 Simin Liang
Simin Liang Xiaojia Zhou2
Xiaojia Zhou2 Fernando Rodrigues-Lima
Fernando Rodrigues-Lima Li Wang
Li Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol., 09 September 2021
Sec. Cell Death and Survival
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.685954
This article is part of the Research TopicCell Death and Targeted Cancer TherapiesView all 16 articles
Chidamide (CDM), a novel histone deacetylase inhibitor, is currently used for patients with peripheral T-cell lymphoma. Aspirin (ASA), an anti-inflammatory drug, has been shown to exert anticancer activity. Herein, we investigated the effect of CDM combined with ASA on myelodysplastic syndromes-derived acute myeloid leukemia (AML-MDS) cells and explored the underlying mechanism. The putative targets of CDM and ASA were predicted by network pharmacology approach. GO functional and KEGG pathway enrichment analyses were performed by DAVID. Furthermore, experimental validation was conducted by Cell Counting Kit-8 assay, Flow cytometry and Western blotting. Network pharmacology analysis revealed 36 AML-MDS-related overlapping genes that were targets of CDM and ASA, while 10 hub genes were identified by the plug-in cytoHubba in Cytoscape. Pathway enrichment analysis indicated CDM and ASA significantly affected PI3K/AKT signaling pathway. Functional experiments demonstrated that the combination of CDM and ASA had a remarkable synergistic anti-proliferative effect by blocking the cell cycle in G0/G1 phase and inducing apoptosis. Mechanistically, the combination treatment significantly down-regulated the phosphorylation levels of PI3K and AKT. In addition, insulin-like growth factor 1 (IGF-1), an activator of PI3K/AKT pathway, reversed the effects of the combination treatment. Our findings suggested that CDM combined with ASA exerted a synergetic inhibitory effect on cell growth by inactivating PI3K/AKT pathway, which might pave the way for effective treatments of AML-MDS.
Myelodysplastic syndromes (MDS) are a heterogeneous group of myeloid disorders characterized by ineffective hematopoiesis, peripheral blood cytopenia and high risk of transformation to acute myeloid leukemia (AML) with poor prognosis (Cogle et al., 2011; Scalzulli et al., 2020). MDS-derived AML (AML-MDS) shows slower hematologic recovery and poorer outcomes following intensive remission-induction chemotherapy than de novo AML (Boddu et al., 2017; Ramadan et al., 2020). Additionally, allogeneic stem cell transplantation (alloSCT) remains the only curative option for patients with AML-MDS. But unfortunately, it is only suitable for a minority (Schroeder et al., 2019). Therefore, a novel effective treatment strategy with minimal cytotoxicity still needs to be developed for AML-MDS.
Chidamide (CDM), a novel histone deacetylase inhibitor, selectively inhibits HDAC1, 2, 3, and 10, and has been approved for treatment of patients with recurrent or refractory peripheral T cell lymphoma (PTCL) in China (Shi et al., 2015; Lu et al., 2016). Strikingly, a number of studies have suggested that CDM exerts cytotoxic effects on lymphoma (Zhou et al., 2018), multiple myeloma (MM) (Sun et al., 2019), MDS (Liu et al., 2016), and leukemia (Li et al., 2015), as well as non-hematological malignancies, including lung cancer (Wu et al., 2019), colon cancer (Liu et al., 2010) and hepatocellular carcinoma (Wang et al., 2012). Moreover, CDM has been shown to synergize effects with other anti-tumor agents. For example, several studies have showed that CDM combined with hypomethylating agents, including decitabine, resulted in synergistic effects on the proliferation and apoptosis of myeloid leukemia cells (Xu et al., 2019; Li et al., 2020). Co-treatment with CDM and Bortezomib reduced proliferation, invasion and migration of gastric cancer cells (Zhang et al., 2020). Co-treatment with CDM and Rituximab inhibited tumor growth by upregulating CD20 in diffuse large B-cell lymphoma (DLBCL) (Guan et al., 2020).
Aspirin (acetylsalicylic acid, ASA) has been widely used as an anti-inflammatory, analgesic drug, as well as in cardiovascular disease and platelet aggregation. ASA can reduce the morbidity and mortality of several malignancies, including gastric cancer (García Rodríguez et al., 2020), lung cancer (Erickson et al., 2018) and prostate cancer (Hurwitz et al., 2019). Recent studies revealed that ASA combined with other drugs, such as sorafenib and atorvastatin, exhibited strong anti-cancer effects in vitro and in vivo (Pennarun et al., 2013; He et al., 2017). In addition, since ASA could affect histone methylation, we aimed to investigate the potential effects and mechanisms of CDM combined with ASA on AML-MDS.
Recently, network pharmacology has been used to predict the therapeutic targets and efficacy of drugs by constructing drug-drug, drug-target and other networks, using a variety of database resources. Through preliminary experiment, we found that CDM and ASA had synergistic inhibitory effect on cell growth in leukemia cells. In this study, we aimed to investigate the anti-tumor activity of CDM combined with ASA in AML-MDS, explore underlying mechanisms by predicting related targets through the network pharmacology approach, so as to provide theoretical and experimental basis for the treatment of AML-MDS. The flowchart of this study design was presented in Figure 1.
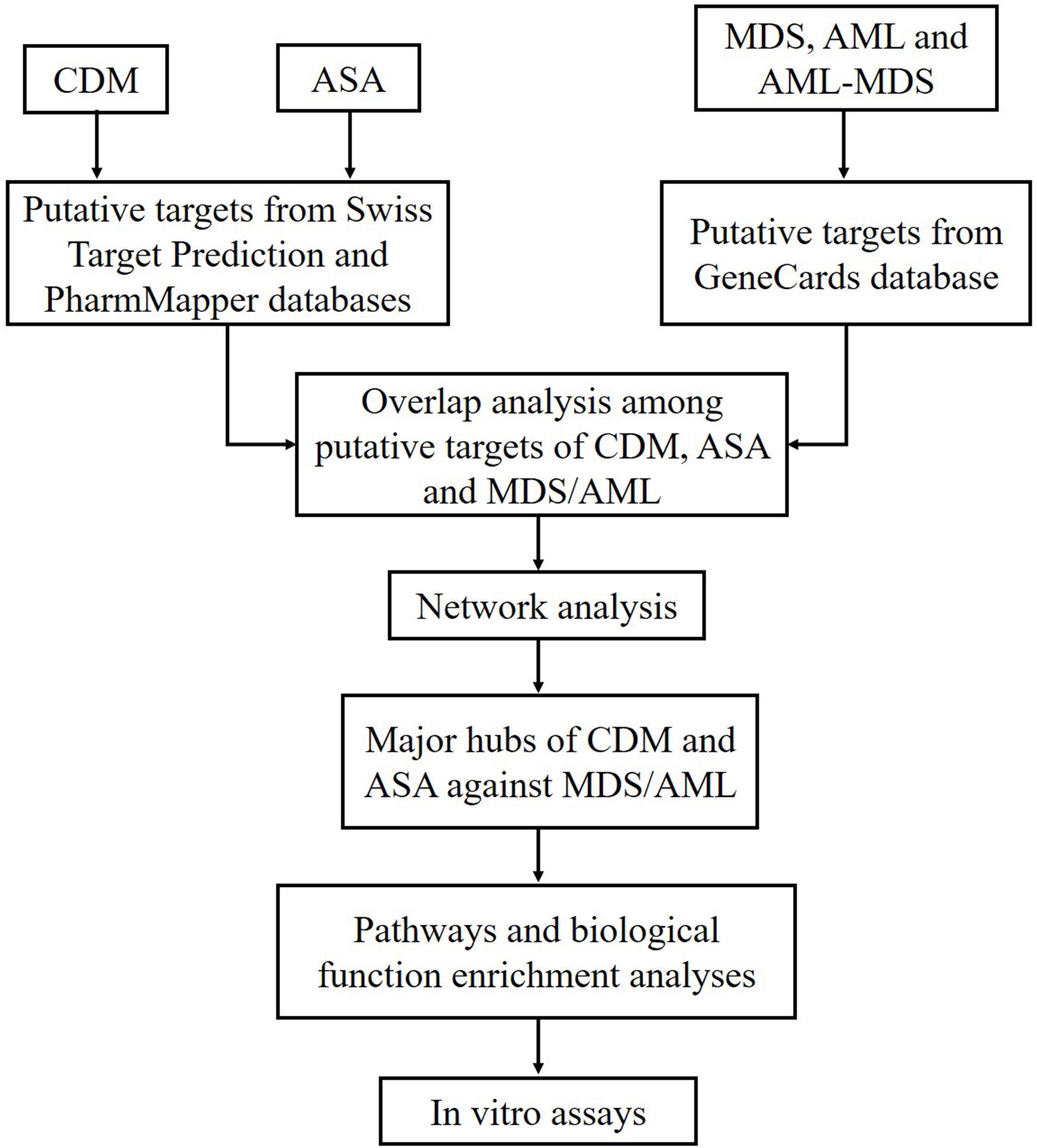
Figure 1. Flowchart of the study design based on network pharmacology approaches for deciphering the mechanisms of CDM and ASA acting on AML-MDS. CDM, Chidamide; ASA, Aspirin; MDS, myelodysplastic syndrome; AML, acute myeloid leukemia; AML-MDS, myelodysplastic syndromes-derived acute myeloid leukemia.
