 Carlos S. Subauste
Carlos S. Subauste
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 09 June 2021
Sec. Membrane Traffic and Organelle Dynamics
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.673813
This article is part of the Research Topic The Cell Biology of Protist Parasite-Host Interfaces View all 12 articles
Toxoplasma gondii is an obligate intracellular protozoan that can cause encephalitis and retinitis in humans. The success of T. gondii as a pathogen depends in part on its ability to form an intracellular niche (parasitophorous vacuole) that allows protection from lysosomal degradation and parasite replication. The parasitophorous vacuole can be targeted by autophagy or by autophagosome-independent processes triggered by autophagy proteins. However, T. gondii has developed many strategies to preserve the integrity of the parasitophorous vacuole. Here, we review the interaction between T. gondii, autophagy, and autophagy proteins and expand on recent advances in the field, including the importance of autophagy in the regulation of invasion of the brain and retina by the parasite. We discuss studies that have begun to explore the potential therapeutic applications of the knowledge gained thus far.
Intracellular pathogens have developed a broad range of strategies to survive within host cells. Toxoplasma gondii is an important example of these pathogens. T. gondii secretes proteins during its interaction with host cells that: (i) Enable the formation of a parasitophorous vacuole (PV) that avoids the classical pathway of phagolysosomal fusion; (ii) Activate host cell signaling cascades that negatively regulate autophagic targeting of the PV. As a result, the parasite avoids lysosomal degradation mediated by constitutive autophagy and prevents its complete eradication when autophagy is stimulated by CD40; (iii) Impair the ability of interferon-gamma (IFN-γ) to activate autophagosome-independent effector mechanisms directed against the PV. We provide a brief overview of early work on the role of macroautophagy (referred herein as autophagy) and autophagy proteins in T. gondii infection and discuss in more detail recent discoveries that include the role of autophagy in the regulation of invasion of the brain and retina by T. gondii, identification of a signaling pathway that causes sustained blockade of autophagic targeting of the parasite, identification of a molecular link between IFN-γ and autophagy proteins (ATG) in human cells as well as the role of ATG8 orthologs in the cell-autonomous control of T. gondii induced by IFN-γ. We also discuss the potential implications for new therapeutic approaches against toxoplasmosis.
Toxoplasma gondii is an apicomplexan protozoan of worldwide distribution. T. gondii is believed to remain in the infected host for life. One-third of the world population is chronically infected with T. gondii (Montoya and Liesenfeld, 2004). Although the infection is usually asymptomatic, T. gondii can cause ocular toxoplasmosis in immunocompetent and immunosuppressed patients and encephalitis in immunocompromised individuals (Montoya and Liesenfeld, 2004). In addition, pregnant women acutely infected with T. gondii can transmit the infection to the unborn baby (Montoya and Liesenfeld, 2004). Congenital toxoplasmosis can result in severe ocular and neurologic sequelae as well as abortion (Montoya and Liesenfeld, 2004). Most of the strains of T. gondii isolated in North America and Europe fall into three clonal lineages: I, II, and III. Atypical strains that can have a higher propensity to cause disease have been reported in South America (Gilbert et al., 2008).
There are various forms of the parasite during its life cycle that include: (i) the tachyzoite that infects almost any nucleated cell of the host; (ii) the tissue cyst (containing bradyzoites) that persists in tissues of infected hosts during the chronic phase of the infection; and (iii) the oocyst (containing sporozoites) that is produced in the intestine of felines (the definitive host) during the sexual cycle of the parasite. Humans and other intermediary hosts become infected by ingesting tissue cysts or oocysts. This is followed by the release of bradyzoites or sporozoites into the intestinal lumen, invasion of intestinal epithelial cells, transformation into tachyzoites, and dissemination via the blood and lymphatic.
Tachyzoites of T. gondii invade mammalian cells and form a PV within these cells. This process is dependent on the secretion of contents from parasite organelles: (i) Micronemes that secrete micronemal proteins (MICs), adhesins that bind host cell membrane receptors (Carruthers and Tomley, 2008). (ii) Rhoptries that secrete rhoptry neck proteins that form the moving junction (Bradley and Sibley, 2007). This structure anchors the parasite to the host cell cytoskeleton and excludes host cell type I transmembrane proteins from the membrane surrounding the parasite (Mordue et al., 1999). The exclusion of these proteins could explain the first strategy identified for parasite survival: avoiding the classical pathway of phagolysosomal fusion (Mordue et al., 1999). Rhoptries also inject rhoptry kinases (ROPs) into the host cell cytoplasm that manipulate host cell signaling (Hakimi et al., 2017). (iii) Dense granules that release GRA proteins that maintain the intravacuolar network, anchor to the PV membrane (PVM), and interact with the host cytosol (Cesbron-Delauw et al., 2008; Hakimi et al., 2017). As explained later, proteins from these organelles are also instrumental in setting in motion signaling cascades that subvert cell autonomous mechanisms of parasite eradication.
Studies in humans and mice revealed that CD40 and its main ligand CD154 are central for protection against cerebral and ocular toxoplasmosis (Subauste et al., 1999; Reichmann et al., 2000; Portillo et al., 2010). Although studies in patients with congenital deficiency of CD154 (X-linked hyper immunoglobulin M syndrome) linked defective T cell priming and type 1 cytokine production to susceptibility to toxoplasmosis (Subauste et al., 1999), work done in CD40–/– and CD154–/– mice showed that these animals had increased susceptibility to cerebral and ocular toxoplasmosis despite seemingly unimpaired expression of IFN-γ (Reichmann et al., 2000; Portillo et al., 2010). The likely mechanisms of protection against cerebral and ocular toxoplasmosis identified in mice are as follows: (i) CD40-induced toxoplasmacidal activity in macrophages/microglia (Reichmann et al., 2000; Portillo et al., 2010); (ii) recently identified induction of toxoplasmacidal activity in neural endothelial cells accompanied by reduced invasion of the brain and retina by the parasite (Portillo et al., 2019); (iii) reduced anti-T. gondii antibody production (Portillo et al., 2021); (iv) protection against CD8+ T cell exhaustion (Bhadra et al., 2011). CD8+ T cells, important mediators of protection against T. gondii, undergo exhaustion (loss of functional capacities) during the chronic phase of T. gondii infection, a process that is mediated by program death-1 (Bhadra et al., 2011). Blockade of program death-1 rescues CD8+ T cells, an effect that is dependent on the CD40-CD154 pathway (Bhadra et al., 2011). However, it is important to note that cerebral and ocular toxoplasmosis in CD40–/– and CD154–/– mice precede the development of CD8+ T cell exhaustion.
CD40 kills a type I strain of T. gondii in human and mouse macrophages, a process independent of IFN-γ and effector responses downstream of this cytokine, but is instead caused by autophagy-mediated degradation of the parasite (Andrade et al., 2006). The molecular events involved in autophagy, including the role of this process in T. gondii infection, have been reviewed elsewhere (Mizushima et al., 2010; Besteiro, 2019; Subauste, 2019; Melia et al., 2020; Nakatogawa, 2020). A schematic summary of autophagy is shown in Figure 1.
