
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 19 August 2021
Sec. Epigenomics and Epigenetics
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.685775
This article is part of the Research TopicEpigenetic Regulation in Cardiovascular DiseasesView all 16 articles
 Jiamin Li1†
Jiamin Li1† Yunxiang Zhou2†
Yunxiang Zhou2† Hui Wang3
Hui Wang3 Jianyao Lou4Cameron Lenahan5,6
Jianyao Lou4Cameron Lenahan5,6 Shiqi Gao7
Shiqi Gao7 Xiaoyu Wang7Yongchuan Deng2*
Xiaoyu Wang7Yongchuan Deng2* Han Chen1*
Han Chen1* Anwen Shao7*
Anwen Shao7*The recently discovered ferroptosis is a new kind of iron-regulated cell death that differs from apoptosis and necrosis. Ferroptosis can be induced by an oxidative stress response, a crucial pathological process implicated in cardiovascular diseases (CVDs). Accordingly, mounting evidence shows that oxidative stress-induced ferroptosis plays a pivotal role in angio-cardiopathy. To date, the inhibitors and activators of ferroptosis, as well as the many involved signaling pathways, have been widely explored. Among which, epigenetic regulators, molecules that modify the package of DNA without altering the genome, emerge as a highly targeted, effective option to modify the signaling pathway of ferroptosis and oxidative stress, representing a novel and promising therapeutic potential target for CVDs. In this review, we will briefly summarize the mechanisms of ferroptosis, as well as the role that ferroptosis plays in various CVDs. We will also expound the epigenetic regulators of oxidative stress-induced ferroptosis, and the promise that these molecules hold for treating the intractable CVDs.
Cardiovascular diseases (CVDs), including heart conditions and vascular disorders, are the leading cause of mortality around the world, and comprise approximately one-third of annual deaths [World Health Organization (WHO), 2017]. Moreover, CVDs carry an enormous economic burden to every country, especially China and India [The Institute for Health Metrics and Evaluation (IHME), 2018]. The most prevalent CVDs include hypertension, coronary heart disease, atrial fibrillation, and valvular heart disease, but most CVDs develop into heart failure at the advanced or terminal stages. In 2017, update of the guidelines for the management of heart failure released by ACC/AHA/HFSA (American College of Cardiology Foundation/American Heart Association/Heart Failure Society of America), angiotensin receptor–neprilysin inhibitors (ARNI) (sacubitril/valsartan) and sinoatrial node modulators (ivabradine) were classified as the therapy for stage C heart failure in the evidence of level B-R, but require further high-quality randomized clinical trials to be conducted (Dixon et al., 2012; Yancy et al., 2017). The current treatment of CVDs is unsatisfactory, and the underlying mechanisms are not fully understood. As such, it is imperative that new mechanisms and corresponding therapeutic targets are explored.
Ferroptosis was first introduced by Dixon et al. (2012), and was featured as an iron-dependent lipid peroxidation, a regulated cell death that is different from apoptosis and necrosis. Currently, ferroptosis was defined as a unique iron-dependent form of non-apoptotic cell death triggered by erastin, an oncogenic RAS-selective lethal small molecule, and inhibited by ferrostatin-1 in cancer cells or glutamate-induced cell death in organotypic rat brain slices (Dixon et al., 2012). Mitochondria are crucial in ferroptosis, tricarboxylic acid (TCA) cycle participates in cysteine-deprivation induced ferroptosis and that the electron transport chain (ETC) regulates the process (Gao et al., 2019). Mitochondria participate in metabolism, and are the main source of reactive oxygen species (ROS) (Tang D. et al., 2021). Oxidative stress occurs when the antioxidant defense systems, such as GSH, coenzyme Q10, and tetrahydrobiopterin (BH4), cannot find equilibrium of ROS (Dixon et al., 2012; Bersuker et al., 2019; Doll et al., 2019; Kraft et al., 2020; Soula et al., 2020). GSH, the major antioxidant in mammalian cells, is tightly tuned intracellularly and extracellularly for homeostasis (Gao et al., 2018), and is also the key component in ferroptosis. Ferroptosis has been found in many diseases, such as cancer, CVD, neurological disease, and ischemia/reperfusion injuries in kidney, liver, lung, and skeletal muscle (Stamenkovic et al., 2019). Ferroptosis may be a potential mechanism underlying CVDs as many studies pointed out that ferroptosis have been implicated in CVDs (Birnbaum et al., 1996; Shiomi et al., 2004; Dabkowski et al., 2008; Fang et al., 2019).
The biological processes are regulated by genetics and epigenetics. Epigenetics is known as the unchanged nucleotide sequence of the gene that is modulated by several environmental factors while genetics irreversibly change the gene code via mutation (Borrelli et al., 2008). Epigenetics act on DNA or chromatin by DNA methylation, histone modifications, chromatin remodeling and noncoding RNAs (Prasher et al., 2020; Wu et al., 2020). Based on epidemiological studies, alteration of lifestyle and environment can reduce the risk of developing CVDs (Wang et al., 2013). It has been suggested that ferroptosis can be regulated by epigenetic, transcriptional, and post-translational mechanisms (Chen et al., 2020). Accumulating evidence indicates that a series of epigenetic regulators are involved in the processes of ferroptosis. In the present review, we will elaborate on the mechanism of ferroptosis, the roles of ferroptosis in CVDs, as well as the roles of epigenetic regulators in oxidative stress-induced ferroptosis, and we will offer an option for the therapeutic application of ferroptosis in CVDs.
Cell death is frequently required to maintain the normal functions of the body/system, either under physiological conditions or pathophysiological circumstances. Two major classifications of cell death are apoptosis and necrosis. Other patterns of “non-classical” cell death, such as autophagy, pyroptosis, and necroptosis, reportedly also have important roles in cell survival and body function (Dixon et al., 2012).
Dolma et al. (2003) found a novel compound that can kill tumor cells without damaging isogenic normal cell counterparts. They named it “erastin,” and it induces nonapoptotic cell death in a RASV12- and small T(ST)-dependent manner (Dolma et al., 2003). Furthermore, Yang and Stockwell (2008) found that two small molecules, RSL (ras-selective-lethal compound) 3 and RSL5, were lethal to tumors with oncogenic RAS, similar to the function of erastin. RSL3- or RSL5-induced cell death is considered iron-dependent as it could be inhibited by either iron chelation or decreased iron uptake, with increased levels of ROS (Yang and Stockwell, 2008). Ferroptotic cells cannot be restrained by inhibitors of necrosis, apoptosis, or autophagy and exhibit morphological changes in mitochondria, such as decreased size, increased membrane density, and reduction or disappearance of cristae (Xie et al., 2016). In 2012, the team of Dixon SJ conducted further research to support and extend this newly discovered form of regulated cell death. They proposed the concept of ferroptosis for the first time, and defined it as the regulatory cell death induced by the accumulation of lipid peroxides and ROS, which can be inhibited by lipid peroxide inhibitors and iron chelators (Dixon et al., 2012). Outer mitochondrial membrane (OMM) rupture was observed in immortalized fibroblasts and glutathione peroxidase 4 (GPX4)-inactivated kidney tissue (Angeli et al., 2014).
Iron is an important essential microelement in the human body, and plays a key role in maintaining homeostasis of the internal environment, and ensuring the normal physiological functions of cells. Iron in the human body is mostly distributed in the hemoglobin of red blood cells and the myoglobin of muscles, but a small amount exists in enzymes, such as cytochrome oxidase, peroxidase, and catalase. There are two types of iron ions: ferrous and ferric. Ferric ions bind to transferrin, and are transported into the cell, entering via the transferrin receptor 1 (TFR1) on the cell membrane (Gao M. et al., 2015). Ferrous ions reduced to ferric ions in the cell, and are then transported and released into the cytoplasmic iron pool. Ferrous ion can react with oxygen, and generates ROS, such as hydroxyl radical (•OH) and hydrogen peroxide (H2O2), in a process known as the Fenton reaction. Iron overload leads to an increase of ROS, which cause harm to DNA, protein, and lipids. The Haber–Weiss reaction provides •OH from the substrates of H2O2 and superoxide (•O2–): (1) Fe3++•O2–→Fe2++O2; (2) Fe2++H2O2→Fe3++OH–+•OH (Fenton reaction); (3) •O2–+H2O2→•OH+OH–+O2 (Gao M. et al., 2015). Cellular iron overload can impair mitochondrial oxidative phosphorylation and produce a large amount of ROS, even exceeding the scavenging ability of the body’s antioxidant system [e.g., glutathione (GSH) and GPX4], thereby oxidizing cell membranes, as well as the unsaturated fatty acids on cell and organelle membranes, forming lipid peroxides, destroying cell structure and function, and causing cell damage or death (Dixon et al., 2012; Ooko et al., 2015; Hassan et al., 2016).
It is widely accepted that ferroptosis is regulated by the cystine/glutamate antiporter system (system Xc–) and GPX) (Dixon et al., 2012). System Xc– is an amino acid antiporter, which mainly includes SLC7A11 (solute carrier family 7 member 11) and SLC3A2 (solute carrier family 3 member 2), which causes the exchange of cysteine and glutamate into and out of the cell, respectively, at a 1:1 ratio (Lewerenz et al., 2013). Glutathione is an important antioxidant and free radical scavenger in vivo, and can be categorized as either reduced (GSH) or oxidized (GSSG). GPx4 converts GSH to GSSG, GSH/GSSG constitutes an antioxidant system and provides reducing equivalents to eliminate oxidative species (Xie et al., 2016; Yang W. S. et al., 2014). The synthesis of GSH depends on the cysteine, which is made by cystine, and glutamate-cysteine ligase (GCL). As a member of the glutathione peroxidase family, GPX4 inhibits ferroptosis by decreasing the level of lipid peroxides (Liang et al., 2009). While erastin and RSL-3 are both inducers of ferroptosis, erastin depends on VDAC2/VDAC3 or downregulation of GSH. However, RSL-3 does not require the above-mentioned molecules. Lipid oxidation is observed in both erastin and RSL3-induced cell death. Further investigation verified that GPX4 is the target of RSL-3 through a binding site (Yang W. S. et al., 2014). Many inducers (e.g., erastin, RSL3, RSL5, buthioninesulfoximine, acetaminophen, fin, lanperisone, sulfasalazine, sorafenib, and artesunate) and inhibitors (e.g., ferrostatin, liproxstatin-1, and zileuton) of ferroptosis have been identified, but the specific mechanisms and pathways are diverse (Xie et al., 2016). In summary, ferroptosis is a complex process, and more pathways will be discussed in the following sections (Figure 1).
