 Ziye Rong
Ziye Rong Peipei Tu2
Peipei Tu2 Yan Sun
Yan Sun Yanan Yang
Yanan Yang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol. , 12 May 2021
Sec. Cell Death and Survival
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.669379
This article is part of the Research Topic The Role of Different Subcellular Organelles in DNA Damage Response View all 11 articles
Mitochondria are double membrane organelles in eukaryotic cells that provide energy by generating adenosine triphosphate (ATP) through oxidative phosphorylation. They are crucial to many aspects of cellular metabolism. Mitochondria contain their own DNA that encodes for essential proteins involved in the execution of normal mitochondrial functions. Compared with nuclear DNA, the mitochondrial DNA (mtDNA) is more prone to be affected by DNA damaging agents, and accumulated DNA damages may cause mitochondrial dysfunction and drive the pathogenesis of a variety of human diseases, including neurodegenerative disorders and cancer. Therefore, understanding better how mtDNA damages are repaired will facilitate developing therapeutic strategies. In this review, we focus on our current understanding of the mtDNA repair system. We also discuss other mitochondrial events promoted by excessive DNA damages and inefficient DNA repair, such as mitochondrial fusion, fission, and mitophagy, which serve as quality control events for clearing damaged mtDNA.
The mitochondria provide most of the cell energy in the form of adenosine triphosphate (ATP) through oxidative phosphorylation (OXPHOS), which is executed by the electron transport chain (ETC) within the mitochondria. In addition, mitochondria provide other key metabolic intermediates to many other basic cellular processes, such as lipid biogenesis and the synthesis of amino acids (Zong et al., 2016).
In the 1960s, nucleic acids and DNA were discovered in the mitochondria (Saki and Prakash, 2017). The mitochondrial DNA (mtDNA) is circular, and its length is varied in different species, from 75∼85 kb in Saccharomyces cerevisiae to around 16.5 kb in mammalian cells (Anderson et al., 1981; Foury et al., 1998). Although most of the mitochondrial proteins are transcribed in the nucleus, synthesized in the cytosol, and then transported into the mitochondria (Gray, 2012), the mtDNA is not non-sense. For example, in human cells, it encodes for 22 tRNAs and two ribosomal RNAs, as well as 13 polypeptides that comprise core subunits of the ETC Complex I, III, IV, and V, which are essential for the OXPHOS activity.
In mammalian cells, mtDNA is tightly linked with proteins to form complexes called nucleoids, which localize to the mitochondrial inner membrane (Chen and Butow, 2005; Holt et al., 2007). Some replication- and transcription-related proteins, such as mitochondrial transcription factor A (TFAM), DNA polymerase gamma (POLG), mitochondrial single-strand binding protein (mtSSB), and mtDNA helicase Twinkle, have been reported to interact with mtDNA (Garrido et al., 2003; Wang and Bogenhagen, 2006; Farge and Falkenberg, 2019).
Similar to nuclear DNA, the mtDNA is constantly assaulted by both exogenous and endogenous stresses, such as ultraviolet (UV) radiation and reactive oxygen species (ROS) (Alexeyev et al., 2013). However, evidence suggests that the mtDNA is more susceptible to certain stress-induced damages (e.g., oxidized DNA damages) than nuclear DNA due to its proximity to the sites of oxidative phosphorylation and lack of the protection by histones (Yakes and Van Houten, 1997; Druzhyna et al., 2008).
Excessive mtDNA damages, if not repaired efficiently, may increase ROS production, which in turn leads to mitochondrial dysfunction and provokes the pathogenesis of many human diseases, including Kearns-Sayre Syndrome, Alzheimer’s, Parkinson’s disease (PD), cancer, and diabetes (Wallace, 2005; Nakabeppu et al., 2007; Tsang et al., 2018; Llanos-Gonzalez et al., 2020). Therefore, accurate maintenance of the mtDNA is essential for a healthy life. In this review, we discuss how mitochondria respond to DNA damages, with an emphasis on how such damages are repaired. We also discuss a series of mitochondrial quality control events, including fission, fusion, and mitophagy, which are important for clearing damaged mtDNA that cannot be efficiently repaired.
Compared with nuclear DNA repair mechanisms, which have been extensively studied, the mtDNA repair mechanisms are much less understood. Unlike nuclear DNA, mtDNA is multi-copied (Yasukawa and Kang, 2018). Due to such nature, it was previously thought that DNA repair mechanisms might not be present or necessary in the mitochondria. For many years, this had led to a hypothesis that damaged mtDNA was simply degraded without being repaired, while the remained undamaged mtDNA served as the template for mtDNA synthesis (Druzhyna et al., 2008). This hypothesis was primarily based on the experiments showing that pyrimidine dimers induced by UV were not repaired in mammalian mitochondria (Clayton et al., 1974). Given that the nucleotide base repair (NER) is a highly efficient nuclear repair pathway for correcting a variety of DNA damages caused by UV radiation and many other environmental insults (Lee and Kang, 2019), these data suggest that the NER is absent for mtDNA damage repair.
However, more and more studies have shown that several other DNA repair pathways, including base excision repair (BER), direct reversal (DR), mismatch repair (MMR), and possibly double-strand break repair (DSBR), exist in the mammalian mitochondria (Kazak et al., 2012; Saki and Prakash, 2017; Figure 1). Key components of these pathways are encoded by nuclear genes and transported to the mitochondria after being synthesized in the cytosol.
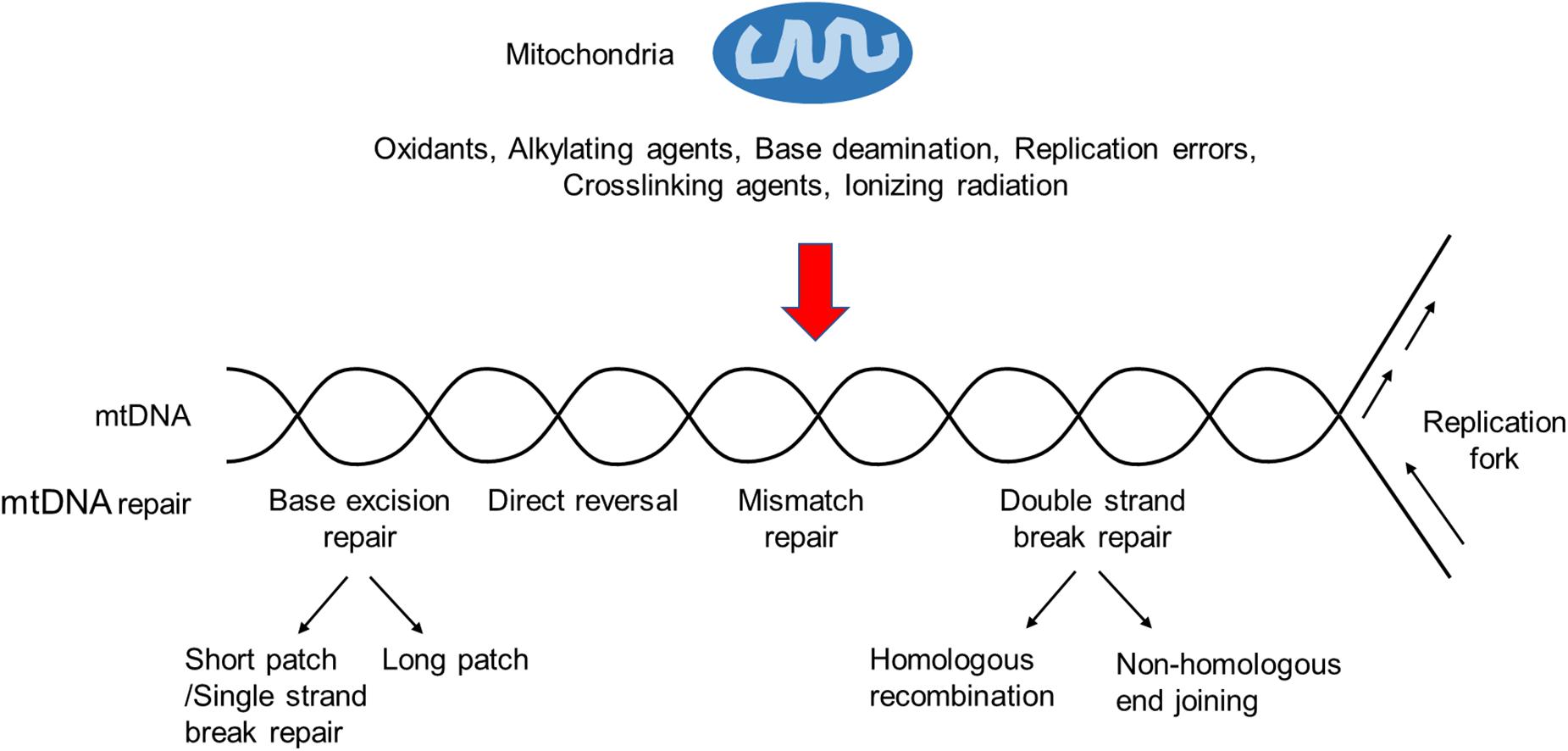
Figure 1. An overview of the mtDNA damage repair. Ionizing radiation, base deamination, replication errors, and DNA damaging agents, such as oxidants, alkylating agents, and crosslinking agents, can cause mtDNA damages. BER is the primary mtDNA damage repair pathway, whereas NER is not observed in mitochondria. In addition, other repair pathways, such as DR, MMR and DSBR, may exist in the mitochondria.