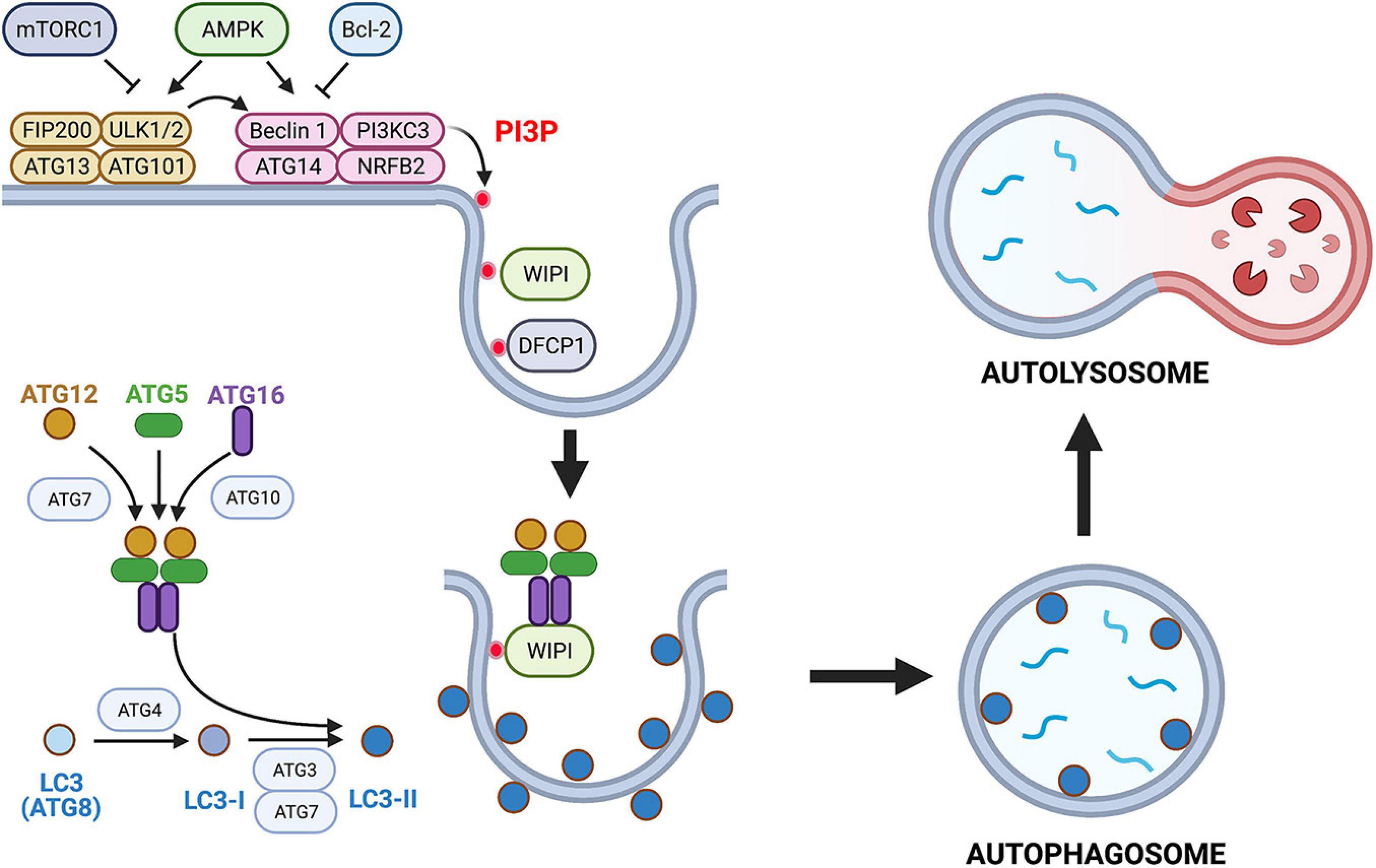
Figure 1. Overview of the autophagy pathway. Autophagy is a constitutive and well-conserved process of lysosomal degradation. Autophagy is triggered by activation of ULK1 that forms a complex with ATG13, ATG101, and FIP200. ULK1 is under control of AMPK and mechanistic target of rapamycin complex 1 (mTORC1). A decrease in energy status activates ULK1 via AMPK leading to autophagy stimulation, whereas nutrient-rich conditions inhibit ULK1 via mTORC1. Membrane-associated ULK1 complex recruits and activates the PI3KC3 complex that also contains Beclin 1, ATG14, nuclear receptor binding factor 2 (NRBF2), as well as p150 and activating molecule in BECN1-regulated autophagy protein 1 (AMBRA1) (not shown). Bcl-2 inhibits pro-autophagic effect of Beclin 1. Activation of PI3KC3 complex causes local production of PI3P that recruits PI3P-binding proteins double FYVE domain-containing protein 1 (DFCP1) and WD-repeat protein interacting with phosphoinositides (WIPI). These scaffold proteins mediate membrane remodeling. Active PI3KC3 complex and WIPI promote expansion of isolation membrane through recruitment of ATG proteins. These proteins function as two ubiquitin-like conjugation systems: (i) ATG7 and ATG10 that conjugate ATG5 to ATG12 and ATG16. (ii) ATG3 and ATG7 together with ATG12–ATG5–ATG16 complex induce lipidation (phosphatidylethanolamine conjugation) of LC3 (ATG8). Lipidated LC3 (LC3-II) binds to several autophagy receptors promoting substrate uptake. Sequestration of the cargo by autophagosomes is followed by fusion with lysosomes resulting in formation of an autolysosome and lysosomal degradation of the cargo. This figure is a simplified overview of some of central components of autophagy pathway in mammalian cells. Figure was generated using BioRender.com.
CD40 stimulates autophagy through events that include: (i) Activation of Unc-51-like kinase 1 (ULK1), the upstream kinase that stimulates autophagy, through a process dependent on calcium/calmodulin-dependent kinase kinase-β (a sensor of intracellular Ca2+) and AMP-activated protein kinase (an energy and nutrient sensor) (Figure 2); (ii) dissociation of the autophagy protein Beclin 1 (ATG6) from its negative regulator Bcl-2; (iii) upregulation of Beclin 1; and (iv) activation of protein kinase double-stranded RNA-dependent (PKR) and the α subunit of eukaryotic initiation factor 2 α, proteins that stimulate autophagy (Ogolla et al., 2013; Liu et al., 2016; Figure 2). As a result, the autophagy protein LC3 (ATG8) is recruited around the PV, a process that is followed by vacuole–lysosomal fusion (VLF) as assessed by an accumulation of the late endosomal and lysosomal markers Rab7, mannose-6-phosphate receptor, CD63, lysosomal-associated membrane protein 1 (LAMP-1), and cathepsin D (Andrade et al., 2006). Parasite killing is dependent on ULK1, Beclin 1, and its interacting partner phosphatidylinositol 3-kinase catalytic subunit type 3 (PI3KC3), the autophagy proteins ATG5 and ATG7, and requires the activity of lysosomal enzymes, indicating that CD40 kills T. gondii in macrophages via autophagy (Andrade et al., 2006; Portillo et al., 2010; Liu et al., 2016). These findings have in vivo relevance because Beclin 1-deficient mice (BECN1±), mice with deficiency of ATG7 in myeloid cells (Atg7flox/flox-Lyz-M Cre mice) and mice deficient in PKR (PKR–/–), exhibited increased susceptibility to cerebral and ocular toxoplasmosis (Portillo et al., 2010; Ogolla et al., 2013).
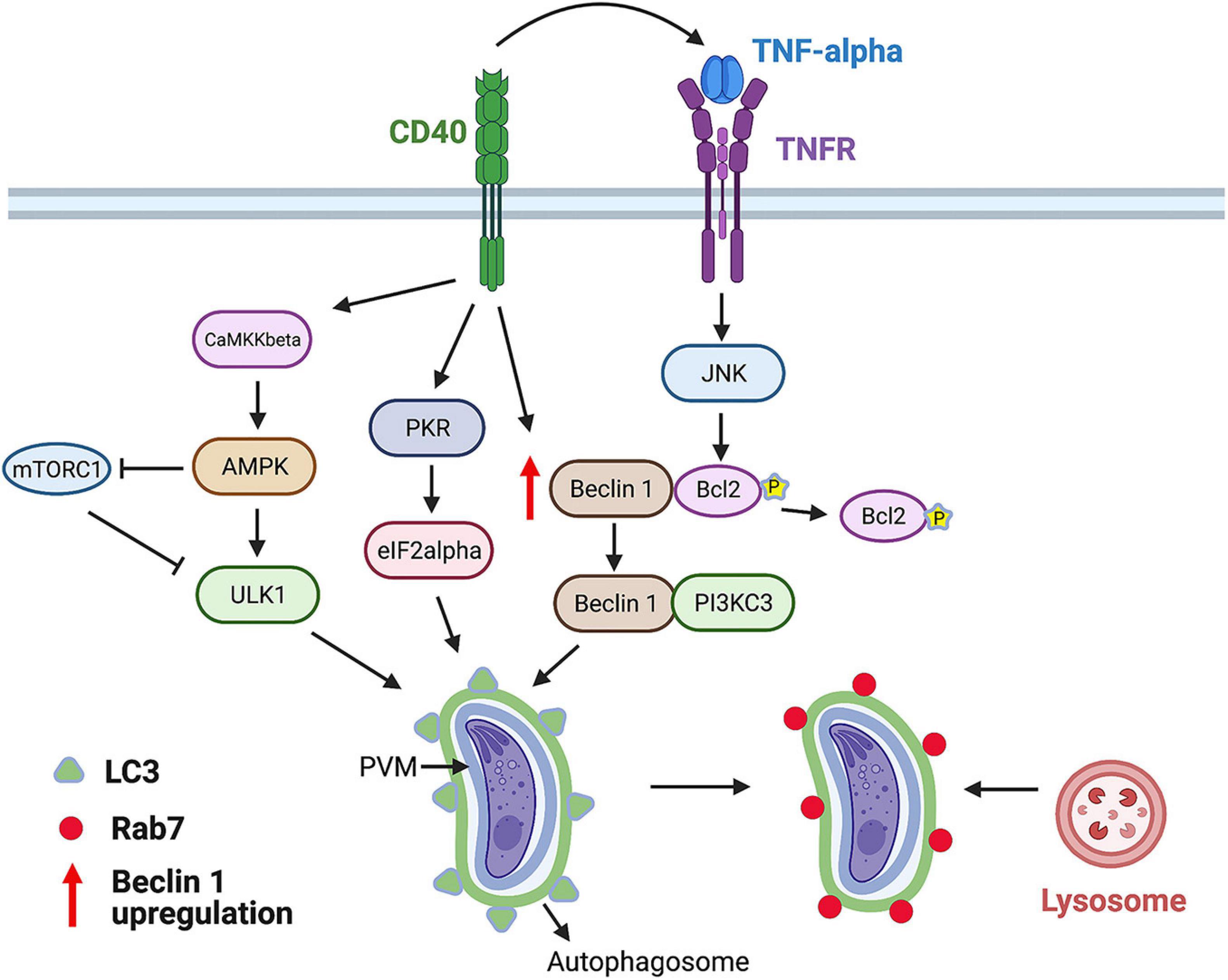
Figure 2. CD40 induces killing of T. gondii via autophagy. CD40 ligation stimulates autophagy by activating ULK1, upstream kinase in the autophagy pathway. This process is dependent on calcium sensor CaMKKβ that activates energy/nutrient sensor AMPK. Besides activating ULK1, AMPK inhibits mTORC1, an inhibitor of autophagy. In addition, CD40 stimulates autophagy by triggering activation of pro-autophagy proteins PKR and eIF2α, upregulating autophagy protein Beclin 1, and releasing Beclin 1 from its negative regulator Bcl-2 after Bcl-2 phosphorylation induced by TNF receptor (TNFR)-JNK signaling. CD40 induces recruitment of LC3 around parasitophorous vacuole membrane (PVM) followed by Rab7-dependent fusion with lysosomes and killing of parasite. This process requires ULK1, Beclin 1, PI3KC3, ATG5, ATG7, and lysosomal enzymes. Figure was generated using BioRender.com.
