
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 07 October 2021
Sec. Molecular and Cellular Pathology
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.671564
This article is part of the Research Topic The Role of Cellular Senescence in Health and Disease View all 16 articles
 Jiawen Zhou1†
Jiawen Zhou1† Chenxing Hou1†
Chenxing Hou1† Haiyun Chen2†
Haiyun Chen2† Ziyue Qin1†
Ziyue Qin1† Zi’an Miao1
Zi’an Miao1 Jingyu Zhao1
Jingyu Zhao1 Qiuyi Wang1
Qiuyi Wang1 Min Cui1
Min Cui1 Chunfeng Xie3
Chunfeng Xie3 Rong Wang1
Rong Wang1 Qing Li4
Qing Li4 Guoping Zuo1
Guoping Zuo1 Dengshun Miao1,2
Dengshun Miao1,2 Jianliang Jin1*
Jianliang Jin1*This study aimed to determine whether Bmi-1 deficiency leads to intestinal epithelial barrier destruction and microbiota dysfunction, which members of the microbial community alter barrier function with age, and whether p16INK4a deletion could reverse the damage of intestinal epithelial barrier and microbial dysbiosis. Intestines from Bmi-1–deficient (Bmi-1–/–), Bmi-1 and p16INK4a double-knockout (Bmi-1–/–p16INK4a–/–), and wild-type mice were observed for aging and inflammation. Duolink Proximity Ligation Assay, immunoprecipitation, and construction of p16INK4a overexpressed adenovirus and the overexpressed plasmids of full-length, mutant, or truncated fragments for occludin were used for analyzing the interaction between p16INK4a and occludin. High-throughput sequencing of V4 region amplicon of 16S ribosomal RNA was conducted using intestinal microbiota. We found Bmi-1 deficiency destructed barrier structure, barrier function, and tight junction (TJ) in intestinal epithelium; decreased the TJ proteins; increased tumor necrosis factor α (TNF-α)–dependent barrier permeability; and up-regulated proinflammatory level of macrophages induced by intestinal microbial dysbiosis. The transplantation of fecal microbiota from wild-type mice ameliorated TJ in intestinal epithelium of Bmi-1–/– and Bmi-1–/–p16INK4a–/– mice. Harmful bacteria including Desulfovibrio, Helicobacter, and Oscillibacter were at a higher level in Bmi-1–/– mice. More harmful bacteria Desulfovibrio entered the epithelium and promoted macrophages-secreted TNF-α and caused TNF-α–dependent barrier permeability and aging. Accumulated p16INK4a combined with occludin at the 1st–160th residue in cytoplasm of intestinal epithelium cells from Bmi-1–/– mice, which blocked formation of TJ and the repair of intestinal epithelium barrier. P16INK4a deletion could maintain barrier function and microbiota balance in Bmi-1–/– mice through strengthening formation of TJ and decreasing macrophages-secreted TNF-α induced by Desulfovibrio entering the intestinal epithelium. Thus, Bmi-1 maintained intestinal TJ, epithelial barrier function, and microbiota balance through preventing senescence characterized by p16INK4a accumulation. The clearance of p16INK4a-positive cells in aging intestinal epithelium would be a new method for maintaining barrier function and microbiota balance. The residues 1–160 of occludin could be a novel therapeutic target for identifying small molecular antagonistic peptides to prevent the combination of p16INK4a with occludin for protecting TJ.
Mammalian intestine is the critical site of digestion, absorption and assimilation, and a highly immune-active ecosystem that harbors and preserves a large abundance of commensal microorganisms. Intestinal epithelium is the first defensive barrier against environmental and microbial attacks by maintaining a tight physical barrier and executing several critical innate immune functions (Patankar and Becker, 2020).
Aging is an intrinsic physiological process characterized by a gradual function decline in the organs, including intestine and its microbiota (Lin et al., 2019). With aging-dependent decrease in the intestinal barrier function, microbial productions including proinflammatory factors enter the bloodstream, triggering systemic inflammation (Thevaranjan et al., 2017). Besides, aging-related microbial imbalance increases intestinal permeability. Age-related microbial imbalance and intestinal barrier dysfunction have been associated with various senescence-associated intestinal and systemic diseases including inflammatory bowel diseases, celiac disease, type 1 diabetes, obesity, and Alzheimer disease (Lee, 2015). Previous study shows that tumor necrosis factor α (TNF-α) is the main proinflammatory cytokine that causes the destruction of epithelial tight junction (TJ), increases intestinal epithelial permeability, and aggravates senescence-associated systemic inflammation; however, which members of the microbial community alter barrier function with age have not been yet identified (Thevaranjan et al., 2017). The tumor suppressor protein p16INK4a (hereafter referred to as p16), which is encoded by INK4a locus, is often transcriptionally activated in senescent cells and seen as a classical aging marker. P16 is up-regulated in multiple tissues during aging and contributes to senescence-associated decline in tissue function and regenerative capacity (Helman et al., 2016). However, whether accumulated p16 plays a critical role in damaging intestinal epithelial barrier and microbial homeostasis in inflammaging process is unclear.
B-cell–specific Moloney murine leukemia virus insertion region 1 (Bmi-1) is associated with senescence and cell cycle regulation. It regulates cell cycle and delays cell aging by inhibiting INK4a/ARF gene locus (Chen et al., 2020). Previous study on pig suggests that Bmi-1 is one of the markers of fast-cycling and quiescent intestinal stem cells (ISCs) that drives self-renewal of intestinal epithelial cells, increasing intestinal epithelial cell proliferation by stimulating WNT/β-catenin signaling (Li et al., 2018). Bmi-1–positive ISCs are critical for intestinal epithelium to maintain its function as a primary barrier (Smith et al., 2018). Loss of Bmi-1 in mice reduces proliferation in the ISC compartment accompanied by p16 accumulation (Lopez-Arribillaga et al., 2015). Several lines of evidence demonstrate that Bmi-1–deficient mouse is a stress-induced premature senescence (SIPS) model that appears frail with malnutrition and shortened life span (Liu et al., 2009; Zhang et al., 2010; Jin et al., 2014, 2017; Xie et al., 2015; Chen et al., 2020). However, whether Bmi-1 deficiency could cause damage of intestinal epithelial barrier and microbial dysbiosis is unclear. It is also unknown if p16 deletion could ameliorate the damage of intestinal epithelial barrier and microbial dysbiosis in Bmi-1–/– mice.
Herein we report that Bmi-1 maintained intestinal TJ, epithelial barrier function, and microbiota balance through preventing senescence characterized by p16 accumulation. Accumulated p16 combined with occludin at the 1st to 160th residue in the cytoplasm of aging intestinal epithelium cells, which blocked the repair of intestinal epithelium barrier. P16 deletion could maintain barrier function and microbiota balance in Bmi-1–/– mice through strengthening formation of TJ and decreasing macrophages-secreted TNF-α induced by Desulfovibrio entering the intestinal epithelium.
Bmi-1 and p16 were widely expressed in epithelial cells of jejunum, ileum, and colon (Supplementary Figures 1, 2). To examine if p16 deletion ameliorated the damage of barrier structure and dysfunction in intestinal epithelium in Bmi-1–/– mice, body size and weight, and intestinal length, histological structure and secretory function were observed in 7-week-old Bmi-1–/– mice, Bmi-1–/–p16–/– mice, and wild-type (WT) mice. Results showed that besides the smaller body size and weight, Bmi-1–/– mice also showed shorter intestinal length (especially the ileum) in comparison with WT mice. Deletion of p16 significantly rescued the body size, weight, and intestinal length caused by Bmi-1 deficiency (Figures 1A–D). Significant decreases were observed in the villus length, the ratio of villus length to crypt, the number of Paneth cells, and the number of acid mucin and glycoprotein in Bmi-1–/– mice compared with WT mice. P16 deletion significantly rescued the abnormalities in the villus length, the ratio of villus length to crypt, the number of Paneth cells, and the number of acid mucin and glycoprotein observed in Bmi-1–/– mice (Figures 1E–K). Ki67-positive cells significantly decreased after Bmi-1 deletion, which was rescued by p16 knockout (Supplementary Figure 3). P53 significantly increased in jejunum, ileum, and colon of Bmi-1–/– mice and then down-regulated by p16 deletion (Figures 1L–N). These results demonstrated that p16 deletion ameliorated the damage of barrier structure and dysfunction in intestinal epithelium caused by Bmi-1 deficiency.
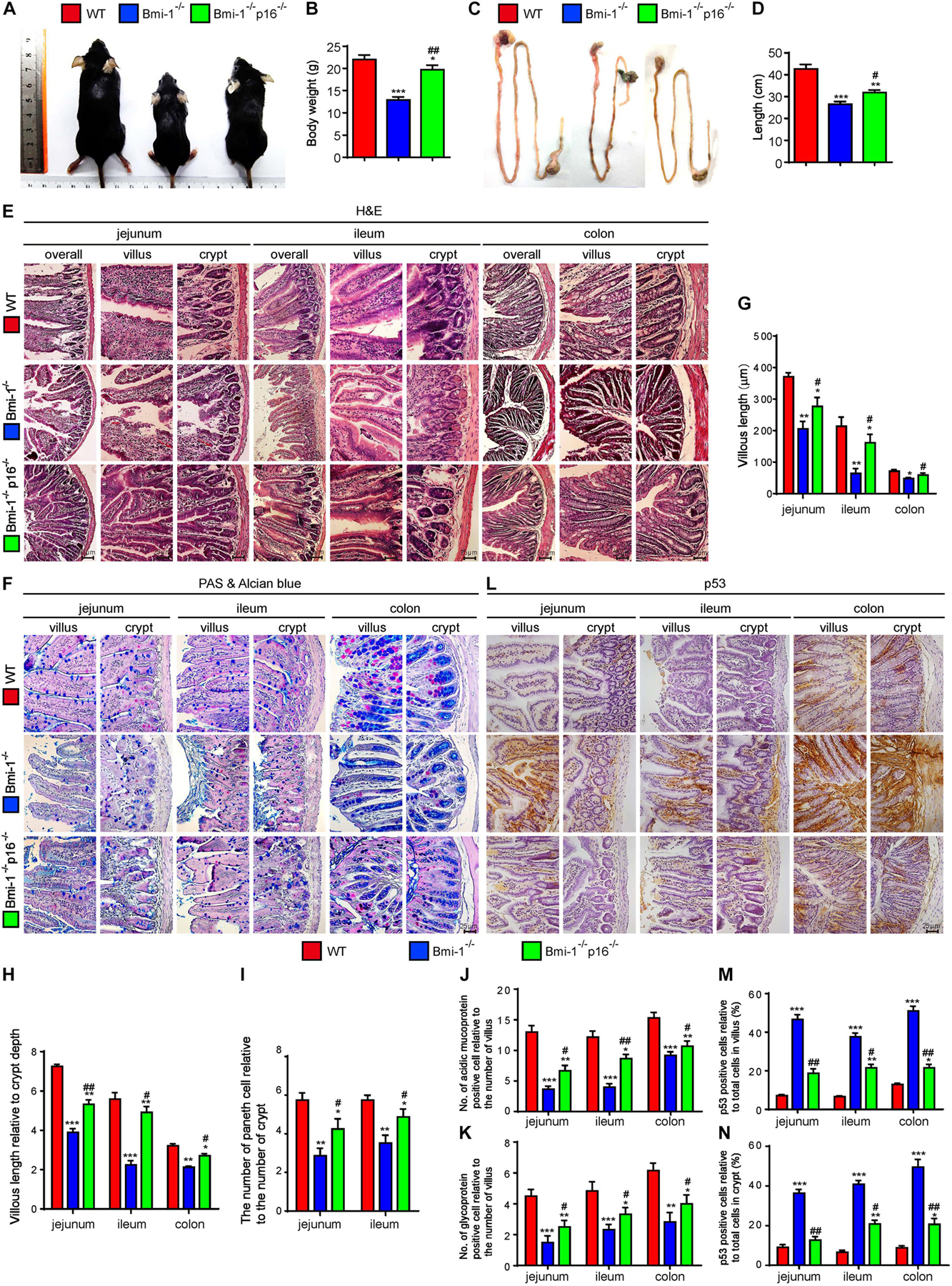
Figure 1. P16 deletion improved damage of barrier structure and dysfunction in intestinal epithelium in Bmi-1–/– mice. The experiments were carried out on the 7-week-old Bmi-1–/–, Bmi-1–/–p16–/–, and WT mice. (A) Representative appearances. (B) Body weight (g). (C) Whole view of the intestine. (D) Intestinal length (cm). (E,F) H&E and AB-PAS staining of jejunum, ileum, and colon. (G) Villous length (μm). (H) Villous length relative to crypt depth. (I) The number of Paneth cells relative to the number of crypts. (J) The number of acidic mucoprotein positive cells. (K) Glycoprotein-positive cells relative to the number of villus. (L) Representative micrographs of paraffin-embedded intestinal sections immunohistochemical staining for p53, with hematoxylin staining the nucleus. (M,N) Percentage of p53-positive cells relative to the total cells. Six mice per group were used for experiments. Statistical analysis was performed with one-way ANOVA test. Values are mean ± SEM from six determinations per group, *p < 0.05, **p < 0.01, ***p < 0.001 compared with the WT group; #p < 0.05, ##p < 0.01 compared with the Bmi-1–/– group.
To investigate if p16 deletion ameliorated the destruction of TJ and the increase of barrier permeability in Bmi-1–/– mice, the key TJ protein ZO-1, tight junctional structure, and several primary TJ proteins including claudin-1, occludin, and claudin-2 in the epithelia of jejunum, ileum, and colon were detected and analyzed. Moreover, peripheral blood was collected and detected fluorescein isothiocyanate (FITC) fluorescence for evaluating intestinal permeability at the fourth hour after oral gavage of FITC–dextran. In Bmi-1–/– mice compared with WT mice, significant decreases were observed in ZO-1– and occludin-positive areas (Figures 2A–C and Supplementary Figures 4A,B) and protein levels of claudin-1, occludin, and claudin-2 in epithelia (Figures 2F,G); meanwhile, a significant increase was observed in FITC fluorescence for evaluating intestinal permeability (Figure 2D). It was also observed in electron micrographs that the bottom of the epithelial TJ was cracked in epithelia of jejunum, ileum, and colon (Figure 2E). P16 deletion significantly increased the expressions of TJ proteins, maintained the tight junctional structure, and decreased the intestinal permeability (Figure 2). These results demonstrated that p16 deletion improved the destruction of TJ and decreased barrier permeability in intestinal epithelium caused by Bmi-1 deficiency.