Base excision repair is highly conserved from bacteria to human and has been considered as the primary repair pathway in the mitochondria, responsible for the removal of non-bulky DNA lesions caused predominantly by oxidation, alkylation, deamination, and methylation (Kazak et al., 2012; Krokan and Bjoras, 2013). Its basic process includes damaged DNA base recognition and removal, leaving an apurinic/apyrimidinc (AP) site (or called an abasic site), and subsequently the cleavage of the resulted AP site, end processing, gap filling, and ligation to complete the BER (Fortini et al., 2003).
At the initiation step of the BER, DNA glycosylases play important roles in damaged DNA recognition and removal (Jacobs and Schar, 2012). Seven of eleven known DNA glycosylases have mitochondrial targeting signal and have been identified in mammalian mitochondria (Sharma et al., 2019). They are categorized into two groups, namely the mono-functional glycosylases and bi-functional DNA glycosylases (Mullins et al., 2019). Mono-functional glycosylases include alkyladenine DNA glycosylase (AAG) (van Loon and Samson, 2013), MutY glycosylase homolog (MUTYH) (Ohtsubo et al., 2000), and uracil N-glycosyalse (UNG) (Slupphaug et al., 1993). Bi-functional DNA glycosylases include 8-oxoguanine DNA glycosylase-1 (OGG1) (Nishioka et al., 1999), Nei-like 1 (NEIL1) and Nei-like 2 (NEIL2) (Han et al., 2019), and Nth-Like 1 (NTHL1) (Imai et al., 1998).
Mono-functional glycosylases lack lyase activity and cleave the N-glycosidic bonds to generate AP sites, which are further processed by apurinic/apyrimidinic endonuclease 1 (APE1). Bi-functional DNA glycosylases with intrinsic lyase activity are capable of cleaving the abasic sites, and the DNA strand on the 3′ end of the AP site (Kazak et al., 2012; Stein and Sia, 2017). During the end processing, APE1 processes the ends generated by OGG1 and NTHL1 glycosylases (Hegde et al., 2008). The polynucleotide kinase/phosphatase (PNKP) processes the ends generated by the NEIL glycosylases (Tahbaz et al., 2012).
During short-patch BER (SP-BER), 5′-deoxyribose phosphate moiety created by APEI is excised by polymerase γ (Pol γ) with its lyase activity (Longley et al., 1998). Next, Pol γ inserts a single nucleotide to fill the gap. In long patch BER (LP-BER), 2–6 nucleotides are incorporated at the nick by Pol γ through its strand displacement synthesis activity (Longley et al., 1998; Akbari et al., 2008). Then, the 5′ flap is possibly cleaved by flap endonuclease 1 (FEN1) (Liu et al., 2008). Previous reports have shown that other enzymes, including DNA replication helicase/nuclease 2 (DNA2) and 5′-exo/endonuclease (EXOG) are important for removing 5′ flaps during mitochondrial LP-BER (Duxin et al., 2009; Tann et al., 2011). In both SP-BER and LP-BER, DNA ligase III seals the nick to complete the final ligation step of the mitochondrial BER (Simsek et al., 2011).
The single strand break repair (SSBR) pathway is also likely to be present in the mitochondria (Hegde et al., 2012). SSBR can be considered as a form of BER, because many of the proteins involved in BER are important for SSBR. The two pathways have common gap filling and ligation steps. Previously, poly-ADP-ribose polymerase 1 (PARP1), which is required for recognizing single-strand breaks, has been reported to localize to the mitochondria (Rossi et al., 2009). However, the exact role for PARP1 in mammalian mtDNA SSBR is still unclear, and conflicting results have been reported (Szczesny et al., 2014).
During the end processing step, besides of APE1 and PNKP, tyrosyl DNA phosphodiesterase 1 (TDP1) and aprataxin (APTX) have been found to be involved in the mitochondrial SSBR pathway (Hegde et al., 2012; Meagher and Lightowlers, 2014). In the mitochondria, TDP1 typically hydrolyzes the phosphodiester bond between a tyrosyl moiety and a 3′-DNA end. It can also hydrolyze other 3′-end DNA alterations including 3′-phosphoglycolate and 3′-abasic sites. Therefore, TDP1 may function as a general 3′-end-processing DNA repair enzyme to remove a variety of adducts that are poor substrates for APE1 (Das et al., 2010). APTX belongs to the histidine triad (HIT) superfamily of nucleotide hydrolases and transferasese (Kijas et al., 2006). It removes 5′-adenylate (5′-AMP) group from DNA (Zheng et al., 2019). 5′-AMP group is an abortive DNA ligation product during DNA repair and replication, and it must be removed for the later DNA ligation. Previously, a slow rate of 5′-AMP removal was reported in the mitochondrial extracts compared with nuclear extracts (Akbari et al., 2015). Biochemical and cell biological analysis indicated it due to the lack of back up enzymes or repair mechanisms in mtDNA (Caglayan et al., 2017). All these results suggest that APTX is more critical in mtDNA repair than in the nuclear DNA repair.
Notably, mtDNA modifications are also involved in BER. For example, an enrichment of N6-methyldeoxyadenosine (m6A) in mammalian mtDNA has been recently reported (Hao et al., 2020). M6A could repress DNA binding and bending by TFAM, which possesses a greater affinity for oxidized lesions and inhibits the activity of OGG1, UNG, and APE1 (Canugovi et al., 2010). Increased m6A in the mtDNA affect the binding between TFAM and DNA so that the BER glycosylases are stimulated.
Direct reversal is responsible for the repair of DNA O-alkylated and N-alkylated damages caused by DNA alkylating agents (Fu et al., 2012). This repair process does not require excision, synthesis, and ligation. In mammalian cells, DR is performed by O6-methylguanine DNA methyltransferase (MGMT) or ALKBH (AlkB homolog) proteins in the nucleus (Ragg et al., 2000; Ahmad et al., 2015). MGMT repairs the O6-methylguanine lesions by removing methyl groups, whereas ALKBH repairs certain N-alkyl lesions via dealkylation. According to previous studies, mitochondrial localization of ALKBH has not been confirmed, and a protein with similar molecular weight to MGMT may localize to mammalian mitochondria (Myers et al., 1988). However, different groups have reported conflicting results, and whether DR is present in the mammalian mitochondria remain to be further clarified (Cai et al., 2005).
In 2003, MMR activity in rat liver mitochondrial extracts was reported (Mason et al., 2003). Notably, unlike nuclear MMR, there is no evidence that such activity relies on any well-known nuclear MMR proteins, such as MSH and MLH. Instead, it may depend on the Y-box binding protein YB-1 (de Souza-Pinto et al., 2009; Lyabin et al., 2014). This is supported by the facts that depleting YB-1 leads to both a decrease of MMR activity in the mitochondrial extracts and an increase of mtDNA mutations in YB-1-depleted cells. While these findings strongly suggest the presence of YB-1-dependent MMR in the mitochondria, more investigations are needed for better characterizing its detailed mechanism, especially the mitochondrial YB-1-associated cofactor(s) involved in this process.