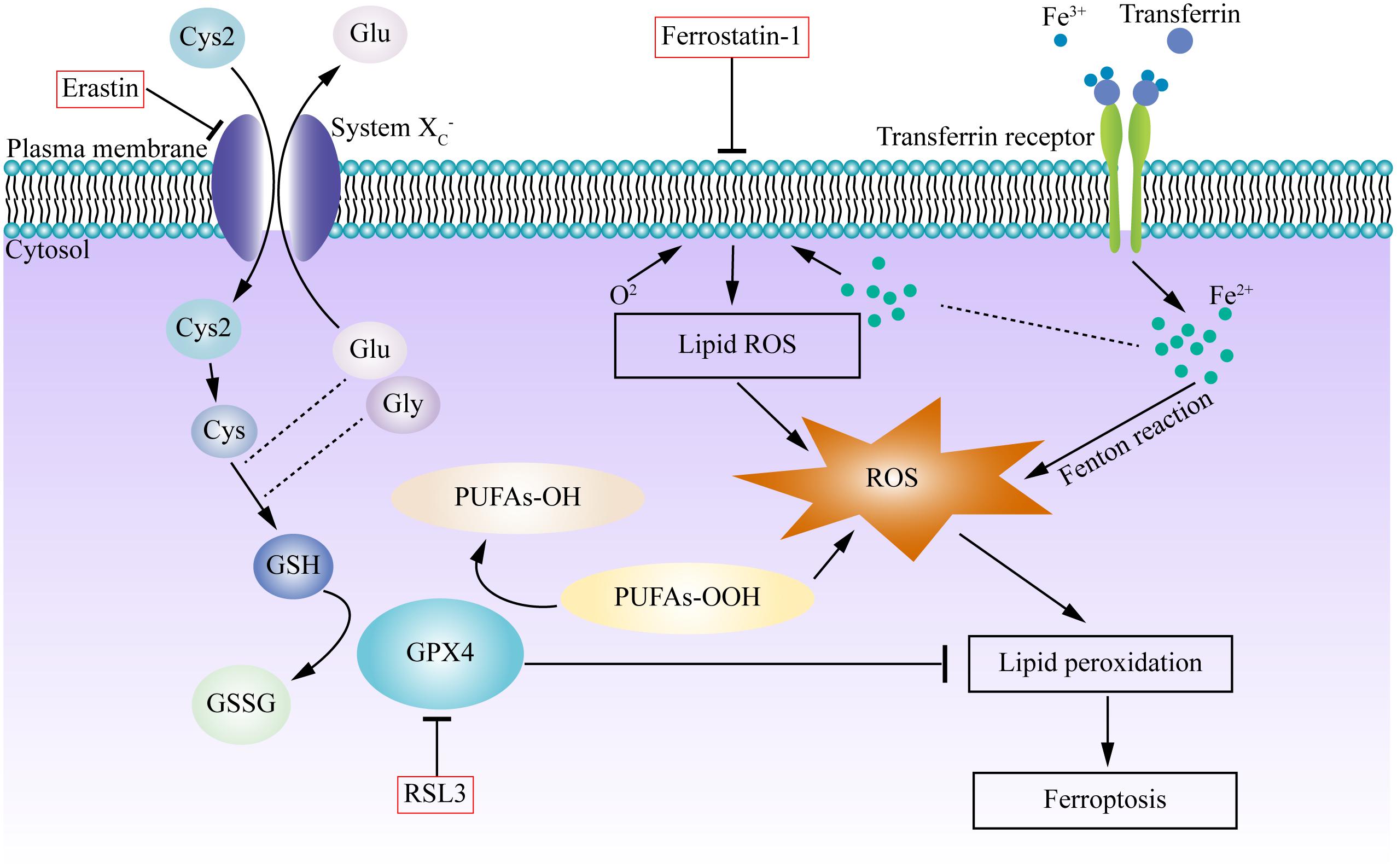
Figure 1. Graphic mechanisms of ferroptosis. Dysregulation of intracellular iron metabolism or glutathione peroxidation pathways leads to accumulation of lipid ROS and eventually causes ferroptosis. Various inducers and inhibitors are shown in the red box. Arrows indicate promotion; blunt-ended lines indicate inhibition. Excessive free irons are the foundation for ferroptosis execution. Circulated Fe3+ was combined with transferrin, and then entered into cells by transferrin receptor. Iron in Fe3+ form was deoxidized into iron in Fe2+, the latter generated ROS by Fenton reaction. System Xc- is an antiporter that imports cystine and exports glutamate, providing cysteine for glutathione synthesis, can be inhibited by erastin. GPX4 is a redox enzyme which reduces reactive aldehydes (PUFAs-OOH) to their alcohol form (PUFAs-OH), reduces ROS accumulation, plays important roles in the regulation of ferroptosis. RSL3 directly inhibit GPX4, which inhibits lipid peroxidation and prevent cell death, triggering the accumulation of ROS and boost ferroptosis. Ferrostatin-1 reduced the production of lipid ROS and attenuated ferroptosis. Cys, cysteine; Cys2, cystine; Glu, glutamate; Gly, glycine; GSH, glutathione; GSSH, glutathione disulfide; GPX4, glutathione peroxidase 4; PUFA, polyunsaturated fatty acid.
The heart is one of the most important organs, and is responsible for pumping blood throughout the body, providing energy and oxygen to nourish tissues and organs, and removing metabolites, such as carbon dioxide. There are complex regulatory mechanisms involved in maintaining normal cardiac function.
Inflammation is an important molecular trigger in CVD. Considerable evidence has shown the close relationship between inflammation and atherosclerosis (Ross, 1999; Libby and Ridker, 2006; Wong et al., 2012), and some anti-inflammatory drugs, such as statins, work to prevent cardiovascular events (Shepherd et al., 1995; Ridker et al., 2001, 2005).
Endothelial dysfunction arises when endothelial cells (EC) are injured, or if there is an imbalance between vasoconstriction and vasodilation (Chatzizisis et al., 2007). The gathering of low-density lipoprotein (LDL) in the subendothelial layer is thought to be the initial event of atherogenesis (Russell et al., 1989; Williams and Tabas, 1995; Tabas et al., 2007). Oxidative-LDLs (Ox-LDLs) induce proinflammatory expression and formation of foam cells, which lead to endothelial dysfunction (Berliner et al., 2009; Golia et al., 2014), including the release of proinflammatory cytokines, such as interleukin (IL), interferon-γ (IFN-γ), and tumor necrosis factor (TNF; Ait-Oufella et al., 2006; Moriya, 2019). Many autoimmune diseases (e.g., system lupus erythematosus, psoriasis, and rheumatoid arthritis), are found to correlate with increased cardiovascular risk (Kiss et al., 2006; Hak et al., 2009; Vena et al., 2010; Dougados et al., 2014). When anti-inflammatory therapy is applied to systemic lupus erythematosus patients, the mortality of CVD is lower. The mortality is also lower when anti-inflammatory therapy is given to patients with psoriasis (Leonardi et al., 2012; Papp et al., 2012) and rheumatoid arthritis (Liuzzo et al., 1999; Pasceri and Yeh, 1999). Moreover, inflammatory responses, including the monocyte-macrophages, neutrophils, T-cell subsets, and oxidative stress, also contribute to the initiation and development of heart failure (Tanai and Frantz, 2016).
Substrate metabolism is essential for normal cellular physiological function, carbohydrates (e.g., glucose and lactate), and fatty acids are the general cellular energy substrates (Ussher et al., 2016). The production of ATP in the heart is derived mainly from mitochondrial oxidative phosphorylation (OXPHOS), the others come from glycolysis (Bertero and Maack, 2018). When the cardiac supply cannot satisfy the demand, the heart will shift from one substrate to another. The glucose metabolism produces much more phosphates, but less ATP than lipids, which means that glucose metabolism expends less oxygen compared to fatty acid oxidation (FAO) when synthesizing equivalent ATP (Nagoshi et al., 2011). As is shown in the Randle cycle, the lipid metabolism correlates with glucose metabolism in a competitive manner (Randle et al., 1963; Randle, 1998; Sugden, 2007).
Mechanistically, calcium overload regulates the cardiomyocytes, especially in ischemia/reperfusion. When the blood supply decreases, anaerobic metabolism will be upregulated, but cellular pH and ATP production will decline. Accordingly, the Na+/H+ exchanger (NHE) excretes hydrogen ions in exchange for sodium ions (Pike et al., 1993; Sanada et al., 2011). Ca2+ efflux deficiency, and constriction of the reuptake by the endoplasmic reticulum (ER) due to the lack of ATP, will result in calcium overload. Subsequently, the mitochondrial permeability transition (MPT) pore will open, and the mitochondrial membrane potential will change, further weaken the production of energy. After the blood supply is re-established, a cascade of events will be triggered to aggravate the injury.
Many kinds of cell death were found to be engaged in CVDs. Kuwana et al. (2002) provide compelling evidence that the permeabilization of the OMM is involved in apoptosis. When Ca2+ gets access into the mitochondria, and opens the mitochondrial permeability transition pore (mPTP), water flows into the mitochondria, causing it to swell and undergo necrosis (Baines et al., 2005). Matsui et al. (2007) reported that autophagy was mediated by AMP-activated protein kinase (AMPK)-dependent pathway in the heart during ischemia/reperfusion injury. Kanamori et al. (2011) verified that autophagy can protect cardiomyocytes from death when suffering from ischemia.
