 Lijie Yao
Lijie Yao Liqing Xu
Liqing Xu Weihao Zou
Weihao Zou Hongjuan Peng
Hongjuan Peng
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol., 26 May 2021
Sec. Molecular and Cellular Pathology
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.685913
This article is part of the Research TopicCellular and Molecular Basis in Parasitic Diseases Control: Research TrendsView all 42 articles
Toxoplasma gondii is an intracellular pathogen that exerts its virulence through inhibiting host’s innate immune responses, which is mainly related to the type II interferon (IFN-γ) response. IFN-γ inducible tripartite motif 21 (TRIM21), an E3 ligase, plays an important role in anti-infection responses against the intracellular pathogens including bacteria, virus, and parasite. We found that T. gondii virulence factor ROP18 of the type I RH strain (TgROP18I) interacted with human TRIM21, and promoted the latter’s phosphorylation, which subsequently accelerated TRIM21 degradation through lysosomal pathway. Furthermore, TRIM21 protein level was found to be upregulated during RH and CEP strains of T. gondii infection. TRIM21 knocking down reduced the ubiquitin labeling on the parasitophorous vacuole membrane (PVM) [which led to parasitophorous vacuole (PV) acidification and death of CEP tachyzoites], and relieved the inhibition of CEP proliferation induced by IFN-γ in human foreskin fibroblast (HFF) cells which was consistent with the result of TRIM21 overexpression. On the other hand, TRIM21 overexpression enhanced the inhibition of CEP proliferation, and inhibited the binding of IκB-α with p65 to activate the IFN-γ-inducible NF-κB pathway, which might be resulted by TRIM21-IκB-α interaction. In brief, our research identified that in human cells, IFN-γ-inducible TRIM21 functioned in the innate immune responses against type III T. gondii infection; however, TgROP18I promoted TRIM21 phosphorylation, leading to TRIM21 degradation for immune escape in type I strain infection.
Toxoplasma gondii is an obligate intracellular protozoon, and about 30% of the world population is serum positive with its antibodies (Montoya and Liesenfeld, 2004). Human infections are mostly asymptomatic, but bring a lifelong threat to the infected population because of the latent stage (bradyzoites) in the tissues and organs (Harker et al., 2015). T. gondii infection can cause life-threatening symptoms such as encephalitis and retinochoroiditis in immunocompromised individuals, and adverse pregnancy outcomes such as stillbirth, abortion, and tetras in pregnant women in the primary infection (Montoya and Liesenfeld, 2004; Elmore et al., 2010; Torgerson and Mastroiacovo, 2013; Hide, 2016).
During infection, T. gondii rhoptries discharge many effectors to the host cytoplasm and the parasitophorous vacuole (PV) to modulate the homeostasis between the host cell and the parasite (Bradley et al., 2005; Boothroyd and Dubremetz, 2008; Hakimi et al., 2017). Among these effectors, TgROP18I is considered to be a key determinant related to the high mortality phenotype of type I strains (Saeij et al., 2006; Taylor et al., 2006). As a protein kinase of the ROP2 subfamily, TgROP18I is discharged into the host cell and mediates some host proteins for successful parasitism (Bradley et al., 2005; Sinai, 2007). On the other hand, host cells have to trigger a rapid recognition and defense immunity to control the multiplication of the intracellular pathogens (Bianchi and van den Bogaart, 2020). The cytokine gamma interferon (IFN-γ) is induced in the early infection of the intracellular pathogens including T. gondii (MacMicking, 2012), which is crucial to restrain both the acute infection and chronic infection (Suzuki et al., 1988; Yap and Sher, 1999). The transcription of genes related to host immune system can be upregulated by IFN-γ treatment (Platanias, 2005); T. gondii can also manipulate some of these genes’ transcription in infected cells to maintain its replication (Platanias, 2005; Kim et al., 2007).
NF-κB signaling pathway, as one of the major IFN-γ-inducible signaling cascades involved in host’s innate immunity, could be regulated by T. gondii (Ge et al., 2014). In infected human foreskin fibroblasts (HFFs), type I T. gondii prohibits NF-κB activation by inhibiting p65/RelA phosphorylation and translocation to the nucleus (Shapira et al., 2005). Moreover, it has been confirmed that T. gondii inhibits IκB-α degradation and p65 phosphorylation, which results in an inhibition of caspase-1 cleavage in the infected neutrophils (Lima et al., 2018), but these phenomena are not found in the infected human monocytes (Gov et al., 2013, 2017). These reports imply that T. gondii regulates host NF-κB pathway through varied ways in different host cells.
Tripartite motif (TRIM) proteins have emerged as an important family of E3 ligases that play a versatile role in innate immunity against infection (Giraldo et al., 2020; Koepke et al., 2020; Lee et al., 2020; Wang and Hur, 2021). For instance, TRIM31 promotes the K63-linked polyubiquitination of mitochondrial antiviral signaling protein (MAVS) by inducing expression of interferons (IFNs) to against viral infection (Liu et al., 2017). TRIM 21, also known as Ro52/SS-A, is involved in innate and acquired immunity against viruses and bacteria via the ubiquitination of interferon regulatory factors IRF3, IRF5, IRF7, and IRF8, but little is known about its role in parasitic infection (Lazzari et al., 2014; Liu et al., 2018). Following Salmonella typhimurium infection, TRIM21 is also essential to promote cell death through conferring p62 ubiquitination and proteasomal degradation (Pan et al., 2016; Hos et al., 2020). Furthermore, it has been reported that TRIM21 mediates NF-κB activation to aggravate the inflammatory response in psoriasis (Yang et al., 2020).
In this research, based on the finding of the interaction of TgROP18I with human TRIM21, we investigated the role of TRIM21 in T. gondii infection.
All T. gondii strains used in this study were maintained by serial passage of tachyzoites in HFF monolayers cultured at the condition of 37°C and 5% CO2. The T. gondii strains used in our study included Type-I strain RH, type-III strain CEP, the ROP18 knockout strain (RH-Δrop18), and the recombinant CEP expressing type I ROP18 (CEP-rop18I). HFF and human embryonic kidney 293T (HEK293T) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen, Thermo Fisher Scientific, Shanghai, China) supplemented with 10% fetal bovine serum (FBS, Invitrogen, Thermo Fisher Scientific, Shanghai, China), and 50 mg/ml of penicillin and streptomycin.
Human foreskin fibroblast cells were cultured in 12-well plates and infected with the RH or CEP tachyzoites at Multiplication of Infection (MOI) three for 1 h or 24 h, or left uninfected. The TRIzolTM Reagent (Invitrogen, Thermo Fisher Scientific, Shanghai, China) was used for total RNA extraction from HFFs (1.0 × 106 cells per well), and cDNA was synthesized with Random Primer (N9) using TransScript® All-in-One First-Strand cDNA Synthesis SuperMix for qPCR (One-Step gDNA Removal) (Transgen Biotech, Beijing, China) and subjected to Real-time PCR with the primers shown in Supplementary Table 1. For protein level detection, HFF cells were harvested and subjected to Western blotting.
Human foreskin fibroblast cells were seeded in 12-well plate and stimulated with 100 U/ml IFN-γ for 24 h, or transfected with 0.5 μg pcDNA3.1-TRIM21-HA for 24 h, or 80 nmol of si-TRIM21 (combining of si-TRIM21-1, si-TRIM21-2, and si-TRIM21-3) or si-NC for 48 h (the sequence of si-TRIM21 are shown in Supplementary Table 2), and then stimulated with 100 U/ml IFN-γ for 24 h. After these treatments, the HFF cells were infected with RH tachyzoites (MOI = 3) for 18 h, or CEP tachyzoites (MOI = 3) for 24 h. Subsequently, cell monolayers were subjected to immunofluorescence assay (IFA) to determine the average number of parasites within 100 PVs or the number of PVs containing 1, 2, 4, or 8 tachyzoites from 25 separated fields of view (He et al., 2017). Anti-SAG1 mouse monoclonal antibody (Abcam, Cambridge, United Kingdom), and Goat anti-Mouse IgG (H + L) highly cross-adsorbed secondary antibody and Alexa Fluor Plus 488 (Invitrogen, United States) were used. Experiments were repeated three times for statistical analysis.
HEK293T cells were seeded in T25 flasks to 90% confluence, and then transfected with pcDNA3.1-ROP18I-FLAG, pcDNA3.1-ROP18I-KD-FLAG, and pcDNA3.1-TRIM21-HA (or the truncation mutants of TRIM21) alone or in combination for 48 h, using Lipofectamine® 3000 reagent (Thermo Fisher Scientific). The cell lysates were subjected to immunoprecipitation with anti-HA-Tag rabbit monoclonal antibody (Abcam, Cambridge, United Kingdom) or anti-FLAG M2 mouse monoclonal antibody (Sigma, St. Louis, MO, United States) followed by Western blotting.
HEK293T cells were seeded in T25 flasks to 90% confluence, and transfected with pcDNA3.1-ROP18I-FLAG using Lipofectamine® 3000 reagent (Thermo Fisher Scientific) for 24 h. The cell lysates were subjected to immunoprecipitation with anti-TRIM21 rabbit polyclonal antibody (Proteintech, IL, United States) followed by Western blotting.
HEK293T cells were seeded in 6-well plate to 90% confluence, and transfected with 0, 0.6, or 1.2 μg of pcDNA3.1-ROP18I-FLAG for 24 h; or co-transfected with 1 μg pcDNA3.1-TRIM21-HA and the increased amounts of pcDNA3.1-ROP18I-FLAG (0, 0.5, or 1.0 μg) for 24 h; or co-transfected with 1 μg pcDNA3.1-TRIM21-HA and the increased amounts of pcDNA3.1-ROP18I-FLAG (0, 0.25, 0.5, or 1.0 μg) for 24 h. The cells were then treated with 10 μM MG132 or Leupeptin for 12 h, or left untreated. After that, the cells were harvested and the cell lysates were subjected to Western blotting for TRIM21 detection.
Human foreskin fibroblasts were grown on coverslips in 12-well plate to 30–50% confluence. The cells were then transfected for 48 h with either 80 nmol of non-targeting small interfering RNA (si-NC) or siRNA specific for TRIM21 (si-TRIM21) (which were synthetized in RiboBio, Guangzhou, China; the sequences are shown in Supplementary Table 2), using Lipofectamine® 3000 Transfection Reagent (Thermo Fisher Scientific) following the manufacturer’s instruction. The efficiency of TRIM21 knockdown was verified by Western blotting. After the following stimulation with 100 U/ml IFN-γ, the HFF cells were infected with CEP tachyzoites for 6 h, and then subjected to IFA with anti-ubiquitin FK-2 mouse monoclonal antibody (Enzo Life Sciences, NY, United States), and Goat anti-Mouse IgG (H + L) highly cross-adsorbed secondary antibody, Alexa Fluor Plus 594 (Invitrogen, United States).
Human foreskin fibroblasts were grown on the coverslips in 12-well plate to 90% confluence, and stimulated with 100 U/ml IFN-γ for 24 h or not, followed by CEP infection for 2 h. HFFs were then treated with 50 mM LysoTracker® Deep Red dye (Invitrogen, United States) for another 2.5 h, which specifically stained the acidic organelles in live cells including lysosomes. The infected cells were then washed with phosphate buffered saline (PBS) for three times, rinsed with ddH2O and mounted with mounting-oil containing DAPI. The PV labeling was observed under a fluorescence microscope (ECLIPSE Ni, NA = 1.4, Nikon, Tokyo, Japan).
HEK293T cells were seeded in T25 flasks to 90% confluence, and then transfected with pcDNA3.1-TRIM21-HA for 24 h, or treated with 100 U/ml IFN-γ for 0, 12, or 24 h. The cells were then harvested and the cell lysates were subjected to immunoprecipitation with anti-TRIM21 rabbit polyclonal antibody (Proteintech, IL, United States) or anti-p65 mouse monoclonal antibody (Cell Signaling Technology, MA, United States). The immunoprecipitates were further analyzed by Western blotting.