Double-strand breaks (DSBs) are the most cytotoxic DNA lesions if not efficiently repaired (Stein and Sia, 2017). DSBs can be induced by ROS, replication stalling, or radiation. In the nucleus, DSBs are mainly repaired by two pathways, including homologous recombination (HR) and non-homologous end joining (NHEJ) pathways (Pannunzio et al., 2018; Wright et al., 2018). HR is considered as an “error-free” repair pathway due to its use of homologous partner as the template to repair DNA lesions. Several lines of evidence support the presence of HR in the mitochondria. For instance, the recombination intermediates containing four-way junctions can be detected in human mitochondria (Pohjoismaki et al., 2009), and Rad51, a RecA homolog, has been reported to execute HR in mammalian mitochondria (Sage et al., 2010). In addition, Rad51 related proteins, e.g., XRCC3, also exist in human mitochondria and may participate in HR (Mishra et al., 2018).
Compared with HR, NHEJ is more error-prone and results in large deletions. DNA end-joining activity has been observed in mitochondrial extracts prepared from mammalian cells, where both cohesive and blunt-ended DNA molecules can be repaired (Lakshmipathy and Campbell, 1999). Breast cancer type 1 susceptibility protein (BRCA1) and p53-binding protein 1 (53BP1) oppositely affect the extent of DNA end resection and are key determinants for the choice of HR or NHEJ (Panier and Boulton, 2014). During G2/S phase, BRCA1 promotes DNA end resection and thus induces HR. During G1 phase, 53BP1 protects the broken DNA end and inhibits long-range end resection, thereby promoting NHEJ. Both BRCA1 and 53BP1 have been identified in the mammalian mitochondria (Coene et al., 2005; Youn et al., 2017).
Taken together, previous findings suggest that both DSB and its related proteins are present in the mitochondria (Coffey and Campbell, 2000; Coene et al., 2005; Sage et al., 2010). However, whether DSB plays a major role in the mtDNA damage repair and whether the mitochondrial HR and NHEJ exert pathophysiological functions in disease contexts remain to be further elucidated.
To maintain a healthy mitochondrial network, mammalian cells have developed several mechanisms to respond to unrepaired DNA damages, including trans-lesion synthesis, mitochondrial fusion and fission, and mitophagy. The interactions between some of them have also been reported (Figure 2).
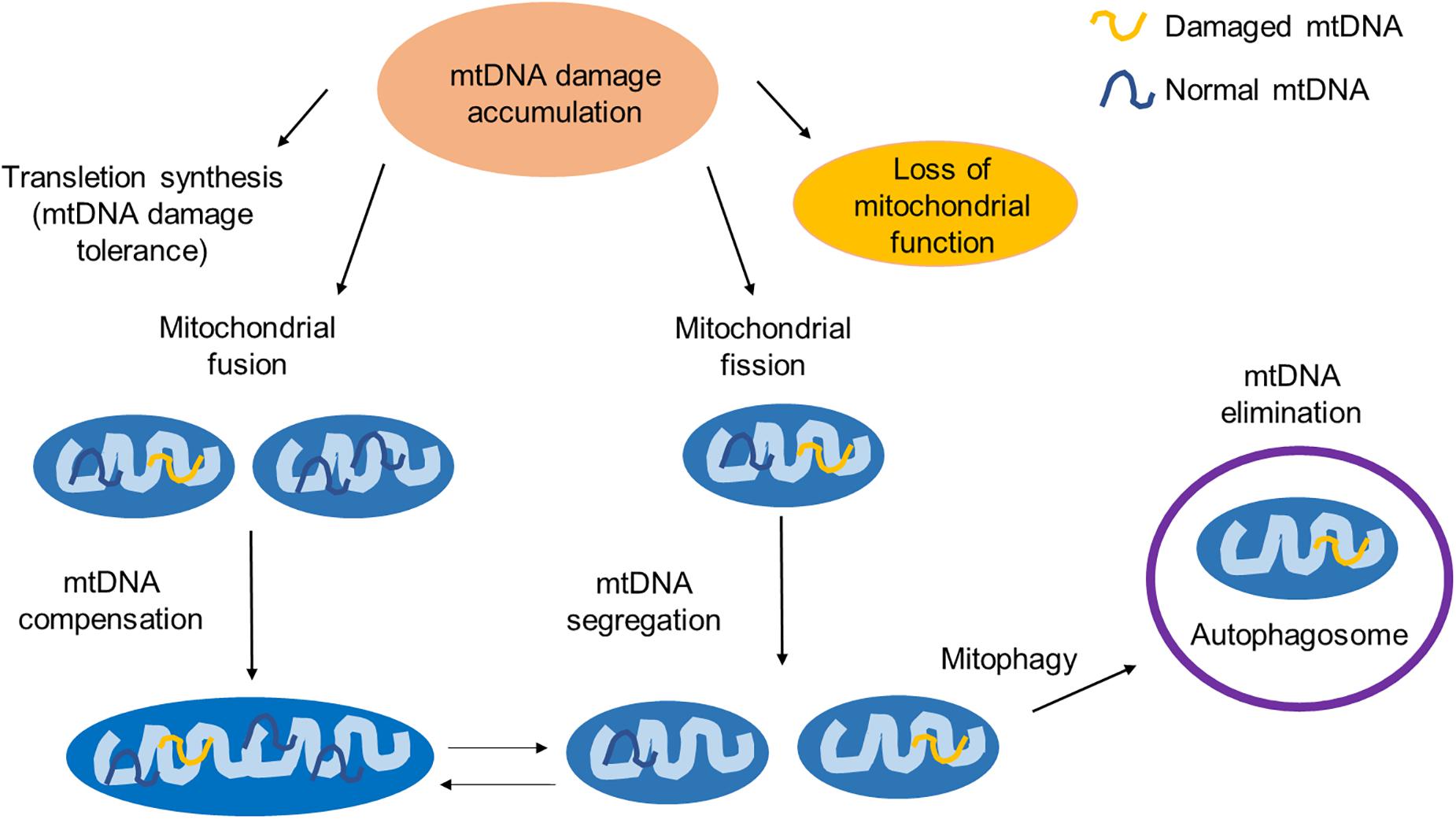
Figure 2. The mitochondrial response to unrepaired DNA damages. Unrepaired mtDNA damages can be accumulated and lead to the mitochondrial dysfunction. To maintain its function, the mitochondria have developed several quality control events. TLS is an mtDNA damage tolerance pathway to bypass the mtDNA replication block. Mitochondrial fusion provides mtDNA complementation by mixing damaged and normal mtDNA. Mitochondrial fission segregates the part of a mitochondrion with damaged mtDNA and maintains the integrity of the healthy part of a mitochondrion. Mitochondria with excessive damaged mtDNA are selectively degraded through mitophagy. The interaction between the mitophagy and the mitochondrial fission/fusion has also been reported.
Trans-lesion synthesis (TLS) can bypass the DNA replication block caused by DNA damages (Waters et al., 2009; Sale, 2013). It is an error-prone process, in which the polymerases with low fidelity are involved (Ma X. et al., 2020). In TLS, nucleotides are incorporated to repair DNA lesions but convert them to DNA mutations. It requires DNA polymerase selection and switching (Friedberg et al., 2005). A group of specialized DNA polymerases termed TLS polymerases are utilized in this process, which are categorized into several families (Sale, 2013; Ma X. et al., 2020). Rev1, Polκ, Polη, and Polι belong to the best characterized Y-family polymerases, while polymerase zeta (Polζ) is a B-family polymerase (Yang and Gao, 2018). In mammalian mitochondria, there are evidence that REV3, the catalytic subunit of Polζ localizes to the mitochondria (Singh et al., 2015). REV3 associates with POLG and prevents the mtDNA damage. PrimPol is another TLS polymerase that has been detected in the mammalian mitochondria (Garcia-Gomez et al., 2013; Rudd et al., 2014). This enzyme has both DNA primase activity and DNA polymerase activity. It is capable of bypassing DNA lesions such as 8-oxoG (Guilliam et al., 2015).
If mtDNA damages cannot be repaired, they will accumulate and subsequently evoke mtDNA quality control events, including mitochondrial fusion, fission, and mitophagy (Youle and van der Bliek, 2012).
Mitochondrial fusion and fission are essential for cell metabolic activities as well as re-distribution of mtDNA (Tilokani et al., 2018). Both fusion and fission are mediated by GTPases (Hoppins et al., 2007). Specifically, Mitofusin 1 (Mfn1), Mitofusin 2 (Mfn2), and Optic Atrophy 1 (OPA1) are involved in the regulation of mitochondrial fusion (Delettre et al., 2000; Chen et al., 2003; Olichon et al., 2003; Detmer and Chan, 2007), and Drp1 is involved in mitochondrial fission (Fonseca et al., 2019).