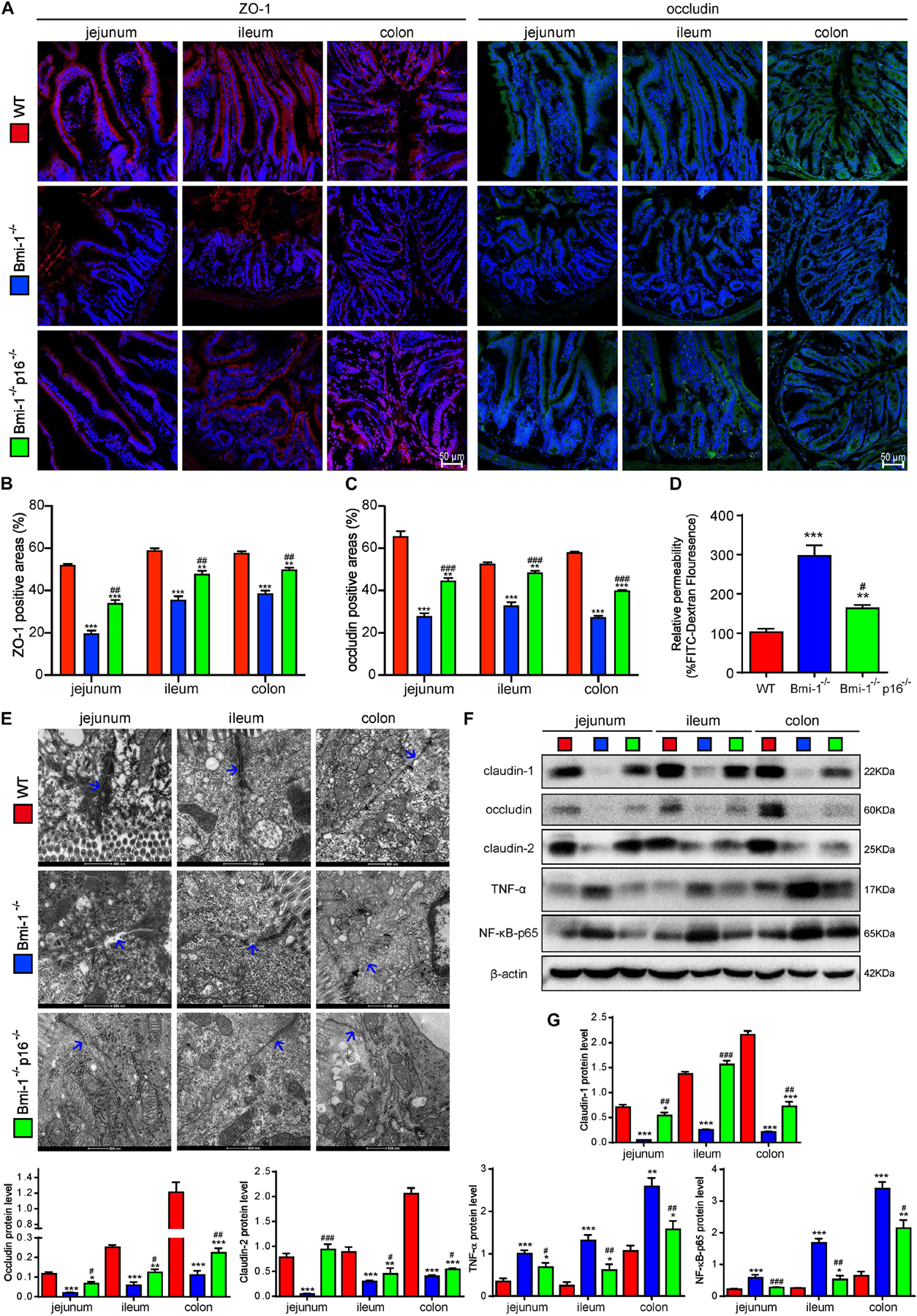
Figure 2. P16 deletion improved destruction of TJ and increasing barrier permeability in intestinal epithelium in Bmi-1–/– mice. The experiments were carried out on the 7-week-old Bmi-1–/–, Bmi-1–/–p16–/–, and WT mice. (A) Representative micrographs showing immunofluorescence for ZO-1 and occludin, with DAPI staining the nuclei. (B) Percentage of ZO-1–positive areas relative to the total area. (C) Percentage of occludin-positive areas relative to the total area. (D) Intestinal FITC–dextran transmittance showing intestinal permeability. (E) Representative micrographs showing transmission electron microscope for the epithelial cells of jejunum, ileum, and colon, with blue arrow showing the bottom of the epithelial TJ. (F) Western blots for claudin-1, occludin, claudin-2, TNF-α, and NF-κB–p65 in the epithelial cells of jejunum, ileum, and colon; β-actin was used as the loading control. (G) Protein levels relative to β-actin were assessed by densitometric analysis. Six mice per group were used for experiments. Statistical analysis was performed with one-way ANOVA test. Values are mean ± SEM from six determinations per group, *p < 0.05, **p < 0.01, ***p < 0.001 compared with the WT group; #p < 0.05, ##p < 0.01, ###p < 0.001compared with the Bmi-1–/– group.
To investigate how p16 deletion ameliorated intestinal epithelium barrier function in Bmi-1–/– mice, we conducted immunoprecipitation of p16 and occludin, and validated p16 could combine with occludin (Figure 3A). P16 expression in Bmi-1–/– ileum was higher than WT ileum; however, occludin expression in Bmi-1–/– ileum was lower than WT ileum in input and Immunoprecipitation (IP) samples (Figure 3A). The results of immunofluorescence colocalization and Duolink Proximity Ligation Assay showed p16 bound to occludin in the cytoplasm of intestinal epithelium cells in Bmi-1–/– mice (Figures 3B,C). Our results showed that p16 expressed in the nucleus and cytoplasm, especially more concentrated in the cytoplasm, and interacted with occludin in the cytoplasm (Figures 3B,C). Compared with the Bmi-1–/– mice, p16 expression was obviously decreased in epithelium cells of Bmi-1–/–p16–/– and WT mice (Figure 3B and Supplementary Figure 2). It has been reported that N-terminal of p16 bound to the N-terminal region of JNK1 (also known as MAPK8) or the N-terminal region of JNK3 (also known as MAPK10), which contain the glycine-rich site (Choi et al., 2005). Analysis with https://www.uniprot.org/align showed an extremely similar domain among human occludin (residues 107–139), MAPK8 (residues 16–49), and MAPK10 (residues 54–100) and also showed an extremely similar domain among mouse occludin (residues 107–139), MAPK8 (residues 24–72), and MAPK10 (residues 62–110). Besides, the 107th–139th residues are also a glycine-rich site of occludin (Figure 3D and Supplementary Information 2, 3).
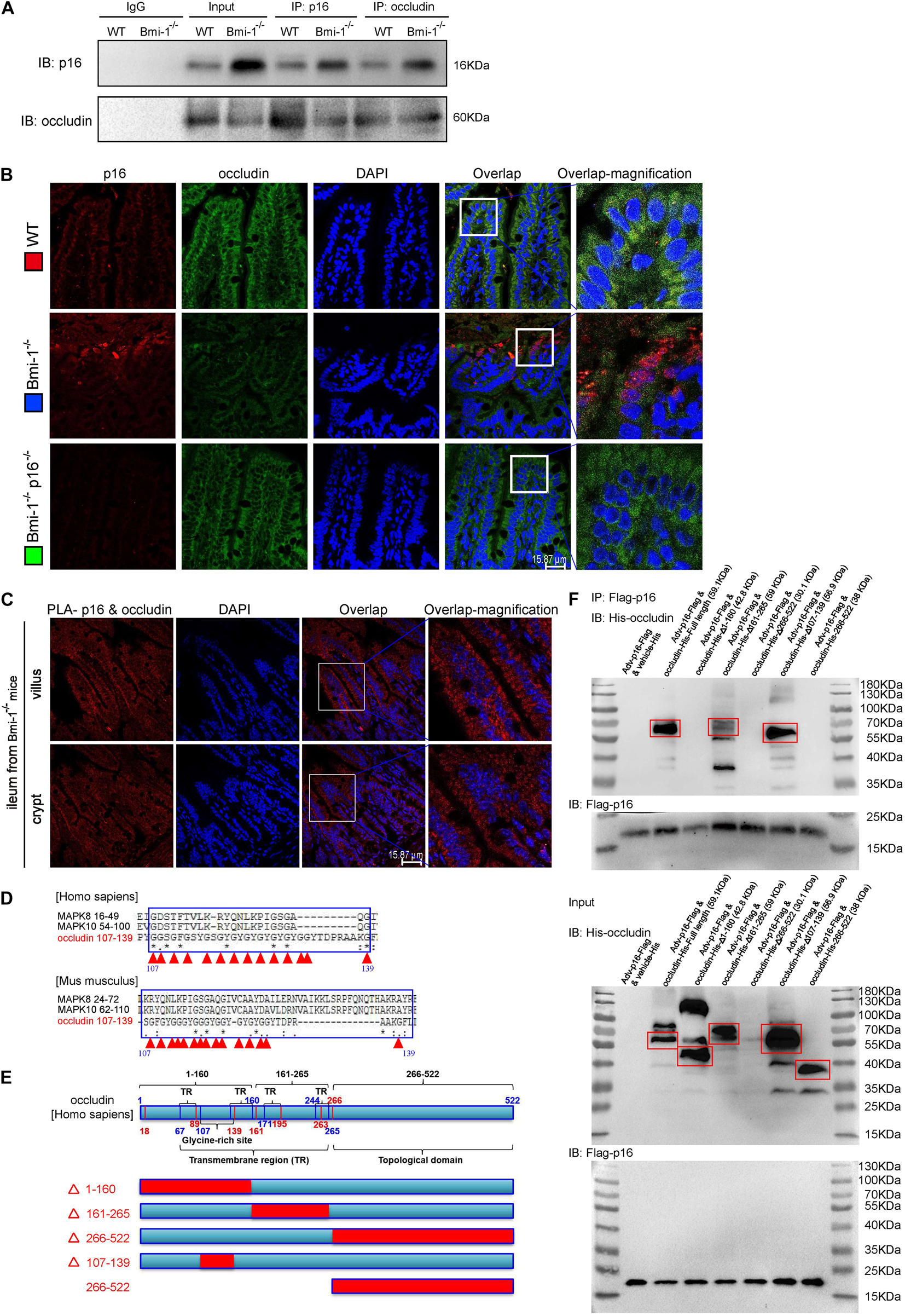
Figure 3. P16 combined with occludin in the cytoplasm of intestinal epithelium cells in Bmi-1–/– mice for blocking reparation of TJ. The experiments were carried out on the 7-week-old Bmi-1–/–, and WT mice. (A) Intestinal epithelial proteins from WT and Bmi-1–/– mice were extracted for anti-p16 or anti-occludin immunoprecipitation. Western blots were used for detecting p16 and occludin. (B) Representative micrographs showing immunofluorescence double-labeling of p16 and occludin in ileum from 7-week-old Bmi-1–/–, Bmi-1–/–p16–/– and WT mice. (C) Representative micrographs of Duolink PLA for interaction between p16 and occludin in villus and crypt of ileum from Bmi-1–/– mice, with DAPI staining nuclei. (D) Analysis with https://www.uniprot.org/align showed an extremely similar domain among occludin, MAPK8, and MAPK10 in human and mouse, with red triangle showing glycine site, “*” indicating a single and fully conserved residue, “:” indicating residue with very similar properties, and “.” indicating residue that is weakly similar. (E) Protein domain of human occludin. (F) The 239T cells were transfected with Flag-p16 (human) overexpressed adenovirus and the His-tagged plasmid of full length, 1–160–mutant (△1–160), 161–265–mutant (△161–265), 266–522–mutant (△266–522), 107–139–mutant (△107–139), or 266–522–truncated fragment for human occludin. Cell proteins were extracted for anti–Flag-Tag immunoprecipitation and detected anti-Flag and anti-His antibodies with Western blots. The input proteins also detected anti-Flag and anti-His antibodies with Western blots.
To determine which domain of occludin combined with p16, 293T cells were transfected with green fluorescent protein (GFP)-labeled Flag-p16 overexpressed adenovirus and then with vehicle–His-tagged vector (negative control), His-tagged overexpressed plasmids of full-length, mutant, or truncated fragments for occludin, respectively. Cell extracts were immunoprecipitated with anti-Flag antibody, and the precipitated proteins were detected by immunoblotting with anti-His or anti-Flag antibody. As was shown from the results of immunoprecipitation, p16 combined with occludin at the residues 1–160 rather than limited to residues 107–139. Because the 266th–522nd mutant plasmid did not express protein, we constructed the 266th–522nd truncated fragment and found that p16 did not combine with this domain (Figures 3E,F). Thus, these results suggested that accumulated p16 combined with occludin at the residues 1–160 in the cytoplasm of aging intestinal epithelium cells in Bmi-1–/– mice for blocking the repair of intestinal epithelium barrier.
To further determine if the p16–occludin interaction blocks the repair of TJ, Cacao-2 cells were transfected with GFP-labeled Flag-p16 overexpressed adenovirus and then were given TNF-α stimulation to damage the TJ. Immunofluorescence staining and Western blot were used to detect the expression levels of occludin and ZO-1. Immunofluorescence staining was also used to observe the interaction of p16 and occludin. Our results showed that in comparison with vehicle treatment, TNF-α treatment or p16 overexpression damaged TJ and decreased the expressions of occludin and ZO-1 (Figures 4A–F). Compared with vehicle–adenovirus and TNF-α treatment, p16 overexpressed adenovirus, and TNF-α treatment further damaged TJ and decreased the expressions of occludin and ZO-1 (Figures 4A–F). We also observed that when the TJ between cells was destroyed, more occludin expression was found in cytoplasm and colocalized with p16 (Figure 4G).
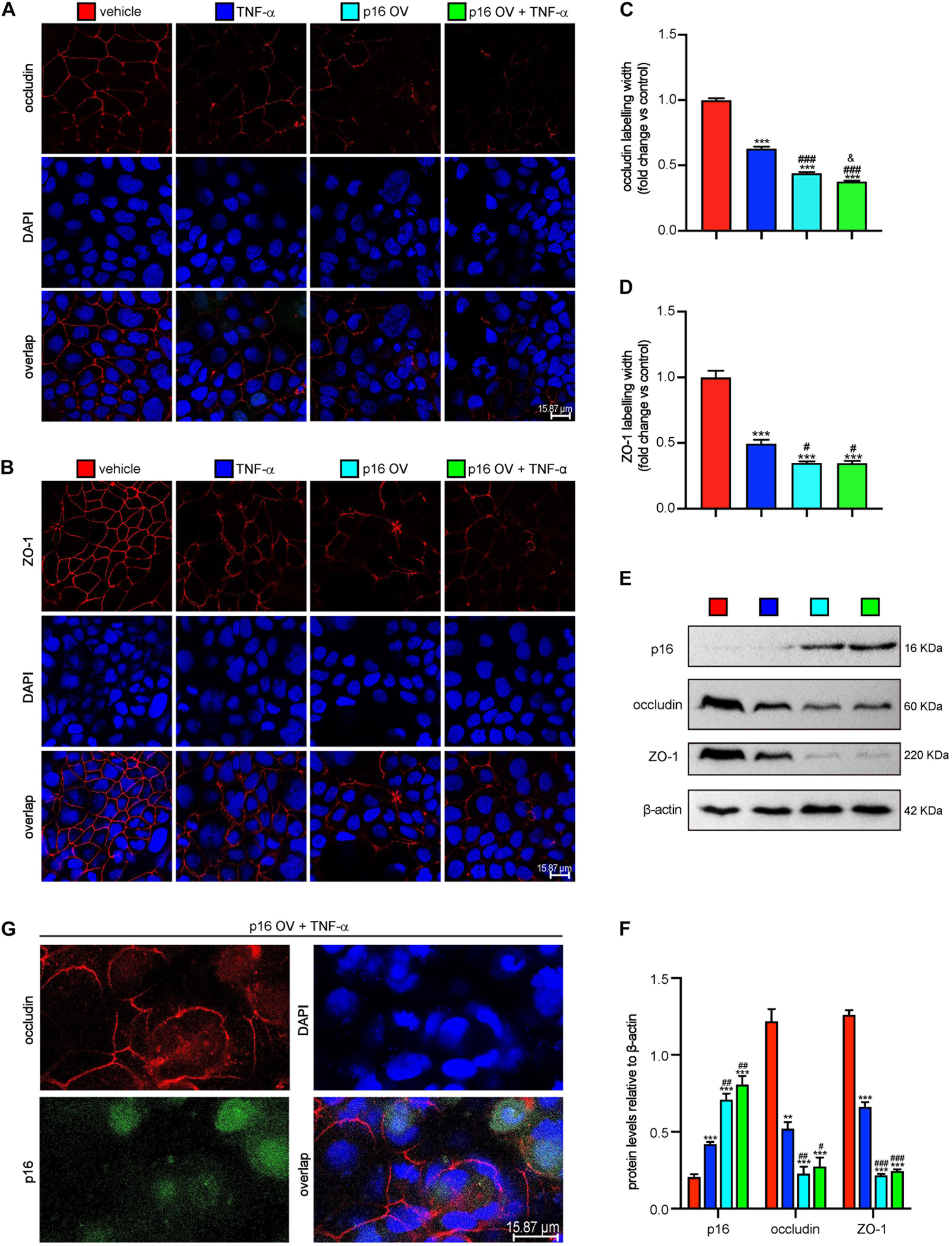
Figure 4. Accumulated p16 in Cacao-2 cells blocked the repair of TJ after TJ was damaged by TNF-α. The Cacao-2 cells were transfected with GFP-labeled Flag-p16–overexpressed (p16-OV) or vehicle adenovirus and then treated with or without 10 ng/mL TNF-α for 24 h. Immunofluorescence staining was introduced to detect the expressions of occludin and ZO-1. (A) Representative micrographs showing immunofluorescence for occludin, with DAPI staining the nuclei. (B) Representative micrographs showing immunofluorescence for ZO-1, with DAPI staining the nuclei. (C) Occludin positive width relative to control group. (D) ZO-1 positive width relative to control group. (E) Western blots for p16, occludin, and ZO-1 in Cacao-2 cells, and β-actin was used as the loading control. (F) Protein levels relative to β-actin were assessed by densitometric analysis. Cell experiments were performed with three biological repetitions per group. Statistical analysis was performed with one-way ANOVA test. Values are mean ± SEM from six determinations per group, **p < 0.01, ***p < 0.001 compared with the vehicle group; #p < 0.05, ##p < 0.01, ###p < 0.001 compared with TNF-α–treated group, &P < 0.05 compared with the p16-OV group. (G) Representative micrographs showing occludin and GFP-labeled p16 in p16-OV and TNF-α–treated cells, with DAPI staining the nuclei.
