 Wei Wang
Wei Wang Brittany M. Jack
Brittany M. Jack Henry H. Wang
Henry H. Wang Matthew A. Kavanaugh
Matthew A. Kavanaugh Robin L. Maser
Robin L. Maser Pamela V. Tran
Pamela V. Tran
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 19 May 2021
Sec. Cell Adhesion and Migration
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.661350
This article is part of the Research TopicThe Cytoskeleton and Cellular Compartmentation: Cilia as Specialized Cellular DomainsView all 18 articles
Primary cilia are small, antenna-like organelles that detect and transduce chemical and mechanical cues in the extracellular environment, regulating cell behavior and, in turn, tissue development and homeostasis. Primary cilia are assembled via intraflagellar transport (IFT), which traffics protein cargo bidirectionally along a microtubular axoneme. Ranging from 1 to 10 μm long, these organelles typically reach a characteristic length dependent on cell type, likely for optimum fulfillment of their specific roles. The importance of an optimal cilia length is underscored by the findings that perturbation of cilia length can be observed in a number of cilia-related diseases. Thus, elucidating mechanisms of cilia length regulation is important for understanding the pathobiology of ciliary diseases. Since cilia assembly/disassembly regulate cilia length, we review the roles of IFT in processes that affect cilia assembly/disassembly, including ciliary transport of structural and membrane proteins, ectocytosis, and tubulin posttranslational modification. Additionally, since the environment of a cell influences cilia length, we also review the various stimuli encountered by renal epithelia in healthy and diseased states that alter cilia length and IFT.
Cilia or flagella are evolutionarily conserved organelles that protrude from a wide range of eukaryotic cells, from single-celled protists, like Chlamydomonas reinhardtii, to almost every vertebrate cell. Cilia are classified by function and structure into two general categories: motile and non-motile. Motile cilia not only generate motion but also contain receptors that provide sensory function (Jain et al., 2012). Motile cilia propel C. reinhardtii, as well as sperm, and sweep fluid and particles along the mammalian brain ventricles and respiratory and reproductive tracts. Non-motile cilia, also termed primary cilia, have chemo- and mechanosensory roles and are present on the sensory neurons of Caenorhabditis elegans and on most vertebrate cells. Primary cilia transduce light and mechanical and chemical cues (Poole et al., 1985; R Ferreira et al., 2019), mediate signaling pathways (Huangfu et al., 2003; Corbit et al., 2008; Wheway et al., 2018), and regulate cell cycle (Pan et al., 2013), cell differentiation (Ezratty et al., 2011; Forcioli-Conti et al., 2015), cell–cell communication (Viau et al., 2018), and autophagy (Pampliega et al., 2013; Orhon et al., 2016). The multiplicity and importance of these functions render primary cilia vital for organismal development and homeostasis (Badano et al., 2006).
A cilium is comprised of a microtubule-based axoneme that extends from a modified centriole, the basal body, and is ensheathed by a ciliary membrane (Figure 1). In most motile cilia, nine doublet peripheral microtubules are arranged in a circle around two single central microtubules, while in motile cilia of the embryonic node as well as in non-motile cilia, only nine doublet peripheral microtubules comprise the axoneme, forming “9 + 2” and “9 + 0” conformations, respectively (Satir, 2005). Distal to the basal body is the ciliary gate, consisting of transition fibers that join the basal body to the base of the ciliary membrane, and the transition zone, comprised of modules and has Y-links that tether the most proximal part of the axoneme to the ciliary membrane. The transition fibers and transition zone regulate ciliary entry and exit of proteins, allowing for compartmentalization and formation of a unique environment, such that the cilioplasm and ciliary membrane are composed of proteins, second messengers, and phospholipids, distinct from that of the cytosol and plasma membrane (Deane et al., 2001; Stephan et al., 2007; Garcia-Gonzalo et al., 2011; Williams et al., 2011; Garcia-Gonzalo and Reiter, 2017; Gonçalves and Pelletier, 2017).
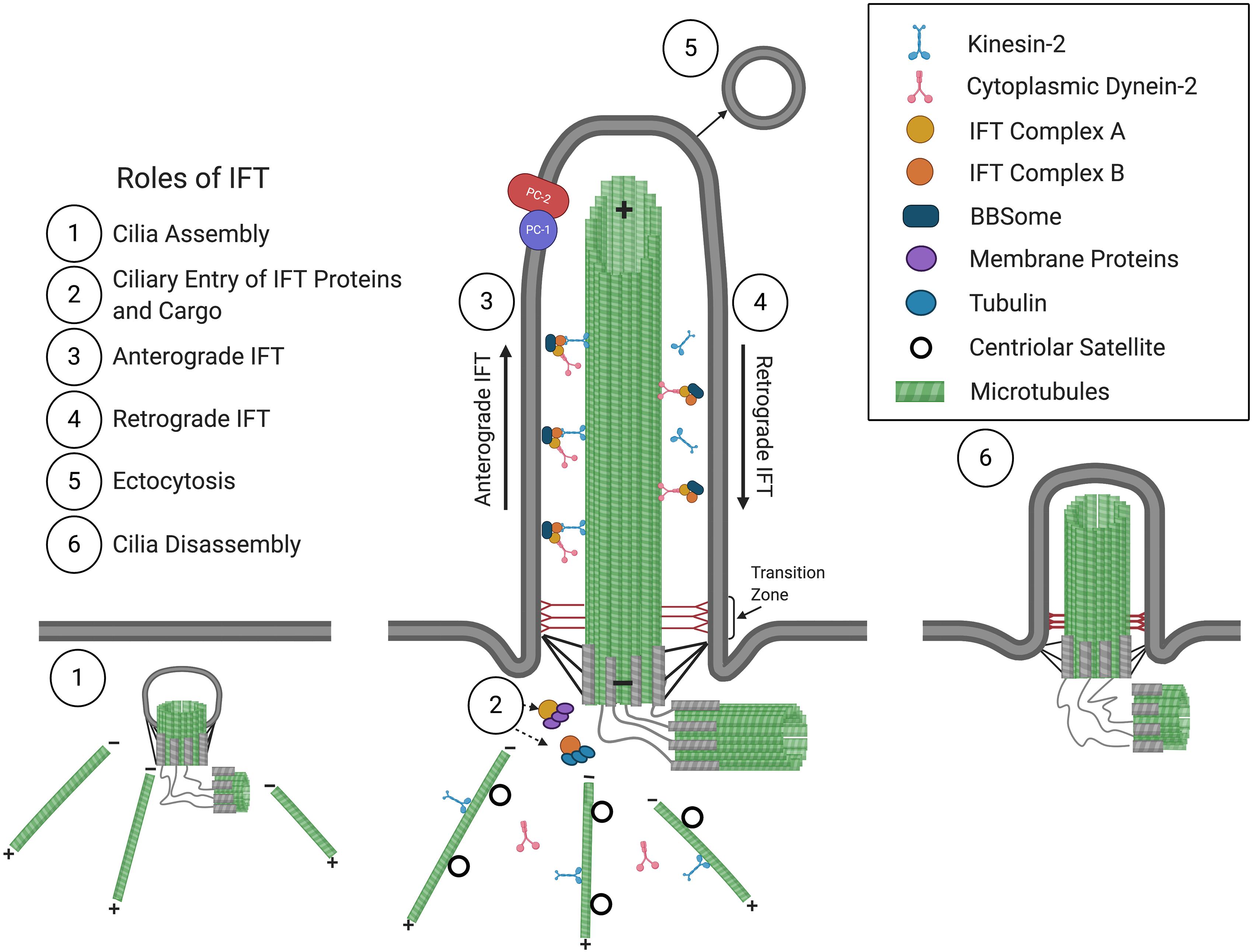
Figure 1. The primary cilium and roles of IFT in cilium assembly/disassembly. Consisting of a 9 + 0 arrangement of a microtubular axoneme ensheathed by a specialized ciliary membrane, the primary cilium assembles initially within the cytoplasm at the modified centriole (1), which becomes the basal body that forms the base of the primary cilium at the plasma membrane. Extension and maintenance of the cilium, along with the entry and exit of structural and functional components and the BBSome, are mediated by IFT (2–4). Roles for IFT in ectocytosis (5) and cilia disassembly (6) have also been implicated (see text for details). The positive (+) and negative (–) ends of axonemal and cytoplasmic microtubules are indicated. The polycystins, PC1 and PC2, are localized at the ciliary membrane and are mutated in ADPKD.