Ohara et al. (1993) first pictured the hallmark of oxidative stress in CVDs in a hypercholesterolemia model. The redox crosstalk contributes to many diseases, such as atherosclerosis. Endothelial dysfunction initiates the process of atherosclerosis. Oxidized LDL (oxLDL) leads to the release bioactive phospholipids that can activate ECs and promote the pathogenesis of atherosclerosis (Hansson et al., 2006). Judkins et al. (2010) discovered the elevated expression of NOX2, an isoform of NADPH oxidase, in ECs and macrophages of lipoprotein deficient ApoE-/- mice, which leads to the formation of atherosclerotic lesions and increased aortic superoxide production. Two studies led by Nishida et al. (2000, 2002) indicate that activation of G-protein coupled receptors (GPCR) can generate ROS. Experiments on neonatal rat ventricular myocytes verified the function of ROS in activating hypertrophic growth signaling via G-proteins (Dai et al., 2011). In hypertension, ROS elevate the concentration of intracellular Ca2+ as a second messenger, causing vasoconstriction (Brito et al., 2015). The NOX signaling pathway is important in vascular processes, and lack of NO (nitric oxide), but increased oxidative stress, can be observed in hypertension (Touyz, 2004). Dudley et al. (2005) found increased oxidative stress and O2⋅– production relating to NADPH oxidase in an atrial fibrillation model. Aforementioned articles show evidence suggesting the function of oxidative stress in various kinds of CVDs, but further research on the mechanisms of oxidative stress may produce some unexpected breakthroughs.
Cardiomyocytes account for approximately 75% of the heart’s volume, and are rich in mitochondria. They are the main source of cardiac energy metabolism, and are the main site for the production of reactive oxidative species (ROS). Different kinds of cell death, including apoptosis, necrosis, pyroptosis, and ferroptosis, have been shown to be involved in the pathophysiologic process of various CVDs. Studies that describe the roles of ferroptosis in CVDs are listed in Table 1.
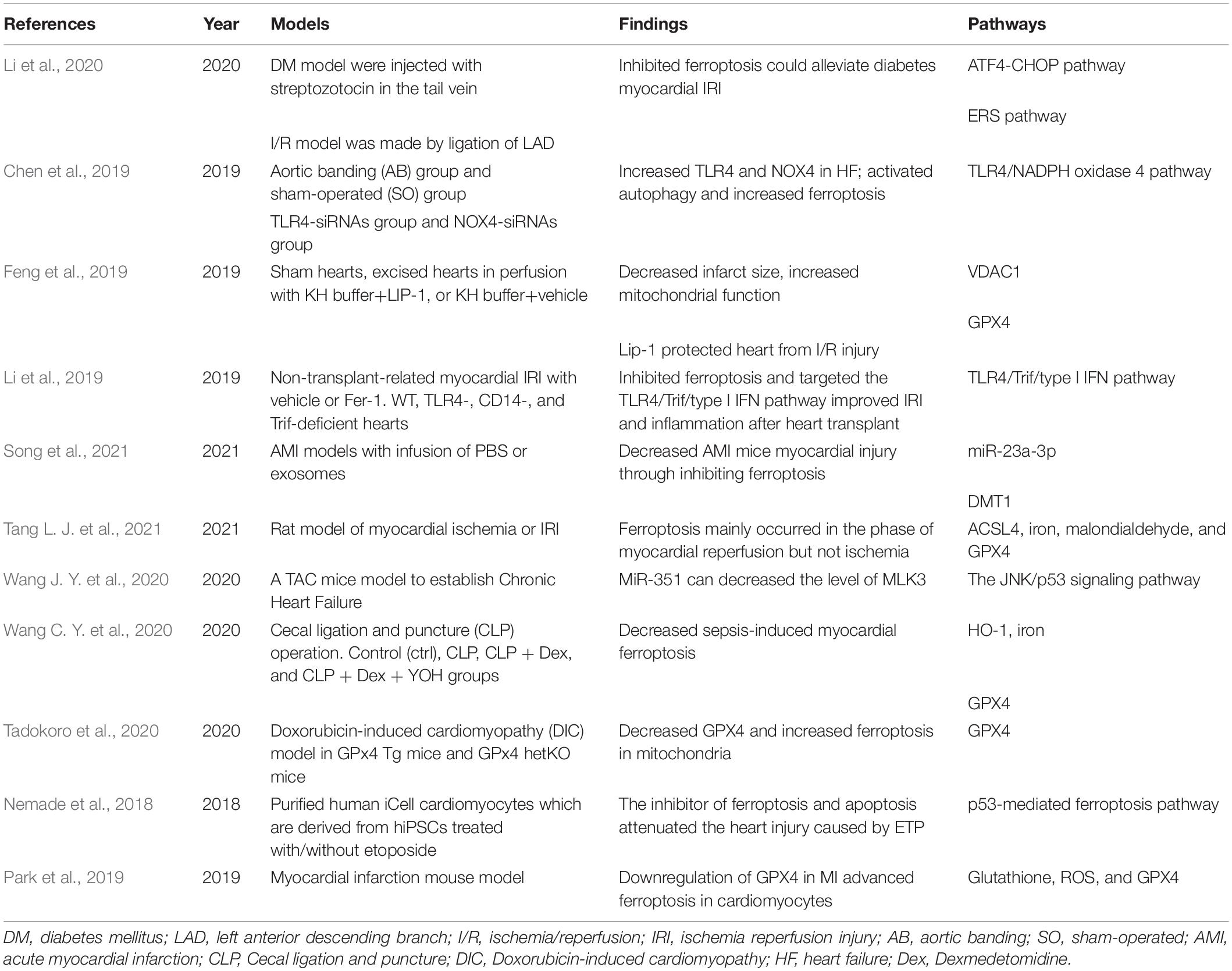
Table 1. Existing studies suggesting the roles of ferroptosis in cardiovascular diseases.
With the development of international research on ferroptosis, various types of iron death inducers and inhibitors were invented, but the specific mechanisms remain unknown. Myocardium iron overload is detected in mice I/R model, and the treatment of ferroptosis inhibitors can greatly improve cardiac function after I/R (Bulluck et al., 2016; Fang et al., 2019). Proteomic studies found that the down-regulation of myocardial GPX4 expression was detected in the early-stage (1 day) and mid-term (1 week) of myocardial infarction in mice, and inhibition of GPX4 expression or function in an in vitro model can significantly increase ferroptosis of myocardial cells (Park et al., 2019). Park et al. (2019) revealed that ROS and GPX4 is downregulated in the progression of MI regarding the involvement of the glutathione metabolic pathway. Li et al. found that severe myocardial damage is observed in DM rat with I/R and cell in high-glucose reoxygenation. Ferrostatin-1, the inhibitor of ferroptosis, reduces the endoplasmic reticulum stress (ERS) and myocardial injury in diabetes mellitus (DM) rats with I/R, whereas erastin shows the opposite effect (Li et al., 2020). Ferroptosis is thought to contribute in the progression of heart failure. Chen et al. (2019) found that toll-like receptor 4 (TLR4) and NADPH oxidase 4 (NOX4) were up-regulated and differentially expressed genes (DEGs) in myocardium resulting from heart failure. The HF rats with knock-down of TLR4 and NOX4 by lentivirus siRNA were detected with attenuated autophagy and ferroptosis, improved heart function, and decreased death of myocytes. Friedmann Angeli et al. (2014) showed that liprostatin-1 suppresses ferroptosis in human cells. Feng et al. (2019) provided evidence suggesting that Lip-1 reduced the size of MI and preserved the mitochondrial function via the I/R model reperfused with Lip-1, further study suggested that Lip-1 treatment reduced VDAC1 level and oligomerization, increased antioxidant GPX4 protein level and decreased mitochondrial ROS production. Tang et al. concluded that ferroptosis participates in the phase of reperfusion rather than ischemia (Tang L. J. et al., 2021). The addition of ferrostatin-1 leads to reduced size of MI, and it improved systolic function. It is proposed that ferroptosis and the TLR4/Trif/type I IFN signaling pathway initiate the inflammation which is involved in the adhesion of neutrophils and endothelium after cardiac transplantation (Li et al., 2019). Human umbilical cord blood (HUCB) mesenchymal stem cells (MSC)-derived exosomes inhibited ferroptosis, and exhibited cardioprotective effects on myocardial injury of acute myocardial injury mice, which may be related to the reduced divalent metal transporter 1 (DMT1) expression caused by miR-23a-3p (Song et al., 2021). Mixed lineage kinase 3 (MLK3) regulates oxidative stress through the JNK/p53 signaling pathway, inducing ferroptosis in the pathophysiologic process of myocardial fibrosis under pressure overload (Wang J. Y. et al., 2020).
Research conducted by Wang C. Y. et al. (2020) found that dexmedetomidine can promote sepsis-related myocardial ferroptosis and heart injury, acting through the decline of Ho-1 overexpression, iron levels, and GPX4 activity. Doxorubicin (DOX) is a traditional anthracycline chemotherapeutic with dose-dependent cardiac toxicity. In a study conducted by Tadokoro et al., DOX induced ferroptosis via downregulation of GPX4 and lipid peroxidation in mitochondria (Tadokoro et al., 2020). The other anti-cancer drug, etoposide (ETP), also causes cardiotoxicity. Human pluripotent stem cell-derived cardiomyocytes (hPSC-CMs) treated with liproxstatin-1 had increased function after the addition of ETP. The activation of the p53-mediated ferroptosis pathway by ETP is the key toward ETP-induced cardiotoxicity (Nemade et al., 2018). In summary, ferroptosis can be a target for protection against many CVDs, such as autotaxin (ATX), ferritin H, rapamycin, apart from ferroptosis inhibitors, such as ferrostatin-1 and liproxstatin-1 (Baba et al., 2017; Bai et al., 2018; Fang et al., 2019, 2020).
In 1942, Waddington CH first proposed the name “epigenotype” and used the term “epigenetics” as the branch of biology emphasizing the relation between genes and their products (Waddington, 2012). Owing to the technological advances and new discoveries, the definition of epigenetics has evolved. Nowadays the most common definition is “the study of mitotically and/or meiotically heritable changes in gene function that cannot be explained by changes in DNA sequence” (Bonasio et al., 2010). One hallmark of epigenetics is the fixed nucleotide sequence (Goldberg et al., 2007). It is a new direction for the therapy of some related diseases. Epigenetics is a bridge that links genotype and phenotype. Currently, the epigenetic process can be clarified into DNA methylation, histone modification (including methylation, acetylation, phosphorylation, ubiquitination, and SUMOylation) and RNA-based mechanism [including long non-coding RNAs (lncRNAs) and microRNAs (miRNAs)] (Prasher et al., 2020).