To determine if p16 deletion ameliorated the TNF-α–dependent epithelial barrier destruction in Bmi-1–/– mice, TNF-α–positive cells or areas; TNF-α and F4/80 double-positive macrophages; TNF-α protein levels in jejunum, ileum, and colon; and serous TNF-α protein levels of mice were detected and analyzed. Results showed that TNF-α–positive cells; TNF-α and F4/80 double-positive macrophages; TNF-α protein levels in jejunum, ileum, and colon; and serous TNF-α protein levels were significantly increased in Bmi-1–/– mice compared with WT mice; however, they were significantly reduced in Bmi-1–/–p16–/– mice compared with the Bmi-1–/– mice (Figures 5A–E). Then, nuclear factor κB (NF-κB)–p65–positive cells and NF-κB–p65 and F4/80 double-positive macrophages were detected in jejunum, ileum, and colon, and results showed that they were significantly increased in Bmi-1–/– mice compared with WT mice, while significantly reduced in Bmi-1–/–p16–/– mice compared with the Bmi-1–/– mice (Figures 5F–H). It suggested that the proinflammatory level of macrophages in intestines was up-regulated in Bmi-1–/– mice compared with WT mice and ameliorated by p16 deletion.
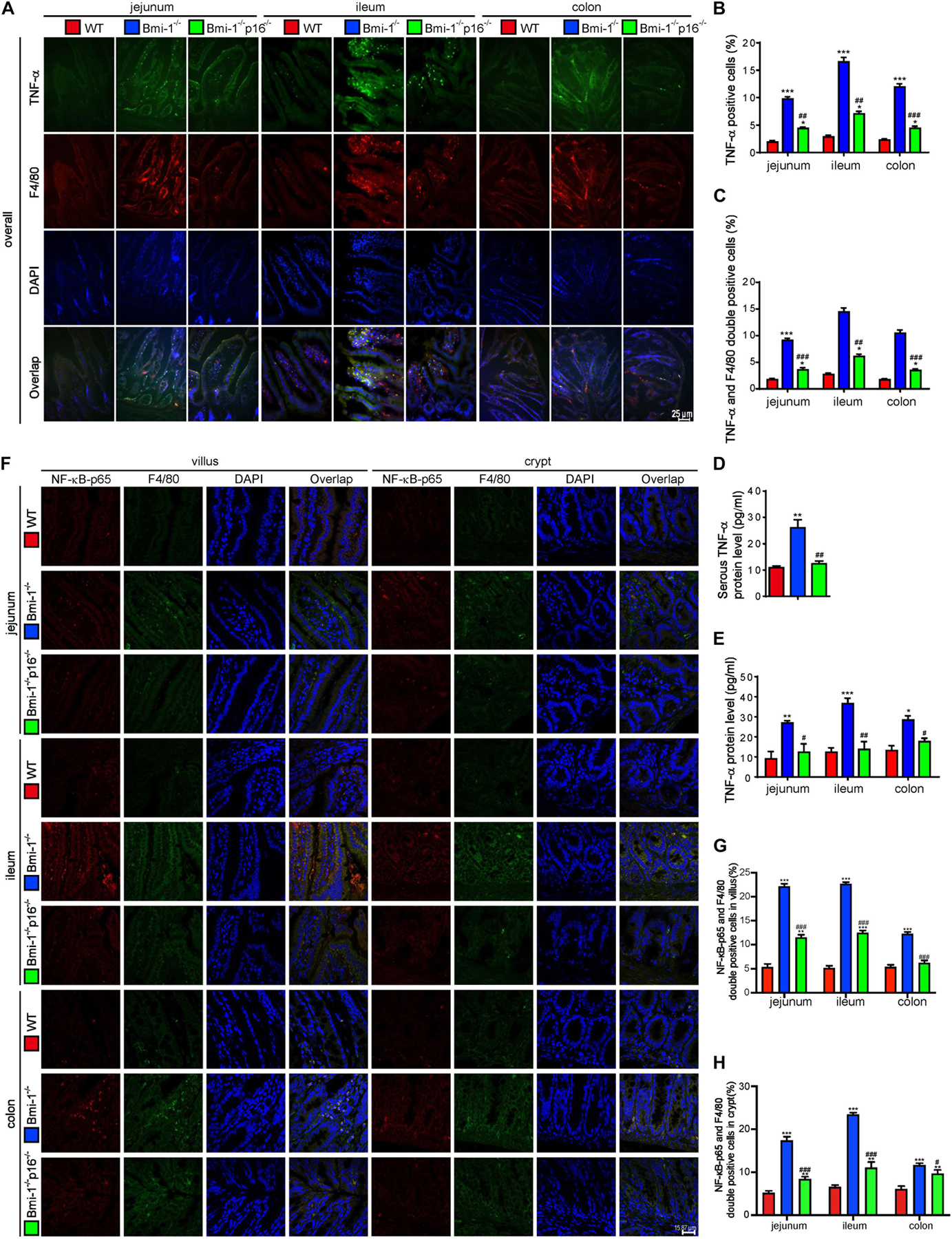
Figure 5. P16 deletion improved TNF-α–dependent epithelial barrier destruction in Bmi-1–/– mice. The experiments were carried out on the 7-week-old Bmi-1–/–, Bmi-1–/–p16–/–, and WT mice. (A) Representative micrographs showing immunofluorescence for TNF-α and F4/80, with DAPI staining the nuclei. (B) Percentage of TNF-α–positive cells. (C) Percentage of TNF-α and F4/80 double-positive cells. (D) Serous TNF-α protein level (pg/mL) was detected with ELISA assay. (E) TNF-α protein levels in jejunum, ileum, and colon (pg/mL) were detected with ELISA assay. (F) Representative micrographs showing immunofluorescence for NF-κB–p65 and F4/80, with DAPI staining the nuclei. (G,H) Percentage of NF-κB–p65 and F4/80 double-positive cells or areas in villus and crypt. Six mice per group were used for experiments. Statistical analysis was performed with one-way ANOVA test. Values are mean ± SEM from six determinations per group, *p < 0.05, **p < 0.01, ***p < 0.001 compared with the WT group; #p < 0.05, ##p < 0.01, ###p < 0.001 compared with the Bmi-1–/– group.
To investigate if Bmi-1 deficiency and/or p16 deletion changed the phagocytosis of macrophages, DiI (1,1’-dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine perchlorate)–labeled fecal mixed bacteria from WT mice (WT-FB) were used to infect bone marrow–derived macrophages (BMDMs) from Bmi-1–/–, Bmi-1–/–p16–/–, and WT mice. Immunofluorescence staining of macrophage marker F4/80 proved that there was no difference in phagocytosis of BMDMs from WT, Bmi-1–/–, and Bmi-1–/–p16–/– mice (Figures 6A,B). This result suggested that Bmi-1 and/or p16 did not affect the phagocytosis of macrophages.
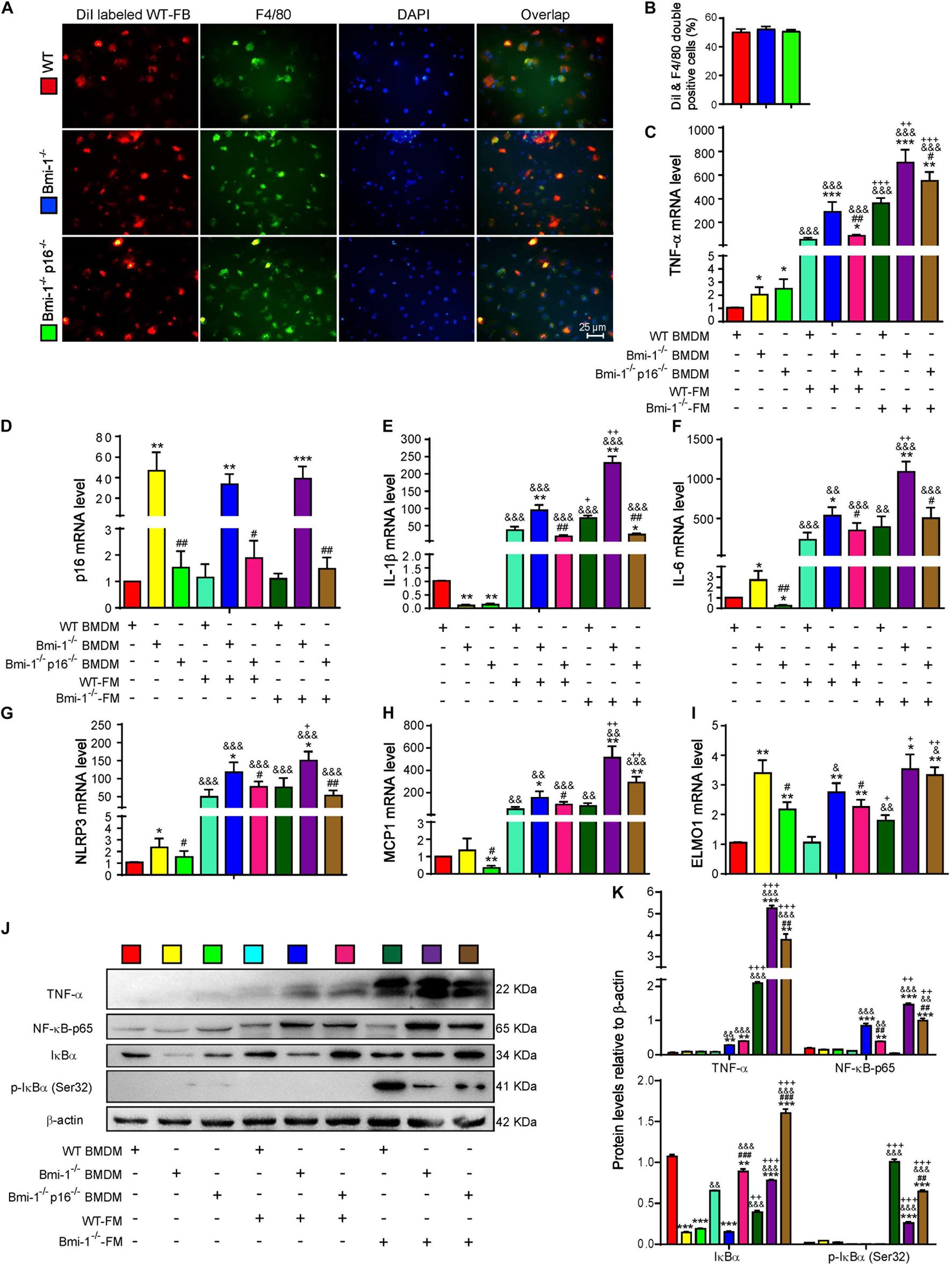
Figure 6. P16 deletion improved up-regulated proinflammatory level of macrophages induced by change of intestinal microbiota ameliorated in Bmi-1–/– mice. Fecal mixed bacteria (FB) were prepared using the fecal of 7-week-old WT and Bmi-1–/– mice to infect BMDMs from WT, Bmi-1–/–, and Bmi-1–/–p16–/– mice. (A) Representative micrographs showing immunofluorescence for F4/80 after Dil-labeled WT-FM–infected BMDMs, with DAPI staining the nuclei, red dots showing Dil-labeled WT-FB, and green dots showing F4/80. (B) Percentage of DiI and F4/80 double-positive cells. (C–I) Tumor necrosis factor (TNF-α), p16, interleukin (IL-1β), IL-6, NACHT, LRR, and PYD domain–containing protein 3 (NLRP3) and monocyte chemoattractant protein 1 (MCP1) and engulfment and cell motility protein 1 (ELMO1) mRNA levels in BMDMs by real-time RT-PCR, calculated as the ratio to β-actin mRNA and expressed relative to WT. (J) Western blots of BMDM extracts showing TNF-α, NF-κB–p65, IκBα, and p-IκBα (Ser32); β-actin was used as the loading control. (K) Protein levels relative to β-actin were assessed by densitometric analysis. Cell experiments were performed with three biological repetitions per group. Statistical analysis was performed with one-way ANOVA test. Values are mean ± SEM from six determinations per group, *p < 0.05, **p < 0.01, ***p < 0.001 compared with the WT group at the same treatment; #p < 0.05, ##p < 0.01, ###p < 0.001 compared with the Bmi-1–/– group at the same treatment; &p < 0.05, &&p < 0.01, &&&p < 0.001 compared with the same genotyped BMDMs of vehicle-FM group; +p < 0.05, ++p < 0.01, +++p < 0.001 compared with the same genotyped BMDMs of the WT-FM group.
To further make clear if the up-regulated proinflammatory level of macrophages was associated with the change of intestinal microbiota in Bmi-1–/– mice, WT–fecal microbiota (FM), Bmi-1–/––FM, and vehicle were administered to BMDMs from WT, Bmi-1–/–, and Bmi-1–/–p16–/– mice. In comparison with the WT-FM–treated BMDMs of the same genotype, an increase was observed in the mRNA levels of TNF-α, interleukin (IL)-1β, IL-6, NACHT, LRR, and PYD domain–containing protein 3 (NLRP3), monocyte chemoattractant protein 1 (MCP1), and engulfment and cell motility protein1 (ELMO1) and in the protein levels of TNF-α, NF-κB–p65, IκBα, and p-IκBα (Ser32) in Bmi-1–/––FM treated BMDMs, especially in Bmi-1–null BMDMs. Compared with vehicle of the same genotype, WT-FM and Bmi-1–/––FM treatments significantly increased the above mRNA levels and protein levels, except the ELMO1 mRNA level and IκBα and p-IκBα (Ser32) protein level (Figures 6C–K). These results suggested that the up-regulated proinflammatory level of macrophages was associated with the change of intestinal microbiota in Bmi-1–/– mice.
