 Mary Jo Talley
Mary Jo Talley Diana Nardini2
Diana Nardini2 Ronald R. Waclaw
Ronald R. Waclaw
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Cell Dev. Biol., 16 June 2021
Sec. Epigenomics and Epigenetics
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.673995
This article is part of the Research TopicRole of Genetics and Epigenetics in Major Structural MalformationsView all 28 articles
The leucine zipper-like transcriptional regulator 1 (Lztr1) is a BTB-Kelch domain protein involved in RAS/MAPK pathway regulation. Mutations in LZTR1 are associated with cancers and Noonan syndrome, the most common RASopathy. The expression and function of Lztr1 in the developing brain remains poorly understood. Here we show that Lztr1 is expressed in distinct regions of the telencephalon, the most anterior region of the forebrain. Lztr1 expression was robust in the cortex, amygdala, hippocampus, and oligodendrocytes in the white matter. To gain insight into the impact of Lztr1 deficiency, we generated a conditional knockout (cKO) restricted to the telencephalon using Foxg1IREScre/+. Lztr1 cKOs are viable to postnatal stages and show reduced Lztr1 expression in the telencephalon. Interestingly, Lztr1 cKOs exhibit an increase in MAPK pathway activation in white matter regions and subsequently show an altered expression of stage-specific markers in the oligodendrocyte lineage with increased oligodendrocyte progenitor cells (OPCs) and decreased markers of oligodendrocyte differentiation. Moreover, Lztr1 cKOs also exhibit an increased expression of the astrocyte marker GFAP. These results highlight the generation of a new mouse model to study Lztr1 deficiency in the brain and reveal a novel role for Lztr1 in normal oligodendrocyte and astrocyte development in the telencephalon.
LZTR1 was originally identified as one of the candidate genes deleted in some 22q11.2 deletion syndrome patients (Kurahashi et al., 1995). It is a unique member of the BTB-Kelch superfamily due to its localized expression to the golgi network via the C-terminal BTB/POZ domain (Nacak et al., 2006). LZTR1 is also thought to be a tumor suppressor as loss-of-function mutations have been associated with schwannomatosis and glioma (Frattini et al., 2013; Piotrowski et al., 2014). The function of Lztr1 has been elusive, but the protein contains six N-terminal Kelch motifs (Nacak et al., 2006) which are proposed to mediate protein–protein interactions and are associated with Cullin3 ubiquitination complexes (Shi et al., 2019). LZTR1 mutations have also been associated with Noonan syndrome (NS), a type of RASopathy affecting multiple organ systems, including growth, craniofacial, cardiovascular, and neurocognitive abnormalities (Yamamoto et al., 2015). Interestingly, mutations in LZTR1 are associated with both autosomal dominant and biallelic recessive forms of NS. Missense mutations in the Kelch domain that do not affect protein stability are associated with dominant NS, while loss-of-function or inactivating mutations are associated with biallelic recessive NS (Yamamoto et al., 2015; Johnston et al., 2018; Motta et al., 2019). LZTR1 is the first NS gene described to be inherited in this unique manner. While the NS clinical phenotype can be variable, Johnston et al. (2018) showed examples of early lethality and also cancers associated with the biallelic recessive mutations, which suggests that the LZTR1 loss-of-function or inactivating mutations might align with a more severe type of NS.
RASopathies, including NF1, NS, Costello syndrome, and cardiofaciocutaneous syndrome, are a group of distinct diseases that are caused by mutations that upregulate the RAS/MAPK pathway (reviewed in Tartaglia and Gelb, 2010). The identification of LZTR1 mutations with NS implicates LZTR1 as a regulator of the RAS/MAPK pathway. However, the function of LZTR1 in the RAS/MAPK pathway is still an active area of research. Recent studies have suggested that LZTR1 plays a key role in the ubiquitination of RAS proteins (Bigenzahn et al., 2018; Steklov et al., 2018). Indeed LZTR1 has been referred to as a “RAS killer protein” through the polyubiquitination and degradation of RAS-GTPases, thereby controlling the appropriate activation levels of the RAS/MAPK pathway (Abe et al., 2020). In addition to the regulation of RAS ubiquitination, LZTR1 has also been suggested to regulate other levels of the RAS/MAPK pathway through phosphorylation of RAF1 or stabilization of RIT1 to impact the activity of the MAPK pathway (Castel et al., 2019; Umeki et al., 2019). Given its association with human disease and the multiple lines of evidence suggesting that LZTR1 function is key to controlling MAPK pathway activation, the development of new approaches and models to modulate gene function is essential to understand the pathophysiology of LZTR1-associated diseases.
Neurocognitive abnormalities are observed in NS patients (Pierpont et al., 2009; Alfieri et al., 2011), making it necessary to understand how gene mutations associated with NS impact the developing brain. While progress continues to be made in determining the molecular role of LZTR1 in the RAS/MAPK pathway, the impact of LZTR1 loss, specifically in the brain, has not yet been examined. Heterozygous loss of Lztr1 in mice (Lztr1+/–) reproduces some of the craniofacial, cardiovascular, and growth-associated NS phenotypes in the heterozygous state (Steklov et al., 2018). Lethality in Lztr1–/– mouse embryos (Steklov et al., 2018) prevents the analysis of the biallelic loss-of-function mutations on cellular populations in the postnatal brain. Our study will be useful for understanding Lztr1 biology by developing a cell type-specific genetic mouse model to determine the impact of Lztr1 loss of function in the developing brain.
LZTR1 mutations are associated with NS and 22q11.2 deletion syndrome, both of which show a variety of phenotypes, including neurological dysfunction (Pierpont et al., 2009; Fiksinski et al., 2021). In this study, we examine a role for Lztr1 in the anterior forebrain, a region associated with higher neural functions, like cognition and emotion (Rubenstein, 2011). Our studies reveal that Lztr1 is highly expressed in the cortex, amygdala, hippocampus, and white matter regions. Utilizing a conditional knockout strategy restricted to the telencephalon that results in postnatal viability, we have identified a novel role for Lztr1 in the normal generation of oligodendrocyte progenitor cells (OPCs) and the balanced establishment of oligodendrocyte and astrocyte differentiation markers. Our results suggest that Lztr1 gene function is crucial for the normal generation of glial cells in the telencephalon.
The animal protocols for experiments using mice were approved by the Cincinnati Children’s Hospital Medical Center Institutional Animal Care and Use Committee and carried out in accordance with the National Institutes of Health guidelines. Lztr1tm1a/+ knock-out mice were first purchased from EMMA/EUCOMM (EM #06794), and the mouse generation strategy was as described in Skarnes et al. (2011). These mice were converted to a lacZ reporter line (Lztr1tm1b/+, also called Lztr1lacZ/+) using EIIA germline cre transgenic mice (JAX#003724). The Lztr1tm1a/+ mice were also converted to the conditional allele (Lztr1tm1c/+, also called Lztr1fx/+) using germline Flpase transgenic mice (JAX#005703). Foxg1IREScre/+ (JAX #029690) mice were used to delete Lztr1 in the telencephalon. Note that this cre is inserted downstream of the FoxG1 coding region in the UTR, preserving FoxG1 expression (Kawaguchi et al., 2016). The mice obtained from Jax were genotyped from published protocols on the Jax website. The Lztr1fx/+ mice have loxP sites flanking exon7 of Lztr1 variant 1 and were genotyped with the following primers: lztr1-loxP5′: TCTGAGCTCCTGTGTTGCAG and lztr1-loxP3′: GGGGTAATGAGTCGCCTGAG. The WT allele results in a 503-bp product, and the Lztr1-loxP allele results in a 583-bp product. Lztr1lacZ/+ was genotyped with the following β-gal-specific primers, β-gal-5′: AACTTAATCGCCTTGCAGCA and β-gal-3′: GTAACCGTGCATCTGCCAGT, that produced a 218-bp product. Embryonic and postnatal brains were collected, fixed, and processed for histology as previously described (Waclaw et al., 2006, 2010; Ehrman et al., 2014). For all the stages analyzed, at least three embryos or postnatal brains were included for each genotype. Lztr1 conditional knockouts (Lztr1fx/lacZ; Foxg1IREScre/+) were compared to controls (Lztr1+/+ or Lztr1fx/+).