SwissTargetPrediction1 and PharmMapper2 were used to establish the targets of CDM and ASA. Genomic targets of MDS, AML and AML-MDS were obtained from GeneCards3 and overlapping genes were collected. Subsequently, CDM- and ASA-associated targets were mapped to these overlapping disease-targets, followed by therapeutic targets of CDM and ASA against AML-MDS were obtained. The STRING database4 was used to obtain interactions among potential targets of CDM, ASA and the aforementioned diseases. Protein interactions with a combined score > 0.4 were selected. Cytoscape 3.2.1 was used to construct and analyze the protein-protein interaction (PPI) network. DAVID database5 was used to perform Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses.
CDM (CS055, purity > 95%) was supplied by Chipscreen Biosciences (Shenzhen, China), while ASA was obtained from Maclin (A800349, Shanghai, China). IGF-1 was purchased from MedChemExpress (MCE, Shanghai, China). The following primary antibodies were used: rabbit anti-Bcl-2 (YT0470), cleaved Caspase-3 (YC0006) from ImmunoWay (Texas, United States). PI3K (bsm-33219M), p-PI3K (AB1235888), and Caspase-3 (bs-0081R) from Bioss (Beijing, China). AKT (4691T), p-AKT (4060T), p21CIP1 (2947T) from Cell Signaling Technology (Danvers, United States). CDK2 (H08211543) and CDK4 (H10082274) from Wanleibio (Shenyang, China). Mouse anti-β-actin (KM9001) from Sungene (Tianjin, China). Horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (A0239) and anti-mouse IgG (A0258) were purchased from Beyotime Biotechnology (Shanghai, China).
The human AML-MDS cell line, SKM-1, was a gift from Professor Jianfeng Zhou working in Tongji Medical College of Huazhong University of Science and Technology (Wuhan, China), while the T cell acute lymphoblastic leukemia (T-ALL) cell line Molt-4 was provided by the Children’s Hospital of Chongqing Medical University (Chongqing, China). The cells were maintained in RPMI-1640 (Gibco, Thermo Fisher Scientific, MA, United States) supplemented with 10% fetal bovine serum (PAN seratech, Germany) and 100 U/ml penicillin and 100 μg/ml streptomycin (1 × P/S).
Cells in the logarithmic growth phase were seeded at a density of 1,500 cells/well and cultured overnight. Drugs were administered at 0.1 μL per well. After a 72-h incubation, cell viability was measured using Cell Titer-Glo luminescent cell viability assay kit (Promega, Madison, United States) and luminescence was quantified using Envision Plate-Reader.
Cell viability was also measured by Cell Counting kit-8 (CCK-8) assay (MCE, Shanghai, China). Briefly, cells were seeded at 3,000 cells/100 μL and treated with different concentrations of CDM and ASA for 24, 48, and 72 h. CCK-8 reagent was added and incubated for 3 h. The absorbance at 450 nm was measured using a Multiskan Go Microplate Spectrophotometer (Thermo Fisher Scientific, United States). Cell proliferation inhibition rate was calculated based on the formula: absorbance of (control group − experimental group)/absorbance of (control group – blank group) × 100%.
CDM, at a concentration of 0.5 μM, or ASA, at a concentration of 1 mM, was added to 1 million cells for 48 h. For cell cycle analysis, cells were fixed with ice-cold 75% ethanol overnight at 4°C and then incubated with 50 mg/ml of propidium iodide (PI) for 30 min at room temperature. For apoptosis, cells were incubated with 5 μl of Annexin V-FITC and 10 μl of PI, at 4°C for 15 min in the dark. Cell cycle and apoptosis were analyzed using a flow cytometer (CytoFLEX, Beckman Coulter, United States).
Total RNA was extracted from cells using TRIzol reagent (Beyotime, China) according to the manufacturer’s instructions. cDNA was synthesized using PrimeScript Reverse Transcription reagent kit (Takara, Japan). Quantitative PCR (qPCR) was performed using a CFX96 TouchTM Real-Time PCR Detection System (Bio-Rad, Hercules, CA, United States). The following RT-qPCR parameters were used: 95°C for 30 s; 95°C for 5 s, and 60°C for 30 s repeated over 40 cycles. All primers were synthesized by Tsingke (Beijing, China) and the sequences were as follows: P21 forward: 5′-CTGCCTTAGTCTCAGTTTGTGT-3′; P21 reverse: 5′-AACCTCTCATTCAACCGCCTA-3′; Bcl-2 forward: 5′-CTGCACCTGACGCCCTTC-3′; Bcl-2 reverse: 5′- ACACATGACCCCACCGAAC-3′; caspase-3 forward: 5′-TGC TGAAACAGTATGCCGACA-3′; caspase-3 reverse: 5′-CAAAT TCTGTTGCCACCTTTCG-3′; β-actin forward: 5′-CCCAAA GTTCACAATGTGGC-3′; β-actin reverse: 5′-GACTTCCTGT AACAACGCATC-3′. Transcript levels were normalized to β-actin expression and the target gene expression was calculated using the formula 2–ΔΔCt.
Total protein from the cells was harvested using RIPA lysis buffer supplemented with 1 μM PMSF (Beyotime, Shanghai, China) and 30 μg protein was separated on a 10% SDS-polyacrylamide gradient gel. The proteins were transferred onto PVDF membranes and blocked with 5% non-fat milk in Tris Buffered Saline with Tween-20 (TBST) for 2 h at room temperature. The blots were then incubated with primary antibodies overnight at 4°C. Membranes were then washed 3 times with TBST and incubated with secondary antibodies for 1 h at room temperature. Protein bands were visualized with an ECL kit (Advansta, United States) and the band intensity was analyzed using Vilber Fusion software (Fusion, FX5 Spectra, France). β-actin was used as a loading control.
All data was presented as means ± standard deviation (SD) and statistical analyses were performed using GraphPad Prism 5.01 (GraphPad Software Inc., San Diego, CA, United States). The results were analyzed using one-way and two-way ANOVA followed by the Bonferroni post hoc test. A value of p < 0.05 was considered as statistically significant. All experiments were performed in triplicates.
A total of 522 possible targets of CDM and ASA were predicted by Swiss Target Prediction and PharmMapper (Supplementary Table 1), and 607 overlapping targets of MDS, AML and AML-MDS were obtained from the GeneCards database (Supplementary Table 2). Ultimately, 36 targets of CDM and ASA against AML-MDS were collected (Figure 2A and Supplementary Table 3). A PPI network of these predicted targets was analyzed using STRING database and constructed by Cytoscape, and the network contained 35 nodes and 245 edges (Figure 2B). Clustering subnetworks were produced using the MCODE algorithm (Figure 2C). Specifically, 10 nodes were identified as hub genes by the cytoHubba plugin in Cytoscape and grouped together, including AKT1, ALB, CASP3, SRC, MMP9, IL2, HRAS, CCNA2, STAT1, HSP90AA1 (Figure 2D and Supplementary Table 4).