To determine if p16 deletion ameliorated proinflammatory level of macrophages activated by aging-associated intestinal microbiota in Bmi-1–/– mice, WT-FM, Bmi-1–/––FM, and vehicle were administered to BMDMs from Bmi-1–/–, Bmi-1–/–p16–/–, and WT mice. Results showed that whatever the treatment was with WT-FM or Bmi-1–/––FM, an increase was observed in the mRNA levels of TNF-α, IL-1β, IL-6, NLRP3, MCP1, and ELMO1 and in the protein levels of TNF-α and NF-κB–p65 in Bmi-1–/– BMDMs compared with WT BMDMs; however, they were significantly reduced in Bmi-1–/–p16–/– mice compared with the Bmi-1–/– mice (Figures 6C–K). These results suggested that the proinflammatory level of macrophages activated by intestinal microbiota was ameliorated by p16 deletion in Bmi-1–/– mice.
To determine if the damage of TJ in intestinal epithelium was associated with microbial dysbiosis, FM from 3- to 4-week-old WT mice were transplanted (WT-FMT) to WT, Bmi-1–/–, and Bmi-1–/–p16–/– mice by gavage every other day and lasted for 21 days. Results showed that compared with the WT mice without or with FMT, ZO-1 and occludin expression in ileum epithelium were obviously decreased in Bmi-1–/– mice without or with WT-FMT; however, they were significantly increased in Bmi-1–/–p16–/– mice without or with WT-FMT compared with the Bmi-1–/– mice without or with WT-FMT, respectively (Supplementary Figures 4A–D). Compared with the Bmi-1–/– mice or Bmi-1–/–p16–/– mice without WT-FMT, ZO-1 and occludin expressions in ileum epithelium were obviously increased in Bmi-1–/– mice or Bmi-1–/–p16–/– mice with WT-FMT, respectively (Supplementary Figures 4A–D). These results suggested that the destructive degree of TJ was regulated by intestinal microbiota.
To investigate if p16 deletion could ameliorate the dysbiosis in the species and abundance of intestinal microbiota in Bmi-1–/– mice, principal coordinates analysis of microbial DNA encoding 16S ribosomal RNA (rRNA) was used to detect intestinal microbiota changes. The analysis was carried out at the level of the phylum. Differences were observed between WT and Bmi-1–/– mice, and the abundance of Bacteroidetes decreased in Bmi-1–/– mice when compared with those in WT mice, leading to intestinal microbiota ratio imbalance (Figures 7A,B). P16 deletion obviously decreased the abundance of Firmicutes and Proteobacteria and increased the abundance of Bacteroidetes in Bmi-1–/– mice. After validating that the sequencing sample size meets the requirements of diversity (Figures 7C,D), α-diversity was then assessed to reflect within-sample diversity. The richness of species at the phylum level was lower in the Bmi-1–/– group than that in the WT group (Figure 7E). Venn diagram displayed 329 same operational taxonomic units (OTUs) share between the WT and DKO groups, whereas there were only 110 same OTUs between the WT and KO groups (Figure 7F). The results indicated that p16 deletion could rescue intestinal microbial dysbiosis induced by Bmi-1 deficiency.
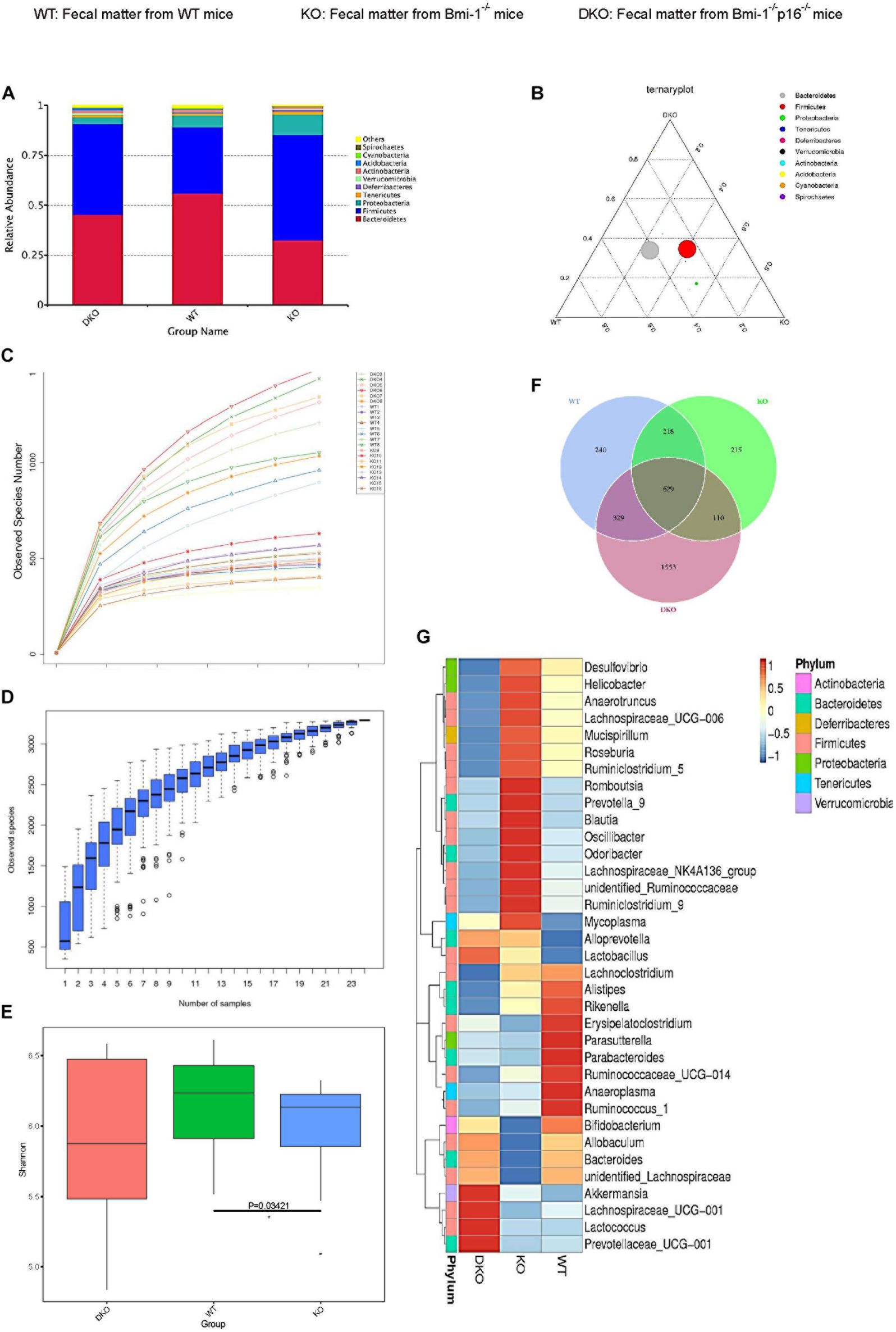
Figure 7. P16 deletion improved intestinal microbial dysbiosis in Bmi-1–/– mice. High-throughput sequencing of V4 region amplicon of 16S rRNA from intestinal microbiota was conducted using the fecal of 7-week-old WT, Bmi-1–/– (KO), and Bmi-1–/–p16–/– (DKO) mice. (A) Principal components at the phylum level in the intestinal microbiota of WT, Bmi-1–/–, and Bmi-1–/–p16–/– mice. Relative contribution of the top 10 phylum of each sample was shown; “others” represents the sum of abundance except the top 10 phylum, n = 8. (B) Proportion of intestinal microbiota in 7-week-old WT, KO, and DKO mice at the phylum level. Circle represents a taxonomy at the level of phylum; the size of the circle represents the abundance of the microbiota at the phylum level. (C) Abundance display curve of intestinal microbiota in WT, KO, and DKO mice. Abscissa represents sequencing depth; ordinate represents the number of species found. (D) Box plot showing abundance dilution. The end of the curve is close to gentle, indicating that the sample size of sequencing meets the requirements of diversity. (E) The observed species number and α-diversity index of intestinal bacteria were examined. Significant differences are indicated: Wilcoxon rank sum test, n = 8 per group, *p < 0.05, compared with the WT group. (F) Venn diagrams demonstrate the number of species shared among WT, KO, and DKO groups. (G) Heat map showing distribution of intestinal microbiota at genus level from 7-week-old WT, Bmi-1–/–, and Bmi-1–/–p16–/– mice; the phylum of each genus is listed in the note, n = 8. The heat map is colored based on row Z scores. The mice with the highest and lowest bacterial levels are in red and blue, respectively.
To further clarify the specific changes of intestinal microbiota among Bmi-1–/–, Bmi-1–/–p16–/–, and WT mice, bioinformatics analysis was conducted. Compared with WT mice, Bmi-1–/– mice had less diversity of intestinal dominant microbiota, most of which affiliated to Firmicutes including Anaerobtruncus, Lachnospiraceae, Roseburia, and Ruminiclostridium, together with Blautia and Ruminococcaceae, which were closely related to intestinal inflammation. Moreover, the abundance of some intestinal harmful bacteria including Desulfovibrio, Helicobacter, and Oscillibacter was at a higher level in Bmi-1–/– mice, whereas the abundance of probiotics such as Bifidobacterium was at a lower level. In comparison with Bmi-1–/– mice, the abundance of the intestinal harmful bacteria such as Desulfovibrio and Helicobacter was at a lower level, whereas the diversity of dominant genus elevated, and the abundance of intestinal probiotics including Bifidobacterium and Lactobacillus was at a higher level in Bmi-1–/–p16–/– mice (Figure 7G). These results demonstrated that p16 deletion ameliorated intestinal microbial dysbiosis including increasing intestinal microbiota diversity and probiotic abundance and decreasing the abundance of harmful microbiota in Bmi-1–/– mice.
To determine if p16 deletion could ameliorate intestinal microbial function in Bmi-1 mice, Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUST) sequencing analysis was carried out based on principal coordinates analysis of microbial DNA encoding 16S rRNA. Linear discriminant analysis (LDA) sequence was used to make clear species with significant differences among samples. The results illustrated that there were 5, 11, and 9 bacterial biomarkers, respectively, in the intestinal microbiota of WT, Bmi-1–/–, and Bmi-1–/–p16–/– mice (Figure 8A). Distribution of marker bacteria in three genotypes of mice is shown in the cladogram (Figure 8B). These results could help understand the biomarkers of intestinal microbial changes in WT, Bmi-1–/–, and Bmi-1–/–p16–/– mice.
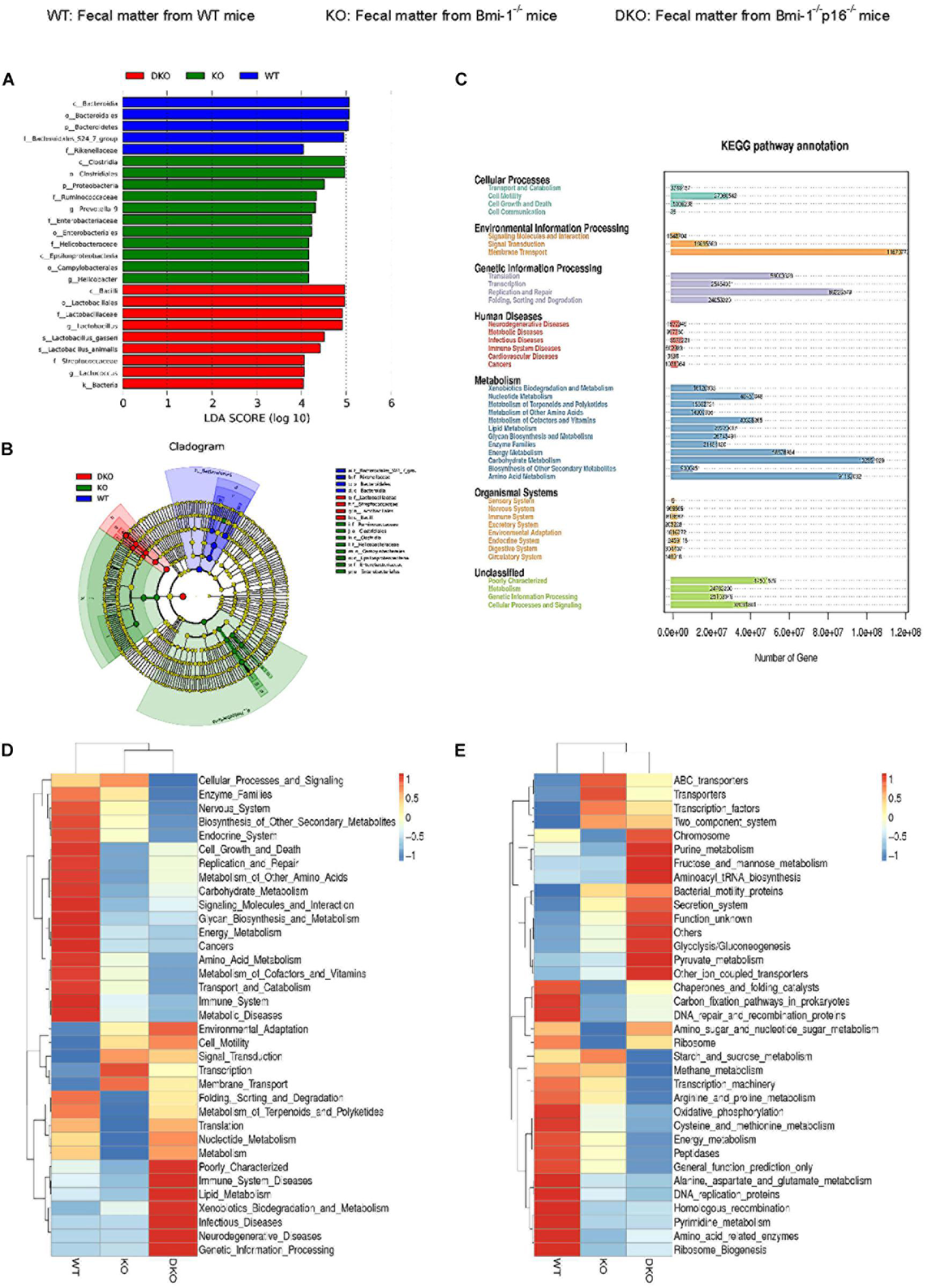
Figure 8. P16 deletion improved intestinal microbial function ameliorated in Bmi-1–/– mice. High-throughput sequencing of V4 region amplicon of 16S rRNA from intestinal microbiota was conducted using the fecal of 7-week-old WT, Bmi-1–/–, and Bmi-1–/–p16–/– mice. (A) LEfSe results represented significant differences in bacterial taxa enriched in the WT, KO, and DKO groups. Only taxa with LDA > 4.0 are shown. (B) The evolutionary branch map obtained from LEfSe sequence of WT, Bmi-1–/–, and Bmi-1–/–p16–/– mice. Radiation circle represents the classification level from phylum to genus. Yellow circle indicates no difference; red, green, and blue represent species with significant difference in each group. (C) Functional enrichment analysis of KEGG pathway. (D,E) Heat map of functional analysis based on the average abundance of KEGG pathway in the three groups, n = 8. The heat map is colored based on row Z scores. The mice with the highest and lowest functional enrichment are in red and blue, respectively.
