 Kai Murk
Kai Murk Marta Ornaghi
Marta Ornaghi Juliane Schiweck
Juliane Schiweck
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 12 August 2021
Sec. Cell Adhesion and Migration
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.681122
This article is part of the Research Topic Evolution, Emerging Functions and Structure of Actin-Binding Proteins View all 32 articles
Profilins are small actin binding proteins, which are structurally conserved throughout evolution. They are probably best known to promote and direct actin polymerization. However, they also participate in numerous cell biological processes beyond the roles typically ascribed to the actin cytoskeleton. Moreover, most complex organisms express several profilin isoforms. Their cellular functions are far from being understood, whereas a growing number of publications indicate that profilin isoforms are involved in the pathogenesis of various diseases. In this review, we will provide an overview of the profilin family and “typical” profilin properties including the control of actin dynamics. We will then discuss the profilin isoforms of higher animals in detail. In terms of cellular functions, we will focus on the role of Profilin 1 (PFN1) and Profilin 2a (PFN2a), which are co-expressed in the central nervous system. Finally, we will discuss recent findings that link PFN1 and PFN2a to neurological diseases, such as amyotrophic lateral sclerosis (ALS), Fragile X syndrome (FXS), Huntington’s disease and spinal muscular atrophy (SMA).
Over forty years ago, the first profilin was identified as a small actin monomer binding protein with the ability to inhibit actin polymerization in vitro (Carlsson et al., 1977). From there, the profilin field has expanded enormously: Originally described as actin sequestering protein, a wealth of literature established profilin as driving and directing force for actin polymerization and network homeostasis. Besides canonical actin dynamics, miscellaneous profilin ligands and numerous functional studies have linked profilin to diverse fields of cell biology including membrane trafficking, signaling, synaptic scaffolding, nuclear export, mRNA splicing, and transcription (see Supplementary Table 1). The (co-)expression of different profilin isoforms generates another level of complexity and raises the question as to whether these proteins are functionally unique or if they are redundant. Moreover, recent studies show the association of profilin isoforms with the onset and/or progression of several diseases through either mutation in their ligands or in the profilin isoforms themselves. In this review article, we begin by summarizing main aspects of profilin properties and functions. We introduce profilins with their typical structural and biochemical features including their function as actin regulators. Furthermore, we will discuss the functional diversity of profilin isoforms of higher animals in cellular processes. We will then focus on overlapping and unique roles of PFN1 and PFN2a in the central nervous system (CNS). Finally, we will give insight into the biomedical relevance of these profilin isoforms through their emerging roles in neurological diseases.
The identification of 172 paralogous and orthologous profilin genes in lower eukaryotes, fungi, plants and animals supports the conservation of profilins throughout evolution (Pandey and Chaudhary, 2017). Even cyanobacterial and viral profilin genes exist, which likely were hijacked from eukaryotic cells by horizontal gene transfer (Blasco et al., 1991; Guljamow et al., 2007). Most lower eukaryotes usually have one profilin gene (Pandey and Chaudhary, 2017). Exceptions to the rule are the free-living amoeba Dictyostelium discoideum and the slime mold Physarum polycephalum, which express, either constitutively or temporally, additional profilin isoforms (Binette et al., 1990; Arasada et al., 2007). Most studied multicellular organisms express several profilin isoforms some of which are generated by alternative splicing and/or are expressed in a tissue-specific manner. In silico analyses of profilin amino acid sequences from various origins show degrees of homologies down to less than 25% between profilins from lower eukaryotes and animals (Figure 1A). But also profilin isoforms in the same organism may substantially differ in their amino acid composition (Figure 1B; Polet et al., 2007). In contrast to this variability in amino acid sequences, the secondary and tertiary structures of profilins are conserved. Seven beta-strands form a compact core surrounded by four alpha-helices. N- and C-termini of profilins are part of alpha-helices and are positioned adjacent to each other (Cedergren-Zeppezauer et al., 1994; Mahoney et al., 1997; Haikarainen et al., 2009). A typical profilin protein contains functional binding sites for G-actin, poly-proline-motifs, and phosphoinositides: the G-actin binding site of profilin occupies a relatively large surface area involving residues of the beta-strands 4,5 and 6, the alpha-helices 3 and 4 as well as interspacing loop regions (Schutt et al., 1993). The interaction with profilin induces a conformational change in G-actin toward a wider nucleotide binding pocket and thereby largely accelerates the nucleotide exchange from ADP to ATP (Perelroizen et al., 1996; Selden et al., 1999). In addition, profilin uses the actin binding site to interact with two other ligands: the actin related protein 2 (Arp2), a subunit of the Arp2/3 complex (Mullins et al., 1998), and gephyrin, which is involved in cellular metabolism through its role in molybdenum cofactor synthesis, but also serves as a postsynaptic scaffolding protein at inhibitory synapses in neurons (Stallmeyer et al., 1999; Giesemann et al., 2003). On the opposite side of the profilin molecule, aromatic amino acids in N- and C-terminal areas form the binding groove for poly-proline stretches (Björkegren et al., 1993; Haarer et al., 1993; Kaiser and Pollard, 1996). Typical poly-proline motifs for profilin binding are continuous sequences of five to ten prolines interspaced by single glycines (Mahoney et al., 1997). Numerous proteins contain such poly-proline motifs and were described as profilin ligands. Many profilin interacting proteins clearly belong in the category of actin regulators like Ena/VASP proteins, formins, and WASP/WAVE. Other PLP-ligands link profilin to diverse cellular processes such as membrane trafficking, signaling, synaptic scaffolding, nuclear export, mRNA splicing, and transcription (see Supplementary Table 1). In addition, profilin also interacts with phospholipids, while it has the highest affinity to phosphoinositide-4,5-bisphosphate (PIP2) (Lassing and Lindberg, 1985, 1988). The major phosphoinositide-binding pocket with its conserved basic residues overlaps with the actin binding site (Sohn et al., 1995; Behnen et al., 2009). Thus, PIP2-bound profilin is unable to interact with actin monomers (Lassing and Lindberg, 1988). Biochemical analyses also show the presence of an additional PIP2 binding site at the profilin C-terminus (Lambrechts et al., 2002; Skare and Karlsson, 2002). The competition of PIP2 with other profilin ligands serves as a regulative mechanism of profilin activity downstream of phospholipid-based signal transduction (see for review Davey and Moens, 2020).
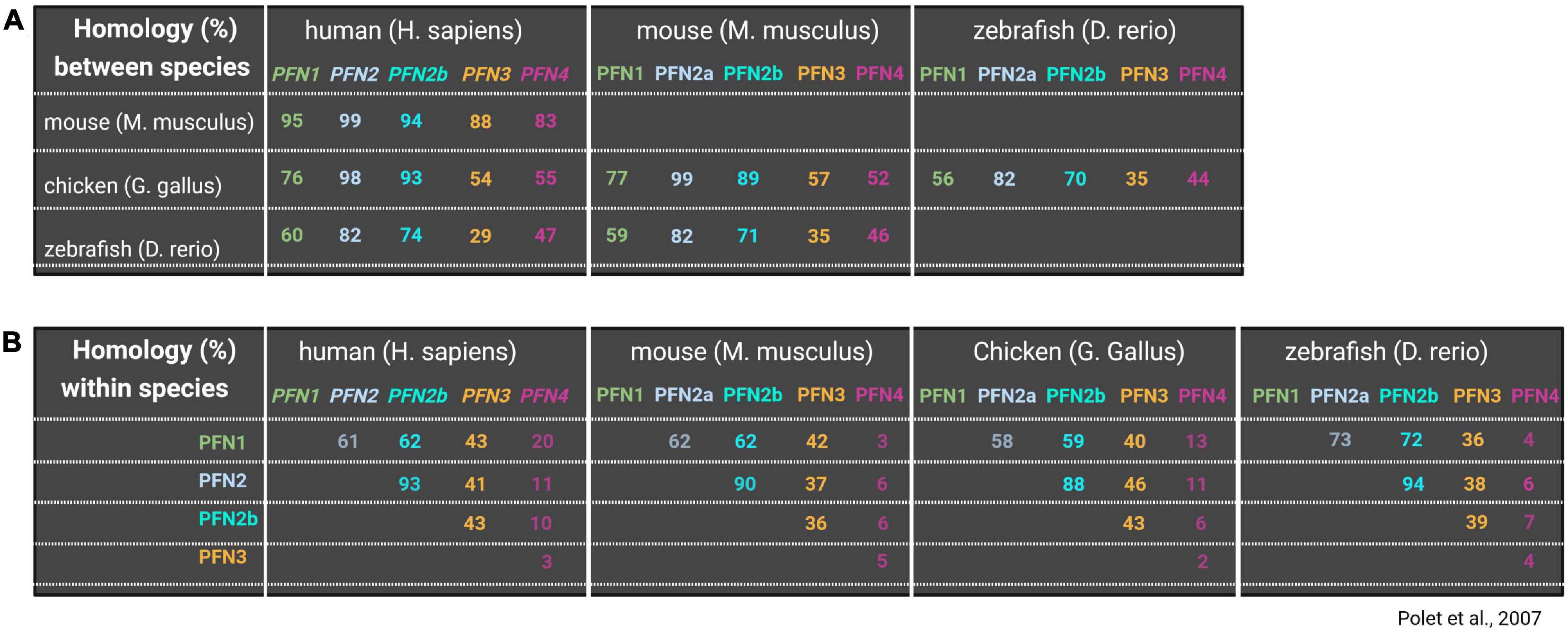
Figure 1. Comparison of profilin isoform amino acid sequences between and within organisms. Upper panel (A) shows homologies (%) of all profilins between different species including human, mouse, chicken and zebrafish. Lower panel (B) shows homology (%) of different profilins within the same species (lower panel). Blank squares were left blank to avoid redundant information. Information on profilin homologies are based on Polet et al. (2007).
The roles of profilin in actin dynamics have been intensively investigated in vitro and in cells, mostly by studying either yeast profilin or mammalian profilin 1 (PFN1): in line with its first description as a protein that can keep actin in a “pro-filamentous state,” profilin was initially considered to maintain the pool of free actin monomers (Carlsson et al., 1977). However, numerous studies identified profilin instead as an actin polymerization-driving force by the following mechanisms: (1) profilin accelerates the nucleotide exchange of G-actin (Perelroizen et al., 1996; Selden et al., 1999), (2) ATP-G-actin-bound profilin can transiently bind to growing barbed ends of actin filaments (Jégou et al., 2011; Courtemanche and Pollard, 2013; Pernier et al., 2016) and (3) profilin delivers G-actin to actin-nucleating and/or polymerizing proteins through the interaction with poly-proline motifs (Ferron et al., 2007; Paul et al., 2008; Funk et al., 2019). Prominent classes of such actin regulators are formins and Ena/VASP proteins, which act as processive actin polymerases by continuously associating with profilin-actin complexes, thereby creating linear actin filaments (Romero et al., 2004; Breitsprecher et al., 2008; Hansen and Mullins, 2010; Winkelman et al., 2014; Brühmann et al., 2017). On the other hand, profilin also interacts with Arp2/3 complex-specific nucleation promoting factors (NPFs) like (N-)WASP and WAVE proteins (Miki et al., 1998; Suetsugu et al., 1998). These proteins activate the Arp2/3 complex, which then binds already existing filaments and initiates there the growth of actin branches (see for review Pollard and Cooper, 2009). In view of this promiscuous binding nature, one could interpret profilins as housekeeping proteins that equally provide polymerization-competent actin monomers to the different actin networks. However, recent studies demonstrate that profilin directs as master switch the competition of branched and linear actin networks for free actin monomers. In yeast, high profilin concentrations blocked Arp2/3-dependent actin networks, while formin-dependent actin polymerization is greatly enhanced. Profilin yeast mutants show increased Arp2/3-dependent actin patches and cytokinesis defects through formin dysfunctions (Figure 2; Suarez et al., 2015). In line with these findings are results obtained in fibroblasts, where acute profilin depletion in wildtype cells cause increased F-actin levels and formation of Arp2/3-rich lamellipodia (Rotty et al., 2015). In contrast, actin dynamics of ArpC2-deficient cells mostly rely on profilin and Ena/VASP proteins (Figure 2). These studies indicate an obvious supportive role of profilin for formin and Ena/VASP-based over Arp2/3-dependent actin networks. From a mechanistic point of view, profilin can inhibit branched actin networks indirectly by re-routing G-actin away from Arp2/3-specific NPFs toward formins and Ena/VASP. On the other hand, a direct inhibition of actin branching is conceivable, as profilin can principally interact with Arp2 and, thus, sterically hinder the Arp2/3 complex itself (Mullins et al., 1998) and/or associated NPFs like WASP (Suarez et al., 2015). However, it is important to note that other studies also showed stimulatory effects of profilin on N-WASP and WAVE1 as well as Arp2/3-dependent actin networks (Bieling et al., 2018; Skruber et al., 2020). Profilin 1-deficient CAD cells, which are of neural origin, show Arp2/3 network-deprived leading edges, while Ena/VASP proteins are non-functional at the same time. Rescue experiments with tightly titrated profilin levels reveal discrete stages of either competition or collaboration between the different actin networks: Low profilin levels in CAD cells favor exclusively linear filaments and filopodia formation at leading edges through Ena/VASP proteins. In contrast, high profilin levels evoke the Arp2/3-dependent branched actin arrays in conjunction with actin bundles resembling filopodia-precursor structures. However, no filopodia are generated in high profilin settings (Figure 2; Skruber et al., 2020). Therefore, the regulative function of profilin on different actin networks in CAD neuroblastoma cells is dosage-dependent. In fibroblasts, Rotty et al. show a clear preference of profilin in driving the formin and Ena/VASP dependent actin machinery. The discrepancies between both studies can be explained by differences in the used substrates and cell types: Primary fibroblasts on fibronectin show prominent stress fibers and several lamellipodia at the leading edge (Rotty et al., 2015). CAD cells on laminin form one continuous and large lamellipodium, where F-actin appears veil-like or in linear arrays of short actin bundles. In contrast to fibroblasts, CAD cells do not form stress fibers (Lee et al., 2013; Skruber et al., 2020). A major source of variations may be the used extracellular matrix proteins: cultivating the same cell type on either fibronectin or laminin can already cause profound differences in its cellular morphology and behavior (Junker and Heine, 1987; Siddiqui et al., 2021). Moreover, several studies demonstrate that neurons can use entirely different modes of migration than fibroblasts (Strasser et al., 2004; Yang et al., 2012; San Miguel-Ruiz and Letourneau, 2014; Santos et al., 2020): For instance, neuronal axons move in an amoeboid manner independent of adhesions and actomyosin mediated forces, while fibroblasts use contractility-based mesenchymal migration when cultivated in 3D matrices (Santos et al., 2020). Thus, the profilin-dependent mechanisms to control actin network architectures could vary between fibroblasts and neuron-derived CAD cells as well. In summary, profilin is a major driving force of actin polymerization and director of the different actin networks. However, the precise mechanisms in how profilin coordinates the different actin networks, appear to vary in a context- and cell type-dependent manner.