Primary antibodies were used at the following concentrations: Gt-β-gal (1:1,000, from Biogenesis, #2282), Rbt-GFAP (1:5,000, from DAKO, #Z0334), Rbt-Olig2 (1:1,000, from Millipore-Sigma, #AB9610), Rbt-Nkx2.2 (1:1,000, from Abcam, ab191077), Rbt-Tcf7l2 (1:500, from Cell Signaling, #2569), Gt-Pdgfra (1:500 from R&D Systems, #AF1062), and Rbt-p-ERK1/2 (1:500 from Cell Signaling, #9101 and #4370). In situ hybridization was completed as described (Kohli et al., 2018). The plasmids to generate Plp1 anti-sense probe were provided by Dr. Q. Richard Lu (CCHMC). The Lztr1 anti-sense probe was generated with the following primers that amplify a product 3′ of the loxP deletion: Lztr1-5′: GCTGTGTGCCGGGATAAGA and Lztr1-3′ with T3: ATTAACCCTCACTAAAGG GCTCAGCGCCAACTTATACAC (966-bp probe). The LacZ anti-sense probe was generated with the following primers, LacZ-5′: AACTCGGCTCACAGT ACGC and LacZ-3′ with T3: ATTAACCCTCACTAAAGGAG TAAGGCGGTCGGGATAGT (656-bp probe). The Enpp6 anti-sense probe was generated from the following primers: Enpp6-5′: TAGCATCCTTGCCTGGCTTC and Enpp6-3′ with T3: ATTAA CCCTCACTAAAGG CCGCTCGGCCACTCTTTAAT (417-bp probe).
Brightfield pictures were captured on a Leica DM2500 microscope with either a Leica DFC500 or DMC6200 camera using Leica Acquisition Software (LAS-X). Fluorescent images were captured on a Nikon C2 confocal microscope using Nikon Elements software. Raw images were processed using CellProfiler for quantification, and numbers are presented as cells/mm2 or arbitrary intensity units, as indicated by graphs. The labeled cells in P21 or P30 mice were counted in two serial sections in both hemispheres at the indicated levels and magnification for a total of four images per brain. For all quantifications, at least three different controls or experimental samples (cKO) were used. Unless otherwise stated, statistics were performed between control and Lztr1 cKO using Student’s unpaired t-test. Graphs were generated using GraphPad Prism. Images were brightened in Adobe Photoshop equally between control and Lztr1 cKOs.
LZTR1 is a human disease gene associated with NS and also deleted in some cases of 22q11.2 deletion syndrome (Kurahashi et al., 1995; Yamamoto et al., 2015; Johnston et al., 2018). Patients of these syndromes show a variety of phenotypes, including developmental delay and neurocognitive dysfunction (Pierpont et al., 2009; Fiksinski et al., 2021). However, little is known about the expression or role of Lztr1 in the developing brain. Early reports suggest a wide expression of Lztr1 in a variety of organ systems (Kurahashi et al., 1995; Nacak et al., 2006). We describe here the expression of Lztr1 in the mouse telencephalon, a region of the brain that contains key brain structures necessary for higher executive function, including movement, learning, memory, and emotion (Rubenstein, 2011). We analyzed the late embryonic (E18.5) and postnatal (1 month) stages for Lztr1 gene expression. In situ hybridization for Lztr1 revealed enriched expression in the mantle zone of the developing cortex at the rostral and caudal levels in E18.5 embryos (Figures 1A,B). A weak expression was detected around the telencephalic ventricles that was more apparent at the rostral levels (Figure 1A). Lztr1 expression was also detected outside of the telencephalon, in the dorsal thalamus of the diencephalon and the trigeminal nerve (Figure 1B). The expression of Lztr1 showed a similar pattern in postnatal brains, with a widespread expression in the cortex at both levels (Figures 1C,D). In addition, we detected enriched expression in the basolateral amygdala complex and hippocampus (Figure 1D). The expression of Lztr1 was also maintained in the mature thalamus of the diencephalon (Figure 1D). A recent report characterized a Lztr1 KO mouse from the European conditional knockout consortium (Skarnes et al., 2011; Steklov et al., 2018; Sewduth et al., 2020); however, the lacZ expression, which is knocked into the Lztr1 locus, was not analyzed. We also generated a variant of this line (see section “Methods”) and analyzed the lacZ activity and gene expression. LacZ activity was specific to Lztr1lacZ/+ heterozygous mice (compare Figure 1E to Figure 1F). In addition, we detected a robust lacZ activity in the postnatal cortex, hippocampus, and thalamus in the same pattern as was detected with the Lztr1 gene expression (Figure 1G). High magnification examination revealed isolated lacZ + cells in the corpus callosum where oligodendrocytes and astrocytes reside (Figure 1H). Given the isolated nature of the cells and the punctate appearance of lacZ + cells, we also performed lacZ gene expression and β-galactosidase immunohistochemistry, which revealed positive cells in the white matter (Figures 1I,J). We confirmed this result with high magnification of the Lztr1 gene expression in the white matter (Figure 1K). The β-galactosidase antibody expression was the most robust method for detecting these cells, likely from the ABC signal amplification used during immunohistochemistry. Our expression analysis suggests that Lztr1 expression is enriched in distinct cell types of the cortex, amygdala, hippocampus, and white matter in the telencephalon.
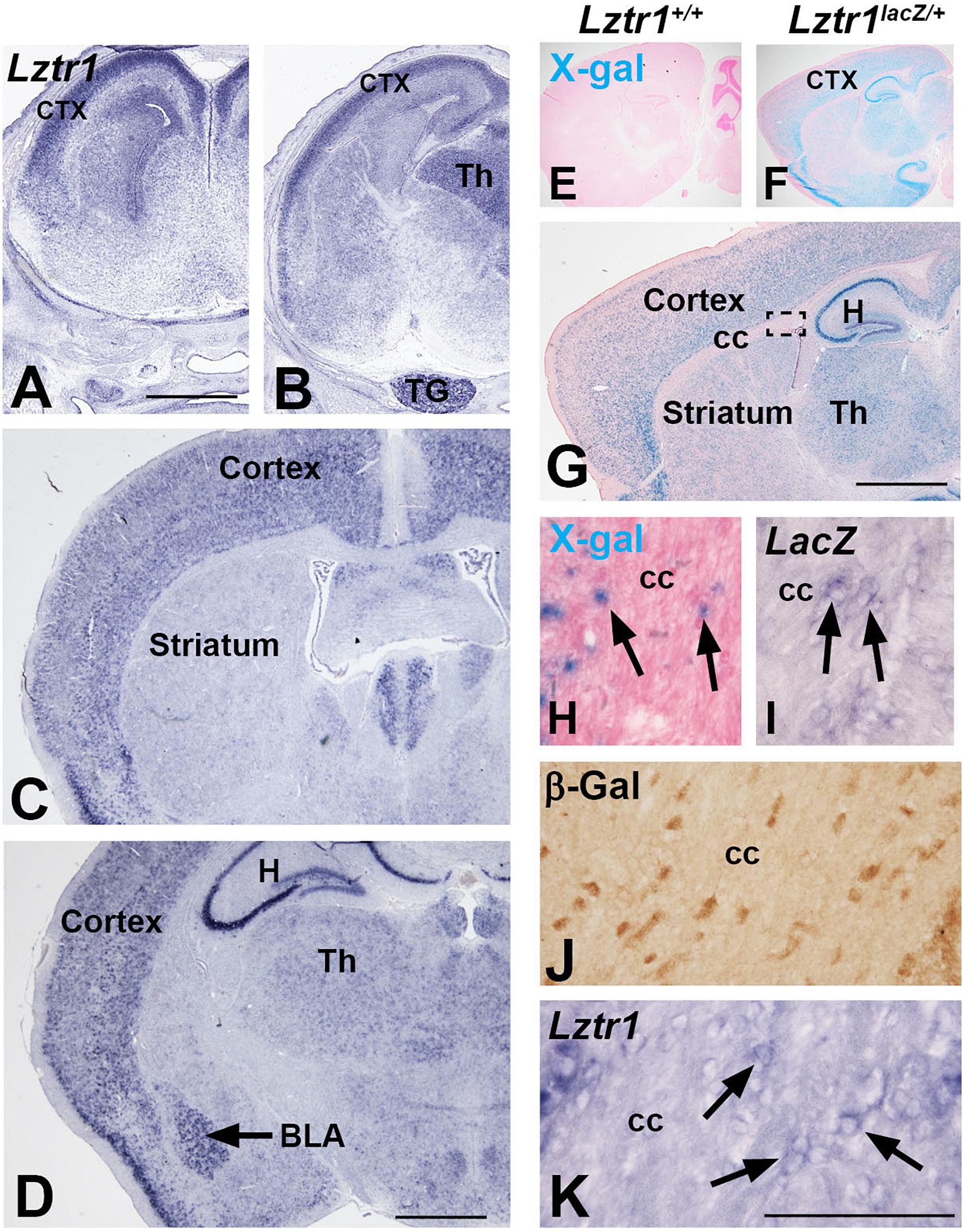
Figure 1. Lztr1expression in the developing telencephalon. Lztr1 gene expression in the cortex and thalamus at both E18.5 (A,B) and postnatal (1 month old) stages (C–K). Lztr1 is also observed in the trigeminal nerve at E18.5 (B) and the hippocampus and basolateral amygdala in the postnatal brain (black arrow, D). X-gal reaction in postnatal brains shows lacZ reporter activity from the Lztr1 locus (compare E to F). Sagittal view of the postnatal forebrain shows that lacZ activity is similar to Lztr1 gene expression (compare G to C,D). High magnification of the corpus callosum reveal LacZ-positive cells (H), lacZ gene expression (I), and β-gal immunohistochemistry (J). This correlates to the Lztr1 expression observed in the corpus callosum (K). Scale bars = 1 mm in for (A–D,G). Scale bar is 100 μm for (H–K). ctx, cortex; Th, thalamus; TG, trigeminal nerve; H, hippocampus; BLA, basolateral amygdala; CC, corpus callosum.