CD40 induces killing of type I and II strains of T. gondii in various human and mouse non-hematopoietic cells, including endothelial cells, through a mechanism dependent on the same components of the autophagy pathway identified in macrophages (Van Grol et al., 2013). As described later, consistent with the evidence that endothelial cells are the portal of entry into the central nervous system (CNS), CD40-driven autophagic killing in endothelial cells has been recently linked to regulation of invasion of the brain and eye, the two main organs affected by toxoplasmosis (Portillo et al., 2019).
Regardless of the route of infection, T. gondii circulates in the blood within infected leukocytes (including CD11b+ monocytes and dendritic cells) and as extracellular tachyzoites, a process that enables the parasite to invade the CNS (Courret et al., 2006; Lambert et al., 2006; Konradt et al., 2016). Neural endothelial cells become infected, and parasite replication within these cells followed by parasite egress into the neural parenchyma is likely the main mechanism of invasion of the CNS (Konradt et al., 2016). CD40–/– mice exhibited lower parasite load in the brain and retina since the initial stages of infection with T. gondii, suggesting that CD40 restricts invasion of these organs (Portillo et al., 2019). CD40–/– transgenic mice where CD40 expression is rescued in endothelial cells were used to examine further the role of endothelial CD40 during the development of cerebral and ocular toxoplasmosis. The presence of CD40 in these cells resulted in diminished parasite load and histopathology in the brain and retina after T. gondii infection (Portillo et al., 2019). Moreover, a lower parasite load was also detected after intravenous injection of T. gondii-infected CD11b+ monocytes or dendritic cells, indicating that endothelial cell CD40 reduced parasite invasion of the brain and retina (Portillo et al., 2019). The protective effect of endothelial CD40 was dependent on the presence of circulating infected leukocytes because no reduction in parasite load in the brain and retina was noted after challenge with extracellular tachyzoites (Portillo et al., 2019). Rather than reducing transmigration of infecting leukocytes across endothelial cells, CD40 likely conferred protection against cerebral and ocular toxoplasmosis because the presence of CD40 reduced the number of parasite foci in neural endothelial cells, diminishing invasion of the CNS (Portillo et al., 2019). In vitro studies revealed that endothelial cells acquired toxoplasmacidal activity upon interaction with T. gondii-infected dendritic cells or macrophages (Portillo et al., 2019). This effect was dependent on the expression of CD40, ULK1, and Beclin 1 in endothelial cells and led to the recruitment of LC3 and LAMP-1 around the PV of infected endothelial cells. During the interaction between infected leukocytes and endothelial cells, CD154 did not seem to be the major trigger for the autophagic killing of T. gondii in CD40+ endothelial cells (Portillo et al., 2019). Rather, the expression on infected leukocytes of inducible heat shock protein 70, a protein reported to function as a ligand for CD40 (Becker et al., 2002; Shevtsov et al., 2014), seemed to induce toxoplasmacidal activity in endothelial cells (Portillo et al., 2019). Finally, the reduction in parasite invasion of the brain and retina was dependent not only on the expression of CD40 by endothelial cells but also on the expression of Beclin 1 and the expression of inducible heat shock protein 70 in dendritic cells (Portillo et al., 2019). These results indicate that CD40 and autophagy enhance protection against cerebral and ocular toxoplasmosis also by restricting parasite invasion of neural tissue.
Immune mechanisms of protection against pathogens, including T. gondii, do not fully overlap in humans and mice. For example, protection against T. gondii in mice requires IFN-γ and its downstream molecule signal transducer and activator of transcription 1 (STAT1) (Gavrielescu et al., 2004; Lieberman et al., 2004). A markedly different phenotype occurs in patients with an autosomal dominant defect in IFN-γR1 that results in deletion of the STAT1 binding site (Janssen et al., 2002). These patients are not susceptible to toxoplasmosis, although their macrophages are unable to display anti-T. gondii activity in response to IFN-γ (Janssen et al., 2002). Incubation with high concentrations of tumor necrosis factor-alpha (TNF-α) partially restored antimicrobial activity of macrophages from these patients, suggesting a protective role for TNF-α in these patients (Janssen et al., 2002). CD40 may contribute to IFN-γ-independent mechanisms of protection in humans (Subauste, 2009), as CD40 induces killing of T. gondii independently of IFN-γ, STAT1, and effector molecules downstream of IFN-γ [immunity-related GTPases (IRGs); nitric oxide synthase 2] and CD40 induces TNF-α production (Andrade et al., 2005; Subauste and Wessendarp, 2006).
Under basal conditions, T. gondii-infected cells accumulate LC3+ structures around the PV without leading to autophagic targeting of the parasite (Wang et al., 2009), indicating that the parasite avoids targeting by autophagy. Indeed, T. gondii activates epidermal growth factor receptor (EGFR) and Src signaling during the invasion of an infected host cell to prevent its degradation by autophagy. MIC3 and MIC6, parasite proteins that contain EGF-like domains, cause rapid autophosphorylation of EGFR in human and mouse host cells followed by activation of phosphoinositide 3-kinase (PI3K), production of phosphatidylinositol 3,4,5 triphosphate around the PV, and activation of Akt, a negative regulator of autophagy (Muniz-Feliciano et al., 2013; Figure 3A). Blockade of any of the components of this cascade caused accumulation of LC3 and LAMP-1 around the PV, entrapment of the PV by a double membrane structure with characteristics compatible with those of an autophagosome, and killing of type I and II strains of T. gondii in human and rodent hematopoietic and non-hematopoietic cells that were dependent on the autophagy proteins ULK1, Beclin 1, ATG7, and lysosomal enzymes (Muniz-Feliciano et al., 2013).
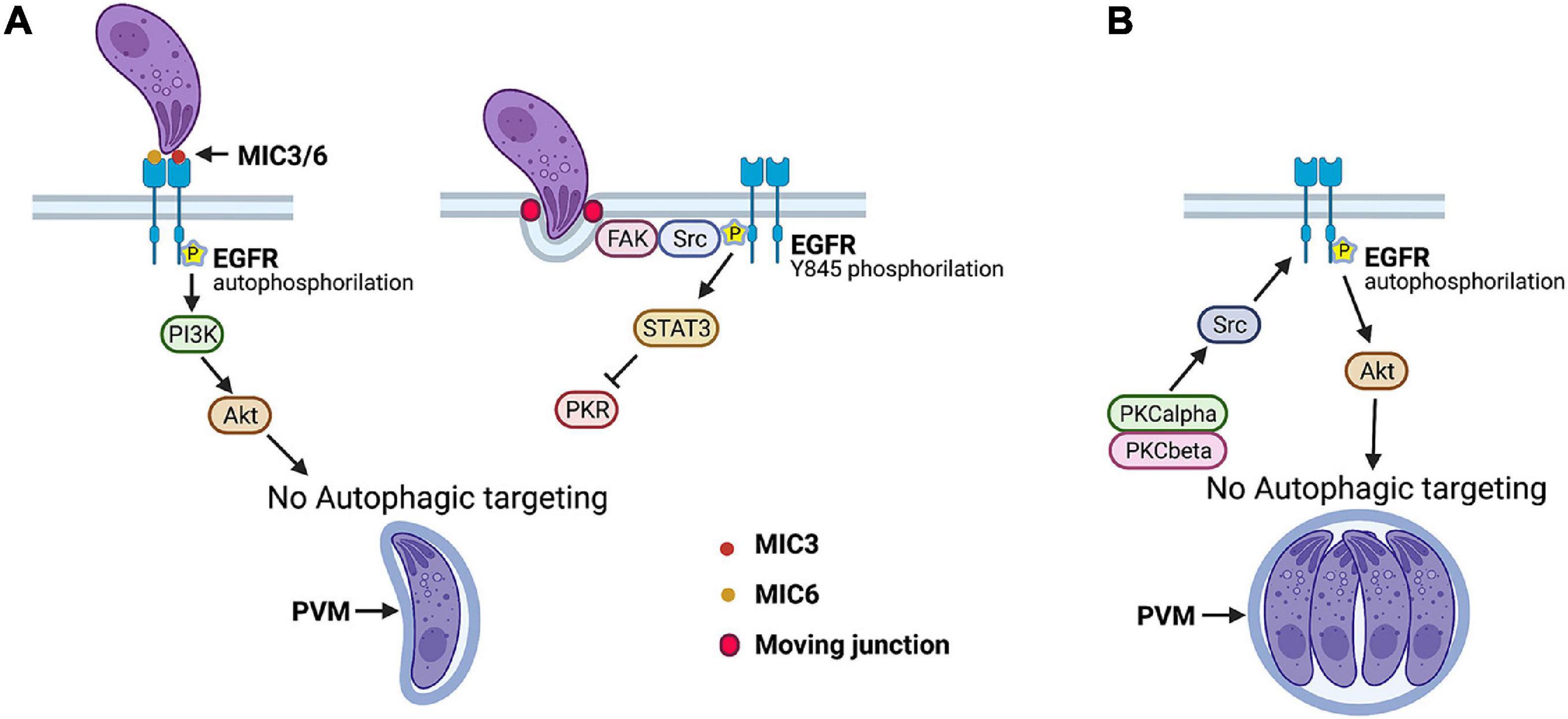
Figure 3. Toxoplasma gondii manipulates host cell signaling cascades that prevent autophagic targeting of parasite. (A) Parasite adhesins MIC3 and MIC6 induce EGFR autophosphorylation that triggers PI3K-dependent activation of Akt, a negative regulator of autophagy. FAK, a cytoplasmic non-receptor tyrosine kinase, is activated during invasion of host cell at level of moving junction. This causes Src-dependent transactivation of EGFR (Y845 phosphorylation) and STAT3 signaling that prevents activation of pro-autophagy proteins PKR and eIF2α. (B) T. gondii maintains a blockade of autophagic targeting during its intracellular state by activating serine/threonine kinases PKCα/PKCβ that in turn maintain Src signaling, EGFR autophosphorylation, and Akt activation. Blockade of any of signaling molecules activated by T. gondii results in autophagic targeting of PV and killing of parasite. This process is dependent on ULK-1, Beclin 1, ATG7, and lysosomal enzymes. Figure was generated using BioRender.com.