Quantitative reverse transcription PCR (qRT-PCR) was performed with Hieff® qPCR SYBR® Green Master Mix (Low Rox Plus) (Yeasen, China) according to the manufacturer’s instruction. Real-time PCR was carried out on QuantStudio 6 Real-Time PCR System (Thermo Fisher Scientific, United States) using iQ SYBR Green Supermix (Bio-Rad, CA, United States), primers used were shown in Supplementary Table 1 and all primers were synthesized in GENERAY, Shanghai, China. The specificity of the PCR amplification was verified by a dissociation curve analysis. Each sample was run in triplicate and relative quantitation was determined using comparative Ct method with data normalized to the housekeeping gene, β-actin. Real-time PCR results were presented as fold change compared to the uninfected sample, and data were derived from four independent experiments.
The transfected or infected HEK293T, or HFF cells, were harvested and boiled in 1 × loading buffer (0.08 M Tris, pH 6.8, with 2.0% SDS, 10% glycerol, 0.1 M dithiothreitol, and 0.2% bromophenol blue). The cell lysates were loaded onto a 12% polyacrylamide gel for separation, and the proteins were then transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was blocked in blocking buffer [PBS containing 5% Bovine Serum Albumin (BSA) and 0.05% Tween-20] at room temperature for 2 h, and then incubated in the primary antibodies, followed by incubation in the secondary antibodies conjugated with horseradish peroxidase (HRP) at room temperature for 2 h, following the manufacturer’s instruction. Specific proteins on membranes were visualized by luminescence generated by using ClarityTM Western ECL Substrate (Bio-Rad, CA, United States) and photographed with a ChemiDocTM Touch Imaging System (Bio-Rad, CA, United States).
The antibodies used in these experiments were anti-TRIM21 rabbit polyclonal antibody (Proteintech, IL, United States), anti-HA-Tag rabbit monoclonal antibody (Abcam, Cambridge, United Kingdom), anti-p65 rabbit monoclonal antibody (Abcam, Cambridge, United Kingdom), anti-p-p65 Ser536 rabbit monoclonal antibody (Cell Signaling Technology, Danvers, MA, United States), anti-IκB-α rabbit polyclonal antibody (GeneTex, Irvine, CA, United States), anti-ubiquitin FK-2 monoclonal antibody (Enzo Life Sciences, NY, United States), anti-ubiquitin (linkage-specific K63) rabbit monoclonal antibody (Abcam, Cambridge, United Kingdom), anti-Phospho-(Ser/Thr) rabbit monoclonal antibody (Abcam, Cambridge, United Kingdom), anti-DDDDK-Tag rabbit polyclonal antibody (Abcam, Cambridge, United Kingdom), anti-SAG1 rabbit polyclonal antibody (Abcam, Cambridge, United Kingdom), anti-GAPDH mouse monoclonal antibody (Enzo Life Sciences, NY, United States), anti-β-actin rabbit monoclonal antibody (Cell Signaling Technology, Danvers, MA, United States), and goat anti-rabbit or goat anti-mouse IgG-HRP (ABclonal Technology, Wuhan, China).
Statistical analyses were performed using GraphPad Prism v5 software. Differences between groups were assessed by two-tailed unpaired Student t test or one-way ANOVA with repeated measures when more than two groups were analyzed. The significance of T. gondii proliferation difference between groups was analyzed by two-way ANOVA. Differences were labeled ∗ (p < 0.05), ∗∗ (p < 0.01), ∗∗∗ (p < 0.001), and no significant difference was presented as “ns.” All experiments were repeated at least three times for statistical analysis.
Our co-immunoprecipitation (Co-IP) assay identified the interaction of TgROP18I with human TRIM21 (Figure 1A). Meanwhile, we found that the kinase dead mutant ROP18-KD still interacted with TRIM21 (Figure 1B). Furthermore, as a potent proteasome inhibitor, MG132 treatment promoted the interaction of TgROP18I-TRIM21 (Figures 1B,C). As we know, TRIM21 contains four domains: RING, B box, CC (coiled-coil domain), and PRY-SPRY. We therefore further identified the domains of TRIM21 required for TgROP18I interaction. We successfully constructed the truncated mutants of TRIM21 as shown in the schematic diagram (Figure 1D) using the primers shown in Supplementary Table 3. The Co-IP results showed that PRY-SPRY domain deletion resulted in TgROP18I loss (Figure 1E), indicating that the PRY-SPRY domain of TRIM21 is responsible for the interaction with TgROP18I. Together, these results suggested that TRIM21 interacted with TgROP18I via its PRY-SPRY domain.
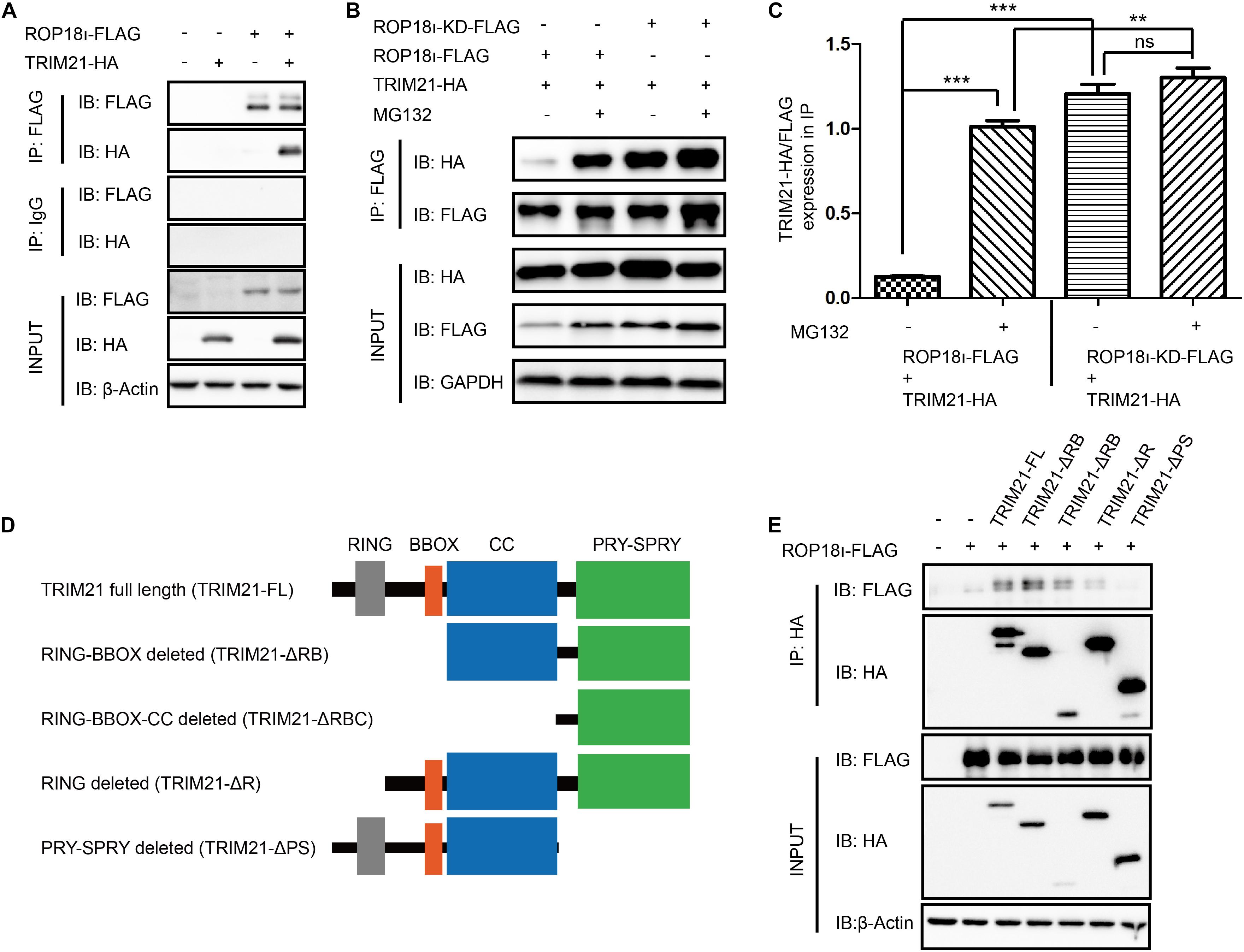
Figure 1. TgROP18I interacted with TRIM21’s PRY-SPRY domain. (A) The Co-IP result indicated the interaction of TgROP18I and TRIM21. (B,C) The Co-IP result indicated that ROP18-KD still bound with TRIM21. (D) Sketch map of TRIM21 WT and truncation mutants. (E) The Co-IP results showed the PRY-SPRY domain of TRIM21 was indispensable for TgROP18I-TRIM21 interaction. The experiments were repeated three times. The values were analyzed using the one-way ANOVA. Data were expressed as the mean ± SEM (**p < 0.01; ***p < 0.001; IP, immunoprecipitation).
Our immunoprecipitation (IP) result indicated that TgROP18I overexpression in the HEK293T cells elevated the phosphorylation level of TRIM21; the more pcDNA3.1-ROP18I-FLAG was transfected, the more phosphorylated TRIM21 was precipitated, and the differences between groups were significant (Figures 2A,B). A consistent result was observed in CEP and CEP-rop18I infection to HFF cells. The phosphorylation level of TRIM21 in CEP infected cells was not significantly different from that in the uninfected cells (Figures 2C,D). However, the TRIM21 phosphorylation level in CEP-rop18I infected HFF cells was significantly higher than that in the uninfected cells and the CEP infected cells (Figures 2C,D).
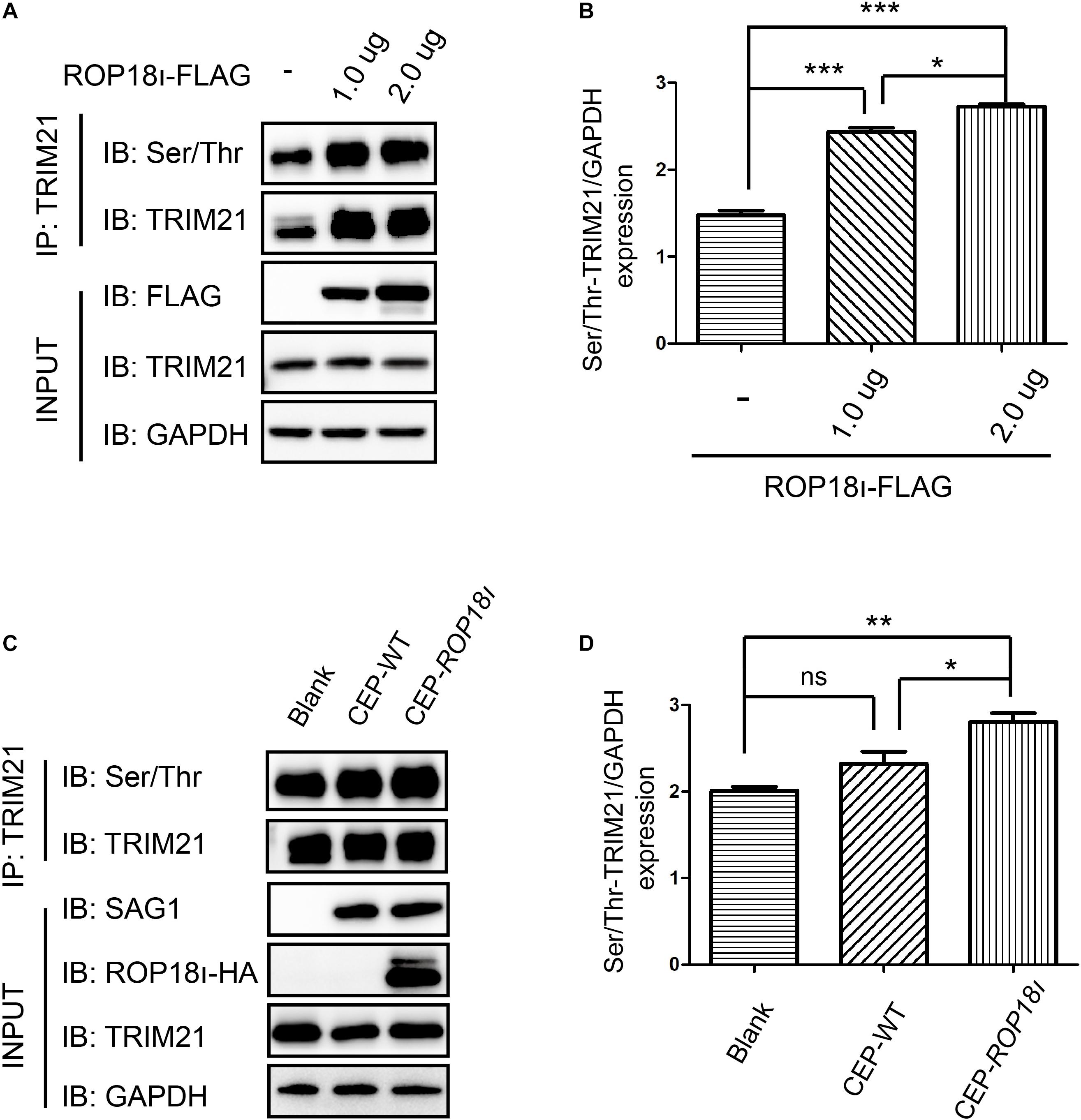
Figure 2. TgROP18I promoted the phosphorylation of TRIM21. (A,B) HEK293T cells were transfected with different amount of pcDNA3.1-ROP18I-FLAG as indicated, and the cell lysates were subjected to TRIM21 IP and Western blotting. The more plasmids were transfected to HEK293T cells, the higher levels of phosphorylated TRIM21 were observed, and the differences between groups were significant. (C,D) HFF cells were infected with CEP or CEP-rop18I, and then the total protein was extracted and subjected to TRIM21 IP and Western blotting. The results showed that more phosphorylated TRIM21 was detected in the CEP-rop18I infected cells than which detected in the CEP infected cells and uninfected cells, while the phosphorylation levels of TRIM21 were not significantly different between the CEP infected and uninfected cells. The experiments were repeated three times. The values were analyzed using the one-way ANOVA. Data were expressed as the mean ± SEM (*p < 0.05; **p < 0.01; ***p < 0.001).