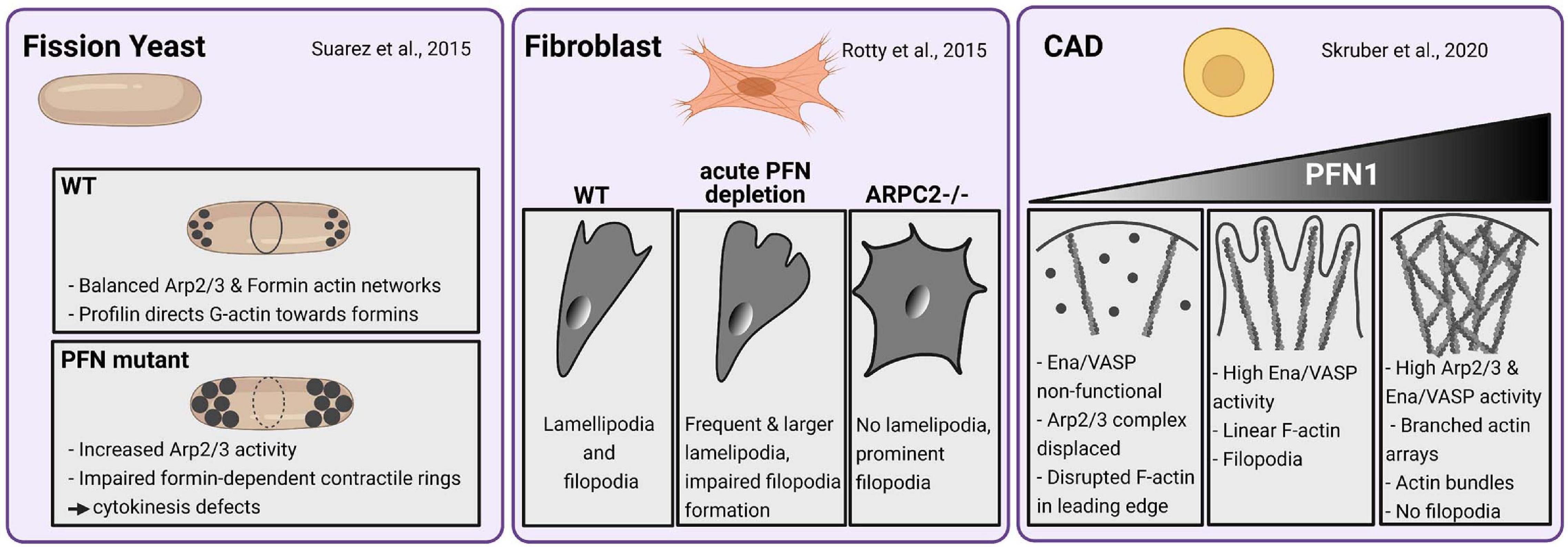
Figure 2. Profilin-dependent mechanisms in coordinating different actin networks vary in a cell type-specific manner. Fission yeast (left panel): Profilin balances ARP2/3 and Formin-dependent actin networks by directing G-Actin toward formins enabling normal contractile ring development and, thus, normal cytokinesis. Under semi-permissive temperatures, PFN mutants show large actin patches by increased ARP2/3 activity and impaired contractile ring formation and cytokinesis defects according to formin dysfunctions (Suarez et al., 2015). Mouse Fibroblasts (center panel): Wildtype fibroblasts form normal lamellipodia and filopodia. An acute depletion of PFN leads to increased F-Actin levels and formation of prominent ARP2/3-rich lamellipodia. Filopodia formation is impaired in PFN1-depleted fibroblasts. ARP2-deficient fibroblasts exhibit prominent filopodia but no lamellipodia. Actin dynamics in ArpC2-deficient cells rely on PFN1 and Ena/VASP-proteins (Rotty et al., 2015). CAD cells (right panel): PFN deficiency disrupts the actin networks in leading edges of CAD cells by rendering Mena/VASP proteins inactive and displacing the Arp2/3 complex. Rescue experiments with low to medium levels of PFN1 mediate exclusively linear actin filaments and filopodia formation by high Mena/VASP-activity. High levels of profilin in these settings evoke both high Arp2/3 and Mena/VASP activity. Thus, dense branched actin arrays are formed along with linear actin bundles. The latter resembled filopodia precursors while actual filopodia were not formed (Skruber et al., 2020).
In the previous chapters, we referred to “typical profilin properties” of which particularly the ability to bind actin and poly-proline motifs is widely used to classify proteins with corresponding structure as profilins. When we reviewed the regulation of actin dynamics by “profiling,” we discussed studies using either yeast profilin or mammalian profilin 1. In the following we shine some light on profilins in higher animals: So far, 4 profilin genes have been identified in vertebrates, where the alternative splicing of pfn2 transcript produces two isoforms, PFN2a and PFN2b. PFN1 is ubiquitously expressed and essential for normal development, as early embryos die during the first cell divisions in PFN1-deficient settings or subsequent to injecting anti-PFN1 antibodies into zygotes (Witke et al., 2001; Rawe et al., 2006). In the CNS, PFN1 constitutes only 25% of total profilin protein levels. The majority is represented by the co-expressed PFN2a, which is, according to its expression pattern in mice, categorized as the CNS-specific profilin isoform (Witke et al., 1998). However, PFN2a is more broadly expressed in other organisms. For instance, PFN2a is the ubiquitous isoform in chicken (Murk et al., 2009). In addition, PFN2a is also present in non-neuronal cells and tissues of humans (Mouneimne et al., 2012). The other PFN2 splice form PFN2b is only expressed in kidney (Di Nardo et al., 2000). PFN3 and PFN4 are testis-specific isoforms, while PFN4 shows the lowest level of homology to the other isoforms (Figures 1, 3; Hu et al., 2001; Obermann et al., 2005).
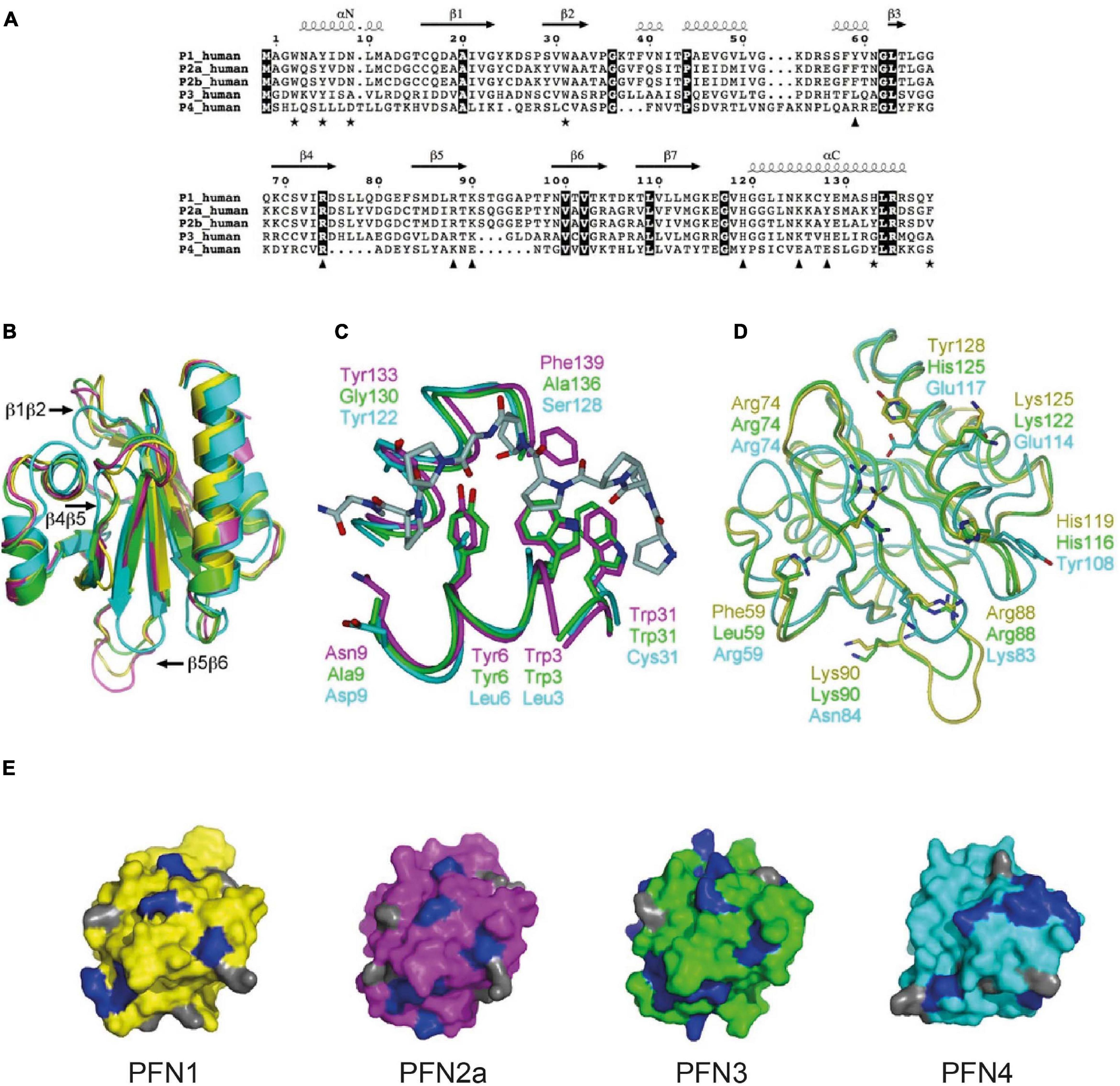
Figure 3. Structural features reflect the different biochemical properties of the mammalian profilin isoforms. (A) Sequence alignment between human profilins. Fully conserved residues are on dark background. Key residues of PLP binding are indicated by asterisks, those involved in binding actin with triangles. Secondary structures are derived from bovine PFN1 crystal structure. For compatibility with most publications, the first methionine is not considered in sequence numbering. (B) Superposition of bovine PFN1 (yellow), mouse PFN2a (magenta), human PFN3 (green), and human PFN4 (cyan). PFN1 structure is from the profilactin complex (PDB core 1HLU) and PFN2a from the complex with a VASP peptide (PDB code 2V8C). Loops variable in length in PFN3 and/or PFN4 are marked. (C) Comparison of the PLP-binding sites of PFN2a, PFN3, and PFN4. Shown is also the PFN2a-VASP complex, coloring as in 3B. Only half of the binding site is conserved in PFN3, no conservation is seen in PFN4. Key residues for peptide binding are indicated. (D) Actin-binding sites of PFN1, PFN3, and PFN4, coloring as in 3B. For clarity, actin is not shown; view is from the direction of actin onto the actin-binding surface on profilin, side chains of key profilin residues are shown. (E) Comparison of PtdIns(4,5)P2 binding surfaces of PFN1, 2a, 3, and 4 (left to right, respectively). Profilins are colored as in 3A, and all arginine residues, crucial for PtdIns(4,5)P2 binding, are highlighted in blue; lysine residues are shown in gray. This figure is used in this review with permission from Behnen et al. (2009).
Structural comparisons show that generally all isoforms possess the typical profilin folding. PFN4, however, is ten amino acids shorter than PFN1 and PFN2a, which reduces the lengths of three interspacing loops (β1β2, β4β5, and β5β6, Figures 3A,B). PFN4 interacts neither with G-actin nor poly-proline stretches, as it lacks all the necessary key residues to bind these typical profilin ligands (Figures 3A–C). Another difference is a shifted phospholipid binding site in PFN4, which enables it to bind PtdIns(4)P and phosphatidic acid in vitro (Figure 3E; Behnen et al., 2009). Analogous to PFN4, PFN2b does not have a significant affinity to G-actin and poly-proline motifs either (Di Nardo et al., 2000). Although the cellular functions of these profilin isoforms are currently unknown, it is conceivable that they fulfill different tasks than controlling actin dynamics. The other profilin isoforms vary more subtly in the typical profilin properties: The testis-specific PFN3 isoform interacts both with G-actin and poly-proline motifs but with significantly lower affinity than “the” PFN1. This different binding behavior is structurally reflected by conservative amino acid sequence differences in the actin binding sites of PFN1 and PFN3 (Figure 3D). In addition, the two key residues His133 and Phe139, which mediate high-affinity binding of PFN1 to poly-proline, are not present in PFN3 (Figures 3A,C). The putative phospholipid binding site of PFN3 is structurally comparable to PFN1 and PFN2a (Figure 3E; Behnen et al., 2009).
PFN2a, the predominant splice form of pfn2, has actin-binding properties comparable to PFN1 (Lambrechts et al., 1995, 2000), while it binds in vitro with higher affinity to synthetic poly-proline peptides (Lambrechts et al., 1995). These experimental findings are supported by structural data showing all residues central to actin binding are conserved between PFN1 and PFN2a (Figures 3A,D). The poly-proline binding domain of PFN2a is highly similar to PFN1, while the aromatic residues Tyr29 and Tyr133 in PFN2a further enhance its affinity to poly-proline (Figures 3A,C; Nodelman et al., 1999; Haikarainen et al., 2009; Walter et al., 2020). The phospholipid binding sites are well conserved between PFN1 and PFN2a (Figure 3E). However, the greater binding strength of PFN2a to poly-prolines prevents, in contrast to PFN1, a competitive binding between synthetic poly-proline peptides and PIP2 (Lambrechts et al., 1997). Both similarities and differences between PFN1 and PFN2a in vitro raise the question as to whether the isoforms are functionally exchangeable or fulfill different tasks in cellular settings.
To discuss the functional diversity of PFN1 and PFN2a, we here elaborate their binding properties to different ligands as well as the phenotypes, caused by manipulating PFN1 and/or PFN2a in cells and animals. For the latter, we mainly focus on neuronal cells, where both isoforms are co-expressed.