To further analyze the function of intestinal microbiota, we carried out the PICRUST sequencing analysis and functional enrichment analysis of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway on the microbiota with significant difference between WT and Bmi-1–/– groups. The results showed that the number of genes related to human functional diseases, including 502,983 functional genes related to human immune system diseases, 3,638 functional genes related to cardiovascular disease, and 148,016 functional genes related to the circulatory system. Environmental information processing, genetic information processing, and metabolism were found as the three aspects with the highest gene abundance (Figure 8C).
Function prediction heat map was drawn based on the functional annotation and abundance information of all intestinal microbial samples in KEGG, showing the top 35 bacteria in abundance ranking. An obvious decline was shown in intestinal microbiota energy metabolism; cell growth and death regulation; replication and repair; carbohydrate, glycan, and nucleotide metabolism; terpenoid and polyketide metabolism; protein folding sequencing degradation; and translation function in Bmi-1–/– mice compared with WT mice. Among them, the metabolic function mainly focuses on cell motility, membrane transport, and transcription. In comparison with Bmi-1–/– mice, the lipid and xenobiotics metabolism functions were enhanced in Bmi-1–/–p16–/– mice (Figure 8D).
Moreover, the genetic information function of intestinal microbiota was predicted and analyzed. In Bmi-1–/– mice compared with WT mice, a significant decrease was shown in recombination proteins, peptidase, DNA replication proteins, and amino acid–related enzymes of the intestine; a significant decline was shown in DNA repair, the function of amino sugar and nucleic acid sugar metabolism, cystine and methionine metabolism, pyrimidine metabolism, and oxidative phosphorylation. The function of purine metabolism, fructose, and mannose metabolism, pyruvate metabolism, aminoacyl tRNA biosynthesis, secretion system, and glycolysis were enhanced in intestinal microbiota of Bmi-1–/–p16–/– mice compared with that of Bmi-1–/– mice (Figure 8E).
These results demonstrated that metabolism and biosynthesis of intestinal microbiota were ameliorated by p16 deletion in Bmi-1–/– mice. It suggested that the disorder and dysfunction of intestinal microbiota mentioned above might help explain how Bmi-1–/– mice developed premature aging and aging-related diseases.
To determine if p16 deletion ameliorated the effect of Desulfovibrio on promoting TNF-α secretion in intestines of Bmi-1–/– mice, DiI-labeled Desulfovibrio was administered by oral gavage into Bmi-1–/–, Bmi-1–/–p16–/–, and WT mice. The number of DiI-positive dots per area, TNF-α–positive cells, and DiI- and TNF-α double-positive cells were analyzed after immunofluorescence staining for TNF-α in intestines. In Bmi-1–/– mice compared with WT mice, a significant increase was shown in the number of DiI-positive dots per area, TNF-α–positive cells, and DiI and TNF-α double-positive cells in jejunum, ileum, and colon; however, they were significantly reduced in Bmi-1–/–p16–/– mice compared with the Bmi-1–/– mice (Figures 9A–D). These results demonstrated that more Desulfovibrio entered the intestinal epithelium and produced lots of TNF-α in Bmi-1–/– mice, which also confirmed the serious destruction of epithelial barrier for this mouse.
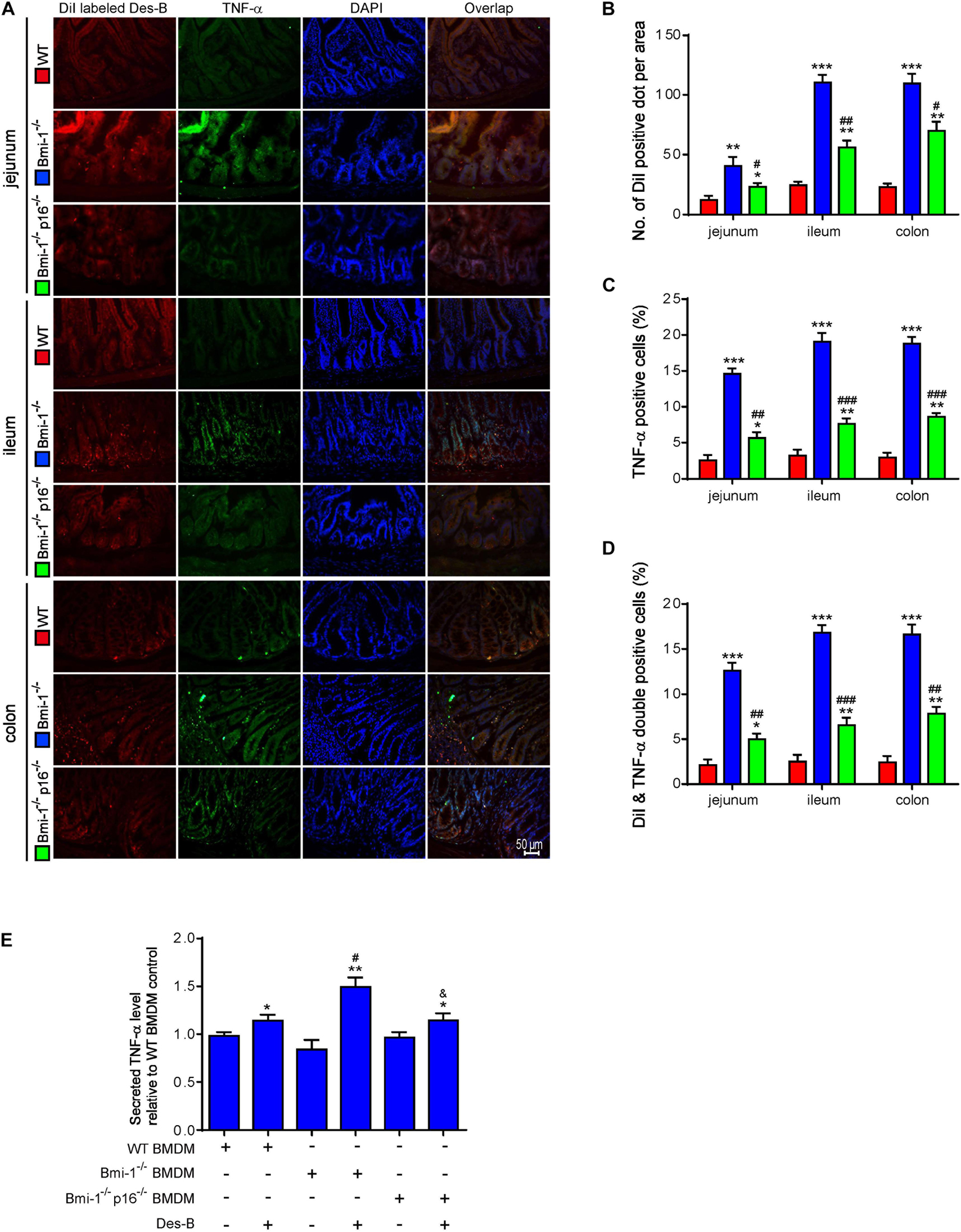
Figure 9. P16 deletion ameliorated the increase in macrophages-secreted TNF-α resulting from Bmi-1 deficiency. DiI-labeled Desulfovibrio (Des-B) was administered by oral gavage into 7-week-old Bmi-1–/–, Bmi-1–/–p16–/–, and WT mice. (A) Representative micrographs showing immunofluorescence for TNF-α in jejunum, ileum, and colon, with DAPI staining the nuclei. (B) Number of DiI-positive dot per area. (C) TNF-α–positive cells. (D) Dil-labeled Des-B and TNF-α double-positive cells. Six mice per group were used for experiments. Values are mean ± SEM from six determinations per group, *p < 0.05, **p < 0.01, ***p < 0.001 compared with the WT group; #p < 0.05, ##p < 0.01, ###p < 0.001 compared with the Bmi-1–/– group. BMDMs was isolated and cultured from bone marrow of WT, Bmi-1–/–, and Bmi-1–/–p16–/– mice. (E) Secreted TNF-α level was detected in the supernatant of BMDMs treated with Des-B or vehicle using ELISA. Cell experiments were performed with three biological repetitions per group. Statistical analysis was performed with one-way ANOVA test. Values are mean ± SEM from six determinations per group, *p < 0.05, **p < 0.01 compared with the Des-B vehicle group; #p < 0.05, ##p < 0.01, ###p < 0.001 compared with the same treatment of WT-BMDM group; &p < 0.05 compared with the same treatment of Bmi-1–/––BMDMs.
To further investigate if p16 deletion ameliorated the effect of Desulfovibrio on promoting macrophages-secreted TNF-α in Bmi-1–/– mice, Desulfovibrio or vehicle was administered to BMDMs of Bmi-1–/–, Bmi-1–/–p16–/–, and WT mice. Secreted TNF-α level was detected in the supernatant of BMDMs with enzyme-linked immunosorbent assay (ELISA). Results showed that an obvious increase was observed in secreted TNF-α level in Desulfovibrio-treated BMDMs compared with vehicle-treated BMDMs at the same genotype. After Desulfovibrio treatment, the elevation of secreted TNF-α level was obviously more than WT BMDMs in Bmi-1–/– BMDMs; however, it was significantly reduced in Bmi-1–/–p16–/– BMDMs compared with the Bmi-1–/– BMDMs (Figure 9E).
To further investigate if Desulfovibrio promoted the TNF-α secretion and activated the NF-κB signaling in BMDMs, primary BMDM cells from WT, Bmi-1–/–, and Bmi-1–/–p16–/– mice were treated with Desulfovibrio (10 μg/mL for 12 h). Immunofluorescence staining was also used to observe the expression level of TNF-α. Western blot was used to detect the expression levels of NF-κB-p65, p-p65 (Ser536), IκBα, and p-IκBα (Ser32). Our results showed that Bmi-1 deficiency caused BMDMs to increase the secretion of TNF-α, and the expressions of NF-κB-p65, p-p65 (Ser536), IκBα, and p-IκBα (Ser32); however, p16 deletion partially corrected this process (Figures 10A–D). Desulfovibrio treatment promoted the secretion of TNF-α (Figures 10A,B), and the expressions of NF-κB-p65, p-p65 (Ser536), IκBα, and p-IκBα (Ser32) in WT, Bmi-1–/–, and Bmi-1–/–p16–/– BMDMs compared with the same genotype BMDMs without Desulfovibrio treatment (Figures 10C,D).
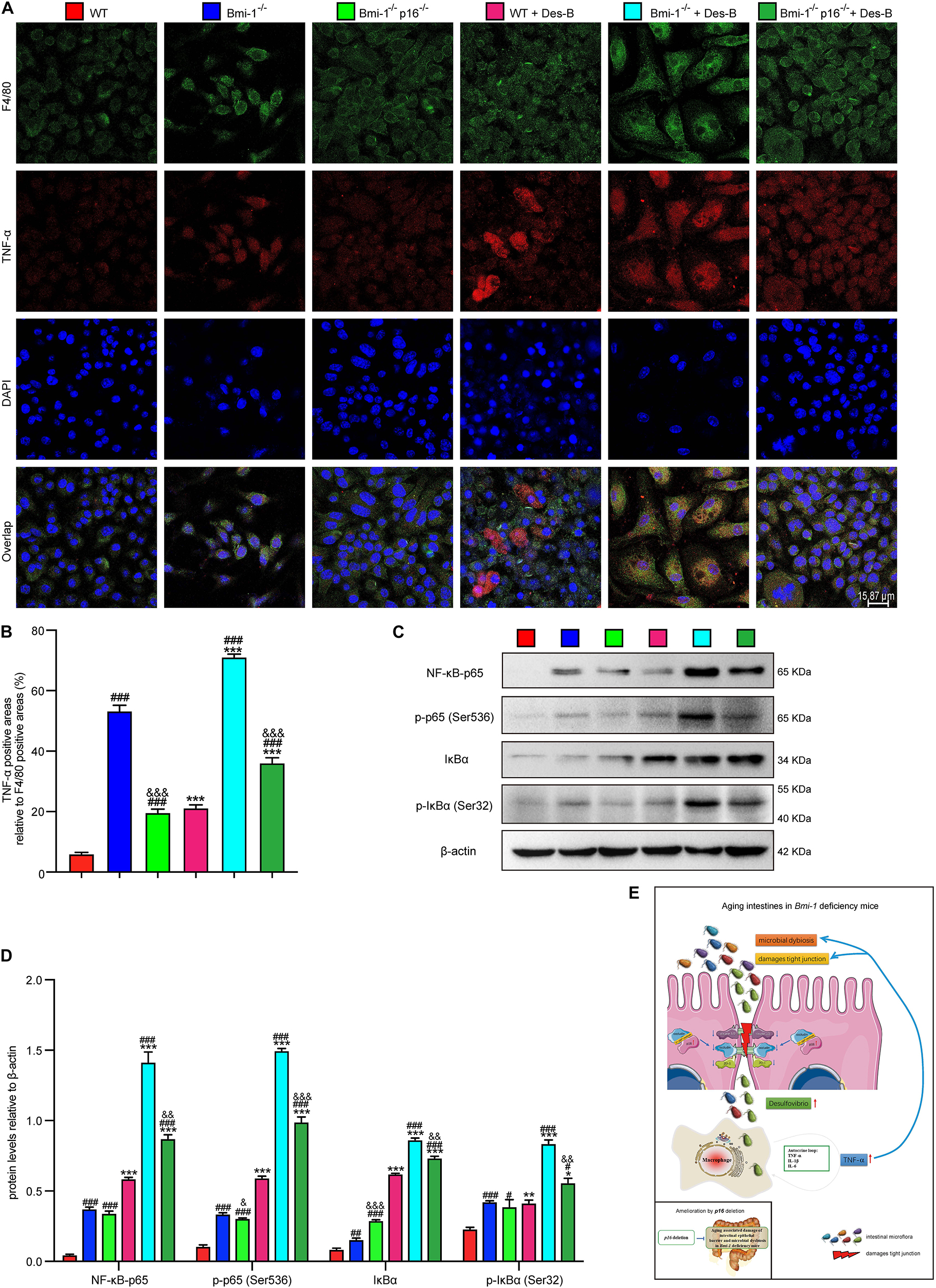
Figure 10. Desulfovibrio promoted the TNF-α secretion and activated the NF-κB signaling in BMDMs. BMDMs from WT, Bmi-1–/–, and Bmi-1–/–p16–/– mice were pretreated with Desulfovibrio (10 μg/mL) for 12 h. (A) Representative micrographs showing immunofluorescence for F4/80 and TNF-α, with DAPI staining the nuclei. (B) Percentage of TNF-α–positive areas relative to F4/80 (%). (C) Western blots for NF-κB–p65, p-p65 (Ser536), IκB-α, and p-IκBα (Ser32) in BMDM cells from WT, Bmi-1–/–, and Bmi-1–/–p16–/– mice; β-actin was used as the loading control. (D) Protein levels relative to β-actin were assessed by densitometric analysis. Statistical analysis was performed with one-way ANOVA test. Values are mean ± SEM from six determinations per group, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 compared with the same genotype without Desulfovibrio treatment group; #p < 0.05, ##p < 0.01, ###p < 0.001 compared with WT-BMDM group at the same treatment; &p < 0.05, &&p < 0.01, &&&p < 0.001 compared with the Bmi-1–/––BMDM group at the same treatment. (E) Graphical abstract about aging intestines in Bmi-1–deficient mice. Macrophages-secreted TNF-α resulting from Bmi-1 deficiency not only stimulated macrophages to secrete more inflammatory factors, but also accelerated the intestinal microbiota disorders and destruction of TJ. Harmful bacteria including Desulfovibrio cross TJ to further trigger systemic inflammations. Up-regulated p16 in the intestine of Bmi-1–deficient mice prevented occludin from forming and repairing intercellular TJ by combining with occludin protein at the 1st–160th residue in the cytoplasm, which exacerbated destruction of TJ. However, p16 deletion improved intestinal microbial function and maintained the intestinal epithelial barrier in Bmi-1–deficient mice.