We further investigated the result of TRIM21-TgROP18I interaction. We found that the endogenous TRIM21 protein level was decreased with the increased TgROP18I level (Figure 3A). Co-transfection of a stable amount of pcDNA3.1-TRIM21-HA with the increased amounts of pcDNA3.1-ROP18I-FLAG in HEK293T cells showed that, the over-expressed TRIM21 was decreased by TgROP18I in a dose-dependent manner (Figure 3B). Furthermore, compared to ROP18 overexpression, the ROP18-KD overexpression resulted in less degradation of TRIM21 (Figure 3C). This result indicated that the kinase activity of TgROP18I was required for TgROP18I-mediated TRIM21 degradation. This phenomenon was further verified by the parasitic infection experiment. HFFs were infected with RH and RH-△rop18 strains. Comparing to RH-△rop18 infection, RH infection led to a significant degradation of TRIM21 (Figure 3D). To identify the pathway that involved in TRIM21 degradation, HEK293T cells were co-transfected with a stable amount of pcDNA3.1-TRIM21-HA and increased amounts of pcDNA3.1-ROP18I-FLAG as indicated in Figure 3E. Before cell harvest, the cells were treated with MG132 or the lysosome inhibitor Leupeptin. From the results, we found that MG132 treatment had no effect on the TRIM21 degradation mediated by TgROP18I; however, Leupeptin treatment resulted in a stable TRIM21 protein level regardless of TgROP18I existence or not (Figure 3E). In conclusion, our experiments demonstrated that TgROP18I targeted human TRIM21 and resulted in TRIM21 degradation through lysosomal pathway, which was dependent on its kinase activity.
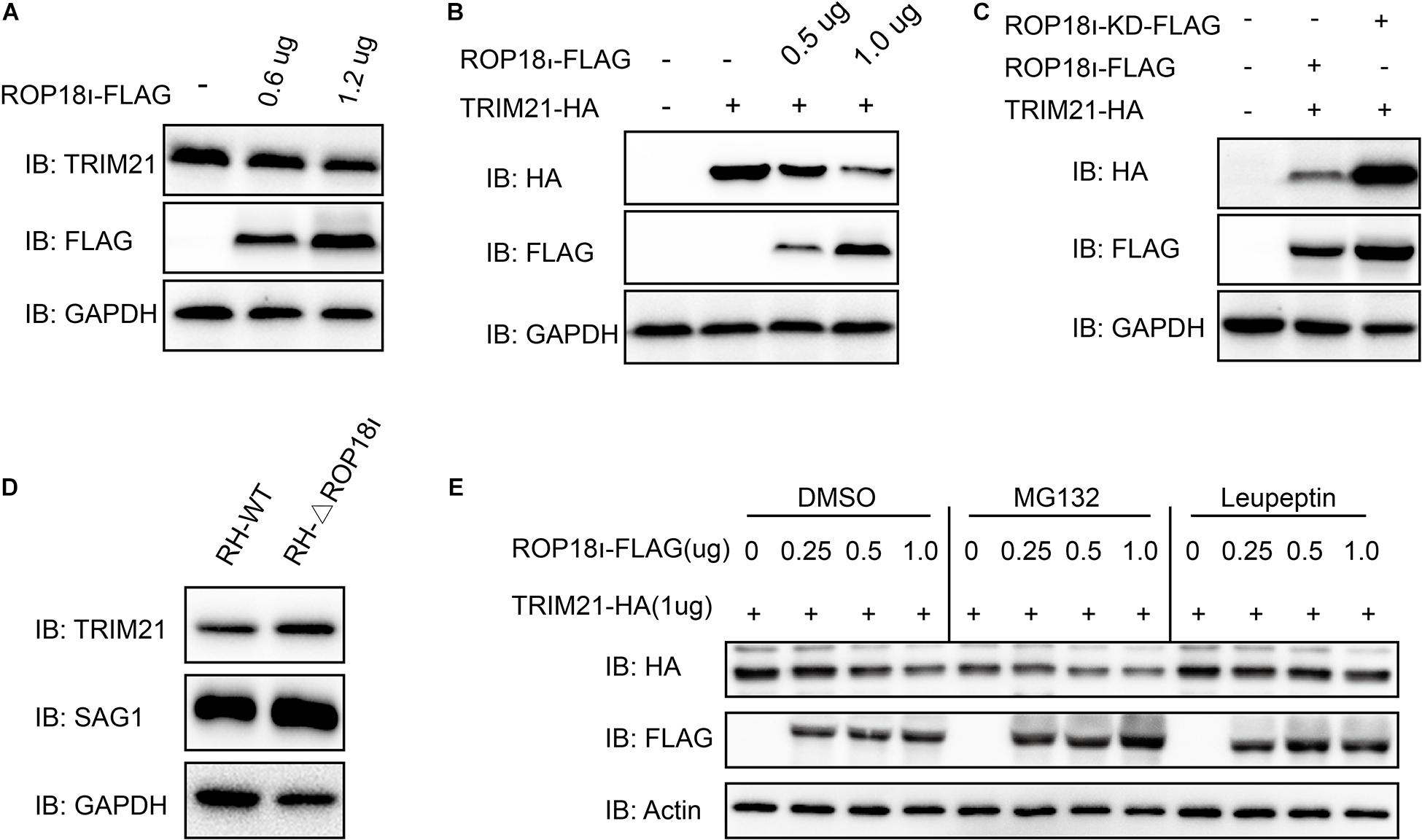
Figure 3. TgROP18I promoted TRIM21 degradation through lysosomal pathway. (A) HEK293T cells were transfected with the increased amounts of pcDNA3.1-ROP18I-FLAG as indicated. (B) HEK293T cells were co-transfected with a stable amount of pcDNA3.1-TRIM21-HA and the increased amounts of pcDNA3.1-ROP18I-FLAG as indicated. The endogenous or overexpressed TRIM21 level was decreased with the increased ROP18 level. (C) HEK293T cells were co-transfected with pcDNA3.1-TRIM21-HA and pcDNA3.1-ROP18I-FLAG or pcDNA3.1-ROP18I-KD-FLAG as indicated. The results of Western blotting detection with the cell lysates indicated that much more TRIM21 was detected in the ROP18-KD overexpression group than in the ROP18 overexpression group. (D) Lysates of HFFs infected with RH or RH-△rop18 was detected by Western blotting, and more TRIM21 was detected in the RH-△rop18 infection group than in the RH infection group. (E) HEK293T cells were co-transfected with 1mg of pcDNA3.1-TRIM21-HA and increased amounts of pcDNA3.1-ROP18I-FLAG. The cells were treated with MG132 or Leupeptin, or left untreated. Cell lysates were subjected to Western blotting, and the results showed that TRIM21’s level was decreased with the increased amount of TgROP18I in the cells treated with MG132 or DMSO. However, TRIM21’s level was kept stable in the Leupeptin treated group. All the experiments were repeated three times. IB, immunoblot.
To investigate whether the IFN-γ inducible TRIM21 expression is regulated by T. gondii infection, we infected HFFs with RH or CEP strains, and evaluated the relative transcription and translation level of TRIM21. The qRT-PCR result indicated that, compared to the normal HFFs, T. gondii RH and CEP infection led to significantly upregulated transcription of TRIM21 at 1 h post-infection; however, with the infection going on, only T. gondii RH infection kept the significantly upregulated transcription of TRIM21 at 24 h post-infection, CEP infection led to a similar transcription level as in the uninfected cells (Figure 4A). Similarly, compared with the uninfected cells, a significantly higher TRIM21 protein level was detected in the HFFs infected with either RH or CEP, at 1–12 h post infection (Figures 4B,C). These results demonstrated that transcription and translation level of TRIM21 were upregulated during T. gondii infection in HFFs.
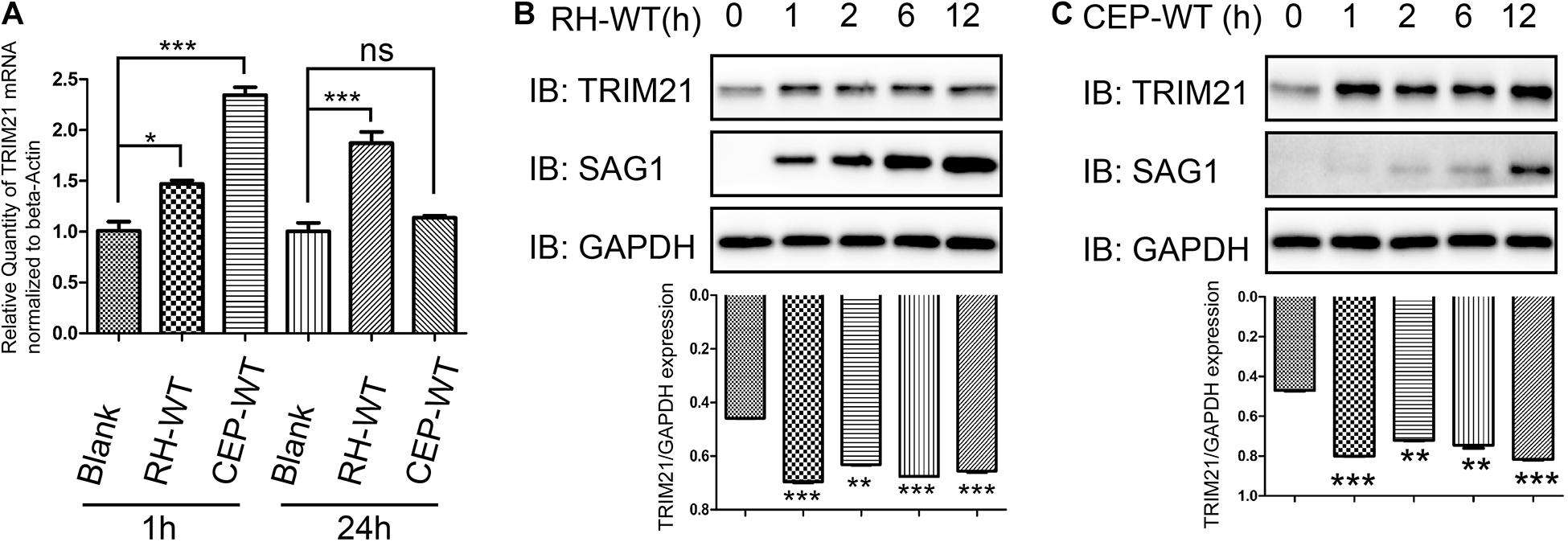
Figure 4. TRIM21 transcription and translation levels were upregulated following T. gondii infection. HFF cells infected with RH or CEP were harvested for detection of TRIM21 transcription and translation level. (A) Comparison of the TRIM21 transcription levels between the indicated groups with qRT-PCR. The CEP infection resulted in the highest TRIM21 transcription level, followed by RH infection, and uninfection at 1 h post infection, the differences between groups were significant. At 24 h post infection, the RH infection resulted in a significant higher TRIM21 transcription level than in CEP infection or uninfection groups between which no significant difference was observed in TRIM21 transcription level. (B,C) Comparison of the translation level of TRIM21 in the HFF cells infected with RH or CEP for the indicated time with Western blot (up panels). The densitometrical analysis for the intensity of TRIM21 bands normalized to its corresponding GAPDH intensity showed that, both CEP and RH infection resulted in significant higher transcription levels of TRIM21 than in uninfected cells (down panels). All the experiments were repeated four times. The values were analyzed using the one-way ANOVA. Data were expressed as the mean ± SEM (*p < 0.05; **p < 0.01; ***p < 0.001).