It has previously been shown that germline Lztr1 KO does not survive to postnatal stages (Steklov et al., 2018; Sewduth et al., 2020). This limitation prevents modeling of the recessive hypomorphic or loss-of-function human mutations in organ systems, including the brain. Therefore, we established a conditional allele for Lztr1 (Skarnes et al., 2011; Sewduth et al., 2020). We developed a strategy to model Lztr1 deficiency during embryonic and postnatal development using an Lztr1 conditional allele (see section “Methods”) and the Foxg1IRES–cre/+ telencephalon-specific cre line (Kawaguchi et al., 2016). Unlike germline Lztr1 mutants (Steklov et al., 2018), conditional mutants (Lztr1fx/lacZ; Foxg1IRES–cre/+) were viable to postnatal stages and showed reduced Lztr1 expression in the cortex and hippocampus at P21 (compare Figure 2B to Figure 2A). Given that Lztr1 is a RASopathy gene and implicated in regulating RAS and the MAPK pathway, we analyzed MAPK pathway activation in the brain using p-ERK1/2 expression. Lztr1 cKOs showed an enhanced immunoreactivity for p-ERK1/2 in the corpus callosum of the white matter (compare Figures 2D,F to Figures 2C,E). This result was observed at all levels of the corpus callosum, including the lateral, medial, and caudal regions (Figures 2D,F and data not shown). The increase in staining was observed generally throughout the white matter and in distinct cell bodies. In fact, Lztr1 cKOs exhibited a 30.1% increase in p-ERK1/2 + cell bodies within the white matter (compare Figure 2F to Figure 2E, graph in Figure 2I, p = 0.0319, n = 3). We did not detect a difference in total ERK1/2 immunoreactivity in the corpus callosum (Figures 2G,H, graph in Figure 2J), further supporting pathway activation using p-ERK1/2 expression in the Lztr1 cKO. Interestingly, we did not detect enhanced p-ERK1/2 immunoreactivity in the cortex despite a robust Lztr1 expression in this region (Figure 1), which suggests a potential regional difference in pathway activation in Lztr1 cKOs. However, this might be due to limitations of the immunohistochemistry technique requiring a certain threshold of change to view the difference. Based on the robust difference in the Lztr1 cKO white matter, we performed immunofluorescence on Lztr1 reporter mice with β-gal and oligodendrocyte markers. Olig2 is a marker of the entire oligodendrocyte lineage, and CC1 labels maturing oligodendrocytes during postnatal development (Emery and Lu, 2015). We found an overlap of β-gal cells with both Olig2 and CC1 (Figures 2K,L). While we detected a nearly complete overlap of β-gal with CC1, there were many Olig2 cells only (red arrows in Figure 2K), suggesting that Lztr1-driven β-gal is enriched in maturing oligodendrocyte but does not label the entire lineage. This is supported by online gene expression databases (Marques et al., 2016) which show that Lztr1 is expressed in the entire oligodendrocyte lineage with a higher expression at mature stages (data not shown). Our results describe a genetic approach to study Lztr1 deficiency in postnatal brain and suggest that Lztr1 is required for normal control of MAPK pathway activation in the white matter regions.
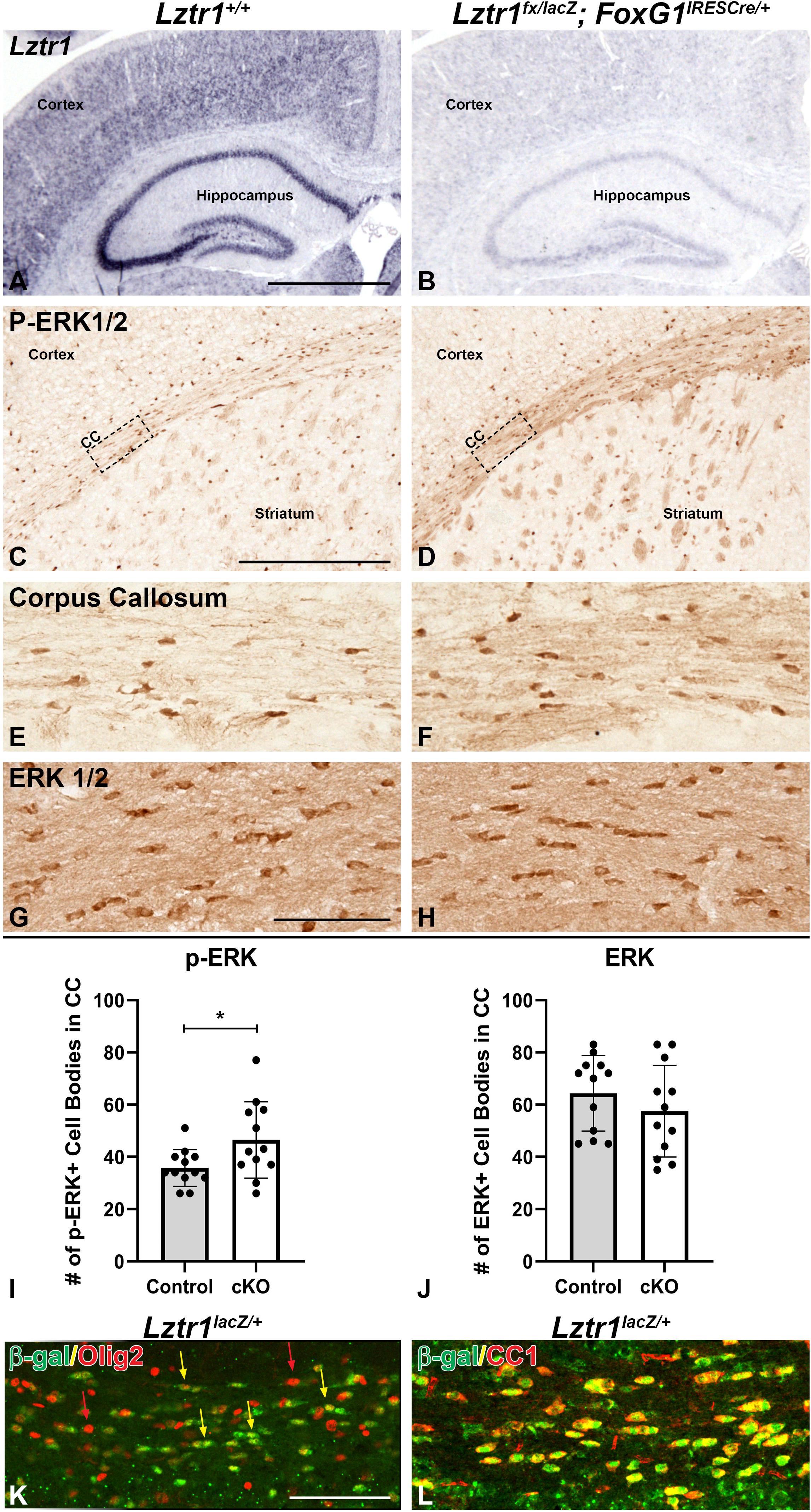