Primary cilia are dynamic structures that assemble and disassemble in coordination with the cell cycle. Cilia form when cells become quiescent, in G1 and G0, and begin disassembly before cells re-enter the cell cycle (Nigg and Stearns, 2011; Paridaen et al., 2013; Malicki and Johnson, 2017). The assembly and maintenance of cilia require intraflagellar transport (IFT), which was first observed in C. reinhardtii (Kozminski et al., 1993) and mediates the bidirectional transport of structural and signaling molecules along the microtubular axoneme. IFT is mediated by multiprotein complexes that can be dissociated biochemically into IFT complexes B (IFT-B) and A (IFT-A), consisting of 10 and 6 subunits, respectively. These IFT complexes form linear arrays or “trains” that are transported from the base to the tip of the cilium in anterograde IFT, powered by the kinesin-2 motor (Cole et al., 1998), then returned to the ciliary base in retrograde IFT, driven by cytoplasmic dynein-2 (Pazour et al., 1998, 1999; Porter et al., 1999; Signor et al., 1999). Another multiprotein complex, the BBSome, acts like an adaptor connecting IFT complexes to signaling molecules and is required for the ciliary export of activated signaling receptors (Nachury, 2018; Ye et al., 2018).
Primary cilia typically obtain a characteristic length for a cell type (Table 1), likely to achieve optimal function. In humans, mutation of ciliary genes results in disease syndromes, termed ciliopathies, which can manifest craniofacial defects, skeletal dysplasia, brain and cognitive defects, retinal degeneration, obesity, and fibrocystic disease of the liver, pancreas, and kidney (Badano et al., 2006; Hildebrandt et al., 2011). These mutations can cause signaling defects as well as cilia length differences (Bredrup et al., 2011; Halbritter et al., 2013; Alazami et al., 2014; Zhang et al., 2016; Duran et al., 2017; Shaheen et al., 2020). Additionally, in complex diseases and conditions not caused by a primary cilia genetic lesion, such as in obesity and type 2 diabetes and kidney injury, cilia lengths have also been reported to be shortened or lengthened on affected cells (Verghese et al., 2008; Han et al., 2014; Ritter et al., 2018; Yu et al., 2019). Thus, understanding cilia length regulation is critical to understanding the pathobiology of cilia-related disease.
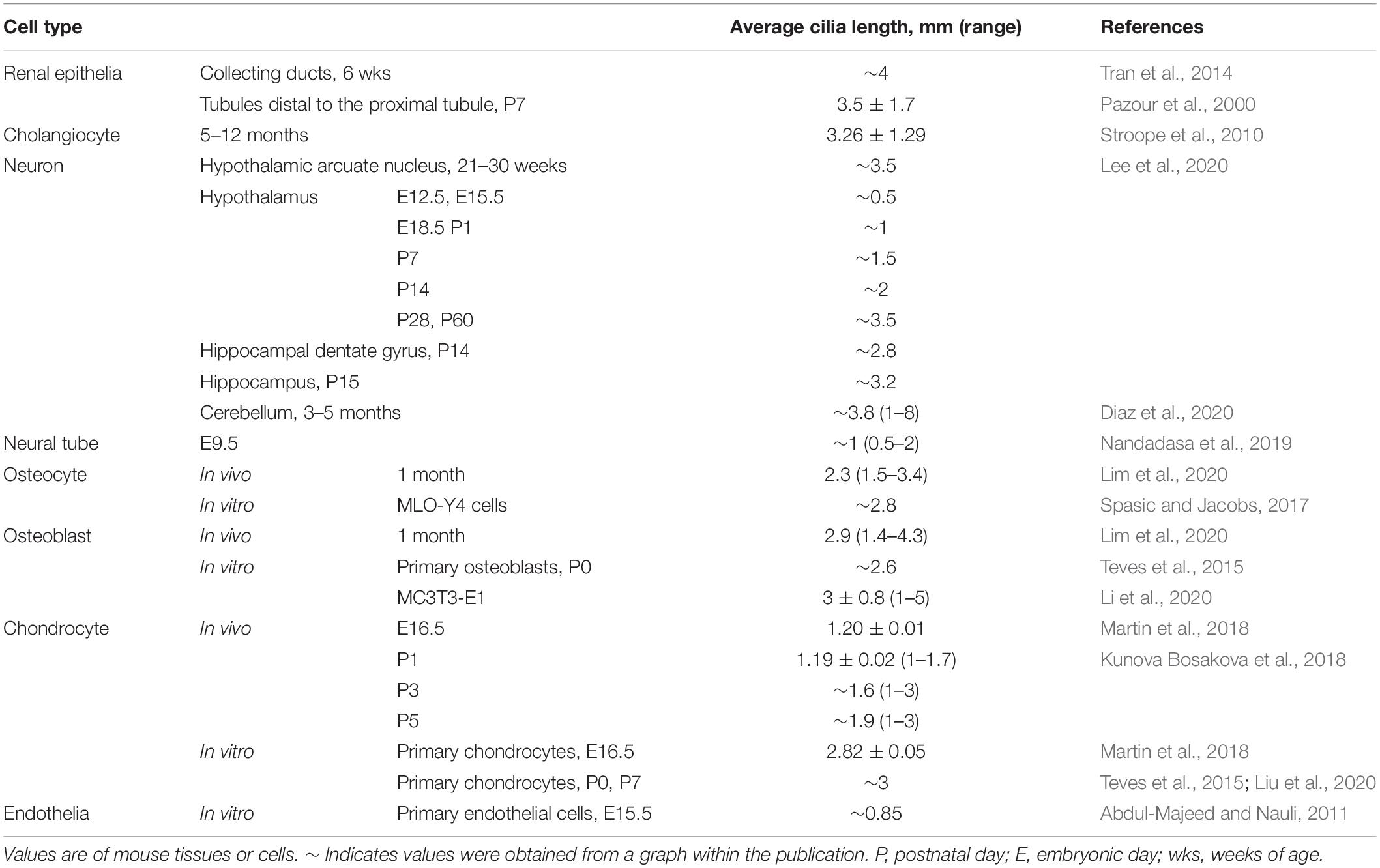
Table 1. Mammalian cilia lengths.
Cilia length is determined by the balance of cilia assembly and disassembly (Marshall and Rosenbaum, 2001; Marshall et al., 2005). In C. reinhardtii, live imaging has revealed that frequency of IFT train ciliary entry, IFT train size and speed, and cargo loading vary with cilia length, which has led to various models of cilia length control (Marshall et al., 2005; Wren et al., 2013; Chien et al., 2017; Fai et al., 2019; Wemmer et al., 2020). However, primary cilia length regulation in mammalian cells has been much less studied. Here, we review the primary cilia phenotypes of IFT-B and IFT-A mammalian mutants to glean mechanisms by which IFT proteins influence cilia assembly/disassembly. This includes roles of IFT in ciliary trafficking of tubulin and membrane-associated proteins and in influencing ectocytosis. Since posttranslational modification of axonemal tubulin can promote cilia assembly or disassembly, we also review the effects of glutamylation and O-GlcNAcylation on IFT. In addition to these intrinsic ciliary factors, primary cilia lengths are modulated by changes in the extracellular environment. In the kidney, cilia lengths change in healthy and diseased states, including in polycystic kidney disease and during kidney injury and repair. Thus, we also review the effects of chemical and mechanical signals in the renal environment on cilia length and IFT.