Several studies have investigated the effect of some epigenetic molecules in ferroptosis in recent years. In Jiang et al.’s study, lymphocyte-specific helicase (LSH), a DNA methylation modifier, can interact with WDR76 to inhibit ferroptosis at the transcriptional level. WDR76 induce lipid metabolic gene and ferroptosis related gene expression in DNA methylation and histone modification via LSH and chromatin modification, the process is affected by lipid ROS and iron concentration (Tao et al., 2017). LncRNAs are made up of over 200 nucleotides but the ability to code protein is relatively low. Abnormal expression of LncRNAs have been shown to be associated with tumorigenesis. For example, LncRNAs participate in the pathophysiology of non-small cell lung cancers (NSCLC) by regulating ferroptosis (Wu et al., 2020). LncRNA function analysis showed that the ferroptosis pathway is associated with SLC7A11 which was downregulated in XAV939-treated NCI-H1299 cells, giving a potential therapeutic target for NSCLC (Yu et al., 2019). Different LncRNAs play different roles in ferroptosis. P53RRA activates the p53 pathway and influences gene transcription to promote ferroptosis, whereas LINC0336 decreases iron concentration and lipid ROS by interacting with ELAV-like RNA binding protein 1 (ELAVL1) to inhibit ferroptosis (Mao et al., 2018; Wang M. et al., 2019). Deubiquitinase is encoded by BRCA1-associated protein (BAP1). Several studies have revealed that BAP1 can inhibit ubiquitinated histone 2A (H2Aub) occupancy on the SLC7A11 promoter (Zhang et al., 2018). Experiments confirm that the downregulated SLC7A11 leads to cystine starvation and GSH depletion to block ferroptosis (Fan et al., 2018). Monoubiquitination of histone H2B on lysine 120 (H2Bub1) promotes the expression of SLC7A11 and regulates many metabolic-related genes, but the p53-USP7-H2Bub1 axis regulates ferroptosis (Wang Y. F. et al., 2019). Selenium-induced selenome gene augmentation can inhibit ferroptosis and protect neuronal cells at the epigenetic level (Alim et al., 2019). Research shows low DNA methylation and elevated levels of H3K4me3 and H3K27ac upstream of GPX4, indicating that high levels of GPX4 may be related with epigenetic regulation (Zhang et al., 2020). Wang et al. reported that overexpression of KDM3B (a histone H3 lysine 9 demethylase) led to decreased histone H3 lysine 9 methylation, but increased the expression of SLC7A11 with the transcription factor, ATF4 (Wang Y. S. et al., 2020).
There are also some epigenetic factors that affect oxidative stress response, but have not yet been proven to be directly related to ferroptosis, owing to the close contact between ferroptosis and oxidative stress, a brief introduction is made here. Experimental studies exploring epigenetic regulators of the oxidative stress response are shown in Table 2.
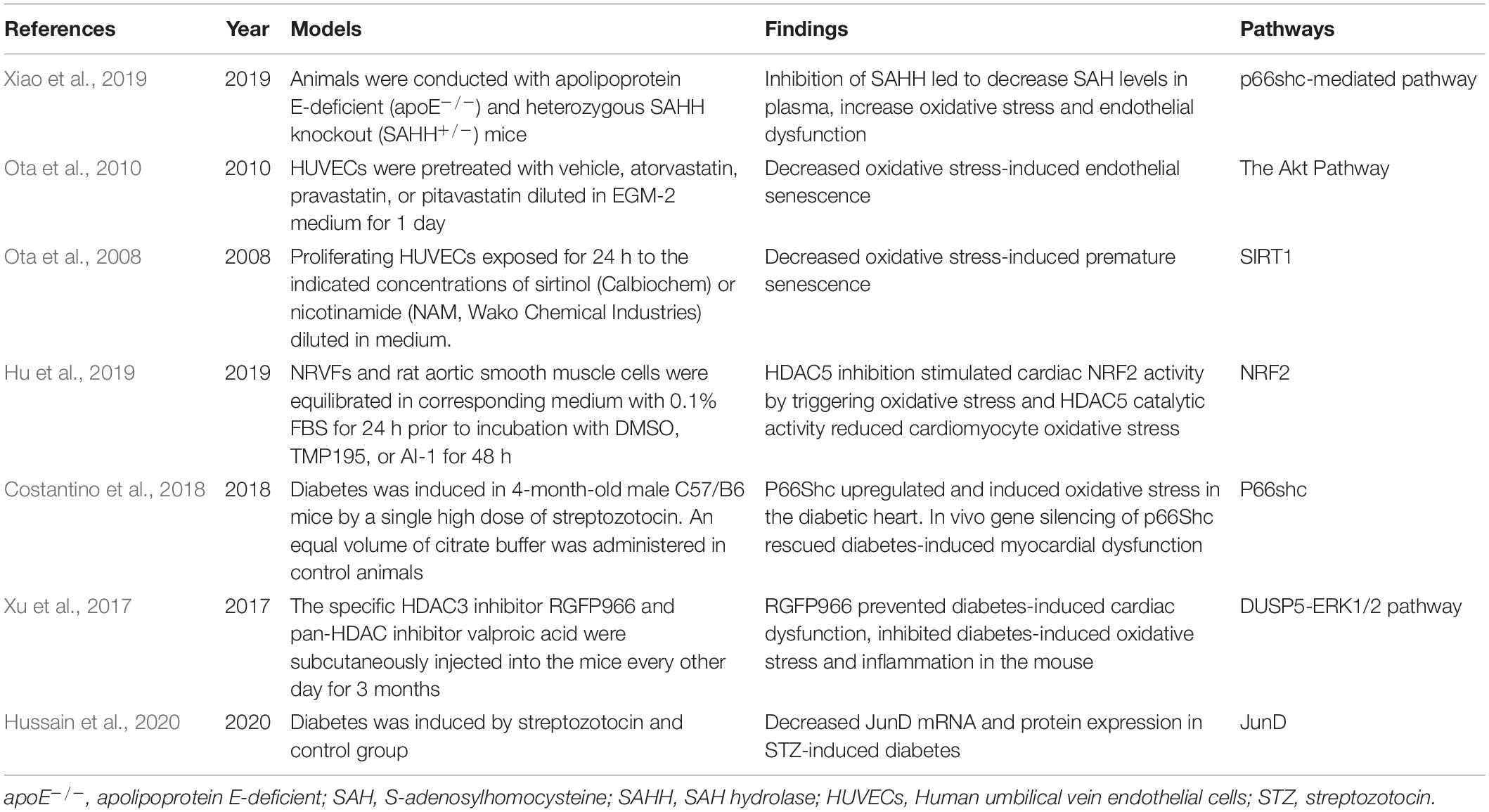
Table 2. Experiments on the epigenetic regulators of oxidative stress response.
Many studies regarding epigenetic regulation have been conducted in cancers, mental illnesses, and immune diseases in recent years. Epigenetic regulation also plays a strong part in CVDs. In a study conducted by Xiao et al. (2019), they found that S-adenosylhomocysteine (SAH) levels in plasma were positively correlated with oxidative stress, and were inversely correlated with flow-mediated dilation and methylation of p66shc promoter in CAD (coronary artery disease) patients and normal subjects. Further research indicates that inhibition of SAH hydrolase results in the increased level of SAH and oxidative stress by epigenetic regulation of p66shc expression, leading to the endothelium injury that may accelerate the progression of atherosclerosis (Xiao et al., 2019).
Nitric oxide is a fundamental molecule that can regulate vasodilatation and prevent vascular inflammation (Tsutsui et al., 2006). SIRT1, a class III histone deacetylase involved in the aging of mice fibroblasts, human ECs, and tumor cells (Ota et al., 2006), may be relevant to the production of ROS and oxidative stress (Hwang et al., 2013). Ota et al. conducted a series of studies on the effects of SIRT1 in ECs. The elevated level of NO strengthens the SIRT1 activity and delays endothelial senescence, but the accumulation of oxidative stress and decreased production of NO in aging will lead to SIRT1 inactivation (Ota et al., 2008). Cilostazol, a selective inhibitor of PDE3, protects ECs from ischemic damage by producing NO. Ota and his colleagues observed cells treated with H2O2 or Cilostazol, and evaluated the expression of senescence-associated beta-galactosidase assay (SA-betagal). They found that cilostazol increased phosphorylation of Akt at Ser473, as well as eNOS at Ser1177, but the phosphorylation increased SIRT1 expression in a dose-dependent manner (Ota et al., 2008). A similar experiment was conducted by Ota et al. to determine the mechanisms underlying the vascular protective effects of statins. Statins prevent the endothelium from aging by enhancing SIRT1 through the Akt pathway (Ota et al., 2010). Hu et al. found that HDAC5 catalytic activity inhibits cardiomyocyte oxidative stress via NRF2 stimulation. The selective class IIa HDAC inhibitors, TMP195 or TMP269, or shRNA-mediated knockdown of HDAC5 can lead to NRF2-mediated transcription (Hu et al., 2019).
In the diabetic heart, the expression of p66shc increases, but 3-week intensive glycemic control cannot reverse it. Further experiments, which silence the gene of p66shc in vivo, lead to the inhibition of ROS and promotion of cardiac function. Upregulation of miR-218 and miR-34a results in changes of the DNMT3b/SIRT1 axis in the diabetic heart, which may be a potential target to cure diabetic cardiomyopathy (Costantino et al., 2018). Another study, conducted by Hussain et al. (2020), found that JunD (a member of AP-1 transcript family) mRNA and protein are shown to have decreased expression in STZ-induced diabetes, which is relevant to oxidative stress, and is regulated by DNA hypermethylation, post-translational modification of histone markers, and translational inhibition by miRNA. Xu et al. treated diabetic mice with HDAC3 inhibitor, RGFP966, and found improved heart dysfunction, hypertrophy, fibrosis, and diminished oxidative stress. Furthermore, increased phosphorylated extracellular signal-regulated kinases (ERK) 1/2 and decreased dual specificity phosphatase 5 (DUSP5) were observed, but RGFP966 can reverse this. Elevated histone H3 acetylation plays an important role DUSP5 gene promoter in diabetic cardiomyopathy (Xu et al., 2017).
Over the past two decades, mounting efforts have been made to uncover new ways for cardiac repair, such as drug development (e.g., diuretics and ARNI), cardiac devices [e.g., pacemakers and implantable cardiac defibrillators (ICD)], and operations [e.g., electrical defibrillations and transcatheter aortic valve replacement (TAVR)]. In addition, the prognosis of CVD is not satisfied, and further investigation exploring fundamental mechanisms of impaired cardiomyocytes is needed. Epigenetic regulators provide a potential kind of therapy to treat CVDs, which will lay the foundation for individualized medical care.