PFN1 and PFN2a share a large number of mutual ligands but also bind some ligands exclusively (Supplementary Table 1). For instance, affinity chromatography based screens identified tubulin among the exclusively PFN1-bound proteins (Witke et al., 1998). This finding was confirmed and extended by recent studies showing a tubulin binding site in PFN1 close to its actin binding domain. The proximity of both binding sites prevents PFN1 to interact with tubulin monomers in in vitro settings, when G-actin is present (Henty-Ridilla et al., 2017). However, PFN1 associates in different cell lines with tubulin, cytoplasmic microtubules, mitotic spindles and centromers (Grenklo et al., 2004; Nejedla et al., 2016; Henty-Ridilla et al., 2017; Nejedlá et al., 2021.) Moreover, the manipulation of cellular PFN1 levels also affects microtubule dynamics: Depletion of PFN1 evokes increased microtubule growth in B16f-melanoma cells (Nejedla et al., 2016). In neuronal cells, PFN1 mediates the opposite effect: The velocity of growing microtubules is three-fold increased in PFN1-overexpressing N2A neuroblastoma cells (Henty-Ridilla et al., 2017). Increased PFN1 levels promote axon growth and regeneration of neurons in cell culture and in vivo by accelerating microtubule growth, while depletion of PFN1 curbs these processes (Pinto-Costa et al., 2020). These findings are in line with in vitro assays showing the direct enhancement of microtubule growth by PFN1 (Henty-Ridilla et al., 2017). In addition, PFN1 coordinates microtubules with actin filaments in axons via formins (Pinto-Costa et al., 2020), which serve as a link between both filament systems (see for review Breitsprecher and Goode, 2013). In summary, PFN1 specifically balances microtubule dynamics in yet to be determined cellular settings and via so far unknown molecular mechanisms.
Loss of function studies show PFN1 to control distinct aspects in CNS physiology, which apparently are not compensated by the co-expressed PFN2a: The CNS-specific deletion of the pfn1 gene causes specific anatomical changes in the mouse brain. PFN1-deficient mice exhibit smaller brains, where in particular the cortex with 25% and the cerebellum with up to 50% of size reduction are affected (Figure 4). The hypoplasia of the cerebellum develops postnatally, when the neuron subtypes of cerebellar granular neurons (CGNs) and Purkinje cells move toward their final positions. Firstly, CGNs and Purkinje cells migrate tangentially in the external granular layer. Then they switch to radial migration and move along the processes of Bergmann Glia to cross the molecular layer and reach their destinations, the Purkinje or internal granule layer inside the cerebellum (Chédotal, 2010). Pfn1-/- CGNs, however, fail to adhere and migrate along Bergmann glia fibers, which cause their ectopic accumulation in the intermediate molecular layer (Kullmann et al., 2011, 2015). Also, the co-migrating Purkinje cells show analogous migration defects. However, this phenotype turned out not to be cell autonomous, as the Purkinje cell-specific pfn1 deletion does not reproduce the migration defect observed in mice with the CNS-wide ablation of pfn1 (Kullmann et al., 2012). Thus, defective migration of PFN1-deprived cerebellar granule cells is somehow translated to migrating Purkinje cells. PFN1 deletion in the rodent CNS evokes impaired motor coordination, which is typical for disturbed cerebellum functions (Kullmann et al., 2012).
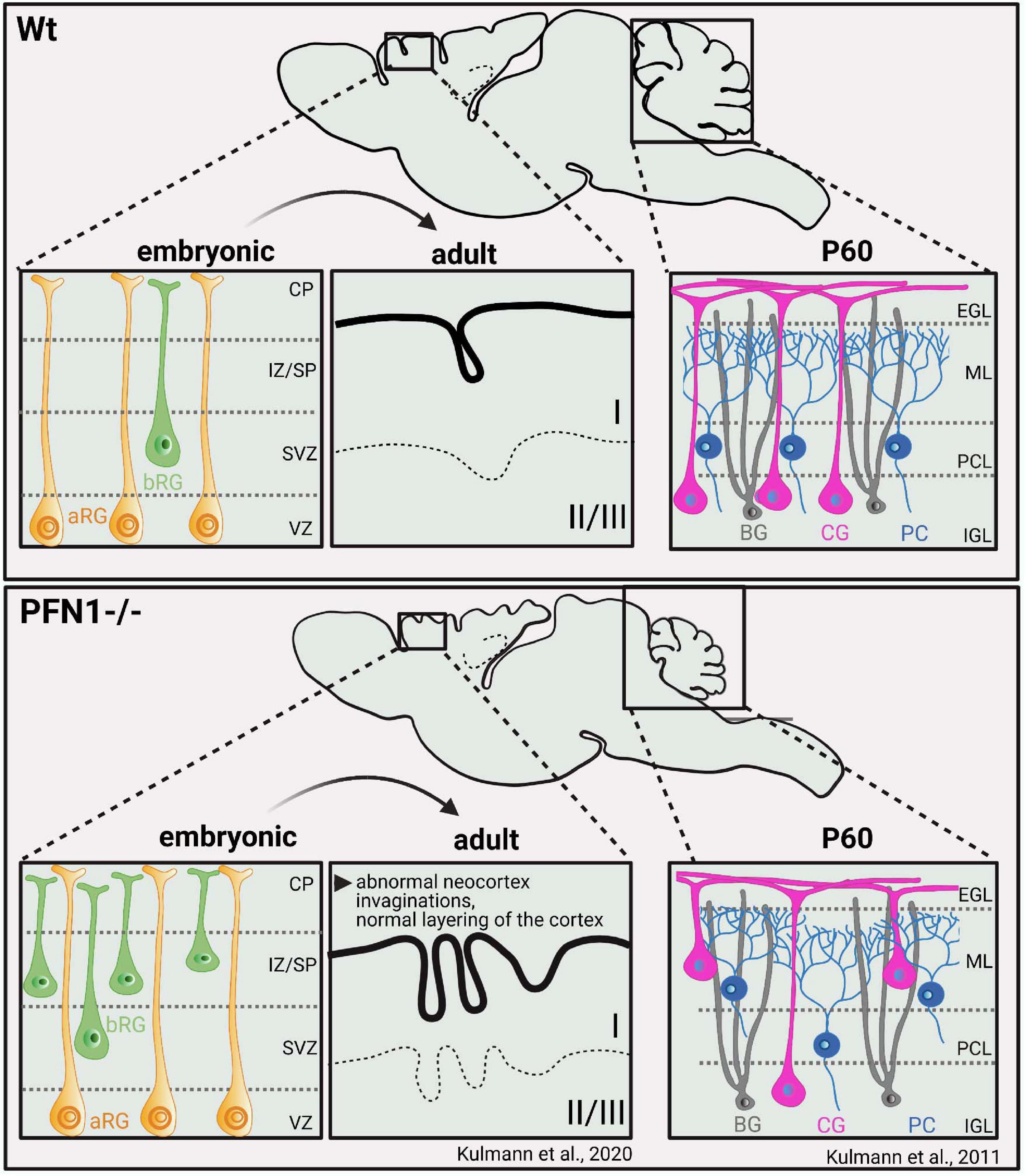
Figure 4. PFN1 loss perturbs brain development and causes malformations of cortex and cerebellum through altered cellular compositions. Pfn1–/– mice exhibit smaller brains, with size reductions, particularly of the cortex (–25%) and cerebellum (–50%). Defects in cortical development are caused prenatally by a distinct subset of neural progenitor cells, known as basal radial glia. PFN1 loss affects the orientation of basal radial glia cell divisions during development, causing transiently ectopic neurogenesis. Adult mice show abnormal invaginations of the neocortex while cortical layering is principally preserved (Kullmann et al., 2020). Cerebellar hypoplasia develops postnatally by migration defects of cerebellar granule cells and Purkinje cells (Kullmann et al., 2011, 2012, 2015). Impaired adhesion and migration of cerebellar granule cells along the processes of Bergmann glia are causative for this phenotype. bRG, basal radial glia; aRG, apical radial Glia; CP, cortical plate; IZ/SP, intermediate zone/subplate; SVZ, subventiruclar zone; VZ, ventricular zone; BG- Bergmann Glia; CG- cerebellar granule cells; PC- purkinje cells; EGL, external granular layer; ML, Molecular layer; PCL, Purkinje cell layer; ICL, Internal granular layer; P60, postnatal day 60.
Defects in the cortex development of pfn1-/- mice occur prenatally (Figure 4; Kullmann et al., 2020). This finding is in line with a wealth of developmental studies showing that the cerebral cortex with its typical six horizontal layers and specialized neuron subtypes forms between embryonic day 10 and birth. The different neuronal cells in the cortex originate from radial glia cells in the ventricular and subventricular zone (Götz et al., 2002; Noctor et al., 2004). From there, radial glia cells and already differentiated neurons migrate and create the cortical layers in a stepwise manner (Gilmore and Herrup, 1997; López-Bendito and Molnár, 2003). In contrast to the developing cerebellum, the migration of radial glia cells and thus, cortical layering in PFN1 deficient brains is unaffected. Pfn1-/- brains display smaller and abnormal invagination-bearing cerebral cortices, a phenotype that can be attributed to cell division defects in basal radial glia cells, a rare subset of neural progenitor cells. Basal radial glia cells are highly neurogenic and can be found in the developing cortex of gyrencephalic species, where they are associated with the development of the cortical folds, known as gyri and sulci (Penisson et al., 2019).
In pfn1-/- brains, alterations in basal radial glia cell division result in locally increased and ectopic neurogenesis at embryonic day 14.5. Later in development, the atypical invaginations form in the cortex periphery after the ectopic basal radial glia cell-derived neuronal cells reached their final positions and differentiate in cortical layer I (Kullmann et al., 2020). In contrast to the distinct CNS deformities in this pfn1 knockout mouse model, excitatory synapses were structurally and functionally intact (Görlich et al., 2012). However, it is noteworthy that the acute depletion of PFN1 in cultured hippocampal neurons and organotypic slices by RNAi affects dendritic spines, which harbor the majority of excitatory postsynapses (see for review Borovac et al., 2018). The PFN1 knockdown reduces the overall densities as well as the content of mature subtypes of dendritic spines (Figure 5B; Michaelsen-Preusse et al., 2016). Thus, PFN1 controls distinct aspects of brain development, while further research is required to clarify its role in synapses.
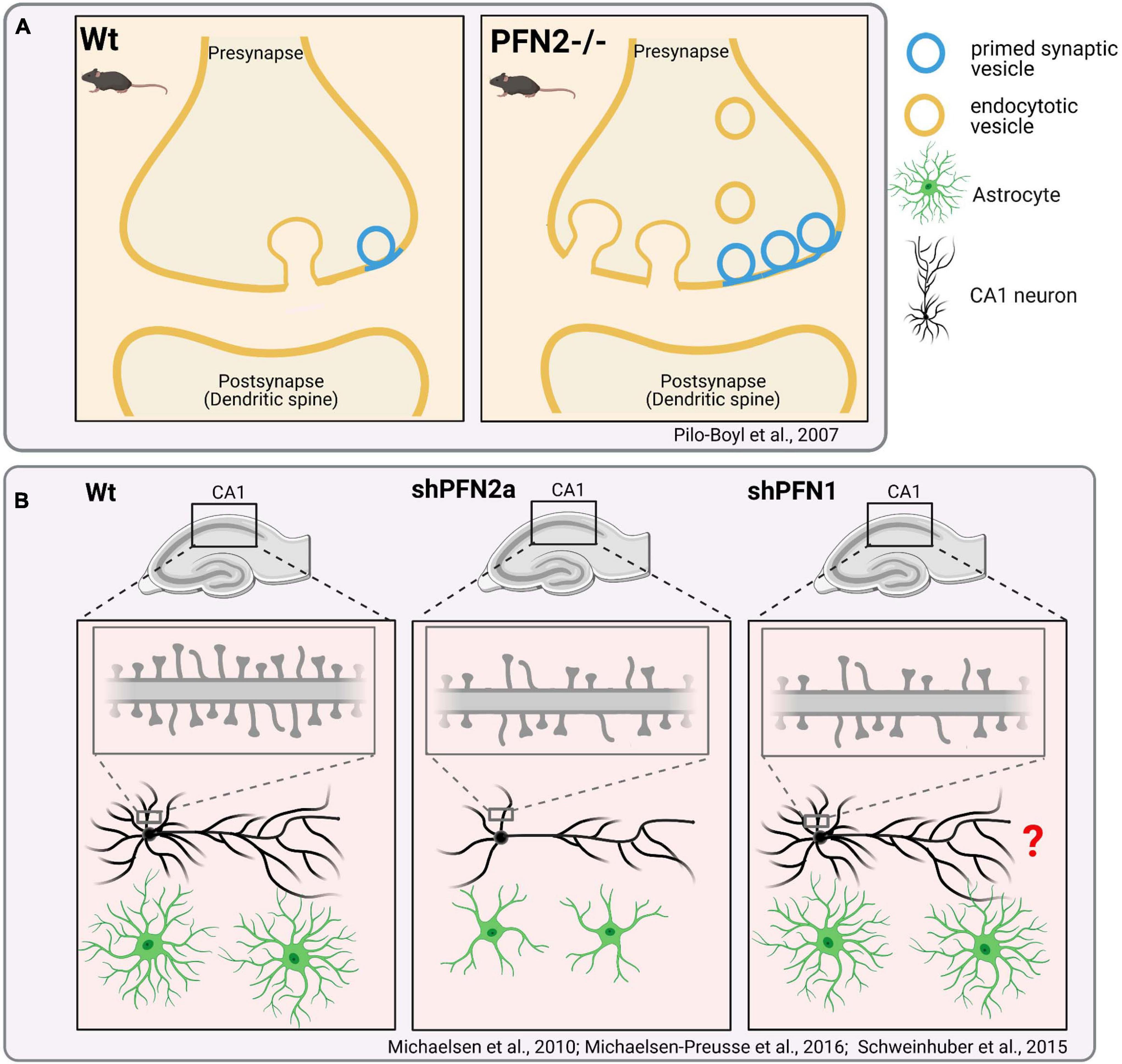
Figure 5. Distinct effects of PFN1- or PFN2- “loss-of-function” in neuronal cells. (A) Genetic ablation of Profilin2 and impact on synapses: Presynapses in pfn2-deficient mice show an increased number of primed synaptic vesicles. Increased exocytosis of synaptic vesicles cause hyperactivity and changed complex behaviors in pfn2-/- mice, such as increased novelty-seeking (Pilo Boyl et al., 2007). Further studies also indicate increased endocytosis in pfn2-/-mice (Gareus et al., 2006; Luscieti et al., 2017). Pfn2 –/– animals display no gross changes in brain anatomy. (B) shRNA-mediated acute depletion of PFN2a in organotypic slice cultures reduces dendritic complexity and spine densities of hippocampal CA1 neurons (Michaelsen et al., 2010). shPFN1 treatment of neurons reduces the overall densities of mature dendritic spines (Michaelsen-Preusse et al., 2016). It was not described, if PFN1 knockdown also affects the dendritic arborization of CA1 neurons (labeled with “?”). Rescue experiments indicate that PFN1 is unable to compensate PFN2a-specific defects in dendritic complexity. Depletion of PFN2a but not PFN1 affects the overall volume of astrocytes in organotypic slice cultures (Schweinhuber et al., 2015).