TRIM21 protein level was upregulated after stimulation with IFN-γ regardless of the concentrations (Figure 5A). Calculating for the number of tachyzoites per vacuole (Figure 5B) and the percentages of the vacuoles containing 1, 2, 4, and 8 tachyzoites (Figure 5C), the results showed the proliferation of both RH and CEP strains was significantly inhibited following IFN-γ treatment (Figures 5B,C). We therefore wanted to know what the role of TRIM21 on T. gondii proliferation. HFF cells were transfected with pcDNA3.1-TRIM21-HA, and the overexpression of TRIM21 was detected by Western blot (Figure 5D); the proliferation of the parasites was determined by IFA. We found that TRIM21 overexpression resulted in the inhibition of CEP replication, but did not affect RH replication (Figures 5E,F). We next wondered if TRIM21 loss affected CEP replication, we examined the CEP replication in the HFFs with TRIM21 knockdown. We found that si-TRIM21 transfection resulted in a significantly reduced TRIM21 level compared to that in the control group (Figure 5G and Supplementary Figure 1A). Though the replication of CEP was significantly inhibited in the IFN-γ treated HFFs, but it was not affected in the TRIM21 knockdown groups regardless of IFN-γ treatment or not (Figures 5H,I and Supplementary Figures 1B,C).
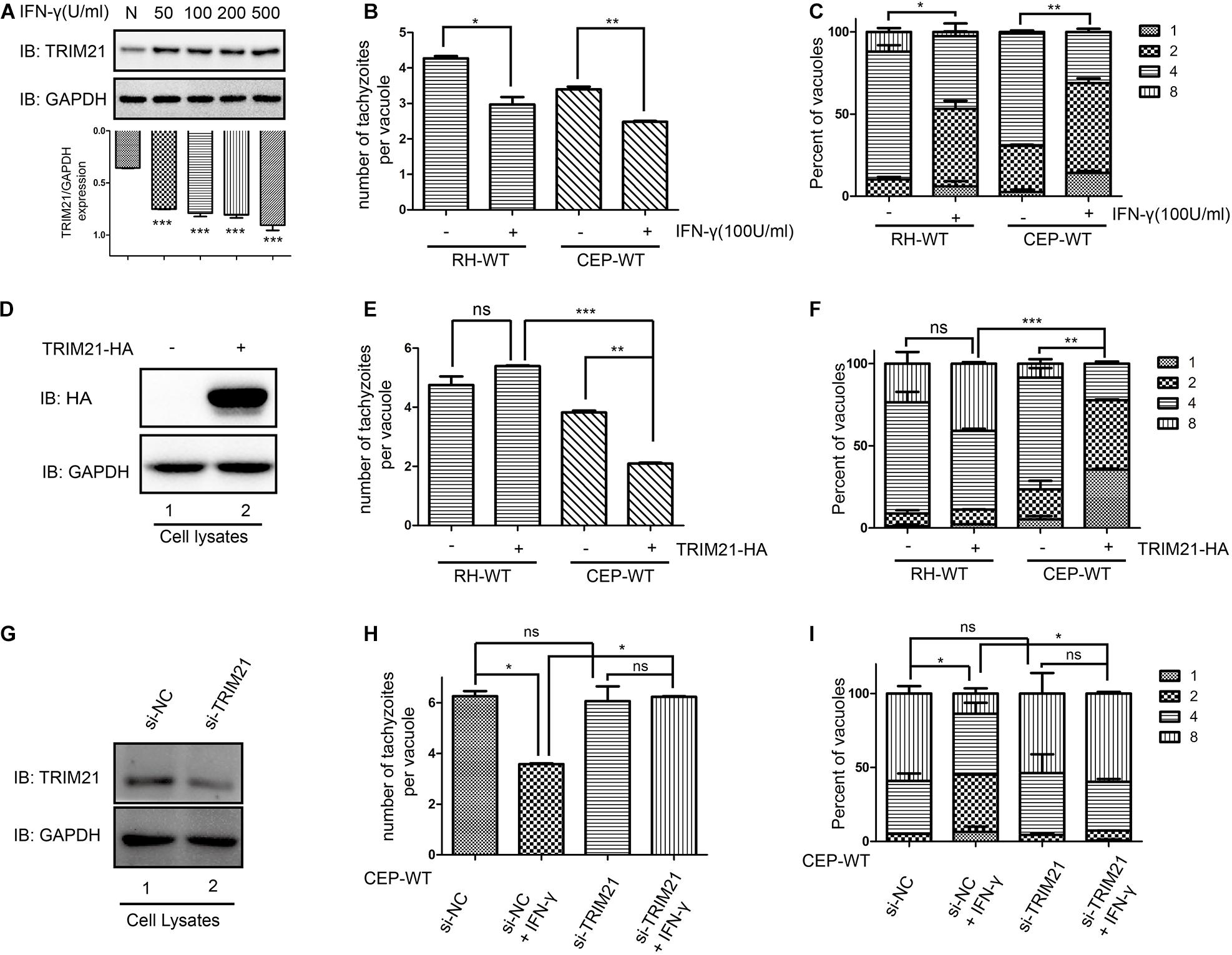
Figure 5. TRIM21 is involved in the IFN-γ induced inhibition of CEP proliferation (but not RH proliferation) in HFFs. The proliferation of RH and CEP tachyzoies in the HFFs was evaluated after stimulation with IFN-γ for 24 h (A–C), or transfection with pcDNA3.1-TRIM21-HA for 24 h (E–F); and proliferation of CEP-WT was evaluated after transfection with si-TRIM21 for 48 h in HFFs followed by IFN-γ treatment for 24 h (H–I). TRIM21 protein levels were measured by Western blotting (A,D,G). The average number of tachyzoites in 100 parasitophorous vacuoles (PVs) was counted (B,E,H), and the percentage of the PVs containing 1, 2, 4, or 8 parasites was determined by immune fluorescence assay (C,F,I). (A) The TRIM21 protein level was significantly up-regulated under IFN-γ stimulation in a dose-dependent manner, with the response concentration ranged from 50 to 500 U/ml. (B,C) The proliferation of the RH and CEP tachyzoites in HFFs was significantly inhibited after IFN-γ stimulation. (D) The overexpression of TRIM21 was detected. (E,F) The overexpression of TRIM21 significantly inhibited the CEP proliferation, but not affected RH proliferation. (G) The si-TRIM21 transfection significantly inhibited the TRIM21 translation. (H,I) The IFN-γ induced inhibition of CEP proliferation was relieved by TRIM21 knockdown. All the experiments were repeated three times. The values were analyzed using the one-way ANOVA and two-way ANOVA. Data were expressed as the mean ± SEM (*p < 0.05; **p < 0.01; ***p < 0.001).
To determine whether TRIM21 was involved in IFN-γ-induced ubiquitin labeling on CEP PVM, we conducted an IFA with anti-FK-2 antibody. We found that IFN-γ induced ubiquitin labeling on the CEP PVM, but this labeling was relieved by TRIM21 knockdown regardless of IFN-γ simulation or not (Figures 6A,B). Furthermore, we investigated the outcome of the CEP tachyzoites in the PVs labeled by ubiquitin with LysoTracker® staining, finding that IFN-γ induced acidification of CEP’s PV (Figures 6C,D). These results indicated that TRIM21 knockdown in HFFs relieved the ubiquitin labeling on CEP’s PV induced by IFN-γ, which would result in PV acidification to kill the parasite in it.
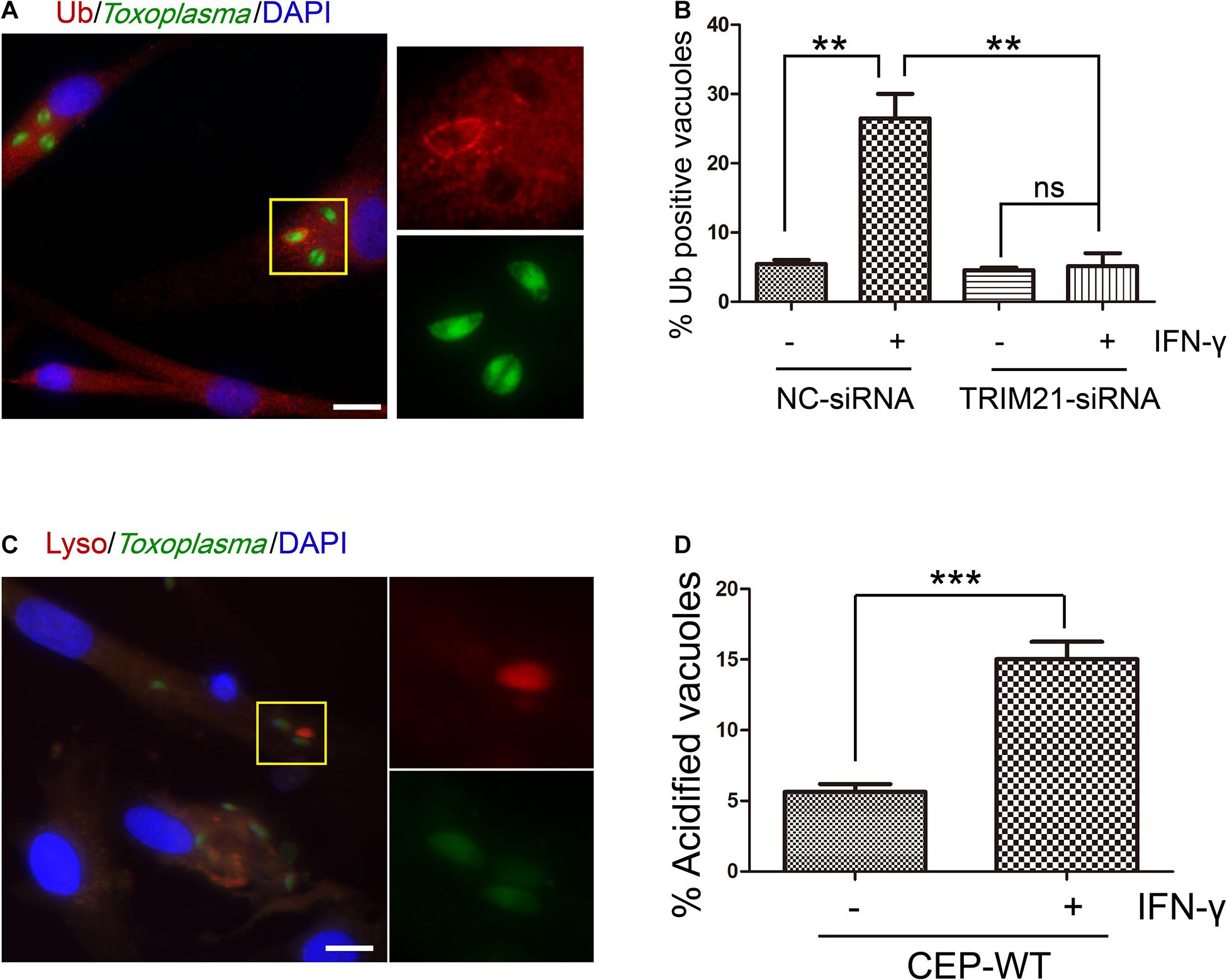
Figure 6. TRIM21 knockdown relieves the IFN-γ-induced ubiquitin labeling on CEP parasitophorous vacuole membrane (PVM) in HFFs. (A,B) HFFs were transfected with negative control siRNA (si-NC) or siRNA specific against TRIM21 (si-TRIM21), and stimulated with IFN-γ or not as indicated. After T. gondii infection, HFFs were subjected to immunofluorescence assay (IFA). IFN-γ induced ubiquitin labeling on the CEP PVM, but this labeling was relieved by TRIM21 knockdown regardless of IFN-γ simulation or not. (C,D) HFFs were stimulated with IFN-γ or not, and infected with CEP. The cells were then treated with LysoTracker® (acidic dye) and subjected to immunofluorescence assay (IFA). The result showed us that IFN-γ induced acidification of CEP. On the left, a representative fluorescent image is shown for the T. gondii CEP strain expressing GFP. The yellow box inside each representative image is shown as magnified pictures nearby (A,C). The percentage of vacuoles stained red with ubiquitin labeling or LysoTracker® was shown in the right bar diagram (B,D). Scale bar is 10 μm. The experiments were repeated three times. The values were analyzed using the one-way ANOVA or two-tailed unpaired Student t test. Data were expressed as the mean ± SEM (**p < 0.01; ***p < 0.001).