The formation of the moving junction triggers a second signaling cascade that prevents autophagic targeting of T. gondii: activation of the cytoplasmic non-receptor tyrosine kinase focal adhesion kinase and its interacting partner Src that in turn causes ligand-independent transactivation of EGFR (Y845 phosphorylation of EGFR) (Portillo et al., 2017; Figure 3A). This pathway is responsible for the early phase of activation of STAT3, a negative regulator of autophagy (Portillo et al., 2017; Figure 3A). STAT3 acts by blocking the activation of the pro-autophagy protein PKR and its downstream signaling molecule eIF2α (Portillo et al., 2017). Inhibition of the focal adhesion kinase–Src–pY845 EGFR–STAT3 pathway results in the autophagic killing of type I, type II, and atypical strains of T. gondii in hematopoietic and non-hematopoietic human and rodent cells (Portillo et al., 2017).
Studies in transgenic mice that express a dominant-negative (DN) mutant of EGFR that impairs EGFR autophosphorylation and EGFR transactivation revealed the importance of manipulating host cell signaling in the development of cerebral and ocular toxoplasmosis. Expression of DN EGFR restricted to endothelial cells was accompanied by a reduction in parasite load and histopathology in the brain and retina after T. gondii infection (Lopez Corcino et al., 2019b). A lower parasite load in the CNS was also noted after intravenous challenge with infected dendritic cells or extracellular tachyzoites. The presence of DN EGFR reduced the number of foci of infection in neural endothelial cells (Lopez Corcino et al., 2019b). Moreover, DN EGFR in these cells led to spontaneous recruitment of LC3 around T. gondii, VLF, and parasite killing dependent on ULK1 and lysosomal enzymes (Lopez Corcino et al., 2019b). Autophagy likely explains the protective role of DN EGFR, as in vivo administration of the autophagy inhibitor 3-methyl adenine prevented DN EGFR mice from exhibiting reduced CNS invasion (Lopez Corcino et al., 2019b). Thus, EGFR enhances T. gondii invasion of the CNS likely by promoting parasite survival in endothelial cells through avoidance of autophagic targeting. Moreover, these studies together with those on mice that express CD40 restricted to endothelial cells support the central role of autophagy as a process that regulates T. gondii invasion of the CNS.
The survival strategies described earlier are operative during the invasion of host cells by T. gondii. However, autophagy is a constitutive process, indicating that the parasite likely uses additional mechanisms to maintain a blockade of autophagic targeting. Indeed, T. gondii causes prolonged EGFR autophosphorylation in mammalian cells that is functionally relevant, as the addition of EGFR tyrosine kinase inhibitors (TKIs) 6 h after challenge with T. gondii resulted in the killing of the parasite (Lopez Corcino et al., 2019a). This process involved entrapment of the PV by a double-membrane structure compatible with an autophagosome, accumulation of LC3 and LAMP-1 around the PV, and pathogen killing dependent on ULK1, Beclin 1, and lysosomal enzymes (Lopez Corcino et al., 2019a). T. gondii induces prolonged EGFR autophosphorylation and activation of its downstream molecule Akt through a cascade that consists of the cytosolic serine/threonine kinases protein kinase C α (PKCα) and PKCβ that cooperate to sustain Src activation that drives prolonged EGFR autophosphorylation (Lopez Corcino et al., 2019a; Figure 3B). All these signaling molecules promote parasite survival, as not only inhibition of EGFR but also of PKCα, PKCβ, Src, and Akt in cells previously infected with T. gondii led to parasite killing (Lopez Corcino et al., 2019a). Thus, whereas T. gondii MIC3 and MIC6 act as an early switch for EGFR autophosphorylation, PKCα/β–Src signaling maintains EGFR autophosphorylation, ensuring the non-fusogenic nature of the PV and parasite survival.
IFN-γ activates mechanisms that target the PV triggering cell-autonomous control of the parasite. In the case of mouse macrophages and fibroblasts (but not in human cells), certain autophagy proteins (ATG3, ATG5, ATG7, ATG12, and ATG16L) are required for recruitment to the PVM of effector GKS subfamily of IRGs (Zhao et al., 2008; Khaminets et al., 2010; Choi et al., 2014; Ohshima et al., 2014; Figure 4). This process induces deposition of ubiquitin and p62 on the PVM, followed by p62-dependent recruitment of guanylate-binding proteins (GBPs) (Haldar et al., 2015; Figure 4). The end result is vesiculation and rupture of the PVM and death of susceptible strains (types II and III) of T. gondii (Martens et al., 2005; Ling et al., 2006; Khaminets et al., 2010; Haldar et al., 2015). Type I strains avoid this process because ROP18, ROP5, ROP17, and GRA7 from these virulent strains prevent IRG recruitment to the PVM (Fentress et al., 2010; Fleckenstein et al., 2012; Etheridge et al., 2014; Reese et al., 2014). Disruption of the PVM in cells infected with type II and III strains of T. gondii does not represent bona fide autophagy, as it is not dependent on ULK1, ATG9, and ATG14L (Choi et al., 2014) and is not mediated by lysosomal degradation (Andrade et al., 2006; Van Grol et al., 2013; Choi et al., 2014).
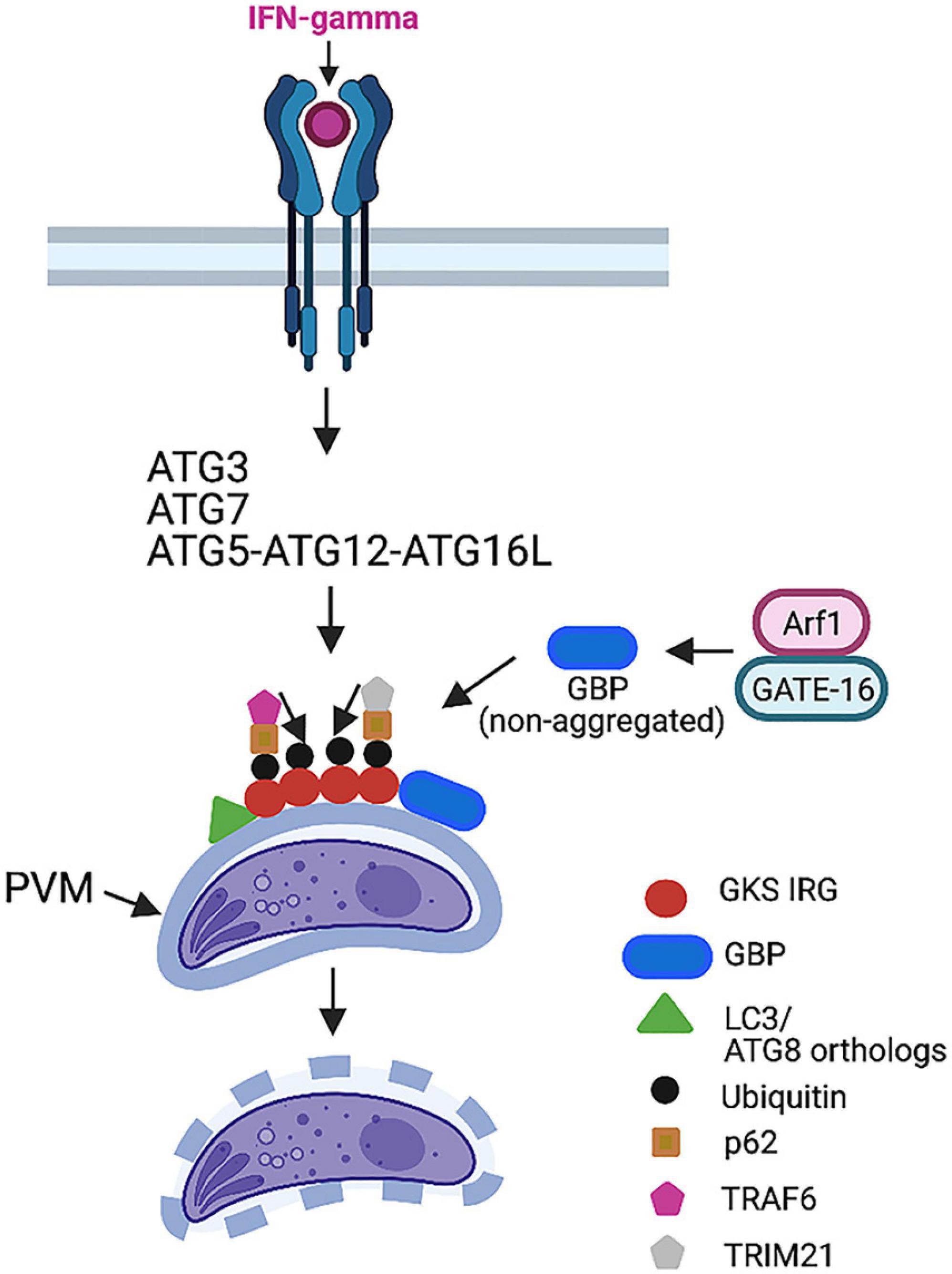
Figure 4. IFN-γ-induces killing of type II and III strains of T. gondii in mouse cells. IFN-γ triggers recruitment of GKS IRGs to parasitophorous vacuole membrane (PVM), a process that requires ATG3, ATG7, and ATG12–ATG5–ATG16L. LC3 and ATG8 orthologs are recruited to PVM. IRGs promote deposition of ubiquitin, p62, and the E3 ubiquitin ligase TRAF6. p62 and TRAF6 further promote sustained decoration of PVM with ubiquitin. This process leads to delivery of GBPs to PVM. The end result is disruption of PVM and death of parasite. TRIM21 is another E3 ubiquitin ligase recruited by p62 that promotes ubiquitination. However, it does not seem to drive vesiculation of PVM. GATE-16 activates Arf1, enabling GBP to remain in a non-aggregated form, thus promoting GBP loading onto PVM. This process does not occur in cells infected with type I strains of T. gondii because ROP5/ROP18/GRA7 inactivate IRGs preventing their loading onto PVM. Figure was generated using BioRender.com.