In contrast to PFN1, PFN2a binds membrane- and synaptic vesicle-associated proteins like synapsins and dynamin I in ligand screenings (Witke et al., 1998). Cell biological and in vivo studies with pfn2-deficient mice reveal accordingly functions of PFN2a in synaptic membrane trafficking: PFN2a is able to inhibit endocytosis through its interaction with dynamin I (Gareus et al., 2006). Moreover, PFN2a-deficient mice show increased synaptic exocytosis upon depolarization, which correlates with altered synaptic F-actin levels and larger number of primed synaptic vesicles in pfn2-/- synapses when compared to wildtype synapses (Figure 5A; Pilo Boyl et al., 2007). Interestingly, pfn2-/- mice do not show gross changes in brain anatomy. Instead they exhibit behavioral abnormalities such as hyperactivity and increased novel object seeking. This phenotype correlates with electrophysiological analyses showing increased synaptic excitability of glutamatergic neurons in the striatum of pfn2-/- mice. The findings support the hypothesis that PFN2a tunes the presynaptic neurotransmitter release of neurons via actin dynamics. In turn, PFN2 loss creates the over-activation of neural circuits like the cortico−striatal glutamatergic pathway, which relays its increased synaptic input to basal ganglia and thereby causes hyperactivity and altered complex behaviors. Thus, studies in pfn2-/- mice reveal a prominent role of PFN2a in presynapses. However, these findings do not rule out postsynaptic functions of PFN2a. NMDA receptor stimulation of cultured hippocampal neurons expressing PFN2a-GFP evokes the translocation of PFN2a into dendritic spines, where actin-dependent changes in spine shape are subsequently blocked (Ackermann and Matus, 2003). Fear conditioning of rats induces the translocation of endogenous profilin into dendritic spines of the amygdala (Lamprecht et al., 2006). Also, localization studies with a PFN2-specific antiserum confirm the presence of PFN2a in dendritic spines of cultured neurons depending on synaptic activity and Rho signaling (Schubert et al., 2006). Finally, acute PFN2a depletion in organotypic slice cultures reduces the dendritic complexity and spine densities of hippocampal CA1 neurons (Figure 5B). The concomitant expression of exogenous PFN1 rescues defects in dendritic spine density but not in dendrite arborization of PFN2a-depleted CA1 neurons (Michaelsen et al., 2010). These results demonstrate functions of PFN2a in postsynaptic and dendritic compartments, which partially can be compensated for by upregulated PFN1.
In addition, PFN1 and PFN2a are also involved in extra-synaptic functions in the CNS. Both isoforms are expressed in astrocytes, the most prevalent and highly arborized glia cells in the CNS, which actively support and protect neurons (see for review Schiweck et al., 2018). Depletion of PFN2a, but not PFN1, reduces the total volume that astrocytes occupy in organotypic brain slices (Figure 5B; Schweinhuber et al., 2015). PFN1 knockdown in cultured astrocytes instead affects the number and motility of filopodia-like protrusions, which resemble dynamic perisynaptic processes of astrocytes in vivo (Molotkov et al., 2013). Another recent study shows that PFN2a is involved in iron homeostasis. The pfn2 transcripts contain an iron response element in their 3′-UTR, which positively regulates pfn2 mRNA stability upon binding by iron response proteins. PFN2 deficiency leads to excessive iron accumulation specifically in the brain, possibly through increased transferrin uptake by deregulated endocytosis (Luscieti et al., 2017). Thus, PFN1 and PFN2a possess mutual as well as isoform-specific functions in the CNS, which regulate multiple processes including membrane trafficking and shaping of the plastic morphology of neuronal cells.
However, the question remains whether PFN2a can also fulfill tasks of PFN1 in non-neuronal cells. To our knowledge, no direct gene replacement of endogenous PFN1 with PFN2a in such settings has been published. However, chicken ubiquitously express PFN2a (chPFN2a), which, surprisingly, differs from murine PFN2a by a single amino acid substitution at position 38 (Murk et al., 2009). In chicken fibroblasts, PFN2a is responsible to control actin dynamics in general. Chicken PFN2a is co-expressed with a PFN1-like isoform (chPFN1). Sequence alignments with mouse PFN1 showed, however, that chPFN1 differs largely in residues, which are otherwise conserved between mammalian PFN1 isoforms and are crucial for actin binding (Schlüter et al., 1998). Functional assays with chicken fibroblasts after single and double knockdown of chPFN1 and/or chPFN2a demonstrate that chPFN1 loss has no significant impact on actin-dependent processes like cell adhesion, spreading, and migration (Murk et al., 2009). Thus, PFN2a can principally exert functions in non-neuronal cells so far ascribed to PFN1. Still, it is currently unknown if PFN2a has additional functions in chicken fibroblasts, such as the regulation of membrane trafficking events.
Growing numbers of publications, studying patients and/or using cell or animal disease models, link profilins to various diseases: in cancers, PFN1 as well as PFN2 are reported to either act as tumor suppressors or possess oncogenic potential, depending on the studied cancer cell type (for review Pimm et al., 2020). In addition, some studies link PFN1 to a degenerative bone pathology known as Paget Disease (Merlotti et al., 2020; Scotto di Carlo et al., 2020), while others implicate this profilin isoform in vascular inflammation and atherosclerosis (see for review Pae and Romeo, 2014). Particularly, studies on neurological diseases emphasize both the biomedical relevance and the functional diversity of PFN1 and PFN2a isoforms. Therefore, in the following we will focus on the roles of PFN1 and/or PFN2a in a subset of prominent neurological pathologies comprising Amyotrophic lateral sclerosis (ALS), Fragile X syndrome (FXS), Huntington’s disease (HD), and spinal muscular atrophy (SMA).
Amyotrophic lateral sclerosis (ALS) is a major neurodegenerative disease, which is caused by mutated Pfn1 alleles (Wu et al., 2012; Smith et al., 2015). This disease is characterized by late-onset progressive degeneration of motor neurons in the brain and spinal cord, leading to impaired motor coordination, paralysis and, ultimately, to death by respiratory failure usually within 3 or 5 years from diagnosis. ALS cases are divided into two major groups - sporadic (sALS) and familial (fALS), while familial cases of ALS represent roughly 10% of total ALS cases (see for review (Hulisz, 2018). Exome sequencing studies reveal 8 mutations in the PFN1 gene in both familial and sporadic ALS cases (C71G, G118V, M114T, E117G, T109M, R136W, A20T, Q139L) (Figure 6.1; Wu et al., 2012; Chen et al., 2013; Ingre et al., 2013; Smith et al., 2015; Alkam et al., 2017). The PFN1-associated ALS pathology is reproducible in several mouse and rat models (Yang et al., 2016; Fil et al., 2017; Barham et al., 2018; Brettle et al., 2019; Yuan et al., 2020). The expression of ALS-PFN1 mutants evokes cytoskeletal and morphological defects in primary neurons, such as abnormally low ratios of F-/G-actin, shorter dendrites and integrity-impaired axons, which undergo Wallerian degeneration over time (Figure 6.2; Wu et al., 2012; Yang et al., 2016; Fil et al., 2017). Different scenarios are currently discussed in the field on how PFN1 mutations could evoke ALS: in view of PFN1’s central role in actin dynamics, one hypothesis is the perturbation of G-actin binding of PFN1 by ALS-associated mutations. The initially discovered mutations C71G, G118V, M114T, and E117G are in close proximity to the PFN1 G-actin binding site. Co-immunoprecipitation assays first indicated an impaired G-actin binding of the PFN1 C71G, PFN1 G118V, and PFN1 M114T mutants (Wu et al., 2012). However, subsequent analyses using recombinant proteins in pyrene-based actin polymerization assays demonstrate that none of those four PFN1 mutants is significantly affected in its binding to G-actin (Boopathy et al., 2015). In addition, the later discovered PFN1 T109M mutant also exhibits a normal affinity to G-actin (Freischmidt et al., 2015). Thus, the direct interaction of ALS-PFN1 mutants with G-actin is unlikely to cause the ALS pathology. Several analyses show that ALS-associated PFN1 mutants are prone to aggregate like the other ALS-causing proteins, such as SOD1, TDP43, and FUS (see for review Saberi et al., 2015). Some ALS-linked PFN1 mutations (C71G, M114T, G118V) form insoluble and ubiquitinated protein aggregates and inclusions in primary motor neurons and N2A cells, while control wildtype PFN1 is diffusely distributed across the cytoplasm (Figure 6.3; Wu et al., 2012). Also, the more recently identified PFN1 A20T variant causes the formation of insoluble aggregates in HEK293T cells and ALS patients-derived fibroblasts (Smith et al., 2015). Comprehensive biochemical analyses demonstrate the mutation-induced destabilization of protein folding in PFN1 C71G, PFN1 M114T, and PFN1 G118V variants (Boopathy et al., 2015; Del Poggetto et al., 2015a, b, 2016, 2018). These studies support the hypothesis that ALS-PFN1 mutants acquire different, partially or alternatively folded conformations, which promote the formation of PFN1 inclusions in the cytosol. X-ray crystallography reveals corresponding structural perturbations in aggregation-prone ALS-PFN1 mutants: the M114T mutation turns a surface pocket of wildtype PFN1 into a cleft, which extends into the protein core. Accordingly, this structural change exposes in wildtype PFN1 buried hydrophobic residues to solvent and, thereby, destabilizes the overall protein stability. Computer modeling suggests also a cavity deep within the PFN1 C71G mutant (Boopathy et al., 2015). Such internal voids are structurally more disruptive than peripheral, solvent-exposed cavities, which explains the particularly high instability and aggregation propensity of PFN1 C71G among the biochemically characterized ALS-PFN1 mutants (Eriksson et al., 1992b, a; Del Poggetto et al., 2015a; Xue et al., 2019). In line with the structural and biochemical findings is the particularly high susceptibility of PFN1 C71G to proteasomal degradation in cells (Schmidt et al., 2021). Interestingly, the PFN1 E117G and PFN1 Q139L mutants are less prone to aggregate (Smith et al., 2015). Structural comparisons also reveal a folding of PFN1 E117G highly similar to wildtype PFN1 (Boopathy et al., 2015). Both PFN1 E117G and PFN1 Q139L have in common to cause only mild phenotypes in cell models (Wu et al., 2012; Figley et al., 2014). PFN1 E117G is also under debate of rather being a benign polymorphism or a potential ALS risk factor, as it is also present in control patients (Fratta et al., 2014; Smith et al., 2015). Therefore, the mutation-dependent destabilization of PFN1 toward aggregation-prone conformations is required to drive the full-scale ALS pathology.
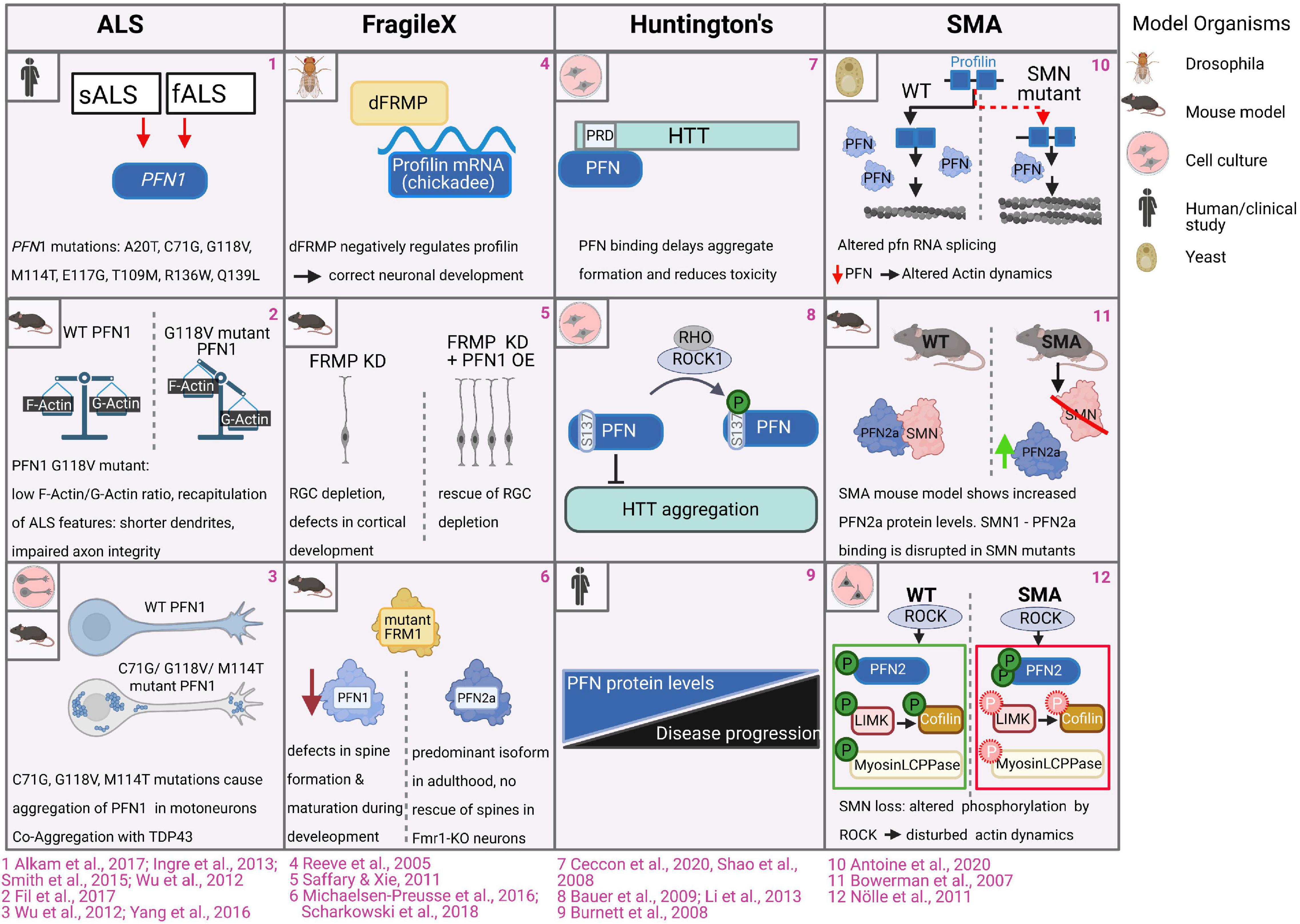
Figure 6. Profilins in CNS pathologies. 1. Exome sequencing studies revealed 8 mutations in the Pfn1 gene in familial and sporadic ALS patients (Ingre et al., 2013; Wu et al., 2012; Chen et al., 2013; Smith et al., 2015; Alkam et al., 2017). 2. Motoneurons expressing ALS mutant PFN1 G188V exhibit abnormally low ratios of F-/G-actin. Motor neurons of ALS-PFN1-mutant expressing mouse model exhibited cytoskeletal disruptions and impaired axon integrity followed by Wallerian degeneration (Yang et al., 2016). 3. In cell culture and mice, wildtype PFN1 is diffusely distributed across the cytoplasm of motor neurons and N2A cells, while PFN1 C71G, PFN1 M114T and PFN1 G118V PFN mutations form protein aggregates and inclusions. PFN1 co-aggregates with TDP43 (Wu et al., 2012; Yang et al., 2016). 4. FRMP directly binds the mRNA of Drosophila profilin (encoded by chickadee), which is required for correct neuronal development in Drosophila (Reeve et al., 2005). 5. FRMP knockdown in mice causes depletion of radial glia cells (RGC) and defects in cortical development, while overexpression of PFN1 in these settings restores the radial glial dependent developmental defects (Saffary and Xie, 2011). 6. Levels of PFN1, but not PFN2a, are reduced in FRM1 mutant mice. Depletion of PFN1 causes defective dendritic spine formation and maturation during development analogous to defects in Fmr1-KO mice. Overexpressed PFN1, but not PFN2a, can rescue dendritic spine defects in Fmr1-KO neurons (Michaelsen et al., 2010; Scharkowski et al., 2018). 7. Binding of PFN to the proline-rich domain of huntingtin reduces huntingtin aggregation and toxicity (Shao et al., 2008; Ceccon et al., 2020). 8. Rho kinase-dependent phosphorylation of PFN1 at serine 137/8 enhances huntingtin aggregation. Pharmacological inhibition of ROCK inhibits huntingtin aggregation and toxicity (Bauer et al., 2009; Li et al., 2013). 9. In humans, reduced profilin protein levels correlate with disease progression of Huntington disease in patients (Burnett et al., 2008). 10. smn-deficient fission yeast displays splicing defects in the profilin gene, resulting in altered actin network homeostasis, perturbed cytokinesis and endocytosis (Antoine et al., 2020). 11. Loss of SMN in SMA settings is associated with the upregulation of PFN2a protein levels (Bowerman et al., 2007). 12. Levels of phosphorylated PFN2a are increased in SMA settings while ROCK-dependent phosphorylation of myosin light chain protein phosphatase (Myosin LCPPase) and the cofilin-regulating LIM-kinase, are reduced, causing disturbed actin dynamics (Nölle et al., 2011).
