 Adebayo Oluwafemi Adekunle1†
Adebayo Oluwafemi Adekunle1† Gabriel Komla Adzika1†
Gabriel Komla Adzika1† Richard Mprah1
Richard Mprah1 Marie Louise Ndzie Noah1
Marie Louise Ndzie Noah1 Joseph Adu-Amankwaah1
Joseph Adu-Amankwaah1 Ruqayya Rizvi2
Ruqayya Rizvi2 Nazma Akhter1
Nazma Akhter1 Hong Sun1,2*
Hong Sun1,2*- 1Department of Physiology, Xuzhou Medical University, Xuzhou, China
- 2Xuzhou Medical University, Xuzhou, China
Heart failure (HF) remains a public health concern as it is associated with high morbidity and death rates. In particular, heart failure with preserved ejection fraction (HFpEF) represents the dominant (>50%) form of HF and mostly occurring among postmenopausal women. Hence, the initiation and progression of the left ventricular diastolic dysfunctions (LVDD) (a typically clinical manifestation of HFpEF) in postmenopausal women have been attributed to estrogen deficiency and the loss of its residue cardioprotective effects. In this review, from a pathophysiological and immunological standpoint, we discuss the probable multiple pathomechanisms resulting in HFpEF, which are facilitated by estrogen deficiency. The initial discussions recap estrogen and estrogen receptors (ERs) and β-adrenergic receptors (βARs) signaling under physiological/pathological states to facilitate cardiac function/dysfunction, respectively. By reconciling these prior discussions, attempts were made to explain how the loss of estrogen facilitates the disruptions both ERs and βARs-mediated signaling responsible for; the modulation of intra-cardiomyocyte calcium homeostasis, maintenance of cardiomyocyte cytoskeletal and extracellular matrix, the adaptive regulation of coronary microvascular endothelial functions and myocardial inflammatory responses. By scaffolding the disruption of these crucial intra- and extra-cardiomyocyte physiological functions, estrogen deficiency has been demonstrated to cause LVDD and increase the incidence of HFpEF in postmenopausal women. Finally, updates on the advancements in treatment interventions for the prevention of HFpEF were highlighted.
Introduction
Heart failure (HF) remains a public health concern as it is associated with high rates of morbidity and deaths that ultimately result from cardiovascular diseases (Roger, 2013). Left ventricular ejection fraction (LVEF) is a crucial index in ascertaining the extent of cardiac dysfunction in HF patients. Hence, by the characterization of the state of the left ventricular’s functionality in ejection fraction, HF is classified into three subtypes, namely; Heart failure with reduced ejection fraction (HFrEF) (LVEF ≤ 40%), heart failure with mid-range ejection fraction (HFmEF) (LVEF 40–49%), and heart failure with preserved ejection fraction (HFpEF) (LVEF ≥ 50%) (Masri et al., 2017; Özlek et al., 2019). Both HFrEF and HFmEF are the predominant forms of HF in men, while HFpEF represents the dominant form of HF, mostly among postmenopausal women (Özlek et al., 2019). Even so, HFpEF accounts for nearly 50% of the HF recorded according to clinical demographics (Lam et al., 2011; Ma et al., 2020). The typical clinical presentation of HFpEF is a left ventricular diastolic dysfunction (LVDD) with an intact systolic function along with other HF signs and symptoms such as shortness of breath, fatigue, and chest pain, among others (Ma et al., 2020).
Circulating catecholamines facilitates the heart’s inotropy and chronotropic functions by signaling via β-adrenergic receptors (βARs) under physiological state. However, hyperactivity of the sympathetic nervous system (SNS) during stress upregulates circulating catecholamines, which overstimulates βARs, causing the receptors to become dysfunctional, leading to their desensitization and downregulation (Adzika et al., 2019). Unfortunately, the calcium handling proteins, such as the L-type calcium channel (LTCC), which ensure homeostatic levels of Ca2+ in cardiomyocytes for proper cardiac contractions, are downstream of the βARs (Aikawa et al., 2017; Ahmad and Gerson, 2018; Morimoto et al., 2020). Hence, the dysfunctionalities of the receptors during chronic catecholamine stress disrupts the intracellular Ca2+ and cardiac contractility. Also implicated in the pathogenesis and exacerbation of HFpEF are the alterations in myocardial extracellular matrix (ECM), cardiac cytoskeletal proteins, and coronary microvascular endothelial (CME) dysfunctionalities, plus maladaptive inflammatory responses in the myocardia. Regardless, estrogen (E2) and estrogen receptors (ERs) have been shown to adaptively modulate the expressions, responses, and activities of the aforementioned factors implicated in the initiation and progression of HFpEF (Sophonsritsuk et al., 2013; Michalson et al., 2018). Hence, a lower incidence of the disease condition is observed in premenopausal women than postmenopausal women and males of all age cohorts (Michalson et al., 2018; Sabbatini and Kararigas, 2020; Ndzie Noah et al., 2021).
This review seeks to discuss the probable mechanisms aggravating the incidence of HFpEF in postmenopausal women from a pathophysiological and immunological standpoint. The initial discussion recaps E2 and ERs and βARs signaling under physiological/pathological states to facilitate cardiac function/dysfunction, respectively. Finally, by reconciling these prior discussions, we explain the possible pathomechanisms of how E2 deficiency scaffolds signaling cascades that hastens the incidence of HFpEF in postmenopausal women and provided updates on the advancements in treatment interventions.
Physiological and Pathophysiological Interplays Between E2 and ERs, and βARs in Cardiac Function Modulation
Physiological Interplays
ERs (ERα, ERβ, and GPR30) and βARs (mainly, β1AR, and β2AR) are both predominate seven transmembrane-spanning receptors expressed in cardiomyocytes, cardiac fibroblast, and vascular tissues, and mediates signaling crucial for their cellular functions (Mahmoodzadeh et al., 2010). Typically, ERs mediate estrogenic responses, which are classified as rapid/non-genomic (via GPR30) and non-rapid/genomic (via ERα and ERβ) (Asokan Shibu et al., 2017; Ndzie Noah et al., 2021). Recently, it has been suggested that GPR30 could indirectly induce genomic effects through interactions with ERα and ERβ to facilitate physiological functions (Fuentes and Silveyra, 2019). Meanwhile, under physiological conditions, β1AR and β2AR mediates catecholamine responses rapidly via Gαs/cAMP/PKA and/or Gαi/PI3K/Akt to facilitate cardiac inotropy or prevent cardiac insults (Adzika et al., 2019). Similar to β2AR, GPR30 is capable of pleiotropic signaling via Gαs/cAMP/PKA and/or Gαi/PI3K/Akt in cardiomyocytes; hence, this is suggestive of their possible multi-interplays in signaling to facilitate proper cardiac functions, as demonstrated (Hou et al., 2018; Machuki et al., 2018; Möller et al., 2018; Ndzie Noah et al., 2021) (Figure 1). In cardiomyocytes, the activation of cAMP/PKA downstream GPR30 and βARs results in the phosphorylation of various calcium handling proteins such as LTCC, ryanodine receptor (RyR), phospholamban (PLB), sarcoplasmic reticulum Ca2+ ATPase (SERCA), and sodium-calcium exchanger (NCX), as well as myofilament proteins [cardiac troponin I (cTnI), myosin binding protein-C (MyBP-C and titin)] to facilitate inotropy, chronotropic, and relaxation (Tilley, 2011; Najafi et al., 2016). While this signaling is prototypic of βARs activation by catecholamine under physiological conditions, E2 and ERs have been shown in our previous studies to adaptively modulate the cascade by enhancing βARs expression and activities (Hou et al., 2018; Machuki et al., 2019; Ndzie Noah et al., 2021). Furthermore, the cardiac cytoskeletal proteins (such as titin) and ECM [comprising of matrix metalloproteinases (MMPs) and tissue inhibitors of matrix metalloproteinases (TIMPs)] perform essential functions for maintaining the myocardial architecture. Also, other cellular constituents; namely; fibroblasts and resident immune cells, which make up the myocardial architecture and ensure proper cardiac functions, have been shown to be adaptively modulated by the direct or indirect interactive signaling of βARs and ERs (Kim et al., 2002; Chancey et al., 2005; Rienks et al., 2014; El Hajj et al., 2017; Ambhore et al., 2019).
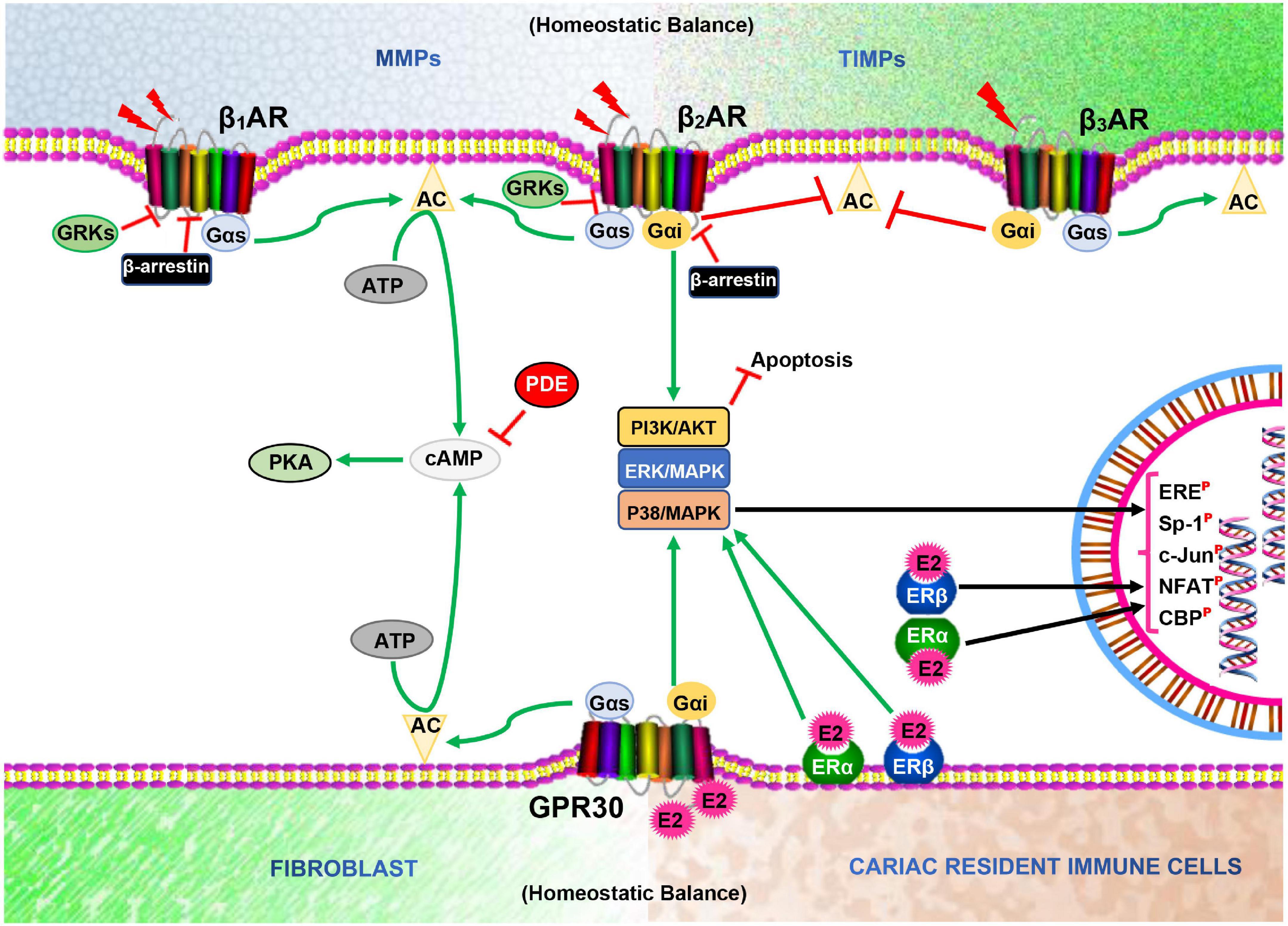
Figure 1. Illustration of homeostatic state between MMPs and TIMPs, and Fibroblast and Cardiac resident immune cells as well as β-adrenergic receptors (βARs) and Estrogen receptors (ERs) signaling interplays in the cardiomyocytes. AC, Adenylyl cyclase; cAMP, Cyclic adenosine monophosphate; PKA, Protein kinase A; GRKs, G protein-coupled receptor kinases; E2, Estrogen; PDE, Phosphodiesterase; ERE, Estrogen receptor element; Sp-1, Stimulating protein-1; NFAT, Nuclear factor of activated T cells;  , Phosphorylated;
, Phosphorylated;  , Inhibit;
, Inhibit;  , Catecholamine.