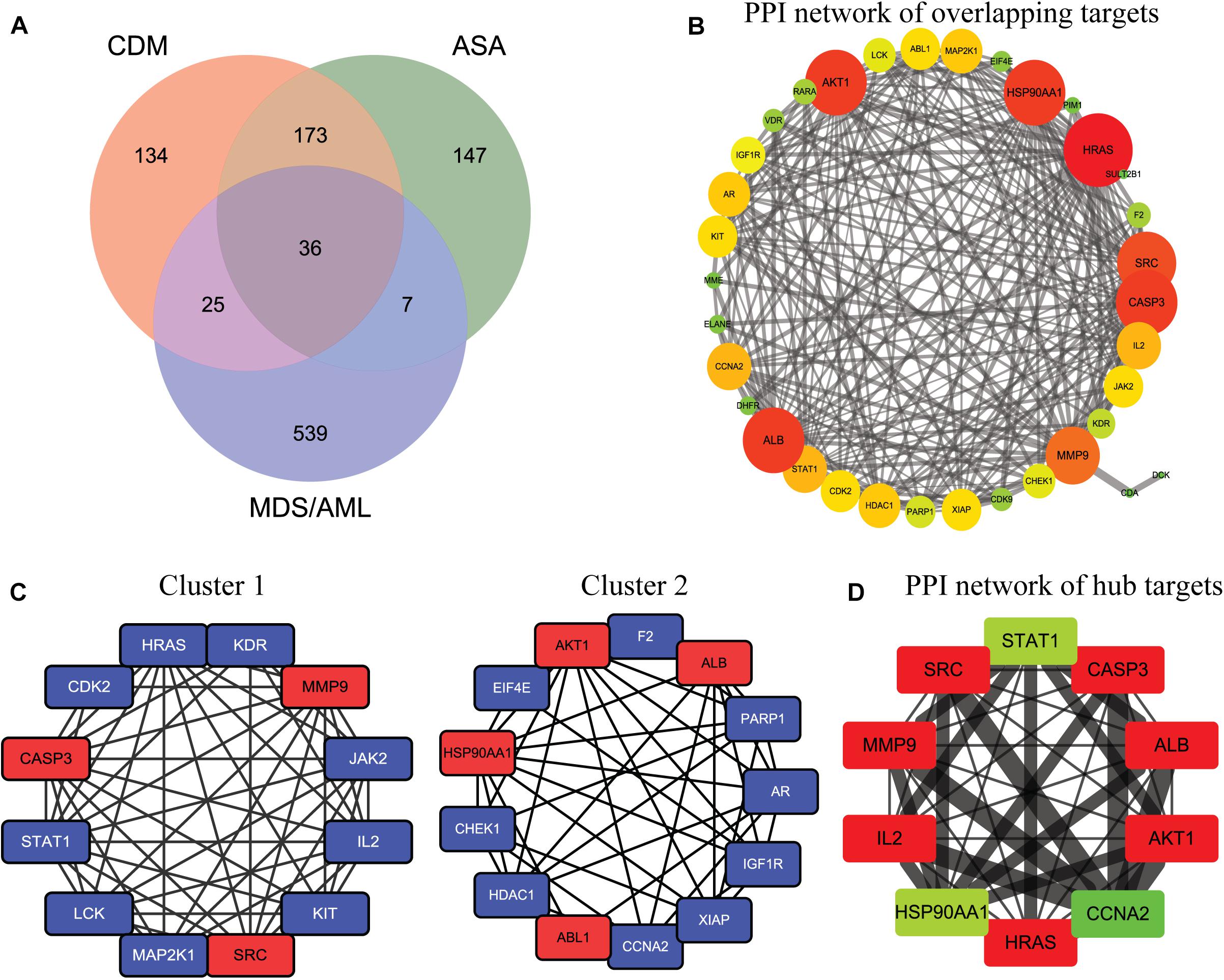
Figure 2. The network pharmacology of CDM and ASA against AML-MDS and cluster analysis. (A) The Venn diagram of the potential targets of CDM and ASA against AML-MDS. (B) A PPI network of the overlapping targets of CDM and ASA against AML-MDS by Cytoscape software. (C) Clusters of interacted proteins by use of MCODE algorithm. (D) Hub targets of the PPI network by use of cytoHubba. The size of node and edge was mapped to the degree and edge betweenness, respectively. The color of the node represents the size of the degree value. The redder the color, the larger the node and the more important it is in the network. Conversely, the greener the color, the smaller the node and the less important it is in the network.
GO and KEGG pathway enrichment analyses of these hub genes were performed using the DAVID database. GO analysis showed that these targets were associated with negative regulation of apoptotic processes and cell proliferation (Figure 3A). Enrichment in cellular component and molecular function was presented in Figures 3B,C, respectively. Additionally, KEGG analysis revealed that these 10 hub targets were involved in 42 pathways, which were mainly enriched in cancer-related pathways, especially PI3K/AKT and VEGF signaling pathways (Supplementary Table 5 and Figure 3D).
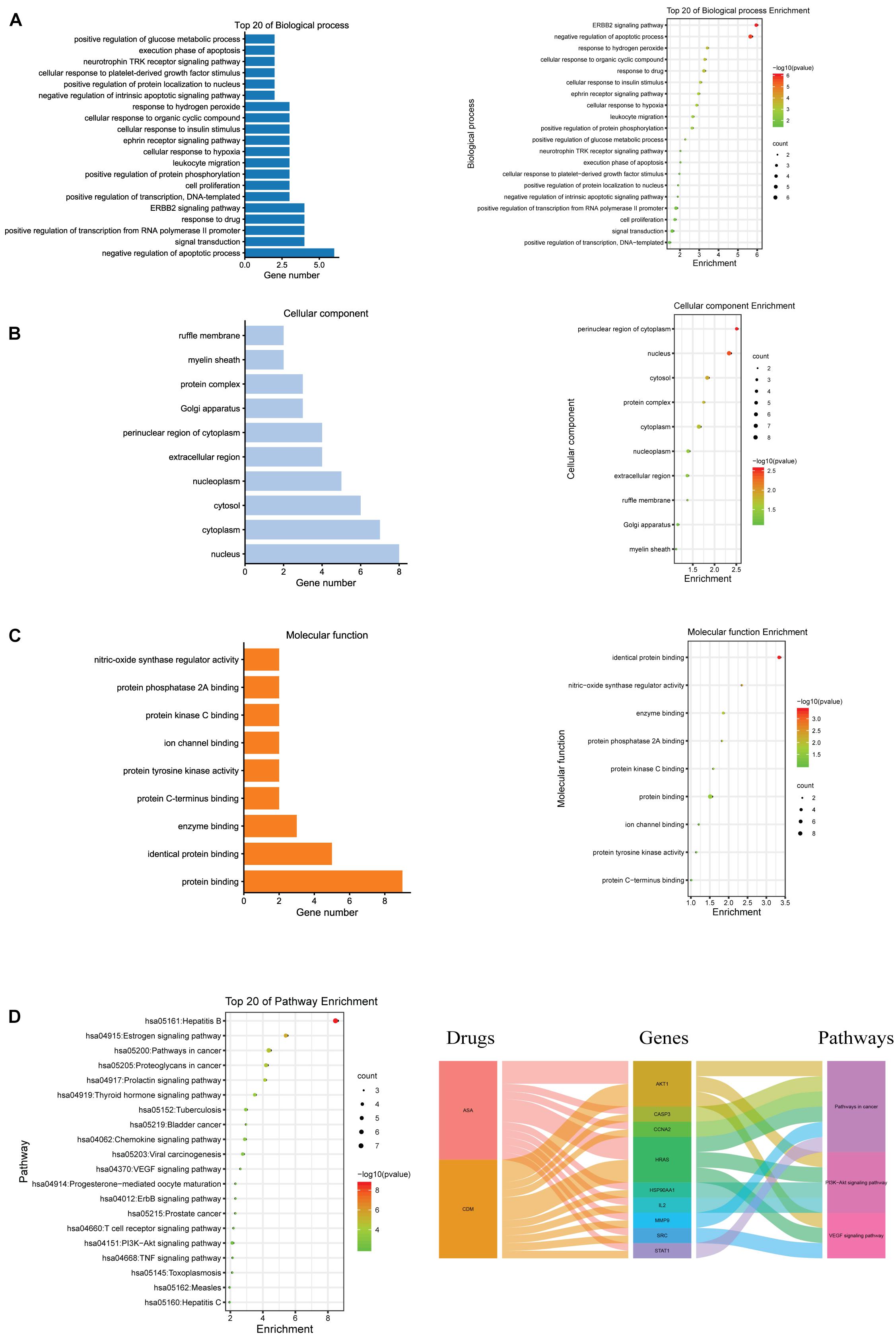
Figure 3. GO and KEGG pathway enrichment analyses for the hub genes of CDM and ASA against AML-MDS. (A) Biological process (BP); (B) cellular component (CC); (C) molecular function (MF); (D) the top 20 of KEGG enrichment analysis (left panel), alluvial plot of interaction among drugs, major hub genes and three main pathways (right panel).
Then sensitivity of SKM-1 cells to the drugs was determined according to the converting plasma concentrations. The results showed that SKM-1 cells were sensitive to CDM and ASA (Figure 4A). For further validation, SKM-1 and Molt-4 cells were treated with different concentrations of the two drugs alone. The results of CCK-8 assay showed that both CDM and ASA inhibited cells viability in a dose- and time-dependent manner (Figures 4B,C). At 48 h, the half-maximal inhibitory concentration (IC50) of CDM on SKM-1 and Molt-4 cells was (19.54 ± 3.34) μM and (1.69 ± 0.08) μM, respectively, and IC50 value of ASA was (1.69 ± 0.06) mM and (1.84 ± 0.08) mM, respectively. Moreover, to evaluate the synergistic effect of co-treatment on cell viability, these cells were treated with a low-dose CDM (0.5 μM) combined with different concentrations of ASA for 48 h. When combined with ASA, low-dose CDM decreased the IC50 of ASA to (0.63 ± 0.06) mM and (0.94 ± 0.05) mM in SKM-1 and Molt-4 cells, respectively. The combination index (CI) demonstrated that CDM combined with ASA had a distinct synergistic effect calculated by CompuSyn software (Figure 4D). As shown in Figure 4E, CDM combined with ASA significantly enhanced the inhibitory effect. Therefore, a combination of 0.5 μM CDM and 1 mM ASA, a value close to the IC50, was selected for subsequent experiments.