Moreover, ALS-PFN1 mutants frequently co-aggregate with TDP43, which on its own can cause ALS and Fronto-temporal dementia when aggregated (see for review Suk and Rousseaux, 2020). Co-aggregated mutated PFN1 and TDP43 possess prion-like properties and act as seeds that trigger the conversion of endogenous TDP43 into toxic conformational states when taken up by wildtype cells (Tanaka et al., 2016). In Drosophila melanogaster, ectopic PFN1 C71G and PFN1 M114T provoke increased neurodegeneration of retinal photoreceptor neurons via co-expressed TDP43 (Matsukawa et al., 2016). Thus, PFN1 aggregations exacerbate the course of ALS by increasing the toxicity of TDP43.
While the findings showing the toxic PFN1 aggregates are compelling, is it still unclear if also monomeric mutant PFN1 proteins contribute to the ALS pathology. In fact, one study reports accelerated motor neuron degeneration in PFN1 C71G expressing mice prior to the occurrence of PFN1 inclusions (Yang et al., 2016). One potential mechanism via monomeric PFN1 mutants could be the regulation of microtubule dynamics: unlike wildtype PFN1, four ALS PFN1 mutants (C71G, M114T, E117G, G118V) are unable to accelerate the growth of microtubules in vitro and in N2A cells (Henty-Ridilla et al., 2017). These findings correlate with the impaired axon integrity and subsequent Wallerian degeneration in mutant PFN1-expressing motor neurons (Yang et al., 2016; Fil et al., 2017). Another possibility could be changes in the activity of specific-PFN1 binding partners involved in actin dynamics: PFN1 M144T and PFN1 G118V mutants bind, with higher affinity than wildtype PFN1, the formin proteins DIAPH1 and 2 and FMNL1. Moreover, PFN1 M144T and PFN1 G118V significantly enhance the actin assembly rate of those formins. The augmenting function of both mutations relies on a heightened flexibility in the PFN1 α4 helix, which contacts the actin as well as the poly-proline binding site (Schmidt et al., 2021). In addition, disturbances in nuclear pore complexes and nuclear-cytoplasmic transport occur in ALS PFN1 mutants expressing neurons (Giampetruzzi et al., 2019). Whether this phenomenon is a primary effect of mutant PFN1 or caused indirectly, is currently unknown. In summary, ALS-associated PFN1 mutants possess toxic gain-of-function of which particularly the formation of amyloid-like protein aggregates with TDP43, but potentially, also additional properties of monomeric PFN1 mutants, cause the ALS pathology. Nevertheless, many open questions remain regarding the molecular pathomechanisms and the reason why specifically motor neurons degenerate through mutations in a ubiquitous actin binding protein.
Animal models show the association of profilin with the Fragile-X syndrome (FXS), one of the most prevalent neurodevelopmental disorders. Cardinal symptoms in the spectrum of FXS are considerable intellectual disabilities, hyperactivity and repetitive behaviors. Frequently, clinical signs of autism may occur in FXS patients, such as reduced social interactions, and impairments of speech. Causative for this X-chromosome-linked disease are mutations in the FMR1 gene affecting the expression and/or function of the Fragile X mental retardation protein (FMRP). As a RNA-binding protein, FMRP binds and regulates the localization, stability and translation of multiple transcripts including those of key regulators controlling neuronal development, morphology, and synaptic plasticity (see for review Bagni and Zukin, 2019). The initial findings connecting FXS to profilin were obtained in Drosophila: FRMP directly binds the mRNA of Drosophila profilin (encoded by chickadee), while FMRP mutant flies expressed significantly increased levels of profilin (Figure 6.4; Reeve et al., 2005). The FMRP-dependent regulation of profilin expression occurs in flies at a late developmental stage, when sensory-input leads to a refinement of neural circuits through limiting of axonal growth and pruning of axonal branches (Tessier and Broadie, 2008). While findings in Drosophila indicate a negative role of FMRP in regulating profilin expression, studies in mice came to opposite conclusions: overexpression of PFN1 in radial glial cells largely rescues developmental defects during corticogenesis in FMRP-depleted mice (Figure 6.5; Saffary and Xie, 2011). In view of profilin isoform specificity, protein levels of PFN1, but not PFN2a, are reduced in FMR1-mutant mice. Depletion of PFN1 also leads to defective dendritic spine formation in hippocampal neurons, which correlate with corresponding structural alterations in FMRP-deficient mice (Figure 6.6; Michaelsen-Preusse et al., 2016; Scharkowski et al., 2018). While PFN1 protein expression is apparently affected in FMR1-mutant mice, it is not clear, whether these effects are caused by a direct regulation of mouse PFN1 transcripts by FMRP or are indirect: one study reports the direct interaction between FMRP and the PFN1 transcript (Michaelsen-Preusse et al., 2016), while large screens did not detect PFN1 transcript as FMRP target (Brown et al., 2001; Miyashiro et al., 2003; Darnell et al., 2011). Whereas the discussed studies demonstrate the implication of PFN1 in animal models, it is to our knowledge unknown, if PFN1 is also affected in FXS patients.
Huntington’s disease (HD) is an autosomal dominant, progressive neurodegenerative disease caused by an expanded CAG repeat/polyglutamine (polyQ) tract beyond 35 repeats within the first exon of the huntingtin (HTT) gene. The physiological functions of the wildtype huntingtin protein are not yet fully understood, as it participates in various processes during development and adulthood, like cell survival, membrane trafficking, ciliogenesis, autophagy and transcription (see for review Saudou and Humbert, 2016). However, mutated huntingtin proteins with N-terminal elongated polyglutamine sites are prone to form toxic aggregates of oligomers and fibers. By these means, mutant huntingtin also impairs the correct function of co-expressed wildtype huntingtin protein to maintain survival and normal activity of neurons. The consequence of carrying toxic huntingtin mutations is comprehensive neurodegeneration particularly in the striatum, which affects neuronal circuits innervating basal ganglia and the cortex. Typical HD symptoms therefore comprise adult-onset progressive motor dysfunctions, personality changes with progressive emotional and psychiatric disturbances as well as cognitive decline (Zuccato et al., 2010). Huntingtin contains two prolin-rich domains (PRDs), which are binding sites for PFN1 and PFN2 (Figure 6.7; Shao et al., 2008). The interaction with profilins significantly reduces huntingtin aggregation and toxicity (Posey et al., 2018; Ceccon et al., 2020): profilins maintain huntingtin in soluble states and, thus, delay its clustering into first nucleation seeds. On one hand, binding of PFN1 to the PRD region of huntingtin abrogates its capability to form aggregation-prone tetramers. On the other hand, PFN1 also stabilizes huntingtin monomers and dimers, which don’t oligomerize further (Ceccon et al., 2020). However, profilins lose their attenuating effect on huntingtin toxicity, once larger aggregates are formed. The interaction with huntingtin is negatively regulated by the Rho kinase-dependent phosphorylation of PFN1 at serine 138 within its poly-proline binding area (Figure 6.8; Shao et al., 2008; Shao and Diamond, 2012). Pharmacological inhibition of ROCK accordingly inhibits huntingtin aggregation and toxicity in cell culture and animal models (Bauer et al., 2009; Li et al., 2013), which alters profilin activity along with other ROCK target proteins (Narayanan et al., 2016). These biochemical and cell biological findings are in line with a comparative study showing the correlation of reduced profilin levels with the progression of Huntington disease in patients (Figure 6.9; Burnett et al., 2008).
The survival of motoneuron 1 protein (SMN1) is another ligand that links profilin to a major neurological disorder known as spinal muscular atrophy (SMA). SMN is probably best known for its roles in RNA splicing by assembling and guiding small nuclear riboproteins (snRNPs). However, they broadly participate in various processes, such as RNA metabolism, mRNA trafficking and localization, cell signaling, and also cytoskeletal dynamics (see for review Singh et al., 2017). Deletions or mutations in the human SMN1 gene cause SMA, leading to progressive loss of lower motoneurons followed by muscle atrophy. SMA varies in its onset and progression, while severe forms develop prenatally and are frequently fatal for infants. Patients with less severe SMA vary in their symptoms ranging from cases, who experience muscle weakness to never being able to walk. The pathogenesis and severity of SMA relies on the type of mutations in the SMN1 gene and the homologous SMN2. The SMN2 isoform is rather unstable through the exclusion of exon7 and cannot fully compensate SMN1. However, increased SMN2 protein levels can render SMA to its milder courses (see for review Kolb and Kissel, 2015). SMN protein interacts with cytoskeletal proteins (Fallini et al., 2012), suggesting that cytoskeleton dynamics might be perturbed in SMA. In particular, SMN1 interact with both PFN1 and PFN2a, while it binds stronger to PFN2a (Giesemann et al., 1999). Pathogenic missense mutations or deletions of exon 5 or 7 in SMN1 disrupt the interaction with PFN2a (Sharma et al., 2005). Mapping the SMN protein shows C-terminal poly-proline motifs being critical for the interaction with profilins. Moreover, endogenous PFN1 and PFN2a co-localize with SMN in neurite-like processes of PC12 cells and nuclei of motoneurons (Giesemann et al., 1999; Sharma et al., 2005). In vitro, SMN attenuates PFN2a’s ability to inhibit actin polymerization assays using pyrene-labeled G-actin (Sharma et al., 2005). These findings implicate a direct regulative role of SMN on profilin functions in actin dynamics. Moreover, other studies indicate a direct regulative role of SMN on profilin protein levels and function: Analyses of SMN-deficient fission yeast show splicing defects in the profilin gene. The SMN-dependent splice defect decreases profilin protein levels resulting in tilted actin network homeostasis, which translate into perturbed cytokinesis and endocytosis in fisson yeast (Figure 6.10; Antoine et al., 2020). However, it is noteworthy that fission yeast expresses only one profilin isoform, which may differ in its regulation by SMN from the mammalian isoforms. Accordingly, depletion of SMN in rat PC12 cells causes an increase in PFN2a protein (Figure 6.11; Bowerman et al., 2007). Furthermore, reduced SMN levels affect post-translational modifications of PFN2a: This profilin isoform is hyper-phosphorylated at multiple sites within its PLP-binding site, when SMN is depleted in cell lines and in spinal cords of a SMA mouse model (Nölle et al., 2011). These findings implicate that the lack of SMN-PFN2a complexes increases the accessibility of PFN2a to its upstream Rho kinase (ROCK). While levels of phosphorylated PFN2a are increased in SMA settings, the phosphorylation of other ROCK targets, such as the myosin light chain protein phosphatase (Myosin LCPPase) and the cofilin-regulating LIM-kinase, are reduced (Figure 6.12). Such changes in ROCK-dependent phosphorylation implicate misbalanced activities of PFN2a, myosins, and cofilin in SMN-deficient cells. The consequences are likely perturbed cytoskeletal dynamics in line with observed changes in F/G-actin ratio, formation of stress-related actin rods and impaired neurite outgrowth in SMN-depleted PC12-cells and SMA motoneurons (Nölle et al., 2011; Walter et al., 2021). Thus, erratic PFN2a levels and activity correlate with the pathogenesis of SMA but further investigations are necessary to clarify the molecular mechanism involved.
In this article, we provided an overview of general profilin features and reviewed the substantial progress made in studying the structural, biochemical, and functional properties of the different profilin isoforms in mammals. We focused on the functional diversity of PFN1 and PFN2a in the central nervous system under healthy and pathological conditions. However, our review also shows that we are still at the beginning of understanding the diverse roles that profilin and its isoforms play in cellular contexts. For many described profilin ligands we still do not know the impact that the interaction with profilins has on the corresponding cellular processes. Moreover, the functions of three out of five mammalian profilin isoforms are unknown, while discrepancies regarding the isoform-specific roles of PFN1 and PFN2a wait to be resolved. In view of profilin-associated diseases, more research is required to understand the involved molecular mechanisms behind the pathologies. Future research that tackles these open questions will immensely profit from newly developed methods and tools. The increasingly improving CRISPR/Cas-based methods will open new avenues to study profilins by loss of function, gene-tagging, and gain of function experiments in a great variety of cell types and organisms (see for review (Pickar-Oliver and Gersbach, 2019). Particularly tagging of endogenous PFN1 will be feasible by new profilin constructs internally fused to fluorescent proteins (Nejedla et al., 2017), as they circumvent the impaired ligand binding of traditional GFP-fused PFN1 variants (Geese et al., 2000; Wittenmayer et al., 2000). A better insight into the functions of profilin and its isoforms will help us to understand fundamental cell biological mechanisms in health and disease.
KM, MO, and JS wrote the manuscript. JS created the figures. All authors contributed to the article and approved the submitted version.