As we had identified that IFN-γ treatment resulted in the inhibition of RH and CEP replication, which also induced TRIM21 expression in HFFs (Figure 5). However, TRIM21 overexpression resulted in the inhibition of CEP replication, but did not affect RH replication (Figure 5); furthermore, TRIM21 knockdown relieved the IFN-γ induced inhibition of CEP (but not RH) proliferation (Figure 5). We therefore deduced that TRIM21 only functioned in CEP infection, but not in RH infection. As CEP strain does not express ROP18 as RH strain does, we speculated that the “loss of function” for TRIM21 during RH infection might be resulted by TgROP18I which degraded TRIM21 through lysosomal pathway (Figure 3).
To assess whether upregulated TRIM21 can be reversed by TgROP18I during RH infection, we conducted a proliferation assay with RH-△rop18 and CEP-rop18I strains. The result showed that TRIM21 overexpression resulted in the inhibition of RH-△rop18 proliferation (Figures 7A,B), but had no effect on CEP-rop18I proliferation (Figures 7C,D). These results confirmed that TRIM21 medicated inhibition of T. gondii proliferation was relieved by TgROP18I, and was irrelevant to T. gondii strain types.
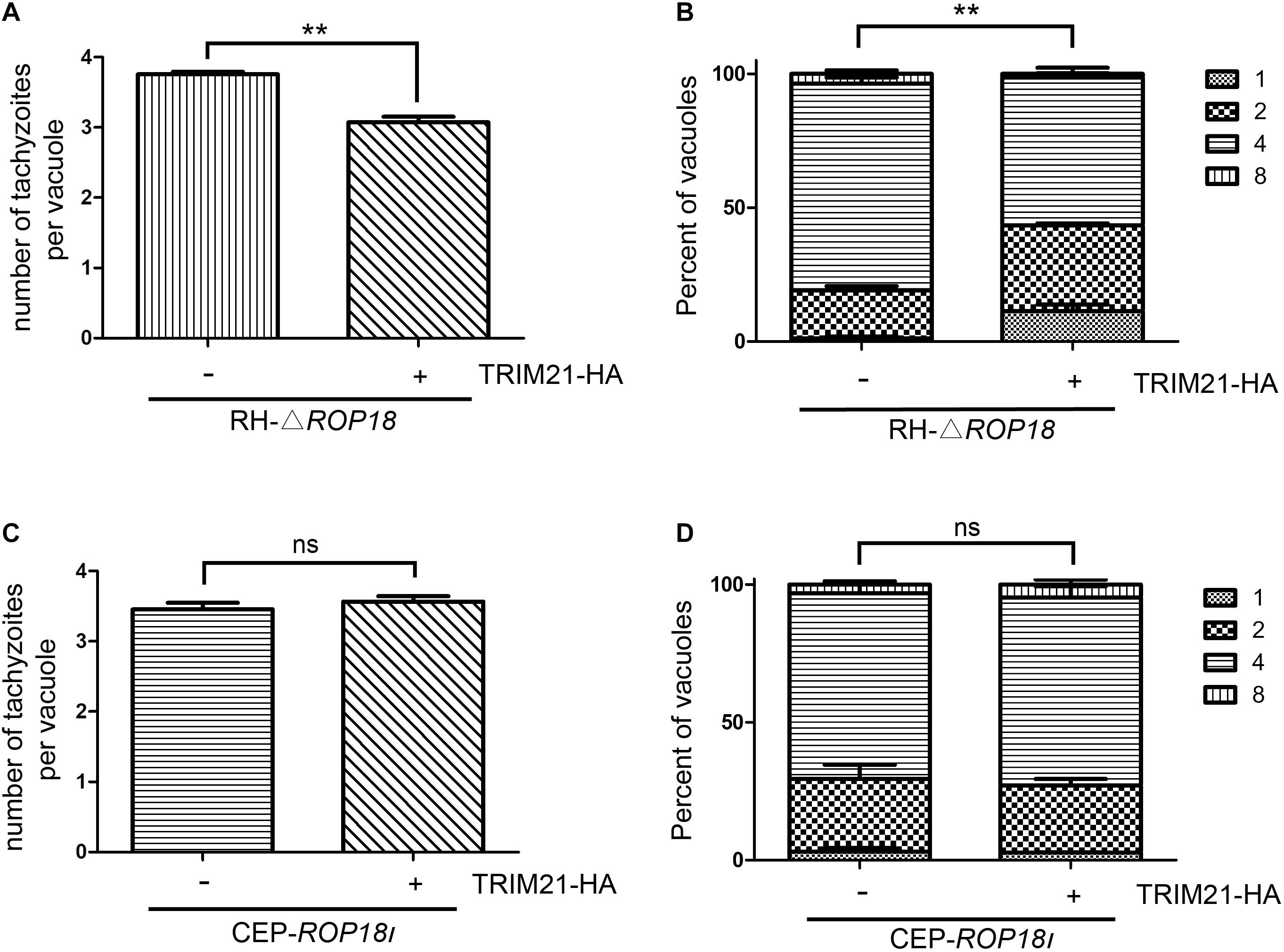
Figure 7. TgROP18I relieved TRIM21 mediated inhibition of T. gondii proliferation regardless of strain types. (A–D) HFFs were transfected with pcDNA3.1-TRIM21-HA or pcDNA3.1(+) for control, and infected with RH-△rop18 (A,B) or CEP-rop18I (C,D) parasites as indicated. Parasitic proliferation was measured at 18 h (RH-△rop18) or 24 h (CEP-rop18I) post-infection. The average number of tachyzoites in 100 vacuoles (A,C) or the number of vacuoles containing 1, 2, 4, or 8 parasites (B,D) was determined by fluorescence microscopy. The results indicated that TRIM21 overexpression inhibited the RH-△rop18 multiplication, but had no significant effect on CEP-rop18I multiplication. The experiments were repeated three times. The values were analyzed using the two-tailed unpaired Student t test and two-way ANOVA. Data were expressed as the mean ± SEM (**p < 0.01).
As p65 phosphorylation represents the activation of NF-κB, both p-p65 (Ser536) and TRIM21 levels were upregulated by IFN-γ treatment in HFF cells, compared to which in the untreated cells (Figures 8A,B). To further reveal the relation between TRIM21 and NF-κB, we overexpressed TRIM21 in HFFs to detect p65 (S536) phosphorylation. The result showed us that the p-p65 (S536) level was significantly upregulated (representing significant NF-κB activation) by TRIM21 overexpression in dose-dependent manner (Figures 8C,D). After that, HFFs were transfected with si-TRIM21, and the TRIM21 knockdown was verified in si-TRIM21 transfected cells (Figure 8E). The result indicated that the phosphorylation of p65 (Ser536) was downregulated by TRIM21 knockdown (Figures 8E,F). We therefore concluded that TRIM21 functioned in NF-κB activation.
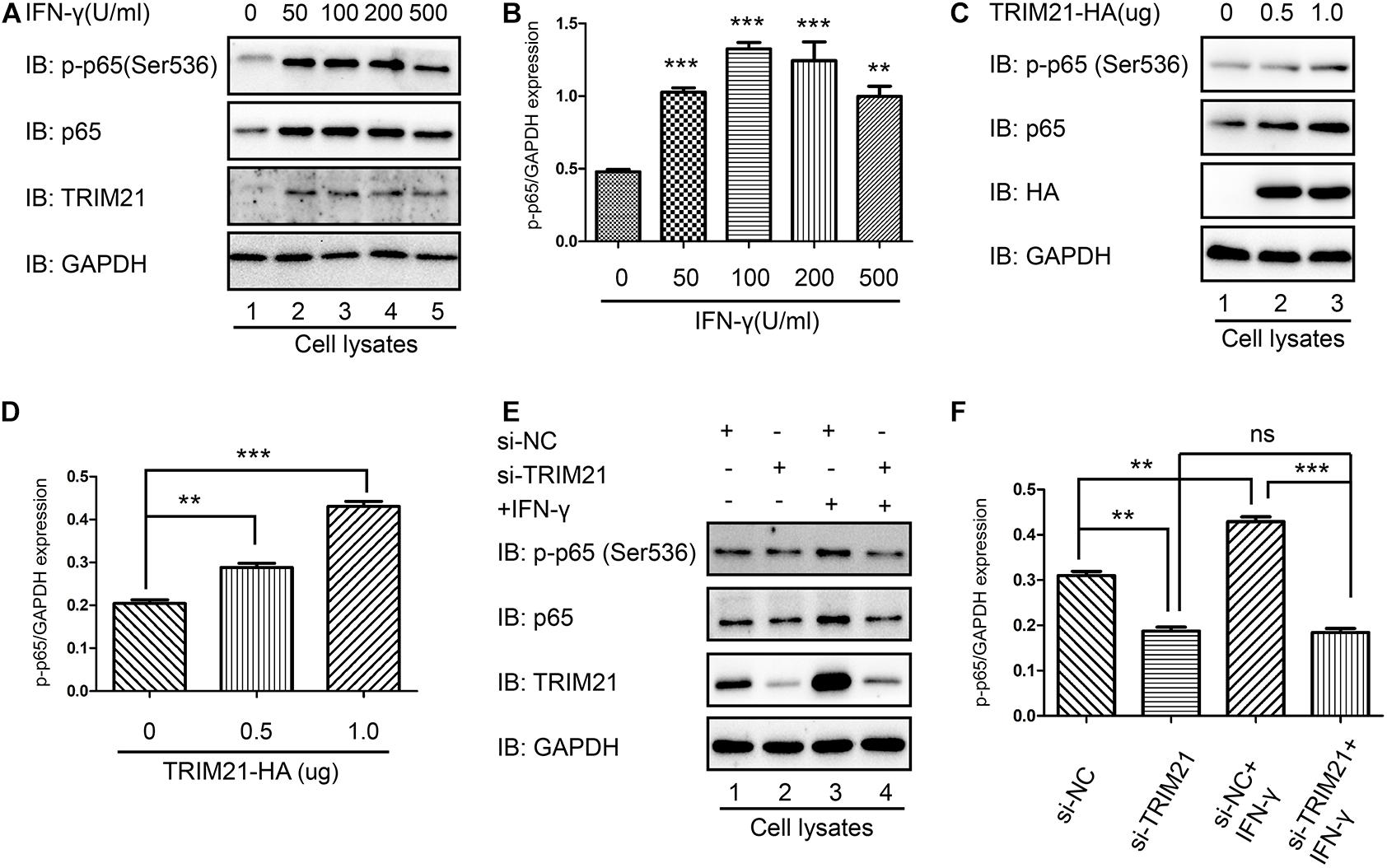
Figure 8. TRIM21 mediated NF-κB activation. (A,B) In the HFF cells stimulated with the indicated concentrations of IFN-γ for 24 h, the p-p65 (S536) level was significantly higher than that in the untreated group. (C,D) Western-blot detection of p-p65 (S536) level showed that TRIM21 overexpression significantly elevated p-p65 (S536) phosphorylation in dose-dependent manner. (E,F) In the siRNA-TRIM21 transfected groups, TRIM21 expression was suppressed, but no significant difference was found in the p-p65 (S536) levels between the TRIM21 knockdown group with or without IFN-γ induction. The experiments were repeated three times. The values were analyzed using the one-way ANOVA and the data were expressed as the mean ± SEM (**p < 0.01; ***p < 0.001).