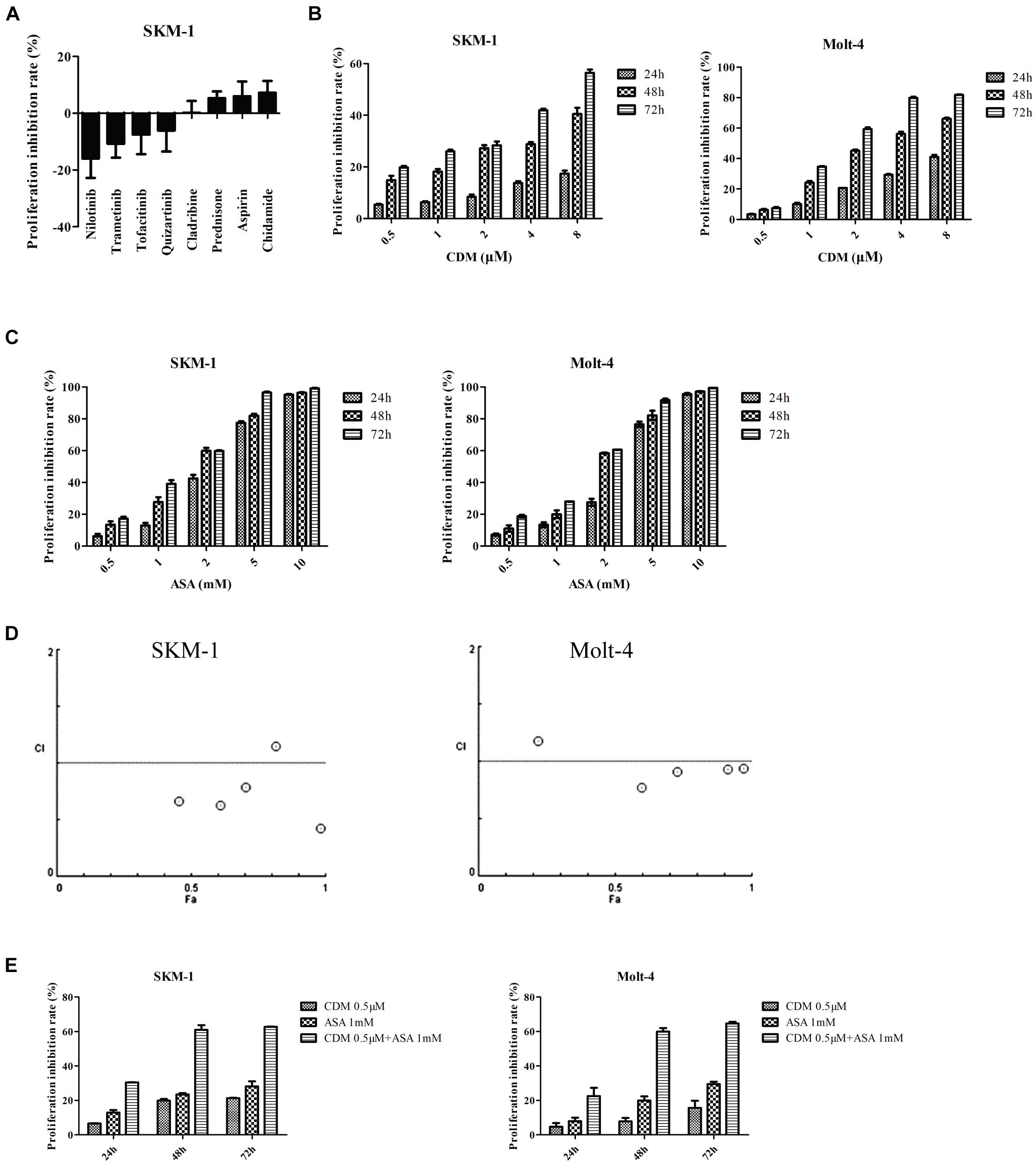
Figure 4. CDM in combination with ASA inhibited the proliferation of AML-MDS cells. (A) Cell viability detected by Cell Titer-Glo luminescent cell viability assay in SKM-1 cells treated with 8 drugs. (B,C) SKM-1 and Molt-4 cells were exposed to CDM (0.5, 1, 2, 4, and 8 μM) or ASA (0.5, 1, 2, 5, and 10 mM) alone. (D) Combination index values were calculated with CompuSyn software. CI < 1 indicates synergy; CI = 1 is additive; and CI > 1 means antagonism. CI, combination index; Fa, effect levels. (E) SKM-1 and Molt-4 cells were treated with 0.5 μM CDM combined with 1 mM ASA for 24, 48, and 72 h. The cell viability was determined by CCK-8 assay. Data are mean ± SD of three independent experiments.
To investigate the efficacy of the combination treatment on cell cycle, cell cycle distribution of each group was detected by flow cytometry with PI staining assay. As shown in Figure 5A, the combination of CDM and ASA resulted in a significant increase in the proportion of G0/G1 phase cells compared with the two drugs alone. A high mRNA expression of p21 in the combined treatment group (Figure 5B) was observed, but there was no statistical difference between the two mono-treatment groups. Also, Western blotting indicated that after combined treatment, the protein expression of CDK2 and CDK4 was down-regulated while p21 was up-regulated (Figure 5C).
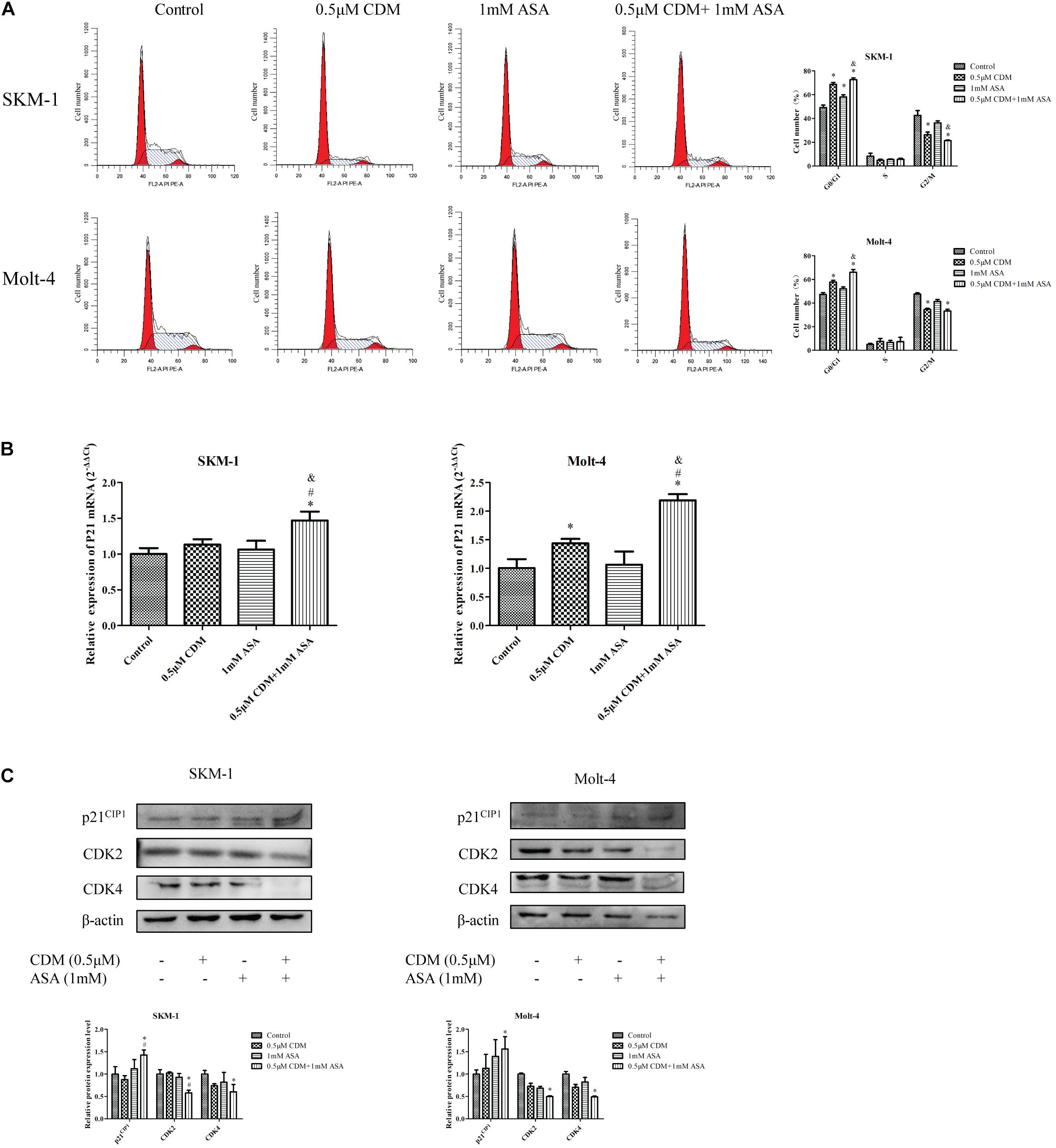
Figure 5. CDM in combination with ASA arrested the cell cycle at G0/G1 phase in AML-MDS cells. (A) Cell cycle distribution was detected by flow cytometry after treatment with 0.5 μM CDM and 1 mM ASA for 48 h. The red color areas on the left and right of the images represent the proportion of cells in the G0/G1 and G2/M phases, respectively. (B) Relative expression of P21 mRNA compared with the control group. (C) Expressions of cell cycle related protein (CDK2, CDK4, p21) were detected by western blot. β-actin was used as a loading control. Data are mean ± SD of three independent experiments. “∗” indicates a significant difference relative to the control group (∗p < 0.05), “#” indicates a significant difference relative to CDM-treated group (#p < 0.05), “&” indicates a significant difference relative to ASA-treated group (&p < 0.05).