We acknowledge support from the Open Access Publication Fund of the Charité—Universitätsmedizin Berlin.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank James E. Bear (University of North Carolina) for his expert opinion and critically reading of our revised manuscript. We thank Britta Eickholt (Charité Universitätsmedizin Berlin) for her great support and interest in our work. This article relies on a wealth of data from many different laboratories. We have cited the appropriate original publications wherever possible, and we apologize to all researchers whose work on profilins was not included in this article. The figures were created with BioRender.com.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.681122/full#supplementary-material
Ackermann, M., and Matus, A. (2003). Activity-induced targeting of profilin and stabilization of dendritic spine morphology. Nat. Neurosci. 6, 1194–1200. doi: 10.1038/nn1135
Alkam, D., Feldman, E. Z., Singh, A., and Kiaei, M. (2017). Profilin1 biology and its mutation, actin(g) in disease. Cell. Mol. Life Sci. 74, 967–981. doi: 10.1007/s00018-016-2372-1
Antoine, M., Patrick, K. L., Soret, J., Duc, P., Rage, F., Cacciottolo, R., et al. (2020). Splicing defects of the profilin gene alter actin dynamics in an S. pombe SMN mutant. iScience 23, 100809. doi: 10.1016/j.isci.2019.100809
Arasada, R., Gloss, A., Tunggal, B., Joseph, J. M., Rieger, D., Mondal, S., et al. (2007). Profilin isoforms in Dictyostelium discoideum. Biochim. Biophys. Acta 1773, 631–641. doi: 10.1016/j.bbamcr.2007.03.009
Bagni, C., and Zukin, R. S. (2019). A synaptic perspective of fragile X syndrome and autism spectrum disorders. Neuron 101, 1070–1088. doi: 10.1016/j.neuron.2019.02.041
Barham, C., Fil, D., Byrum, S. D., Rahmatallah, Y., Glazko, G., and Kiaei, M. (2018). RNA-seq analysis of spinal cord tissues from hPFN1G118V transgenic mouse model of ALS at pre-symptomatic and end-stages of disease. Sci. Rep. 8:13737. doi: 10.1038/s41598-018-31132-y
Bauer, P. O., Wong, H. K., Oyama, F., Goswami, A., Okuno, M., Kino, Y., et al. (2009). Inhibition of Rho kinases enhances the degradation of mutant huntingtin. J. Biol. Chem. 284, 13153–13164. doi: 10.1074/jbc.M809229200
Behnen, M., Murk, K., Kursula, P., Cappallo-Obermann, H., Rothkegel, M., Kierszenbaum, A. L., et al. (2009). Testis-expressed profilins 3 and 4 show distinct functional characteristics and localize in the acroplaxome-manchette complex in spermatids. BMC Cell Biol. 10:34. doi: 10.1186/1471-2121-10-34
Bieling, P., Hansen, S. D., Akin, O., Li, T.-D., Hayden, C. C., Fletcher, D. A., et al. (2018). WH2 and proline-rich domains of WASP-family proteins collaborate to accelerate actin filament elongation. EMBO J. 37, 102–121. doi: 10.15252/embj.201797039
Binette, F., Bénard, M., Laroche, A., Pierron, G., Lemieux, G., and Pallotta, D. (1990). Cell-specific expression of a profilin gene family. DNA Cell Biol. 9, 323–334. doi: 10.1089/dna.1990.9.323
Björkegren, C., Rozycki, M., Schutt, C. E., Lindberg, U., and Karlsson, R. (1993). Mutagenesis of human profilin locates its poly(L-proline)-binding site to a hydrophobic patch of aromatic amino acids. FEBS Lett. 333, 123–126. doi: 10.1016/0014-5793(93)80388-b
Blasco, R., Cole, N. B., and Moss, B. (1991). Sequence analysis, expression, and deletion of a vaccinia virus gene encoding a homolog of profilin, a eukaryotic actin-binding protein. J. Virol. 65, 4598–4608. doi: 10.1128/JVI.65.9.4598-4608.1991
Boopathy, S., Silvas, T. V., Tischbein, M., Jansen, S., Shandilya, S. M., Zitzewitz, J. A., et al. (2015). Structural basis for mutation-induced destabilization of profilin 1 in ALS. Proc. Natl. Acad. Sci. U.S.A. 112, 7984–7989. doi: 10.1073/pnas.1424108112
Borovac, J., Bosch, M., and Okamoto, K. (2018). Regulation of actin dynamics during structural plasticity of dendritic spines: signaling messengers and actin-binding proteins. Mol. Cell. Neurosci. 91, 122–130. doi: 10.1016/j.mcn.2018.07.001
Bowerman, M., Shafey, D., and Kothary, R. (2007). Smn depletion alters profilin II expression and leads to upregulation of the RhoA/ROCK pathway and defects in neuronal integrity. J. Mol. Neurosci. 32, 120–131. doi: 10.1007/s12031-007-0024-5
Breitsprecher, D., and Goode, B. L. (2013). Formins at a glance. J. Cell Sci. 126, 1–7. doi: 10.1242/jcs.107250
Breitsprecher, D., Kiesewetter, A. K., Linkner, J., Urbanke, C., Resch, G. P., Small, J. V., et al. (2008). Clustering of VASP actively drives processive, WH2 domain-mediated actin filament elongation. EMBO J. 27, 2943–2954. doi: 10.1038/emboj.2008.211
Brettle, M., Stefen, H., Djordjevic, A., Fok, S. Y. Y., Chan, J. W., van Hummel, A., et al. (2019). Developmental expression of mutant PFN1 in motor neurons impacts neuronal growth and motor performance of young and adult mice. Front. Mol. Neurosci. 12:231. doi: 10.3389/fnmol.2019.00231
Brown, V., Jin, P., Ceman, S., Darnell, J. C., O’Donnell, W. T., Tenenbaum, S. A., et al. (2001). Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 107, 477–487. doi: 10.1016/s0092-8674(01)00568-2
Brühmann, S., Ushakov, D. S., Winterhoff, M., Dickinson, R. B., Curth, U., and Faix, J. (2017). Distinct VASP tetramers synergize in the processive elongation of individual actin filaments from clustered arrays. Proc. Natl. Acad. Sci. U.S.A. 114, E5815–E5824. doi: 10.1073/pnas.1703145114
Burnett, B. G., Andrews, J., Ranganathan, S., Fischbeck, K. H., and Di Prospero, N. A. (2008). Expression of expanded polyglutamine targets profilin for degradation and alters actin dynamics. Neurobiol. Dis. 30, 365–374. doi: 10.1016/j.nbd.2008.02.007
Carlsson, L., Nyström, L. E., Sundkvist, I., Markey, F., and Lindberg, U. (1977). Actin polymerizability is influenced by profilin, a low molecular weight protein in non-muscle cells. J. Mol. Biol. 115, 465–483. doi: 10.1016/0022-2836(77)90166-8
Ceccon, A., Tugarinov, V., Ghirlando, R., and Clore, G. M. (2020). Abrogation of prenucleation, transient oligomerization of the Huntingtin exon 1 protein by human profilin I. Proc. Natl. Acad. Sci. U.S.A. 117, 5844–5852. doi: 10.1073/pnas.1922264117
Cedergren-Zeppezauer, E. S., Goonesekere, N. C., Rozycki, M. D., Myslik, J. C., Dauter, Z., Lindberg, U., et al. (1994). Crystallization and structure determination of bovine profilin at 2.0 A resolution. J. Mol. Biol. 240, 459–475. doi: 10.1006/jmbi.1994.1461
Chédotal, A. (2010). Should I stay or should I go? Becoming a granule cell. Trends Neurosci. 33, 163–172. doi: 10.1016/j.tins.2010.01.004
Chen, Y., Zheng, Z.-Z., Huang, R., Chen, K., Song, W., Zhao, B., et al. (2013). PFN1 mutations are rare in Han Chinese populations with amyotrophic lateral sclerosis. Neurobiol. Aging 34:1922.e1–e5. doi: 10.1016/j.neurobiolaging.2013.01.013
Courtemanche, N., and Pollard, T. D. (2013). Interaction of profilin with the barbed end of actin filaments. Biochemistry 52, 6456–6466. doi: 10.1021/bi400682n
Darnell, J. C., Van Driesche, S. J., Zhang, C., Hung, K. Y. S., Mele, A., Fraser, C. E., et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. doi: 10.1016/j.cell.2011.06.013
Davey, R. J., and Moens, P. D. (2020). Profilin: many facets of a small protein. Biophys. Rev. 12, 827–849. doi: 10.1007/s12551-020-00723-3
Del Poggetto, E., Bemporad, F., Tatini, F., and Chiti, F. (2015a). Mutations of profilin-1 associated with amyotrophic lateral sclerosis promote aggregation due to structural changes of its native state. ACS Chem. Biol. 10, 2553–2563. doi: 10.1021/acschembio.5b00598
Del Poggetto, E., Chiti, F., and Bemporad, F. (2015b). The folding process of human profilin-1, a novel protein associated with familial amyotrophic lateral sclerosis. Sci. Rep. 5:2332. doi: 10.1038/srep12332
Del Poggetto, E., Gori, L., and Chiti, F. (2016). Biophysical analysis of three novel profilin-1 variants associated with amyotrophic lateral sclerosis indicates a correlation between their aggregation propensity and the structural features of their globular state. Biol. Chem. 397, 927–937. doi: 10.1515/hsz-2016-0154
Del Poggetto, E., Toto, A., Aloise, C., Di Piro, F., Gori, L., Malatesta, F., et al. (2018). Stability of an aggregation-prone partially folded state of human profilin-1 correlates with aggregation propensity. J. Biol. Chem. 293, 10303–10313. doi: 10.1074/jbc.RA118.002087
Di Nardo, A., Gareus, R., Kwiatkowski, D., and Witke, W. (2000). Alternative splicing of the mouse profilin II gene generates functionally different profilin isoforms. J. Cell Sci. 113(Pt 21), 3795–3803. doi: 10.1242/jcs.113.21.3795
Eriksson, A. E., Baase, W. A., Wozniak, J. A., and Matthews, B. W. (1992a). A cavity-containing mutant of T4 lysozyme is stabilized by buried benzene. Nature 355, 371–373. doi: 10.1038/355371a0
Eriksson, A. E., Baase, W. A., Zhang, X. J., Heinz, D. W., Blaber, M., Baldwin, E. P., et al. (1992b). Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science 255, 178–183. doi: 10.1126/science.1553543
Fallini, C., Bassell, G. J., and Rossoll, W. (2012). Spinal muscular atrophy: the role of SMN in axonal mRNA regulation. Brain Res. 1462, 81–92. doi: 10.1016/j.brainres.2012.01.044
Ferron, F., Rebowski, G., Lee, S. H., and Dominguez, R. (2007). Structural basis for the recruitment of profilin-actin complexes during filament elongation by Ena/VASP. EMBO J. 26, 4597–4606. doi: 10.1038/sj.emboj.7601874
Figley, M. D., Bieri, G., Kolaitis, R.-M., Taylor, J. P., and Gitler, A. D. (2014). Profilin 1 associates with stress granules and ALS-linked mutations alter stress granule dynamics. J. Neurosci. 34, 8083–8097. doi: 10.1523/JNEUROSCI.0543-14.2014
Fil, D., DeLoach, A., Yadav, S., Alkam, D., MacNicol, M., Singh, A., et al. (2017). Mutant profilin1 transgenic mice recapitulate cardinal features of motor neuron disease. Hum. Mol. Genet. 26, 686–701. doi: 10.1093/hmg/ddw429
Fratta, P., Charnock, J., Collins, T., Devoy, A., Howard, R., Malaspina, A., et al. (2014). Profilin1 E117G is a moderate risk factor for amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 85, 506–508. doi: 10.1136/jnnp-2013-306761
Freischmidt, A., Schöpflin, M., Feiler, M. S., Fleck, A.-K., Ludolph, A. C., and Weishaupt, J. H. (2015). Profilin 1 with the amyotrophic lateral sclerosis associated mutation T109M displays unaltered actin binding and does not affect the actin cytoskeleton. BMC Neurosci. 16:77. doi: 10.1186/s12868-015-0214-y
Funk, J., Merino, F., Venkova, L., Heydenreich, L., Kierfeld, J., Vargas, P., et al. (2019). Profilin and formin constitute a pacemaker system for robust actin filament growth. eLife 8:e50963. doi: 10.7554/eLife.50963
Gareus, R., Di Nardo, A., Rybin, V., and Witke, W. (2006). Mouse profilin 2 regulates endocytosis and competes with SH3 ligand binding to dynamin 1. J. Biol. Chem. 281, 2803–2811. doi: 10.1074/jbc.M503528200
Geese, M., Schlüter, K., Rothkegel, M., Jockusch, B. M., Wehland, J., and Sechi, A. S. (2000). Accumulation of profilin II at the surface of Listeria is concomitant with the onset of motility and correlates with bacterial speed. J. Cell Sci. 113(Pt 8), 1415–1426. doi: 10.1242/jcs.113.8.1415
Giampetruzzi, A., Danielson, E. W., Gumina, V., Jeon, M., Boopathy, S., Brown, R. H., et al. (2019). Modulation of actin polymerization affects nucleocytoplasmic transport in multiple forms of amyotrophic lateral sclerosis. Nat. Commun. 10:3827. doi: 10.1038/s41467-019-11837-y
Giesemann, T., Rathke-Hartlieb, S., Rothkegel, M., Bartsch, J. W., Buchmeier, S., Jockusch, B. M., et al. (1999). A role for polyproline motifs in the spinal muscular atrophy protein SMN. Profilins bind to and colocalize with smn in nuclear gems. J. Biol. Chem. 274, 37908–37914. doi: 10.1074/jbc.274.53.37908
Giesemann, T., Schwarz, G., Nawrotzki, R., Berhörster, K., Rothkegel, M., Schlüter, K., et al. (2003). Complex formation between the postsynaptic scaffolding protein gephyrin, profilin, and Mena: a possible link to the microfilament system. J. Neurosci. 23, 8330–8339. doi: 10.1523/jneurosci.23-23-08330.2003
Gilmore, E. C., and Herrup, K. (1997). Cortical development: layers of complexity. Curr. Biol. 7, R231–R234. doi: 10.1016/s0960-9822(06)00108-4
Görlich, A., Zimmermann, A.-M., Schober, D., Böttcher, R. T., Sassoè-Pognetto, M., Friauf, E., et al. (2012). Preserved morphology and physiology of excitatory synapses in profilin1-deficient mice. PLoS One 7:e30068. doi: 10.1371/journal.pone.0030068
Götz, M., Hartfuss, E., and Malatesta, P. (2002). Radial glial cells as neuronal precursors: a new perspective on the correlation of morphology and lineage restriction in the developing cerebral cortex of mice. Brain Res. Bull. 57, 777–788. doi: 10.1016/s0361-9230(01)00777-8
Grenklo, S., Johansson, T., Bertilson, L., and Karlsson, R. (2004). Anti-actin antibodies generated against profilin:actin distinguish between non-filamentous and filamentous actin, and label cultured cells in a dotted pattern. Eur. J. Cell Biol. 83, 413–423. doi: 10.1078/0171-9335-00400
Guljamow, A., Jenke-Kodama, H., Saumweber, H., Quillardet, P., Frangeul, L., Castets, A. M., et al. (2007). Horizontal gene transfer of two cytoskeletal elements from a eukaryote to a cyanobacterium. Curr. Biol. 17, R757–R759. doi: 10.1016/j.cub.2007.06.063
Haarer, B. K., Petzold, A. S., and Brown, S. S. (1993). Mutational analysis of yeast profilin. Mol. Cell. Biol. 13, 7864–7873. doi: 10.1128/mcb.13.12.7864
Haikarainen, T., Chen, W. Q., Lubec, G., and Kursula, P. (2009). Structure, modifications and ligand-binding properties of rat profilin 2a. Acta Crystallogr. D Biol. Crystallogr. 65, 303–311. doi: 10.1107/S0907444909000699
Hansen, S. D., and Mullins, R. D. (2010). VASP is a processive actin polymerase that requires monomeric actin for barbed end association. J. Cell Biol. 191, 571–584. doi: 10.1083/jcb.201003014
Henty-Ridilla, J. L., Juanes, M. A., and Goode, B. L. (2017). Profilin directly promotes microtubule growth through residues mutated in amyotrophic lateral sclerosis. Curr. Biol. 27, 3535–3543.e4. doi: 10.1016/j.cub.2017.10.002
Hu, E., Chen, Z., Fredrickson, T., and Zhu, Y. (2001). Molecular cloning and characterization of profilin-3: a novel cytoskeleton-associated gene expressed in rat kidney and testes. Exp. Nephrol. 9, 265–274. doi: 10.1159/000052621
Hulisz, D. (2018). Amyotrophic lateral sclerosis: disease state overview. Am. J. Manag. Care 24, S320–S326.