Iron metabolism homeostasis is strictly regulated by multiple genes, including divalent metal transport-1 (DMT1), TFR1, TFR2, ferroportin (FPN), hepcidin (HAMP), hemojuvelin (HJV), and Ferritin H (Duan et al., 2020). Moreover, epigenetic regulators, such as DNA methylation, histone acetylation, and microRNA participate in iron metabolism homeostasis.
The therapies of CVDs targeting epigenetics are relatively rare. As mentioned above, SIRT1 expression has positive effects in many diseases, including cancer, CVDs, chronic obstructive pulmonary disease (COPD), and type 2 diabetes (Satoh et al., 2011). Resveratrol, a SIRT1 activator, has been suggested to improve heart function via vasodilation, antioxidant activity, and platelet aggregation (Baur and Sinclair, 2006). Resveratrol regulate the vasorelaxant activity through Ca2+-activated K+ channels (Li et al., 2000) and NO signaling in the endothelium (Orallo et al., 2002). Das et al. suggested resveratrol upregulates both endothelial and inducible NO synthase (eNOS and iNOS) (Das et al., 2005). Cilostazol protects ECs from ischemic injury by increasing SIRT1-dependent eNOS phosphorylation, producing substantial NO, and the inhibition of SIRT1 leads to inactivation of cilostazol on premature senescence (Ota et al., 2008). Currently, cilostazol is a common clinical drug aiming at ameliorating damage from ischemic injury. A novel SIRT activator, 1,4-dihydropyridine derivatives (DHPs), shows enhanced mitochondrial activity involving PGC-1α (Mai et al., 2009).
Noncoding RNAs are reportedly a potential target for therapeutics in CVDs (Lucas et al., 2018). In acute myocardial infarction, several families of miRNAs kick in, miR-34 can promote telomere erosion, and can regulate the target gene PNUTS (Bernardo et al., 2012; Boon et al., 2013), miR-24 target sirtuin 1, and can regulate EC apoptosis (Fiedler et al., 2011). Inhibition of miR-25 promotes heart function pertaining to the calcium uptake pump, SERCA2a (sarco/ER Ca2+-ATPase 2a) (Wahlquist et al., 2014). The related therapies include antisense oligonucleotides, siRNAs, antagomiRNAs, and antimiRNA application.
Yang K. C. et al. (2014) described the myocardial RNA sequence, suggested that the expression profiles of lncRNAs, but not mRNAs or miRNAs, can predict the different pathology of failing heart, indicating the important role of lncRNAs in CVDs. The lncRNA Mhrt (myosin heavy chain-associated RNA transcript) was found repressed under pathological stress condition such as pressure overload-induced hypertrophy and showed cardioprotective effects when restore the physiological concentration (Han et al., 2014). Piccoli et al. (2017) provided evidence suggesting that Inhibition of lncRNA Meg3, which is rich in cardiac fibroblasts, can prevent cardiac fibrosis and diastolic dysfunction. Mutations of the lncRNA H19 have been found related to coronary artery disease (Gao W. et al., 2015). The expression of H19 is highly reduced in atherosclerotic plaques or vascular injury, indicating the important role in cardiovascular system (Kim et al., 1994; Han et al., 1996). LncRNA therapeutics are also promising, in addition to inhibit lncRNA by antimiRs, the function of lncRNA can be blocked by shRNAs including siRNAs, modifed ASOs (antisense oligonucleotides), and gapmers (Lucas et al., 2018).
A growing number of experiments show evidence suggesting the involvement of epigenetics in cancer, CVDs, and metabolic diseases, which provides new ideas on the therapy of refractory diseases. Precision medicine and personalized therapy is the trend, as the development of medicine, genomics, and epigenetics will be the most important tools of the new generation of doctors.
Ferroptosis is a novel programmed cell death involving inhibition of enzyme GPX4 and lipid hydroperoxides, which was first widely studied in oncology. Oxidative stress-induced ferroptosis have been found to be extensively involved in the biogenesis and development of CVDs, and the inducers, the inhibitors, and the pathways of ferroptosis have been widely explored. Nowadays, the understanding of the role of epigenetics in ferroptosis have greatly increased. However, epigenetic mechanisms, such as lncRNAs, histone monoubiquitination, and DNA methylation are showed to engage in the ferroptosis process involved with CVDs.
Research on epigenetic drugs for CVDs has made great achievements, such as resveratrol, cilostazol, and miRNA family, which reveal the potential of epigenetic therapy for CVDs. However, the current epigenetic molecular mechanism of ferroptosis and the study of cardiac ferroptosis still need to be studied in depth. Thorough research in both basic research and clinical studies, are necessary to fully elucidate the relationship between ferroptosis and epigenetics in CVDs. Some biology approaches including total RNA-sequencing (RNA-seq), single cell RNA-seq, chromatin-immunoprecipitation-sequencing (ChIP-seq), and DNA methylation profiling can help us to further explore the epigenetic regulation with ferroptosis (Xu et al., 2018). And modern biotechnologies such as CRISPR/Cas9, Cre-loxp, proteomics, metabolism omics can comprehensively study the specific mechanisms of epigenetic regulation of ferroptosis for different genes and different stages of iron homeostasis. Hopefully, therapy against epigenetic targets will be promising for treating CVDs in the future.
YZ and JLi participated in the design of the review. JLi, YZ, HW, and JLo drafted the manuscript and made the original figures. HC, CL, and YD critically revised the texts and the figures. AS, HC, and YD supervised the research and led the discussion. All authors read and approved the final manuscript.
This work was funded by the National Natural Science Foundation of China (81701144).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ait-Oufella, H., Salomon, B. L., Potteaux, S., Robertson, A. K., Gourdy, P., Zoll, J., et al. (2006). Natural regulatory T cells control the development of atherosclerosis in mice. Nat. Med. 12, 178–180. doi: 10.1038/nm1343
Alim, I., Caulfield, J. T., Chen, Y. X., Swarup, V., Geschwind, D. H., Ivanova, E., et al. (2019). Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell 177, 1262.e–1279.e. doi: 10.1016/j.cell.2019.03.032
Angeli, J. P. F., Schneider, M., Proneth, B., Tyurina, Y. Y., Tyurin, V. A., Hammond, V. J., et al. (2014). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 16, 1180–1191. doi: 10.1038/ncb3064
Baba, Y., Higa, J. K., Shimada, B. K., Horiuchi, K. M., Suhara, T., Kobayashi, M., et al. (2017). Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 314, H659–H668. doi: 10.1152/ajpheart.00452.2017
Bai, Y. T., Chang, R., Wang, H., Xiao, F. J., Ge, R. L., and Wang, L. S. (2018). ENPP2 protects cardiomyocytes from erastin-induced ferroptosis. Biochem. Biophys. Res. Commun 499, 44–51. doi: 10.1016/j.bbrc.2018.03.113
Baines, C. P., Kaiser, R. A., Purcell, N. H., Blair, N. S., Osinska, H., Hambleton, M. A., et al. (2005). Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434, 658–662. doi: 10.1038/nature03434
Baur, J. A., and Sinclair, D. A. (2006). Therapeutic potential of resveratrol: the in vivo evidence. Nat. Rev. Drug Discov. 5, 493–506. doi: 10.1038/nrd2060
Berliner, J. A., Leitinger, N., and Tsimikas, S. (2009). The role of oxidized phospholipids in atherosclerosis. J. Lipid Res. 50(Suppl.), S207–S212. doi: 10.1194/jlr.R800074-JLR200
Bernardo, B. C., Gao, X. M., Winbanks, C. E., Boey, E. J., Tham, Y. K., Kiriazis, H., et al. (2012). Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc. Natl. Acad. Sci. U.S.A. 109, 17615–17620. doi: 10.1073/pnas.1206432109
Bersuker, K., Hendricks, J. M., Li, Z., Magtanong, L., Ford, B., Tang, P. H., et al. (2019). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692. doi: 10.1038/s41586-019-1705-2
Bertero, E., and Maack, C. (2018). Metabolic remodelling in heart failure. Nat. Rev. Cardiol. 15, 457–470. doi: 10.1038/s41569-018-0044-6
Birnbaum, Y., Hale, S. L., and Kloner, R. A. (1996). The effect of coenzyme Q10 on infarct size in a rabbit model of ischemia/reperfusion. Cardiovasc. Res. 32, 861–868.