To initiate ciliogenesis, the mother centriole matures into the basal body and migrates and docks at the plasma membrane. During migration of the mother centriole, preciliary vesicles derived from the Golgi and recycling endosome attach to the subdistal appendages of the maturing mother centriole and fuse into a larger ciliary vesicle (Sorokin, 1962). Centriolar coiled coil protein 110 (CP110) localizes to the distal end of the mother centriole and regulates the start of cilium extension (Chen et al., 2002; Yadav et al., 2016). Rab8a is recruited to the mother centriole and activated by Rab11 and Rabin8 to enable ciliary membrane assembly (Westlake et al., 2011). This, together with the recruitment of IFT and transition zone proteins to the cilia base, allows for cilium extension (Deane et al., 2001; Rosenbaum and Witman, 2002; Wang and Dynlacht, 2018).
To extend the axoneme, α- and β-tubulin are imported into primary cilia and are added to the distal plus ends of microtubules at the cilia tip (Witman, 1975; Johnson and Rosenbaum, 1992). Live imaging of green fluorescent protein (GFP)-tagged tubulin in C. reinhardtii demonstrates that both diffusion and IFT allow for tubulin ciliary import (Craft et al., 2015; Craft Van De Weghe et al., 2020). In cells with growing cilia, anterograde transport of tubulin was increased, and in flagella length mutants, tubulin transport was dysregulated (elevated and reduced in long flagella2-1 and short flagella2 mutants, respectively, relative to steady-state wild-type cilia), suggesting a possible link between IFT-mediated transport of tubulin and regulation of cilia length (Craft et al., 2015; Wemmer et al., 2020).
In most IFT-B mutants, cilia are shortened or even lost (Table 2), indicating that the IFT-B complex is essential for ciliogenesis. The IFT-B complex consists of a 10-subunit core subcomplex and a 6-subunit peripheral subcomplex (IFT38, IFT57, IFT80, IFT20, IFT172, IFT54) (Katoh et al., 2016; Taschner et al., 2016). The core can be separated further into Core 1 (IFT25, IFT27, IFT74, IFT81, IFT22) and Core 2 (IFT56, IFT46, IFT52, IFT88, IFT70) subcomplexes (Nakayama and Katoh, 2020). Except for IFT74, all IFT-B components have been knocked out or mutated in mammalian cells with primary cilia (Table 2). Generally, loss of most Core 2 or peripheral subunits, with exception of IFT56, results in severely shortened or absent primary cilia. In contrast, deletion of Core 1 subunits has much milder effects, with many mutants lacking overt cilia length defects, although ciliary localization of signaling molecules and membrane-associated proteins is aberrant. Mammalian IFT70A and IFT70B constitute the orthologs of C. reinhardtii IFT70. Loss of both IFT70A and IFT70B in retinal pigment epithelial (RPE) cells causes the absence of cilia, while re-expression of either IFT70A or IFT70B restores ciliogenesis, revealing redundancy between IFT70A and IFT70B (Takei et al., 2018). In certain cases, cilia phenotypes are more severe in C. reinhardtii than in mammalian cells. For instance, IFT56-deficient C. reinhardtii have shortened cilia (Ishikawa et al., 2014), but Ift56-null mice do not (Xin et al., 2017). This may reflect greater functional redundancy among mammalian IFT proteins. Furthermore, deletion versus deficiency of an IFT protein can result in different ciliary phenotypes. Depletion of IFT80 in C3H10T1/2 mesenchymal cells causes a lack of cilia (Yang and Wang, 2012), while hypomorphic mutation of Ift80 in mice results in normal cilia morphology (Rix et al., 2011), suggesting a threshold of IFT deficiency which can be tolerated.
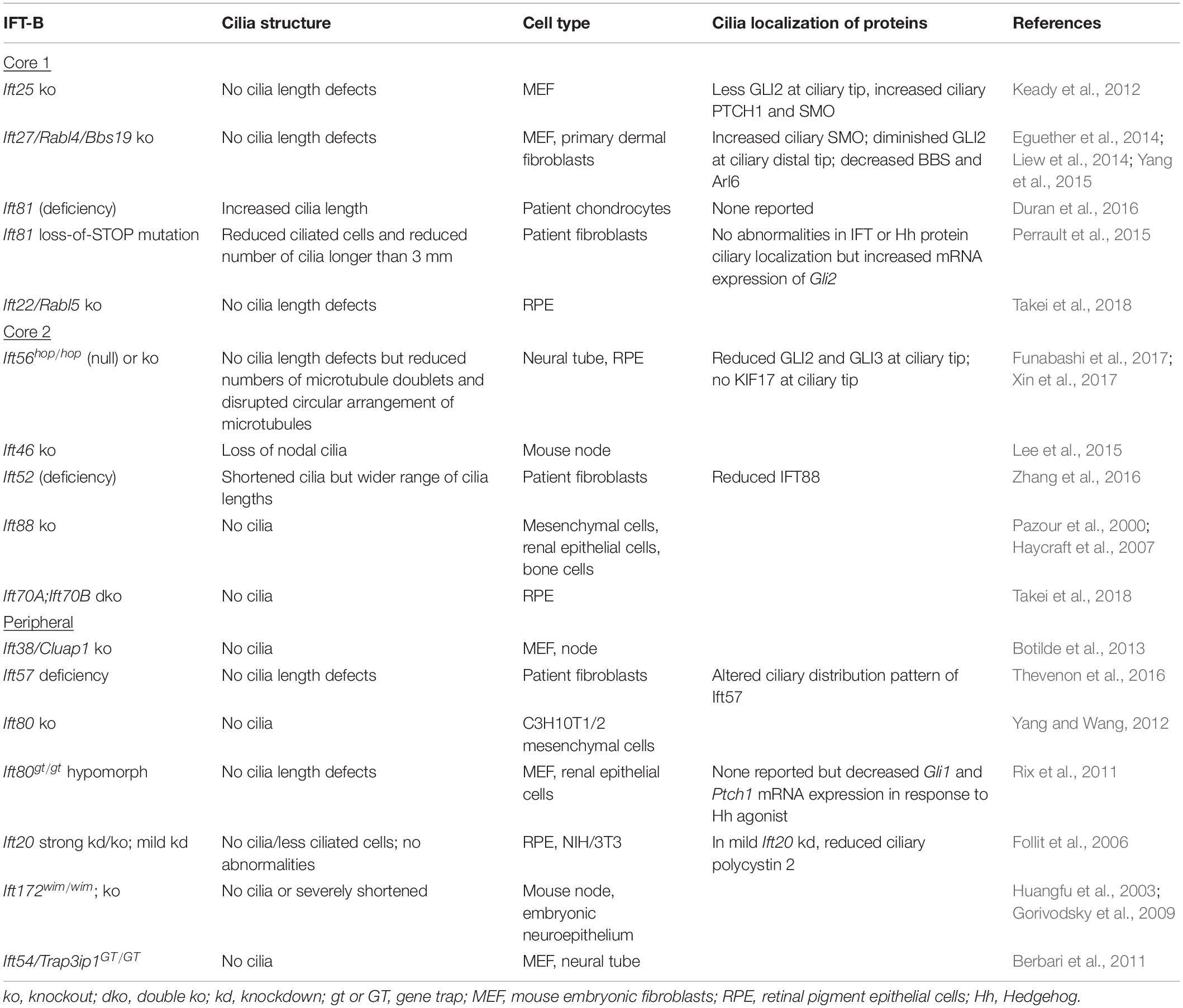
Table 2. Mammalian IFT-B ciliary phenotypes.
Given that axonemal elongation requires anterograde transport of tubulin (Craft et al., 2015), the ciliogenesis defects of IFT-B mutants could result from the lack of IFT-based transport of tubulin. In C. reinhardtii, the N-termini of IFT74 and IFT81 dimerize and bind α- and β-tubulin as cargo of anterograde IFT (Bhogaraju et al., 2013; Kubo et al., 2016). Additionally, IFT proteins are designed to form protein–protein interactions, and the loss of an IFT-B subunit can cause destabilization of the IFT-B complex. The molecular architecture of the IFT-B complex appears conserved between C. reinhardtii and mammalian cells (Katoh et al., 2016). IFT88, together with IFT52, connects the IFT-B core and peripheral complexes (Taschner et al., 2016). In fibroblasts of a ciliopathy patient with short rib polydactyly, mutation of IFT52 greatly reduced IFT52 protein levels, leading to a destabilized anterograde IFT complex, demonstrated by reduced cellular levels of IFT88, IFT74, IFT81, and ADP ribosylation factor like GTPase 13B (ARL13B), a ciliary membrane protein, as well as reduced IFT88 in cilia (Zhang et al., 2016). This anterograde IFT defect caused the presence of less ciliated cells and irregular distribution of ciliary lengths in patient cells.