, Catecholamine.
Pathophysiological Interplays
Over the recent decades, attempts to elucidate the pathomechanisms expediting the incidence of cardiovascular diseases (including HFpEF) in postmenopausal women have revealed that the synergy of βARs hyperstimulation and E2 deficiency permits maladaptive signaling cascades which drives the adverse cardiac remodeling (Sherwood et al., 2010; Fares et al., 2013; Machi et al., 2016; Hou et al., 2018; Ndzie Noah et al., 2021). Apparently, the loss of E2 during menopause permits an increase in SNS activities, hence, increases the level of circulating catecholamines which overstimulates the βARs and causes their desensitization, depletion, and dysregulation. This implicates βARs (mainly, β2AR) in the pathogenesis of LVDD and, ultimately, HFpEF (Brooks and Conrad, 2009; Wang et al., 2019; Wu et al., 2019). Meanwhile, the presence of E2 would have averted the initiations of the cardiac dysfunction via multiple mechanisms such as: (1) Modulating the activities of the SNS to keep the levels of circulating catecholamines normal to ensure proper cardiac health. (2) Mitigating the dysregulation of βARs by regulating G-protein-coupled receptor kinases (GRKs) and other kinases that disrupt the receptor’s physiological functions (Abraham et al., 2018; Adzika et al., 2019). (3) Signaling via GPR30/Gai/PI3K/Akt to compliment β2AR/Gai/PI3K/Akt signaling to reenforce the attenuation of any adverse cardiac remodeling that may distort the myocardial architecture (ECM and cardiac cytoskeletal proteins) and ultimately induce HFpEF (El Hajj et al., 2017; Ndzie Noah et al., 2021). (4) Preventing βARs-induced Ca2+ hypersensitivity of the cardiac contractile apparatus, which results in LVDD (Thawornkaiwong et al., 2007; Hamdani et al., 2013). (5) Sustaining the bioavailability of nitric oxide (NO) for CME vasodilatory function via GPR30/Gai-induced Akt, ERK1/2, or CaMK-II phosphorylation of endothelial nitric oxide synthase (eNOS) at Ser1177 when β2AR/Gai/PI3K/Akt-induced NO production is dysregulated, thereby, reduces the initiation or progression toward HFpEF (Fulton et al., 1999; Kolluru et al., 2010; Sabbatini and Kararigas, 2020; Ndzie Noah et al., 2021). (6) Downregulating inflammatory cytokines; interleukin (IL)-6 and tumor necrosis factor-alpha (TNFα) as well as NFκB to prevent excess fibroblast activation and proliferation, which distorts the ECM homeostasis and hastens LVDD/HFpEF progression (McLarty et al., 2013; Ambhore et al., 2019; Sabbatini and Kararigas, 2020). However, unlike premenopausal women, postmenopausal women lose all these multiple cardioprotective mechanisms mediated and exerted by E2 and ERs directly or indirectly via βARs modulations.
Heart Failure With Preserved Ejection Fraction Pathomechanism Scaffolds Facilitated by Estrogen Deficiency
Herein, the discussions will be centered on explaining the probable pathomechanisms involved in the initiation and progression of HFpEF in postmenopausal women, mostly due to the synergy of E2 deficiency and βARs dysfunctionalities.
Intra-Cardiomyocyte Maladaptive Alterations
Disruption of Ca2+ Handling Homeostatic Functions
Focusing on individual calcium handling proteins to assess the extent of their alterations and dysfunctionalities resulting from E2 deficiency reveals multiple vulnerabilities in the Ca2+ cycle that might explain the predisposition of postmenopausal women to HFpEF.
In cardiomyocytes, Ca2+ regulates both mitochondrial and contractile activities; hence, it plays a crucial role in sustaining the energy demands and supplies of the myocardium. Although primarily regulated by signaling via βARs, E2 and ERs have also been shown to influence the expression of the receptor and its downstream proteins (PKA, LTCC, PLB, SERCA, RyR, and NCX) involved in maintaining the intracellular calcium homeostasis (Michalson et al., 2018; Lai et al., 2019; Machuki et al., 2019; Maslov et al., 2019). As depicted in Figure 2, the initial phase of the PKA-dependent calcium cycle involves LTCC allowing entry of Ca2+ into cardiomyocytes upon phosphorylation and the influx of Ca2+ into the cytosol from the sarcoplasmic reticulum (SR) via RyR to facilitate contraction. The latter phase, which ensures relaxation, requires that the excess Ca2+ be taken back into the SR via SERCA and/or expelled out of the myocyte via NCX (Fearnley et al., 2011; Machuki et al., 2018; Jiao et al., 2020). Hence, the inability to effectively carry out the cascades required for the latter results in LVDD, a typical characteristic of HFpEF (Kilfoil et al., 2020).
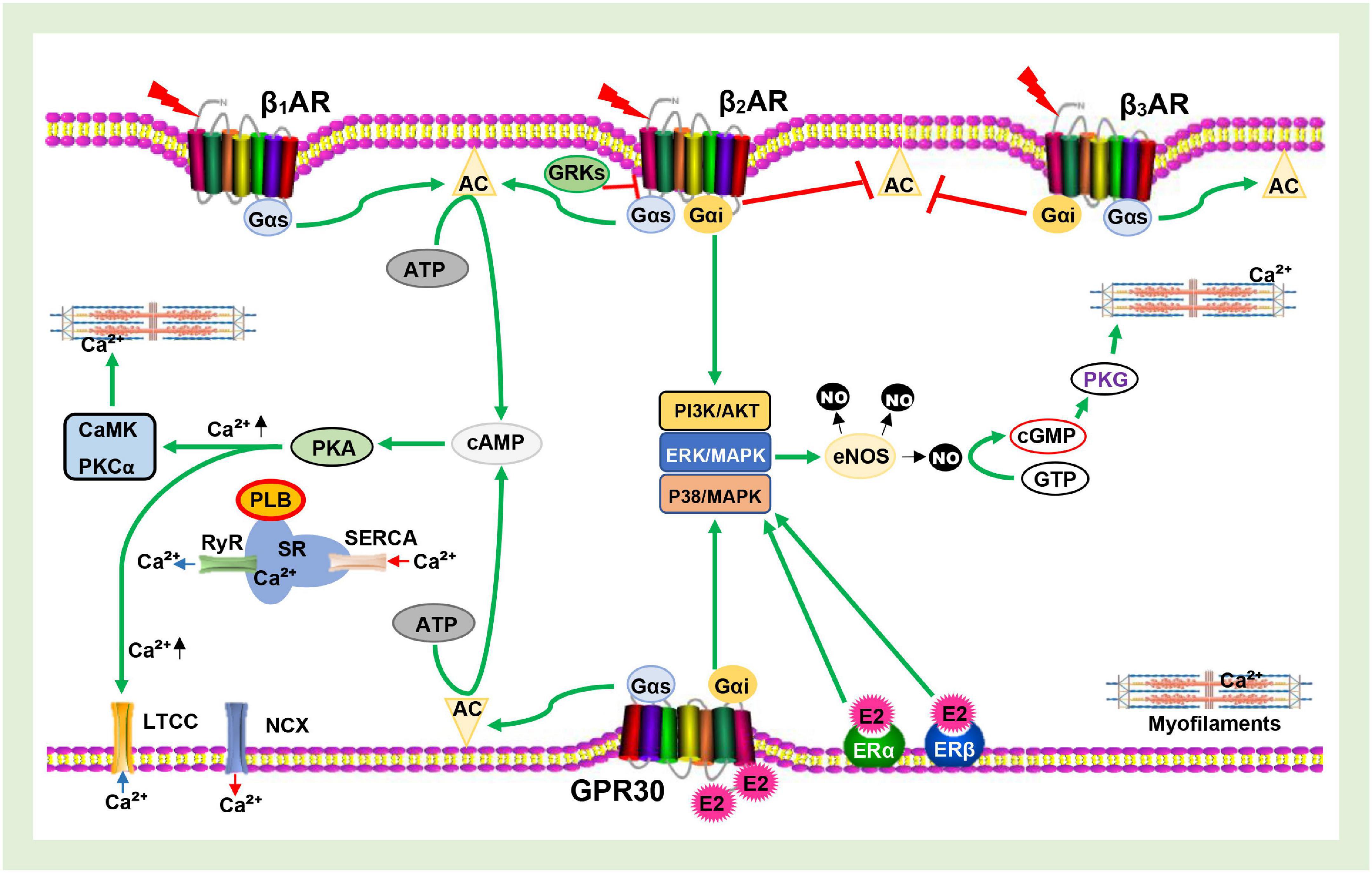
Figure 2. Schematics of Calcium channel cascades and Myofilament activation being facilitated by β-adrenergic receptors (βARs) and Estrogen receptors (ERs signaling. AC, Adenylyl cyclase; cAMP, Cyclic adenosine monophosphate; PKA, Protein kinase A; PKCα, Protein kinase C alpha; GRKs, G protein-coupled receptor kinases; E2, Estrogen; eNOS, Endothelial nitric oxide synthase; NO, Nitric oxide; PDE, Phosphodiesterase; SR, Sarcoplasmic reticulum; PLB, Phospholamban; RyR, Ryanodine receptor; SERCA, Sarcoplasmic reticulum Ca2+ ATPase; LTCC, L-type calcium channel; NCX, Sodium-calcium exchanger; CaMK, Calmodulin- dependent kinase; cGMP, Cyclic guanosine monophosphate; GTP, Guanosine triphosphate; PKG, Protein kinase G;  , Phosphorylated;
, Phosphorylated;  , Inhibit;
, Inhibit;  , Upregulate;
, Upregulate;  , Ca2+ entry/release;
, Ca2+ entry/release;  , Ca2+ uptake/expulsion;
, Ca2+ uptake/expulsion;  , Catecholamine.
, Catecholamine.
In their attempt to investigate the disease mechanism, Kilfoil et al. demonstrated that βARs are mostly desensitized in HFpEF models, therefore, might account for the dysregulation in calcium handling downstream (Kilfoil et al., 2020). Even so, the phosphorylation of cTnI and MyBP-C by PKA does reduce the affinity of troponin C for Ca2+ and the acceleration of the cross-bridge detachment to hasten myocardial relaxation (Matsuba et al., 2009). However, in HFpEF, these two proteins are less phosphorylated (Hamdani et al., 2013). E2 being able to prevent βARs desensitization by inhibiting GRKs would have impeded dysfunctionalities of the receptor and enhanced the phosphorylation of cTnI and MyBP-C by PKA. This would have ensured proper calcium handling to circumvent the LVDD as suggested (Maslov et al., 2019); however, E2 deficiency in postmenopausal women leaves them vulnerable.
E2 has been extensively shown to decrease LTCC expression as well as regulate its gating activities to ensure only a sufficient amount of Ca2+ enters the cardiomyocyte (Kurata et al., 2001; Chu et al., 2006; Brewer et al., 2009; Sribnick et al., 2009; Harvey, 2012; Lai et al., 2019). Contrarily, ovariectomy (mimicking E2 loss) resulted in the upregulation of the LTCC and increased Ca2+ entry (Chu et al., 2006; Brewer et al., 2009; Lai et al., 2019). Also, it has been demonstrated that E2 stabilizes the Ca2+ holding and Ca2+ gating activities of the SR and RyR, respectively. As such, the deficiency of E2 was observed to induce SR Ca2+ overload, increased cardiomyocyte contraction, and the amplitude of Ca2+ transient (Parks et al., 2017; Yang et al., 2017; Jiao et al., 2020). In addition, RyR was found to be highly phosphorylated at Ser2808, causing an increased release of Ca2+ besides being “leaky” in ovariectomy animal models (Ai et al., 2005; Kravtsov et al., 2007; Györke and Carnes, 2008; Fares et al., 2012; Kilfoil et al., 2020). However, the replacement of E2 reverted these abnormalities in SR and RyR Ca2+ handling functions (Kravtsov et al., 2007; Parks et al., 2017; Yang et al., 2017). Although it might be argued that these initial phase cascades (Ca2+ entry and Ca2+ release) of the Ca2+ cycle are responsible for contraction, any inefficiencies occurring here may indirectly impact negatively on the latter phase cascade (Ca2+ uptake and Ca2+ expulsion), as the amount of Ca2+ influx in the cytosol might exceed the Ca2+ hold capacity plus Ca2+ efflux capability. Consistent with these inferences, Miranda et al. suggested that SR Ca2+ release and SR Ca2+ uptake are impaired during HFpEF (Miranda-Silva et al., 2020).