Next, flow cytometry was used to detect the apoptotic effects of the combination treatment on SKM-1 and Molt-4 cells, and apoptosis rate was calculated as the sum of percentage of Annexin+ cells. As shown in Figure 6A, the co-treatment remarkably induced apoptosis compared with CDM and ASA mono-treatment. Furthermore, Western blotting showed that the expression of apoptosis-related protein cleaved caspase-3 was up-regulated and the expression of Bcl-2 was down-regulated, while no change in caspase-3 was observed. The observation was consistent with the results of RT-qPCR (Figures 6B,C).
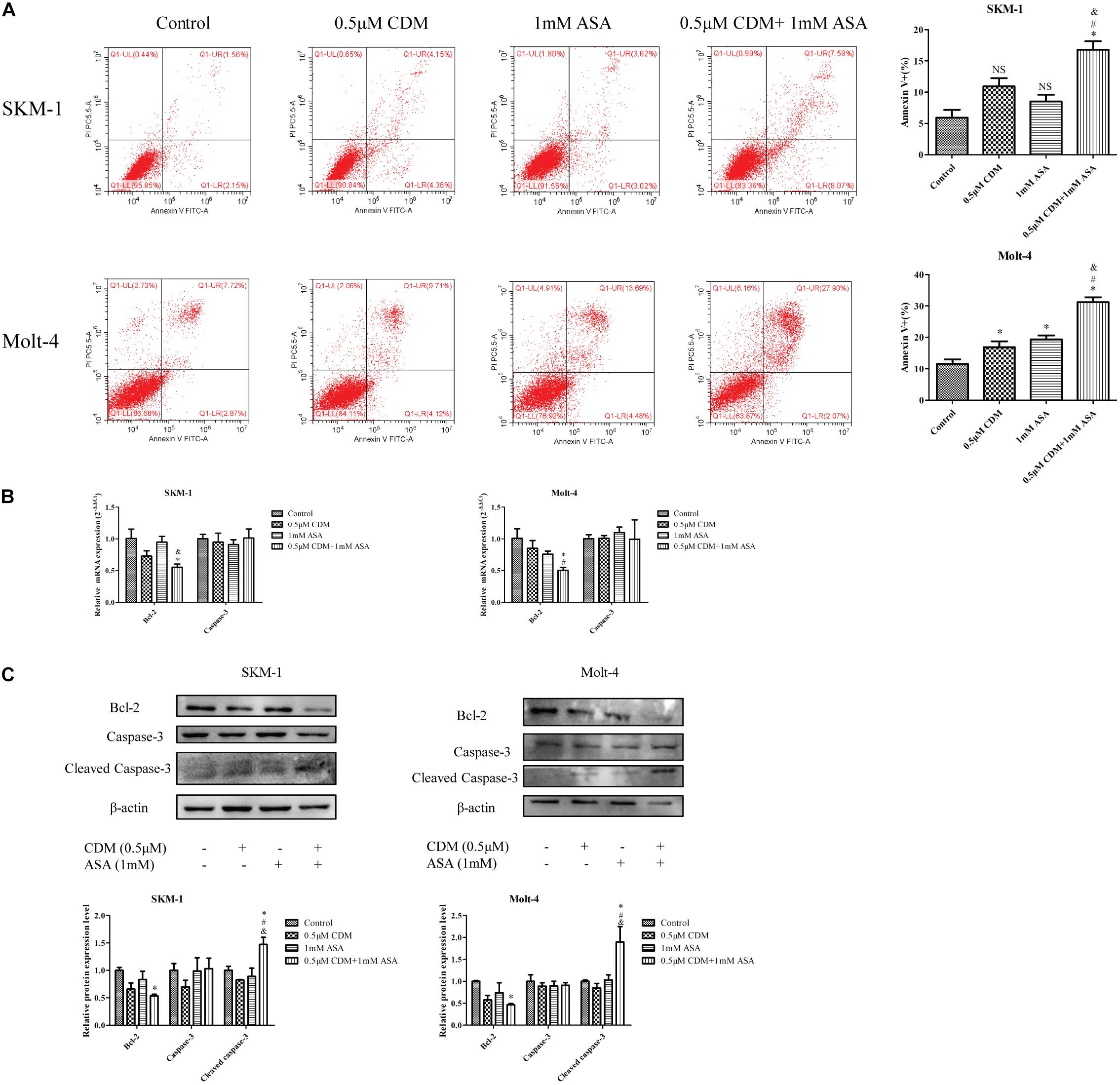
Figure 6. CDM in combination with ASA induced cell apoptosis in AML-MDS cells. (A) Cell apoptotic rate was detected by flow cytometry after treatment of the combination of 0.5 μM CDM and 1 mM ASA for 48 h. (B) Relative mRNA expression of Bcl-2 and caspase-3 compared with the control group. (C) Expressions of the apoptosis-related protein (Bcl-2, caspase-3, cleaved caspase-3) were detected by western blot. β-actin was used as a loading control. Data are mean ± SD of three independent experiments. “∗” indicates a significant difference relative to the control group (∗p < 0.05), “#” indicates a significant difference relative to CDM-treated group (#p < 0.05), “&” indicates a significant difference relative to ASA-treated group (&p < 0.05), “NS” indicates no significant difference relative to ASA-treated group or CDM-treated group.
Based on the results of GO and KEGG analyses, we focused on PI3K/AKT signaling pathway and hypothesized that the combination of CDM and ASA could synergistically inhibit the activation of PI3K/AKT pathway. The results of Western blotting demonstrated that the expression levels of p-PI3K and p-AKT in the combination treatment group were distinctly lower than those in each drug alone and control group, while the level of total PI3K and AKT remained constant (Figure 7A). Intriguingly, IGF-1, an agonist of PI3K/AKT signaling pathway, reversed the effects of the combination treatment on cell cycle and apoptosis-related proteins (Figures 7B–D), indicating that PI3K/AKT pathway was involved in the process induced by the combination treatment of CDM and ASA.