Bonasio, R., Tu, S., and Reinberg, D. (2010). Molecular signals of epigenetic states. Science 330, 612–616. doi: 10.1126/science.1191078
Boon, R. A., Iekushi, K., Lechner, S., Seeger, T., Fischer, A., Heydt, S., et al. (2013). MicroRNA-34a regulates cardiac ageing and function. Nature 495, 107–110. doi: 10.1038/nature11919
Borrelli, E., Nestler, E. J., Allis, C. D., Murakami, K., Hayashidani, S., Ikeuchi, M., et al. (2008). Decoding the epigenetic language of neuronal plasticity. Neuron 60, 961–974. doi: 10.1016/j.neuron.2008.10.012
Brito, R., Castillo, G., Gonzalez, J., Valls, N., and Rodrigo, R. (2015). Oxidative stress in hypertension: mechanisms and therapeutic opportunities. Exp. Clin. Endocrinol. Diabetes 123, 325–335. doi: 10.1055/s-0035-1548765
Bulluck, H., Rosmini, S., Abdel-Gadir, A., White, S. K., Bhuva, A. N., Treibel, T. A., et al. (2016). Residual myocardial iron following intramyocardial hemorrhage during the convalescent phase of reperfused ST-segment-elevation myocardial infarction and adverse left ventricular remodeling. Circ. Cardiovasc. Imaging 9:e004940. doi: 10.1161/CIRCIMAGING.116.004940
Chatzizisis, Y. S., Coskun, A. U., Jonas, M., Edelman, E. R., Feldman, C. L., and Stone, P. H. (2007). Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling: molecular, cellular, and vascular behavior. J. Am. Coll. Cardiol. 49, 2379–2393. doi: 10.1016/j.jacc.2007.02.059
Chen, X., Li, J., Kang, R., Klionsky, D. J., and Tang, D. (2020). Ferroptosis: machinery and regulation. Autophagy 1–28. doi: 10.1080/15548627.2020.1810918
Chen, X. Q., Xu, S. D., Zhao, C. X., and Liu, B. (2019). Role of TLR4/NADPH oxidase 4 pathway in promoting cell death through autophagy and ferroptosis during heart failure. Biochem. Biophys. Res. Commun. 516, 37–43. doi: 10.1016/j.bbrc.2019.06.015
Costantino, S., Paneni, F., Mitchell, K., Mohammed, S. A., Hussain, S., Gkolfos, C., et al. (2018). Hyperglycaemia-induced epigenetic changes drive persistent cardiac dysfunction via the adaptor p66(Shc). Int. J. Cardiol. 268, 179–186. doi: 10.1016/j.ijcard.2018.04.082
Dabkowski, E. R., Williamson, C. L., and Hollander, J. M. (2008). Mitochondria-specific transgenic overexpression of phospholipid hydroperoxide glutathione peroxidase (GPx4) attenuates ischemia/reperfusion-associated cardiac dysfunction. Free Radic. Biol. Med. 45, 855–865. doi: 10.1016/j.freeradbiomed.2008.06.021
Dai, D. F., Johnson, S. C., Villarin, J. J., Chin, M. T., Nieves-Cintrón, M., Chen, T., et al. (2011). Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ. Res. 108, 837–846. doi: 10.1161/CIRCRESAHA.110.232306
Das, S., Alagappan, V. K., Bagchi, D., Sharma, H. S., Maulik, N., and Das, D. K. (2005). Coordinated induction of iNOS-VEGF-KDR-eNOS after resveratrol consumption: a potential mechanism for resveratrol preconditioning of the heart. Vascul. Pharmacol. 42, 281–289. doi: 10.1016/j.vph.2005.02.013
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. doi: 10.1016/j.cell.2012.03.042
Doll, S., Freitas, F. P., Shah, R., Aldrovandi, M., da Silva, M. C., Ingold, I., et al. (2019). FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698. doi: 10.1038/s41586-019-1707-0
Dolma, S., Lessnick, S. L., Hahn, W. C., and Stockwell, B. R. (2003). Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 3, 285–296. doi: 10.1016/S1535-6108(03)00050-3
Dougados, M., Soubrier, M., Antunez, A., Balint, P., Balsa, A., Buch, M. H., et al. (2014). Prevalence of comorbidities in rheumatoid arthritis and evaluation of their monitoring: results of an international, cross-sectional study (COMORA). Ann. Rheum. Dis. 73, 62–68. doi: 10.1136/annrheumdis-2013-204223
Duan, L., Yin, X., Meng, H., Fang, X., Min, J., and Wang, F. (2020). [Progress on epigenetic regulation of iron homeostasis]. Zhejiang Da Xue Xue Bao Yi Xue Ban 49, 58–70.
Dudley, S. C., Hoch, N. E., McCann, L. A., Honeycutt, C., Diamandopoulos, L., Fukai, T., et al. (2005). Atrial fibrillation increases production of superoxide by the left atrium and left atrial appendage–role of the NADPH and xanthine oxidases. Circulation 112, 1266–1273. doi: 10.1161/Circulationaha.105.538108
Fan, L. H., Yin, S. T., Zhang, E. X., and Hu, H. (2018). Role of p62 in the regulation of cell death induction. Apoptosis 23, 187–193. doi: 10.1007/s10495-018-1445-z
Fang, X., Wang, H., Han, D., Xie, E., Yang, X., Wei, J., et al. (2019). Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. U.S.A. 116, 2672–2680. doi: 10.1073/pnas.1821022116
Fang, X. X., Cai, Z. X., Wang, H., Han, D., Cheng, Q., Zhang, P., et al. (2020). Loss of cardiac ferritin H facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circ. Res. 127, 486–501. doi: 10.1161/Circresaha.120.316509
Feng, Y. S., Madungwe, N. B., Aliagan, A. D. I., Tombo, N., and Bopassa, J. G. (2019). Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem. Biophys. Res. Commun. 520, 606–611. doi: 10.1016/j.bbrc.2019.10.006
Fiedler, J., Jazbutyte, V., Kirchmaier, B. C., Gupta, S. K., Lorenzen, J., Hartmann, D., et al. (2011). MicroRNA-24 regulates vascularity after myocardial infarction. Circulation 124, 720–730. doi: 10.1161/CIRCULATIONAHA.111.039008
Friedmann Angeli, J. P., Schneider, M., Proneth, B., Tyurina, Y. Y., Tyurin, V. A., Hammond, V. J., et al. (2014). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180–1191. doi: 10.1038/ncb3064
Gao, H., Bai, Y. S., Jia, Y. Y., Zhao, Y., Kang, R., Tang, D., et al. (2018). Ferroptosis is a lysosomal cell death process. Biochem. Biophys. Res. Commun. 503, 1550–1556. doi: 10.1016/j.bbrc.2018.07.078
Gao, M., Monian, P., Quadri, N., Ramasamy, R., and Jiang, X. (2015). Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell 59, 298–308. doi: 10.1016/j.molcel.2015.06.011
Gao, M., Yi, J., Zhu, J., Minikes, A. M., Monian, P., Thompson, C. B., et al. (2019). Role of mitochondria in ferroptosis. Mol. Cell 73, 354.e3–363.e3. doi: 10.1016/j.molcel.2018.10.042
Gao, W., Zhu, M., Wang, H., Zhao, S., Zhao, D., Yang, Y., et al. (2015). Association of polymorphisms in long non-coding RNA H19 with coronary artery disease risk in a Chinese population. Mutat. Res. 772, 15–22. doi: 10.1016/j.mrfmmm.2014.12.009
Goldberg, A. D., Allis, C. D., and Bernstein, E. (2007). Epigenetics: a landscape takes shape. Cell 128, 635–638. doi: 10.1016/j.cell.2007.02.006
Golia, E., Limongelli, G., Natale, F., Fimiani, F., Maddaloni, V., Pariggiano, I., et al. (2014). Inflammation and cardiovascular disease: from pathogenesis to therapeutic target. Curr. Atheroscler. Rep. 16:435. doi: 10.1007/s11883-014-0435-z
Hak, A. E., Karlson, E. W., Feskanich, D., Stampfer, M. J., and Costenbader, K. H. (2009). Systemic lupus erythematosus and the risk of cardiovascular disease: results from the nurses’ health study. Arthritis Rheum. 61, 1396–1402. doi: 10.1002/art.24537
Han, D. K., Khaing, Z. Z., Pollock, R. A., Haudenschild, C. C., and Liau, G. (1996). H19, a marker of developmental transition, is reexpressed in human atherosclerotic plaques and is regulated by the insulin family of growth factors in cultured rabbit smooth muscle cells. J. Clin. Invest. 97, 1276–1285. doi: 10.1172/JCI118543
Han, P., Li, W., Lin, C. H., Yang, J., Shang, C., Nuernberg, S. T., et al. (2014). A long noncoding RNA protects the heart from pathological hypertrophy. Nature 514, 102–106. doi: 10.1038/nature13596
Hansson, G. K., Robertson, A. K. L., and Soderberg-Naucler, C. (2006). Inflammation and atherosclerosis. Annu. Rev. Pathol. Mech. 1, 297–329. doi: 10.1146/annurev.pathol.1.110304.100100
Hassan, W., Noreen, H., Khalil, S. U., Hussain, A., Rehman, S., Sajjad, S., et al. (2016). Ethanolic extract of Nigella sativa protects Fe(II) induced lipid peroxidation in rat’s brain, kidney and liver homogenates. Pak. J. Pharm. Sci. 29, 231–237.