As the interaction of p65 and IκB-α was essential for NF-κB activation, we next sought to find out whether their interaction regulated by TRIM21. We firstly performed immunoprecipitation with anti-TRIM21 antibody. The result showed that TRIM21 interacted with IκB-α and TRIM21 overexpression promoted the interaction of TRIM21 and IκB-α, this phenomenon was also confirmed with IFN-γ stimulation (Figures 9A,B). Furthermore, their interaction was promoted with the prolonged IFN-γ treating time (Figure 9C). Moreover, the complex of IκB-α-TRIM21-p65 was detected after immunoprecipitation with anti-p65 antibody, and TRIM21 overexpression resulted in inhibition of IκB-α and p65 interaction in the HEK293T cells, for less IκB-α was detected in the over-expression cells than in the normal cells (Figure 9D). Since TRIM21 is an E3 ubiquitin ligase, we next examined the p65 ubiquitination level in the TRIM21 overexpressed HEK293T cells. An increased ubiquitinated p65 level was observed in the TRIM21 overexpression cells (Figure 9E). Moreover, the result showed us that the p65-ubiquitin was mainly of K63 linkage type (Figure 9E). Together, these results suggested that TRIM21 interacted with p65 and IκB-α, so as to inhibit the interaction of p65 and IκB-α and promote p65-K63 ubiquitination. As a result, NF-κB activation was inhibited.
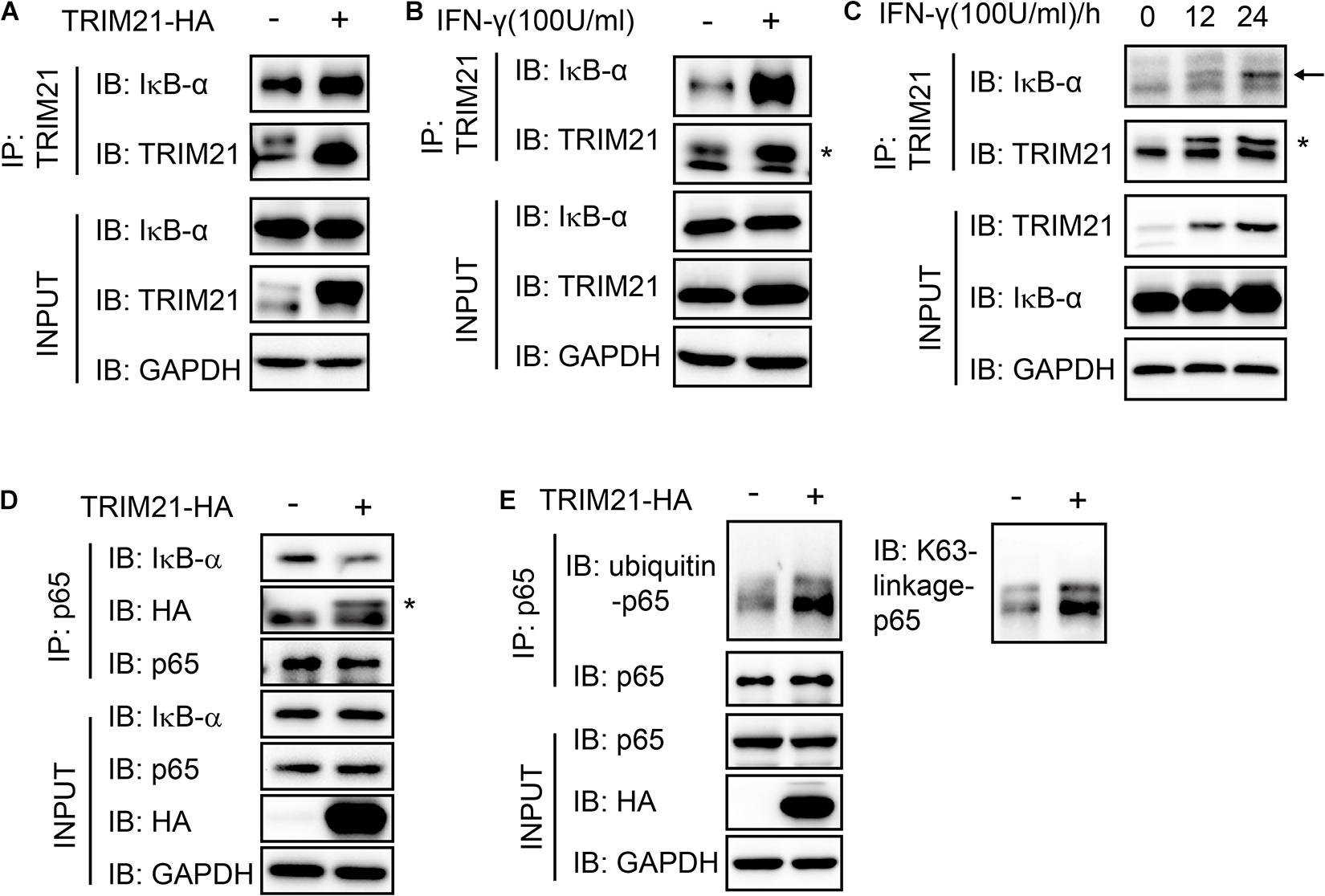
Figure 9. The interaction of p65 and IκB-α was suppressed with TRIM21 overexpression. Interaction of TRIM21 and IκB-α in the total cell lysates of HEK293T cells were analyzed by IP and Western-blot after either treatment with IFN-γ or transfection with pcDNA3.1-TRIM21-HA for the indicated time. (A–C) The IP with the anti-TRIM21 antibody identified the interaction of IκB-α with the overexpressed and the endogenous TRIM21. TRIM21 overexpression promoted TRIM21-IκB-α interaction (A). IFN-γ induction elevated TRIM21 production and promoted TRIM21-IκB-α interaction (B). The TRIM21 production induced by IFN-γ increased with the prolonged treating time and promoted TRIM21-IκB-α interaction (C). (D,E) The IP with the anti-p65 antibody identified the complex of IκB-α-TRIM21-p65 (D), and the interaction of p65-ubiquitin (E). The experiments were repeated three times. The TRIM21 band is indicated with “*”, and the IκB-α band is indicated with a black arrow.
TRIM21 was originally identified as an autoantigen in autoimmune diseases, including rheumatoid arthritis, Systemic lupus erythematosus, and Sjogren’s syndrome (Schulte-Pelkum et al., 2009). TRIM21 acts as an important factor involved in the innate immune responses against microbial infection, including bacterial, viral, and parasite (Hos et al., 2020; Mu et al., 2020; Xie et al., 2020), however, little about TRIM21 is known for parasites infection. In this study, we reported that T. gondii RH and CEP infection upregulated the expression of TRIM21, thus restricted parasite replication through NF-κB activation. Moreover, TRIM21 suppressed the p65-IκB-α interaction to activate NF-κB pathway through binding with IκB-α.
Interferon gamma (IFN-γ) is the major cytokine responsible for controlling T. gondii infection (Suzuki et al., 1988; Yap and Sher, 1999; MacMicking, 2012). Previous studies reported that TRIM21 can be upregulated by pathogens and suppress pathogens by interacting with the unique host factor, causing the ubiquitination and degradation of the pathogens through the proteasome pathway (Vaysburd et al., 2013; Rhodes and Isenberg, 2017). As an interferon stimulating gene (Rhodes et al., 2002), TRIM21 level was upregulated after stimulation with IFN-γ or T. gondii infection. We found that T. gondii proliferation was inhibited by IFN-γ stimulation, which is consistent with the other reports (Suzuki et al., 1988; Yap and Sher, 1999).
Our research revealed one of the mechanisms for the different outcomes when human cells were infected by type I and type III T. gondii. The virulence factor discharged by intruder has become one of the most effective ways for immune escape (Ashida et al., 2010; Sanada et al., 2012; Zhou and Zhu, 2015). During S. typhimurium infection, SPI-1-encoded effector protein SopA discharged by bacteria promoted TRIM65 and TRIM56 ubiquitination and degradation (Fiskin et al., 2017). TgROP18I, as the key virulence factor during T. gondii infection (El Hajj et al., 2007), is a Ser/Thr kinase of ROP2 subfamily, which interacted and phosphorylated host immune factor, including p65 (Du et al., 2014), p53, p38, UBE2N and Smad1 (Yang et al., 2017). In this study, we found the interaction of TgROP18I with human TRIM21 on the PRY-SPRY domain of TRIM21, and it promoted TRIM21 phosphorylation, and resulted in TRIM21 degradation through lysosomal way. We identified that IFN-γ treatment resulted in the inhibition of RH and CEP replication, and induced TRIM21 expression in HFFs. However, TRIM21 overexpression resulted in the inhibition of CEP replication, but did not affect RH replication; on the other hand, TRIM21 knockdown relieved the IFN-γ induced inhibition of CEP (but not RH) proliferation. Therefore, we deduced that TRIM21 only functioned in CEP infection, but not in RH infection. TgROP18I may be the reason for the different responses of host cells mediated by TRIM21 during RH and CEP infection.
Furthermore, our proliferation assay conducted with RH-△rop18 and CEP-rop18I strains showed that TRIM21 overexpression resulted in the inhibition of RH-△rop18 proliferation, but had no effect on CEP-rop18I proliferation. A recent study reported that deletion of TRIM21 gene promoted T. gondii replication (Foltz et al., 2017), while the effect of TRIM21 overexpression on T. gondii infection needed further investigations. Therefore, we concluded that TRIM21 medicated inhibition of T. gondii proliferation was relieved by TgROP18I, and was irrelevant to strain types.
TRIM family members have been regarded as key factors of innate immunity (Giraldo et al., 2020; Koepke et al., 2020). Accumulating studies have been reported that some members of the TRIM family positively regulated the NF-κB pathway (Kawai and Akira, 2011; Li et al., 2011; Hu et al., 2014). For instance, a recent study suggested that TRIM13 interfered TNF receptor associated factor 6 (TRAF6) to upregulate NF-κB activity (Huang and Baek, 2017). TRIM21 has been shown to stimulate NF-κB pathway activation (Yang et al., 2020). Moreover, IκB-α binds p65 subunit, which inhibits NF-κB activation and the IκB proteins are degraded, which promotes p65 entering the nucleus to activate NF-κB (Scheidereit, 2006). In consistence with these reports, we found that TRIM21 overexpression suppressed the p65-IκB-α interaction to promote the activation of NF-κB, which resulted in the inhibition of CEP proliferation. On the other hand, a suppressed NF-κB activation was observed in TRIM21 knockdown HFFs after CEP infection, but had no effects on CEP proliferation.
We also found that the IFN-γ-induced ubiquitin labeling of PVM was tightly controlled by TRIM21. Ubiquitin contains seven lysine residues that can form polyubiquitin chains (Komander and Rape, 2012). The formation of ubiquitin chains at different lysine residues leads to distinct outcomes (Zhao and Ulrich, 2010). Lysine 63 (K63) polyubiquitination to substrates is important for activation of signal transduction (Zhao and Ulrich, 2010; Komander and Rape, 2012). In our study, we identified that TRIM21 interacted with p65 and increased the p65 K63-ubiquitination level to promote NF-κB activation, which may be a possible mechanism for TRIM21 enhancing NF-κB activation to inhibit the proliferation of T. gondii. Ubiquitin recognition of intracellular bacteria and virus has become the hallmark event to mediate cellular immune response for restriction of the intruders (Heaton et al., 2016; Kuo et al., 2018; Wang et al., 2018). The coating of intracellular pathogens with ubiquitin is considered as a conserved defense mechanism from fruit flies to humans (Boyle and Randow, 2013; Manzanillo et al., 2013). Ubiquitin recognition of the substrates depends on the specificity of E3 ubiquitin ligases, and it’s important to screen the specific E3 ubiquitin ligases responsible for the interaction of host and pathogens. For instance, Salmonella was directly recognized by an E3 ubiquitin ligase, leucine rich repeat and sterile alpha motif containing 1 (LRSAM1), which led to the ubiquitination and autophagy of the bacterium (Huett et al., 2012). During T. gondii infection, K63-ubiquitin recognition of PVM conferred it to acidic destruction and proliferation obstacle in human umbilical vein endothelial cells (HUVEC) (Clough et al., 2016). We also found that TRIM21 knockdown relieved the ubiquitin labeling on PVM in IFN-γ primed HFFs, which indicated that TRIM21, as an E3 ubiquitin ligase, contributed to the IFN-γ-induced ubiquitin immune response against T. gondii infection.