Beyond the role of IFT-B subunits to form the IFT-B complex, IFT-B proteins also connect the IFT-B and IFT-A complexes, as well as anterograde and retrograde IFT. In C. reinhardtii, IFT74 was shown to associate IFT-B and IFT-A particles at the flagellar base and to be essential for flagellar import of IFT-A. Loss of the IFT74 residues required to bind IFT-A caused stunted cilia, thus revealing a role for the interdependence between IFT-B and IFT-A in ciliogenesis (Brown et al., 2015). Additionally, in mammalian retinal pigment epithelial (RPE) cells, expression of a truncated form of IFT88 on a CRISPR/Cas9-mediated IFT88 knockout background produced a ciliary phenotype similar to IFT-A knockout cells (Kobayashi et al., 2021). In C. reinhardtii and in mice, Ift54-null mutants lack cilia (Berbari et al., 2011; Zhu et al., 2017b), and a recent study showed that in C. reinhardtii and mammalian cells, IFT54 interacts with both the kinesin-2 and dynein motors (Zhu et al., 2020). Deletion of the IFT54 residues required to bind kinesin-2 reduced anterograde IFT, causing IFT motors and proteins to accumulate in the proximal region of cilia, while deletion of the residues that bind dynein impaired retrograde IFT, causing accumulation of IFT proteins at the distal tip (Zhu et al., 2020). Thus, IFT-B and IFT-A as well as anterograde and retrograde transport are interconnected, and these interconnections are integral to ciliogenesis.
The IFT-A complex consists of three core subunits (IFT122/IFT140/IFT144) and three peripheral subunits (IFT42/IFT121/IFT139) (Nakayama and Katoh, 2020). In mammals, the IFT139 homolog consists of two paralogs, THM1/TTC21B and THM2/TTC21A (Tran et al., 2008; Wang et al., 2020). With the exception of THM2, loss of any IFT-A core or peripheral subunit can result in shortened cilia with bulbous distal tips (Table 3). The severity of cilia phenotypes varies with cell type and/or in vitro or in vivo contexts. Additionally, as observed with IFT56, the occurrence of a more severe phenotype – loss of cilia – in ift140-null C. reinhardtii mutants (Picariello et al., 2019) compared to shortened cilia in mammalian cells (Liem et al., 2012; Hirano et al., 2017) supports that there may be greater redundancy among IFT proteins in mammalian cells or that other compensatory mechanisms exist.
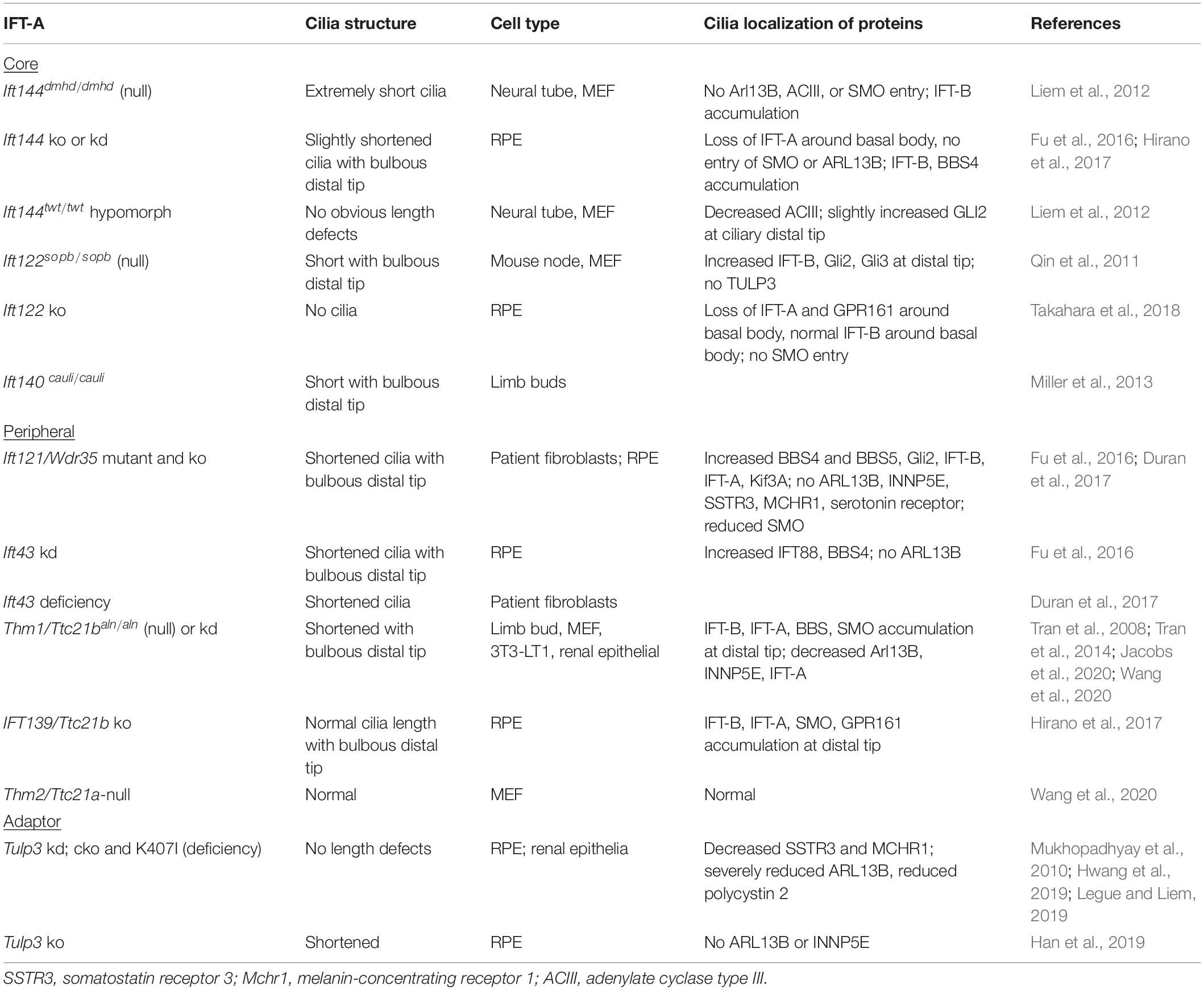
Table 3. Mammalian IFT-A ciliary phenotypes.
Live imaging of short hairpin RNA (shRNA)-mediated Thm1 knockdown inner medullary collecting duct (IMCD) cells expressing IFT88-eYFP demonstrates that a mammalian IFT-A subunit is required for retrograde IFT (Tran et al., 2008). Consistent with a general role for IFT-A in retrograde IFT, bulbous distal tips with sequestered IFT-B, IFT-A, BBS, and signaling proteins in mutants of both the IFT-A core and peripheral complexes indicate defective retrograde IFT (Table 3). Since retrograde IFT is required to bring back IFT proteins to the base for their recycling and re-entry into cilia, this could be a contributing mechanism by which IFT-A loss or deficiency decreases cilia assembly and, in turn, cilia length.