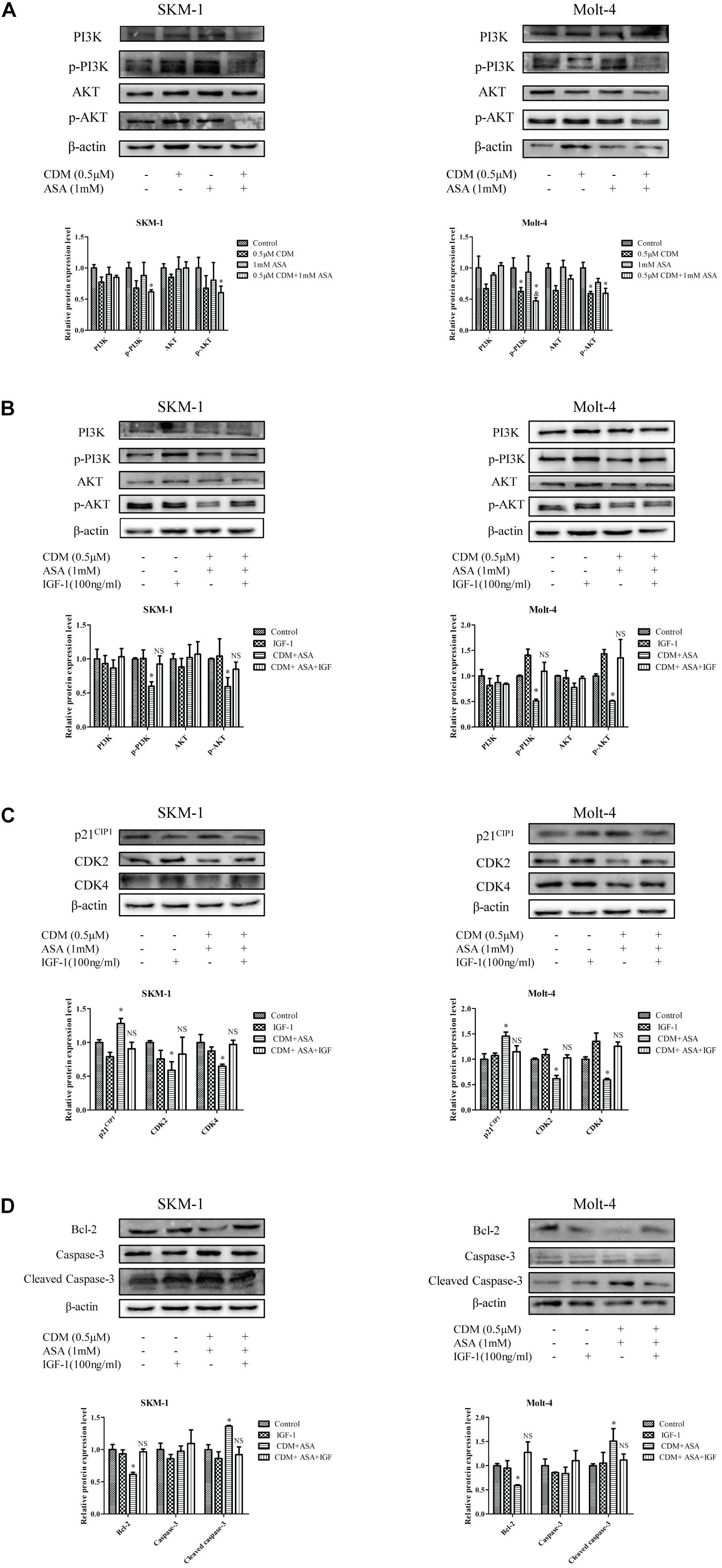
Figure 7. CDM combined with ASA inhibited the activation of PI3K/AKT pathway in AML-MDS cells. (A) Western blot analysis of PI3K, p-PI3K AKT, p-AKT. (B) Western blotting of PI3K, p-PI3K, AKT and p-AKT. (C) Western blotting of CDK2, CDK4 and p21. (D) Western blotting of Bcl-2, caspase-3 and cleaved caspase-3. IGF-1 reversed the effect of CDM combined with ASA on AML-MDS cells. β-actin served as a loading control. Data are mean ± SD of three independent experiments. “∗” indicates a significant difference relative to the control group (∗p < 0.05), “&” indicates a significant difference relative to ASA-treated group (&p < 0.05), “NS” indicates no significant difference relative to the control group.
Drug combination has been proposed as a promising therapeutic strategy with fewer side effects and lower toxicity, which could be used to improve the efficacy of single-agent treatment. There was evidence that ASA synergized with HDAC inhibitors (FK228) to inhibit growth of COX-1 positive ovarian cancer cells (Son et al., 2010). Zhang H. et al. (2018) also reported that in the presence of CDM, ASA significantly suppressed tumor growth of natural killer/T-cell lymphoma (NKTCL). In line with previous reports, our study showed that the efficiency of co-treatment was superior to CDM or ASA mono-treatment alone in inhibiting growth of AML-MDS cells, suggesting that a small dose of drug combination may be an effective therapy. However, the underlying molecular mechanisms remain unclear.
To identify the mechanisms in which CDM and ASA affected cell viability, we examined the cell cycle distribution and apoptotic rate. Dysregulation of cell cycle progression is a hallmark of cancer that enables limitless cell division. It has been reported that CDM and ASA inhibit tumor cell proliferation by inducing cell cycle arrest in G0/G1 and G2/M phases (Liu et al., 2016; Fan et al., 2017; He et al., 2018; Zhang X. et al., 2018). Our data showed that the efficiency of inducing cell cycle arrest was significantly improved when CDM was combined with ASA. The interactions of cyclin, cyclin-dependent kinases (CDKs) and CDK inhibitors play indispensable roles in controlling cell cycle. CDK2 is necessary for transition from G1 phase to S phase, while CDK4 controls G1 phase, both of which are positive regulators of the cell cycle (Lim and Kaldis, 2013). On the contrary, p21, a putative tumor-suppressor protein, is negative regulator that inhibits the CDKs/cyclin complexes in the G1 phase (Rodriguez-Cupello et al., 2020). Our study manifested that the combination of CDM and ASA synergistically down-regulated CDK2 and CDK4, and up-regulated p21, leading to G0/G1 arrest.
Dysregulation of apoptosis causes excessive cell proliferation or excessive apoptosis, resulting in disease. As a signaling pathway that regulates cell apoptosis and survival, the Bcl-2/Cleaved caspase-3 apoptotic pathway has been implicated in many cancers including leukemia (Tian et al., 2019; Zhang et al., 2019). Bcl-2 is a member of anti-apoptotic Bcl-2 family proteins, which plays an important role in maintaining the integrity of the outer mitochondrial membrane (OMM), while the pro-apoptotic protein Bax inserts into the OMM and facilitates the release of inter-membrane space (IMS) protein, leading to the activation of caspases (Renault and Chipuk, 2014; Delbridge et al., 2016). Cleaved caspase-3 is an activated form of caspase-3, a major effector protease in apoptosis that triggers the apoptotic cascade (Braunstein et al., 2020). We found that CDM combined with ASA significantly accelerated cell apoptosis by downregulation of Bcl-2 and activation of caspase-3, indicating that the combination treatment might be a potential strategy for the treatment of leukemia.
Based on the network pharmacology approach, we collected 36 putative targets of CDM and ASA against AML-MDS, and revealed that AKT1 was one of the hub genes. Through KEGG enrichment analysis, PI3K/AKT signaling pathway was highlighted as a potential target. The expression of PI3K/AKT signaling pathway is often dysregulated in various cancers and activated PI3K/AKT pathway is implicated in a variety of processes, including inducing tumor cell proliferation, inhibiting apoptosis and promoting invasion and metastasis (Yang et al., 2019). Previous studies have confirmed that blocking PI3K/AKT pathway induces cell death by regulating cell proliferation, apoptosis and cell cycle in leukemia (Bertacchini et al., 2015; Banerjee et al., 2016; Cheng et al., 2019). CDM was able to increase the acetylation levels of histone H3 and inhibit PI3K/AKT signaling pathway, resulting in arresting colon cancer cells at G1 phase and accelerating cell apoptosis (Liu et al., 2010). ASA was shown to inhibit cell proliferation by blocking cell cycle by suppressing the activation of the phosphorylation of AKT (Zhang X. et al., 2018). Consistently, our data showed that the expression levels of p-PI3K and p-AKT were remarkably downregulated by the combination of CDM and ASA, leading to the inactivation of the PI3K/AKT pathway. To confirm this, cells were treated with IGF-1, a PI3K/AKT agonist. The results showed that IGF-1 reversed the inhibitory effect of the combination treatment on PI3K/AKT pathway. The above results suggested that the combination of CDM and ASA inhibited cell proliferation, induced cell cycle arrest and promoted apoptosis in AML-MDS cells partially through suppressing the PI3K/AKT pathway (Figure 8).