Hu, T. J., Schreiter, F. C., Bagchi, R. A., Tatman, P. D., Hannink, M., and McKinsey, T. A. (2019). HDAC5 catalytic activity suppresses cardiomyocyte oxidative stress and NRF2 target gene expression. J. Biol. Chem. 294, 8640–8652. doi: 10.1074/jbc.RA118.007006
Hussain, S., Khan, A. W., Akhmedov, A., Suades, R., Costantino, S., Paneni, F., et al. (2020). Hyperglycemia induces myocardial dysfunction via epigenetic regulation of JunD. Circ. Res. 127, 1261–1273. doi: 10.1161/Circresaha.120.317132
Hwang, J. W., Yao, H. W., Caito, S., Sundar, I. K., and Rahman, I. (2013). Redox regulation of SIRT1 in inflammation and cellular senescence. Free Radic. Biol. Med. 61, 95–110. doi: 10.1016/j.freeradbiomed.2013.03.015
Judkins, C. P., Diep, H., Broughton, B. R. S., Mast, A. E., Hooker, E. U., Miller, A. A., et al. (2010). Direct evidence of a role for Nox2 in superoxide production, reduced nitric oxide bioavailability, and early atherosclerotic plaque formation in ApoE(-/-) mice. Am. J. Physiol. Heart Circ. Physiol. 298, H24–H32. doi: 10.1152/ajpheart.00799.2009
Kanamori, H., Takemura, G., Goto, K., Maruyama, R., Ono, K., Nagao, K., et al. (2011). Autophagy limits acute myocardial infarction induced by permanent coronary artery occlusion. Am. J. Physiol. Heart Circ. Physiol. 300, H2261–H2271. doi: 10.1152/ajpheart.01056.2010
Kim, D. K., Zhang, L., Dzau, V. J., and Pratt, R. E. (1994). H19, a developmentally regulated gene, is reexpressed in rat vascular smooth muscle cells after injury. J. Clin. Invest. 93, 355–360. doi: 10.1172/JCI116967
Kiss, E., Soltesz, P., Der, H., Kocsis, Z., Tarr, T., Bhattoa, H., et al. (2006). Reduced flow-mediated vasodilation as a marker for cardiovascular complications in lupus patients. J. Autoimmun. 27, 211–217. doi: 10.1016/j.jaut.2006.09.008
Kraft, V. A. N., Bezjian, C. T., Pfeiffer, S., Ringelstetter, L., Müller, C., Zandkarimi, F., et al. (2020). GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Central Sci. 6, 41–53. doi: 10.1021/acscentsci.9b01063
Kuwana, T., Mackey, M. R., Perkins, G., Ellisman, M. H., Latterich, M., Schneiter, R., et al. (2002). Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111, 331–342. doi: 10.1016/s0092-8674(02)01036-x
Leonardi, C., Matheson, R., Zachariae, C., Cameron, G., Li, L., Edson-Heredia, E., et al. (2012). Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N. Engl. J. Med. 366, 1190–1199. doi: 10.1056/NEJMoa1109997
Lewerenz, J., Hewett, S. J., Huang, Y., Lambros, M., Gout, P. W., Kalivas, P. W., et al. (2013). The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 18, 522–555. doi: 10.1089/ars.2011.4391
Li, H. F., Chen, S. A., and Wu, S. N. (2000). Evidence for the stimulatory effect of resveratrol on Ca(2+)-activated K+ current in vascular endothelial cells. Cardiovasc. Res. 45, 1035–1045. doi: 10.1016/s0008-6363(99)00397-1
Li, W., Feng, G., Gauthier, J. M., Lokshina, I., Higashikubo, R., Evans, S., et al. (2019). Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J. Clin. Invest. 129, 2293–2304. doi: 10.1172/JCI126428
Li, W. Y., Li, W., Leng, Y., Xiong, Y., and Xia, Z. (2020). Ferroptosis is involved in diabetes myocardial ischemia/reperfusion injury through endoplasmic reticulum stress. DNA Cell Biol. 39, 210–225. doi: 10.1089/dna.2019.5097
Liang, H. Y., Yoo, S. E., Na, R., Walter, C. A., Richardson, A., and Ran, Q. (2009). Short form glutathione peroxidase 4 is the essential isoform required for survival and somatic mitochondrial functions. J. Biol. Chem. 284, 30836–30844. doi: 10.1074/jbc.M109.032839
Libby, P., and Ridker, P. M. (2006). Inflammation and atherothrombosis–from population biology and bench research to clinical practice. J. Am. Coll. Cardiol. 48, A33–A46. doi: 10.1016/j.jacc.2006.08.011
Liuzzo, G., Kopecky, S. L., Frye, R. L., O’Fallon, W. M., Maseri, A., Goronzy, J. J., et al. (1999). Perturbation of the T-cell repertoire in patients with unstable angina. Circulation 100, 2135–2139. doi: 10.1161/01.cir.100.21.2135
Lucas, T., Bonauer, A., and Dimmeler, S. (2018). RNA therapeutics in cardiovascular disease. Circ. Res. 123, 205–220. doi: 10.1161/CIRCRESAHA.117.311311
Mai, A., Valente, S., Meade, S., Carafa, V., Tardugno, M., Nebbioso, A., et al. (2009). Study of 1,4-dihydropyridine structural scaffold: discovery of novel sirtuin activators and inhibitors. J. Med. Chem. 52, 5496–5504. doi: 10.1021/jm9008289
Mao, C., Wang, X., Liu, Y. T., Wang, M., Yan, B., Jiang, Y., et al. (2018). A G3BP1-interacting lncRNA promotes ferroptosis and apoptosis in cancer via nuclear sequestration of p53. Cancer Res. 78, 3484–3496. doi: 10.1158/0008-5472.Can-17-3454
Matsui, Y., Takagi, H., Qu, X. P., Abdellatif, M., Sakoda, H., Asano, T., et al. (2007). Distinct roles of autophagy in the heart during ischemia and reperfusion–roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 100, 914–922. doi: 10.1161/01.Res.0000261924.76669.36
Moriya, J. (2019). Critical roles of inflammation in atherosclerosis. J. Cardiol. 73, 22–27. doi: 10.1016/j.jjcc.2018.05.010
Nagoshi, T., Yoshimura, M., Rosano, G. M., Lopaschuk, G. D., and Mochizuki, S. (2011). Optimization of cardiac metabolism in heart failure. Curr. Pharm. Des. 17, 3846–3853. doi: 10.2174/138161211798357773
Nemade, H., Chaudhari, U., Acharya, A., Hescheler, J., Hengstler, J. G., Papadopoulos, S., et al. (2018). Cell death mechanisms of the anti-cancer drug etoposide on human cardiomyocytes isolated from pluripotent stem cells. Arch. Toxicol. 92, 1507–1524. doi: 10.1007/s00204-018-2170-7
Nishida, M., Maruyama, Y., Tanaka, R., Kontani, K., Nagao, T., and Kurose, H. (2000). G alpha(i) and G alpha(o) are target proteins of reactive oxygen species. Nature 408, 492–495. doi: 10.1038/35044120
Nishida, M., Schey, K. L., Takagahara, S., Kontani, K., Katada, T., Urano, Y., et al. (2002). Activation mechanism of Gi and Go by reactive oxygen species. J. Biol. Chem. 277, 9036–9042. doi: 10.1074/jbc.M107392200
Ohara, Y., Peterson, T. E., and Harrison, D. G. (1993). hypercholesterolemia increases endothelial superoxide anion production. J. Clin. Invest. 91, 2546–2551. doi: 10.1172/Jci116491
Ooko, E., Saeed, M. E. M., Kadioglu, O., Sarvi, S., Colak, M., Elmasaoudi, K., et al. (2015). Artemisinin derivatives induce iron-dependent cell death (ferroptosis) in tumor cells. Phytomedicine 22, 1045–1054. doi: 10.1016/j.phymed.2015.08.002
Orallo, F., Alvarez, E., Camina, M., Leiro, J. M., Gómez, E., and Fernández, P. (2002). The possible implication of trans-resveratrol in the cardioprotective effects of long-term moderate wine consumption. Mol. Pharmacol. 61, 294–302. doi: 10.1124/mol.61.2.294
Ota, H., Eto, M., Kano, M. R., Kahyo, T., Setou, M., Ogawa, S., et al. (2010). Induction of endothelial nitric oxide synthase, SIRT1, and catalase by statins inhibits endothelial senescence through the Akt pathway. Arterioscler. Thromb. Vasc. Biol. 30, 2205–2211. doi: 10.1161/Atvbaha.110.210500
Ota, H., Eto, M., Kano, M. R., Ogawa, S., Iijima, K., Akishita, M., et al. (2008). Cilostazol inhibits oxidative stress-induced premature senescence via upregulation of Sirt1 in human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 28, 1634–1639. doi: 10.1161/Atvbaha.108.164368
Ota, H., Tokunaga, E., Chang, K., Hikasa, M., Iijima, K., Eto, M., et al. (2006). Sirt1 inhibitor, sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene 25, 176–185. doi: 10.1038/sj.onc.1209049
Papp, K. A., Leonardi, C., Menter, A., Ortonne, J. P., Krueger, J. G., Kricorian, G., et al. (2012). Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N. Engl. J. Med. 366, 1181–1189. doi: 10.1056/NEJMoa1109017
Park, T. J., Park, J. H., Lee, G. S., Lee, J.-Y., Shin, J. H., Kim, M. W., et al. (2019). Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. 10:835. doi: 10.1038/s41419-019-2061-8
Pasceri, V., and Yeh, E. T. (1999). A tale of two diseases: atherosclerosis and rheumatoid arthritis. Circulation 100, 2124–2126. doi: 10.1161/01.cir.100.21.2124
Piccoli, M. T., Gupta, S. K., Viereck, J., Foinquinos, A., Samolovac, S., Kramer, F. L., et al. (2017). Inhibition of the cardiac fibroblast-enriched lncRNA Meg3 prevents cardiac fibrosis and diastolic dysfunction. Circ. Res. 121, 575–583. doi: 10.1161/CIRCRESAHA.117.310624
Pike, M. M., Luo, C. S., Clark, M. D., Kirk, K. A., Kitakaze, M., Madden, M. C., et al. (1993). NMR measurements of Na+ and cellular energy in ischemic rat heart: role of Na(+)-H+ exchange. Am. J. Physiol. 265(6 Pt 2), H2017–H2026. doi: 10.1152/ajpheart.1993.265.6.H2017
Prasher, D., Greenway, S. C., and Singh, R. B. (2020). The impact of epigenetics on cardiovascular disease. Biochem. Cell Biol. 98, 12–22. doi: 10.1139/bcb-2019-0045
Randle, P. J. (1998). Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes Metab. Rev. 14, 263–283. doi: 10.1002/(Sici)1099-0895(199812)14:4<263::Aid-Dmr233<3.0.Co;2-C
Randle, P. J., Garland, P. B., Hales, C. N., and Newsholme, E. A. (1963). The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1, 785–789. doi: 10.1016/s0140-6736(63)91500-9
Ridker, P. M., Cannon, C. P., Morrow, D., Rifai, N., Rose, L. M., McCabe, C. H., et al. (2005). C-reactive protein levels and outcomes after statin therapy. N. Engl. J. Med. 352, 20–28. doi: 10.1056/NEJMoa042378
Ridker, P. M., Rifai, N., Clearfield, M., Downs, J. R., Weis, S. E., Miles, J. S., et al. (2001). Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N. Engl. J. Med. 344, 1959–1965. doi: 10.1056/NEJM200106283442601
Ross, R. (1999). Atherosclerosis–an inflammatory disease. N. Engl. J. Med. 340, 115–126. doi: 10.1056/NEJM199901143400207
Russell, P., Garland, D., and Epstein, D. L. (1989). Analysis of the proteins of calf and cow trabecular meshwork: development of a model system to study aging effects and glaucoma. Exp. Eye Res. 48, 251–260. doi: 10.1016/s0014-4835(89)80074-0
Sanada, S., Komuro, I., and Kitakaze, M. (2011). Pathophysiology of myocardial reperfusion injury: preconditioning, postconditioning, and translational aspects of protective measures. Am. J. Physiol. Heart Circ. Physiol. 301, H1723–H1741. doi: 10.1152/ajpheart.00553.2011
Satoh, A., Stein, L., and Imai, S. (2011). The role of mammalian sirtuins in the regulation of metabolism, aging, and longevity. Handb. Exp. Pharmacol. 206, 125–162. doi: 10.1007/978-3-642-21631-2_7
Shepherd, J., Cobbe, S. M., Ford, I., Isles, C. G., Lorimer, A. R., MacFarlane, P. W., et al. (1995). Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland coronary prevention study group. N. Engl. J. Med. 333, 1301–1307. doi: 10.1056/NEJM199511163332001
Shiomi, T., Tsutsui, H., Matsusaka, H., Murakami, K., Hayashidani, S., Ikeuchi, M., et al. (2004). Overexpression of glutathione peroxidase prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation 109, 544–549. doi: 10.1161/01.CIR.0000109701.77059.E9
Song, Y. F., Wang, B. C., Zhu, X. L., Hu, J., Sun, J., Xuan, J., et al. (2021). Human umbilical cord blood-derived MSCs exosome attenuate myocardial injury by inhibiting ferroptosis in acute myocardial infarction mice. Cell Biol. Toxicol. 37, 51–64. doi: 10.1007/s10565-020-09530-8
Soula, M., Weber, R. A., Zilka, O., Alwaseem, H., La, K., Yen, F., et al. (2020). Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat. Chem. Biol. 16, 1351–1360. doi: 10.1038/s41589-020-0613-y
Stamenkovic, A., Pierce, G. N., and Ravandi, A. (2019). Phospholipid oxidation products in ferroptotic myocardial cell death. Am. J. Physiol. Heart Circ. Physiol. 317, H156–H163. doi: 10.1152/ajpheart.00076.2019
Sugden, M. C. (2007). In appreciation of Sir Philip Randle: the glucose-fatty acid cycle. Br. J. Nutr. 97, 809–813. doi: 10.1017/S0007114507659054
Tabas, I., Williams, K. J., and Boren, J. (2007). Subendothelial lipoprotein retention as the initiating process in atherosclerosis–update and therapeutic implications. Circulation 116, 1832–1844. doi: 10.1161/Circulationaha.106.676890
Tadokoro, T., Ikeda, M., Ide, T., Deguchi, H., Ikeda, S., Okabe, K., et al. (2020). Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 5:e132747. doi: 10.1172/jci.insight.132747
Tanai, E., and Frantz, S. (2016). Pathophysiology of heart failure. Compr. Physiol. 6, 187–214. doi: 10.1002/cphy.c140055
Tang, D., Chen, X., Kang, R., and Kroemer, G. (2021). Ferroptosis: molecular mechanisms and health implications. Cell Res. 31, 107–125. doi: 10.1038/s41422-020-00441-1
Tang, L. J., Luo, X. J., Tu, H., Chen, H., Xiong, X. M., Li, N. S., et al. (2021). Ferroptosis occurs in phase of reperfusion but not ischemia in rat heart following ischemia or ischemia/reperfusion. Naunyn. Schmiedebergs Arch. Pharmacol. 394, 401–410. doi: 10.1007/s00210-020-01932-z
Tao, Y. G., Liu, S., and Jiang, Y. Q. (2017). EGLN1/c-Myc induced lymphoid-specific helicase inhibits ferroptosis through lipid metabolic gene expression changes. Cancer Res. 77, 3293–3305. doi: 10.1158/1538-7445.Am2017-4317
The Institute for Health Metrics and Evaluation (IHME) (2018). GBD Compare | Viz Hub. Washington, DC: IHME.