Damaged and undamaged mtDNAs yield a heteroplasmic mixture of normal and mutant mitochondrial genomes within the same cell (Wonnapinij et al., 2008; Aryaman et al., 2018). Mitochondrial fusion allows the mitochondria with normal mtDNA to compensate for defects in the mitochondria with damaged mtDNA (Nakada et al., 2001; Ono et al., 2001; Yang et al., 2015). Through fusion, essential functional and structural components (e.g., proteins and lipids) in the two types of mitochondria can be diffused and shared, thereby rescuing the mitochondria with damaged mtDNA and mitigating the effects of environmental stresses on the mitochondria if the stress level is below a critical threshold (Stewart and Chinnery, 2015).
Mitochondrial fission allows mtDNAs to be divided into two mitochondria and provides a chance for a mitochondrion to re-fuse with another mitochondrion (Tilokani et al., 2018). It is suggested that fission divides a mitochondrion into two parts and distributes the damaged mtDNA and other harmful components asymmetrically to eliminate the part with most seriously damaged mtDNA but preserve the healthier part with normal mtDNA (Meyer et al., 2017).
Homoplasmy in mtDNA is rarely observed because of inherited or sporadic mutations that result in heteroplasmy (Wei et al., 2020). A broad range of heteroplasmy among different cell types and tissues are observed. The dynamics of heteroplasmy is one of the most challenging aspects of mtDNA disease. The mechanisms regulating heteroplasmy shifts are still needed to be elucidated. In T cell lineage, purifying selection occurs and heteroplasmy ratio is dramatically reduced (Walker et al., 2020). This process may enrich for certain mtDNA sequences and eliminate the pathogenic mutant mtDNA.
Irreparably damaged mtDNA may trigger mitophagy, a process through which depolarized or damaged mitochondria are selectively degraded by autophagy (Lemasters, 2005; Pickles et al., 2018). During autophagy, unwanted cellular materials are sequestered by double-membrane autophagosomes and delivered to lysosomes for degradation (Wollert, 2019). In normal cells, autophagy is utilized to clear and recycle almost all types of cellular materials, including proteins, lipids, damaged organelles, and etc. Mitophagy is an important mitochondrial quality control mechanism to maintain a healthy mitochondrial network (Ma K. et al., 2020). In mammalian cells, two pathways of mitophagy have been described. The first is an ubiquitin-mediated pathway, in which PINK1 (PTEN-induced putative protein kinase 1) has been shown to play an important role by recruiting the E3 ubiquitin ligase Parkin from the cytosol to the mitochondria with damaged DNA (Narendra et al., 2008; Matsuda et al., 2010; Pickrell and Youle, 2015). The other pathway is a receptor-mediated pathway, in which mitophagy receptors on the mitochondria with at least one LC3-interacting region have been identified. Some examples are BCL2 interacting protein 3 (BNIP3) (Chourasia and Macleod, 2015), FUN14 domain containing 1 (FUNDC1) (Chen M. et al., 2016), and mitochondria inner membrane protein Prohibitin 2 (PHB2) (Wei et al., 2017). These receptors mediate mitophagy by binding with LC3 to recruit the autophagosome (Yoo and Jung, 2018).
Previous reports also suggest that mitophagy may be linked to mitochondrial fission and fusion (Shirihai et al., 2015; Yoo and Jung, 2018). For example, overexpression of Opa1 or dominant-negative mutant of Drp1 can inhibit mitophagy (Twig et al., 2008). An interaction between the PINK1/Parkin pathway and the fission/fusion machinery has also been reported (Yu et al., 2011; Murata et al., 2020). It is proposed that fission is prerequisite for mitophagy by segregating the damaged part of a mitochondrion and targeting it for autophagic degradation. It is suspected that the damaged mtDNA and unwanted debris should distribute unevenly in the mitochondria to achieve such asymmetric fission (Zorov et al., 2017). However, the underlying mechanism remains largely unknown.
As above-discussed, the UV-radiation induced mtDNA damages cannot be repaired by mtDNA repair mechanisms. However, the mitochondria with photo-damaged mtDNA undergoes mitophagy (Kim and Lemasters, 2011). It was confirmed that mitophagy can be induced by different types of mtDNA damage stimuli, and it at least partially depends on DNA damage response pathways (Furda et al., 2012). It should be noted that the modulation of mitophagy after DNA damage is independent of the type of mtDNA damage stimuli. Recently, it has been suggested that Spata18, a p53 inducible protein, is critical in mitophagy after mtDNA damage (Dan et al., 2020). Knocking down Spata18 suppresses mitophagy, promotes mtDNA damage, and attenuates mtDNA repair. To better understand the mitophagy process in response to mtDNA damage, it is important to identify related proteins and figure out how a mitochondrion, or its part, with damaged mtDNA can be selectively removed.
The interplay of mitophagy with mitochondrial dynamics is critical for maintaining mitochondrial homeostasis in normal and stressed conditions. Damaged mitochondria may either induce innate immunity or trigger cell death to drive aging-related diseases when mtDNA damages are beyond repairable (Ma K. et al., 2020).
Previous reports suggest that the mtDNA is implicated in the regulation of inflammation and innate immunity, and a major mediator for these processes is the “cGAS-STING” pathway (West et al., 2015; Chen Q. et al., 2016). For instance, internalized bacterial endotoxin lipopolysaccharide (LPS) can trjgger the mitochondrial membrane localization of active GSDMD, which in turn stimulates the release of mtDNA into the cytosol (Huang et al., 2020). The released mtDNA can be detected by the cyclic AMP-GMP synthase (cGAS), which generates cyclic GMP-AMP (cGAMP) to activate the pro-inflammatory stimulator of interferon genes (STING), eventually leading to increased production of type 1 interferons (IFNs).
In cancer cells, the mitochondrial outer membrane permeabilization (MOMP) has been shown to promote the cytosolic release of mtDNA, the activation of the cGAS-STING pathway, and the production of type I IFNs, which collectively facilitate an effective radiation therapy (Yamazaki et al., 2020); whereas Bcl2-dependent mitophagy can deplete MOMP, thereby limiting the release of mtDNA and associated immune responses.
Age-related accumulation of mtDNA mutations in human somatic and germ cells have also been reported (Arbeithuber et al., 2020). Replication errors are likely the main sources of de novo mtDNA mutations. Accumulation of certain types of mtDNA mutations may trigger cell death and lead to age-related diseases, such as PD, Cockayne syndrome (CS), and Werner syndrome (WS) (Palikaras et al., 2015; Fang et al., 2019; Lopes et al., 2020).
Parkinson’s disease is a progressive neurodegenerative disorder with movement problems due to the loss of dopamine-producing neurons (Kalia and Lang, 2015). Interestingly, most of PD-related proteins have been shown to lead to dysfunctional mitochondria and/or accumulated damaged mitochondria (Martin-Jimenez et al., 2020). Some of them, including PINK1 and Parkin and LRRK2, play important roles in the execution of mitophagy (Vives-Bauza et al., 2010; Wallings et al., 2015). These facts not only establish a strong link between mitophagy and the pathogenesis of PD, but also suggest that manipulating mitophagy may have diagnostic/therapeutic values.
Werner syndrome is a human premature aging disease caused by mutations in the gene encoding for Werner protein (WRN), which is an important DNA helicase involved in DNA repair (Fang et al., 2019). WRN regulates the transcription of nicotinamide nucleotide adenylyltransferase 1 and NAD+ biosynthesis. Accordingly, decreased NAD+ levels and impaired mitophagy have been reported in WS patient samples.
In CS patient samples, there is a reduction in the activation of AMP-activated kinase (AMPK) and its downstream target, autophagy protein unc-51 like autophagy activating kinase 1 (ULK1), suggesting dysfunctional mitochondrial dynamics and impaired mitophagy (Okur et al., 2020). Reduced NAD+ metabolism level was discovered in all three diseases. NAD+ precursors can be used for treatment by promoting mitophagy via increasing the levels of key mitophagy-related proteins, such as PINK1, ULK1, and DCT-1 (Aman et al., 2020). Mitochondrial quality is improved and mitochondrial defects are reversed. These results further implicate that promoting mitophagy can be a promising approach to treat some age-related diseases.
We have known that cells are continuously exposed to both internal and external stresses, which may cause mtDNA damages. In response to such damages, the mitochondria have developed multiple ways to maintain its DNA integrity and function. Nevertheless, we still lack a comprehensive understanding of how distinct pathways/mechanisms act together to maintain a functional mitochondrial genome.