Figure 2. Conditional deletion of Lztr1 using FoxG1IRES– Cre/+ results in increased p-ERK1/2 expression in postnatal animals at postnatal day 21. Lztr1 gene expression shows a reduction of expression in the cKO, especially in the cortex and hippocampus (A,B). Lztr1 cKO shows increased p-ERK1/2 expression (D,F) compared to controls (C,E). Increased p-ERK1/2 expression is especially observable in major white matter tracts of the corpus callosum (C–F). Total ERK1/2 staining is similar in Lztr1 cKO compared to control in the corpus callosum (G,H). Quantification of p-ERK1/2 and ERK1/2 by number of cell bodies per field in the CC (I,J). β-gal reporter expression in Lztr1lacZ/+ mice shows co-expression with oligodendrocyte lineage marker Olig2 (K) and mature oligodendrocyte marker CC1 (L). Scale bars = 1 mm for (A,B), 500 μm for (C,D), and 100 μm for (E–H,K,L). BLA, basolateral amygdala; CC, corpus callosum. The asterisk in (I) refers to a p < 0.05 as detected by Student’s t-test.
Previous work has shown that the MAPK pathway activation plays a key role at multiple stages of oligodendrocyte development (Li et al., 2012; Fyffe-Maricich et al., 2013; Ishii et al., 2013; Li et al., 2014). In addition, RASopathy mutations have been shown to impact both early and mature stages of oligodendrogenesis (Bennett et al., 2003; Dasgupta and Gutmann, 2005; Zhu et al., 2005; Mayes et al., 2013; Ehrman et al., 2014; Lopez-Juarez et al., 2017; Titus et al., 2017; Aoidi et al., 2018; Holter et al., 2019). To determine if Lztr1 plays a role in oligodendrocyte development, we analyzed stage-specific markers that label oligodendrocyte progression at 1 month of age when developmental myelination is largely complete. We did not detect dramatic changes in Olig2 and Plp gene expression which labels the entire lineage and maturing oligodendrocytes, respectively (Figures 3A,B,I,J,K). However, Lztr1 cKOs exhibited alterations in stage-specific markers that label distinct transition points from progenitors to differentiated oligodendrocytes. Pdgfrα labels OPCs and is increased in the Lztr1 cKO corpus callosum (Figures 3C,D,K; 40.5% increase, p = 0.000581, n = 3). This is in line with other RASopathy mouse models that show increased OPC markers (Bennett et al., 2003; Dasgupta and Gutmann, 2005; Ehrman et al., 2014; Holter et al., 2019). Tcf7l2 is a key factor in oligodendrocyte differentiation (Fancy et al., 2009; Ye et al., 2009; Hammond et al., 2015; Zhao et al., 2016). Lztr1 cKOs show a reduction in Tcf7l2 + cells in the corpus callosum (Figures 3E,F,K; 26.0% decrease, p = 0.00587, n = 3). Enpp6 is a transient marker enriched in newly formed oligodendrocytes (Zhang et al., 2014; Xiao et al., 2016). Similar to Tcf7l2 expression, Lztr1 cKOs also showed a reduction in Enpp6 expression in the white matter (Figures 3G,H,K; 58.6% decrease, p = 0.006622, n = 3). These results suggest that Lztr1 deficiency results in changes in the stage-specific expression of oligodendrocyte markers, with increased OPCs and decreased expression of markers that label newly formed oligodendrocytes. Despite this, the total oligodendrocytes labeled by Olig2 and mature oligodendrocytes labeled by Plp were not affected.
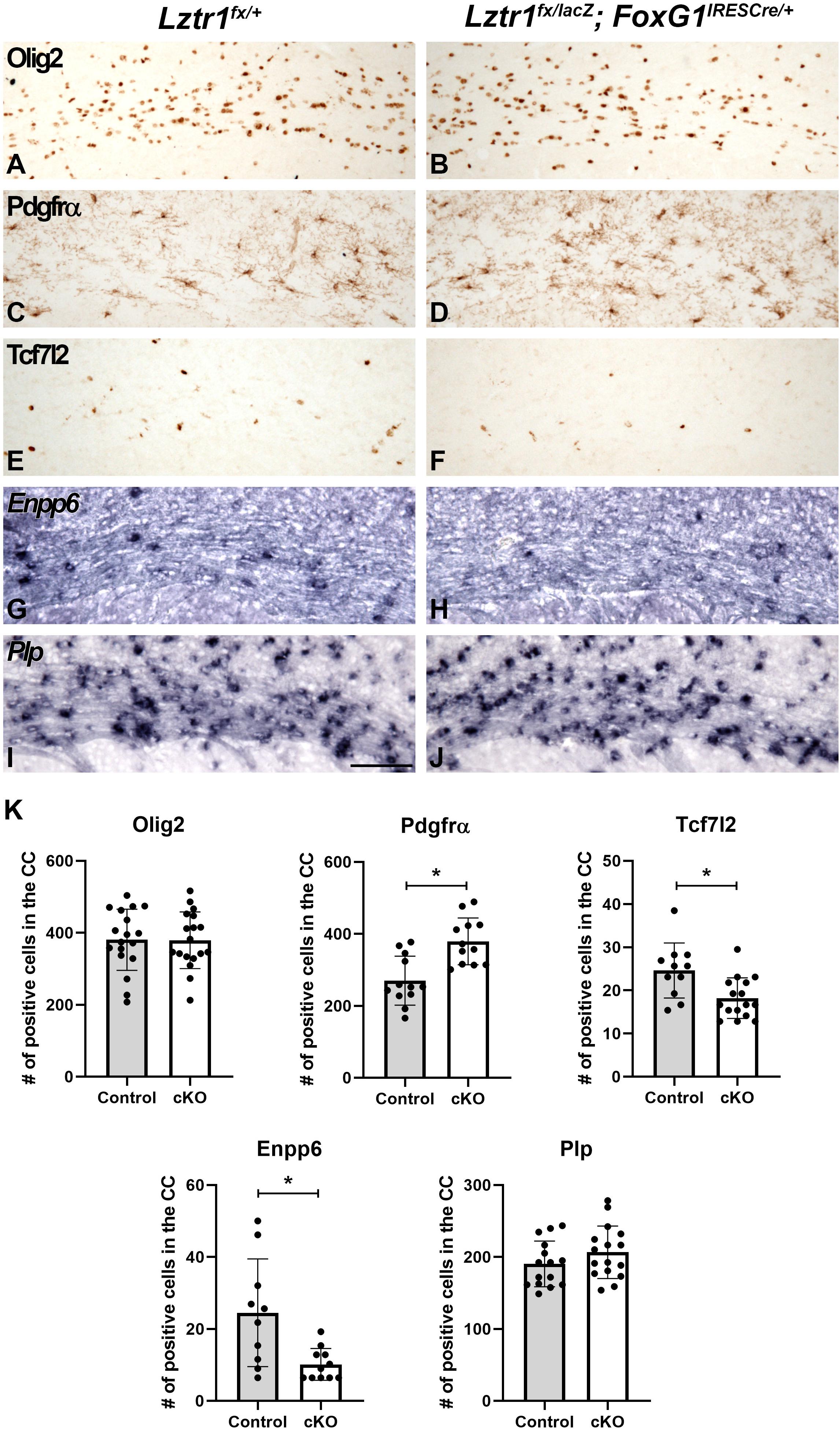
Figure 3. Loss of Lztr1 impacts distinct stages of oligodendrocyte maturation in the corpus callosum of postnatal animals at 1 month of age. Olig2 marks the entire oligodendrocyte lineage, and no difference between the number of Olig2 + cells is observed in controls (A) and cKOs (B). The OPC marker Pdgfrα was increased in the corpus callosum in cKOs (D) compared to controls (C). Tcf7l2 and Enpp6 expression observed in differentiating and newly formed oligodendrocytes (E,G) are decreased in cKOs (F,H). The mature oligodendrocyte marker Plp appears similar in controls and cKOs (I,J). Quantification of oligodendrocyte markers by the number of positive cells per field in the CC (K). Scale bar = 100 μm for (A–J). The asterisk in (K) refers to a p < 0.05 as detected by Student’s t-test.