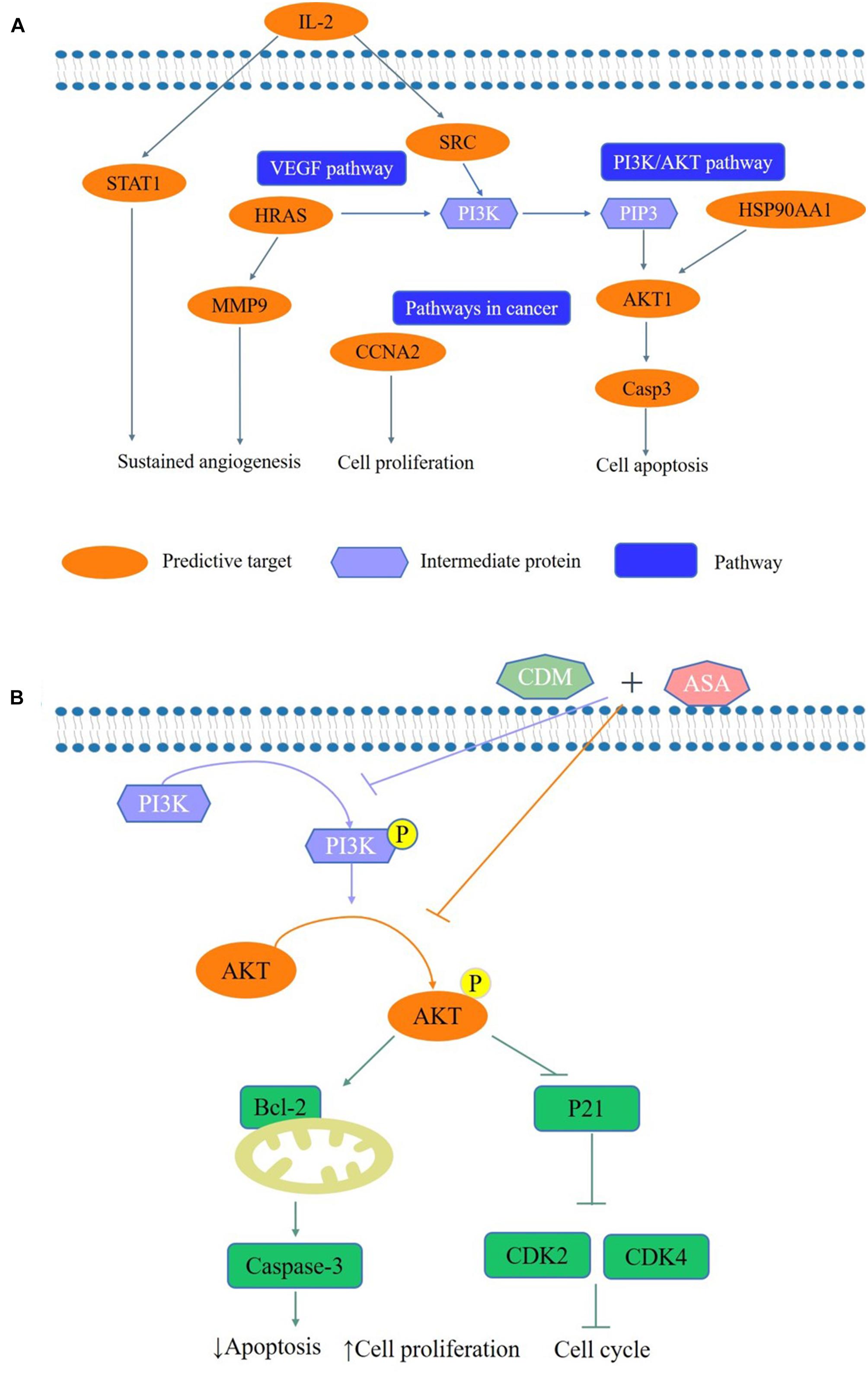
Figure 8. A schematic representation of the proposed pathway responsible for CDM combined with ASA in AML-MDS cells. (A) The predictive pathways of CDM and ASA in AML-MDS through network pharmacology. (B) CDM combined with ASA could inhibited the activation of PI3K/AKT signaling pathways, and then affected the expression of cell cycle and apoptosis-related proteins to induce cell cycle arrest and apoptosis in AML-MDS cells through experimental validation.
Our study demonstrated that CDM and ASA exerted synergistic effect on G0/G1 arrest and apoptosis by inhibiting the PI3K/AKT pathway in vitro. This provides a promising chemotherapeutic strategy for AML-MDS in combination with low dose agents. Future studies should focus on the in vivo efficacy of the combination treatment and the determination of the optimal combination regimens.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
SL and XZ conceived and designed the experiments. SL, XZ, and DC performed the experiments. LW contributed as regards the reagents, materials, and analysis tools. SL, XZ, DC, FR-L, JC, and LW participated to the analysis and interpretation of the results. SL wrote the manuscript. All authors reviewed and approved the final manuscript.
This study was supported by Natural Science Foundation Project of Chongqing (cstc2018jcyjAX0688), the Chongqing Science and Health joint project (2018ZDXM001), the Chongqing Education Commission Foundation (KJ1702017), and the Science and Technology Planning Project of Yuzhong District of Chongqing City (20190121).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the Chongqing Key Laboratory of Translational Medicine in Major Metabolic Diseases (The First Affiliated Hospital of Chongqing Medical University, Chongqing, China) for providing laboratory facilities.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.685954/full#supplementary-material
Banerjee, K., Das, S., Sarkar, A., Chatterjee, M., Biswas, J., and Choudhuri, S. K. (2016). A copper chelate induces apoptosis and overcomes multidrug resistance in T-cell acute lymphoblastic leukemia through redox imbalance and inhibition of EGFR/PI3K/Akt expression. Biomed. Pharmacother. 84, 71–92. doi: 10.1016/j.biopha.2016.08.056
Bertacchini, J., Heidari, N., Mediani, L., Capitani, S., Shahjahani, M., Ahmadzadeh, A., et al. (2015). Targeting PI3K/AKT/mTOR network for treatment of leukemia. Cell. Mol. Life Sci. 72, 2337–2347. doi: 10.1007/s00018-015-1867-5
Boddu, P., Kantarjian, H., Garcia-Manero, G., Ravandi, F., Verstovsek, S., Jabbour, E., et al. (2017). Treated secondary acute myeloid leukemia: a distinct high-risk subset of AML with adverse prognosis. Blood Adv. 1, 1312–1323. doi: 10.1182/bloodadvances.2017008227
Braunstein, I., Engelman, R., Yitzhaki, O., Ziv, T., Galardon, E., and Benhar, M. (2020). Opposing effects of polysulfides and thioredoxin on apoptosis through caspase persulfidation. J. Biol. Chem. 295, 3590–3600. doi: 10.1074/jbc.RA119.012357
Cheng, J. Y., Li, Y., Liu, S. Q., Jiang, Y. J., Ma, J., Wan, L., et al. (2019). CXCL8 derived from mesenchymal stromal cells supports survival and proliferation of acute myeloid leukemia cells through the PI3K/AKT pathway. FASEB J. 33, 4755–4764. doi: 10.1096/fj.201801931R
Cogle, C. R., Craig, B. M., Rollison, D. E., and List, A. F. (2011). Incidence of the myelodysplastic syndromes using a novel claims-based algorithm: high number of uncaptured cases by cancer registries. Blood 117, 7121–7125. doi: 10.1182/blood-2011-02-337964
Delbridge, A. R., Grabow, S., Strasser, A., and Vaux, D. L. (2016). Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 16, 99–109. doi: 10.1038/nrc.2015.17
Erickson, P., Gardner, L. D., Loffredo, C. A., St George, D. M., Bowman, E. D., Deepak, J., et al. (2018). Racial and Ethnic Differences in the Relationship between Aspirin Use and Non-Small Cell Lung Cancer Risk and Survival. Cancer Epidemiol. Biomarkers Prev. 27, 1518–1526. doi: 10.1158/1055-9965.epi-18-0366
Fan, W. S., Li, J. H., Chen, J. F., Zhu, L., Wang, Y. M., Sun, B. L., et al. (2017). Aspirin inhibits the proliferation of synovium-derived mesenchymal stem cells by arresting the cell cycle in the G0/G1 phase. Am. J. Transl. Res. 9, 5056–5062.
García Rodríguez, L. A., Soriano-Gabarró, M., Vora, P., and Cea Soriano, L. (2020). Low-dose aspirin and risk of gastric and oesophageal cancer: A population-based study in the United Kingdom using The Health Improvement Network. Int. J. Cancer 147, 2394–2404. doi: 10.1002/ijc.33022
Guan, X. W., Wang, H. Q., Ban, W. W., Chang, Z., Chen, H. Z., Jia, L., et al. (2020). Novel HDAC inhibitor Chidamide synergizes with Rituximab to inhibit diffuse large B-cell lymphoma tumour growth by upregulating CD20. Cell Death Dis. 11:20. doi: 10.1038/s41419-019-2210-0
He, J. S., Chen, Q. X., Gu, H. Y., Chen, J., Zhang, E. F., Guo, X., et al. (2018). Therapeutic effects of the novel subtype-selective histone deacetylase inhibitor chidamide on myeloma-associated bone disease. Haematologica 103, 1369–1379. doi: 10.3324/haematol.2017.181172
He, Y., Huang, H. R., Farischon, C., Li, D. L., Du, Z. Y., Zhang, K., et al. (2017). Combined effects of atorvastatin and aspirin on growth and apoptosis in human prostate cancer cells. Oncol. Rep. 37, 953–960. doi: 10.3892/or.2017.5353
Hurwitz, L. M., Joshu, C. E., Barber, J. R., Prizment, A. E., Vitolins, M. Z., Jones, M. R., et al. (2019). Aspirin and Non-Aspirin NSAID Use and Prostate Cancer Incidence, Mortality, and Case Fatality in the Atherosclerosis Risk in Communities Study. Cancer Epidemiol. Biomarkers Prev. 28, 563–569. doi: 10.1158/1055-9965.epi-18-0965
Li, Q., Huang, J. C., Liao, D. Y., and Wu, Y. (2020). Chidamide plus decitabine synergistically induces apoptosis of acute myeloid leukemia cells by upregulating PERP. Am. J. Transl. Res. 12, 3461–3475.