In this review, we have mainly focused on the mitochondrial response that repairs, attenuates, or eliminate mtDNA damages. Compared with nuclear DNA repair pathways, the pathways for repairing mtDNA damages are less understood. Many nuclear DNA repair proteins, especially those found in the nuclear BER pathway, are also found in the mitochondria (Supplementary Table 1). Previous data suggest that BER is the major mtDNA repair pathway, whereas NER has not been confirmed in the mitochondria. DR, MMR and DSBR may exist in the mitochondria. However, key components involved in these pathways have not been fully elucidated. To that end, further efforts are needed to identify the proteins/enzymes participating in distinct mtDNA damage repair processes.
Mitochondria have multiple quality control pathways to maintain its function in response to stresses. Herein, we have briefly discussed several such pathways, including TLS, mitochondrial fission, fusion, and mitophagy. TLS can also be considered as a post-replication repair pathway. It utilizes damaged mtDNA as template to replicate with low-fidelity polymerases. Mitochondrial fusion and fission are dynamic to balance mtDNA damage compensation and elimination, and mitophagy is primarily responsible for the elimination of mitochondria with damaged mtDNA. Mis-regulation of such mtDNA damage responses has been linked to the immunity and pathogenesis of human diseases, including neurodegenerative disorders and cancer. In-depth understanding of these processes will provide insights for developing novel treatment strategies.
ZR and YY wrote the manuscript. All authors discussed and approved the contents.
This work was partly supported by the National Natural Science Foundation of China (NSFC) Project 82073257 (to YY) and University Natural Science Research Project of Anhui Province (China) (KJ2020A0159) (to PT).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.669379/full#supplementary-material
Supplementary Table 1 | Enzymes/proteins in mtDNA repair. Important enzymes/proteins in mammalian mitochondria in different mtDNA damage repair mechanisms are present. Functions of each enzyme/protein are also listed.
Ahmad, A., Nay, S. L., and O’Connor, T. R. (2015). Direct reversal repair in mammalian cells. Adv. DNA Repair 95–128. doi: 10.5772/60037
Akbari, M., Sykora, P., and Bohr, V. A. (2015). Slow mitochondrial repair of 5’-AMP renders mtDNA susceptible to damage in APTX deficient cells. Sci. Rep. 5:12876. doi: 10.1038/srep12876
Akbari, M., Visnes, T., Krokan, H. E., and Otterlei, M. (2008). Mitochondrial base excision repair of uracil and AP sites takes place by single-nucleotide insertion and long-patch DNA synthesis. DNA Repair (Amst.) 7, 605–616. doi: 10.1016/j.dnarep.2008.01.002
Alexeyev, M., Shokolenko, I., Wilson, G., and LeDoux, S. (2013). The maintenance of mitochondrial DNA integrity–critical analysis and update. Cold Spring Harb. Perspect. Biol. 5:a012641. doi: 10.1101/cshperspect.a012641
Aman, Y., Frank, J., Lautrup, S. H., Matysek, A., Niu, Z., Yang, G., et al. (2020). The NAD(+)-mitophagy axis in healthy longevity and in artificial intelligence-based clinical applications. Mech. Ageing Dev. 185:111194. doi: 10.1016/j.mad.2019.111194
Anderson, S., Bankier, A. T., Barrell, B. G., de Bruijn, M. H., Coulson, A. R., Drouin, J., et al. (1981). Sequence and organization of the human mitochondrial genome. Nature 290, 457–465. doi: 10.1038/290457a0
Arbeithuber, B., Hester, J., Cremona, M. A., Stoler, N., Zaidi, A., Higgins, B., et al. (2020). Age-related accumulation of de novo mitochondrial mutations in mammalian oocytes and somatic tissues. PLoS Biol. 18:e3000745. doi: 10.1371/journal.pbio.3000745
Aryaman, J., Johnston, I. G., and Jones, N. S. (2018). Mitochondrial heterogeneity. Front. Genet. 9:718. doi: 10.3389/fgene.2018.00718
Caglayan, M., Prasad, R., Krasich, R., Longley, M. J., Kadoda, K., Tsuda, M., et al. (2017). Complementation of aprataxin deficiency by base excision repair enzymes in mitochondrial extracts. Nucleic Acids Res. 45, 10079–10088. doi: 10.1093/nar/gkx654
Cai, S., Xu, Y., Cooper, R. J., Ferkowicz, M. J., Hartwell, J. R., Pollok, K. E., et al. (2005). Mitochondrial targeting of human O6-methylguanine DNA methyltransferase protects against cell killing by chemotherapeutic alkylating agents. Cancer Res. 65, 3319–3327. doi: 10.1158/0008-5472.CAN-04-3335
Canugovi, C., Maynard, S., Bayne, A. C., Sykora, P., Tian, J., de Souza-Pinto, N. C., et al. (2010). The mitochondrial transcription factor A functions in mitochondrial base excision repair. DNA Repair (Amst.) 9, 1080–1089. doi: 10.1016/j.dnarep.2010.07.009
Chen, H., Detmer, S. A., Ewald, A. J., Griffin, E. E., Fraser, S. E., and Chan, D. C. (2003). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 160, 189–200. doi: 10.1083/jcb.200211046
Chen, M., Chen, Z., Wang, Y., Tan, Z., Zhu, C., Li, Y., et al. (2016). Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 12, 689–702. doi: 10.1080/15548627.2016.1151580
Chen, Q., Sun, L., and Chen, Z. J. (2016). Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 17, 1142–1149. doi: 10.1038/ni.3558
Chen, X. J., and Butow, R. A. (2005). The organization and inheritance of the mitochondrial genome. Nat. Rev. Genet. 6, 815–825. doi: 10.1038/nrg1708
Chourasia, A. H., and Macleod, K. F. (2015). Tumor suppressor functions of BNIP3 and mitophagy. Autophagy 11, 1937–1938. doi: 10.1080/15548627.2015.1085136
Clayton, D. A., Doda, J. N., and Friedberg, E. C. (1974). The absence of a pyrimidine dimer repair mechanism in mammalian mitochondria. Proc. Natl. Acad. Sci. U. S. A. 71, 2777–2781. doi: 10.1073/pnas.71.7.2777
Coene, E. D., Hollinshead, M. S., Waeytens, A. A., Schelfhout, V. R., Eechaute, W. P., Shaw, M. K., et al. (2005). Phosphorylated BRCA1 is predominantly located in the nucleus and mitochondria. Mol. Biol. Cell 16, 997–1010. doi: 10.1091/mbc.e04-10-0895
Coffey, G., and Campbell, C. (2000). An alternate form of Ku80 is required for DNA end-binding activity in mammalian mitochondria. Nucleic Acids Res. 28, 3793–3800. doi: 10.1093/nar/28.19.3793
Dan, X., Babbar, M., Moore, A., Wechter, N., Tian, J., Mohanty, J. G., et al. (2020). DNA damage invokes mitophagy through a pathway involving Spata18. Nucleic Acids Res. 48, 6611–6623. doi: 10.1093/nar/gkaa393
Das, B. B., Dexheimer, T. S., Maddali, K., and Pommier, Y. (2010). Role of tyrosyl-DNA phosphodiesterase (TDP1) in mitochondria. Proc. Natl. Acad. Sci. U. S. A. 107, 19790–19795. doi: 10.1073/pnas.1009814107