Ubiquitin is a key component of the machinery that mediates the killing of T. gondii in IFN-γ-activated cells. Deposition of ubiquitin in the PVM was shown to be mediated not only by IRGs but also by p62 and the E3 ubiquitin ligase TNF receptor-associated factor 6 (TRAF6) (Haldar et al., 2015). Ubiquitin binds p62 that, in turn, recruits TRAF6 (Haldar et al., 2015; Figure 4). Both p62 and TRAF6 establish a positive feedback loop that contributes to the rapid and sustained decoration of the PV with ubiquitin, resulting in the delivery of GBPs to the PV and parasite killing (Haldar et al., 2015). The tripartite motif protein 21 (TRIM21) is another E3 ubiquitin ligase that was recruited to the PV and promoted ubiquitin deposition in IFN-γ-treated cells (Haldar et al., 2015; Foltz et al., 2017; Figure 4). Whereas TRIM21 localized to GBP1-positive PV, TRIM21 did not promote vesiculation of the PVM (Foltz et al., 2017). However, TRIM21 was still able to restrict parasite replication (Foltz et al., 2017). In addition, studies in TRIM21–/– mice revealed that TRIM21 played an in vivo role in protection against T. gondii (Foltz et al., 2017). It is possible that the in vivo protective effect of this molecule is also mediated by the modulation of cytokine production (Foltz et al., 2017).
Mammalian cells express various ATG8 orthologs, including LC3A, LC3B, LC3C, gamma-aminobutyric (GABA)-A-receptor-associated protein (GABARAP), GABARAP-like1 (GABARAPL1), and GABARAPL2 (GATE-16). However, the PV is heavily decorated with LC3 in IFN-γ-activated mammalian cells; studies in mouse fibroblasts that lacked LC3A and LC3B revealed that LC3 proteins were dispensable for the control of T. gondii induced by IFN-γ (Sasai et al., 2017). In contrast, mouse fibroblasts that lacked GATE-16, GABARAP, and GABARAPL1 showed a marked defect in the recruitment of Irga6, ubiquitin, p62, and GBP1-5 to the PV as well as impaired parasite clearance after stimulation with IFN-γ (Sasai et al., 2017). The recruitment of GBP was dependent on the small GTPase ADP ribosylation factor 1 (Arf1) (Sasai et al., 2017). GATE-16 associated with Arf1 promoted its activation and, in turn, the recruitment of GBP (Sasai et al., 2017; Figure 4). Similar to mice with deficiency in ATG5, ATG7, or ATG16L targeted to phagocytes (Zhao et al., 2008; Choi et al., 2014), GATE-16-deficient mice died during the acute phase of T. gondii infection (Sasai et al., 2017). Thus, GATE-16 is central for resistance against T. gondii infection.
Although IFN-γ activates cell-autonomous control of T. gondii in mouse and human cells, there are significant differences in the effects of this cytokine between these two species. In this regard, IRGs and GBPs do not mediate anti-T. gondii activity in IFN-γ-stimulated human cells (Bekpen et al., 2005; Niedelman et al., 2013; Ohshima et al., 2014). Ubiquitination and some autophagy proteins are involved in the control of T. gondii induced by IFN-γ in human cells. However, these effector mechanisms are cell-type specific. In the case of human epithelial cells infected with type II or III (but not type I) strains of T. gondii, IFN-γ induces deposition of ubiquitin, p62, nuclear domain 10 protein 52 (NDP52), and LC3 around the PV (Selleck et al., 2015; Figure 5A). Although this process is dependent on ATG16L and ATG7, it does not represent autophagy, as it occurs independently of Beclin 1 and does not lead to VLF (Selleck et al., 2015). Although the integrity of the PV is maintained, a multilayer structure is formed around the PV that seems to explain the reduced growth rate of T. gondii (Selleck et al., 2015; Figure 5A).
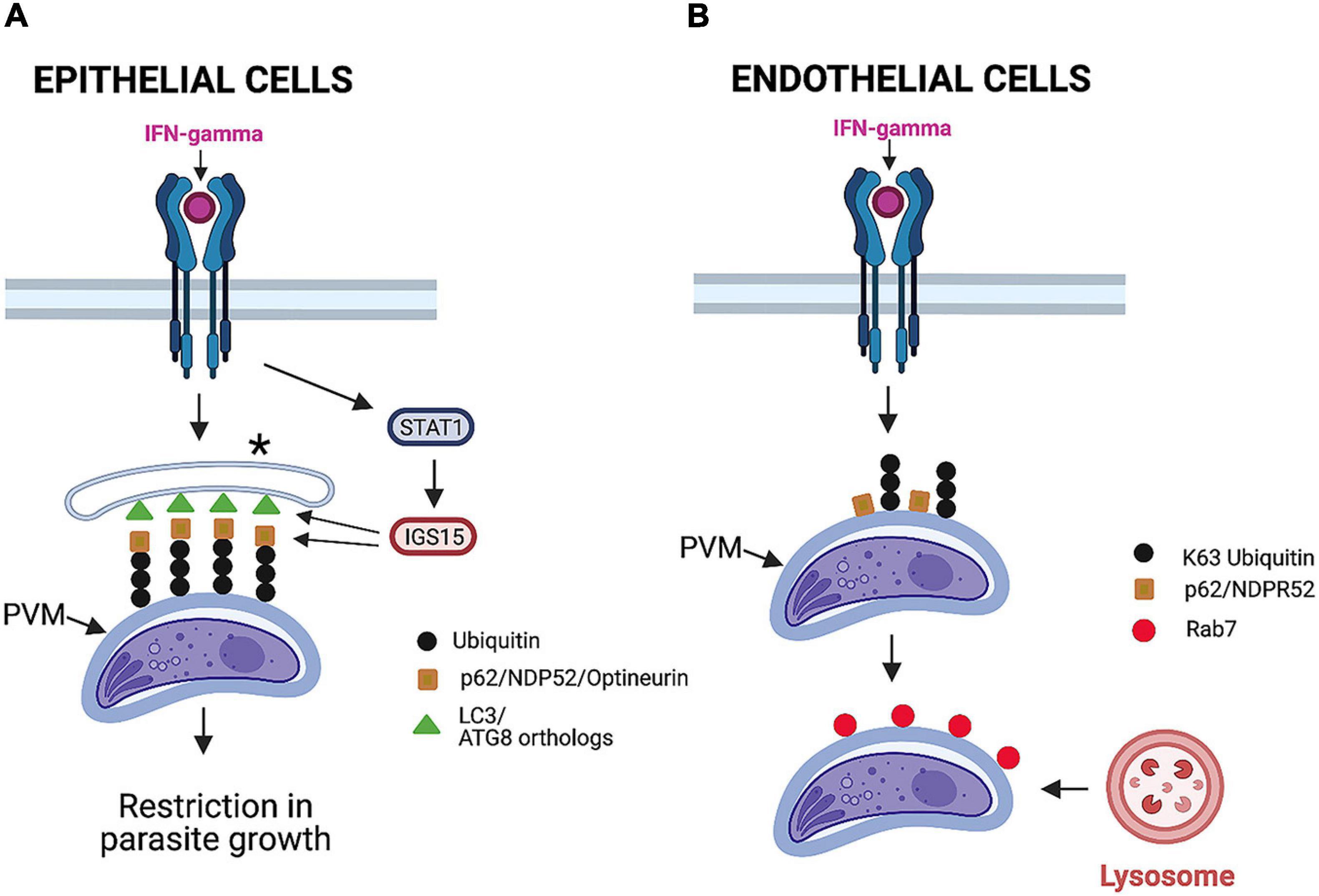
Figure 5. Mechanisms induced by IFN-γ to restrict T. gondii in human cells. (A) In epithelial cells, IFN-γ promotes deposition of ubiquitin, adaptor proteins (p62, NDPR52, and optineurin), and LC3 around parasitophorous vacuole membrane (PVM). STAT1-dependent upregulation of ISG15 stimulates deposition of adaptor proteins around PVM. This process is independent of Beclin 1 and does not cause vacuole lysosomal fusion. Instead, it results in entrapment of parasitophorous vacuole by multilayer membrane structure (*) and inhibition of parasite growth. (B) In endothelial cells infected with a type II strain of T. gondii, IFN-γ induces deposition of ubiquitin, p62, and NDPR52. This is followed by recruitment of Rab7, fusion with lysosomes, and parasite killing. Importantly, LC3 is not recruited around PVM, and this process is not dependent on key autophagy molecule ATG16L. Figure was generated using BioRender.com.