Furthermore, Ca2+ uptake via SERCA aids partly in restoring intracellular Ca2+ levels to ensure the relaxation of cardiomyocytes. Nonetheless, in HFpEF models, the expression and function of SERCA are reportedly downregulated (Miranda-Silva et al., 2020). This phenomenon is consistent with observations in ovariectomized animal models (Bupha-Intr and Wattanapermpool, 2006). Also, in conformity with the finding from ovariectomized animal models, Miranda et al. and others have observed lower SERCA/PLB ratio and decreased phosphorylated PLB in HFpEF models (Dupont et al., 2012; Rouhana et al., 2019; Miranda-Silva et al., 2020). Regardless, E2 and ERs have been widely demonstrated to promote the functions of SERCA by upregulating its expression, maintaining SERCA/PLB ratio, and enhancing PLB phosphorylation to increase Ca2+ uptake (Ren et al., 2003; Chu et al., 2006; Turdi et al., 2015; Machuki et al., 2019). In addition, the functionality of NCX is crucial in complimenting SERCA/PLB in restoring intracellular Ca2+ levels. Although the effect of E2 on NCX expressions and activities remains inconclusive (Müller-Ehmsen et al., 2001; Kravtsov et al., 2007; Chen et al., 2011; Yang et al., 2017), it is suggested that E2 normalizes its expression and current density to enhance Ca2+ expulsion. If not so, NCX might fail to ensure calcium homeostasis either by ineffective expulsion of Ca2+ or switch into the reverse mode to increase the accumulation of intracellular calcium along with increased intracellular Na+ as reported in HFpEF (Louch et al., 2010). Besides, E2 and ERs have been shown to decrease the sensitivity of myofilaments to Ca2+ so as to enhance myocardial relaxation and prevent HFpEF (Fares et al., 2013; Hamdani et al., 2013; Pandit et al., 2014). In contrast, Pandit et al. and others have demonstrated that ovariectomy aggravated myofilaments Ca2+ sensitivity which slows down myocardial relaxation and exacerbates HFpEF (Fares et al., 2013; Pandit et al., 2014).
Taken together, the obvious cardioprotective roles of E2 exerted through the adaptive modulation of calcium handling proteins fortifies premenopausal women against the initiation and progression of HFpEF while E2 deficiency and loss of its residual protection during the postmenopausal period scaffolds a derangement in calcium homeostasis in cardiomyocytes; hence the higher incidence of HFpEF in postmenopausal women.
Alterations in Cardiac Cytoskeletal Titin
The cardiomyocyte cytoskeleton consists of microtubules, cytoplasmic actin, intermediate filaments, and titin. However, besides titin which provides myocardial elasticity and facilitates contractile functions, the prior mentioned cardiomyocyte cytoskeleton constituents are non-contractile (Sequeira et al., 2014). The dysregulation in myocardial contractility has been hallmarked as an underlying factor contributing to LVDD and HFpEF (Rao and Burkhoff, 2021). As such, the maladaptive alteration in titin (the contractile cytoskeleton) will be centered on, rather than the entire cardiomyocyte cytoskeleton. Titin is a giant cytoskeletal protein that plays a significant role in the elastic force of the sarcomere. The elasticity properties of titin are embedded in the I-band region, which is made up of three segments; tandem immunoglobulin (Ig) repeats, unique N2B element, and the PEVK region (LeWinter and Granzier, 2010). Titin primarily exists in two isoforms, N2B isoform (stiffer isoform) and N2BA (compliance isoform), and modulates myocardial passive stiffness either by switching isoform posttranslational modification, as well as via oxidative regulation (Linke and Hamdani, 2014). Titin can be directly phosphorylated at N2B by PKA and/or PKG as well as indirectly phosphorylated at both N2B and PEVK via βARs/PKA/CaMK in a calcium-dependent manner, besides ERK and PKCα phosphorylations (Krüger et al., 2009; Raskin et al., 2012; Hamdani et al., 2013; Hidalgo et al., 2013).
In HFpEF, reduced phosphorylation of titin by both PKA and PKG has been described as the leading cause of the increase in passive stiffness (Borbely et al., 2009; LeWinter and Granzier, 2010; van Heerebeek et al., 2012). Primarily, the dysregulation of βARs due to hyperstimulation might account for the hypophosphorylations of the N2B and PEVK spring elements of titin. βARs signaling disruption impedes the direct phosphorylation of N2B by PKA, thereby, increases the stiffness in the cardiomyocyte cytoskeleton. Also, with the signaling of βARs/PKA, which regulates intracellular Ca2+ being hampered, CaMK is unable to phosphorylate neither N2B nor PEVK region (Borbely et al., 2005; van Heerebeek et al., 2012). Hence, the disruption of both direct and indirect regulation of the titin aggravates myocardial stiffness, as seen in HFpEF patients. Interestingly, E2 can mitigate the dysregulation of βARs and also signal via GPR30/Gαs/PKA (Machuki et al., 2018; Ndzie Noah et al., 2021). Thus, these cascades suggest that E2 might mask and/or complement for induction of PKA, PKC, and CaMK to phosphorylate the N2B and PEVK elements. In addition, E2, by upregulating NO, could further induce the phosphorylation of N2B via PKG (Krüger et al., 2009; Ndzie Noah et al., 2021). By these multiple mechanisms facilitated by E2, the passive stiffness of titin is normalized, thereby enhancing its physiological functions or delaying the progression of pathological stiffening of the myocardial. However, the deficiency of E2 and inhibition of ERs might abolish these bypasses to sustain the titin functions. This is further proved by relating Michalson et al. and Zile et al.’s findings. It could be inferred from their works that the deficiency of E2 and impediment in ERs signaling might contribute to the maladaptive alteration in cardiomyocyte titin (Zile et al., 2015; Michalson et al., 2018).
Extra-Cardiomyocyte Maladaptive Alterations
Disruption of Extracellular Matrix Homeostasis Functions
The healthy cardiac ECM consists of non-structural and structural matricellular proteins, which are kept in a homeostatic balance by MMPs and TIMPs to preserve the myocardial architecture and its functions (Figure 1). In-depth discussions on the roles of MMPs and TIMPs have been reviewed elsewhere (Ma et al., 2014; Arpino et al., 2015; Wang and Khalil, 2018; Raeeszadeh-Sarmazdeh et al., 2020). Notably, patients presenting HFpEF are found to have stiffer myocardia resulting from excessive collagen deposition. Elucidation of the pathomechanisms resulting in the LVDD mainly implicates the dysregulation and aggravation of fibroblast activation.
Intriguingly, βARs and ERs are well expressed in fibroblasts; hence, circulating catecholamine and E2 can modulate their proliferation, differentiation to myofibroblast, and migration. As such, a typical dysregulation of the βARs in fibroblast by chronic sustained stimulation induces cascades that contribute to the adverse ECM remodeling, as reported (Jaffré et al., 2012; Lv et al., 2016; Tanner et al., 2020). Also, the deficiency of E2 and inhibition of ERs have been largely demonstrated to permit series of maladaptive cascades aggravating ECM remodeling, LV stiffness, and HFpEF. Michalson et al. in their recent study, showed that ovariectomy induced LV diastolic dysfunction in cynomolgus monkeys by increasing ECM deposition via upregulating genes encoding ECM components (COL1A2, MFAP5, and MGP) and ECM deposition promoters (CXCL12, THBS1, TIMP1, S100A4, and RPS29) (Michalson et al., 2018). Together with others, they found that during ova riectomy, synthetic myofibroblasts upregulate the synthesis of fibrillar collagen, ECM, and TIMPs while downregulating the degradative MMPs, thereby causing an imbalanced ECM turnover and exacerbating fibrosis (Dubey et al., 1998; Michalson et al., 2018). Also, cardiac collagen I and III genes were shown to increase due to E2 deficiency (Spinale et al., 1999; Dworatzek et al., 2019). Consistent with the findings of Michalson et al., El Hajj et al. also found that by antagonizing ERs, the exacerbation of cardiac structure and function were hastened in female rats (El Hajj et al., 2017). Furthermore, E2 deficiency and ERs inhibition have been shown to increase fibroblast activation and ECM depositions by; increasing NF-kB pathway activity, upregulating proinflammatory cytokine (TNFα and IL-6) secretions, and reducing NO bioavailability (McLarty et al., 2013; Vettel et al., 2014; Ambhore et al., 2019).
Within all the aforementioned studies demonstrating the adverse remodeling of the ECM due to E2 deficiency or ERs inhibition, E2 replacement and/or normalizations of ERs expression reverted all the maladaptive cascades, facilitated all the homeostatic functions that sustain the ECM (including mitigating βARs dysregulation) and improved myocardial contraction to attenuate HFpEF. Altogether, it has been suggested and shown that E2 by signaling via non-genomic (GPR30) and/or genomic (ERα and ERβ) could directly or indirectly exert these cardioprotective effects to prevent HFpEF by preserving the ECM and cardiovascular function (Watanabe et al., 2003; Wang et al., 2015; El Hajj et al., 2017). Hence, E2 deficiency in postmenopausal women increases the incidence of HFpEF by scaffolding the disruptions in homeostasis functions sustaining cardiac ECM, thereby aggravating LV stiffness and LVDD.
Coronary Microvascular Endothelial Dysfunctionalities
Physiological functions of coronary vessels (blood circulation in the myocardia) are dependent on functionalities of their intima layer made of endothelial cells, which in turn depend on NO bioavailability. The production of NO by eNOS is crucial for the regulation of several cardiovascular physiology such as; endothelial cell proliferation and migration, ECM degradation, angiogenesis, platelet function, vascular inflammation modulation, and vascular tone regulation (Naseem, 2005; Kolluru et al., 2010; Tousoulis et al., 2012; Ndzie Noah et al., 2021). Again, βARs and ERs are prevalent on endothelial cells; hence E2 and catecholamines are capable of indirectly regulating coronary microvessels through the facilitation of their endothelial functions (Ahmad et al., 1990; Michalson et al., 2018). In evidence to these, E2 via GPR30 was shown to activate eNOS by inducing PI3K/Phosphokinase B–mediated phosphorylation of serine 1177 (Deschamps and Murphy, 2009). Also, ERα and ERβ were shown to upregulate transcription activities that enhance endothelial nitric oxide synthase production by binding to estrogen response elements (Michalson et al., 2018). In addition, β2AR has been demonstrated to induce NO production via Gai/PI3K/Akt (Hu et al., 2020). Nonetheless, during chronic catecholamine stress, β2AR desensitization and dysregulation disrupt eNOS activation; hence, this affects the NO bioavailability, thereby causing CME dysfunction.
Disruption of NO-cGMP-PKG signaling in coronary endothelial cells delays the onset of myocardial relaxation and increases diastolic LV stiffness (Paulus, 2001; Brutsaert, 2003; LeWinter and Granzier, 2010; Paulus and Tschöpe, 2013). Therefore, the chronic inhibition of eNOS due to chronic hyperstimulation of β2AR might induce LVDD, as Matsubara et al. demonstrated in their study (Matsubara et al., 1998). However, it is suggested that E2 and ERs may directly enhance eNOS-NO production and as well mitigate the extent of β2AR dysfunctionalities to ensure the bioavailability of NO, which might help in the attenuation of LVDD/HFpEF (Huang et al., 2000; Westermann et al., 2009; Barton and Meyer, 2020; Ndzie Noah et al., 2021).
Recently, efforts to elucidate the underlying mechanisms of HFpEF are focused on coronary microvascular inflammation, besides LV afterload excess (Paulus and Tschöpe, 2013; Franssen et al., 2016; Sickinghe et al., 2019). Under proinflammatory conditions, CME cells are observed to produce reactive oxygen species (ROS), which affects NO bioavailability. This affects both endothelial cells and adjacent cardiomyocytes’ NO-cGMP-PKG signaling; hence, it delays myocardial relaxation, induces LV hypertrophy, the proliferation of fibroblasts and myofibroblasts, and also increases diastolic LV stiffness (Calderone et al., 1998; Paulus and Tschöpe, 2013; Vettel et al., 2014). On the other hand, HFpEF-induced systemic inflammation has been shown to cause CME dysfunction by increasing vascular cell adhesion molecule and E-selectin expressions. Their expression is characteristic of decreased eNOS activity and NO bioavailability. This encourages the recruitment and trafficking of circulating leukocytes to the subendothelial, which initiates the formation of atherosclerotic lesions formation and the progression of atherosclerosis which hastens the incidence of HFpEF (Venugopal et al., 2002; Kolluru et al., 2010; Westermann et al., 2011; Rush et al., 2021). These aforementioned CME dysfunctionalities have been associated with E2 deficiency. Lack of physiological levels of E2 permitted renin-angiotensin-aldosterone system-induced upregulation of ROS in cardiac and endothelial mitochondria (Alvarez et al., 2002; Rattanasopa et al., 2015). Besides, aging (menopause in women) is also shown to promote endothelial ROS production, thereby causing endothelial mitochondrial dysfunction (Rajapakse et al., 2011; Paulus and Tschöpe, 2013). Consistent with the adaptive roles of E2, its replacement inhibited renin-angiotensin-aldosterone system and ROS production, enhanced coronary blood flow as well as attenuated leukocyte activations via enhancing eNOS activity and NO bioavailability (Lang et al., 1997; Alvarez et al., 2002; Kim et al., 2006; Sabbatini and Kararigas, 2020).