A comprehensive study examining ciliogenesis in Ift121/Wdr35 knockout RPE cells showed that cilia assembly was reduced and delayed due to disruption of the ciliary import and export of the various cargoes of IFT-A (Fu et al., 2016). Rab8 localization near centrioles was reduced, suggesting that early formation of cilium membrane assembly is disrupted. Additionally, the localization of centriolar satellite proteins was misregulated. Centriolar satellites regulate protein composition of cilia and are essential for efficient ciliogenesis (Odabasi et al., 2019). Ciliary entry of ciliary membrane proteins, ARL13B and inositol polyphosphate-5-phosphatase E (INNP5E), was also impeded. This could contribute to impaired ciliogenesis, since ARL13B is essential for ciliary membrane extension, which is coupled to axoneme elongation (Lu et al., 2015). Ciliary ARL13B is also lost in Ift144 knockout and Ift43 knockdown cells and reduced in Thm1-null mouse embryonic fibroblasts (MEFs) (Liem et al., 2012; Fu et al., 2016; Wang et al., 2020). Furthermore, ARL13B was shown to bind to IFT43, IFT122, and IFT139/THM1, suggesting that ARL13B is a cargo passenger of IFT-A (Fu et al., 2016).
Fu et al. (2016) proposed a unifying mechanism for the IFT-A mutant defects in ciliary entry of membrane proteins and in retrograde IFT: that IFT-A mutation causes a defect in trafficking cargo to the minus ends of both axonemal and cytoplasmic microtubules. In Ift122 knockout RPE cells, IFT-A localization around the basal body was lost, suggesting that IFT122 transports IFT-A proteins to the cilium base (Takahara et al., 2018). Similarly, in C. reinhardtii, loss of IFT43 resulted in reduced IFT proteins in the peri-basal body region, and both IFT43 and IFT140 were demonstrated to transport ciliary proteins from the cytosol to the peri-basal body region (Zhu et al., 2017a). These data support a defect in transporting cargo to the minus ends of microtubules with loss of IFT-A.
Depletion of IFT-A also causes ciliary localization defects of the transmembrane Hedgehog transducer, Smoothened (SMO). Loss of IFT-A core components, IFT144 and IFT122, results in absence of SMO in cilia (Liem et al., 2012; Hirano et al., 2017; Takahara et al., 2018), while loss of peripheral subunit IFT121 results in reduced SMO in cilia (Fu et al., 2016). In contrast, loss of THM1 causes increased ciliary SMO and its accumulation at the distal tip (Wang et al., 2020). While ciliary localization defects of SMO do not overtly affect cilia length, these differential phenotypes may reflect differences in cargoes or protein interactions of core versus peripheral subcomplexes or of individual IFT proteins. Like IFT121, THM1 is part of the peripheral subcomplex, and except for the opposing SMO ciliary localization defect, Thm1-null MEF show ciliary protein localization defects as well as reduced and delayed ciliogenesis (Wang et al., 2020), similar to Ift121-depleted RPE cells (Fu et al., 2016). However, while contrasting from other IFT-A mutants, the increased ciliary SMO in Thm1-null MEF could be due to a similar mechanism that causes increased ciliary SMO in Ift25- and Ift27-null IFT-B Core 1 mutants. IFT25 and IFT27 form a heterodimer and connect IFT to the BBSome, which exports signaling molecules out of primary cilia (Keady et al., 2012; Eguether et al., 2014; Liew et al., 2014).
In mammalian cells, the IFT-A core also binds to the tubby-related protein 3 (TULP3), which acts like an adaptor to import a subset of G protein-coupled receptors (GPCRs) into primary cilia. These GPCRs include melanin-concentrating hormone receptor (MCHR1), somatostatin receptor subtype 3 (SSTR3), GPR161, neuropeptide Y receptor 2 (NPY2R), and free fatty acid receptor 4 (FFAR4) (Mukhopadhyay et al., 2010, 2013; Badgandi et al., 2017; Hilgendorf et al., 2019). Consistent with the requirement of the IFT-A core for ciliary entry of TULP3, TULP3 was absent from cilia of an Ift122-null mutant mouse (Qin et al., 2011). Tulp3 knockdown RPE cells, a Tulp3 kidney-specific knockout mouse and a Tulp3 hypomorphic mutant mouse showed a lack of ciliary ARL13B but normal cilia length (Mukhopadhyay et al., 2010; Hwang et al., 2019; Legue and Liem, 2019). However, Tulp3 knockout RPE cells showed absent ciliary ARL13B as well as shortened cilia lengths (Han et al., 2019). Thus, cell specificity, extent of Tulp3 depletion, and in vitro versus in vivo contexts, which would expose cilia to various extracellular factors, likely contribute to affecting cilia length. Unlike the loss of IFT-A core and peripheral subunits, Tulp3 knockdown did not cause accumulation of ciliary IFT-B proteins (Mukhopadhyay et al., 2010), indicating Tulp3 is not required for retrograde IFT.
Since loss of many IFT-B proteins results in absence of primary cilia, the role of IFT-B in ciliary entry of membrane-associated proteins is comparatively less explored. IFT25 and IFT27, which are components of IFT-B Core 1, are dispensible for cilia formation but are required for ciliary removal of the BBsome and associated cargo (Keady et al., 2012; Eguether et al., 2014; Liew et al., 2014). Conversely, since ciliary entry of SSTR3 or SMO is not impeded in Ift25- or Ift27-null mutants, this may indicate that ciliary import of these receptors may not require IFT-B (Eguether et al., 2018; Ye et al., 2018). However, the role of other IFT-B proteins in SSTR3 and SMO ciliary import has not been investigated. Knockdown of Ift57 and Ift172 in IMCD cells severely reduced ciliogenesis, but in cells retaining primary cilia, D1-type dopaminergic receptors in the ciliary membrane was reduced, suggesting that IFT-B is required for ciliary import of certain membrane-associated signaling receptors (Leaf and Von Zastrow, 2015). This requirement for IFT-B in ciliary membrane import of D1-type dopaminergic receptors also involves Rab23 and Kif17. Supporting a role for IFT172 in associating with the membrane, C. reinhardtii IFT172 was shown to interact with and remodel membrane, and in mammalian (RPE) cells, IFT172 localized to the ciliary membrane (Wang et al., 2018).
Primary cilia can release small vesicles or ectosomes, containing components from the ciliary membrane in a process termed ectocytosis (Long and Huang, 2019). This secretion function of cilia was first discovered in C. reinhardtii and now has been observed in multiple species, including in C. elegans, Trypanosoma brucei, and in mammalian cells (Wood et al., 2013; Wang et al., 2014; Long et al., 2016; Szempruch et al., 2016; Nager et al., 2017; Phua et al., 2017). Shedding of ciliary ectosomes or extracellular vesicles regulates ciliary compartmentalization and homeostasis, signaling, and organismal and intercellular communication.
There is some variation in the release of ciliary vesicles and their contents across organisms. In C. reinhardtii, ectosomes are released from the ciliary tip in a process that requires the endosomal sorting complex required for transport (ESCRT) pathway. C. reinhardtii ectosomes contain ciliary membrane proteins, enzymes, ubiquitinated proteins, and ESCRT proteins (Long et al., 2016). In C. elegans, sensory neurons release extracellular vesicles from the cilia base in a process that is ESCRT independent but requires IFT-B and IFT-A (Wang et al., 2014). These extracellular vesicles contain LOV-1 and PKD-2, the C. elegans polycystin orthologs, and regulate communication and mating-related behavior. In mammalian cells, ectocytosis occurs from the ciliary tip upon stimulation with growth factors, which changes the phospholipid content of the ciliary membrane via INNP5E (Phua et al., 2017). Mammalian ectosomes contain ciliary membrane proteins, GPCRs (Nager et al., 2017), as well as IFT-B proteins, which are not present in ciliary vesicles of C. reinhardtii and C. elegans (Phua et al., 2017). Ectocytosis is linked to cilia resorption and disassembly in C. reinhardtii and mammalian cells (Long et al., 2016; Phua et al., 2017), and the containment of IFT-B proteins in mammalian ectosomes may be a mechanism by which primary cilia dispose of IFT-B proteins and become primed for disassembly (Phua et al., 2017).