IFN-γ also induces ubiquitination of the PV and recruitment of p62 and NDP52 in human endothelial cells (Clough et al., 2016; Figure 5B). However, in contrast to epithelial cells, LC3, GABARAP, and ATG16L are not recruited around the PV, and the induction of anti-T. gondii activity in endothelial cells is not dependent on ATG16L (Clough et al., 2016). Importantly, recruitment of ubiquitin and the adaptors p62 and NDP52 is followed by acidification of the PV and parasite killing (Clough et al., 2016; Figure 5B). This mechanism is operative against type II but not type I strains of T. gondii (Clough et al., 2016).
A recent study revealed that the presence of the ATG8 ortholog GABARAPL2 was key for the restriction of T. gondii replication in IFN-γ-activated HeLa cells (Zhang et al., 2020). GABARAPL2 accumulated around the PV in these cells in response to IFN-γ (Zhang et al., 2020). Similar to previous studies, IFN-γ induced deposition around the PV of ubiquitin and the adaptor proteins p62, NDP52, and optineurin (Zhang et al., 2020). These events are functionally relevant, as ubiquitination inhibitors or knockdown of the adaptor proteins impaired the recruitment of GABARAPL2 (Zhang et al., 2020). Moreover, knockdown of p62, NDP52, and optineurin impaired the ability of IFN-γ to reduce T. gondii replication. Finally, the recruitment of GABARAPL2 was dependent on the presence of ATG5, suggesting that lipidation of GABARAPL2 is required for its localization around the PV (Zhang et al., 2020). Thus, these studies, similar to those conducted in mice (Sasai et al., 2017), revealed the importance of GABARAPL2 in the control of T. gondii induced by IFN-γ.
Although IFN-γ sets in motion cell-autonomous immune responses to control T. gondii that is dependent on autophagy proteins, IFN-γ does not seem to upregulate ATG proteins. Recent studies were conducted to uncover the link between IFN-γ and the ATG pathway (Bhushan et al., 2020). IFN-γ upregulates IFN-stimulated genes (ISGs) (MacMicking, 2012). Work done in human epithelial cells revealed that ISG15 represents a molecular link between IFN-γ and the ATG pathway (Bhushan et al., 2020). ISG15 was markedly upregulated in IFN-γ-treated HeLa and A549 epithelial cells (Bhushan et al., 2020). Deletion of ISG15 partially inhibited the recruitment of p62, NDP52, and LC3 around the PV in IFN-γ-treated A549 cells without impairing ubiquitination of the vacuole (Bhushan et al., 2020; Figure 5A). The effect of ISG15 was selective, as the lack of this protein did not affect autophagy flux induced by rapamycin plus bafilomycin A1 (Bhushan et al., 2020). Moreover, ISG15 played an important role in the induction of IFN-γ-dependent restriction in T. gondii replication (Bhushan et al., 2020). Interestingly, ISG15–/– mice did not have increased susceptibility to T. gondii (Napolitano et al., 2018). This may be explained by the lack of a known role for ISG15 in the recruitment of IRGs and GBPs, the major mechanism that mediates cell-autonomous control of T. gondii in mice.
Current antibiotic regimens against toxoplasmosis have significant adverse effects and are not proven to be effective in the case of ocular toxoplasmosis. Compounds that stimulate autophagy could become part of treatment regimens against T. gondii. Treatment of T. gondii-infected mammalian cells with rapamycin, a mTORC1 inhibitor and inducer of autophagy, triggered the killing of the parasite in a manner dependent on the presence of Beclin 1 (Andrade et al., 2006). Other studies evaluated the effects of docosahexaenoic acid (DHA), an omega-3 polyunsaturated fatty acid (Choi et al., 2019). DHA likely stimulated autophagy flux in mouse bone marrow-derived macrophages (BMDM) (Choi et al., 2019). In addition, DHA induced anti-T. gondii activity in BMDM that seemed to be mediated by autophagy, as DHA promoted the recruitment of LC3 around the PV, and 3-methyl adenine inhibited the anti-T. gondii activity induced by DHA (Choi et al., 2019). Similar findings were observed in BMDM from Fat-1 transgenic mice that synthesize omega-3 polyunsaturated fatty acid from ω6-PUFA (Choi et al., 2019). Moreover, Fat-1 mice infected with a type II strain of T. gondii exhibited lower tissue cyst counts in the brain (Choi et al., 2019).
More recent studies evaluated 4-hydroxybenzaldehyde (4-HBA) (Lee et al., 2020). 4-HBA-induced anti-T. gondii activity in BMDM seemed to be mediated by autophagy (Lee et al., 2020). 4-HBA seems to act via sirtuin-1 (SIRT1), a nicotine adenine dinucleotide-dependent protein deacetylase that can stimulate autophagy (Lee et al., 2020). 4-HBA upregulated SIRT1 protein levels, and SIRT1 inhibitors impaired the anti-T. gondii activity induced by 4-HBA. Although drugs that stimulate autophagy may facilitate the elimination of T. gondii, there are caveats with this approach. Autophagy regulates a broad range of homeostatic responses, including those in T cells, raising the concern that global stimulation of autophagy may have unintended negative consequences.
As part of an interesting approach, high-throughput screening of diverse small molecule libraries was performed in search of compounds that would enhance the anti-T. gondii activity of IFN-γ (Radke et al., 2018). A number of small molecules were identified that inhibited the growth of T. gondii at micromolar concentrations and exhibited increased potency in the presence of low concentrations of IFN-γ (Radke et al., 2018). Interestingly, some of these compounds increased the recruitment of LC3 around the PV, suggesting that they may act by potentiating the autophagy protein-mediated anti-T. gondii effects of IFN-γ (Radke et al., 2018). Compounds that synergize with immune mechanisms of control of T. gondii may improve the efficacy and tolerability of treatment regimens against toxoplasmosis.
The discovery that T. gondii manipulates host cell signaling to persistently block autophagic killing indicates that host-derived therapy may represent an attractive approach to improve the treatment of toxoplasmosis. Addition of Gefitinib, an EGFR TKI currently being used against cancer, to a broad range of human and rodent cells previously infected with T. gondii induced autophagic killing of type I and II strains of T. gondii (Lopez Corcino et al., 2019a). Importantly, Gefitinib was effective at concentrations up to ∼2 logs lower than those required for cytostatic activity against cancer cells (Lopez Corcino et al., 2019a). Gefitinib acted “on target” (required the presence of EGFR) and was active in the absence of immune activation of host cells. Moreover, in vivo administration of a relatively low dose of Gefitinib (∼1/4th to 1/12th of those used in cancer models) to mice with preestablished ocular and cerebral toxoplasmosis resulted in control of the disease (Lopez Corcino et al., 2019a). The protective effect of Gefitinib was not mediated by enhanced cellular or humoral immunity against T. gondii but was dependent on the normal expression of Beclin 1 (Lopez Corcino et al., 2019a). These findings, together with the demonstration that inhibition of EGFR or the upstream inducers of EGFR signaling results in autophagic killing of type I, II, or atypical strains of T. gondii in various human and mouse cells (Muniz-Feliciano et al., 2013; Portillo et al., 2017; Lopez Corcino et al., 2019a), identified EGFR as a therapeutic target against toxoplasmosis. A combination of low-dose EGFR TKI with antibiotics may lead to an improved regimen against toxoplasmosis.
Maintaining the integrity of the PV is essential for the survival of T. gondii. CD40 stimulates autophagy that targets the PV resulting in the killing of T. gondii. Blocking the counterregulatory signaling induced by the parasite is sufficient to enable constitutive autophagy to target the PV and kill T. gondii (without the need for immune stimulation of autophagy). Studies to date indicate that in both scenarios, autophagic killing is not restricted to cell type, host species, or parasite strain. In contrast, IFN-γ triggers various autophagosome-independent mechanisms that attack the PV. The mechanisms activated by IFN-γ that have been identified thus far are not operative across species or parasite strains. Moreover, those uncovered in humans are not operative across cell types. Although much has been learned, further studies are needed to uncover the full spectrum of effector mechanisms induced by IFN-γ, especially in human cells, and identify the pathways of evasion utilized by T. gondii. This knowledge may contribute to the development of new approaches to improve the treatment of toxoplasmosis. The constitutive and conserved nature of autophagy already indicates that drugs that target components of the counterregulatory signaling pathway activated by T. gondii may become part of the novel and improved therapy against toxoplasmosis.