Ingre, C., Landers, J. E., Rizik, N., Volk, A. E., Akimoto, C., Birve, A., et al. (2013). A novel phosphorylation site mutation in profilin 1 revealed in a large screen of US, Nordic, and German amyotrophic lateral sclerosis/frontotemporal dementia cohorts. Neurobiol. Aging 34:1708.e1–e6. doi: 10.1016/j.neurobiolaging.2012.10.009
Jégou, A., Niedermayer, T., Orbán, J., Didry, D., Lipowsky, R., Carlier, M.-F., et al. (2011). Individual actin filaments in a microfluidic flow reveal the mechanism of ATP hydrolysis and give insight into the properties of profilin. PLoS Biol. 9:e1001161. doi: 10.1371/journal.pbio.1001161
Junker, J. L., and Heine, U. I. (1987). Effect of adhesion factors fibronectin, laminin, and type IV collagen on spreading and growth of transformed and control rat liver epithelial cells. Cancer Res. 47, 3802–3807.
Kaiser, D. A., and Pollard, T. D. (1996). Characterization of actin and poly-L-proline binding sites of Acanthamoeba profilin with monoclonal antibodies and by mutagenesis. J. Mol. Biol. 256, 89–107. doi: 10.1006/jmbi.1996.0070
Kolb, S. J., and Kissel, J. T. (2015). Spinal muscular atrophy. Neurol. Clin. 33, 831–846. doi: 10.1016/j.ncl.2015.07.004
Kullmann, J. A., Meyer, S., Pipicelli, F., Kyrousi, C., Schneider, F., Bartels, N., et al. (2020). Profilin1-dependent F-actin assembly controls division of apical radial glia and neocortex development. Cereb. Cortex 30, 3467–3482. doi: 10.1093/cercor/bhz321
Kullmann, J. A., Neumeyer, A., Gurniak, C. B., Friauf, E., Witke, W., and Rust, M. B. (2011). Profilin1 is required for glial cell adhesion and radial migration of cerebellar granule neurons. EMBO Rep. 13, 75–82. doi: 10.1038/embor.2011.211
Kullmann, J. A., Neumeyer, A., Wickertsheim, I., Böttcher, R. T., Costell, M., Deitmer, J. W., et al. (2012). Purkinje cell loss and motor coordination defects in profilin1 mutant mice. Neuroscience 223, 355–364. doi: 10.1016/j.neuroscience.2012.07.055
Kullmann, J. A., Wickertsheim, I., Minnerup, L., Costell, M., Friauf, E., and Rust, M. B. (2015). Profilin1 activity in cerebellar granule neurons is required for radial migration in vivo. Cell Adhes. Migr. 9, 247–253. doi: 10.4161/19336918.2014.983804
Lambrechts, A., Braun, A., Jonckheere, V., Aszodi, A., Lanier, L. M., Robbens, J., et al. (2000). Profilin II is alternatively spliced, resulting in profilin isoforms that are differentially expressed and have distinct biochemical properties. Mol. Cell. Biol. 20, 8209–8219. doi: 10.1128/mcb.20.21.8209-8219.2000
Lambrechts, A., Jonckheere, V., Dewitte, D., Vandekerckhove, J., and Ampe, C. (2002). Mutational analysis of human profilin I reveals a second PI(4,5)-P2 binding site neighbouring the poly(L-proline) binding site. BMC Biochem. 3:12. doi: 10.1186/1471-2091-3-12
Lambrechts, A., van Damme, J., Goethals, M., Vandekerckhove, J., and Ampe, C. (1995). Purification and characterization of bovine profilin II. Actin, poly(L-proline) and inositolphospholipid binding. Eur. J. Biochem. 230, 281–286. doi: 10.1111/j.1432-1033.1995.tb20561.x
Lambrechts, A., Verschelde, J. L., Jonckheere, V., Goethals, M., Vandekerckhove, J., and Ampe, C. (1997). The mammalian profilin isoforms display complementary affinities for PIP2 and proline-rich sequences. EMBO J. 16, 484–494. doi: 10.1093/emboj/16.3.484
Lamprecht, R., Farb, C. R., Rodrigues, S. M., and LeDoux, J. E. (2006). Fear conditioning drives profilin into amygdala dendritic spines. Nat. Neurosci. 9, 481–483. doi: 10.1038/nn1672
Lassing, I., and Lindberg, U. (1985). Specific interaction between phosphatidylinositol 4,5-bisphosphate and profilactin. Nature 314, 472–474. doi: 10.1038/314472a0
Lassing, I., and Lindberg, U. (1988). Specificity of the interaction between phosphatidylinositol 4,5-bisphosphate and the profilin:actin complex. J. Cell. Biochem. 37, 255–267. doi: 10.1002/jcb.240370302
Lee, C. W., Vitriol, E. A., Shim, S., Wise, A. L., Velayutham, R. P., and Zheng, J. Q. (2013). Dynamic localization of G-actin during membrane protrusion in neuronal motility. Curr. Biol. 23, 1046–1056. doi: 10.1016/j.cub.2013.04.057
Li, M., Yasumura, D., Ma, A. A. K., Matthes, M. T., Yang, H., Nielson, G., et al. (2013). Intravitreal administration of HA-1077, a ROCK inhibitor, improves retinal function in a mouse model of huntington disease. PLoS One 8:e56026. doi: 10.1371/journal.pone.0056026
López-Bendito, G., and Molnár, Z. (2003). Thalamocortical development: how are we going to get there? Nat. Rev. Neurosci. 4, 276–289. doi: 10.1038/nrn1075
Luscieti, S., Galy, B., Gutierrez, L., Reinke, M., Couso, J., Shvartsman, M., et al. (2017). The actin-binding protein profilin 2 is a novel regulator of iron homeostasis. Blood 130, 1934–1945. doi: 10.1182/blood-2016-11-754382
Mahoney, N. M., Janmey, P. A., and Almo, S. C. (1997). Structure of the profilin-poly-L-proline complex involved in morphogenesis and cytoskeletal regulation. Nat. Struct. Biol. 4, 953–960. doi: 10.1038/nsb1197-953
Matsukawa, K., Hashimoto, T., Matsumoto, T., Ihara, R., Chihara, T., Miura, M., et al. (2016). Familial amyotrophic lateral sclerosis-linked mutations in profilin 1 exacerbate TDP-43-induced degeneration in the retina of Drosophila melanogaster through an increase in the cytoplasmic localization of TDP-43. J. Biol. Chem. 291, 23464–23476. doi: 10.1074/jbc.M116.729152
Merlotti, D., Materozzi, M., Bianciardi, S., Guarnieri, V., Rendina, D., Volterrani, L., et al. (2020). Mutation of PFN1 gene in an early onset, polyostotic paget-like disease. J. Clin. Endocrinol. Metab 105:dgaa252. doi: 10.1210/clinem/dgaa252
Michaelsen, K., Murk, K., Zagrebelsky, M., Dreznjak, A., Jockusch, B. M., Rothkegel, M., et al. (2010). Fine-tuning of neuronal architecture requires two profilin isoforms. Proc. Natl. Acad. Sci. U.S.A. 107, 15780–15785. doi: 10.1073/pnas.1004406107
Michaelsen-Preusse, K., Zessin, S., Grigoryan, G., Scharkowski, F., Feuge, J., Remus, A., et al. (2016). Neuronal profilins in health and disease: relevance for spine plasticity and fragile X syndrome. Proc. Natl. Acad. Sci. U.S.A. 113, 3365–3370. doi: 10.1073/pnas.1516697113
Miki, H., Suetsugu, S., and Takenawa, T. (1998). WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. EMBO J. 17, 6932–6941. doi: 10.1093/emboj/17.23.6932
Miyashiro, K. Y., Beckel-Mitchener, A., Purk, T. P., Becker, K. G., Barret, T., Liu, L., et al. (2003). RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron 37, 417–431. doi: 10.1016/s0896-6273(03)00034-5
Molotkov, D., Zobova, S., Arcas, J. M., and Khiroug, L. (2013). Calcium-induced outgrowth of astrocytic peripheral processes requires actin binding by Profilin-1. Cell Calcium 53, 338–348. doi: 10.1016/j.ceca.2013.03.001
Mouneimne, G., Hansen, S. D., Selfors, L. M., Petrak, L., Hickey, M. M., Gallegos, L. L., et al. (2012). Differential remodeling of actin cytoskeleton architecture by profilin isoforms leads to distinct effects on cell migration and invasion. Cancer Cell 22, 615–630. doi: 10.1016/j.ccr.2012.09.027
Mullins, R. D., Kelleher, J. F., Xu, J., and Pollard, T. D. (1998). Arp2/3 complex from Acanthamoeba binds profilin and cross-links actin filaments. Mol. Biol. Cell 9, 841–852. doi: 10.1091/mbc.9.4.841
Murk, K., Buchmeier, S., Jockusch, B. M., and Rothkegel, M. (2009). In birds, profilin-2a is ubiquitously expressed and contributes to actin-based motility. J. Cell Sci. 122, 957–964. doi: 10.1242/jcs.041715
Narayanan, K. L., Chopra, V., Rosas, H. D., Malarick, K., and Hersch, S. (2016). Rho kinase pathway alterations in the brain and leukocytes in Huntington’s disease. Mol. Neurobiol. 53, 2132–2140. doi: 10.1007/s12035-015-9147-9
Nejedlá, M., Klebanovych, A., Sulimenko, V., Sulimenko, T., Dráberová, E., Dráber, P., et al. (2021). The actin regulator profilin 1 is functionally associated with the mammalian centrosome. Life Sci. Alliance 4:e202000655. doi: 10.26508/lsa.202000655
Nejedla, M., Li, Z., Masser, A. E., Biancospino, M., Spiess, M., Mackowiak, S. D., et al. (2017). A fluorophore fusion construct of human profilin i with non-compromised Poly(L-Proline) binding capacity suitable for imaging. J. Mol. Biol. 429, 964–976. doi: 10.1016/j.jmb.2017.01.004
Nejedla, M., Sadi, S., Sulimenko, V., de Almeida, F. N., Blom, H., Draber, P., et al. (2016). Profilin connects actin assembly with microtubule dynamics. Mol. Biol. Cell 27, 2381–2393. doi: 10.1091/mbc.E15-11-0799
Noctor, S. C., Martínez-Cerdeño, V., Ivic, L., and Kriegstein, A. R. (2004). Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat. Neurosci. 7, 136–144. doi: 10.1038/nn1172
Nodelman, I. M., Bowman, G. D., Lindberg, U., and Schutt, C. E. (1999). X-ray structure determination of human profilin II: a comparative structural analysis of human profilins. J. Mol. Biol. 294, 1271–1285. doi: 10.1006/jmbi.1999.3318
Nölle, A., Zeug, A., van Bergeijk, J., Tönges, L., Gerhard, R., Brinkmann, H., et al. (2011). The spinal muscular atrophy disease protein SMN is linked to the Rho-kinase pathway via profilin. Hum. Mol. Genet. 20, 4865–4878. doi: 10.1093/hmg/ddr425
Obermann, H., Raabe, I., Balvers, M., Brunswig, B., Schulze, W., and Kirchhoff, C. (2005). Novel testis-expressed profilin IV associated with acrosome biogenesis and spermatid elongation. Mol. Hum. Reprod. 11, 53–64. doi: 10.1093/molehr/gah132
Pae, M., and Romeo, G. R. (2014). The multifaceted role of profilin-1 in adipose tissue inflammation and glucose homeostasis. Adipocyte 3, 69–74. doi: 10.4161/adip.26965
Pandey, D. K., and Chaudhary, B. (2017). Evolutionary expansion and structural functionalism of the ancient family of profilin proteins. Gene 626, 70–86. doi: 10.1016/j.gene.2017.05.024
Paul, A. S., Paul, A., Pollard, T. D., and Pollard, T. (2008). The role of the FH1 domain and profilin in formin-mediated actin-filament elongation and nucleation. Curr. Biol. 18, 9–19. doi: 10.1016/j.cub.2007.11.062
Penisson, M., Ladewig, J., Belvindrah, R., and Francis, F. (2019). Genes and mechanisms involved in the generation and amplification of basal radial glial cells. Front. Cell. Neurosci. 13:381. doi: 10.3389/fncel.2019.00381
Perelroizen, I., Didry, D., Christensen, H., Chua, N. H., and Carlier, M. F. (1996). Role of nucleotide exchange and hydrolysis in the function of profilin in action assembly. J. Biol. Chem. 271, 12302–12309. doi: 10.1074/jbc.271.21.12302
Pernier, J., Shekhar, S., Jegou, A., Guichard, B., and Carlier, M.-F. (2016). Profilin interaction with actin filament barbed end controls dynamic instability, capping, branching, and motility. Dev. Cell 36, 201–214. doi: 10.1016/j.devcel.2015.12.024
Pickar-Oliver, A., and Gersbach, C. A. (2019). The next generation of CRISPR-Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 20, 490–507. doi: 10.1038/s41580-019-0131-5
Pilo Boyl, P., Di Nardo, A., Mulle, C., Sassoè-Pognetto, M., Panzanelli, P., Mele, A., et al. (2007). Profilin2 contributes to synaptic vesicle exocytosis, neuronal excitability, and novelty-seeking behavior. EMBO J. 26, 2991–3002. doi: 10.1038/sj.emboj.7601737
Pimm, M. L., Hotaling, J., and Henty-Ridilla, J. L. (2020). Profilin choreographs actin and microtubules in cells and cancer. Int. Rev. Cell Mol. Biol. 355, 155–204. doi: 10.1016/bs.ircmb.2020.05.005
Pinto-Costa, R., Sousa, S. C., Leite, S. C., Nogueira-Rodrigues, J., Ferreira da Silva, T., Machado, D., et al. (2020). Profilin 1 delivery tunes cytoskeletal dynamics toward CNS axon regeneration. J. Clin. Invest. 130, 2024–2040. doi: 10.1172/JCI125771
Polet, D., Lambrechts, A., Vandepoele, K., Vandekerckhove, J., and Ampe, C. (2007). On the origin and evolution of vertebrate and viral profilins. FEBS Lett. 581, 211–217. doi: 10.1016/j.febslet.2006.12.013