Li, Y., Chen, K., Zhou, Y., Xiao, Y. R., Deng, M. M., Jiang, Z. W., et al. (2015). A New Strategy to Target Acute Myeloid Leukemia Stem and Progenitor Cells Using Chidamide, a Histone Deacetylase Inhibitor. Curr. Cancer Drug Targets 15, 493–503. doi: 10.2174/156800961506150805153230
Lim, S., and Kaldis, P. (2013). Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development 140, 3079–3093. doi: 10.1242/dev.091744
Liu, L., Chen, B. A., Qin, S. K., Li, S. Y., He, X. M., Qiu, S. M., et al. (2010). A novel histone deacetylase inhibitor Chidamide induces apoptosis of human colon cancer cells. Biochem. Biophys. Res. Commun. 392, 190–195. doi: 10.1016/j.bbrc.2010.01.011
Liu, Z. Y., Ding, K., Li, L. J., Liu, H., Wang, Y. H., Liu, C. Y., et al. (2016). A novel histone deacetylase inhibitor Chidamide induces G0/G1 arrest and apoptosis in myelodysplastic syndromes. Biomed. Pharmacother. 83, 1032–1037. doi: 10.1016/j.biopha.2016.08.023
Lu, X. P., Ning, Z. Q., Li, Z. B., Cao, H. X., and Wang, X. H. (2016). Development of chidamide for peripheral T-cell lymphoma, the first orphan drug approved in China. Intractable Rare Dis. Res. 5, 185–191. doi: 10.5582/irdr.2016.01024
Pennarun, B., Kleibeuker, J. H., Boersma-van, Ek, W., Kruyt, F. A., Hollema, H., et al. (2013). Targeting FLIP and Mcl-1 using a combination of aspirin and sorafenib sensitizes colon cancer cells to TRAIL. J. Pathol. 229, 410–421. doi: 10.1002/path.4138
Ramadan, S., Suciu, S., Stevens-Kroef, M., Willemze, R., Amadori, S., de Witte, T., et al. (2020). Survival Improvement over Time of 960 s-AML Patients Included in 13 EORTC-GIMEMA-HOVON Trials. Cancers 12:3334. doi: 10.3390/cancers12113334
Renault, T. T., and Chipuk, J. E. (2014). Death upon a kiss: mitochondrial outer membrane composition and organelle communication govern sensitivity to BAK/BAX-dependent apoptosis. Chem. Biol. 21, 114–123. doi: 10.1016/j.chembiol.2013.10.009
Rodriguez-Cupello, C., Dam, M., Serini, L., Wang, S., Lindgren, D., Englund, E., et al. (2020). The STRIPAK Complex Regulates Response to Chemotherapy Through p21 and p27. Front. Cell Dev. Biol. 8:146. doi: 10.3389/fcell.2020.00146
Scalzulli, E., Pepe, S., Colafigli, G., and Breccia, M. (2020). Therapeutic strategies in low and high-risk MDS: What does the future have to offer? Blood Rev. 45:100689. doi: 10.1016/j.blre.2020.100689
Schroeder, T., Wegener, N., Lauseker, M., Rautenberg, C., Nachtkamp, K., Schuler, E., et al. (2019). Comparison between Upfront Transplantation and different Pretransplant Cytoreductive Treatment Approaches in Patients with High-Risk Myelodysplastic Syndrome and Secondary Acute Myelogenous Leukemia. Biol. Blood Marrow Transplant. 25, 1550–1559. doi: 10.1016/j.bbmt.2019.03.011
Shi, Y., Dong, M., Hong, X., Zhang, W., Feng, J., Zhu, J., et al. (2015). Results from a multicenter, open-label, pivotal phase II study of chidamide in relapsed or refractory peripheral T-cell lymphoma. Ann. Oncol. 26, 1766–1771. doi: 10.1093/annonc/mdv237
Son, D. S., Wilson, A. J., Parl, A. K., and Khabele, D. (2010). The effects of the histone deacetylase inhibitor romidepsin (FK228) are enhanced by aspirin (ASA) in COX-1 positive ovarian cancer cells through augmentation of p21. Cancer Biol. Ther. 9, 928–935. doi: 10.4161/cbt.9.11.11873
Sun, Y. F., Li, J., Xu, Z., Xu, J. D., Shi, M. J., and Liu, P. (2019). Chidamide, a novel histone deacetylase inhibitor, inhibits multiple myeloma cells proliferation through succinate dehydrogenase subunit A. Am. J. Cancer Res. 9, 574–584.
Tian, Y., Jia, S. X., Shi, J., Gong, G. Y., Yu, J. W., Niu, Y., et al. (2019). Polyphyllin I induces apoptosis and autophagy via modulating JNK and mTOR pathways in human acute myeloid leukemia cells. Chem. Biol. Interact. 311:108793. doi: 10.1016/j.cbi.2019.108793
Wang, H. J., Guo, Y., Fu, M., Liang, X., Zhang, X. Y., Wang, R. Z., et al. (2012). Antitumor activity of Chidamide in hepatocellular carcinoma cell lines. Mol. Med. Rep. 5, 1503–1508. doi: 10.3892/mmr.2012.858
Wu, Y. F., Ou, C. C., Chien, P. J., Chang, H. Y., Ko, J. L., and Wang, B. Y. (2019). Chidamide-induced ROS accumulation and miR-129-3p-dependent cell cycle arrest in non-small lung cancer cells. Phytomedicine 56, 94–102. doi: 10.1016/j.phymed.2018.09.218
Xu, F. F., Guo, H. G., Shi, M. Y., Liu, S. W., Wei, M., Sun, K., et al. (2019). A combination of low-dose decitabine and chidamide resulted in synergistic effects on the proliferation and apoptosis of human myeloid leukemia cell lines. Am. J. Transl. Res. 11, 7644–7655.
Yang, Q., Jiang, W., and Hou, P. (2019). Emerging role of PI3K/AKT in tumor-related epigenetic regulation. Semin. Cancer Biol. 59, 112–124. doi: 10.1016/j.semcancer.2019.04.001
Zhang, H., Lu, J., Jiao, Y., Chen, Q., Li, M., Wang, Z., et al. (2018). Aspirin Inhibits Natural Killer/T-Cell Lymphoma by Modulation of VEGF Expression and Mitochondrial Function. Front. Oncol. 8:679. doi: 10.3389/fonc.2018.00679
Zhang, X., Feng, H., Li, Z., Guo, J., and Li, M. (2018). Aspirin is Involved in the Cell Cycle Arrest, Apoptosis, Cell Migration, and Invasion of Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 19:2029. doi: 10.3390/ijms19072029
Zhang, W. J., Niu, J. W., Ma, Y. C., Yang, X. W., Cao, H. X., Guo, H. G., et al. (2020). The Synergistic Antitumor Activity of Chidamide in Combination with Bortezomib on Gastric Cancer. Onco Targets Ther. 13, 3823–3837. doi: 10.2147/ott.s240721
Zhang, Y., Yang, X., Ge, X. H., and Zhang, F. Y. (2019). Puerarin attenuates neurological deficits via Bcl-2/Bax/cleaved caspase-3 and Sirt3/SOD2 apoptotic pathways in subarachnoid hemorrhage mice. Biomed. Pharmacother. 109, 726–733. doi: 10.1016/j.biopha.2018.10.161
Keywords: chidamide, aspirin, network pharmacology, myelodysplastic syndromes, PI3K/Akt pathway
Citation: Liang S, Zhou X, Cai D, Rodrigues-Lima F, Chi J and Wang L (2021) Network Pharmacology and Experimental Validation Reveal the Effects of Chidamide Combined With Aspirin on Acute Myeloid Leukemia-Myelodysplastic Syndrome Cells Through PI3K/AKT Pathway. Front. Cell Dev. Biol. 9:685954. doi: 10.3389/fcell.2021.685954
Received: 26 March 2021; Accepted: 23 August 2021;
Published: 09 September 2021.
Edited by:
Ozgur Kutuk, Başkent University, TurkeyReviewed by:
Hou Yuzhu, The College, University of Chicago, United StatesCopyright © 2021 Liang, Zhou, Cai, Rodrigues-Lima, Chi and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Wang, bGl3YW5nbHNAeWFob28uY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.