The author confirms being the sole contributor of this work and has approved it for publication.
CS was funded by NIH-R01 EY018341 and NIH-R01 EY019250.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author thanks all the members of the Subauste lab for their feedback on this manuscript.
Andrade, R. M., Portillo, J.-A. C., Wessendarp, M., and Subauste, C. S. (2005). CD40 signaling in macrophages induces anti-microbial activity against an intracellular pathogen independently of IFN-(and reactive nitrogen intermediates. Infect. Immun. 73, 3115–3123. doi: 10.1128/iai.73.5.3115-3123.2005
Andrade, R. M., Wessendarp, M., Gubbels, M. J., Striepen, B., and Subauste, C. S. (2006). CD40 induces macrophage anti-Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J. Clin. Invest. 116, 2366–2377. doi: 10.1172/jci28796
Becker, T., Hartl, F. U., and Wieland, F. (2002). CD40, an extracellular receptor for binding and uptake of Hsp70-peptide complexes. J. Cell Biol. 158, 1277–1285. doi: 10.1083/jcb.200208083
Bekpen, C., Hunn, J. P., Rohde, C., Parvanova, I., Guethlein, L., Dunn, D. M., et al. (2005). The interferon-inducible p47 (IRG) GTPases in vertebrates: loss of the cell autonomous resistance mechanism in the human lineage. Genome Biol. 6:R92.
Besteiro, S. (2019). The role of host autophagy machinery in controlling Toxoplasma infection. Virulence 10, 438–447. doi: 10.1080/21505594.2018.1518102
Bhadra, R., Gigley, J. P., and Khan, I. A. (2011). Cutting edge: CD40-CD40 ligand pathway plays a critical CD8-intrinsic and -extrinsic role during rescue of exhausted CD8 T cells. J. Immunol. 187, 4421–4425. doi: 10.4049/jimmunol.1102319
Bhushan, J., Radke, J. B., Perng, Y. C., Mcallaster, M., Lenschow, D. J., Virgin, H. W., et al. (2020). ISG15 Connects autophagy and IFN-gamma-dependent control of Toxoplasma gondii infection in human cells. mBio 11:e00852-20.
Bradley, P. J., and Sibley, L. D. (2007). Rhoptries: an arsenal of secreted virulence factors. Curr. Opin. Microbiol. 10, 582–587. doi: 10.1016/j.mib.2007.09.013
Carruthers, V. B., and Tomley, F. M. (2008). Receptor-ligand interaction and invasion: microneme proteins in apicomplexans. Subcell. Biochem. 47, 33–45. doi: 10.1007/978-0-387-78267-6_2
Cesbron-Delauw, M. F., Gendrin, C., Travier, L., Ruffiot, P., and Mercier, C. (2008). Apicomplexa in mammalian cells: trafficking to the parasitophorous vacuole. Traffic 9, 657–664. doi: 10.1111/j.1600-0854.2008.00728.x
Choi, J., Park, S., Biering, S. B., Selleck, E., Liu, C. Y., Zhang, X., et al. (2014). The parasitophorous vacuole membrane of Toxoplasma gondii is targeted for disruption by ubiquitin-like conjugation systems of autophagy. Immunity 40, 924–935. doi: 10.1016/j.immuni.2014.05.006
Choi, J. W., Lee, J., Lee, J. H., Park, B. J., Lee, E. J., Shin, S., et al. (2019). Omega-3 polyunsaturated fatty acids prevent Toxoplasma gondii infection by inducing autophagy via AMPK activation. Nutrients 11:2137. doi: 10.3390/nu11092137
Clough, B., Wright, J. D., Pereira, P. M., Hirst, E. M., Johnston, A. C., Henriques, R., et al. (2016). K63-Linked ubiquitination targets Toxoplasma gondii for endo-lysosomal destruction in IFN-gamma-stimulated human cells. PLoS Pathog 12:e1006027. doi: 10.1371/journal.ppat.1006027
Courret, N., Darche, S., Sonigo, P., Milon, G., Buzoni-Gatel, D., and Tardieux, I. (2006). CD11c- and CD11b-expressing mouse leukocytes transport single Toxoplasma gondii tachyzoites to the brain. Blood 107, 309–316. doi: 10.1182/blood-2005-02-0666
Etheridge, R. D., Alaganan, A., Tang, K., Lou, H. J., Turk, B. E., and Sibley, L. D. (2014). The Toxoplasma pseudokinase ROP5 forms complexes with ROP18 and ROP17 kinases that synergize to control acute virulence in mice. Cell Host Microbe 15, 537–550. doi: 10.1016/j.chom.2014.04.002
Fentress, S. J., Behnke, M. S., Dunay, I. R., Mashayekhi, M., Rommereim, L. M., Fox, B. A., et al. (2010). Phosphorylation of immunity-related GTPases by a Toxoplasma gondii-secreted kinase promotes macrophage survival and virulence. Cell Host Microbe 8, 484–495. doi: 10.1016/j.chom.2010.11.005
Fleckenstein, M. C., Reese, M. L., Konen-Waisman, S., Boothroyd, J. C., Howard, J. C., and Steinfeldt, T. (2012). A Toxoplasma gondii pseudokinase inhibits host IRG resistance proteins. PLoS Biol. 10:e1001358. doi: 10.1371/journal.pbio.1001358
Foltz, C., Napolitano, A., Khan, R., Clough, B., Hirst, E. M., and Frickel, E. M. (2017). TRIM21 is critical for survival of Toxoplasma gondii infection and localises to GBP-positive parasite vacuoles. Sci. Rep. 7:5209.
Gavrielescu, L. C., Butcher, B. A., Del Rio, L., Taylor, G. A., and Denkers, E. Y. (2004). STAT1 is essential for antimicrobial effector function but dispensable for gamma interferon production during Toxoplasma gondii infection. Infect. Immun. 72, 1257–1264. doi: 10.1128/iai.72.3.1257-1264.2004
Gilbert, R. E., Freeman, K., Lago, E. G., Bahia-Oliveira, L. M., Tan, H. K., Wallon, M., et al. (2008). Ocular sequelae of congenital toxoplasmosis in Brazil compared with Europe. PLoS Negl. Trop. Dis. 2:e277. doi: 10.1371/journal.pntd.0000277
Hakimi, M. A., Olias, P., and Sibley, L. D. (2017). Toxoplasma effectors targeting host signaling and transcription. Clin. Microbiol. Rev. 30, 615–645. doi: 10.1128/cmr.00005-17
Haldar, A. K., Foltz, C., Finethy, R., Piro, A. S., Feeley, E. M., Pilla-Moffett, D. M., et al. (2015). Ubiquitin systems mark pathogen-containing vacuoles as targets for host defense by guanylate binding proteins. Proc. Natl. Acad. Sci. U S A. 112, E5628–E5637.
Janssen, R., Van Wengen, A., Verhard, E., De Boer, T., Zomerdijk, T., Ottenhoff, T. H. M., et al. (2002). Divergent role for TNF-(in IFN-(-induced killing of Toxoplasma gondii and Salmonella typhimurium contributes to selective susceptibility of patients with partial IFN-(receptor 1 deficiency. J. Immunol. 169, 3900–3907. doi: 10.4049/jimmunol.169.7.3900
Khaminets, A., Hunn, J. P., Konen-Waisman, S., Zhao, Y. O., Preukschat, D., Coers, J., et al. (2010). Coordinated loading of IRG resistance GTPases on to the Toxoplasma gondii parasitophorous vacuole. Cell Microbiol. 12, 939–961. doi: 10.1111/j.1462-5822.2010.01443.x
Konradt, C., Ueno, N., Christian, D. A., Delong, J. H., Pritchard, G. H., Herz, J., et al. (2016). Endothelial cells are a replicative niche for entry of Toxoplasma gondii to the central nervous system. Nat. Microbiol. 1:16001.
Lambert, H., Hitziger, N., Dellacasa, I., Svensson, M., and Barragan, A. (2006). Induction of dendritic cell migration upon Toxoplasma gondii infection potentiates parasite dissemination. Cell. Microbiol. 8, 1611–1623. doi: 10.1111/j.1462-5822.2006.00735.x
Lee, J., Choi, J. W., Han, H. Y., Kim, W. S., Song, H. Y., Byun, E. B., et al. (2020). 4-Hydroxybenzaldehyde restricts the intracellular growth of Toxoplasma gondii by inducing SIRT1-mediated autophagy in macrophages. Korean J. Parasitol. 58, 7–14. doi: 10.3347/kjp.2020.58.1.7
Lieberman, L. A., Banica, M., Reiner, S. L., and Hunter, C. A. (2004). STAT1 plays a critical role in the regulation of antimicrobial effector mechanisms, but not in the development of Th1-type responses during toxoplasmosis. J. Immunol. 172, 457–463. doi: 10.4049/jimmunol.172.1.457
Ling, Y. M., Shaw, M. H., Ayala, C., Coppens, I., Taylor, G. A., Ferguson, D. J. P., et al. (2006). Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J. Exp. Med. 203, 2063–2071. doi: 10.1084/jem.20061318
Liu, E., Lopez Corcino, Y., Portillo, J.-A. C., Miao, Y., and Subauste, C. S. (2016). Identification of signaling pathways by which CD40 stimulates autophagy and anti-microbial activity against Toxoplasma gondii in macrophages. Infect. Immun. 84, 2616–2626. doi: 10.1128/iai.00101-16
Lopez Corcino, Y., Gonzalez Ferrer, S., Mantilla, L. E., Trikeriotis, S., Yu, J. S., Kim, S., et al. (2019a). Toxoplasma gondii induces prolonged host epidermal growth factor receptor signalling to prevent parasite elimination by autophagy: perspectives for in vivo control of the parasite. Cell Microbiol. 21:e13084.