Besides promoting CME eNOS-NO activities, E2 regulates vascular angiotensin II receptor type 1. The deficiency of E2 results in the upregulation of angiotensin II receptor type 1, which increases oxidative stress and disrupts CME function. Similarly, E2 replacement attenuated the oxidative stress and protected the endothelial cells from its induced apoptosis by signaling via GPR30/Akt (Nickenig et al., 1998; Sudoh et al., 2001; Xue et al., 2014). Also, E2/GPR30 has been shown to facilitate the proper formation of endothelial cell tubes (Zhou et al., 2017); this indirectly ensures coronary vascular functions are sustained.
Overall, E2 and ERs, through these mechanisms, ensure proper CME functions and attenuate the HFpEF development; however, the absence of its physiological levels in postmenopausal women predisposes them to CME dysfunctions and expedites the incidence of HFpEF.
Maladaptive Myocardial Inflammatory Responses
Immune cells being constituents of the myocardia and capable of crosstalk with endothelial cells, fibroblasts, and cardiomyocytes, have been extensive implicated in the exacerbation of cardiovascular diseases (Paulus and Tschöpe, 2013; Franssen et al., 2016; Adzika et al., 2019). Just as the other cellular constituents of the myocardia, cardiac resident immune cells also have profound expressions of β2AR and ERs. Primarily, cardiac resident immune cells have been shown to elicit hyperactive proinflammatory responses, which aggravate fibrosis and LV stiffness and increases the incidence of HFpEF via multiple mechanisms (McLarty et al., 2013; Sandstedt et al., 2019; Tanner et al., 2020). The excessive secretion of proinflammatory cytokines, IL-6 and TNFα, and the activation of NFκB (a prototypic inflammatory pathway) by immune cells induce myofibroblast differentiation, fibroblast proliferation, and migration (Sandstedt et al., 2019). While β2AR is implicated in the mediation of signaling in cardiomyocytes and fibroblasts during the pathological remodeling of the heart (Paur et al., 2012; Adzika et al., 2019), Tanner et al.’s recent study demonstrated that the β2AR upregulation of fibroblast proliferation is dependent on IL-6 secretion (Tanner et al., 2020). Hence, besides dysregulated β2AR, the hypersecretions of IL-6 in the myocardia have the potential of inducing excessive interstitial deposition of collagen, which distorts the myocardial architecture and causes LVDD. Furthermore, TNFα upregulation also activates cardiac fibroblast cascades that contribute to the adverse remodeling of the ECM (McLarty et al., 2013). Nonetheless, E2 and ERs exert their cardioprotective effects by inhibiting the TNFα secretions from inflammatory cells, thereby preventing fibrosis (Chancey et al., 2005; McLarty et al., 2013). Notably, NFκB regulates the expression of IL-6 and TNFα; as such, by repressing NFκB transcriptional activities, E2 and ERs can adaptively modulate inflammatory responses in the myocardia (Galien and Garcia, 1997; Arenas et al., 2005; Michalson et al., 2018; Ambhore et al., 2019). In fact, E2 and ERs have been shown to repress several inflammatory cascade pathways in the myocardia (Sabbatini and Kararigas, 2020). For example, (1) HFpEF patients are found to have elevated levels of monocyte chemoattractant protein 1, which scaffolds infiltration and accumulation of macrophage in the myocardium to promote fibrosis (Kuwahara et al., 2004). Nevertheless, E2 treatment in animal models after ovariectomy-induced LVDD was observed to downregulate sera monocyte chemoattractant protein 1 levels (Sophonsritsuk et al., 2013; Michalson et al., 2018). This implied that E2 is a potent adaptive regulator of cardiac and systemic inflammation to prevent cardiac dysfunction. (2) In the coronary vessels, E2 enhances NO bioavailability. Besides vascular tone regulation, NO exerts anti-inflammatory effects on the vascular wall, thereby attenuates pathological inflammation by preventing leukocyte interaction and transendothelial migration (Kubes et al., 1991; Venugopal et al., 2002; Westermann et al., 2009; Kolluru et al., 2010). As such, E2 indirectly attenuates HFpEF by preventing CME inflammation (Chakrabarti and Davidge, 2012). (3) Also, E2 increases angiotensin Ang 1-7 synthesis, and similar to NO, induces anti-inflammatory and antioxidative effects (Mompeón et al., 2016). (4) Lastly, adipose tissues harbor a significant amount of inflammatory cells and are a major source of inflammatory responses (Ouchi et al., 2011; Renovato-Martins et al., 2017; Srikakulapu and McNamara, 2020). Meanwhile, adipose accumulation in ovariectomy animal models and postmenopausal women is observed to increase (Lee et al., 2015; Kozakowski et al., 2017; Sharma et al., 2017). This suggests that epicardial adipose may increase during E2 deficiency and serve as a source from which inflammatory cells infiltrate the myocardia and exacerbate adverse cardiac remodeling. Consistent with the prior discussion, Mori et al. in the study demonstrated that in ovariectomized rats, cardiac inflammation and fibrosis are enhanced, resulting in diastolic dysfunction just as observed in postmenopausal women (Mori et al., 2011). However, overall, E2 treatments attenuating HFpEF, as demonstrated (Michalson et al., 2018), might involve E2-initiated inhibition of maladaptive inflammatory responses in the myocardial (including coronary vessels).
Conclusions and Perspectives on E2 Replacement Therapy in the Management of Heart Failure With Preserved Ejection Fraction
Heart Failure With Preserved Ejection Fraction Pathomechanism Hastened by E2 Deficiency in Brief
In summary, elucidating the underlying cause of the prevalence of HFpEF in postmenopausal women over the recent decades has revealed that the loss of E2 and its residual cardioprotective effects permits the initiation of maladaptive alterations intra- and extra-cardiomyocytes. These adverse alterations tend to affect the physiological functions of the cardiomyocytes as well as other constituents of the myocardia, such as; fibroblasts, inflammatory cells, coronary vessels, and epicardial adipose (Table 1). The synergy of maladaptive functions scaffolded by E2 deficiencies and ERs inactivity expedite the initiation of LVDD and progression in HFpEF as predominantly manifested in postmenopausal women (Figure 3).
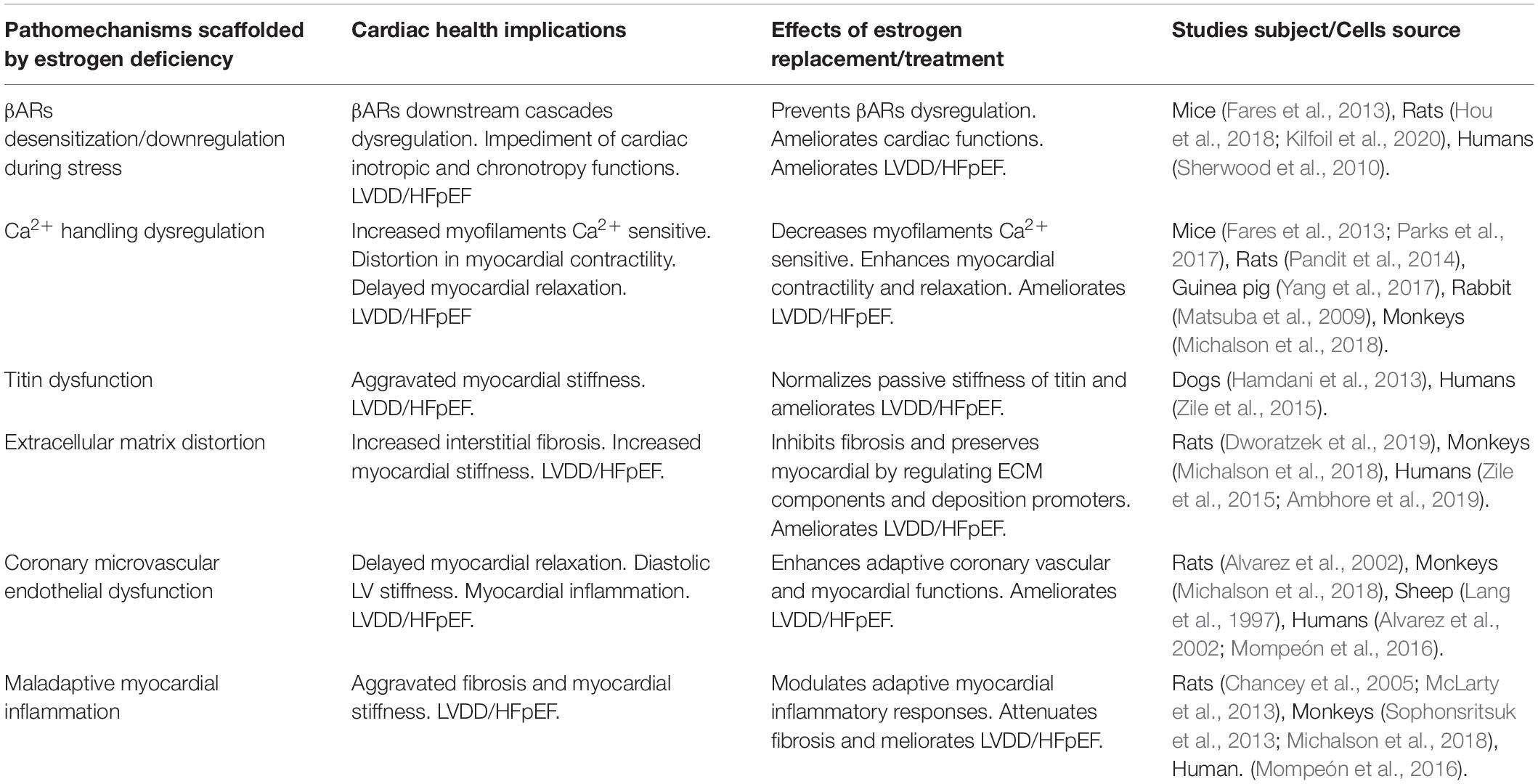
Table 1. Overview of HFpEF underlying pathomechanisms scaffolded by estrogen deficiency and the ameliorative effects of estrogen therapy.