These results demonstrated that Desulfovibrio promoted the TNF-α secretion and activated the NF-κB signaling in BMDMs, and p16 deletion ameliorated the effect of Desulfovibrio on promoting macrophages-secreted TNF-α in Bmi-1–/– mice.
This study demonstrated that in an SIPS model of Bmi-1 deficiency, barrier structure and function in intestinal epithelium were damaged; TNF-α–induced destruction of TJ increased barrier permeability; up-regulated proinflammatory level of macrophages was induced by intestinal microbial dysbiosis and dysfunction; and more Desulfovibrio entered the intestinal epithelium to promote TNF-α secretion and NF-κB signaling activation in macrophages. P16 protein was accumulated in aging intestinal epithelium from Bmi-1–deficient mice, combined with occludin in the cytoplasm, and finally prevented the form and repair of TJ. P16 deletion ameliorated damage of intestinal epithelial barrier and microbial dysbiosis caused by Bmi-1 deficiency. FM transplantation from WT mice ameliorates TJ in intestinal epithelium of Bmi-1 deficiency and Bmi-1 and p16 double-deficient mice (Figure 10E).
Bmi-1 is a member of the polycomb repressor complex 1 that mediates gene silencing by regulating chromatin structure and is indispensable for self-renewal of both normal and cancer stem cells (Bhattacharya et al., 2015). It has been reported that Bmi-1 also plays a crucial role in maintaining self-renewal of ISCs in the crypt of epithelium. When the stem cells responsible for daily maintenance of intestinal epithelium are destroyed, Bmi-1–expressing cells increase in number, acting as a compensatory mechanism (Tian et al., 2011). Several lines of evidence demonstrate that aging leads to the dysfunction of ISCs in intestinal crypts, driving rapid renewal of intestinal epithelium, generating progenitor cells, and differentiating several cell types such as Paneth cells and goblet cells, which are critical to maintain intestinal epithelial function and homeostasis (Henning and von Furstenberg, 2016; Moorefield et al., 2017). Our previous reports demonstrate that Bmi-1–deficient mouse is an SIPS model, induced by imbalanced redox and DNA damage (Zhang et al., 2010; Jin et al., 2014, 2017; Xie et al., 2015; Chen et al., 2020; Sun et al., 2020). We observed that besides the smaller body size and shorter intestinal length, Bmi-1–deficient mice also showed abnormal intestinal morphology, such as shorter villus and down-regulation of glycoprotein and mucin, which made up a relatively significant share of the intestinal mucus. Length of villi influences the absorptive surface involved in digestion (Rao and Fritz-Niggli, 1988). Mucus in small intestine is important for efficient nutritional uptake, whereas the mucus in colon helps keep bacteria away from the epithelium (Johansson and Hansson, 2016). In this study, our results showed TJ destruction in intestinal epithelial cells from Bmi-1–null mice. TJ integrity, closely associated with intestinal epithelium function, contributes to the function of physical intestinal barrier by regulating paracellular transportation (Lee et al., 2018). Previous report finds that intestinal Bmi-1 deficiency inhibits epithelium proliferation and hampers self-renewal of the ISCs accompanied by p16 accumulation (Lopez-Arribillaga et al., 2015). In this study, we found that aging epithelium cells characterized by p16 accumulation inhibited proliferation and hampered repair of intestinal epithelium and further disrupted microbial homeostasis. Moreover, the up-regulated proinflammatory level of intestinal macrophages was associated with the change of intestinal microbiota in Bmi-1–deficient mice. Bmi-1 deficiency resulted in significant decreases in the body weight, villus length, the ratio of villus length to crypt depth, the number of Paneth cells, and the number of acid mucin and glycoprotein. Thus, we speculated that all changes of the intestine caused by Bmi-1 deficiency weakened intestinal immune barrier function, nutrient absorption, and intestinal immunity in mice, which finally contributed to the smaller body size and increased inflammation level of mice.
TJ barrier of the intestine is formed by claudins, occludin, and ZO-1, helping to maintain the selective barrier function of intestinal epithelium (Lee et al., 2018). Among these, occludin is important in the assembly and maintenance of TJ (Raleigh et al., 2011). Occludin that is phosphorylated locates mainly in the membrane, while the less phosphorylated occludin is found in the cytoplasm (Lee et al., 2018). In our study, we observed breakage of cell–cell TJ in the intestinal epithelium of Bmi-1–deficient mice, locating exactly at the occludin section. Also, the occludin is down-regulated in intestinal epithelium of Bmi-1–deficient mice. These conditions were then reversed by p16 deletion. We then investigated how p16 deletion rescues intestinal epithelial barrier dysfunction. Former experiments discovered that N-terminal of p16 (residues 1–80) bound to the N-terminal region of JNK1 (also known as MAPK8) (residues 1–60) or the N-terminal region of JNK3 (also known as MAPK10) (residues 75–100), which contain the glycine-rich site (Choi et al., 2005). Analysis with https://www.uniprot.org/align showed an extremely similar domain between occludin (residues 107–139, which is a glycine-rich site) and MAPK8 (residues 16–38) or MAPK10 (residues 54–76) in human. In mice, an extremely similar domain showed between occludin (residues 107–139) and MAPK8 (residues 24–72) or MAPK10 (residues 62–110). Besides, the 107th-139th residue is also a glycine-rich site of occludin for mouse. As a transmembrane protein, the 90th-135th residue of occludin locates at the extracellular domain. Thus, we speculated that p16 could bind with occludin in cytoplasm, blocking occludin in participating in the formation of intercellular TJ, which further aggravated the destruction of TJ in Bmi-1–deficient mice. Then, we found that p16 and occludin were colocalized and combined in the cytoplasm of intestinal epithelial cells. Accumulated p16 combined with occludin at the 1st–160th residue rather than limited to the 107th–139th residue in the cytoplasm of aging intestinal epithelium cells in Bmi-1–/– mice, inhibiting its translocation from cytoplasm to cell membrane for blocking the TJ formation and repair of intestinal epithelium barrier. Moreover, the premature senescence induced by Bmi-1 deficiency caused destruction of intestinal TJ by TNF-α and reduced levels of TJ proteins, exacerbating the destruction of intercellular TJ and increasing intestinal epithelial permeability.
Several lines of evidence demonstrate that TNF-α stimulates permeability and disrupts integrity by up-regulating myosin light chain kinase (MLCK) aggravating intestinal epithelial barrier dysfunction and immune-mediated colitis, and/or reduces TJ protein expressions through activating NF-κB signaling pathway via TNFR1 and ERK1/2 signaling pathway (Ye et al., 2011; Hall et al., 2017; Drolia et al., 2018). Our results confirmed that TNF-α treatment damaged TJ and decreased the expressions of occludin and ZO-1. It has been reported that TNFR2 plays an important role in stimulating epithelial long MLCK expression, aggravating intestinal epithelial barrier dysfunction and immune-mediated colitis. TNFR2 interferes with programmed cell death by activating NF-κB and JNK pathways or by inhibiting TRAF-2 (Su et al., 2009, 2013). In this study, we detected TNF-α and other related proinflammatory factor levels in ileum, jejunum, and colon from Bmi-1–deficient mice and observed the up-regulation of proinflammatory factor and activation of NF-κB pathway. Besides, we observed no difference in phagocytosis of macrophage between Bmi-1 deficiency and WT mice. However, how Bmi-1 and/or p16 regulates the subtype and function of macrophages remains unknown. Previous observation finds that the expression of p16 and senescence-associated β-galactosidase (SA-β-gal) are markers of their physiological programs of polarization in response to immunomodulatory stimuli in macrophages. This characterization is reversible and p53-independent in macrophages and obviously different from cellular senescence characterized by the constitutive expression of these biomarkers following p53-dependent proliferation arrest in rodent cells at least. Thus, p16 and/or SA-β-gal–positive phases are non-senescent phases in macrophages (Hall et al., 2017). In this study, BMDMs in Bmi-1–null conditions, especially in Bmi-1–null plus Desulfovibrio treatment, showed a bigger size and irregular shapes. We think that this shape might be determined by its physiological programs of polarization in response to immunomodulatory stimuli in macrophages, although not because they are senescent. We will investigate whether Bmi-1 determines the polarization and response of macrophages through inhibiting p16 in the follow-up study. It has been reported that chronic exposure to inflammation alters proinflammatory function of macrophage, thus secreting more proinflammatory factors (Thevaranjan et al., 2017). In this study, we found that the up-regulated proinflammatory level of macrophages was associated with the change of aging associated intestinal microbiota in Bmi-1–null mice.
A former study has reported that intestinal microbiota dysbiosis not only triggers inflammatory bowel disease (Larabi et al., 2020), but also leads to systemic inflammation, obesity, type 2 diabetes, and chronic kidney disease (Sabatino et al., 2017; Thevaranjan et al., 2017; Virtue et al., 2019). Thus, we conducted high-throughput sequencing of V4 region amplicon of 16S rRNA to detect intestinal microbiota changes, which showed the intestinal microbiota ratio imbalance. Besides the diversity of intestinal bacterium, the number of intestinal probiotics also decreased after Bmi-1 deletion, whereas the pathogenic bacteria such as Desulfovibrio, Helicobacter, and Oscillibacter increased, leading to the dysbiosis of microbiota. Moreover, the function of carbohydrate, glycan and nucleotide metabolism, cell growth and death regulation, replication, and repair were all impaired in Bmi-1–deficient mice, which further damaged the intestinal function and nutrition absorption. Not only the pathogenic bacteria itself, but also the harmful substances it secreted can also lead to intestinal diseases and even systemic diseases (Moreto and Perez-Bosque, 2009). Then, DiI-labeled Desulfovibrio, a kind of pathogenic bacteria, was administered by oral gavage into Bmi-1 knockout, Bmi-1 and p16 double-knockout, and WT mice. TNF-α level and the number of Desulfovibrio were increased in the intestinal epithelium of Bmi-1–deficient mice. To further confirm whether up-regulation of TNF-α is secreted by macrophages through activating NF-κB signaling after Desulfovibrio inducing, TNF-α protein level was up-regulated in the supernatant, and NF-κB signaling was activated in BMDMs from the mice, illustrating that Desulfovibrio could induce macrophages secreting more TNF-α and further aggravate the damage of intestinal barrier. We also observed that p16 deletion could ameliorate the microbiota dysbiosis and decrease the inflammation level in Bmi-1–deficient mice, which provides potential targets for the treatment of age-associated intestinal diseases and intestinal microbiota dysfunction.
Taken together, our results demonstrated that Bmi-1 maintained intestinal TJ, epithelial barrier function, and microbiota balance through preventing senescence characterized by p16 accumulation. Clearance of p16-positive cells in aging intestinal epithelium would be a new method for maintaining barrier function and microbiota balance. The 1st to 160th residues of occludin could be a novel therapeutic target for identifying small molecular antagonistic peptides to prevent the interaction of p16 with occludin for protecting TJ.
Bmi-1–/–p16–/–, Bmi-1–/–, and WT SPF mice were prepared as described previously (Jin et al., 2014, 2017). All of them were housed in pathogen-free conditions. For studies of the microbiota, mice were selected from multiple cages and multiple breeding pairs to minimize cage effects or familial transfer of the microbiota as previously described (Thevaranjan et al., 2017). No evidence of cage effects was found in the studies of the microbiota. This study was carried out in strict accordance with the guidelines of the Institute for Laboratory Animal Research of Nanjing Medical University in Nanjing of China. The protocol was approved by the Committee on the Ethics of Animal Experiments of Nanjing Medical University (permit number IACUC-1706001).
Seven-week-old mice were anesthetized with 3% pentobarbital sodium (40 mg/kg). The leg bones were separated, and bone marrow was rushed out with phosphate-buffered saline (PBS). BMDMs were then isolated and cultured in Dulbecco modified eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Gibco, Life Technologies Inc., NY, United States) and 50 ng/mL recombinant mouse macrophage colony-stimulating factor (Novoprotein Scientific Inc., Shanghai, China) at 37°C as previously described (Assouvie et al., 2018).
The Cacao-2 cells (#ZQ0056) (Shanghai Zhong Qiao Xin Zhou Biotechnology Co., Ltd., Shanghai, China) were cultured in 1640 medium with 10% FBS according to the manufacturer’s instructions.
Feces were collected daily from 3- to 4-week-old WT or Bmi-1–/– mice in sterile conditions. Stools from each group were pooled 100 mg and then resuspended in 1 mL of sterile saline. The solution of FM was vigorously mixed before centrifugation at 800 g for 3 min as previously described (Chang et al., 2015). The supernatant bacterial concentration of FM was detected with microplate reader at 600-nm excitation wavelength and should be more than 0.6; 0.6–0.8 is the logarithmic phase of bacterial growth, and 0.8–1.0 is the platform phase of bacterial growth. The concentration of FM was used in both phases.
The supernatant from the WT group was collected and labeled with DiI (Sigma-Aldrich, St. Louis, MO, United States) for tracking according to the manufacturer’s protocol before infecting BMDMs as previously described methods (Zhang et al., 2011; Nagyova et al., 2014; Xie et al., 2015). Then, the DiI-labeled FM was incubated with BMDMs for immunofluorescent staining of cells. The supernatant from WT or Bmi-1–/– group was collected and incubated with BMDMs for real-time reverse transcriptase–polymerase chain reaction (RT-PCR) detection.
Mice were deprived of food 4 h before an oral gavage. FM from 3- to 4-week-old WT mice (fecal samples 100 μL, 100 mg/mL) were transplanted to WT, Bmi-1–/–, and Bmi-1–/–p16–/– mice by gavage every other day and lasted for 21 days.
Mice were anesthetized with 3% pentobarbital sodium (40 mg/kg) at 7 weeks of age. Jejunum, ileum, and colon were separated and washed with PBS. The intestines were cut into several pieces following the annular surface. Then, the intestine samples were fixed with periodate-lysine-paraformaldehyde (PLP) solution for 24 h at 4°C (Zhang et al., 2010). For hematoxylin–eosin (H&E), Alcian blue and periodic acid–Schiff (AB-PAS) staining, immunohistochemistry, and immunofluorescence, the samples were dehydrated in a series of graded ethanol solutions, embedded in paraffin, and cut into 5-μm sections using a rotary microtome (Leica Biosystems Nussloch GmbH, Nussloch, Germany) as previously described (Jin et al., 2017). For immunofluorescence staining, the samples were dehydrated in 15 and 30% sucrose solutions (prepared in 1 × PBS) in turn until the tissue settled to the bottom of the tube. Then, the samples were cut into 5-μm sections using a freezing microtome (Thermo Scientific Cryotome FSE Cryostats, Loughborough, Leicestershire, England) as previously described (Chen et al., 2020).
For H&E, AB-PAS staining, or immunofluorescence, serial paraffin sections were deparaffinized and rehydrated.