Touyz, R. M. (2004). Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: what is the clinical significance? Hypertension 44, 248–252. doi: 10.1161/01.HYP.0000138070.47616.9d
Tsutsui, M., Shimokawa, H., Morishita, T., Nakashima, Y., and Yanagihara, N. (2006). Development of genetically engineered mice lacking all three nitric oxide synthases. J. Pharmacol. Sci. 102, 147–154. doi: 10.1254/jphs.CPJ06015X
Ussher, J. R., Elmariah, S., Gerszten, R. E., and Dyck, J. R. (2016). The emerging role of metabolomics in the diagnosis and prognosis of cardiovascular disease. J. Am. Coll. Cardiol. 68, 2850–2870. doi: 10.1016/j.jacc.2016.09.972
Vena, G. A., Vestita, M., and Cassano, N. (2010). Psoriasis and cardiovascular disease. Dermatol. Ther. 23, 144–151. doi: 10.1111/j.1529-8019.2010.01308.x
Waddington, C. H. (2012). The epigenotype. 1942. Int. J. Epidemiol. 41, 10–13. doi: 10.1093/ije/dyr184
Wahlquist, C., Jeong, D., Rojas-Munoz, A., Kho, C., Lee, A., Mitsuyama, S., et al. (2014). Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature 508, 531–535. doi: 10.1038/nature13073
Wang, C. Y., Yuan, W. L., Hu, A. M., Lin, J., Xia, Z., Yang, C. F., et al. (2020). Dexmedetomidine alleviated sepsis-induced myocardial ferroptosis and septic heart injury. Mol. Med. Rep. 22, 175–184. doi: 10.3892/mmr.2020.11114
Wang, D., He, Y. N., Li, Y. P., Luan, D., Zhai, F., Yang, X., et al. (2013). Joint association of dietary pattern and physical activity level with cardiovascular disease risk factors among Chinese Men: a cross-sectional study. PloS One 8:e66210. doi: 10.1371/journal.pone.0066210
Wang, J. Y., Deng, B., Liu, Q., Huang, Y., Chen, W., Li, J., et al. (2020). Pyroptosis and ferroptosis induced by mixed lineage kinase 3 (MLK3) signaling in cardiomyocytes are essential for myocardial fibrosis in response to pressure overload. Cell Death Dis. 11:574. doi: 10.1038/s41419-020-02777-3
Wang, M., Mao, C., Ouyang, L. L., Liu, Y., Lai, W., Liu, N., et al. (2019). Long noncoding RNA LINC00336 inhibits ferroptosis in lung cancer by functioning as a competing endogenous RNA. Cell Death Differ. 26, 2329–2343. doi: 10.1038/s41418-019-0304-y
Wang, Y. F., Yang, L., Zhang, X. J., Cui, W., Liu, Y., Sun, Q. R., et al. (2019). Epigenetic regulation of ferroptosis by H2B monoubiquitination and p53. EMBO Rep. 20:e47563. doi: 10.15252/embr.201847563
Wang, Y. S., Zhao, Y., Wang, H. H., Zhang, C., Wang, M., Yang, Y., et al. (2020). Histone demethylase KDM3B protects against ferroptosis by upregulating SLC7A11. FEBS Open Bio 10, 637–643. doi: 10.1002/2211-5463.12823
Williams, K. J., and Tabas, I. (1995). The response-to-retention hypothesis of early atherogenesis. Arterioscler. Thromb. Vasc. Biol. 15, 551–561. doi: 10.1161/01.atv.15.5.551
Wong, B. W., Meredith, A., Lin, D., and McManus, B. M. (2012). The biological role of inflammation in atherosclerosis. Can. J. Cardiol. 28, 631–641. doi: 10.1016/j.cjca.2012.06.023
World Health Organization (WHO) (2017). Cardiovascular Diseases (CVDs). Available online at: https://www.who.int/en/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed June 11, 2021).
Wu, Y. Q., Zhang, S. W., Gong, X. X., Tam, S., Xiao, D., Liu, S., et al. (2020). The epigenetic regulators and metabolic changes in ferroptosis-associated cancer progression. Mol. Cancer 19:39. doi: 10.1186/s12943-020-01157-x
Xiao, Y. J., Xia, J. J., Cheng, J. Q., Huang, H., Zhou, Y., Yang, X., et al. (2019). Inhibition of S-adenosylhomocysteine hydrolase induces endothelial dysfunction via epigenetic regulation of p66shc-mediated oxidative stress pathway. Circulation 139, 2260–2277. doi: 10.1161/Circulationaha.118.036336
Xie, Y., Hou, W., Song, X., Yu, Y., Huang, J., Sun, X., et al. (2016). Ferroptosis: process and function. Cell Death Differ. 23, 369–379. doi: 10.1038/cdd.2015.158
Xu, S., Pelisek, J., and Jin, Z. G. (2018). Atherosclerosis is an epigenetic disease. Trends Endocrinol. Metab. 29, 739–742. doi: 10.1016/j.tem.2018.04.007
Xu, Z., Tong, Q., Zhang, Z. G., Wang, S., Zheng, Y., Liu, Q., et al. (2017). Inhibition of HDAC3 prevents diabetic cardiomyopathy in OVE26 mice via epigenetic regulation of DUSP5-ERK1/2 pathway. Clin. Sci. 131, 1841–1857. doi: 10.1042/Cs20170064
Yancy, C. W., Jessup, M., Bozkurt, B., Butler, J., Casey, D. E. Jr., Colvin, M. M., et al. (2017). 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines and the heart failure society of America. J. Card. Fail. 23, 628–651. doi: 10.1016/j.cardfail.2017.04.014
Yang, K. C., Yamada, K. A., Patel, A. Y., Topkara, V. K., George, I., Cheema, F. H., et al. (2014). Deep RNA sequencing reveals dynamic regulation of myocardial noncoding RNAs in failing human heart and remodeling with mechanical circulatory support. Circulation 129, 1009–1021. doi: 10.1161/CIRCULATIONAHA.113.003863
Yang, W. S., SriRamaratnam, R., Welsch, M. E., Shimada, K., Skouta, R., Viswanathan, V. S., et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. doi: 10.1016/j.cell.2013.12.010
Yang, W. S., and Stockwell, B. R. (2008). Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 15, 234–245. doi: 10.1016/j.chembiol.2008.02.010
Yu, H. X., Han, Z. F., Xu, Z. A., An, C., Xu, L., and Xin, H. (2019). RNA sequencing uncovers the key long non-coding RNAs and potential molecular mechanism contributing to XAV939-mediated inhibition of non-small cell lung cancer. Oncol. Lett. 17, 4994–5004. doi: 10.3892/ol.2019.10191
Zhang, X. F., Sui, S. Y., Wang, L. L., Li, H., Zhang, L., Xu, S., et al. (2020). Inhibition of tumor propellant glutathione peroxidase 4 induces ferroptosis in cancer cells and enhances anticancer effect of cisplatin. J. Cell. Physiol. 235, 3425–3437. doi: 10.1002/jcp.29232
Keywords: iron, ferroptosis, oxidative stress, epigenetic regulators, cardiovascular diseases
Citation: Li J, Zhou Y, Wang H, Lou J, Lenahan C, Gao S, Wang X, Deng Y, Chen H and Shao A (2021) Oxidative Stress-Induced Ferroptosis in Cardiovascular Diseases and Epigenetic Mechanisms. Front. Cell Dev. Biol. 9:685775. doi: 10.3389/fcell.2021.685775
Received: 25 March 2021; Accepted: 04 August 2021;
Published: 19 August 2021.
Edited by:
Christoph D. Rau, University of North Carolina at Chapel Hill, United StatesReviewed by:
Douglas J. Chapski, University of California, Los Angeles, United StatesCopyright © 2021 Li, Zhou, Wang, Lou, Lenahan, Gao, Wang, Deng, Chen and Shao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongchuan Deng, ZHljMDAxQHpqdS5lZHUuY24=; Han Chen, Y2hlbmhhbnpqdUB6anUuZWR1LmNu; Anwen Shao, c2hhb2Fud2VuQHpqdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.