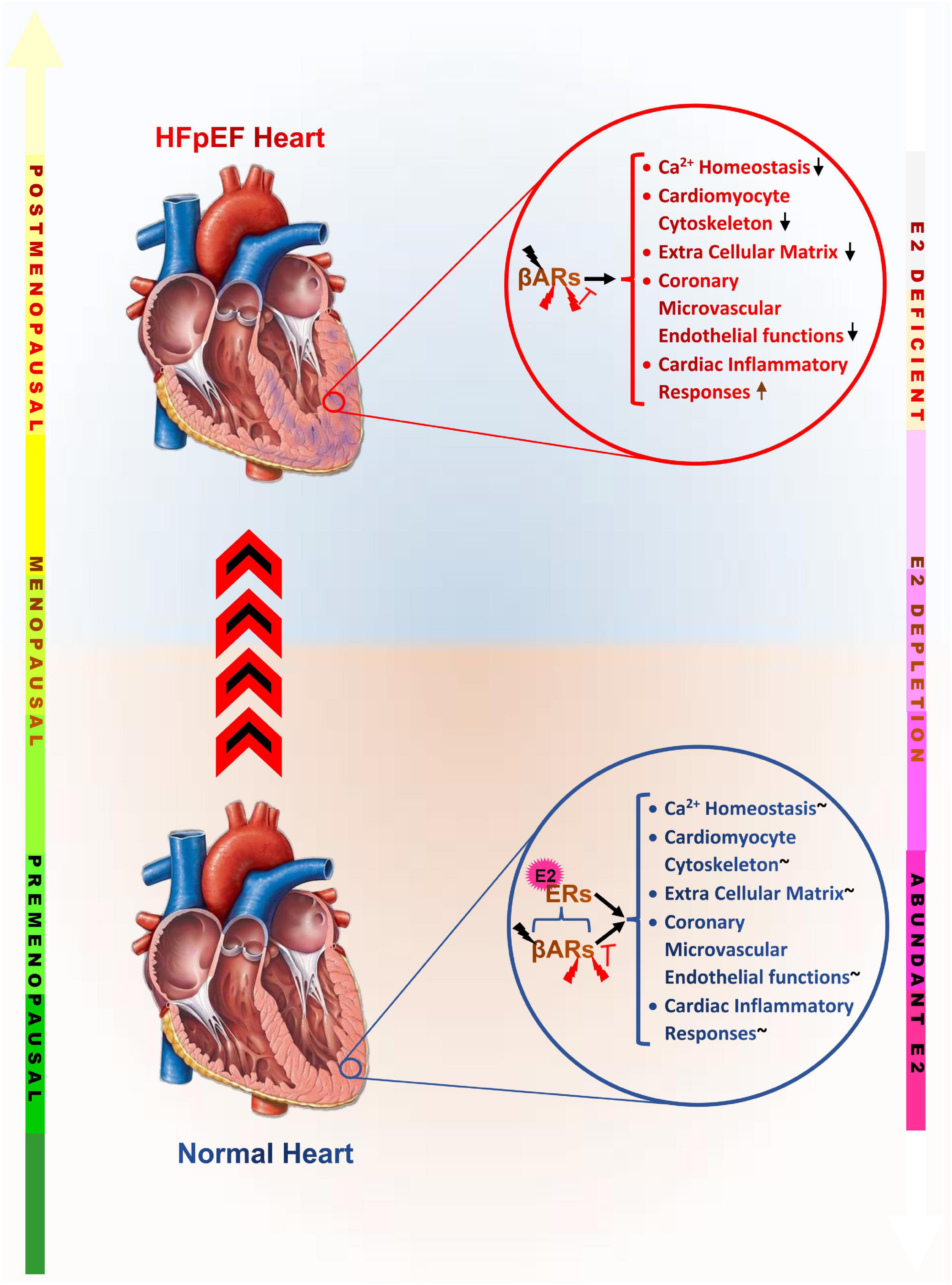
Figure 3. Graphical abstract of the underlying maladaptive alterations scaffolded by estrogen deficiency, which increase the incidence of heart failure with preserved ejection fraction in postmenopausal women. βARs, β-adrenergic receptors; ERs, Estrogen receptors;  , Physiological levels of catecholamine;
, Physiological levels of catecholamine;  , Pathological levels catecholamine;
, Pathological levels catecholamine;  , inhibition;
, inhibition;  , Adaptively modulated; E2, Estrogen;
, Adaptively modulated; E2, Estrogen;  , Disrupted/dysregulated;
, Disrupted/dysregulated;  , Upregulated maladaptively.
, Upregulated maladaptively.
Prospects on Therapeutic Interventions for Heart Failure With Preserved Ejection Fraction Management
Taken together, decades of research works focused on elucidating the underlying cause of increased HFpEF in postmenopausal women have found that E2 exerts cardioprotection via the adaptive modulation of multiple homeostatic functions that sustains proper cardiac health and functions. Therefore, its deficiency during menopause permits dysregulations of cascades and physiological functions crucial for facilitating cardiac inotropy and chronotropic, should there be any stressful event.
Therapeutic interventions for the prevention and management of HFpEF in postmenopausal women has solely been the use of menopausal hormonal treatment until the findings of the Women’s Health Initiative randomized controlled trial and others, reporting the side effects and discouraged their usage significantly (Rossouw et al., 2002; Modena et al., 2005; Schierbeck et al., 2012; Antoine et al., 2016; Iorga et al., 2017; Kohn et al., 2019). Exploration of the mechanisms causing the reported side effects revealed that the late initiation of menopausal hormonal treatment and the usage of the combination of conjugated equine estrogen with medroxyprogesterone acetate as the probable causes (LaCroix et al., 2011; Maria Grazia, 2016; Hickey et al., 2018; Shufelt et al., 2018).
Also, due to the complexity of the disease mechanism of HFpEF, it is suggested that multiple pathomechanisms may be targeted with their appropriate drug combinations to improve treatment outcomes. Meanwhile, employing E2 replacement therapy might circumvent the use of multiple drugs as it has been shown to adaptively modulate these multiple pathways to exert cardioprotection and/or ameliorate adverse cardiac remodeling (Michalson et al., 2018; Sabbatini and Kararigas, 2020; Ndzie Noah et al., 2021).
Clinical data have shown that the severity of LVDD/HFpEF is increased mostly after 6 years of menopause (Gerber et al., 2015; Pfeffer et al., 2019). In line with these reports, it has been shown in ovariectomized monkeys that when E2 replacement therapies were initiated late (4.5 years) after ovariectomy, E2 was ineffective at attenuating maladaptive inflammatory responses, disruption of ECM, and dysregulation of Ca2+ handling. Conversely, when the E2 replacement therapy was initiated early (1 month) after ovariectomy, E2 was potent at inhibiting the aforementioned dysfunctions, which hasten the incidence of HFpEF (Sophonsritsuk et al., 2013; Michalson et al., 2018). These studies have further suggested that E2 replacement therapy initiated early (within 5-6 years of menopause) has more efficacy in improving heart function and preventing LVDD/HFpEF while circumventing any treatment side effects (Shao et al., 2012; Sophonsritsuk et al., 2013; Hodis et al., 2016; Michalson et al., 2018). Therefore, E2RT might not be effective for treating the latter stages of HFpEF. Therefore, it is proposed that E2 replacement therapy targeted at the attenuation of HFpEF in postmenopausal women should be initiated within the critical window of 5-6 years after menopause.
Author Contributions
AOA conceived the review idea. GKA and AOA fine-tuned the review idea and drafted and wrote the manuscript. GKA, AOA, RM, MLNN, JA-A, RR, and NA proofread and revised the manuscript with the supervision of HS. All authors accepted the final version of the manuscript.
Funding
The following grants supported this study: National Natural Science Foundation of China (Nos. 81461138036 and 81370329), Postgraduate Research & Practice Innovation Program of Jiangsu Province, China (KYCX17-1712), and Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abraham, A. D., Schattauer, S. S., and Reichard, K. L. (2018). Estrogen regulation of GRK2 inactivates kappa opioid receptor signaling mediating analgesia, but not aversion. J. Neurosci. 38, 8031–8043. doi: 10.1523/jneurosci.0653-18.2018
Adzika, G. K., Machuki, J. O., Shang, W., Hou, H., Ma, T., Wu, L., et al. (2019). Pathological cardiac hypertrophy: the synergy of adenylyl cyclases inhibition in cardiac and immune cells during chronic catecholamine stress. J. Mol. Med. (Berl) 97, 897–907. doi: 10.1007/s00109-019-01790-0
Ahmad, S. S., and Gerson, M. C. (2018). Sympathetic drive stimulating diastolic dysfunction? J. Nucl. Cardiol. 25, 1110–1113. doi: 10.1007/s12350-017-0809-z
Ahmad, S., Chrétien, P., Daniel, E. E., and Shen, S. H. (1990). Characterization of beta adrenoceptors on cultured endothelial cells by radioligand binding. Life Sci. 47, 2365–2370. doi: 10.1016/0024-3205(90)90276-w
Ai, X., Curran, J. W., Shannon, T. R., Bers, D. M., and Pogwizd, S. M. (2005). Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 97, 1314–1322. doi: 10.1161/01.res.0000194329.41863.89
Aikawa, T., Naya, M., Obara, M., Manabe, O., Tomiyama, Y., Magota, K., et al. (2017). Impaired myocardial sympathetic innervation is associated with diastolic dysfunction in heart failure with preserved ejection fraction: (11)C-Hydroxyephedrine PET study. J. Nucl. Med. 58, 784–790. doi: 10.2967/jnumed.116.178558
Alvarez, A., Hermenegildo, C., Issekutz, A. C., Esplugues, J. V., and Sanz, M. J. (2002). Estrogens inhibit angiotensin II-induced leukocyte-endothelial cell interactions in vivo via rapid endothelial nitric oxide synthase and cyclooxygenase activation. Circ. Res. 91, 1142–1150. doi: 10.1161/01.res.0000046018.23605.3e
Ambhore, N. S., Kalidhindi, R. S. R., Pabelick, C. M., Hawse, J. R., Prakash, Y. S., and Sathish, V. (2019). Differential estrogen-receptor activation regulates extracellular matrix deposition in human airway smooth muscle remodeling via NF-κB pathway. FASEB J. 33, 13935–13950. doi: 10.1096/fj.201901340R
Antoine, C., Ameye, L., Paesmans, M., de Azambuja, E., and Rozenberg, S. (2016). Menopausal hormone therapy use in relation to breast cancer incidence in 11 European countries. Maturitas 84, 81–88. doi: 10.1016/j.maturitas.2015.11.010
Arenas, I. A., Armstrong, S. J., Xu, Y., and Davidge, S. T. (2005). Chronic tumor necrosis factor-alpha inhibition enhances NO modulation of vascular function in estrogen-deficient rats. Hypertension 46, 76–81. doi: 10.1161/01.HYP.0000168925.98963.ef
Arpino, V., Brock, M., and Gill, S. E. (2015). The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. 44-46, 247–254. doi: 10.1016/j.matbio.2015.03.005
Asokan Shibu, M., Kuo, W.-W., Kuo, C.-H., Day, C.-H., Shen, C.-Y., Chung, L.-C., et al. (2017). Potential phytoestrogen alternatives exert cardio-protective mechanisms via estrogen receptors. BioMedicine 7:11. doi: 10.1051/bmdcn/2017070204
Barton, M., and Meyer, M. R. (2020). Heart failure with preserved ejection fraction in women: new clues to causes and treatment. JACC Basic Transl. Sci. 5, 296–299. doi: 10.1016/j.jacbts.2020.02.001
Borbely, A., Falcao-Pires, I., van Heerebeek, L., Hamdani, N., Edes, I., Gavina, C., et al. (2009). Hypophosphorylation of the Stiff N2B titin isoform raises cardiomyocyte resting tension in failing human myocardium. Circ. Res. 104, 780–786. doi: 10.1161/CIRCRESAHA.108.193326
Borbely, A., van der Velden, J., Papp, Z., Bronzwaer, J. G., Edes, I., Stienen, G. J., et al. (2005). Cardiomyocyte stiffness in diastolic heart failure. Circulation 111, 774–781. doi: 10.1161/01.CIR.0000155257.33485.6D
Brewer, L. D., Dowling, A. L., Curran-Rauhut, M. A., Landfield, P. W., Porter, N. M., and Blalock, E. M. (2009). Estradiol reverses a calcium-related biomarker of brain aging in female rats. J. Neurosci. 29, 6058–6067. doi: 10.1523/jneurosci.5253-08.2009
Brooks, W. W., and Conrad, C. H. (2009). Isoproterenol-induced myocardial injury and diastolic dysfunction in mice: structural and functional correlates. Comp. Med. 59, 339–343.