Pollard, T. D., and Cooper, J. A. (2009). Actin, a central player in cell shape and movement. Science 326, 1208–1212. doi: 10.1126/science.1175862
Posey, A. E., Ruff, K. M., Harmon, T. S., Crick, S. L., Li, A., Diamond, M. I., et al. (2018). Profilin reduces aggregation and phase separation of huntingtin N-terminal fragments by preferentially binding to soluble monomers and oligomers. J. Biol. Chem. 293, 3734–3746. doi: 10.1074/jbc.RA117.000357
Rawe, V. Y., Payne, C., and Schatten, G. (2006). Profilin and actin-related proteins regulate microfilament dynamics during early mammalian embryogenesis. Hum. Reprod. (Oxf. Engl.) 21, 1143–1153. doi: 10.1093/humrep/dei480
Reeve, S. P., Bassetto, L., Genova, G. K., Kleyner, Y., Leyssen, M., Jackson, F. R., et al. (2005). The Drosophila fragile X mental retardation protein controls actin dynamics by directly regulating profilin in the brain. Curr. Biol. 15, 1156–1163. doi: 10.1016/j.cub.2005.05.050
Romero, S., Le Clainche, C., Didry, D., Egile, C., Pantaloni, D., and Carlier, M.-F. (2004). Formin is a processive motor that requires profilin to accelerate actin assembly and associated ATP hydrolysis. Cell 119, 419–429. doi: 10.1016/j.cell.2004.09.039
Rotty, J. D., Wu, C., Haynes, E. M., Suarez, C., Winkelman, J. D., Johnson, H. E., et al. (2015). Profilin-1 serves as a gatekeeper for actin assembly by Arp2/3-dependent and -independent pathways. Dev. Cell 32, 54–67. doi: 10.1016/j.devcel.2014.10.026
Saberi, S., Stauffer, J. E., Schulte, D. J., and Ravits, J. (2015). Neuropathology of amyotrophic lateral sclerosis and its variants. Neurol. Clin. 33, 855–876. doi: 10.1016/j.ncl.2015.07.012
Saffary, R., and Xie, Z. (2011). FMRP regulates the transition from radial glial cells to intermediate progenitor cells during neocortical development. J. Neurosci. 31, 1427–1439. doi: 10.1523/JNEUROSCI.4854-10.2011
San Miguel-Ruiz, J. E., and Letourneau, P. C. (2014). The role of Arp2/3 in growth cone actin dynamics and guidance is substrate dependent. J. Neurosci. 34, 5895–5908. doi: 10.1523/JNEUROSCI.0672-14.2014
Santos, T. E., Schaffran, B., Broguière, N., Meyn, L., Zenobi-Wong, M., and Bradke, F. (2020). Axon growth of CNS neurons in three dimensions is amoeboid and independent of adhesions. Cell Rep. 32:107907. doi: 10.1016/j.celrep.2020.107907
Saudou, F., and Humbert, S. (2016). The biology of huntingtin. Neuron 89, 910–926. doi: 10.1016/j.neuron.2016.02.003
Scharkowski, F., Frotscher, M., Lutz, D., Korte, M., and Michaelsen-Preusse, K. (2018). Altered connectivity and synapse maturation of the hippocampal mossy fiber pathway in a mouse model of the fragile X syndrome. Cereb. Cortex 28, 852–867. doi: 10.1093/cercor/bhw408
Schiweck, J., Eickholt, B. J., and Murk, K. (2018). Important shapeshifter: mechanisms allowing astrocytes to respond to the changing nervous system during development, injury and disease. Front. Cell. Neurosci. 12:261. doi: 10.3389/fncel.2018.00261
Schlüter, K., Schleicher, M., and Jockusch, B. M. (1998). Effects of single amino acid substitutions in the actin-binding site on the biological activity of bovine profilin I. J. Cell Sci. 111(Pt 22), 3261–3273. doi: 10.1242/jcs.111.22.3261
Schmidt, E. J., Funes, S., McKeon, J. E., Morgan, B. R., Boopathy, S., O’Connor, L. C., et al. (2021). ALS-linked PFN1 variants exhibit loss and gain of functions in the context of formin-induced actin polymerization. Proc. Natl. Acad. Sci. U.S.A. 118:e2024605118. doi: 10.1073/pnas.2024605118
Schubert, V., Da Silva, J. S., and Dotti, C. G. (2006). Localized recruitment and activation of RhoA underlies dendritic spine morphology in a glutamate receptor-dependent manner. J. Cell Biol. 172, 453–467. doi: 10.1083/jcb.200506136
Schutt, C. E., Myslik, J. C., Rozycki, M. D., Goonesekere, N. C., and Lindberg, U. (1993). The structure of crystalline profilin-beta-actin. Nature 365, 810–816. doi: 10.1038/365810a0
Schweinhuber, S. K., Meßerschmidt, T., Hänsch, R., Korte, M., and Rothkegel, M. (2015). Profilin isoforms modulate astrocytic morphology and the motility of astrocytic processes. PLoS One 10:e0117244. doi: 10.1371/journal.pone.0117244
Scotto di Carlo, F., Pazzaglia, L., Esposito, T., and Gianfrancesco, F. (2020). The loss of profilin 1 causes early onset Paget’s disease of bone. J. Bone Miner. Res. 35, 1387–1398. doi: 10.1002/jbmr.3964
Selden, L. A., Kinosian, H. J., Estes, J. E., and Gershman, L. C. (1999). Impact of profilin on actin-bound nucleotide exchange and actin polymerization dynamics. Biochemistry 38, 2769–2778. doi: 10.1021/bi981543c
Shao, J., and Diamond, M. I. (2012). Protein phosphatase 1 dephosphorylates profilin-1 at Ser-137. PLoS One 7:e32802. doi: 10.1371/journal.pone.0032802
Shao, J., Welch, W. J., Diprospero, N. A., and Diamond, M. I. (2008). Phosphorylation of profilin by ROCK1 regulates polyglutamine aggregation. Mol. Cell. Biol. 28, 5196–5208. doi: 10.1128/MCB.00079-08
Sharma, A., Lambrechts, A., Hao, L. T., Le, T. T., Sewry, C. A., Ampe, C., et al. (2005). A role for complexes of survival of motor neurons (SMN) protein with gemins and profilin in neurite-like cytoplasmic extensions of cultured nerve cells. Exp. Cell Res. 309, 185–197. doi: 10.1016/j.yexcr.2005.05.014
Siddiqui, A. M., Brunner, R., Harris, G. M., Miller, A. L., Waletzki, B. E., Schmeichel, A. M., et al. (2021). Promoting neuronal outgrowth using ridged scaffolds coated with extracellular matrix proteins. Biomedicines 9:479. doi: 10.3390/biomedicines9050479
Singh, R. N., Howell, M. D., Ottesen, E. W., and Singh, N. N. (2017). Diverse role of survival motor neuron protein. Biochim. Biophys. Acta Gene Regul. Mech. 1860, 299–315. doi: 10.1016/j.bbagrm.2016.12.008
Skare, P., and Karlsson, R. (2002). Evidence for two interaction regions for phosphatidylinositol(4,5)-bisphosphate on mammalian profilin I. FEBS Lett. 522, 119–124. doi: 10.1016/s0014-5793(02)02913-7
Skruber, K., Warp, P. V., Shklyarov, R., Thomas, J. D., Swanson, M. S., Henty-Ridilla, J. L., et al. (2020). Arp2/3 and Mena/VASP require profilin 1 for actin network assembly at the leading edge. Curr. Biol. 30, 2651–2664.e5. doi: 10.1016/j.cub.2020.04.085
Smith, B. N., Vance, C., Scotter, E. L., Troakes, C., Wong, C. H., Topp, S., et al. (2015). Novel mutations support a role for Profilin 1 in the pathogenesis of ALS. Neurobiol. Aging 36:1602.e17–e27. doi: 10.1016/j.neurobiolaging.2014.10.032
Sohn, R. H., Chen, J., Koblan, K. S., Bray, P. F., and Goldschmidt-Clermont, P. J. (1995). Localization of a binding site for phosphatidylinositol 4,5-bisphosphate on human profilin. J. Biol. Chem. 270, 21114–21120. doi: 10.1074/jbc.270.36.21114
Stallmeyer, B., Schwarz, G., Schulze, J., Nerlich, A., Reiss, J., Kirsch, J., et al. (1999). The neurotransmitter receptor-anchoring protein gephyrin reconstitutes molybdenum cofactor biosynthesis in bacteria, plants, and mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 96, 1333–1338. doi: 10.1073/pnas.96.4.1333
Strasser, G. A., Rahim, N. A., VanderWaal, K. E., Gertler, F. B., and Lanier, L. M. (2004). Arp2/3 is a negative regulator of growth cone translocation. Neuron 43, 81–94. doi: 10.1016/j.neuron.2004.05.015
Suarez, C., Carroll, R. T., Burke, T. A., Christensen, J. R., Bestul, A. J., Sees, J. A., et al. (2015). Profilin regulates F-actin network homeostasis by favoring formin over Arp2/3 complex. Dev. Cell 32, 43–53. doi: 10.1016/j.devcel.2014.10.027
Suetsugu, S., Miki, H., and Takenawa, T. (1998). The essential role of profilin in the assembly of actin for microspike formation. EMBO J. 17, 6516–6526. doi: 10.1093/emboj/17.22.6516
Suk, T. R., and Rousseaux, M. W. C. (2020). The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 15:45. doi: 10.1186/s13024-020-00397-1
Tanaka, Y., Nonaka, T., Suzuki, G., Kametani, F., and Hasegawa, M. (2016). Gain-of-function profilin 1 mutations linked to familial amyotrophic lateral sclerosis cause seed-dependent intracellular TDP-43 aggregation. Hum. Mol. Genet. 25, 1420–1433. doi: 10.1093/hmg/ddw024
Tessier, C. R., and Broadie, K. (2008). Drosophila fragile X mental retardation protein developmentally regulates activity-dependent axon pruning. Dev. Camb. Engl. 135, 1547–1557. doi: 10.1242/dev.015867
Walter, L. M., Franz, P., Lindner, R., Tsiavaliaris, G., Hensel, N., and Claus, P. (2020). Profilin2a-phosphorylation as a regulatory mechanism for actin dynamics. FASEB J. 34, 2147–2160. doi: 10.1096/fj.201901883R
Walter, L. M., Rademacher, S., Pich, A., and Claus, P. (2021). Profilin2 regulates actin rod assembly in neuronal cells. Sci. Rep. 11:10287. doi: 10.1038/s41598-021-89397-9
Winkelman, J. D., Bilancia, C. G., Peifer, M., and Kovar, D. R. (2014). Ena/VASP enabled is a highly processive actin polymerase tailored to self-assemble parallel-bundled F-actin networks with Fascin. Proc. Natl. Acad. Sci. U.S.A. 111, 4121–4126. doi: 10.1073/pnas.1322093111
Witke, W., Podtelejnikov, A. V., Di Nardo, A., Sutherland, J. D., Gurniak, C. B., Dotti, C., et al. (1998). In mouse brain profilin I and profilin II associate with regulators of the endocytic pathway and actin assembly. EMBO J. 17, 967–976. doi: 10.1093/emboj/17.4.967
Witke, W., Sutherland, J. D., Sharpe, A., Arai, M., and Kwiatkowski, D. J. (2001). Profilin I is essential for cell survival and cell division in early mouse development. Proc. Natl. Acad. Sci. U.S.A. 98, 3832–3836. doi: 10.1073/pnas.051515498
Wittenmayer, N., Rothkegel, M., Jockusch, B. M., and Schlüter, K. (2000). Functional characterization of green fluorescent protein-profilin fusion proteins. Eur. J. Biochem. 267, 5247–5256. doi: 10.1046/j.1432-1327.2000.01600.x
Wu, C.-H., Fallini, C., Ticozzi, N., Keagle, P. J., Sapp, P. C., Piotrowska, K., et al. (2012). Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488, 499–503. doi: 10.1038/nature11280
Xue, M., Wakamoto, T., Kejlberg, C., Yoshimura, Y., Nielsen, T. A., Risør, M. W., et al. (2019). How internal cavities destabilize a protein. Proc. Natl. Acad. Sci. U.S.A. 116, 21031–21036. doi: 10.1073/pnas.1911181116
Yang, C., Danielson, E. W., Qiao, T., Metterville, J., Brown, R. H., Landers, J. E., et al. (2016). Mutant PFN1 causes ALS phenotypes and progressive motor neuron degeneration in mice by a gain of toxicity. Proc. Natl. Acad. Sci. U.S.A. 113, E6209–E6218. doi: 10.1073/pnas.1605964113
Yang, Q., Zhang, X.-F., Pollard, T. D., and Forscher, P. (2012). Arp2/3 complex-dependent actin networks constrain myosin II function in driving retrograde actin flow. J. Cell Biol. 197, 939–956. doi: 10.1083/jcb.201111052
Yuan, G., Cui, S., Chen, X., Song, H., Huang, C., Tong, J., et al. (2020). Detergent-insoluble inclusion constitutes the first pathology in PFN1 transgenic rats. J. Neurochem. 157, 1244–1252. doi: 10.1111/jnc.15139
Keywords: profilin, actin, cytoskeleton, central nervous system, amyotrophic lateral sclerosis, Fragile X syndrome, Huntington’s disease, spinal muscular atrophy
Citation: Murk K, Ornaghi M and Schiweck J (2021) Profilin Isoforms in Health and Disease – All the Same but Different. Front. Cell Dev. Biol. 9:681122. doi: 10.3389/fcell.2021.681122
Received: 15 March 2021; Accepted: 12 July 2021;
Published: 12 August 2021.
Edited by:
Michael Schnoor, Centro de Investigaciones y Estudios Avanzados, Instituto Politécnico Nacional de México (CINVESTAV), MexicoReviewed by:
Darrell Mousseau, University of Saskatchewan, CanadaCopyright © 2021 Murk, Ornaghi and Schiweck. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kai Murk, a2FpLm11cmtAY2hhcml0ZS5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.