In conclusion, our findings are highlighted as follows (Figure 10). 1. Host IFN-γ-induced factor TRIM21 restricted T. gondii replication through NF-κB activation and TRIM21 overexpression suppressed the p65-IκB-α interaction to activate NF-κB pathway. 2. TgROP18I which was discharged by T. gondii, interacted with the PRY-SPRY domain of human TRIM21, promoted TRIM21 phosphorylation, and induced TRIM21 degradation via lysosomal pathway. 3. IFN-γ induced ubiquitin labeling on the CEP PVM which resulted in PV acidification and death of parasites, but this labeling was relieved by TRIM21 knockdown regardless of IFN-γ simulation or not.
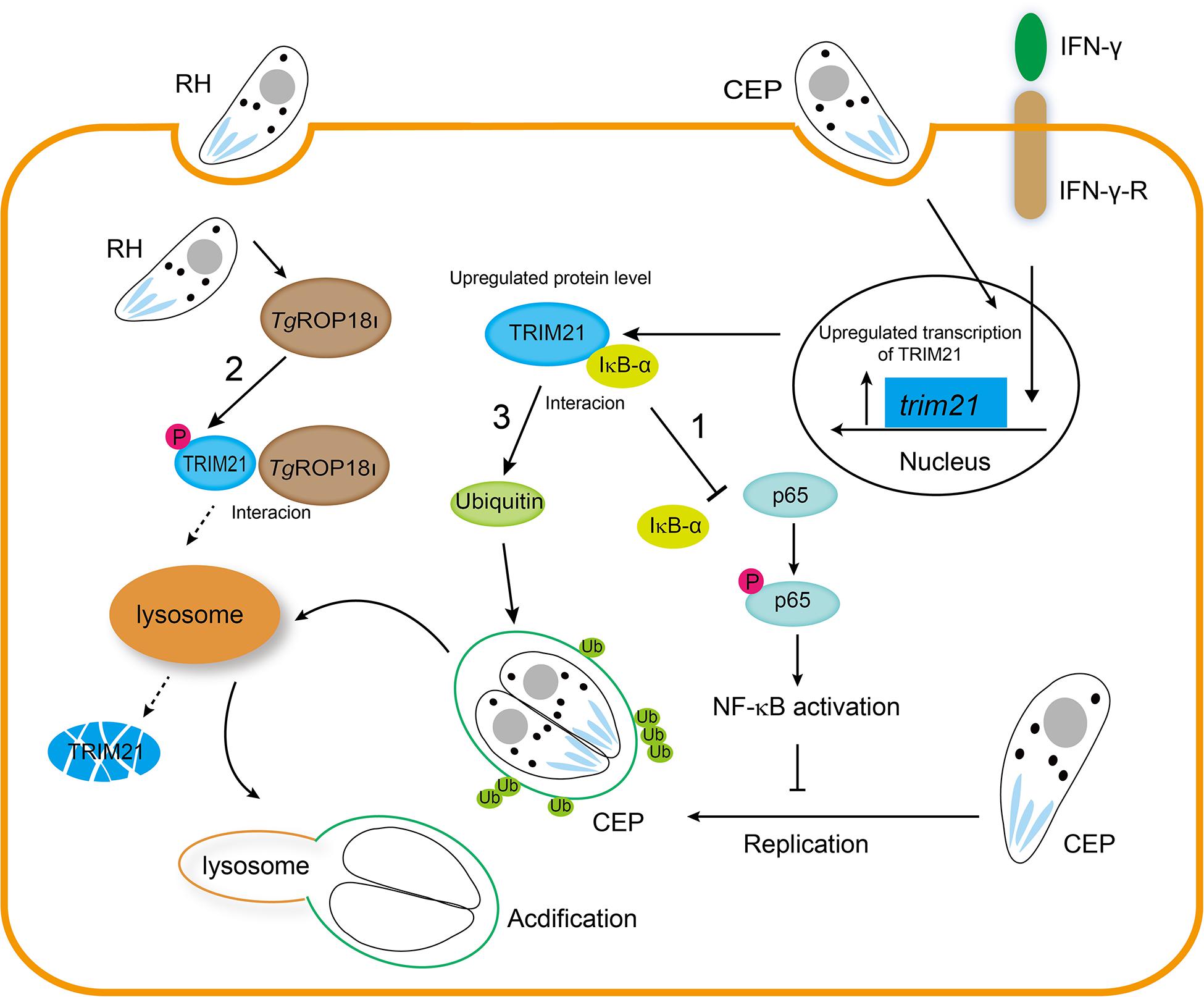
Figure 10. TgROP18I targets host TRIM21 for immune escape. 1. Host IFN-γ-induced factor TRIM21 restricted T. gondii replication through NF-κB activation and TRIM21 overexpression suppressed the p65-IκB-α interaction to activate NF-κB pathway. 2. TgROP18I which was discharged by T. gondii, interacted with the PRY-SPRY domain of human TRIM21, promoted TRIM21 phosphorylation, and induced TRIM21 degradation via lysosomal pathway. 3. IFN-γ induced ubiquitin labeling on the CEP PVM which resulted in PV acidification and death of parasites, but this labeling was relieved by TRIM21 knockdown regardless of IFN-γ simulation or not.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This research was supported by National Key R&D Program of China (2017YFD0500400), National Natural Science Foundation of China (81772217, 201828006, and 81971954), Science and Technology Planning Project of Guangdong Province (2018A050506038), Key Project of Guangzhou Science Research (201904020011), and the Basic Research Project of Key Laboratory of Guangzhou (202102100001) to HP.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.685913/full#supplementary-material
Supplementary Figure 1 | The proliferation of RH was measured after TRIM21 knockdown for 48 h following IFN-γ stimulation for 24 h. (A) TRIM21 protein levels were measured by Western blotting. (B,C) The average number of tachyzoites in 100 parasitophorous vacuoles (PVs) was counted (B), and the percentage of the PVs containing 1, 2, 4, or 8 parasites was determined by fluorescence microscope (C). TRIM21 knockdown following IFN-γ had no significant effect on the proliferation of RH tachyzoites in HFF cells. The experiments were repeated three times. The values were analyzed using the one-way ANOVA. Data were expressed as the mean ± SEM (∗∗p < 0.01).
Supplementary Table 1 | The gene names and sequences used for q-RT-PCR analysis.
Supplementary Table 2 | The siRNA sequences used for TRIM21 knockdown.
Supplementary Table 3 | The primers used for construction of the full length and truncation mutants of TRIM21 with HA-tag.
Ashida, H., Kim, M., Schmidt-Supprian, M., Ma, A., Ogawa, M., and Sasakawa, C. (2010). A bacterial E3 ubiquitin ligase IpaH9.8 targets NEMO/IKKgamma to dampen the host NF-kappaB-mediated inflammatory response. Nat. Cell Biol. 12, 66-73; sup 61-69. doi: 10.1038/ncb2006
Bianchi, F., and van den Bogaart, G. (2020). Vacuolar escape of foodborne bacterial pathogens. J. Cell. Sci. 134:jcs247221. doi: 10.1242/jcs.247221
Boothroyd, J. C., and Dubremetz, J. F. (2008). Kiss and spit: the dual roles of Toxoplasma rhoptries. Nat. Rev. Microbiol. 6, 79–88. doi: 10.1038/nrmicro1800
Boyle, K. B., and Randow, F. (2013). The role of ‘eat-me’ signals and autophagy cargo receptors in innate immunity. Curr. Opin. Microbiol. 16, 339–348. doi: 10.1016/j.mib.2013.03.010
Bradley, P. J., Ward, C., Cheng, S. J., Alexander, D. L., Coller, S., Coombs, G. H., et al. (2005). Proteomic analysis of rhoptry organelles reveals many novel constituents for host-parasite interactions in Toxoplasma gondii. J. Biol. Chem. 280, 34245–34258. doi: 10.1074/jbc.M504158200
Clough, B., Wright, J. D., Pereira, Pm, Hirst, Em, Johnston, Ac, Henriques, R., et al. (2016). K63-linked ubiquitination targets toxoplasma gondii for endo-lysosomal destruction in IFNγ-stimulated human cells. PLoS Pathog. 12:e1006027. doi: 10.1371/journal.ppat.1006027
Du, J., An, R., Chen, L., Shen, Y., Chen, Y., Cheng, L., et al. (2014). Toxoplasma gondii virulence factor ROP18 inhibits the host NF-κB pathway by promoting p65 degradation. J. Biol. Chem. 289, 12578–12592. doi: 10.1074/jbc.M113.544718
El Hajj, H., Lebrun, M., Arold, S. T., Vial, H., Labesse, G., and Dubremetz, J. F. (2007). ROP18 is a rhoptry kinase controlling the intracellular proliferation of Toxoplasma gondii. PLoS Pathog. 3:e14. doi: 10.1371/journal.ppat.0030014
Elmore, S. A., Jones, J. L., Conrad, P. A., Patton, S., Lindsay, D. S., and Dubey, J. P. (2010). Toxoplasma gondii: epidemiology, feline clinical aspects, and prevention. Trends Parasitol. 26, 190–196. doi: 10.1016/j.pt.2010.01.009
Fiskin, E., Bhogaraju, S., Herhaus, L., Kalayil, S., Hahn, M., and Dikic, I. (2017). Structural basis for the recognition and degradation of host TRIM proteins by Salmonella effector SopA. Nat. Commun. 8:14004. doi: 10.1038/ncomms14004
Foltz, C., Napolitano, A., Khan, R., Clough, B., Hirst, E. M., and Frickel, E. M. (2017). TRIM21 is critical for survival of Toxoplasma gondii infection and localises to GBP-positive parasite vacuoles. Sci. Rep. 7:5209. doi: 10.1038/s41598-017-05487-7
Ge, Y., Chen, J., Qiu, X., Zhang, J., Cui, L., Qi, Y., et al. (2014). Natural killer cell intrinsic toll-like receptor MyD88 signaling contributes to IL-12-dependent IFN-γ production by mice during infection with Toxoplasma gondii. Int. J. Parasitol. 44, 475–484. doi: 10.1016/j.ijpara.2014.03.004
Giraldo, M. I., Hage, A., Van Tol, S., and Rajsbaum, R. (2020). TRIM proteins in host defense and viral pathogenesis. Curr. Clin. Microbiol. Rep. 7, 101–114. doi: 10.1007/s40588-020-00150-8
Gov, L., Karimzadeh, A., Ueno, N., and Lodoen, M. B. (2013). Human innate immunity to Toxoplasma gondii is mediated by host caspase-1 and ASC and parasite GRA15. mBio 4, e00255-13. doi: 10.1128/mBio.00255-13
Gov, L., Schneider, C. A., Lima, T. S., Pandori, W., and Lodoen, M. B. (2017). NLRP3 and potassium efflux drive rapid IL-1β release from primary human monocytes during Toxoplasma gondii infection. J. Immunol. 199, 2855–2864. doi: 10.4049/jimmunol.1700245
Hakimi, M. A., Olias, P., and Sibley, L. D. (2017). Toxoplasma effectors targeting host signaling and transcription. Clin. Microbiol. Rev. 30, 615–645. doi: 10.1128/cmr.00005-17
Harker, K. S., Ueno, N., and Lodoen, M. B. (2015). Toxoplasma gondii dissemination: a parasite’s journey through the infected host. Parasite Immunol. 37, 141–149. doi: 10.1111/pim.12163
He, C., Kong, L., Zhou, L., Xia, J., Wei, H., Liu, M., et al. (2017). Host cell vimentin restrains Toxoplasma gondii invasion and phosphorylation of vimentin is partially regulated by interaction with TgROP18. Int. J. Biol. Sci. 13, 1126–1137. doi: 10.7150/ijbs.21247
Heaton, S. M., Borg, N. A., and Dixit, V. M. (2016). Ubiquitin in the activation and attenuation of innate antiviral immunity. J. Exp. Med. 213, 1–13. doi: 10.1084/jem.20151531
Hide, G. (2016). Role of vertical transmission of Toxoplasma gondii in prevalence of infection. Expert Rev. Anti Infect. Ther. 14, 335–344. doi: 10.1586/14787210.2016.1146131
Hos, N. J., Fischer, J., Hos, D., Hejazi, Z., Calabrese, C., Ganesan, R., et al. (2020). TRIM21 is targeted for chaperone-mediated autophagy during Salmonella Typhimurium infection. J. Immunol. 205, 2456–2467. doi: 10.4049/jimmunol.2000048
Hu, M. M., Yang, Q., Zhang, J., Liu, S. M., Zhang, Y., Lin, H., et al. (2014). TRIM38 inhibits TNFα- and IL-1β-triggered NF-κB activation by mediating lysosome-dependent degradation of TAB2/3. Proc. Natl. Acad. Sci. U.S.A. 111, 1509–1514. doi: 10.1073/pnas.1318227111
Huang, B., and Baek, S. H. (2017). Trim13 potentiates toll-like receptor 2-mediated nuclear factor κB activation via K29-linked polyubiquitination of tumor necrosis factor receptor-associated factor 6. Mol. Pharmacol. 91, 307–316. doi: 10.1124/mol.116.106716
Huett, A., Heath, R. J., Begun, J., Sassi, S. O., Baxt, L. A., Vyas, J. M., et al. (2012). The LRR and RING domain protein LRSAM1 is an E3 ligase crucial for ubiquitin-dependent autophagy of intracellular Salmonella Typhimurium. Cell Host Microbe 12, 778–790. doi: 10.1016/j.chom.2012.10.019