Serial paraffin sections of jejunum, ileum, and colon were stained as previously described (Jin et al., 2017).
AB-PAS staining kit (#G1285, Solarbio Life Sciences, Beijing, China) was used according to the manufacturer’s instructions and following methods as previously described (Li et al., 2019).
Serial paraffin sections were generated for antigen retrieval, steamed for 20 min in sodium citrate buffer (10 mM sodium citrate acid, 0.05% Tween-20, pH 6.0) followed by blocking of endogenous peroxidase (3% H2O2) and preincubation with serum (Jin et al., 2014; Xie et al., 2015). Primary antibodies were against Bmi-1 (#5856, Cell Signaling Technology, United States), p16 (#ab211542, Abcam, Cambridge, MA, United States), p53 (#2524, Cell Signaling Technology, United States), and Ki67 (#BS1679, Bioworld Technology Inc., MN, United States). Biotin-labeled second antibody was used. Nuclei were stained with hematoxylin.
Primary antibodies against ZO-1 (sc33725, Santa Cruz Biotechnology Inc., Dallas, TX, United States), TNF-α (sc-52746, Santa Cruz Biotechnology Inc., United States), F4/80 (sc-377009, Santa Cruz Biotechnology Inc., United States), occludin (sc133256, Santa Cruz Biotechnology Inc., United States), and p16 (10883-1-AP, Proteintech Group, Inc., IL, United States) were used, and affinity-purified Alexa Fluor 488–conjugated secondary antibody and 594-conjugated secondary antibody (Life Technologies Corporation, Carlsbad, CA, United States) were used. Nuclei were labeled with DAPI (Sigma-Aldrich, United States) and mounted with medium to prevent quenching (Vector Laboratories Inc., United States) following previously described methods (Jin et al., 2017).
GFP-labeled p16 full-length overexpression adenovirus carrying the Flag-tag was designed and synthesized by Genechem Co., Ltd., in Shanghai, China.
Considering the structural features of occludin as a transmembrane protein, we generated four mutant plasmids based on its intracellular and extracellular domain, which contains mutation of 1–160 residues, 161–265 residues, 266–522 residues, and 107–139 residues and a full-length or a truncated fragment (residues 266–522) of occludin overexpression plasmid all carrying the His-tag with pcDNA3.1 vector plasmid. Production of the above plasmids was by TranSheep Bio Co., Ltd., Shanghai, China.
The above adenovirus was transfected in 293T cells incubated with antibiotic-free complete medium at 40–50% fluence for 8 h. After 24 h, plasmids were transfected into these 293T cells using X-tremeGENE HP DNA Transfection Reagent (cat. no.06366236001, Roche Diagnostics GmbH, Mannheim, Germany). Cells were incubated with antibiotic-free complete medium including the above transfection mixture for 48 h, and protein was harvested. Specific details followed the instruction of X-tremeGENE HP DNA Transfection Reagent.
When the Cacao-2 cell density reached 60%, they were transfected with GFP-labeled Flag-p16–overexpressed (p16-OV) or vehicle adenovirus and then treated with or without 10 ng/mL TNF-α (#C008, Novoprotein Scientific Inc., Shanghai, China) for 24 h. Then, Cacao-2 cells were fixed with PLP for 10 min and washed three times with PBST (PBS with 0.05–0.1% Tween-20) for 10 min each and blocked with 5% bovine serum albumin at room temperature for 1 h to prevent non-specific reactions. Primary antibodies against F4/80 (sc-377009, Santa Cruz Biotechnology Inc., United States), ZO-1 (#21773-1-AP, Proteintech Group, Inc., IL, United States), and occludin (sc133256, Santa Cruz Biotechnology Inc., United States) were used. Affinity-purified Alexa Fluor 488–conjugated secondary antibody (Life Technologies Corporation, United States) was used. Nuclei were labeled with DAPI.
BMDMs were pretreated with Desulfovibrio (10 μg/mL) for 12 h or cultured with DiI-labeled FM preincubated with serum. Cells were reacted with primary antibody after being fixed and blocked. Primary antibodies against F4/80 (sc-377009, Santa Cruz Biotechnology Inc., United States) and TNF-α (NBP1-19532, Novus Biologicals, Centennial, Co., United States) were used, and affinity-purified Alexa Fluor 488–conjugated secondary antibody (Life Technologies Corporation, United States) was used. Nuclei were labeled with DAPI (Sigma-Aldrich, United States) and mounted with medium to prevent quenching (Vector Laboratories Inc., Burlingame, CA, United States) (Gu et al., 2016).
After routinely dewaxing and hydration, serial paraffin sections of ileum were blocked with sheep serum and detected with antibodies against p16 (#ab211542, Abcam, Cambridge, MA, United States) and occludin (#sc-133256, Santa Cruz Biotechnology Inc., United States). Then, Duolink Proximity Ligation Assay (PLA) in situ fluorescence (Sigma-Aldrich, United States) was performed following the manufacturer’s instructions with Duolink in situ PLA probe anti-mouse PLUS (#DUO92001), Duolink in situ PLA probe anti-rabbit MINUS (#DUO92005), Duolink in situ detection reagents Red (#DUO92008), and Duolink in situ wash buffer fluorescence (#DUO82049). The PLA signal (λex 594 nm, λem 624 nm; Texas Red) was analyzed (Chen et al., 2020).
Intestinal samples of mice were fixed with 1% glutaraldehyde in a 0.1 M sodium phosphate buffer (pH 7.4, 4°C, 2 h), postfixed in 2% osmium tetroxide in a 0.1 M phosphate buffer (4°C, 1.5 h), and dehydrated in a graded series of concentrations of ethanol (50, 70, 90, 95, and 4 × 100% each for 15 min) and acetone (15 min). Eventually, the material was embedded in epoxy resin. Sections were cut to stain with uranyl acetate and lead citrate (Jin et al., 2014; Wang et al., 2019). JEOL 1200EX TEMSCAN electron microscope was used to observe the TJ of intestinal epithelial cells.
Tracer FITC–labeled dextran (#46944; Sigma-Aldrich) was used to assess in vivo intestinal permeability as previously described (Thevaranjan et al., 2017). Mice were deprived of food 4 h before an oral gavage using FITC–dextran (600 mg/kg body weight, 125 mg/mL). Blood was collected from heart after 6 h, and fluorescence intensity was measured on fluorescence plates using an excitation wavelength of 493 nm and an emission wavelength of 518 nm (Thevaranjan et al., 2017).
Intestinal epithelium of 7-week-old mice or BMDM cells was dissected and immediately placed into RIPA lysis buffer containing a cocktail of proteinase inhibitors and phosphatase inhibitors (#4906845001, Roche Diagnostics Corp., Basel, Switzerland) and phenylmethanesulfonyl fluoride (#ST506, Beyotime Institute of Biotechnology, Shanghai, China) for protein extraction. Western blots were performed as previously described (Smith et al., 2003; Jin et al., 2017). Primary antibodies against claudin-1 (sc-81796, Santa Cruz Biotechnology Inc., United States), occludin (sc133256, Santa Cruz Biotechnology Inc., United States), claudin-2 (sc-293233, Santa Cruz Biotechnology Inc., United States), TNF-α (NBP1-19532, Novus Biologicals, Centennial, Co., United States), NF-κB–p65 (#8242, Cell Signaling Technology, Beverly, MA, United States), p-p65 (Ser536) (ab76302, Abcam, United States), IκBα (AF1282, Beyotime Biotechnology, China) and p-IκBα (Ser32) (sc-8404, Santa Cruz Biotechnology Inc., United States), ZO-1 (sc33725, Santa Cruz Biotechnology Inc., United States), p16 (#ab211542, Abcam, United States), DYKDDDDK Tag (binds the same epitope as Sigma’s anti-FLAG M2 antibody, #14793, Cell Signaling Technology, United States), and His-Tag (#12698, Cell Signaling Technology, United States) were used. β-Actin (BS6007M, Bioworld Technology, St. Louis Park, MN, United States) was the loading control for the cytoplasmic fraction and total cell protein.
RNA was extracted from BMDMs untreated or treated with fecal bacteria using TRIzol reagent (#15596, Invitrogen Inc., United States) according to the manufacturer’s protocol. Levels of mRNA in cell samples were quantified by real-time RT-PCR as previously described (Jin et al., 2011; Jin et al., 2017). Primers were in Supplementary Table 1.
After extracting total protein from mouse ileum tissues, immunoprecipitation assay was performed using Pierce Co-Immunoprecipitation (Co-IP) Kit (#26149, Pierce Co-IP Kit, Thermo Fisher Scientific, IL, United States) as the manufacturer instructed. Followed by preclearing lysate with the control agarose resin, tissue proteins were mixed with 1 μg of antibody and then incubated overnight, anti–immunoglobulin G (IgG) antibody as a control. The bound antigens were eluted from the agarose resin using elution buffer. Eluted samples were carried out with sodium dodecyl sulfate–polyacrylamide gel electrophoresis gel electrophoresis. Immunoblotting was carried out as previously described (Chen et al., 2018). Clean-BlotTM IP Detection Reagent (horseradish peroxidase) (#21230, Thermo Fisher Scientific) was used as secondary antibody to eliminate IgG bands. Primary antibodies against p16 (#ab211542, Abcam, United States), occludin (#sc-133256, Santa Cruz Biotechnology Inc., United States), and DYKDDDDK Tag (binds the same epitope as Sigma’s Anti-FLAG M2 antibody, #14793, Cell Signaling Technology, United States) were used. The immunoreactive bands were visualized by ECL chemiluminescence (Amersham Pharmacia Biotech, NJ, United States) and analyzed by the Scion image Beta 4.02 (Scion, National Institutes of Health, Bethesda, MD, United States).
The amino acid sequences of occludin, MAPK8, and MAPK10 protein from mouse or human were aligned using online tools (Uniprot/Align)1 (Supplementary Information 4, 5 alignment of occludin, MAPK8 and MAPK10 in human or mouse).
Eight mice were selected from each genotype. Mice feces were collected and then stored at −80°C before use. Total genome DNA was extracted from the samples using DNA magnetic bead extraction kit following the manufacturer’s instructions. After extraction, DNA was treated with DNase-free RNase to remove contaminating RNA. DNA concentration and quality were assessed (Pandit et al., 2018; Zhao et al., 2019). Then, the DNA was stored at −20°C for use.
The V4 region of the 16S rRNA gene was amplified with primers and sequenced as previously described (Caporaso et al., 2010, 2012). Amplified DNA was sequenced using QIIME (Caporaso et al., 2010) software package (Quantitative Insights Into Microbial Ecology, V1.7.0).2
Raw data were analyzed by Novogene Bioinformatic Technology Co., Ltd., In-house Perl scripts were used to analyze α- (within samples) and β- (among samples) diversity. Sequences with ≥ 97% similarity were assigned to the same OTUs. We pick representative sequences for each OTU and use the RDP classifier (Wang et al., 2007) to annotate taxonomic information for each representative sequence. In order to compute α-diversity, we rarify the OTU table and calculate three metrics: Chao1 estimates the species abundance; Observed Species estimates the amount of unique OTUs found in each sample, and Shannon index. Based on Unifrac distance, principal component analysis was conducted using QIIME to analyze β-diversity (Hamady et al., 2010). Biomarkers were selected with LEfSe (LDA effect size) (Segata et al., 2011). In total, species with significant differences were analyzed using Wilcoxon rank sum test. LDA score was used to assess the impact of significantly different species.
PICRUST was used to predict the metabolic pathways from 16S rRNA gene-based microbiota of the intestine. The predicted functions were then collapsed into hierarchical KEGG pathways using the categorize-by-function step in the PICRUST pipeline (Garcia-Amado et al., 2018). Heat maps of function categories based on KEGG pathways and abundance changes of the differential bacteria were generated by R software.
Desulfovibrio freeze-dried powder (#BNCC173631) was obtained from Beijing Beina Chuanglian Biotechnology Research Institute in Beijing of China. According to the manufacturer’s instructions, certain amount of germ-free calf serum was added into the Desulfovibrio freeze-dried powder and mixed. The bacterial solution was dropped into Colombian blood agar plate with triangle coating rod spreading evenly. It was then cultured in anaerobic culture zone at 37°C. After the colony was formed, its shape was confirmed under the microscope. The Desulfovibrio colony was inoculated into LB liquid medium and amplified after passaging two to three times (the whole process was completed in anaerobic culture zone). Every 100 μL of bacterial solution was added into 900 μL of 10% glycerin and stored at −20°C for use.
In vivo, before the oral gavage, the concentration of Desulfovibrio solution was detected with microplate reader at 600-nm excitation wavelength and should be more than 0.6. Desulfovibrio solution was labeled with DiI (Sigma-Aldrich, United States) for tracking according to the manufacturer’s protocol as previously described methods (Xie et al., 2015). After depriving of food 4 h, mice were administered 108 DiI-labeled Desulfovibrio by an oral gavage. Then, after 6 h, intestine was taken for immunofluorescence.
In vitro, for the infection experiment, the Desulfovibrio solution (10 μg/mL) was incubated with BMDMs in α-MEM medium supplemented with 15% FBS (Gibco, United States) at 37°C for 12 h.
BMDMs untreated or infected with Desulfovibrio bacteria (Des-B) were cultured in DMEM (without phenol red; Gibco) without FBS for 6 h. Supernatants were collected and filtered with MILLEX-GP 0.22-μm filters (Merck Millipore Ltd., Co., Cork, Munster, Ireland) to remove cell debris, concentrated to 1% volume with Amicon Ultra-4 centrifugal ultrafiltration tubes (NMWL 3KDa) (Merck Millipore Ltd., Co.) (Xie et al., 2015; Jin et al., 2017; Chen et al., 2020), and detected the concentrations of mouse-derived TNF-α with ELISA kit (#YFXEM00031, Yifeixue Biotechnology, Nanjing, China).
All analyses were performed using GraphPad Prism software (version 6.07; GraphPad Software Inc., San Diego, CA, United States) as previously described (Schafer et al., 2017). Measurement data were described as mean ± SEM fold-change over control and analyzed by Student t-test and one-way analysis of variance (ANOVA) to compare differences among groups. Qualitative data were described as percentages and analyzed using χ2-tests as indicated (Jin et al., 2014, 2017; Chen et al., 2020). p-values were two-sided, and differences were considered significant at p < 0.05.
As in previously described statistical methods (Zhang et al., 2020), to identify differential abundance of phyla, genera, and species between any two groups, Wilcoxon rank-sum test was used, p-value was corrected as false discovery rate with the Benjamini–Hochberg method. p-values were considered significant at p < 0.05. To identify features (taxa and functional modules) differentially represented between any two groups, differentially abundant taxa or functional modules were selected using the LEfSe. Only taxa with LDA score > 4.0 are shown.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: http://www.ncbi.nlm.nih.gov/bioproject/704896. The BioProject ID is PRJNA704896.
The animal study was reviewed and approved by the Committee on the Ethics of Animal Experiments of Nanjing Medical University.