Brutsaert, D. L. (2003). Cardiac endothelial-myocardial signaling: its role in cardiac growth, contractile performance, and rhythmicity. Physiol. Rev. 83, 59–115. doi: 10.1152/physrev.00017.2002
Bupha-Intr, T., and Wattanapermpool, J. (2006). Regulatory role of ovarian sex hormones in calcium uptake activity of cardiac sarcoplasmic reticulum. Am. J. Physiol. Heart Circ. Physiol. 291, H1101–H1108. doi: 10.1152/ajpheart.00660.2005
Calderone, A., Thaik, C. M., Takahashi, N., Chang, D. L., and Colucci, W. S. (1998). Nitric oxide, atrial natriuretic peptide, and cyclic GMP inhibit the growth-promoting effects of norepinephrine in cardiac myocytes and fibroblasts. J. Clin. Invest. 101, 812–818. doi: 10.1172/jci119883
Chakrabarti, S., and Davidge, S. T. (2012). G-protein coupled receptor 30 (GPR30): a novel regulator of endothelial inflammation. PLoS One 7:e52357. doi: 10.1371/journal.pone.0052357
Chancey, A. L., Gardner, J. D., Murray, D. B., Brower, G. L., and Janicki, J. S. (2005). Modulation of cardiac mast cell-mediated extracellular matrix degradation by estrogen. Am. J. Physiol. Heart Circ. Physiol. 289, H316–H321. doi: 10.1152/ajpheart.00765.2004
Chen, G., Yang, X., Alber, S., Shusterman, V., and Salama, G. (2011). Regional genomic regulation of cardiac sodium-calcium exchanger by oestrogen. J. Physiol. 589(Pt 5), 1061–1080. doi: 10.1113/jphysiol.2010.203398
Chu, S. H., Goldspink, P., Kowalski, J., Beck, J., and Schwertz, D. W. (2006). Effect of estrogen on calcium-handling proteins, beta-adrenergic receptors, and function in rat heart. Life Sci. 79, 1257–1267. doi: 10.1016/j.lfs.2006.03.037
Deschamps, A. M., and Murphy, E. (2009). Activation of a novel estrogen receptor, GPER, is cardioprotective in male and female rats. Am. J. Physiol. Heart Circ. Physiol. 297, H1806–H1813. doi: 10.1152/ajpheart.00283.2009
Dubey, R. K., Gillespie, D. G., Jackson, E. K., and Keller, P. J. (1998). 17Beta-estradiol, its metabolites, and progesterone inhibit cardiac fibroblast growth. Hypertension 31(1 Pt 2), 522–528. doi: 10.1161/01.hyp.31.1.522
Dupont, S., Maizel, J., Mentaverri, R., Chillon, J. M., Six, I., Giummelly, P., et al. (2012). The onset of left ventricular diastolic dysfunction in SHR rats is not related to hypertrophy or hypertension. Am. J. Physiol. Heart Circ. Physiol. 302, H1524–H1532. doi: 10.1152/ajpheart.00955.2010
Dworatzek, E., Mahmoodzadeh, S., Schriever, C., Kusumoto, K., Kramer, L., Santos, G., et al. (2019). Sex-specific regulation of collagen I and III expression by 17β-Estradiol in cardiac fibroblasts: role of estrogen receptors. Cardiovasc. Res. 115, 315–327. doi: 10.1093/cvr/cvy185
El Hajj, M. C., Ninh, V. K., El Hajj, E. C., Bradley, J. M., and Gardner, J. D. (2017). Estrogen receptor antagonism exacerbates cardiac structural and functional remodeling in female rats. Am. J. Physiol. Heart Circ. Physiol. 312, H98–H105. doi: 10.1152/ajpheart.00348.2016
Fares, E., Parks, R. J., Macdonald, J. K., Egar, J. M., and Howlett, S. E. (2012). Ovariectomy enhances SR Ca(2)(+) release and increases Ca(2)(+) spark amplitudes in isolated ventricular myocytes. J. Mol. Cell. Cardiol. 52, 32–42. doi: 10.1016/j.yjmcc.2011.09.002
Fares, E., Pyle, W. G., Ray, G., Rose, R. A., Denovan-Wright, E. M., Chen, R. P., et al. (2013). The impact of ovariectomy on calcium homeostasis and myofilament calcium sensitivity in the aging mouse heart. PLoS One 8:e74719. doi: 10.1371/journal.pone.0074719
Fearnley, C. J., Roderick, H. L., and Bootman, M. D. (2011). Calcium signaling in cardiac myocytes. Cold Spring Harb. Perspect. Biol. 3:a004242. doi: 10.1101/cshperspect.a004242
Franssen, C., Chen, S., Unger, A., Korkmaz, H. I., De Keulenaer, G. W., Tschöpe, C., et al. (2016). Myocardial microvascular inflammatory endothelial activation in heart failure with preserved ejection fraction. JACC Heart Fail. 4, 312–324. doi: 10.1016/j.jchf.2015.10.007
Fuentes, N., and Silveyra, P. (2019). Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 116, 135–170. doi: 10.1016/bs.apcsb.2019.01.001
Fulton, D., Gratton, J. P., McCabe, T. J., Fontana, J., Fujio, Y., Walsh, K., et al. (1999). Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 399, 597–601. doi: 10.1038/21218
Galien, R., and Garcia, T. (1997). Estrogen receptor impairs interleukin-6 expression by preventing protein binding on the NF-kappaB site. Nucleic Acids Res. 25, 2424–2429. doi: 10.1093/nar/25.12.2424
Gerber, Y., Weston, S. A., Redfield, M. M., Chamberlain, A. M., Manemann, S. M., Jiang, R., et al. (2015). A contemporary appraisal of the heart failure epidemic in Olmsted County, Minnesota, 2000 to 2010. JAMA Intern. Med. 175, 996–1004. doi: 10.1001/jamainternmed.2015.0924
Györke, S., and Carnes, C. (2008). Dysregulated sarcoplasmic reticulum calcium release: potential pharmacological target in cardiac disease. Pharmacol. Ther. 119, 340–354. doi: 10.1016/j.pharmthera.2008.06.002
Hamdani, N., Bishu, K. G., von Frieling-Salewsky, M., Redfield, M. M., and Linke, W. A. (2013). Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc. Res. 97, 464–471. doi: 10.1093/cvr/cvs353
Harvey, X. M. P. (2012). Estrogen differentially affects expression of calcium handling genes in female and male adult cardiomyocytes. J. Stud. Res. 1, 31–37. doi: 10.47611/jsr.v1i3.93
Hickey, M., McNamara, H. C., and Mishra, G. D. (2018). Menopausal estrogen therapy and breast cancer mortality. JAMA 319, 193. doi: 10.1001/jama.2017.18308
Hidalgo, C. G., Chung, C. S., Saripalli, C., Methawasin, M., Hutchinson, K. R., Tsaprailis, G., et al. (2013). The multifunctional Ca(2+)/calmodulin-dependent protein kinase II delta (CaMKIIδ) phosphorylates cardiac titin’s spring elements. J. Mol. Cell. Cardiol. 54, 90–97. doi: 10.1016/j.yjmcc.2012.11.012
Hodis, H. N., Mack, W. J., Henderson, V. W., Shoupe, D., Budoff, M. J., Hwang-Levine, J., et al. (2016). Vascular effects of early versus late postmenopausal treatment with estradiol. N. Engl. J. Med. 374, 1221–1231. doi: 10.1056/NEJMoa1505241
Hou, H., Zhao, Z., Machuki, J. O., Zhang, L., Zhang, Y., Fu, L., et al. (2018). Estrogen deficiency compromised the β(2)AR-Gs/Gi coupling: implications for arrhythmia and cardiac injury. Pflugers Arch. 470, 559–570. doi: 10.1007/s00424-017-2098-4
Hu, Q., Zhang, T., Li, Y., Feng, J., Nie, R., Wang, X., et al. (2020). β2AR-dependent signaling contributes to in-vivo reendothelialization capacity of endothelial progenitor cells by shear stress. J. Hypertens. 38, 82–94. doi: 10.1097/hjh.0000000000002203
Huang, A., Sun, D., Koller, A., and Kaley, G. (2000). 17beta-estradiol restores endothelial nitric oxide release to shear stress in arterioles of male hypertensive rats. Circulation 101, 94–100. doi: 10.1161/01.cir.101.1.94
Iorga, A., Cunningham, C. M., Moazeni, S., Ruffenach, G., Umar, S., and Eghbali, M. (2017). The protective role of estrogen and estrogen receptors in cardiovascular disease and the controversial use of estrogen therapy. Biol. Sex Differ. 8:33. doi: 10.1186/s13293-017-0152-8
Jaffré, F., Friedman, A. E., Hu, Z., Mackman, N., and Blaxall, B. C. (2012). β-adrenergic receptor stimulation transactivates protease-activated receptor 1 via matrix metalloproteinase 13 in cardiac cells. Circulation 125, 2993–3003. doi: 10.1161/circulationaha.111.066787
Jiao, L., Machuki, J. O., Wu, Q., Shi, M., Fu, L., Adekunle, A. O., et al. (2020). Estrogen and calcium handling proteins: new discoveries and mechanisms in cardiovascular diseases. Am. J. Physiol. Heart Circ. Physiol. 318, H820–H829. doi: 10.1152/ajpheart.00734.2019
Kilfoil, P. J., Lotteau, S., Zhang, R., Yue, X., Aynaszyan, S., Solymani, R. E., et al. (2020). Distinct features of calcium handling and β-adrenergic sensitivity in heart failure with preserved versus reduced ejection fraction. J. Physiol. 598, 5091–5108. doi: 10.1113/jp280425
Kim, J. K., Pedram, A., Razandi, M., and Levin, E. R. (2006). Estrogen prevents cardiomyocyte apoptosis through inhibition of reactive oxygen species and differential regulation of p38 kinase isoforms. J. Biol. Chem. 281, 6760–6767. doi: 10.1074/jbc.M511024200
Kim, J., Eckhart, A. D., Eguchi, S., and Koch, W. J. (2002). Beta-adrenergic receptor-mediated DNA synthesis in cardiac fibroblasts is dependent on transactivation of the epidermal growth factor receptor and subsequent activation of extracellular signal-regulated kinases. J. Biol. Chem. 277, 32116–32123. doi: 10.1074/jbc.M204895200
Kohn, G. E., Rodriguez, K. M., Hotaling, J., and Pastuszak, A. W. (2019). The history of estrogen therapy. Sex Med. Rev. 7, 416–421. doi: 10.1016/j.sxmr.2019.03.006
Kolluru, G. K., Siamwala, J. H., and Chatterjee, S. (2010). eNOS phosphorylation in health and disease. Biochimie 92, 1186–1198. doi: 10.1016/j.biochi.2010.03.020
Kozakowski, J., Gietka-Czernel, M., Leszczyńska, D., and Majos, A. (2017). Obesity in menopause - our negligence or an unfortunate inevitability? Prz. Menopauzalny 16, 61–65. doi: 10.5114/pm.2017.68594
Kravtsov, G. M., Kam, K. W., Liu, J., Wu, S., and Wong, T. M. (2007). Altered Ca(2+) handling by ryanodine receptor and Na(+)-Ca(2+) exchange in the heart from ovariectomized rats: role of protein kinase A. Am. J. Physiol. Cell Physiol. 292, C1625–C1635. doi: 10.1152/ajpcell.00368.2006
Krüger, M., Kötter, S., Grützner, A., Lang, P., Andresen, C., Redfield, M. M., et al. (2009). Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ. Res. 104, 87–94. doi: 10.1161/circresaha.108.184408
Kubes, P., Suzuki, M., and Granger, D. N. (1991). Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc. Natl. Acad. Sci. U.S.A. 88, 4651–4655. doi: 10.1073/pnas.88.11.4651
Kurata, K., Takebayashi, M., Kagaya, A., Morinobu, S., and Yamawaki, S. (2001). Effect of beta-estradiol on voltage-gated Ca(2+) channels in rat hippocampal neurons: a comparison with dehydroepiandrosterone. Eur. J. Pharmacol. 416, 203–212. doi: 10.1016/s0014-2999(01)00880-9
Kuwahara, F., Kai, H., Tokuda, K., Takeya, M., Takeshita, A., Egashira, K., et al. (2004). Hypertensive myocardial fibrosis and diastolic dysfunction: another model of inflammation? Hypertension 43, 739–745. doi: 10.1161/01.HYP.0000118584.33350.7d
LaCroix, A. Z., Chlebowski, R. T., Manson, J. E., Aragaki, A. K., Johnson, K. C., Martin, L., et al. (2011). Health outcomes after stopping conjugated equine estrogens among postmenopausal women with prior hysterectomy: a randomized controlled trial. JAMA 305, 1305–1314. doi: 10.1001/jama.2011.382
Lai, Y. J., Zhu, B. L., Sun, F., Luo, D., Ma, Y. L., Luo, B., et al. (2019). Estrogen receptor α promotes Cav1.2 ubiquitination and degradation in neuronal cells and in APP/PS1 mice. Aging Cell 18, e12961. doi: 10.1111/acel.12961