In addition to oligodendrocytes, the white matter also contains astrocytes (reviewed in Molofsky et al., 2012). In control brains, GFAP is robustly expressed in the white matter and hippocampus but only in isolated cells of the cortex (Figures 4A,C,E). Lztr1 cKOs showed increased GFAP-positive cells in the cortex and hippocampus (Figures 4A–D,G). The increase was measured by total pixel intensity in the lateral cortex and CA region of the hippocampus. There was a 57.8% increase in the cortex (p = 0.000596, n = 3) and a 33.5% increase in the hippocampus (p = 0.0208, n = 3). The GFAP-positive cells in the corpus callosum appeared to have more defined cell bodies in the Lztr1 cKOs compared to controls (Figures 4E,F). However, the staining intensity was not significantly different in the corpus callosum, indicating that the robust increase in astrocytes might be associated more with gray matter areas. GFAP expression labels normal astrocytes but is also expressed in reactive astrocytes in injury and disease models. The increase of GFAP immunoreactivity in the cortex is supported by previous reports that loss of the RASopathy gene NF1 results in a robust increase in GFAP expression in reactive astrocytes (Hewett et al., 1995; Nordlund et al., 1995; Gutmann et al., 1999; Rizvi et al., 1999). This suggests that hyperactivation of the RAS/MAPK pathway through NF1 or Lztr1 loss results in increased GFAP + astrocytes. Overall, our results from telencephalon-specific conditional knockout suggest that Lztr1 plays a role during oligodendrocyte and astrocyte development in the postnatal telencephalon.
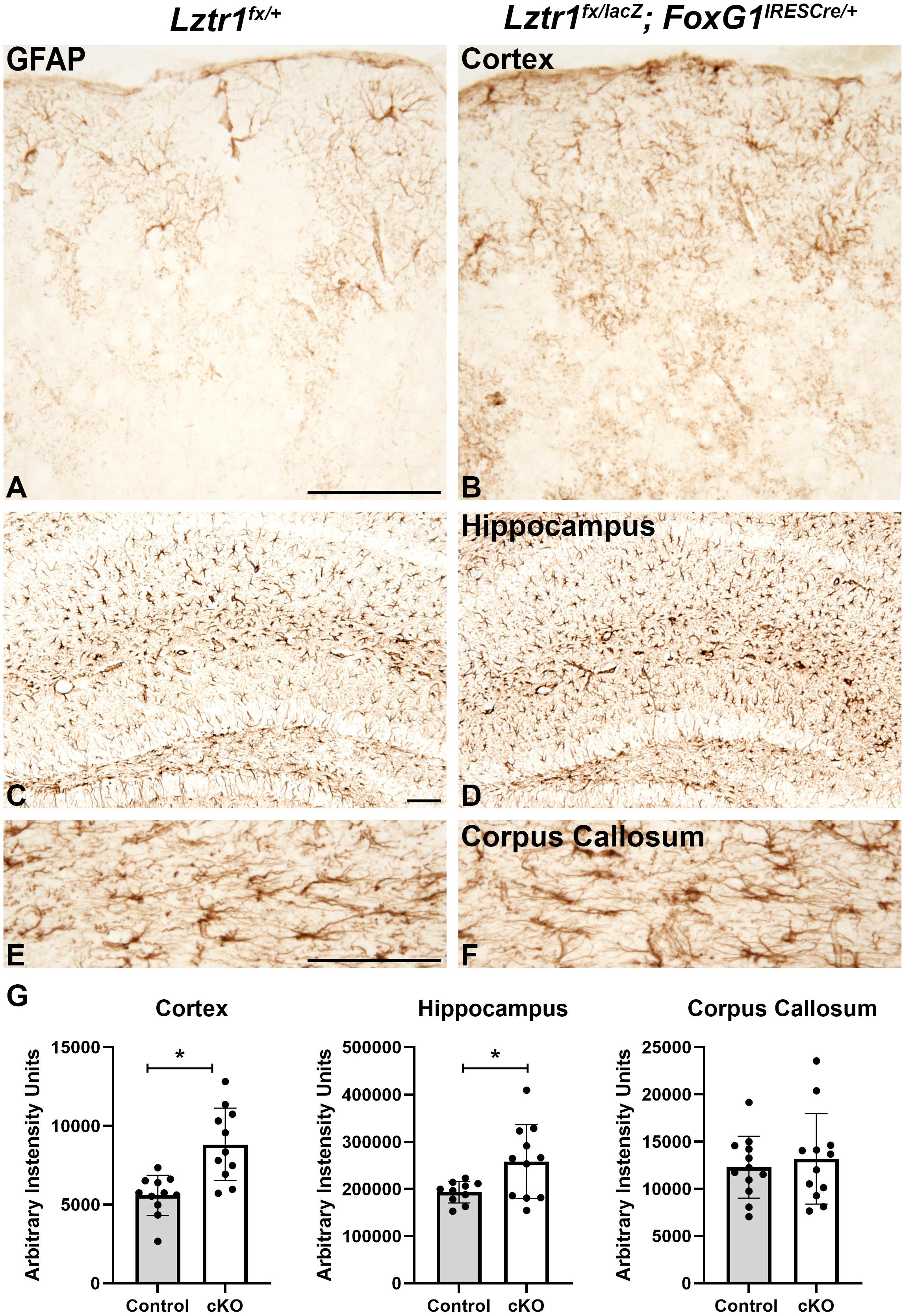
Figure 4. Loss of Lztr1 increases the expression of the astrocyte marker GFAP in postnatal animals at 1 month of age. Increased GFAP as measured by stain intensity (arbitrary intensity units) is observed in the cortex and hippocampus of cKOs compared to controls (A–D,G). The GFAP expression in the corpus callosum was similar in cKOs compared to controls (E,F). Quantification (G). Scale bars = 100 μm for (A–F). The asterisk in (G) refers to a p < 0.05 as detected by Student’s t-test.
In this study, we evaluated Lztr1 in the developing brain by characterizing the expression in the telencephalon. Our results indicate that Lztr1 is highly expressed in the embryonic and postnatal cortex. We detected robust expression in the postnatal hippocampus, amygdala, and oligodendrocytes. To address a role for Lztr1 in the postnatal brain, we developed a conditional approach to specifically delete Lztr1 in the telencephalon using Foxg1IRES–cre/+ and an Lztr1 conditional allele. We found that Lztr1 deletion results in robust MAPK pathway activation and impacts the oligodendrocyte lineage. In addition, we detected ectopic GFAP astrocyte expression in the cortex of conditional mutants. Our results suggest that Lztr1 plays a role in MAPK pathway activation and normal glial cell development in the telencephalon. These findings provide a “roadmap” of Lztr1-enriched areas of the brain that might be affected by Lztr1 mutation or deficiency. In addition, we describe a new genetic approach to address the impact of Lztr1 deficiency in the brain and model loss-of-function mutations.