de Souza-Pinto, N. C., Mason, P. A., Hashiguchi, K., Weissman, L., Tian, J., Guay, D., et al. (2009). Novel DNA mismatch-repair activity involving YB-1 in human mitochondria. DNA Repair (Amst.) 8, 704–719. doi: 10.1016/j.dnarep.2009.01.021
Delettre, C., Lenaers, G., Griffoin, J. M., Gigarel, N., Lorenzo, C., Belenguer, P., et al. (2000). Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 26, 207–210. doi: 10.1038/79936
Detmer, S. A., and Chan, D. C. (2007). Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J. Cell Biol. 176, 405–414. doi: 10.1083/jcb.200611080
Druzhyna, N. M., Wilson, G. L., and LeDoux, S. P. (2008). Mitochondrial DNA repair in aging and disease. Mech. Ageing Dev. 129, 383–390. doi: 10.1016/j.mad.2008.03.002
Duxin, J. P., Dao, B., Martinsson, P., Rajala, N., Guittat, L., Campbell, J. L., et al. (2009). Human Dna2 is a nuclear and mitochondrial DNA maintenance protein. Mol. Cell Biol. 29, 4274–4282. doi: 10.1128/MCB.01834-08
Fang, E. F., Hou, Y., Lautrup, S., Jensen, M. B., Yang, B., SenGupta, T., et al. (2019). NAD(+) augmentation restores mitophagy and limits accelerated aging in Werner syndrome. Nat. Commun. 10:5284. doi: 10.1038/s41467-019-13172-8
Farge, G., and Falkenberg, M. (2019). Organization of DNA in mammalian mitochondria. Int. J. Mol. Sci. 20:2770. doi: 10.3390/ijms20112770
Fonseca, T. B., Sanchez-Guerrero, A., Milosevic, I., and Raimundo, N. (2019). Mitochondrial fission requires DRP1 but not dynamins. Nature 570, E34–E42. doi: 10.1038/s41586-019-1296-y
Fortini, P., Pascucci, B., Parlanti, E., D’Errico, M., Simonelli, V., and Dogliotti, E. (2003). The base excision repair: mechanisms and its relevance for cancer susceptibility. Biochimie 85, 1053–1071. doi: 10.1016/j.biochi.2003.11.003
Foury, F., Roganti, T., Lecrenier, N., and Purnelle, B. (1998). The complete sequence of the mitochondrial genome of Saccharomyces cerevisiae. FEBS Lett. 440, 325–331. doi: 10.1016/s0014-5793(98)01467-7
Friedberg, E. C., Lehmann, A. R., and Fuchs, R. P. (2005). Trading places: how do DNA polymerases switch during translesion DNA synthesis? Mol. Cell 18, 499–505. doi: 10.1016/j.molcel.2005.03.032
Fu, D., Calvo, J. A., and Samson, L. D. (2012). Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 12, 104–120. doi: 10.1038/nrc3185
Furda, A. M., Marrangoni, A. M., Lokshin, A., and Van Houten, B. (2012). Oxidants and not alkylating agents induce rapid mtDNA loss and mitochondrial dysfunction. DNA Repair (Amst.) 11, 684–692. doi: 10.1016/j.dnarep.2012.06.002
Garcia-Gomez, S., Reyes, A., Martinez-Jimenez, M. I., Chocron, E. S., Mouron, S., Terrados, G., et al. (2013). PrimPol, an archaic primase/polymerase operating in human cells. Mol. Cell 52, 541–553. doi: 10.1016/j.molcel.2013.09.025
Garrido, N., Griparic, L., Jokitalo, E., Wartiovaara, J., van der Bliek, A. M., and Spelbrink, J. N. (2003). Composition and dynamics of human mitochondrial nucleoids. Mol. Biol. Cell 14, 1583–1596. doi: 10.1091/mbc.e02-07-0399
Gray, M. W. (2012). Mitochondrial evolution. Cold Spring Harb. Perspect. Biol. 4:a011403. doi: 10.1101/cshperspect.a011403
Guilliam, T. A., Jozwiakowski, S. K., Ehlinger, A., Barnes, R. P., Rudd, S. G., Bailey, L. J., et al. (2015). Human PrimPol is a highly error-prone polymerase regulated by single-stranded DNA binding proteins. Nucleic Acids Res. 43, 1056–1068. doi: 10.1093/nar/gku1321
Han, D., Schomacher, L., Schule, K. M., Mallick, M., Musheev, M. U., Karaulanov, E., et al. (2019). NEIL1 and NEIL2 DNA glycosylases protect neural crest development against mitochondrial oxidative stress. Elife 8:e49044. doi: 10.7554/eLife.49044
Hao, Z., Wu, T., Cui, X., Zhu, P., Tan, C., Dou, X., et al. (2020). N(6)-deoxyadenosine methylation in mammalian mitochondrial DNA. Mol. Cell 78, 382–395.e8. doi: 10.1016/j.molcel.2020.02.018
Hegde, M. L., Hazra, T. K., and Mitra, S. (2008). Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 18, 27–47. doi: 10.1038/cr.2008.8
Hegde, M. L., Mantha, A. K., Hazra, T. K., Bhakat, K. K., Mitra, S., and Szczesny, B. (2012). Oxidative genome damage and its repair: implications in aging and neurodegenerative diseases. Mech. Ageing Dev. 133, 157–168. doi: 10.1016/j.mad.2012.01.005
Holt, I. J., He, J., Mao, C. C., Boyd-Kirkup, J. D., Martinsson, P., Sembongi, H., et al. (2007). Mammalian mitochondrial nucleoids: organizing an independently minded genome. Mitochondrion 7, 311–321. doi: 10.1016/j.mito.2007.06.004
Hoppins, S., Lackner, L., and Nunnari, J. (2007). The machines that divide and fuse mitochondria. Annu. Rev. Biochem. 76, 751–780. doi: 10.1146/annurev.biochem.76.071905.090048
Huang, L. S., Hong, Z., Wu, W., Xiong, S., Zhong, M., Gao, X., et al. (2020). mtDNA activates cGAS signaling and suppresses the YAP-mediated endothelial cell proliferation program to promote inflammatory injury. Immunity 52, 475–486.e5. doi: 10.1016/j.immuni.2020.02.002
Imai, K., Sarker, A. H., Akiyama, K., Ikeda, S., Yao, M., Tsutsui, K., et al. (1998). Genomic structure and sequence of a human homologue (NTHL1/NTH1) of Escherichia coli endonuclease III with those of the adjacent parts of TSC2 and SLC9A3R2 genes. Gene 222, 287–295. doi: 10.1016/s0378-1119(98)00485-5
Jacobs, A. L., and Schar, P. (2012). DNA glycosylases: in DNA repair and beyond. Chromosoma 121, 1–20. doi: 10.1007/s00412-011-0347-4
Kalia, L. V., and Lang, A. E. (2015). Parkinson’s disease. Lancet 386, 896–912. doi: 10.1016/S0140-6736(14)61393-3
Kazak, L., Reyes, A., and Holt, I. J. (2012). Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nat. Rev. Mol. Cell Biol. 13, 659–671. doi: 10.1038/nrm3439
Kijas, A. W., Harris, J. L., Harris, J. M., and Lavin, M. F. (2006). Aprataxin forms a discrete branch in the HIT (histidine triad) superfamily of proteins with both DNA/RNA binding and nucleotide hydrolase activities. J. Biol. Chem. 281, 13939–13948. doi: 10.1074/jbc.M507946200
Kim, I., and Lemasters, J. J. (2011). Mitophagy selectively degrades individual damaged mitochondria after photoirradiation. Antioxid. Redox Signal. 14, 1919–1928. doi: 10.1089/ars.2010.3768
Krokan, H. E., and Bjoras, M. (2013). Base excision repair. Cold Spring Harb. Perspect. Biol. 5:a012583. doi: 10.1101/cshperspect.a012583
Lakshmipathy, U., and Campbell, C. (1999). Double strand break rejoining by mammalian mitochondrial extracts. Nucleic Acids Res. 27, 1198–1204. doi: 10.1093/nar/27.4.1198