JJ and DM: conceptualization and funding acquisition. JWZ, CH, HC, ZQ, ZM, JYZ, QW, MC, CX, RW, QL, GZ, DM, and JJ: methodology. JWZ, CH, HC, ZQ, ZM, CX, RW, QL, and JJ: software. JWZ, CH, HC, ZQ, ZM, and JJ: validation. JWZ, CH, HC, ZQ, ZM, JYZ, QW, MC, CX, RW, QL, GZ, and JJ: data analysis. JWZ, JJ, and DM: writing—original draft with help from the other authors. JJ, HC, and DM: writing—review, and editing with help from the other authors. RW, JJ, and DM: project administration and supervision. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Natural Science Foundation of China (81871097, 81571371 and 81200491 to JJ, 81730066 to DM), the “Qinglan project” funded by Jiangsu Universities (2020-10 to JJ), the Jiangsu Government Scholarship for Overseas Studies (JS-2017-095 to JJ), and the Natural Science Foundation of Jiangsu Province (BK20151554 to JJ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.671564/full#supplementary-material
Bmi-1–/–, Bmi-1–deficient; Bmi-1–/–p16 INK4a –/–, Bmi-1 and p16 INK4a double-knockout; TJ, tight junction; SIPS, stress-induced premature senescence: WT-FB, fecal mixed bacteria from WT mice; Des-B, Desulfovibrio bacteria; ISCs, intestinal stem cells.
Assouvie, A., Daley-Bauer, L. P., and Rousselet, G. (2018). Growing murine bone marrow-derived macrophages. Methods Mol. Biol. 1784, 29–33. doi: 10.1007/978-1-4939-7837-3_3
Bhattacharya, R., Mustafi, S. B., Street, M., Dey, A., and Dwivedi, S. K. (2015). Bmi-1: at the crossroads of physiological and pathological biology. Genes Dis. 2, 225–239. doi: 10.1016/j.gendis.2015.04.001
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Huntley, J., Fierer, N., et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. doi: 10.1038/ismej.2012.8
Chang, C. J., Lin, C. S., Lu, C. C., Martel, J., Ko, Y. F., Ojcius, D. M., et al. (2015). Ganoderma lucidum reduces obesity in mice by modulating the composition of the gut microbiota. Nat. Commun. 6:7489. doi: 10.1038/ncomms8489
Chen, H., Chen, H., Liang, J., Gu, X., Zhou, J., Xie, C., et al. (2020). TGF-beta1/IL-11/MEK/ERK signaling mediates senescence-associated pulmonary fibrosis in a stress-induced premature senescence model of Bmi-1 deficiency. Exp. Mol. Med. 52, 130–151. doi: 10.1038/s12276-019-0371-7
Chen, L., Yang, R., Qiao, W., Yuan, X., Wang, S., Goltzman, D., et al. (2018). 1,25-Dihydroxy vitamin D prevents tumorigenesis by inhibiting oxidative stress and inducing tumor cellular senescence in mice. Int. J. Cancer 143, 368–382. doi: 10.1002/ijc.31317
Choi, B. Y., Choi, H. S., Ko, K., Cho, Y. Y., Zhu, F., Kang, B. S., et al. (2005). The tumor suppressor p16(INK4a) prevents cell transformation through inhibition of c-Jun phosphorylation and AP-1 activity. Nat. Struct. Mol. Biol. 12, 699–707. doi: 10.1038/nsmb960
Drolia, R., Tenguria, S., Durkes, A. C., Turner, J. R., and Bhunia, A. K. (2018). Listeria adhesion protein induces intestinal epithelial barrier dysfunction for bacterial translocation. Cell Host Microbe 23, 470–484.e7. doi: 10.1016/j.chom.2018.03.004
Garcia-Amado, M. A., Shin, H., Sanz, V., Lentino, M., Martinez, L. M., Contreras, M., et al. (2018). Comparison of gizzard and intestinal microbiota of wild neotropical birds. PLoS One 13:e0194857. doi: 10.1371/journal.pone.0194857
Gu, X., Gu, B., Lv, X., Yu, Z., Wang, R., Zhou, X., et al. (2016). 1, 25-Dihydroxy-vitamin D3 with tumor necrosis factor-alpha protects against rheumatoid arthritis by promoting p53 acetylation-mediated apoptosis via Sirt1 in synoviocytes. Cell Death Dis. 7:e2423. doi: 10.1038/cddis.2016.300
Hall, B. M., Balan, V., Gleiberman, A. S., Strom, E., Krasnov, P., Virtuoso, L. P., et al. (2017). p16(Ink4a) and senescence-associated beta-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging 9, 1867–1884. doi: 10.18632/aging.101268
Hamady, M., Lozupone, C., and Knight, R. (2010). Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J. 4, 17–27. doi: 10.1038/ismej.2009.97
Helman, A., Klochendler, A., Azazmeh, N., Gabai, Y., Horwitz, E., Anzi, S., et al. (2016). p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nat. Med. 22, 412–420. doi: 10.1038/nm.4054
Henning, S. J., and von Furstenberg, R. J. (2016). GI stem cells - new insights into roles in physiology and pathophysiology. J. Physiol. 594, 4769–4779. doi: 10.1113/JP271663
Jin, J., Lv, X., Chen, L., Zhang, W., Li, J., Wang, Q., et al. (2014). Bmi-1 plays a critical role in protection from renal tubulointerstitial injury by maintaining redox balance. Aging Cell 13, 797–809. doi: 10.1111/acel.12236
Jin, J., Tao, J., Gu, X., Yu, Z., Wang, R., Zuo, G., et al. (2017). P16 (INK4a) deletion ameliorated renal tubulointerstitial injury in a stress-induced premature senescence model of Bmi-1 deficiency. Sci. Rep. 7:7502. doi: 10.1038/s41598-017-06868-8
Jin, J., Zhao, Y., Tan, X., Guo, C., Yang, Z., and Miao, D. (2011). An improved transplantation strategy for mouse mesenchymal stem cells in an acute myocardial infarction model. PLoS One 6:e21005. doi: 10.1371/journal.pone.0021005
Johansson, M. E., and Hansson, G. C. (2016). Immunological aspects of intestinal mucus and mucins. Nat. Rev. Immunol. 16, 639–649. doi: 10.1038/nri.2016.88
Larabi, A., Barnich, N., and Nguyen, H. T. T. (2020). New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy 16, 38–51. doi: 10.1080/15548627.2019.1635384
Lee, B., Moon, K. M., and Kim, C. Y. (2018). Tight junction in the intestinal epithelium: its association with diseases and regulation by phytochemicals. J. Immunol. Res. 2018:2645465. doi: 10.1155/2018/2645465
Lee, S. H. (2015). Intestinal permeability regulation by tight junction: implication on inflammatory bowel diseases. Intest. Res. 13, 11–18. doi: 10.5217/ir.2015.13.1.11
Li, J., Tan, Y. J., Wang, M. Z., Sun, Y., Li, G. Y., Wang, Q. L., et al. (2019). Loganetin protects against rhabdomyolysis-induced acute kidney injury by modulating the toll-like receptor 4 signalling pathway. Br. J. Pharmacol. 176, 1106–1121. doi: 10.1111/bph.14595
Li, X. G., Wang, Z., Chen, R. Q., Fu, H. L., Gao, C. Q., Yan, H. C., et al. (2018). LGR5 and BMI1 increase pig intestinal epithelial cell proliferation by stimulating WNT/beta-catenin signaling. Int. J. Mol. Sci. 19:1036. doi: 10.3390/ijms19041036
Lin, C., Zhao, S., Zhu, Y., Fan, Z., Wang, J., Zhang, B., et al. (2019). Microbiota-gut-brain axis and toll-like receptors in Alzheimer’s disease. Comput. Struct. Biotechnol. J. 17, 1309–1317. doi: 10.1016/j.csbj.2019.09.008
Liu, J., Cao, L., Chen, J., Song, S., Lee, I. H., Quijano, C., et al. (2009). Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature 459, 387–392. doi: 10.1038/nature08040
Lopez-Arribillaga, E., Rodilla, V., Pellegrinet, L., Guiu, J., Iglesias, M., Roman, A. C., et al. (2015). Bmi1 regulates murine intestinal stem cell proliferation and self-renewal downstream of Notch. Development 142, 41–50. doi: 10.1242/dev.107714
Moorefield, E. C., Andres, S. F., Blue, R. E., Van Landeghem, L., Mah, A. T., Santoro, M. A., et al. (2017). Aging effects on intestinal homeostasis associated with expansion and dysfunction of intestinal epithelial stem cells. Aging 9, 1898–1915. doi: 10.18632/aging.101279
Moreto, M., and Perez-Bosque, A. (2009). Dietary plasma proteins, the intestinal immune system, and the barrier functions of the intestinal mucosa. J. Anim. Sci. 87(Suppl. 14), E92–E100. doi: 10.2527/jas.2008-1381
Nagyova, M., Slovinska, L., Blasko, J., Grulova, I., Kuricova, M., Cigankova, V., et al. (2014). A comparative study of PKH67, DiI, and BrdU labeling techniques for tracing rat mesenchymal stem cells. In Vitro Cell. Dev. Biol. Anim. 50, 656–663. doi: 10.1007/s11626-014-9750-5
Pandit, R. J., Hinsu, A. T., Patel, N. V., Koringa, P. G., Jakhesara, S. J., Thakkar, J. R., et al. (2018). Microbial diversity and community composition of caecal microbiota in commercial and indigenous Indian chickens determined using 16s rDNA amplicon sequencing. Microbiome 6:115. doi: 10.1186/s40168-018-0501-9
Patankar, J. V., and Becker, C. (2020). Cell death in the gut epithelium and implications for chronic inflammation. Nat. Rev. Gastroenterol. Hepatol. 17, 543–556. doi: 10.1038/s41575-020-0326-4
Raleigh, D. R., Boe, D. M., Yu, D., Weber, C. R., Marchiando, A. M., Bradford, E. M., et al. (2011). Occludin S408 phosphorylation regulates tight junction protein interactions and barrier function. J. Cell Biol. 193, 565–582. doi: 10.1083/jcb.201010065
Rao, K. R., and Fritz-Niggli, H. (1988). Alterations in the length of jejunal villi in mice irradiated with graded doses of X rays. Br. J. Radiol. 61, 839–842. doi: 10.1259/0007-1285-61-729-839
Sabatino, A., Regolisti, G., Cosola, C., Gesualdo, L., and Fiaccadori, E. (2017). Intestinal microbiota in type 2 diabetes and chronic kidney disease. Curr. Diab. Rep. 17:16. doi: 10.1007/s11892-017-0841-z
Schafer, S., Viswanathan, S., Widjaja, A. A., Lim, W.-W., Moreno-Moral, A., DeLaughter, D. M., et al. (2017). IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 552, 110–115. doi: 10.1038/nature24676
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Smith, K. S., Chanda, S. K., Lingbeek, M., Ross, D. T., Botstein, D., van Lohuizen, M., et al. (2003). Bmi-1 regulation of INK4A-ARF is a downstream requirement for transformation of hematopoietic progenitors by E2a-Pbx1. Mol. Cell 12, 393–400. doi: 10.1016/s1097-2765(03)00277-6
Smith, N. R., Swain, J. R., Davies, P. S., Gallagher, A. C., Parappilly, M. S., Beach, C. Z., et al. (2018). Monoclonal antibodies reveal dynamic plasticity between Lgr5- and Bmi1-expressing intestinal cell populations. Cell. Mol. Gastroenterol. Hepatol. 6, 79–96. doi: 10.1016/j.jcmgh.2018.02.007
Su, L., Nalle, S. C., Shen, L., Turner, E. S., Singh, G., Breskin, L. A., et al. (2013). TNFR2 activates MLCK-dependent tight junction dysregulation to cause apoptosis-mediated barrier loss and experimental colitis. Gastroenterology 145, 407–415. doi: 10.1053/j.gastro.2013.04.011
Su, L., Shen, L., Clayburgh, D. R., Nalle, S. C., Sullivan, E. A., Meddings, J. B., et al. (2009). Targeted epithelial tight junction dysfunction causes immune activation and contributes to development of experimental colitis. Gastroenterology 136, 551–563. doi: 10.1053/j.gastro.2008.10.081
Sun, H., Qiao, W., Cui, M., Yang, C., Wang, R., Goltzman, D., et al. (2020). The Polycomb protein Bmi1 plays a crucial role in the prevention of 1,25(OH)2 D deficiency-induced bone loss. J. Bone Miner. Res. 35, 583–595. doi: 10.1002/jbmr.3921
Thevaranjan, N., Puchta, A., Schulz, C., Naidoo, A., Szamosi, J. C., Verschoor, C. P., et al. (2017). Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe 21, 455–466.e4. doi: 10.1016/j.chom.2017.03.002
Tian, H., Biehs, B., Warming, S., Leong, K. G., Rangell, L., Klein, O. D., et al. (2011). A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature 478, 255–259. doi: 10.1038/nature10408
Virtue, A. T., McCright, S. J., Wright, J. M., Jimenez, M. T., Mowel, W. K., Kotzin, J. J., et al. (2019). The gut microbiota regulates white adipose tissue inflammation and obesity via a family of microRNAs. Sci. Transl. Med. 11:eaav1892. doi: 10.1126/scitranslmed.aav1892
Wang, J., Yao, Y., Liu, X., Wang, K., Zhou, Q., and Tang, Y. (2019). Protective effects of lycium barbarum polysaccharides on blood-retinal barrier via ROCK1 pathway in diabetic rats. Am. J. Transl. Res. 11, 6304–6315.
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Xie, C., Jin, J., Lv, X., Tao, J., Wang, R., and Miao, D. (2015). Anti-aging effect of transplanted amniotic membrane mesenchymal stem cells in a premature aging model of Bmi-1 deficiency. Sci. Rep. 5:13975. doi: 10.1038/srep13975
Ye, D., Guo, S., Al-Sadi, R., and Ma, T. Y. (2011). MicroRNA regulation of intestinal epithelial tight junction permeability. Gastroenterology 141, 1323–1333. doi: 10.1053/j.gastro.2011.07.005
Zhang, D., Jiang, M., and Miao, D. (2011). Transplanted human amniotic membrane-derived mesenchymal stem cells ameliorate carbon tetrachloride-induced liver cirrhosis in mouse. PLoS One 6:e16789. doi: 10.1371/journal.pone.0016789
Zhang, H. W., Ding, J., Jin, J. L., Guo, J., Liu, J. N., Karaplis, A., et al. (2010). Defects in mesenchymal stem cell self-renewal and cell fate determination lead to an osteopenic phenotype in Bmi-1 null mice. J. Bone Miner. Res. 25, 640–652. doi: 10.1359/jbmr.090812
Zhang, Q., Hu, J., Feng, J. W., Hu, X. T., Wang, T., Gong, W. X., et al. (2020). Influenza infection elicits an expansion of gut population of endogenous Bifidobacterium animalis which protects mice against infection. Genome Biol. 21:99. doi: 10.1186/s13059-020-02007-1
Keywords: inflammaging, TNF-α, Desulfovibrio, occludin, p16INK4a, Bmi-1
Citation: Zhou J, Hou C, Chen H, Qin Z, Miao Z, Zhao J, Wang Q, Cui M, Xie C, Wang R, Li Q, Zuo G, Miao D and Jin J (2021) P16INK4a Deletion Ameliorates Damage of Intestinal Epithelial Barrier and Microbial Dysbiosis in a Stress-Induced Premature Senescence Model of Bmi-1 Deficiency. Front. Cell Dev. Biol. 9:671564. doi: 10.3389/fcell.2021.671564
Received: 24 February 2021; Accepted: 31 August 2021;
Published: 07 October 2021.
Edited by:
Ignacio Palmero, Consejo Superior de Investigaciones Científicas (CSIC), SpainReviewed by:
Tangliang Li, Shandong University, ChinaCopyright © 2021 Zhou, Hou, Chen, Qin, Miao, Zhao, Wang, Cui, Xie, Wang, Li, Zuo, Miao and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianliang Jin, amluamlhbmxpYW5nQG5qbXUuZWR1LmNu
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.