradiol in skeletal muscle cells. J. Steroid Biochem. Mol. Biol. 122, 287–294. doi: 10.1016/j.jsbmb.2010.05.002
Saikia, M., Retnakumari, A. P., Anwar, S., Anto, N. P., Mittal, R., Shah, S., et al. (2018). Heteronemin, a marine natural product, sensitizes acute myeloid leukemia cells towards cytarabine chemotherapy by regulating farnesylation of Ras. Oncotarget 9, 18115–18127. doi: 10.18632/oncotarget.24771
Sastre-Serra, J., Valle, A., Company, M. M., Garau, I., Oliver, J., and Roca, P. (2010). Estrogen down-regulates uncoupling proteins and increases oxidative stress in breast cancer. Free Radic. Biol. Med. 48, 506–512. doi: 10.1016/j.freeradbiomed.2009.11.025
Schumacher, M., Cerella, C., Eifes, S., Chateauvieux, S., Morceau, F., Jaspars, M., et al. (2010). Heteronemin, a spongean sesterterpene, inhibits TNF alpha-induced NF-kappa B activation through proteasome inhibition and induces apoptotic cell death. Biochem. Pharmacol. 79, 610–622. doi: 10.1016/j.bcp.2009.09.027
Tan, D. J., Bai, R. K., and Wong, L. J. (2002). Comprehensive scanning of somatic mitochondrial DNA mutations in breast cancer. Cancer Res. 62, 972–976.
Tian, H., Gao, Z., Wang, G., Li, H., and Zheng, J. (2016). Estrogen potentiates reactive oxygen species (ROS) tolerance to initiate carcinogenesis and promote cancer malignant transformation. Tumour Biol. 37, 141–150. doi: 10.1007/s13277-015-4370-6
Weng, C. J., Yang, Y. T., Ho, C. T., and Yen, G. C. (2009). Mechanisms of apoptotic effects induced by resveratrol, dibenzoylmethane, and their analogues on human lung carcinoma cells. J. Agric. Food Chem. 57, 5235–5243. doi: 10.1021/jf900531m
Wu, J. C., Wang, C. T., Hung, H. C., Wu, W. J., Wu, D. C., Chang, M. C., et al. (2016). Heteronemin is a novel c-Met/STAT3 inhibitor against advanced prostate cancer cells. Prostate 76, 1469–1483. doi: 10.1002/pros.23230
Wu, S. Y., Sung, P. J., Chang, Y. L., Pan, S. L., and Teng, C. M. (2015). Heteronemin, a spongean sesterterpene, induces cell apoptosis and autophagy in human renal carcinoma cells. Biomed. Res. Int. 2015:738241.
Keywords: heteronemin, estrogen, breast cancer, estrogen receptor status, anti-proliferation, mitochondrial ROS
Citation: Yang Y-CSH, Li Z-L, Huang T-Y, Su K-W, Lin C-Y, Huang C-H, Chen H-Y, Lu M-C, Huang H-M, Lee S-Y, Whang-Peng J, Lin H-Y, Davis PJ and Wang K (2021) Effect of Estrogen on Heteronemin-Induced Anti-proliferative Effect in Breast Cancer Cells With Different Estrogen Receptor Status. Front. Cell Dev. Biol. 9:688607. doi: 10.3389/fcell.2021.688607
Received: 08 April 2021; Accepted: 02 July 2021;
Published: 26 July 2021.
Edited by:
Leticia Veras Costa Lotufo, University of São Paulo, BrazilReviewed by:
Glaucia Maria Machado-Santelli, University of São Paulo, BrazilPaula Christine Jimenez, Federal University of São Paulo, Brazil
Copyright © 2021 Yang, Li, Huang, Su, Lin, Huang, Chen, Lu, Huang, Lee, Whang-Peng, Lin, Davis and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hung-Yun Lin, bGluaHlAdG11LmVkdS50dw==