The expression of Lztr1 has been described as widespread (Kurahashi et al., 1995). However, expression data on tissue sections of any organ system, including the brain, have not been reported. Therefore, detecting enriched Lztr1 expression in the cortex, hippocampus, amygdala, and oligodendrocytes is significant. The expression data, combined with the results from our telencephalon-specific conditional mutant showing elevated MAPK pathway activation in the white matter, provide important clues to distinct cell populations impacted by Lztr1 mutations. Indeed previous efforts have not been able to describe the postnatal impact of Lztr1 mutations since germline loss resulted in lethality before birth from severe cardiovascular defects (Steklov et al., 2018; Sewduth et al., 2020), thereby preventing a postnatal analysis of the brain. Our strategy is the first to address the requirement of Lztr1 in the postnatal brain. Given the high cortical expression of Lztr1, we were surprised in the lack of widespread MAPK pathway activation in the cortex of conditional mutants. However, this may be a result of the immunohistochemistry technique on tissue sections requiring a threshold of pathway activation to detect the increase. Alternatively, the loss of Lztr1 might regulate other RAS-regulated pathways such as AKT/mTOR in the cortex or white matter. In addition, it remains unknown if Lztr1 loss might impact specific cortical cell populations, neural activity, or neuronal function and behaviors. Our conditional mutant approach will provide a good opportunity to address these issues. In addition, future experiments to parse cortical-specific vs. oligodendrocyte-specific requirements for Lztr1 will be important for addressing cell type-specific impact on neuronal activity or function, oligodendrocyte myelination, and behaviors. Previous work has elucidated specific behaviors impacted by RAF1 and NF1 RASopathy mutations (Lopez-Juarez et al., 2017; Holter et al., 2019). It will be interesting to compare how these behaviors are impacted in an Lztr1-deficient mouse.
There exist multiple lines of evidence showing that appropriate regulation of RAS signaling and the MAPK pathway is crucial for normal oligodendrocyte development. Evidence from RASopathy animal models show that the dysregulation of the RAS/MAPK pathway has a robust effect on early progenitor neural stem cells or OPCs (Bennett et al., 2003; Dasgupta and Gutmann, 2005; Zhu et al., 2005; Wang et al., 2012; Ehrman et al., 2014; Li et al., 2014; Sun et al., 2015; Holter et al., 2019). Hence, our data which indicate that Lztr1 loss increases OPC populations is in line with other studies on RASopathy mutations at various levels of the pathway. Previous studies have utilized hyperactivation of MEK to show that a normal level of MAPK signaling is crucial at the late stages of oligodendrocyte development and myelination (Fyffe-Maricich et al., 2013; Ishii et al., 2013). In addition, Nf1 loss or HRAS-G12V RASopathy mutations have also been shown to impact mature and myelinating oligodendrocytes (Mayes et al., 2013; Lopez-Juarez et al., 2017; Titus et al., 2017). Therefore, the reduction in the newly formed oligodendrocyte markers observed in the Lztr1 cKO might be from either a role for Lztr1 specifically during this mature stage or a secondary effect of the increased OPC populations that do not transition to the newly formed oligodendrocyte stage in a normal manner. Either result suggests that Lztr1 function is key to oligodendrogenesis. However, future experiments will be important to define if developmental deletion of Lztr1 impacts oligodendrocyte myelination and subsequently results in any behavioral phenotypes. It will also be interesting to parse stage-specific roles for Lztr1 in either OPCs or mature oligodendrocytes and also reveal downstream signaling mechanisms underlying the oligodendrocyte phenotype.
GFAP is one of the most common markers for astrocytes in the postnatal mouse brain, but it does not label all astrocytes and is also a marker of reactive astrocytes after various injuries or defects (reviewed in Molofsky et al., 2012). An increased GFAP expression in reactive astrocytes was identified in NF1 mutant tissue (Hewett et al., 1995; Nordlund et al., 1995; Gutmann et al., 1999, 2001; Rizvi et al., 1999; Hegedus et al., 2007). A recent report identified that a mouse model for the Noonan syndrome mutation RAF1-L613V exhibits an increased GFAP expression in the cortex and hippocampus (Holter et al., 2019). We observed a similar effect in the Lztr1 cKO cortex and hippocampus. However, it should be noted that we analyzed our tissue after the weaning stage (1 month), whereas the study of Holter et al. (2019) examined 2-month-old animals. It will be interesting to determine how the increase in GFAP + cells changes during development and the late adult stages in Lztr1 cKOs. Interestingly, a previous study using activating HRAS mutations has shown that iPSC-derived astrocytes from HRAS-G12S Costello patients and astrocytes from HRAS-G12V mice undergo precocious maturation, suggesting that dysregulation of the RAS/MAPK pathway can affect the timing of astrocyte development and even impact neuronal populations (Krencik et al., 2015). Therefore, it may be that Lztr1 gene function is crucial in both astrocyte and oligodendrocyte development. Alternatively, the astrocyte phenotype may be a readout of the oligodendrocyte defect or an unknown defect in cortical neurons or activity promoting reactive astrogliosis. Future experiments in which Lztr1 is conditionally inactivated in either the astrocyte or oligodendrocyte lineage will address this issue.
Increasing genetic evidence in 22q11.2 deletion syndrome, NS, and glioma tumors point toward the negative health impact of Lztr1 mutation or deletion in multiple organ systems, including the brain (Kurahashi et al., 1995; Frattini et al., 2013; Yamamoto et al., 2015; Johnston et al., 2018). Therefore, studies using animal models are key to understanding the role of Lztr1 in specific cells of the brain. Our results identify enriched regions of Lztr1 expression and describe a new conditional mutant approach to determine what cells are most susceptible to Lztr1 dysregulated signaling in the telencephalon. Our expression results and new mouse model are important first steps in understanding Lztr1 human disease in the brain.
The original contributions presented in the study are included in the article, further inquires can be directed to the corresponding author.
The animal study was reviewed and approved by the Cincinnati Children’s Hospital Medical Center Institutional Animal Care and Use Committee.
RW, CP, and MT performed the conception and experimental design. MT, NS, and DN performed the data collection, analysis, and/or animal work. RW and MT wrote the manuscript. CP, LE, and DN edited the manuscript and provided critical input to the final manuscript. All authors contributed to the article and approved the manuscript.
RW was supported by NIH Grant NS088529.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Q. Richard Lu (CCHMC) for providing Plp1 in situ plasmid.
Abe, T., Umeki, I., Kanno, S. I., Inoue, S. I., Niihori, T., and Aoki, Y. (2020). LZTR1 facilitates polyubiquitination and degradation of RAS-GTPases. Cell Death Differ. 27, 1023–1035. doi: 10.1038/s41418-019-0395-5
Alfieri, P., Cesarini, L., Mallardi, M., Piccini, G., Caciolo, C., Leoni, C., et al. (2011). Long term memory profile of disorders associated with dysregulation of the RAS-MAPK signaling cascade. Behav. Genet. 41, 423–429. doi: 10.1007/s10519-011-9446-5
Aoidi, R., Houde, N., Landry-Truchon, K., Holter, M., Jacquet, K., Charron, L., et al. (2018). Mek1(Y130C) mice recapitulate aspects of human cardio-facio-cutaneous syndrome. Dis. Model. Mech. 11:dmm031278.