Lam, C. S. P., Donal, E., Kraigher-Krainer, E., and Vasan, R. S. (2011). Epidemiology and clinical course of heart failure with preserved ejection fraction. Eur. J. Heart Fail. 13, 18–28. doi: 10.1093/eurjhf/hfq121
Lang, U., Baker, R. S., and Clark, K. E. (1997). Estrogen-induced increases in coronary blood flow are antagonized by inhibitors of nitric oxide synthesis. Eur. J. Obstet. Gynecol. Reprod. Biol. 74, 229–235. doi: 10.1016/s0301-2115(97)00104-8
Lee, J. Y., Hyun, H. S., Park, H. G., Seo, J. H., Lee, E. Y., Lee, J. S., et al. (2015). Effects of hormone therapy on serum lipid levels in postmenopausal Korean women. J. Menopausal Med. 21, 104–111. doi: 10.6118/jmm.2015.21.2.104
LeWinter, M. M., and Granzier, H. (2010). Cardiac titin: a multifunctional giant. Circulation 121, 2137–2145. doi: 10.1161/CIRCULATIONAHA.109.860171
Linke, W. A., and Hamdani, N. (2014). Gigantic business: titin properties and function through thick and thin. Circ. Res. 114, 1052–1068. doi: 10.1161/CIRCRESAHA.114.301286
Louch, W. E., Hougen, K., Mork, H. K., Swift, F., Aronsen, J. M., Sjaastad, I., et al. (2010). Sodium accumulation promotes diastolic dysfunction in end-stage heart failure following Serca2 knockout. J. Physiol. 588(Pt 3), 465–478. doi: 10.1113/jphysiol.2009.183517
Lv, T., Du, Y., Cao, N., Zhang, S., Gong, Y., Bai, Y., et al. (2016). Proliferation in cardiac fibroblasts induced by β1-adrenoceptor autoantibody and the underlying mechanisms. Sci. Rep. 6:32430. doi: 10.1038/srep32430
Ma, C., Luo, H., Fan, L., Liu, X., and Gao, C. (2020). Heart failure with preserved ejection fraction: an update on pathophysiology, diagnosis, treatment, and prognosis. Braz. J. Med. Biol. Res. 53:e9646. doi: 10.1590/1414-431X20209646
Ma, Y., de Castro Brás, L. E., Toba, H., Iyer, R. P., Hall, M. E., Winniford, M. D., et al. (2014). Myofibroblasts and the extracellular matrix network in post-myocardial infarction cardiac remodeling. Pflugers Arch. 466, 1113–1127. doi: 10.1007/s00424-014-1463-9
Machi, J. F., Dias Dda, S., Freitas, S. C., de Moraes, O. A., da Silva, M. B., Cruz, P. L., et al. (2016). Impact of aging on cardiac function in a female rat model of menopause: role of autonomic control, inflammation, and oxidative stress. Clin. Interv. Aging 11, 341–350. doi: 10.2147/CIA.S88441
Machuki, J. O., Zhang, H. Y., Geng, J., Fu, L., Adzika, G. K., Wu, L., et al. (2019). Estrogen regulation of cardiac cAMP-L-type Ca(2+) channel pathway modulates sex differences in basal contraction and responses to β(2)AR-mediated stress in left ventricular apical myocytes. Cell Commun. Signal. 17:34. doi: 10.1186/s12964-019-0346-2
Machuki, J. O., Zhang, H. Y., Harding, S. E., and Sun, H. (2018). Molecular pathways of oestrogen receptors and β-adrenergic receptors in cardiac cells: recognition of their similarities, interactions and therapeutic value. Acta Physiol. (Oxf.) 222:e12978. doi: 10.1111/apha.12978
Mahmoodzadeh, S., Dworatzek, E., Fritschka, S., Pham, T. H., and Regitz-Zagrosek, V. (2010). 17beta-Estradiol inhibits matrix metalloproteinase-2 transcription via MAP kinase in fibroblasts. Cardiovasc. Res. 85, 719–728. doi: 10.1093/cvr/cvp350
Maria Grazia, M. (2016). “Estrogens and the heart: do they help or hurt?” how estrogen impacts the cardiovascular system. SOJ Gynecol. Obstet. Womens Health 2:8. doi: 10.15226/2381-2915/2/1/00108
Maslov, P. Z., Kim, J. K., Argulian, E., Ahmadi, A., Narula, N., Singh, M., et al. (2019). Is cardiac diastolic dysfunction a part of post-menopausal syndrome? JACC Heart Fail. 7, 192–203. doi: 10.1016/j.jchf.2018.12.018
Masri, A., Althouse, A. D., Hickey, G., McKibben, J., Ramani, R., Mathier, M., et al. (2017). Abstract 16188: sex differences and outcomes of heart failure admissions stratified by left ventricle ejection fraction. Circulation 136(suppl. 1):A16188. doi: 10.1161/circ.136.suppl_1.16188
Matsuba, D., Terui, T., O-Uchi, J., Tanaka, H., Ojima, T., Ohtsuki, I., et al. (2009). Protein kinase A-dependent modulation of Ca2+ sensitivity in cardiac and fast skeletal muscles after reconstitution with cardiac troponin. J. Gen. Physiol. 133, 571–581. doi: 10.1085/jgp.200910206
Matsubara, B. B., Matsubara, L. S., Zornoff, L. A., Franco, M., and Janicki, J. S. (1998). Left ventricular adaptation to chronic pressure overload induced by inhibition of nitric oxide synthase in rats. Basic Res. Cardiol. 93, 173–181. doi: 10.1007/s003950050084
McLarty, J. L., Li, J., Levick, S. P., and Janicki, J. S. (2013). Estrogen modulates the influence of cardiac inflammatory cells on function of cardiac fibroblasts. J. Inflamm. Res. 6, 99–108. doi: 10.2147/jir.s48422
Michalson, K. T., Groban, L., Howard, T. D., Shively, C. A., Sophonsritsuk, A., Appt, S. E., et al. (2018). Estradiol treatment initiated early after ovariectomy regulates myocardial gene expression and inhibits diastolic dysfunction in female cynomolgus monkeys: potential roles for calcium homeostasis and extracellular matrix remodeling. J. Am. Heart Assoc. 7:e009769. doi: 10.1161/jaha.118.009769
Miranda-Silva, D., Wüst, R., Conceição, G., Gonçalves-Rodrigues, P., Gonçalves, N., Gonçalves, A., et al. (2020). Disturbed cardiac mitochondrial and cytosolic calcium handling in a metabolic risk-related rat model of heart failure with preserved ejection fraction. Acta Physiol. (Oxf.) 228:e13378. doi: 10.1111/apha.13378
Modena, M. G., Sismondi, P., Mueck, A. O., Kuttenn, F., Lignières, B., Verhaeghe, J., et al. (2005). New evidence regarding hormone replacement therapies is urgently required transdermal postmenopausal hormone therapy differs from oral hormone therapy in risks and benefits. Maturitas 52, 1–10. doi: 10.1016/j.maturitas.2005.05.003
Möller, C., Stiermaier, T., Brabant, G., Graf, T., Thiele, H., and Eitel, I. (2018). Comprehensive assessment of sex hormones in Takotsubo syndrome. Int. J. Cardiol. 250, 11–15. doi: 10.1016/j.ijcard.2017.10.047
Mompeón, A., Lázaro-Franco, M., Bueno-Betí, C., Pérez-Cremades, D., Vidal-Gómez, X., Monsalve, E., et al. (2016). Estradiol, acting through ERα, induces endothelial non-classic renin-angiotensin system increasing angiotensin 1-7 production. Mol. Cell. Endocrinol. 422, 1–8. doi: 10.1016/j.mce.2015.11.004
Mori, T., Kai, H., Kajimoto, H., Koga, M., Kudo, H., Takayama, N., et al. (2011). Enhanced cardiac inflammation and fibrosis in ovariectomized hypertensive rats: a possible mechanism of diastolic dysfunction in postmenopausal women. Hypertens. Res. 34, 496–502. doi: 10.1038/hr.2010.261
Morimoto, A., Kadoya, M., Kakutani-Hatayama, M., Kosaka-Hamamoto, K., Miyoshi, A., Shoji, T., et al. (2020). Subclinical decrease in cardiac autonomic and diastolic function in patients with metabolic disorders: HSCAA study. Metabol. Open 5:100025. doi: 10.1016/j.metop.2020.100025
Müller-Ehmsen, J., Wang, J., Schwinger, R. H., and McDonough, A. A. (2001). Region specific regulation of sodium pump isoform and Na, Ca-exchanger expression in the failing human heart–right atrium vs left ventricle. Cell. Mol. Biol. (Noisy-le-grand) 47, 373–381.
Najafi, A., Sequeira, V., Kuster, D. W., and van der Velden, J. (2016). beta-adrenergic receptor signalling and its functional consequences in the diseased heart. Eur. J. Clin. Invest. 46, 362–374. doi: 10.1111/eci.12598
Naseem, K. M. (2005). The role of nitric oxide in cardiovascular diseases. Mol. Aspects Med. 26, 33–65. doi: 10.1016/j.mam.2004.09.003
Ndzie Noah, M. L., Adzika, G. K., Mprah, R., Adekunle, A. O., Adu-Amankwaah, J., and Sun, H. (2021). Sex–Gender disparities in cardiovascular diseases: the effects of estrogen on eNOS, lipid profile, and NFATs during catecholamine stress. Front. Cardiovasc. Med. 8:15. doi: 10.3389/fcvm.2021.639946
Nickenig, G., Bäumer, A. T., Grohè, C., Kahlert, S., Strehlow, K., Rosenkranz, S., et al. (1998). Estrogen modulates AT1 receptor gene expression in vitro and in vivo. Circulation 97, 2197–2201. doi: 10.1161/01.cir.97.22.2197
Ouchi, N., Parker, J. L., Lugus, J. J., and Walsh, K. (2011). Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 11, 85–97. doi: 10.1038/nri2921
Özlek, B., Özlek, E., Kahraman, S., Tekinalp, M., Zencirkiran Agus, H., Çelik, O., et al. (2019). Gender disparities in heart failure with mid-range and preserved ejection fraction: results from APOLLON study. Anatol. J. Cardiol. 21, 242–252. doi: 10.14744/AnatolJCardiol.2019.71954
Pandit, S., Woranush, W., Wattanapermpool, J., and Bupha-Intr, T. (2014). Significant role of female sex hormones in cardiac myofilament activation in angiotensin II-mediated hypertensive rats. J. Physiol. Sci. 64, 269–277. doi: 10.1007/s12576-014-0316-9
Parks, R. J., Bogachev, O., Mackasey, M., Ray, G., Rose, R. A., and Howlett, S. E. (2017). The impact of ovariectomy on cardiac excitation-contraction coupling is mediated through cAMP/PKA-dependent mechanisms. J. Mol. Cell. Cardiol. 111, 51–60. doi: 10.1016/j.yjmcc.2017.07.118
Paulus, W. J. (2001). The role of nitric oxide in the failing heart. Heart Fail. Rev. 6, 105–118. doi: 10.1023/a:1011453809750
Paulus, W. J., and Tschöpe, C. (2013). A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 62, 263–271. doi: 10.1016/j.jacc.2013.02.092
Paur, H., Wright, P. T., Sikkel, M. B., Tranter, M. H., Mansfield, C., O’Gara, P., et al. (2012). High levels of circulating epinephrine trigger apical cardiodepression in a β2-adrenergic receptor/Gi-dependent manner: a new model of Takotsubo cardiomyopathy. Circulation 126, 697–706. doi: 10.1161/circulationaha.112.111591
Pfeffer, M. A., Shah, A. M., and Borlaug, B. A. (2019). Heart failure with preserved ejection fraction in perspective. Circ. Res. 124, 1598–1617. doi: 10.1161/circresaha.119.313572
Raeeszadeh-Sarmazdeh, M., Do, L. D., and Hritz, B. G. (2020). Metalloproteinases and their inhibitors: potential for the development of new therapeutics. Cells 9:1313. doi: 10.3390/cells9051313
Rajapakse, A. G., Yepuri, G., Carvas, J. M., Stein, S., Matter, C. M., Scerri, I., et al. (2011). Hyperactive S6K1 mediates oxidative stress and endothelial dysfunction in aging: inhibition by resveratrol. PLoS One 6:e19237. doi: 10.1371/journal.pone.0019237
Rao, I. V., and Burkhoff, D. (2021). Cardiac contractility modulation for the treatment of moderate to severe HF. Expert Rev. Med. Devices 18, 15–21. doi: 10.1080/17434440.2020.1853525
Raskin, A., Lange, S., Banares, K., Lyon, R. C., Zieseniss, A., Lee, L. K., et al. (2012). A novel mechanism involving four-and-a-half LIM domain protein-1 and extracellular signal-regulated kinase-2 regulates titin phosphorylation and mechanics. J. Biol. Chem. 287, 29273–29284. doi: 10.1074/jbc.M112.372839
Rattanasopa, C., Phungphong, S., Wattanapermpool, J., and Bupha-Intr, T. (2015). Significant role of estrogen in maintaining cardiac mitochondrial functions. J. Steroid Biochem. Mol. Biol. 147, 1–9. doi: 10.1016/j.jsbmb.2014.11.009