Lee, T. H., and Kang, T. H. (2019). DNA oxidation and excision repair pathways. Int. J. Mol. Sci. 20:6092. doi: 10.3390/ijms20236092
Lemasters, J. J. (2005). Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 8, 3–5. doi: 10.1089/rej.2005.8.3
Liu, P., Qian, L., Sung, J. S., de Souza-Pinto, N. C., Zheng, L., Bogenhagen, D. F., et al. (2008). Removal of oxidative DNA damage via FEN1-dependent long-patch base excision repair in human cell mitochondria. Mol. Cell Biol. 28, 4975–4987. doi: 10.1128/MCB.00457-08
Llanos-Gonzalez, E., Henares-Chavarino, A. A., Pedrero-Prieto, C. M., Garcia-Carpintero, S., Frontinan-Rubio, J., Sancho-Bielsa, F. J., et al. (2020). Interplay between mitochondrial oxidative disorders and proteostasis in Alzheimer’s disease. Front. Neurosci. 13:ARTN1444. doi: 10.3389/fnins.2019.01444
Longley, M. J., Prasad, R., Srivastava, D. K., Wilson, S. H., and Copeland, W. C. (1998). Identification of 5’-deoxyribose phosphate lyase activity in human DNA polymerase gamma and its role in mitochondrial base excision repair in vitro. Proc. Natl. Acad. Sci. U. S. A. 95, 12244–12248. doi: 10.1073/pnas.95.21.12244
Lopes, A. F. C., Bozek, K., Herholz, M., Trifunovic, A., Rieckher, M., and Schumacher, B. (2020). A C. elegans model for neurodegeneration in Cockayne syndrome. Nucleic Acids Res. 48, 10973–10985. doi: 10.1093/nar/gkaa795
Lyabin, D. N., Eliseeva, I. A., and Ovchinnikov, L. P. (2014). YB-1 protein: functions and regulation. Wiley Interdiscip. Rev. RNA 5, 95–110. doi: 10.1002/wrna.1200
Ma, K., Chen, G., Li, W., Kepp, O., Zhu, Y., and Chen, Q. (2020). Mitophagy, mitochondrial homeostasis, and cell fate. Front. Cell Dev. Biol. 8:467. doi: 10.3389/fcell.2020.00467
Ma, X., Tang, T. S., and Guo, C. (2020). Regulation of translesion DNA synthesis in mammalian cells. Environ. Mol. Mutagen. 61, 680–692. doi: 10.1002/em.22359
Martin-Jimenez, R., Lurette, O., and Hebert-Chatelain, E. (2020). Damage in mitochondrial DNA associated with Parkinson’s disease. DNA Cell Biol. 39, 1421–1430. doi: 10.1089/dna.2020.5398
Mason, P. A., Matheson, E. C., Hall, A. G., and Lightowlers, R. N. (2003). Mismatch repair activity in mammalian mitochondria. Nucleic Acids Res. 31, 1052–1058. doi: 10.1093/nar/gkg167
Matsuda, N., Sato, S., Shiba, K., Okatsu, K., Saisho, K., Gautier, C. A., et al. (2010). PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 189, 211–221. doi: 10.1083/jcb.200910140
Meagher, M., and Lightowlers, R. N. (2014). The role of TDP1 and APTX in mitochondrial DNA repair. Biochimie 100, 121–124. doi: 10.1016/j.biochi.2013.10.011
Meyer, J. N., Leuthner, T. C., and Luz, A. L. (2017). Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology 391, 42–53. doi: 10.1016/j.tox.2017.07.019
Mishra, A., Saxena, S., Kaushal, A., and Nagaraju, G. (2018). RAD51C/XRCC3 facilitates mitochondrial DNA replication and maintains integrity of the mitochondrial genome. Mol. Cell Biol. 38:e00489–17. doi: 10.1128/MCB.00489-17
Mullins, E. A., Rodriguez, A. A., Bradley, N. P., and Eichman, B. F. (2019). Emerging roles of DNA glycosylases and the base excision repair pathway. Trends Biochem. Sci. 44, 765–781. doi: 10.1016/j.tibs.2019.04.006
Murata, D., Arai, K., Iijima, M., and Sesaki, H. (2020). Mitochondrial division, fusion and degradation. J. Biochem. 167, 233–241. doi: 10.1093/jb/mvz106
Myers, K. A., Saffhill, R., and O’Connor, P. J. (1988). Repair of alkylated purines in the hepatic DNA of mitochondria and nuclei in the rat. Carcinogenesis 9, 285–292. doi: 10.1093/carcin/9.2.285
Nakabeppu, Y., Tsuchimoto, D., Yamaguchi, H., and Sakumi, K. (2007). Oxidative damage in nucleic acids and Parkinson’s disease. J. Neurosci. Res. 85, 919–934. doi: 10.1002/jnr.21191
Nakada, K., Inoue, K., Ono, T., Isobe, K., Ogura, A., Goto, Y. I., et al. (2001). Inter-mitochondrial complementation: mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat. Med. 7, 934–940. doi: 10.1038/90976
Narendra, D., Tanaka, A., Suen, D. F., and Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803. doi: 10.1083/jcb.200809125
Nishioka, K., Ohtsubo, T., Oda, H., Fujiwara, T., Kang, D., Sugimachi, K., et al. (1999). Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol. Biol. Cell 10, 1637–1652. doi: 10.1091/mbc.10.5.1637
Ohtsubo, T., Nishioka, K., Imaiso, Y., Iwai, S., Shimokawa, H., Oda, H., et al. (2000). Identification of human MutY homolog (hMYH) as a repair enzyme for 2-hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucleic Acids Res. 28, 1355–1364. doi: 10.1093/nar/28.6.1355
Okur, M. N., Fang, E. F., Fivenson, E. M., Tiwari, V., Croteau, D. L., and Bohr, V. A. (2020). Cockayne syndrome proteins CSA and CSB maintain mitochondrial homeostasis through NAD(+) signaling. Aging Cell 19:e13268. doi: 10.1111/acel.13268
Olichon, A., Baricault, L., Gas, N., Guillou, E., Valette, A., Belenguer, P., et al. (2003). Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 278, 7743–7746. doi: 10.1074/jbc.C200677200
Ono, T., Isobe, K., Nakada, K., and Hayashi, J. I. (2001). Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat. Genet. 28, 272–275. doi: 10.1038/90116
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2015). Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521, 525–528. doi: 10.1038/nature14300
Panier, S., and Boulton, S. J. (2014). Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 15, 7–18. doi: 10.1038/nrm3719
Pannunzio, N. R., Watanabe, G., and Lieber, M. R. (2018). Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 293, 10512–10523. doi: 10.1074/jbc.TM117.000374
Pickles, S., Vigie, P., and Youle, R. J. (2018). Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 28, R170–R185. doi: 10.1016/j.cub.2018.01.004
Pickrell, A. M., and Youle, R. J. (2015). The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273. doi: 10.1016/j.neuron.2014.12.007
Pohjoismaki, J. L., Goffart, S., Tyynismaa, H., Willcox, S., Ide, T., Kang, D., et al. (2009). Human heart mitochondrial DNA is organized in complex catenated networks containing abundant four-way junctions and replication forks. J. Biol. Chem. 284, 21446–21457. doi: 10.1074/jbc.M109.016600
Ragg, S., Xu-Welliver, M., Bailey, J., D’Souza, M., Cooper, R., Chandra, S., et al. (2000). Direct reversal of DNA damage by mutant methyltransferase protein protects mice against dose-intensified chemotherapy and leads to in vivo selection of hematopoietic stem cells. Cancer Res. 60, 5187–5195.