Bennett, M. R., Rizvi, T. A., Karyala, S., Mckinnon, R. D., and Ratner, N. (2003). Aberrant growth and differentiation of oligodendrocyte progenitors in neurofibromatosis type 1 mutants. J. Neurosci. 23, 7207–7217. doi: 10.1523/jneurosci.23-18-07207.2003
Bigenzahn, J. W., Collu, G. M., Kartnig, F., Pieraks, M., Vladimer, G. I., Heinz, L. X., et al. (2018). LZTR1 is a regulator of RAS ubiquitination and signaling. Science 362, 1171–1177. doi: 10.1126/science.aap8210
Castel, P., Cheng, A., Cuevas-Navarro, A., Everman, D. B., Papageorge, A. G., Simanshu, D. K., et al. (2019). RIT1 oncoproteins escape LZTR1-mediated proteolysis. Science 363, 1226–1230. doi: 10.1126/science.aav1444
Dasgupta, B., and Gutmann, D. H. (2005). Neurofibromin regulates neural stem cell proliferation, survival, and astroglial differentiation in vitro and in vivo. J. Neurosci. 25, 5584–5594. doi: 10.1523/jneurosci.4693-04.2005
Ehrman, L. A., Nardini, D., Ehrman, S., Rizvi, T. A., Gulick, J., Krenz, M., et al. (2014). The protein tyrosine phosphatase Shp2 is required for the generation of oligodendrocyte progenitor cells and myelination in the mouse telencephalon. J. Neurosci. 34, 3767–3778. doi: 10.1523/jneurosci.3515-13.2014
Emery, B., and Lu, Q. R. (2015). Transcriptional and epigenetic regulation of oligodendrocyte development and myelination in the central nervous system. Cold Spring Harb. Perspect. Biol. 7:a020461. doi: 10.1101/cshperspect.a020461
Fancy, S. P., Baranzini, S. E., Zhao, C., Yuk, D. I., Irvine, K. A., Kaing, S., et al. (2009). Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev. 23, 1571–1585. doi: 10.1101/gad.1806309
Fiksinski, A. M., Schneider, M., Zinkstok, J., Baribeau, D., Chawner, S., and Vorstman, J. A. S. (2021). Neurodevelopmental trajectories and psychiatric morbidity: lessons learned from the 22q11.2 deletion syndrome. Curr. Psychiatry Rep. 23:13.
Frattini, V., Trifonov, V., Chan, J. M., Castano, A., Lia, M., Abate, F., et al. (2013). The integrated landscape of driver genomic alterations in glioblastoma. Nat. Genet. 45, 1141–1149.
Fyffe-Maricich, S. L., Schott, A., Karl, M., Krasno, J., and Miller, R. H. (2013). Signaling through ERK1/2 controls myelin thickness during myelin repair in the adult central nervous system. J. Neurosci. 33, 18402–18408. doi: 10.1523/jneurosci.2381-13.2013
Gutmann, D. H., Loehr, A., Zhang, Y., Kim, J., Henkemeyer, M., and Cashen, A. (1999). Haploinsufficiency for the neurofibromatosis 1 (NF1) tumor suppressor results in increased astrocyte proliferation. Oncogene 18, 4450–4459. doi: 10.1038/sj.onc.1202829
Gutmann, D. H., Wu, Y. L., Hedrick, N. M., Zhu, Y., Guha, A., and Parada, L. F. (2001). Heterozygosity for the neurofibromatosis 1 (NF1) tumor suppressor results in abnormalities in cell attachment, spreading and motility in astrocytes. Hum. Mol. Genet. 10, 3009–3016. doi: 10.1093/hmg/10.26.3009
Hammond, E., Lang, J., Maeda, Y., Pleasure, D., Angus-Hill, M., Xu, J., et al. (2015). The Wnt effector transcription factor 7-like 2 positively regulates oligodendrocyte differentiation in a manner independent of Wnt/beta-catenin signaling. J. Neurosci. 35, 5007–5022. doi: 10.1523/jneurosci.4787-14.2015
Hegedus, B., Dasgupta, B., Shin, J. E., Emnett, R. J., Hart-Mahon, E. K., Elghazi, L., et al. (2007). Neurofibromatosis-1 regulates neuronal and glial cell differentiation from neuroglial progenitors in vivo by both cAMP- and Ras-dependent mechanisms. Cell Stem Cell 1, 443–457. doi: 10.1016/j.stem.2007.07.008
Hewett, S. J., Choi, D. W., and Gutmann, D. H. (1995). Expression of the neurofibromatosis 1 (NF1) gene in reactive astrocytes in vitro. Neuroreport 6, 1565–1568. doi: 10.1097/00001756-199507310-00025
Holter, M. C., Hewitt, L. T., Koebele, S. V., Judd, J. M., Xing, L., Bimonte-Nelson, H. A., et al. (2019). The Noonan syndrome-linked Raf1L613V mutation drives increased glial number in the mouse cortex and enhanced learning. PLoS Genet. 15:e1008108. doi: 10.1371/journal.pgen.1008108
Ishii, A., Furusho, M., and Bansal, R. (2013). Sustained activation of ERK1/2 MAPK in oligodendrocytes and schwann cells enhances myelin growth and stimulates oligodendrocyte progenitor expansion. J. Neurosci. 33, 175–186. doi: 10.1523/jneurosci.4403-12.2013
Johnston, J. J., Van Der Smagt, J. J., Rosenfeld, J. A., Pagnamenta, A. T., Alswaid, A., Baker, E. H., et al. (2018). Autosomal recessive Noonan syndrome associated with biallelic LZTR1 variants. Genet. Med. 20, 1175–1185.
Kawaguchi, D., Sahara, S., Zembrzycki, A., and O’leary, D. D. M. (2016). Generation and analysis of an improved Foxg1-IRES-Cre driver mouse line. Dev. Biol. 412, 139–147. doi: 10.1016/j.ydbio.2016.02.011
Kohli, V., Nardini, D., Ehrman, L. A., and Waclaw, R. R. (2018). Characterization of Glcci1 expression in a subpopulation of lateral ganglionic eminence progenitors in the mouse telencephalon. Dev. Dyn. 247, 222–228. doi: 10.1002/dvdy.24556
Krencik, R., Hokanson, K. C., Narayan, A. R., Dvornik, J., Rooney, G. E., Rauen, K. A., et al. (2015). Dysregulation of astrocyte extracellular signaling in Costello syndrome. Sci. Transl. Med. 7:286ra266.