Ren, J., Hintz, K. K., Roughead, Z. K., Duan, J., Colligan, P. B., Ren, B. H., et al. (2003). Impact of estrogen replacement on ventricular myocyte contractile function and protein kinase B/Akt activation. Am. J. Physiol. Heart Circ. Physiol. 284, H1800–H1807. doi: 10.1152/ajpheart.00866.2002
Renovato-Martins, M., Matheus, M. E., de Andrade, I. R., Moraes, J. A., da Silva, S. V., Citelli Dos Reis, M., et al. (2017). Microparticles derived from obese adipose tissue elicit a pro-inflammatory phenotype of CD16(+), CCR5(+) and TLR8(+) monocytes. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 139–151. doi: 10.1016/j.bbadis.2016.09.016
Rienks, M., Papageorgiou, A.-P., Frangogiannis Nikolaos, G., and Heymans, S. (2014). Myocardial extracellular matrix. Circ. Res. 114, 872–888. doi: 10.1161/CIRCRESAHA.114.302533
Roger, V. L. (2013). Epidemiology of heart failure. Circ. Res. 113, 646–659. doi: 10.1161/CIRCRESAHA.113.300268
Rossouw, J. E., Anderson, G. L., Prentice, R. L., LaCroix, A. Z., Kooperberg, C., Stefanick, M. L., et al. (2002). Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA 288, 321–333. doi: 10.1001/jama.288.3.321
Rouhana, S., Farah, C., Roy, J., Finan, A., Rodrigues de Araujo, G., Bideaux, P., et al. (2019). Early calcium handling imbalance in pressure overload-induced heart failure with nearly normal left ventricular ejection fraction. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 230–242. doi: 10.1016/j.bbadis.2018.08.005
Rush, C. J., Berry, C., Oldroyd, K. G., Rocchiccioli, J. P., Lindsay, M. M., Touyz, R. M., et al. (2021). Prevalence of coronary artery disease and coronary microvascular dysfunction in patients with heart failure with preserved ejection fraction. JAMA Cardiol. e211825. doi: 10.1001/jamacardio.2021.1825
Sabbatini, A. R., and Kararigas, G. (2020). Menopause-Related estrogen decrease and the pathogenesis of HFpEF: JACC review topic of the week. J. Am. Coll. Cardiol. 75, 1074–1082. doi: 10.1016/j.jacc.2019.12.049
Sandstedt, J., Sandstedt, M., Lundqvist, A., Jansson, M., Sopasakis, V. R., Jeppsson, A., et al. (2019). Human cardiac fibroblasts isolated from patients with severe heart failure are immune-competent cells mediating an inflammatory response. Cytokine 113, 319–325. doi: 10.1016/j.cyto.2018.09.021
Schierbeck, L. L., Rejnmark, L., Tofteng, C. L., Stilgren, L., Eiken, P., Mosekilde, L., et al. (2012). Effect of hormone replacement therapy on cardiovascular events in recently postmenopausal women: randomised trial. BMJ 345:e6409. doi: 10.1136/bmj.e6409
Sequeira, V., Nijenkamp, L. L., Regan, J. A., and van der Velden, J. (2014). The physiological role of cardiac cytoskeleton and its alterations in heart failure. Biochim. Biophys. Acta 1838, 700–722. doi: 10.1016/j.bbamem.2013.07.011
Shao, H., Breitner, J. C., Whitmer, R. A., Wang, J., Hayden, K., Wengreen, H., et al. (2012). Hormone therapy and Alzheimer disease dementia: new findings from the Cache County Study. Neurology 79, 1846–1852. doi: 10.1212/WNL.0b013e318271f823
Sharma, R., Sharma, N. K., and Thungapathra, M. (2017). Resveratrol regulates body weight in healthy and ovariectomized rats. Nutr. Metab. (Lond.) 14:30. doi: 10.1186/s12986-017-0183-5
Sherwood, A., Park, S. B., Hughes, J. W., Blumenthal, J. A., Hinderliter, A., Trivedi, R., et al. (2010). Cardiovascular hemodynamics during stress in premenopausal versus postmenopausal women. Menopause 17, 403–409. doi: 10.1097/gme.0b013e3181b9b061
Shufelt, C., Bairey Merz, C. N., Pettinger, M. B., Choi, L., Chlebowski, R., Crandall, C. J., et al. (2018). Estrogen-alone therapy and invasive breast cancer incidence by dose, formulation, and route of delivery: findings from the WHI observational study. Menopause 25, 985–991. doi: 10.1097/gme.0000000000001115
Sickinghe, A. A., Korporaal, S. J. A., den Ruijter, H. M., and Kessler, E. L. (2019). Estrogen contributions to microvascular dysfunction evolving to heart failure with preserved ejection fraction. Front. Endocrinol. (Lausanne) 10:442. doi: 10.3389/fendo.2019.00442
Sophonsritsuk, A., Appt, S. E., Clarkson, T. B., Shively, C. A., Espeland, M. A., and Register, T. C. (2013). Differential effects of estradiol on carotid artery inflammation when administered early versus late after surgical menopause. Menopause 20, 540–547. doi: 10.1097/GME.0b013e31827461e0
Spinale, F. G., Coker, M. L., Krombach, S. R., Mukherjee, R., Hallak, H., Houck, W. V., et al. (1999). Matrix metalloproteinase inhibition during the development of congestive heart failure : effects on left ventricular dimensions and function. Circ. Res. 85, 364–376. doi: 10.1161/01.res.85.4.364
Sribnick, E. A., Del Re, A. M., Ray, S. K., Woodward, J. J., and Banik, N. L. (2009). Estrogen attenuates glutamate-induced cell death by inhibiting Ca2+ influx through L-type voltage-gated Ca2+ channels. Brain Res. 1276, 159–170. doi: 10.1016/j.brainres.2009.04.022
Srikakulapu, P., and McNamara, C. A. (2020). B lymphocytes and adipose tissue inflammation. Arterioscler. Thromb. Vasc. Biol. 40, 1110–1122. doi: 10.1161/atvbaha.119.312467
Sudoh, N., Toba, K., Akishita, M., Ako, J., Hashimoto, M., Iijima, K., et al. (2001). Estrogen prevents oxidative stress-induced endothelial cell apoptosis in rats. Circulation 103, 724–729. doi: 10.1161/01.cir.103.5.724
Tanner, M. A., Thomas, T. P., and Maitz, C. A. (2020). β2-Adrenergic receptors increase cardiac fibroblast proliferation through the Gαs/ERK1/2-dependent secretion of interleukin-6. Int. J. Mol. Sci. 21:8507. doi: 10.3390/ijms21228507
Thawornkaiwong, A., Pantharanontaga, J., and Wattanapermpool, J. (2007). Hypersensitivity of myofilament response to Ca2+ in association with maladaptation of estrogen-deficient heart under diabetes complication. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R844–R851. doi: 10.1152/ajpregu.00365.2006
Tilley, D. G. (2011). G protein-dependent and G protein-independent signaling pathways and their impact on cardiac function. Circ. Res. 109, 217–230. doi: 10.1161/CIRCRESAHA.110.231225
Tousoulis, D., Kampoli, A. M., Tentolouris, C., Papageorgiou, N., and Stefanadis, C. (2012). The role of nitric oxide on endothelial function. Curr. Vasc. Pharmacol. 10, 4–18. doi: 10.2174/157016112798829760
Turdi, S., Huff, A. F., Pang, J., He, E. Y., Chen, X., Wang, S., et al. (2015). 17-β estradiol attenuates ovariectomy-induced changes in cardiomyocyte contractile function via activation of AMP-activated protein kinase. Toxicol. Lett. 232, 253–262. doi: 10.1016/j.toxlet.2014.11.012
van Heerebeek, L., Hamdani, N., Falcao-Pires, I., Leite-Moreira, A. F., Begieneman, M. P., Bronzwaer, J. G., et al. (2012). Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation 126, 830–839. doi: 10.1161/CIRCULATIONAHA.111.076075
Venugopal, S. K., Devaraj, S., Yuhanna, I., Shaul, P., and Jialal, I. (2002). Demonstration that C-reactive protein decreases eNOS expression and bioactivity in human aortic endothelial cells. Circulation 106, 1439–1441. doi: 10.1161/01.cir.0000033116.22237.f9
Vettel, C., Lämmle, S., Ewens, S., Cervirgen, C., Emons, J., Ongherth, A., et al. (2014). PDE2-mediated cAMP hydrolysis accelerates cardiac fibroblast to myofibroblast conversion and is antagonized by exogenous activation of cGMP signaling pathways. Am. J. Physiol. Heart Circ. Physiol. 306, H1246–H1252. doi: 10.1152/ajpheart.00852.2013
Wang, H., Zhao, Z., Lin, M., and Groban, L. (2015). Activation of GPR30 inhibits cardiac fibroblast proliferation. Mol. Cell. Biochem. 405, 135–148. doi: 10.1007/s11010-015-2405-3
Wang, J., Huertas-Vazquez, A., Wang, Y., and Lusis, A. J. (2019). Isoproterenol-Induced cardiac diastolic dysfunction in mice: a systems genetics analysis. Front. Cardiovasc. Med. 6:100. doi: 10.3389/fcvm.2019.00100
Wang, X., and Khalil, R. A. (2018). Matrix metalloproteinases, vascular remodeling, and vascular disease. Adv. Pharmacol. 81, 241–330. doi: 10.1016/bs.apha.2017.08.002
Watanabe, T., Akishita, M., He, H., Miyahara, Y., Nagano, K., Nakaoka, T., et al. (2003). 17 beta-estradiol inhibits cardiac fibroblast growth through both subtypes of estrogen receptor. Biochem. Biophys. Res. Commun. 311, 454–459. doi: 10.1016/j.bbrc.2003.09.232
Westermann, D., Lindner, D., Kasner, M., Zietsch, C., Savvatis, K., Escher, F., et al. (2011). Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ. Heart Fail. 4, 44–52. doi: 10.1161/circheartfailure.109.931451
Westermann, D., Riad, A., Richter, U., Jäger, S., Savvatis, K., Schuchardt, M., et al. (2009). Enhancement of the endothelial NO synthase attenuates experimental diastolic heart failure. Basic Res. Cardiol. 104, 499–509. doi: 10.1007/s00395-009-0014-6
Wu, H., Yang, H., Rhee, J. W., Zhang, J. Z., Lam, C. K., Sallam, K., et al. (2019). Modelling diastolic dysfunction in induced pluripotent stem cell-derived cardiomyocytes from hypertrophic cardiomyopathy patients. Eur. Heart J. 40, 3685–3695. doi: 10.1093/eurheartj/ehz326
Xue, B., Zhang, Z., Beltz, T. G., Guo, F., Hay, M., and Johnson, A. K. (2014). Estrogen regulation of the brain renin-angiotensin system in protection against angiotensin II-induced sensitization of hypertension. Am. J. Physiol. Heart Circ. Physiol. 307, H191–H198. doi: 10.1152/ajpheart.01012.2013
Yang, H. Y., Firth, J. M., Francis, A. J., Alvarez-Laviada, A., and MacLeod, K. T. (2017). Effect of ovariectomy on intracellular Ca(2+) regulation in guinea pig cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 313, H1031–H1043. doi: 10.1152/ajpheart.00249.2017
Zhou, L., Chen, H., Mao, X., Qi, H., Baker, P. N., and Zhang, H. (2017). G-protein-coupled receptor 30 mediates the effects of estrogen on endothelial cell tube formation in vitro. Int. J. Mol. Med. 39, 1461–1467. doi: 10.3892/ijmm.2017.2957
Keywords: HFpEF, estrogen deficiency, β-adrenergic receptors, calcium handling, cardiomyocyte cytoskeleton, extra cellular matrix, coronary microvascular endothelial, inflammation
Citation: Adekunle AO, Adzika GK, Mprah R, Ndzie Noah ML, Adu-Amankwaah J, Rizvi R, Akhter N and Sun H (2021) Predominance of Heart Failure With Preserved Ejection Fraction in Postmenopausal Women: Intra- and Extra-Cardiomyocyte Maladaptive Alterations Scaffolded by Estrogen Deficiency. Front. Cell Dev. Biol. 9:685996. doi: 10.3389/fcell.2021.685996
Received: 26 March 2021; Accepted: 09 September 2021;
Published: 29 September 2021.
Edited by:
Hester Den Ruijter, University Medical Center Utrecht, NetherlandsReviewed by:
Saskia C. A. De Jager, Utrecht University, NetherlandsErnest Diez Benavente, University Medical Center Utrecht, Netherlands
Copyright © 2021 Adekunle, Adzika, Mprah, Ndzie Noah, Adu-Amankwaah, Rizvi, Akhter and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong Sun, c3VuaEB4emhtdS5lZHUuY24=
†These authors have contributed equally to this work