Kawai, T., and Akira, S. (2011). Regulation of innate immune signalling pathways by the tripartite motif (TRIM) family proteins. EMBO Mol. Med. 3, 513–527. doi: 10.1002/emmm.201100160
Kim, S. K., Fouts, A. E., and Boothroyd, J. C. (2007). Toxoplasma gondii dysregulates IFN-γ-inducible gene expression in human fibroblasts: insights from a genome-wide transcriptional profiling. J. Immunol. 178, 5154–5165. doi: 10.4049/jimmunol.178.8.5154
Koepke, L., Gack, M. U., and Sparrer, K. M. (2020). The antiviral activities of TRIM proteins. Curr. Opin. Microbiol. 59, 50–57. doi: 10.1016/j.mib.2020.07.005
Komander, D., and Rape, M. (2012). The ubiquitin code. Annu. Rev. Biochem. 81, 203–229. doi: 10.1146/annurev-biochem-060310-170328
Kuo, C. J., Hansen, M., and Troemel, E. (2018). Autophagy and innate immunity: insights from invertebrate model organisms. Autophagy 14, 233–242. doi: 10.1080/15548627.2017.1389824
Lazzari, E., Korczeniewska, J., Ní Gabhann, J., Smith, S., Barnes, B. J., and Jefferies, C. A. (2014). TRIpartite motif 21 (TRIM21) differentially regulates the stability of interferon regulatory factor 5 (IRF5) isoforms. PLoS One 9:e103609. doi: 10.1371/journal.pone.0103609
Lee, H. R., Lee, M. K., Kim, C. W., and Kim, M. (2020). TRIM proteins and their roles in the influenza virus life cycle. Microorganisms 8:1424. doi: 10.3390/microorganisms8091424
Li, Q., Yan, J., Mao, A. P., Li, C., Ran, Y., Shu, H. B., et al. (2011). Tripartite motif 8 (TRIM8) modulates TNFα- and IL-1β-triggered NF-κB activation by targeting TAK1 for K63-linked polyubiquitination. Proc. Natl. Acad. Sci. U.S.A. 108, 19341–19346. doi: 10.1073/pnas.1110946108
Lima, T. S., Gov, L., and Lodoen, M. B. (2018). Evasion of human neutrophil-mediated host defense during Toxoplasma gondii infection. mBio 9, e02027-17. doi: 10.1128/mBio.02027-17
Liu, B., Zhang, M., Chu, H., Zhang, H., Wu, H., Song, G., et al. (2017). The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat. Immunol. 18, 214–224. doi: 10.1038/ni.3641
Liu, H., Li, M., Song, Y., and Xu, W. (2018). TRIM21 restricts coxsackievirus B3 replication, cardiac and pancreatic injury via interacting with MAVS and positively regulating IRF3-mediated Type-I interferon production. Front. Immunol. 9:2479. doi: 10.3389/fimmu.2018.02479
MacMicking, J. D. (2012). Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat. Rev. Immunol. 12, 367–382. doi: 10.1038/nri3210
Manzanillo, P. S., Ayres, J. S., Watson, R. O., Collins, A. C., Souza, G., Rae, C. S., et al. (2013). The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature 501, 512–516. doi: 10.1038/nature12566
Montoya, J. G., and Liesenfeld, O. (2004). Toxoplasmosis. Lancet 363, 1965–1976. doi: 10.1016/s0140-6736(04)16412-x
Mu, T., Zhao, X., Zhu, Y., Fan, H., and Tang, H. (2020). The E3 ubiquitin ligase TRIM21 promotes HBV DNA polymerase degradation. Viruses 12:346. doi: 10.3390/v12030346
Pan, J. A., Sun, Y., Jiang, Y. P., Bott, A. J., Jaber, N., Dou, Z., et al. (2016). TRIM21 ubiquitylates SQSTM1/p62 and suppresses protein sequestration to regulate redox homeostasis. Mol. Cell 61, 720–733. doi: 10.1016/j.molcel.2016.02.007
Platanias, L. C. (2005). Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5, 375–386. doi: 10.1038/nri1604
Rhodes, D. A., Ihrke, G., Reinicke, A. T., Malcherek, G., Towey, M., Isenberg, D. A., et al. (2002). The 52 000 MW Ro/SS-A autoantigen in Sjögren’s syndrome/systemic lupus erythematosus (Ro52) is an interferon-gamma inducible tripartite motif protein associated with membrane proximal structures. Immunology 106, 246–256. doi: 10.1046/j.1365-2567.2002.01417.x
Rhodes, D. A., and Isenberg, D. A. (2017). TRIM21 and the function of antibodies inside cells. Trends Immunol. 38, 916–926. doi: 10.1016/j.it.2017.07.005
Saeij, J. P., Boyle, J. P., Coller, S., Taylor, S., Sibley, L. D., Brooke-Powell, E. T., et al. (2006). Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Science 314, 1780–1783. doi: 10.1126/science.1133690
Sanada, T., Kim, M., Mimuro, H., Suzuki, M., Ogawa, M., Oyama, A., et al. (2012). The Shigella flexneri effector OspI deamidates UBC13 to dampen the inflammatory response. Nature 483, 623–626. doi: 10.1038/nature10894
Scheidereit, C. (2006). IkappaB kinase complexes: gateways to NF-kappaB activation and transcription. Oncogene 25, 6685–6705. doi: 10.1038/sj.onc.1209934
Schulte-Pelkum, J., Fritzler, M., and Mahler, M. (2009). Latest update on the Ro/SS-A autoantibody system. Autoimmun. Rev. 8, 632–637. doi: 10.1016/j.autrev.2009.02.010
Shapira, S., Harb, O. S., Margarit, J., Matrajt, M., Han, J., Hoffmann, A., et al. (2005). Initiation and termination of NF-kappaB signaling by the intracellular protozoan parasite Toxoplasma gondii. J. Cell Sci. 118, 3501–3508. doi: 10.1242/jcs.02428
Sinai, A. P. (2007). The toxoplasma kinase ROP18: an active member of a degenerate family. PLoS Pathog. 3:e16. doi: 10.1371/journal.ppat.0030016
Suzuki, Y., Orellana, M. A., Schreiber, R. D., and Remington, J. S. (1988). Interferon-gamma: the major mediator of resistance against Toxoplasma gondii. Science 240, 516–518. doi: 10.1126/science.3128869
Taylor, S., Barragan, A., Su, C., Fux, B., Fentress, S. J., Tang, K., et al. (2006). A secreted serine-threonine kinase determines virulence in the eukaryotic pathogen Toxoplasma gondii. Science 314, 1776–1780. doi: 10.1126/science.1133643
Torgerson, P. R., and Mastroiacovo, P. (2013). The global burden of congenital toxoplasmosis: a systematic review. Bull. World Health Organ. 91, 501–508. doi: 10.2471/BLT.12.111732
Vaysburd, M., Watkinson, R. E., Cooper, H., Reed, M., O’connell, K., Smith, J., et al. (2013). Intracellular antibody receptor TRIM21 prevents fatal viral infection. Proc. Natl. Acad. Sci. U.S.A. 110, 12397–12401. doi: 10.1073/pnas.1301918110
Wang, H. T., and Hur, S. (2021). Substrate recognition by TRIM and TRIM-like proteins in innate immunity. Semin. Cell Dev. Biol. 111, 76–85. doi: 10.1016/j.semcdb.2020.09.013
Wang, L., Yan, J., Niu, H., Huang, R., and Wu, S. (2018). Autophagy and ubiquitination in Salmonella infection and the related inflammatory responses. Front. Cell. Infect. Microbiol. 8:78. doi: 10.3389/fcimb.2018.00078
Xie, S., Zhang, L., Dong, D., Ge, R., He, Q., Fan, C., et al. (2020). HDAC6 regulates antibody-dependent intracellular neutralization of viruses via deacetylation of TRIM21. J. Biol. Chem. 295, 14343–14351. doi: 10.1074/jbc.RA119.011006
Yang, L., Zhang, T., Zhang, C., Xiao, C., Bai, X., and Wang, G. (2020). Upregulated E3 ligase tripartite motif-containing protein 21 in psoriatic epidermis ubiquitylates nuclear factor-κB p65 subunit and promotes inflammation in keratinocytes. Br. J. Dermatol. 184, 111–122. doi: 10.1111/bjd.19057
Yang, Z., Hou, Y., Hao, T., Rho, H. S., Wan, J., Luan, Y., et al. (2017). A human proteome array approach to identifying key host proteins targeted by toxoplasma kinase ROP18. Mol. Cell. Proteomics 16, 469–484. doi: 10.1074/mcp.M116.063602
Yap, G. S., and Sher, A. (1999). Effector cells of both nonhemopoietic and hemopoietic origin are required for interferon (IFN)-gamma- and tumor necrosis factor (TNF)-alpha-dependent host resistance to the intracellular pathogen, Toxoplasma gondii. J. Exp. Med. 189, 1083–1092. doi: 10.1084/jem.189.7.1083
Zhao, S., and Ulrich, H. D. (2010). Distinct consequences of posttranslational modification by linear versus K63-linked polyubiquitin chains. Proc. Natl. Acad. Sci. U.S.A. 107, 7704–7709. doi: 10.1073/pnas.0908764107
Keywords: Toxoplasma gondii, TRIM21, TgROP18I, NF-κB, ubiquitin
Citation: Yao L, Xu L, Zhou L, Wu S, Zou W, Chen M, Chen J and Peng H (2021) Toxoplasma gondii Type-I ROP18 Targeting Human E3 Ligase TRIM21 for Immune Escape. Front. Cell Dev. Biol. 9:685913. doi: 10.3389/fcell.2021.685913
Received: 26 March 2021; Accepted: 03 May 2021;
Published: 26 May 2021.
Edited by:
Qingfeng Zhang, Tongji University, ChinaReviewed by:
Gaoqian Feng, Burnet Institute, AustraliaCopyright © 2021 Yao, Xu, Zhou, Wu, Zou, Chen, Chen and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongjuan Peng, ZmxvcmlhcGVuZ0Bob3RtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.