Studies with BBS and IFT mutants shed additional light on the regulation of ectocytosis. In IMCD cells null for regulators or subunits of the BBSome, such as IFT27, ARL6, or BBS2, failure of BBSome-mediated ciliary removal of activated GPCRs caused accumulation of active GPCRs at the ciliary tip, which was followed by ectocytosis (Nager et al., 2017). In Thm1-null MEF, which sequester proteins at the ciliary tip, serum stimulation following starvation caused an increased presence of IFT-B foci that were separate from and distal to the cilia tip (Wang et al., 2020). Such observations likely reflect ectocytosis. In RPE cells depleted of mitogen-activated protein kinase-like kinase, ICK, which binds to IFT-B and localizes at ciliary tips, cilia were lengthened with an accumulation of IFT and signaling proteins at the distal tip, indicative of impaired retrograde IFT. Furthermore, these accumulated proteins were released in extracellular vesicles at the distal tip, indicative of ectocytosis (Nakamura et al., 2020). Collectively, these studies suggest that defective retrograde IFT promotes ectocytosis.
Ectocytosis precedes cilium resorption and disassembly (Phua et al., 2017). Consistent with this pattern, Thm1-null MEF, which exhibit a phenomenon consistent with increased ectocytosis also show enhanced serum-induced cilia loss (Wang et al., 2020). Cilia disassembly can occur via cilia resorption, which shortens cilia length gradually; complete cilia shedding, which severs the entire cilium; and a combination of both. Complete cilia shedding was found to be the predominant mechanism of cilia disassembly in wild-type mammalian cells (Mirvis et al., 2019). Future investigation into the role of IFT in regulating cilia disassembly could deepen our understanding of why one mechanism prevails over another.
Posttranslational modification (PTM) of the axonemal microtubules affects their stability, promoting either cilia assembly or disassembly. Acetylation, glutamylation, and glycylation stabilize the axoneme, favoring cilia assembly (Pugacheva et al., 2007; Shida et al., 2010; Gadadhar et al., 2017; He et al., 2018). In contrast, ubiquitination destabilizes the axoneme, leading to cilia disassembly (Huang et al., 2009; Wang et al., 2019).
Studies also suggest that PTMs regulate IFT. In shortening flagella of Chlamydomonas, IFT139 was identified to be ubiquitinated and to interact with ubiquitylated α-tubulin, suggesting that the IFT-A complex may traffic ubiquitylated axonemal proteins out of flagella, thus contributing to cilia disassembly (Wang et al., 2019). In C. elegans, a ccpp-1 deglutamylase mutant altered stability of the axonemal β-tubules and increased velocity of the kinesin 3 KSP-6 and kinesin 2 SOM-3/KIF17 accessory motors (O’hagan et al., 2011). Also in C. elegans, starvation activated tubulin glutamate ligase 4 (TTLL4), which increased glutamylation, as well as the speed of the kinesin II canonical motor and, hence, of anterograde IFT (Kimura et al., 2018).
Recently, N-acetylglucosamine (GlcNAc), a nutrient sensor and product of the hexosamine biosynthetic pathway that can be linked enzymatically to serine/threonine residues of a multitude of intracellular proteins (Hart et al., 2011), has been shown to regulate cilia length (Tian and Qin, 2019; Yu et al., 2019, 2020). Both tubulin and histone deacetylase 6 (HDAC6, which regulates cilia disassembly) (Pugacheva et al., 2007), are O-GlcNAcylated (Tian and Qin, 2019). In one study, pharmacological and genetic inhibition of O-GlcNAc transferase (OGT), which transfers the GlcNAc moiety onto protein substrates, increased cilia length in RPE and IMCD cells (Tian and Qin, 2019). In another study, tissues (retinal photoreceptors, trachea) of Ogt knockout mice and RPE cells treated with an OGT inhibitor displayed shortened primary cilia with an accumulation of IFT proteins in bulbous distal tips, suggesting impaired retrograde IFT (Yu et al., 2020). While more investigations are required to reconcile these opposing results, these studies indicate that O-GlcNAcylation is another PTM affecting cilia length and IFT.
Together, these studies hint at a role for IFT in mediating the cilium assembly/disassembly associated with a particular PTM. Live imaging of IFT in mammalian cells following a PTM event is required to establish that PTMs affect IFT and to explore how PTMs regulate the IFT machinery.
In a context-dependent manner, primary cilia demonstrate multiple roles. These organelles sense and transduce light, chemical, and mechanical cues and are hubs for multiple signaling pathways, including Hedgehog, Wnt, Notch, transforming growth factor beta (TGFβ), platelet-derived growth factor receptor (PDGFR), and G protein coupled receptor (GPCR) (Huangfu et al., 2003; Schneider et al., 2005; Eggenschwiler and Anderson, 2007; Corbit et al., 2008; Ezratty et al., 2011; Clement et al., 2013; Anvarian et al., 2019). Activation or downregulation of these signaling pathways regulates cell behavioral response, including proliferation, differentiation, and tissue organization. Mutation of ciliary genes reveals importance of primary cilia in multiple organ systems. The presence or absence as well as severity of phenotypes or clinical manifestations reflect the cell-specific and developmental and homeostatic roles of primary cilia.
Among the first revelations that primary cilia are essential to mammalian health was the discovery that mutation of IFT causes renal cysts (Yoder et al., 1995, 2002; Pazour et al., 2000). In renal tubules, cilia extend from the apical surface of most epithelial cells, suggesting that the role of renal tubular epithelial cilia is to sense the environment of the renal tubular lumen and transmit this information into the epithelium and possibly to “downstream” tubule segments (via ectosomes). Analyses of human kidneys reveal that cilia lengths change with nephron segment, as well as with development, being shortest in renal vesicles (0.59 μm) and lengthening as fetal nephrons mature (3.04 μm) (Saraga-Babic et al., 2012). Additionally, mutation of genes required for ciliogenesis usually causes shortened or absent cilia, and less commonly lengthened cilia, and often results in fibrocystic renal disease (Davis et al., 2011; Srivastava et al., 2017). In mice, the mutation and/or deletion of IFT genes models this phenomenon (Yoder et al., 1995, 2002; Pazour et al., 2000; Jonassen et al., 2008, 2012; Patel et al., 2008; Tran et al., 2014). Together, these findings indicate that the presence and architecture of primary cilia regulate kidney development, morphology, and function.
In autosomal dominant polycystic kidney disease (ADPKD), the majority of mutations occur in PKD1 and PKD2, which encode polycystin 1 and polycystin 2, which form a complex and function at the primary cilium (Freedman et al., 2013; Cai et al., 2014; Walker et al., 2019). In renal tissue of ADPKD patients as well as of orthologous mouse models, cilia lengths are increased (Hopp et al., 2012; Liu et al., 2018; Shao et al., 2020). Similarly, in the jck non-orthologous mouse model of ADPKD, renal primary cilia are also lengthened (Smith et al., 2006). Interestingly, deletion of Kif3a, Ift20, and Ift88 in Pkd1 and Pkd2 conditional knockout mice ablated or shortened primary cilia and markedly attenuated the ADPKD cystic phenotype (Ma et al., 2013; Viau et al., 2018; Shao et al., 2020). Similarly, pharmacological shortening of primary cilia (via a CDK5 inhibitor) in jck mice ameliorated the cystic disease (Husson et al., 2016). Conversely, genetic and pharmacological inhibition of cilia disassembly in Pkd1 conditional knockout mice increased renal cilia lengths and worsened ADPKD severity (Nikonova et al., 2014). These data suggest that cilia length may be an important modifier of ADPKD severity.
Renal epithelial cilia lengths also change during acute and chronic kidney injury and repair. A variety of methods have been employed to induce renal injury in both in vitro and in vivo model systems (Table 4). Acute injury, or the exposure to hypoxia to model chronic injury, appears to cause deciliation followed by elongation of cilia and eventual return to normal cilia length. Such alterations in cilia length are proposed to alter cilium sensing function and to form part of an epithelial repair mechanism whereby cilia regulate changes in signaling pathways appropriate for normal repair of injured renal tubules (Verghese et al., 2019). This dynamic modulation of renal cilia lengths during renal injury and repair suggests that cilia lengths are tuned to maintain normal renal tubular structure and function (Verghese et al., 2008, 2009; Han et al., 2017; Park, 2018).