Lopez Corcino, Y., Portillo, J.-A. C., and Subauste, C. S. (2019b). Epidermal growth factor receptor promotes cerebral and retinal invasion by Toxoplasma gondii. Sci. Rep. 9:669.
MacMicking, J. D. (2012). Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat. Rev. Immunol. 12, 367–382. doi: 10.1038/nri3210
Martens, S., Parvanova, I., Zerrahn, J., Griffiths, G., Schell, G., Reichmann, G., et al. (2005). Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47-resistance GTPases. PLoS Pathogens 1:e24. doi: 10.1371/journal.ppat.0010024
Melia, T. J., Lystad, A. H., and Simonsen, A. (2020). Autophagosome biogenesis: from membrane growth to closure. J. Cell Biol. 219:e202002085.
Mizushima, N., Yoshimori, T., and Ohsumi, Y. (2010). The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 27, 107–132. doi: 10.1146/annurev-cellbio-092910-154005
Mordue, D. G., Desai, N., Dustin, M., and Sibley, L. D. (1999). Invasion by Toxoplasma gondii establishes a moving junction that selectively excludes host cell plasma membrane proteins on the basis of their membrane anchoring. J. Exp. Med. 190, 1783–1792. doi: 10.1084/jem.190.12.1783
Muniz-Feliciano, L., Van Grol, J., Portillo, J.-A. C., Liew, L., Liu, B., Carlin, C. R., et al. (2013). Toxoplasma gondii-induced activation of EGFR prevents autophagy protein-mediated killing of the parasite. PLoS Pathog. 9:e1003809. doi: 10.1371/journal.ppat.1003809
Nakatogawa, H. (2020). Mechanisms governing autophagosome biogenesis. Nat. Rev. Mol. Cell Biol. 21, 439–458. doi: 10.1038/s41580-020-0241-0
Napolitano, A., Van Der Veen, A. G., Bunyan, M., Borg, A., Frith, D., Howell, S., et al. (2018). Cysteine-reactive free ISG15 generates IL-1beta-producing CD8alpha(+) dendritic cells at the site of infection. J. Immunol. 201, 604–614. doi: 10.4049/jimmunol.1701322
Niedelman, W., Sprokholt, J. K., Clough, B., Frickel, E. M., and Saeij, J. P. (2013). Cell death of gamma interferon-stimulated human fibroblasts upon Toxoplasma gondii infection induces early parasite egress and limits parasite replication. Infect. Immun. 81, 4341–4349. doi: 10.1128/iai.00416-13
Ogolla, P., Portillo, J.-A. C., White, C. L., Patel, K., Lamb, B., Sen, G. C., et al. (2013). The protein kinase double-stranded RNA-dependent (PKR) enhances protection against disease cause by a non-viral pathogen. PLoS Pathog. 9:e100557. doi: 10.1371/journal.ppat.1003557
Ohshima, J., Lee, Y., Sasai, M., Saitoh, T., Su, Ma, J., et al. (2014). Role of mouse and human autophagy proteins in IFN-gamma-induced cell-autonomous responses against Toxoplasma gondii. J. Immunol. 192, 3328–3335. doi: 10.4049/jimmunol.1302822
Portillo, J.-A. C., Okenka, G., Reed, E., Subauste, A., Van Grol, J., Gentil, K., et al. (2010). The CD40-autophagy pathway is needed for host protection despite IFN-γ-dependent immunity and CD40 induces autophagy via control of p21 levels. PLoS One 5:e14472. doi: 10.1371/journal.pone.0014472
Portillo, J.-A. C., Van Grol, J., Saffo, S., Lopez Corcino, Y., Rodriguez, M., Fox, B., et al. (2019). CD40 in endothelial cells restricts neural tissue invasion by Toxoplasma gondii. Infect. Immun. 87:e00868-18.
Portillo, J.-A. C., Yu, J.-S., Hansen, S., Kern, T. S., Subauste, M. C., and Subauste, C. S. (2021). A cell-penetrating CD40-TRAF2,3 blocking peptide diminishes inflammation and neuronal loss after ischemia/reperfusion. FASEB J. 35:e21412.
Portillo, J. C., Muniz-Feliciano, L., Lopez Corcino, Y., Lee, S. J., Van Grol, J., Parsons, S. J., et al. (2017). Toxoplasma gondii induces FAK-Src-STAT3 signaling during infection of host cells that prevents parasite targeting by autophagy. PLoS Pathog 13:e1006671. doi: 10.1371/journal.ppat.1006671
Radke, J. B., Carey, K. L., Shaw, S., Metkar, S. R., Mulrooney, C., Gale, J. P., et al. (2018). High throughput screen identifies Interferon gamma-dependent inhibitors of Toxoplasma gondii growth. ACS Infect. Dis. 4, 1499–1507. doi: 10.1021/acsinfecdis.8b00135
Reese, M. L., Shah, N., and Boothroyd, J. C. (2014). The Toxoplasma pseudokinase ROP5 is an allosteric inhibitor of the immunity-related GTPases. J. Biol. Chem. 289, 27849–27858. doi: 10.1074/jbc.m114.567057
Reichmann, G., Walker, W., Villegas, E. N., Craig, L., Cai, G., Alexander, J., et al. (2000). The CD40/CD40 ligand interaction is required for resistance to toxoplasmic encephalitis. Infect. Immun. 68, 1312–1318. doi: 10.1128/iai.68.3.1312-1318.2000
Sasai, M., Sakaguchi, N., Ma, J. S., Nakamura, S., Kawabata, T., Bando, H., et al. (2017). Essential role for GABARAP autophagy proteins in interferon-inducible GTPase-mediated host defense. Nat. Immunol. 18, 899–910. doi: 10.1038/ni.3767
Selleck, E. M., Orchard, R. C., Lassen, K. G., Beatty, W. L., Xavier, R. J., Levine, B., et al. (2015). A noncanonical autophagy pathway restricts Toxoplasma gondii growth in a strain-specific manner in IFN-gamma-activated human cells. mBio 6:e01157-15.
Shevtsov, M. A., Yakovleva, L. Y., Nikolaev, B. P., Marchenko, Y. Y., Dobrodumov, A. V., Onokhin, K. V., et al. (2014). Tumor targeting using magnetic nanoparticle Hsp70 conjugate in a model of C6 glioma. Neuro Oncol. 16, 38–49. doi: 10.1093/neuonc/not141
Subauste, C. S. (2009). CD40 and the immune response to parasitic infections. Semin. Immunol. 21, 273–282. doi: 10.1016/j.smim.2009.06.003
Subauste, C. S. (2019). Interplay between Toxoplasma gondii, autophagy, and autophagy proteins. Front. Cell Infect. Microbiol. 9:139. doi: 10.3389/fcimb.2019.00139
Subauste, C. S., and Wessendarp, M. (2006). CD40 restrains the in vivo growth of Toxoplasma gondii independently of gamma interferon. Infect. Immun. 74, 1573–1579. doi: 10.1128/iai.74.3.1573-1579.2006
Subauste, C. S., Wessendarp, M., Sorensen, R. U., and Leiva, L. (1999). CD40 - CD40 ligand interaction is central to cell-mediated immunity against Toxoplasma gondii: patients with hyper IgM syndrome have a defective type-1 immune response which can be restored by soluble CD40L trimer. J. Immunol. 162, 6690–6700.
Van Grol, J., Muniz-Feliciano, L., Portillo, J.-A. C., Bonilha, V. L., and Subauste, C. S. (2013). CD40 induces anti-Toxoplasma gondii activity in non-hematopoietic cells dependent on autophagy proteins. Infect. Immun. 81, 2002–2011. doi: 10.1128/iai.01145-12
Wang, Y., Weiss, L. M., and Orlofsky, A. (2009). Host cell autophagy is induced by Toxoplasma gondii and contributes to parasite growth. J. Biol. Chem. 284, 1694–1701. doi: 10.1074/jbc.m807890200
Zhang, Z., Gu, H., Li, Q., Zheng, J., Cao, S., Weng, C., et al. (2020). GABARAPL2 Is critical for growth restriction of Toxoplasma gondii in HeLa cells treated with gamma interferon. Infect. Immun. 88:e00054-20.
Keywords: cell signaling, CD40, IFN-γ, macrophage, endothelial, epithelial
Citation: Subauste CS (2021) Recent Advances in the Roles of Autophagy and Autophagy Proteins in Host Cells During Toxoplasma gondii Infection and Potential Therapeutic Implications. Front. Cell Dev. Biol. 9:673813. doi: 10.3389/fcell.2021.673813
Received: 28 February 2021; Accepted: 30 April 2021;
Published: 09 June 2021.
Edited by:
Carmen Faso, University of Bern, SwitzerlandReviewed by:
Chandra Ramakrishnan, University of Zurich, SwitzerlandCopyright © 2021 Subauste. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlos S. Subauste, Y2FybG9zLnN1YmF1c3RlQGNhc2UuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.