Kurahashi, H., Akagi, K., Inazawa, J., Ohta, T., Niikawa, N., Kayatani, F., et al. (1995). Isolation and characterization of a novel gene deleted in DiGeorge syndrome. Hum. Mol. Genet. 4, 541–549. doi: 10.1093/hmg/4.4.541
Li, S., Mattar, P., Dixit, R., Lawn, S. O., Wilkinson, G., Kinch, C., et al. (2014). RAS/ERK signaling controls proneural genetic programs in cortical development and gliomagenesis. J. Neurosci. 34, 2169–2190. doi: 10.1523/jneurosci.4077-13.2014
Li, X., Newbern, J. M., Wu, Y., Morgan-Smith, M., Zhong, J., Charron, J., et al. (2012). MEK Is a key regulator of gliogenesis in the developing brain. Neuron 75, 1035–1050. doi: 10.1016/j.neuron.2012.08.031
Lopez-Juarez, A., Titus, H. E., Silbak, S. H., Pressler, J. W., Rizvi, T. A., Bogard, M., et al. (2017). Oligodendrocyte Nf1 controls aberrant notch activation and regulates myelin structure and behavior. Cell Rep. 19, 545–557. doi: 10.1016/j.celrep.2017.03.073
Marques, S., Zeisel, A., Codeluppi, S., Van Bruggen, D., Mendanha Falcao, A., Xiao, L., et al. (2016). Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science 352, 1326–1329. doi: 10.1126/science.aaf6463
Mayes, D. A., Rizvi, T. A., Titus-Mitchell, H., Oberst, R., Ciraolo, G. M., Vorhees, C. V., et al. (2013). Nf1 loss and Ras hyperactivation in oligodendrocytes induce NOS-driven defects in myelin and vasculature. Cell Rep. 4, 1197–1212. doi: 10.1016/j.celrep.2013.08.011
Molofsky, A. V., Krencik, R., Ullian, E. M., Tsai, H. H., Deneen, B., Richardson, W. D., et al. (2012). Astrocytes and disease: a neurodevelopmental perspective. Genes Dev. 26, 891–907. doi: 10.1101/gad.188326.112
Motta, M., Fidan, M., Bellacchio, E., Pantaleoni, F., Schneider-Heieck, K., Coppola, S., et al. (2019). Dominant Noonan syndrome-causing LZTR1 mutations specifically affect the kelch domain substrate-recognition surface and enhance RAS-MAPK signaling. Hum. Mol. Genet. 28, 1007–1022. doi: 10.1093/hmg/ddy412
Nacak, T. G., Leptien, K., Fellner, D., Augustin, H. G., and Kroll, J. (2006). The BTB-kelch protein LZTR-1 is a novel Golgi protein that is degraded upon induction of apoptosis. J. Biol. Chem. 281, 5065–5071. doi: 10.1074/jbc.m509073200
Nordlund, M. L., Rizvi, T. A., Brannan, C. I., and Ratner, N. (1995). Neurofibromin expression and astrogliosis in neurofibromatosis (type 1) brains. J. Neuropathol. Exp. Neurol. 54, 588–600. doi: 10.1097/00005072-199507000-00013
Pierpont, E. I., Pierpont, M. E., Mendelsohn, N. J., Roberts, A. E., Tworog-Dube, E., and Seidenberg, M. S. (2009). Genotype differences in cognitive functioning in Noonan syndrome. Genes Brain Behav. 8, 275–282. doi: 10.1111/j.1601-183x.2008.00469.x
Piotrowski, A., Xie, J., Liu, Y. F., Poplawski, A. B., Gomes, A. R., Madanecki, P., et al. (2014). Germline loss-of-function mutations in LZTR1 predispose to an inherited disorder of multiple schwannomas. Nat. Genet. 46, 182–187. doi: 10.1038/ng.2855
Rizvi, T. A., Akunuru, S., De Courten-Myers, G., Switzer, R. C. III, Nordlund, M. L., and Ratner, N. (1999). Region-specific astrogliosis in brains of mice heterozygous for mutations in the neurofibromatosis type 1 (Nf1) tumor suppressor. Brain Res. 816, 111–123. doi: 10.1016/s0006-8993(98)01133-0
Rubenstein, J. L. (2011). Annual research review: development of the cerebral cortex: implications for neurodevelopmental disorders. J Child Psychol. Psychiatry 52, 339–355. doi: 10.1111/j.1469-7610.2010.02307.x
Sewduth, R. N., Pandolfi, S., Steklov, M., Sheryazdanova, A., Zhao, P., Criem, N., et al. (2020). The Noonan syndrome gene Lztr1 controls cardiovascular function by regulating vesicular trafficking. Circ. Res. 126, 1379–1393. doi: 10.1161/circresaha.119.315730
Shi, X., Xiang, S., Cao, J., Zhu, H., Yang, B., He, Q., et al. (2019). Kelch-like proteins: physiological functions and relationships with diseases. Pharmacol. Res. 148:104404. doi: 10.1016/j.phrs.2019.104404
Skarnes, W. C., Rosen, B., West, A. P., Koutsourakis, M., Bushell, W., Iyer, V., et al. (2011). A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337–342. doi: 10.1038/nature10163
Steklov, M., Pandolfi, S., Baietti, M. F., Batiuk, A., Carai, P., Najm, P., et al. (2018). Mutations in LZTR1 drive human disease by dysregulating RAS ubiquitination. Science 362, 1177–1182. doi: 10.1126/science.aap7607
Sun, G. J., Zhou, Y., Ito, S., Bonaguidi, M. A., Stein-O’Brien, G., Kawasaki, N. K., et al. (2015). Latent tri-lineage potential of adult hippocampal neural stem cells revealed by Nf1 inactivation. Nat. Neurosci. 18, 1722–1724. doi: 10.1038/nn.4159
Tartaglia, M., and Gelb, B. D. (2010). Disorders of dysregulated signal traffic through the RAS-MAPK pathway: phenotypic spectrum and molecular mechanisms. Ann. N. Y. Acad. Sci. 1214, 99–121. doi: 10.1111/j.1749-6632.2010.05790.x
Titus, H. E., Lopez-Juarez, A., Silbak, S. H., Rizvi, T. A., Bogard, M., and Ratner, N. (2017). Oligodendrocyte RasG12V expressed in its endogenous locus disrupts myelin structure through increased MAPK, nitric oxide, and notch signaling. Glia 65, 1990–2002. doi: 10.1002/glia.23209
Umeki, I., Niihori, T., Abe, T., Kanno, S. I., Okamoto, N., Mizuno, S., et al. (2019). Delineation of LZTR1 mutation-positive patients with Noonan syndrome and identification of LZTR1 binding to RAF1-PPP1CB complexes. Hum. Genet. 138, 21–35. doi: 10.1007/s00439-018-1951-7
Waclaw, R. R., Allen, Z. J. II, Bell, S. M., Erdelyi, F., Szabo, G., Potter, S. S., et al. (2006). The zinc finger transcription factor Sp8 regulates the generation and diversity of olfactory bulb interneurons. Neuron 49, 503–516. doi: 10.1016/j.neuron.2006.01.018
Waclaw, R. R., Ehrman, L. A., Pierani, A., and Campbell, K. (2010). Developmental origin of the neuronal subtypes that comprise the amygdalar fear circuit in the mouse. J. Neurosci. 30, 6944–6953. doi: 10.1523/jneurosci.5772-09.2010
Wang, Y., Kim, E., Wang, X., Novitch, B. G., Yoshikawa, K., Chang, L. S., et al. (2012). ERK inhibition rescues defects in fate specification of Nf1-deficient neural progenitors and brain abnormalities. Cell 150, 816–830. doi: 10.1016/j.cell.2012.06.034
Xiao, L., Ohayon, D., Mckenzie, I. A., Sinclair-Wilson, A., Wright, J. L., Fudge, A. D., et al. (2016). Rapid production of new oligodendrocytes is required in the earliest stages of motor-skill learning. Nat. Neurosci. 19, 1210–1217. doi: 10.1038/nn.4351
Yamamoto, G. L., Aguena, M., Gos, M., Hung, C., Pilch, J., Fahiminiya, S., et al. (2015). Rare variants in SOS2 and LZTR1 are associated with Noonan syndrome. J. Med. Genet. 52, 413–421.
Ye, F., Chen, Y., Hoang, T., Montgomery, R. L., Zhao, X. H., Bu, H., et al. (2009). HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction. Nat. Neurosci. 12, 829–838. doi: 10.1038/nn.2333
Zhang, Y., Chen, K., Sloan, S. A., Bennett, M. L., Scholze, A. R., O’keeffe, S., et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947. doi: 10.1523/jneurosci.1860-14.2014
Zhao, C., Deng, Y., Liu, L., Yu, K., Zhang, L., Wang, H., et al. (2016). Dual regulatory switch through interactions of Tcf7l2/Tcf4 with stage-specific partners propels oligodendroglial maturation. Nat. Commun. 7:10883.
Keywords: LZTR1, Noonan syndrome, MAPK, oligodendrocyte, astrocyte
Citation: Talley MJ, Nardini D, Shabbir N, Ehrman LA, Prada CE and Waclaw RR (2021) Generation of a Mouse Model to Study the Noonan Syndrome Gene Lztr1 in the Telencephalon. Front. Cell Dev. Biol. 9:673995. doi: 10.3389/fcell.2021.673995
Received: 28 February 2021; Accepted: 07 May 2021;
Published: 16 June 2021.
Edited by:
Desheng Liang, Central South University, ChinaReviewed by:
Antonius Plagge, University of Liverpool, United KingdomCopyright © 2021 Talley, Nardini, Shabbir, Ehrman, Prada and Waclaw. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ronald R. Waclaw, cm9uYWxkLndhY2xhd0BjY2htYy5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.