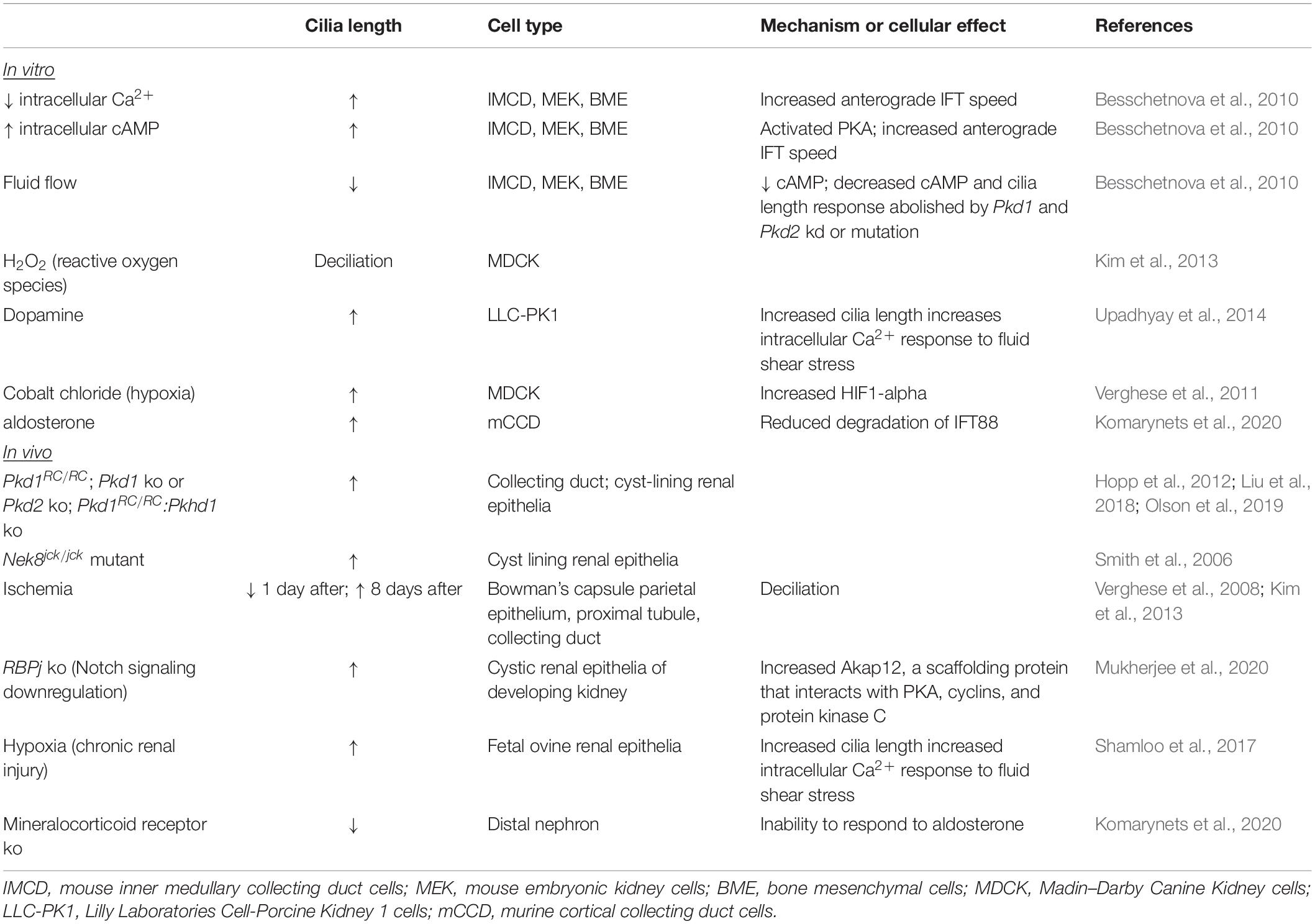
Table 4. Modulators of renal cilia length.
Cellular signaling pathways and signals from the local environment modulate cilium length. As renal primary cilia protrude into the tubular lumen, cilia are exposed to filtrate flow. In response to fluid flow, primary cilia of cultured renal epithelial cells deflect, and intracellular calcium (Ca2+) increases (Praetorius and Spring, 2001, 2003). In cells that are stimulated resulting in lengthened cilia, the increase in intracellular Ca2+ induced by fluid flow is elevated, indicating that increased cilia length increases sensitivity to fluid flow (Upadhyay et al., 2014; R Ferreira et al., 2019). Conversely, in cells mutant for Pkd1 or Pkd2, the increased intracellular Ca2+ response to fluid flow is abrogated (Nauli et al., 2003). In ADPKD cells, intracellular Ca2+ is decreased, and intracellular cyclic AMP (cAMP) is increased (Yamaguchi et al., 2004). Treatments to decrease intracellular Ca2+ and to elevate intracellular cAMP of cultured IMCD cells increased primary cilium lengths (Besschetnova et al., 2010). Using live microscopy, the increase in cilia length coincided with increased velocity of anterograde IFT. Additionally, fluid shear-mediated deflection reduced cilia length, decreased intracellular cAMP, and reduced the intracellular Ca2+ response to fluid flow, indicating the presence of a negative feedback loop. In Pkd1- or Pkd2-deficient cells, the Ca2+ response to fluid flow was abolished, and so was the negative feedback loop. In the absence of fluid flow, cultured Pkd1 MEF and renal epithelia also displayed reduced cilia disassembly via disruption of a centrosomal integrity pathway mediated by p53 (Gerakopoulos et al., 2020). In contrast, Streets et al. reported shortened primary cilia in Pkd1-mutant renal epithelial cells via increased actin polymerization mediated by RhoA (Streets et al., 2020). These varied results may reflect that the altered cilia lengths are captured at a particular time point during dynamic modulation of cilia length by these individual pathways. Together, these studies demonstrate that regulation of cilia length and cilia-mediated function is tightly connected. Furthermore, the disruption of this connection in ADPKD suggests that this connection may be critical to staving off disease.
Extrinsic factors that regulate kidney homeostasis have also been shown to alter cilia length. Dopamine activates the intrarenal dopaminergic pathway to reduce salt and water reabsorption by the kidney, and dysregulation of intrarenal dopaminergic signaling increases risk for essential hypertension (Harris and Zhang, 2012). Treatment of LLC-PK1 porcine kidney epithelial cells with dopamine and fenoldopam to activate ciliary-localized dopamine receptor type-5 increased cilia length (Upadhyay et al., 2014). Fenoldopam caused a greater increase in cilia length than dopamine, and increased cilia length correlated with the sensitivity of the cells to fluid shear stress as measured by the increase in intracellular Ca2+, substantiating the interconnection between cilia length modulation and ciliary signaling. Aldosterone, a mineralocorticoid steroid hormone that is produced by the adrenal cortex, conserves sodium in the kidney. In a mineralocorticoid knockout mouse, cilia of multiple tubule types were shortened. Explaining this result, treatment of cultured cortical collecting duct cells with aldosterone increased primary cilia length, and this increase correlated with intensity of the transepithelial Na+ transport and was demonstrated to occur via reduced degradation of IFT88 (Komarynets et al., 2020). Thus, modulation of cilia length is inherent to kidney homeostasis and physiology.
Primary cilia lengths differ with cell type, developmental stage, disease state, and in response to injury or repair. Multiple intrinsic (IFT and ciliary proteins) and extrinsic factors contribute to modulating cilia length, and these factors are likely intertwined to control cilia length and function. More analyses in mammalian cells are required to determine the extent to which mechanisms discovered in C. reinhardtii and other ciliated organisms are similar or diverge in mammalian cells. The redundancy of IFT proteins in mammalian cells suggests a means of additional species- or cell-specific roles for these proteins in regulating cilia length and function. High-resolution live microscopy of IFT and associated cargo in mammalian cells will be instrumental in obtaining new mechanistic insights.