Rossi, M. N., Carbone, M., Mostocotto, C., Mancone, C., Tripodi, M., Maione, R., et al. (2009). Mitochondrial localization of PARP-1 requires interaction with mitofilin and is involved in the maintenance of mitochondrial DNA integrity. J. Biol. Chem. 284, 31616–31624. doi: 10.1074/jbc.M109.025882
Rudd, S. G., Bianchi, J., and Doherty, A. J. (2014). PrimPol-A new polymerase on the block. Mol. Cell Oncol. 1:e960754. doi: 10.4161/23723548.2014.960754
Sage, J. M., Gildemeister, O. S., and Knight, K. L. (2010). Discovery of a novel function for human Rad51: maintenance of the mitochondrial genome. J. Biol. Chem. 285, 18984–18990. doi: 10.1074/jbc.M109.099846
Saki, M., and Prakash, A. (2017). DNA damage related crosstalk between the nucleus and mitochondria. Free Radic. Biol. Med. 107, 216–227. doi: 10.1016/j.freeradbiomed.2016.11.050
Sale, J. E. (2013). Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb. Perspect. Biol. 5:a012708. doi: 10.1101/cshperspect.a012708
Sharma, N., Pasala, M. S., and Prakash, A. (2019). Mitochondrial DNA: epigenetics and environment. Environ. Mol. Mutagen. 60, 668–682. doi: 10.1002/em.22319
Shirihai, O. S., Song, M., and Dorn, G. W. II (2015). How mitochondrial dynamism orchestrates mitophagy. Circ. Res. 116, 1835–1849. doi: 10.1161/CIRCRESAHA.116.306374
Simsek, D., Furda, A., Gao, Y., Artus, J., Brunet, E., Hadjantonakis, A. K., et al. (2011). Crucial role for DNA ligase III in mitochondria but not in Xrcc1-dependent repair. Nature 471, 245–248. doi: 10.1038/nature09794
Singh, B., Li, X., Owens, K. M., Vanniarajan, A., Liang, P., and Singh, K. K. (2015). Human REV3 DNA polymerase zeta localizes to mitochondria and protects the mitochondrial genome. PLoS One 10:e0140409. doi: 10.1371/journal.pone.0140409
Slupphaug, G., Markussen, F. H., Olsen, L. C., Aasland, R., Aarsaether, N., Bakke, O., et al. (1993). Nuclear and mitochondrial forms of human uracil-DNA glycosylase are encoded by the same gene. Nucleic Acids Res. 21, 2579–2584. doi: 10.1093/nar/21.11.2579
Stein, A., and Sia, E. A. (2017). Mitochondrial DNA repair and damage tolerance. Front. Biosci. (Landmark Ed.) 22:920–943. doi: 10.2741/4525
Stewart, J. B., and Chinnery, P. F. (2015). The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat. Rev. Genet. 16, 530–542. doi: 10.1038/nrg3966
Szczesny, B., Brunyanszki, A., Olah, G., Mitra, S., and Szabo, C. (2014). Opposing roles of mitochondrial and nuclear PARP1 in the regulation of mitochondrial and nuclear DNA integrity: implications for the regulation of mitochondrial function. Nucleic Acids Res. 42, 13161–13173. doi: 10.1093/nar/gku1089
Tahbaz, N., Subedi, S., and Weinfeld, M. (2012). Role of polynucleotide kinase/phosphatase in mitochondrial DNA repair. Nucleic Acids Res. 40, 3484–3495. doi: 10.1093/nar/gkr1245
Tann, A. W., Boldogh, I., Meiss, G., Qian, W., Van Houten, B., Mitra, S., et al. (2011). Apoptosis induced by persistent single-strand breaks in mitochondrial genome: critical role of EXOG (5’-EXO/endonuclease) in their repair. J. Biol. Chem. 286, 31975–31983. doi: 10.1074/jbc.M110.215715
Tilokani, L., Nagashima, S., Paupe, V., and Prudent, J. (2018). Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem. 62, 341–360. doi: 10.1042/EBC20170104
Tsang, S. H., Aycinena, A. R. P., and Sharma, T. (2018). Mitochondrial disorder: kearns-sayre syndrome. Adv. Exp. Med. Biol. 1085, 161–162. doi: 10.1007/978-3-319-95046-4_30
Twig, G., Elorza, A., Molina, A. J., Mohamed, H., Wikstrom, J. D., Walzer, G., et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446. doi: 10.1038/sj.emboj.7601963
van Loon, B., and Samson, L. D. (2013). Alkyladenine DNA glycosylase (AAG) localizes to mitochondria and interacts with mitochondrial single-stranded binding protein (mtSSB). DNA Repair (Amst.) 12, 177–187. doi: 10.1016/j.dnarep.2012.11.009
Vives-Bauza, C., Zhou, C., Huang, Y., Cui, M., de Vries, R. L., Kim, J., et al. (2010). PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. U. S. A. 107, 378–383. doi: 10.1073/pnas.0911187107
Walker, M. A., Lareau, C. A., Ludwig, L. S., Karaa, A., Sankaran, V. G., Regev, A., et al. (2020). Purifying selection against pathogenic mitochondrial DNA in human T cells. N. Engl. J. Med. 383, 1556–1563. doi: 10.1056/NEJMoa2001265
Wallace, D. C. (2005). A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet. 39, 359–407. doi: 10.1146/annurev.genet.39.110304.095751
Wallings, R., Manzoni, C., and Bandopadhyay, R. (2015). Cellular processes associated with LRRK2 function and dysfunction. FEBS J. 282, 2806–2826. doi: 10.1111/febs.13305
Wang, Y., and Bogenhagen, D. F. (2006). Human mitochondrial DNA nucleoids are linked to protein folding machinery and metabolic enzymes at the mitochondrial inner membrane. J. Biol. Chem. 281, 25791–25802. doi: 10.1074/jbc.M604501200
Waters, L. S., Minesinger, B. K., Wiltrout, M. E., D’Souza, S., Woodruff, R. V., and Walker, G. C. (2009). Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol. Mol. Biol. Rev. 73, 134–154. doi: 10.1128/MMBR.00034-08
Wei, W., Pagnamenta, A. T., Gleadall, N., Sanchis-Juan, A., Stephens, J., Broxholme, J., et al. (2020). Nuclear-mitochondrial DNA segments resemble paternally inherited mitochondrial DNA in humans. Nat. Commun. 11:1740. doi: 10.1038/s41467-020-15336-3
Wei, Y., Chiang, W. C., Sumpter, R. Jr., Mishra, P., and Levine, B. (2017). Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell 168(1–2), 224–238.e10. doi: 10.1016/j.cell.2016.11.042
West, A. P., Khoury-Hanold, W., Staron, M., Tal, M. C., Pineda, C. M., Lang, S. M., et al. (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557. doi: 10.1038/nature14156
Wonnapinij, P., Chinnery, P. F., and Samuels, D. C. (2008). The distribution of mitochondrial DNA heteroplasmy due to random genetic drift. Am. J. Hum. Genet. 83, 582–593. doi: 10.1016/j.ajhg.2008.10.007
Wright, W. D., Shah, S. S., and Heyer, W. D. (2018). Homologous recombination and the repair of DNA double-strand breaks. J Biol Chem 293, 10524–10535. doi: 10.1074/jbc.TM118.000372
Yakes, F. M., and Van Houten, B. (1997). Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. U. S. A. 94, 514–519. doi: 10.1073/pnas.94.2.514
Yamazaki, T., Kirchmair, A., Sato, A., Buque, A., Rybstein, M., Petroni, G., et al. (2020). Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat. Immunol. 21, 1160–1171. doi: 10.1038/s41590-020-0751-0
Yang, L., Long, Q., Liu, J., Tang, H., Li, Y., Bao, F., et al. (2015). Mitochondrial fusion provides an ‘initial metabolic complementation’ controlled by mtDNA. Cell Mol. Life Sci. 72, 2585–2598. doi: 10.1007/s00018-015-1863-9
Yang, W., and Gao, Y. (2018). Translesion and repair DNA polymerases: diverse structure and mechanism. Annu. Rev. Biochem. 87, 239–261. doi: 10.1146/annurev-biochem-062917-012405
Yasukawa, T., and Kang, D. (2018). An overview of mammalian mitochondrial DNA replication mechanisms. J. Biochem. 164, 183–193. doi: 10.1093/jb/mvy058
Yoo, S. M., and Jung, Y. K. (2018). A molecular approach to mitophagy and mitochondrial dynamics. Mol. Cells 41, 18–26. doi: 10.14348/molcells.2018.2277
Youle, R. J., and van der Bliek, A. M. (2012). Mitochondrial fission, fusion, and stress. Science 337, 1062–1065. doi: 10.1126/science.1219855
Youn, C. K., Kim, H. B., Wu, T. T., Park, S., Cho, S. I., and Lee, J. H. (2017). 53BP1 contributes to regulation of autophagic clearance of mitochondria. Sci. Rep. 7:45290. doi: 10.1038/srep45290
Yu, W., Sun, Y., Guo, S., and Lu, B. (2011). The PINK1/Parkin pathway regulates mitochondrial dynamics and function in mammalian hippocampal and dopaminergic neurons. Hum. Mol. Genet. 20, 3227–3240. doi: 10.1093/hmg/ddr235
Zheng, J., Croteau, D. L., Bohr, V. A., and Akbari, M. (2019). Diminished OPA1 expression and impaired mitochondrial morphology and homeostasis in Aprataxin-deficient cells. Nucleic Acids Res. 47, 4086–4110. doi: 10.1093/nar/gkz083
Zong, W. X., Rabinowitz, J. D., and White, E. (2016). Mitochondria and cancer. Mol. Cell 61, 667–676. doi: 10.1016/j.molcel.2016.02.011
Keywords: mitochondrial DNA, DNA repair, mitochondrial fusion, mitochondrial fission, mitophagy
Citation: Rong Z, Tu P, Xu P, Sun Y, Yu F, Tu N, Guo L and Yang Y (2021) The Mitochondrial Response to DNA Damage. Front. Cell Dev. Biol. 9:669379. doi: 10.3389/fcell.2021.669379
Received: 18 February 2021; Accepted: 20 April 2021;
Published: 12 May 2021.
Edited by:
Daoxiang Zhang, Washington University in St. Louis, United StatesReviewed by:
Konstantinos Palikaras, National and Kapodistrian University of Athens, GreeceCopyright © 2021 Rong, Tu, Xu, Sun, Yu, Tu, Guo and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ziye Rong, MjAyMTUwMDAwM0BhaG11LmVkdS5jbg==; Yanan Yang, eWFuZy55YW5hbkBhaG11LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.