Signaling events can affect cilia length, which in turn influences sensitivity to external factors, such as fluid flow in the kidney. Expanding our knowledge of the signaling pathways that influence cilia lengths and those that are affected by cilia length would contribute to a regulatory network of cilia length control. Since studies suggest that cell type and in vitro versus in vivo contexts can produce differential results, expansion of cell types and development of in vitro models that more closely mimic the in vivo setting, such as 3D models and cultures that implement multiple cell types, may help to reconcile these differences.
Given that rescue of the increased cilia length in ADPKD mouse models correlates with attenuation of the disease, could cilia length in certain cases be a therapeutic target? In a developmental study, the rescue of cilia length and structure of Dync2h1-null embryos via deletion of one allele of Ift172 mitigated the Hh signaling defects and early embryonic lethality, suggesting that Ift mutations cause their phenotypes primarily by affecting cilia architecture rather than by directly regulating signaling (Ocbina et al., 2011). Since Ift genes have differential roles, determining how to rescue cilia architecture for each Ift gene deletion would require expanding digenic analyses to multiple Ift genes. Additionally, cancer studies show that drugs can have differential effects on cilia lengths of different cell types within a tissue, emphasizing cell-specific control of ciliogenesis (Kiseleva et al., 2019).
However, in ciliopathy patient cells, cilia length is not always altered, or inconsistencies exist in whether a gene mutation causes cilia to be lengthened or shortened. This could suggest that clinical manifestations are due to signaling defects and that cilia length alterations per se may not be the primary cause of perturbed signaling. Rather, IFT proteins may regulate both cilia architecture and signaling pathways. Technologies to target a signaling pathway of a specific cell (Shillingford et al., 2012) or cilia (Pala et al., 2019a, b) of a particular cell type may further help provide the molecular tools to treat cilia-related disease.
WW, RM, and PT wrote the first draft. HW constructed Figure 1. WW, BJ, HW, MK, RM, and PT edited the manuscript and approved the final version.
RM and PT were supported by the NIH (R01DK123590 and R01DK103033).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figure 1 was created with BioRender.com.
Abdul-Majeed, S., and Nauli, S. M. (2011). Dopamine receptor type 5 in the primary cilia has dual chemo- and mechano-sensory roles. Hypertension 58, 325–331. doi: 10.1161/hypertensionaha.111.172080
Alazami, A. M., Seidahmed, M. Z., Alzahrani, F., Mohammed, A. O., and Alkuraya, F. S. (2014). Novel IFT122 mutation associated with impaired ciliogenesis and cranioectodermal dysplasia. Mol. Genet. Genomic Med. 2, 103–106. doi: 10.1002/mgg3.44
Anvarian, Z., Mykytyn, K., Mukhopadhyay, S., Pedersen, L. B., and Christensen, S. T. (2019). Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol. 15, 199–219. doi: 10.1038/s41581-019-0116-9
Badano, J. L., Mitsuma, N., Beales, P. L., and Katsanis, N. (2006). The ciliopathies: an emerging class of human genetic disorders. Annu. Rev. Genomics Hum. Genet. 7, 125–148. doi: 10.1146/annurev.genom.7.080505.115610
Badgandi, H. B., Hwang, S. H., Shimada, I. S., Loriot, E., and Mukhopadhyay, S. (2017). Tubby family proteins are adapters for ciliary trafficking of integral membrane proteins. J. Cell Biol. 216, 743–760. doi: 10.1083/jcb.201607095
Berbari, N. F., Kin, N. W., Sharma, N., Michaud, E. J., Kesterson, R. A., and Yoder, B. K. (2011). Mutations in Traf3ip1 reveal defects in ciliogenesis, embryonic development, and altered cell size regulation. Dev. Biol. 360, 66–76. doi: 10.1016/j.ydbio.2011.09.001
Besschetnova, T. Y., Kolpakova-Hart, E., Guan, Y., Zhou, J., Olsen, B. R., and Shah, J. V. (2010). Identification of signaling pathways regulating primary cilium length and flow-mediated adaptation. Curr. Biol. 20, 182–187. doi: 10.1016/j.cub.2009.11.072
Bhogaraju, S., Cajanek, L., Fort, C., Blisnick, T., Weber, K., Taschner, M., et al. (2013). Molecular basis of tubulin transport within the cilium by IFT74 and IFT81. Science 341, 1009–1012. doi: 10.1126/science.1240985
Botilde, Y., Yoshiba, S., Shinohara, K., Hasegawa, T., Nishimura, H., Shiratori, H., et al. (2013). Cluap1 localizes preferentially to the base and tip of cilia and is required for ciliogenesis in the mouse embryo. Dev. Biol. 381, 203–212. doi: 10.1016/j.ydbio.2013.05.024
Bredrup, C., Saunier, S., Oud, M. M., Fiskerstrand, T., Hoischen, A., Brackman, D., et al. (2011). Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am. J. Hum. Genet. 89, 634–643. doi: 10.1016/j.ajhg.2011.10.001
Brown, J. M., Cochran, D. A., Craige, B., Kubo, T., and Witman, G. B. (2015). Assembly of IFT trains at the ciliary base depends on IFT74. Curr. Biol. 25, 1583–1593. doi: 10.1016/j.cub.2015.04.060
Cai, Y., Fedeles, S. V., Dong, K., Anyatonwu, G., Onoe, T., Mitobe, M., et al. (2014). Altered trafficking and stability of polycystins underlie polycystic kidney disease. J. Clin. Invest. 124, 5129–5144. doi: 10.1172/jci67273
Chen, Z., Indjeian, V. B., Mcmanus, M., Wang, L., and Dynlacht, B. D. (2002). CP110, a cell cycle-dependent CDK substrate, regulates centrosome duplication in human cells. Dev. Cell 3, 339–350. doi: 10.1016/s1534-5807(02)00258-7
Chien, A., Shih, S. M., Bower, R., Tritschler, D., Porter, M. E., and Yildiz, A. (2017). Dynamics of the IFT machinery at the ciliary tip. Elife 6:e28606.
Clement, C. A., Ajbro, K. D., Koefoed, K., Vestergaard, M. L., Veland, I. R., Henriques De Jesus, M. P., et al. (2013). TGF-beta signaling is associated with endocytosis at the pocket region of the primary cilium. Cell Rep. 3, 1806–1814. doi: 10.1016/j.celrep.2013.05.020
Cole, D. G., Diener, D. R., Himelblau, A. L., Beech, P. L., Fuster, J. C., and Rosenbaum, J. L. (1998). Chlamydomonas kinesin-II-dependent intraflagellar transport (IFT): IFT particles contain proteins required for ciliary assembly in Caenorhabditis elegans sensory neurons. J. Cell Biol. 141, 993–1008. doi: 10.1083/jcb.141.4.993
Corbit, K. C., Shyer, A. E., Dowdle, W. E., Gaulden, J., Singla, V., Chen, M. H., et al. (2008). Kif3a constrains beta-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat. Cell Biol. 10, 70–76. doi: 10.1038/ncb1670
Craft, J. M., Harris, J. A., Hyman, S., Kner, P., and Lechtreck, K. F. (2015). Tubulin transport by IFT is upregulated during ciliary growth by a cilium-autonomous mechanism. J Cell Biol 208, 223–237. doi: 10.1083/jcb.201409036
Craft Van De Weghe, J., Harris, J. A., Kubo, T., Witman, G. B., and Lechtreck, K. F. (2020). Diffusion rather than intraflagellar transport likely provides most of the tubulin required for axonemal assembly in Chlamydomonas. J. Cell Sci. 133:jcs249805.
Davis, E. E., Zhang, Q., Liu, Q., Diplas, B. H., Davey, L. M., Hartley, J., et al. (2011). TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 43, 189–196. doi: 10.1038/ng.756
Deane, J. A., Cole, D. G., Seeley, E. S., Diener, D. R., and Rosenbaum, J. L. (2001). Localization of intraflagellar transport protein IFT52 identifies basal body transitional fibers as the docking site for IFT particles. Curr. Biol. 11, 1586–1590. doi: 10.1016/s0960-9822(01)00484-5
Diaz, J., Gerard, X., Emerit, M. B., Areias, J., Geny, D., Degardin, J., et al. (2020). YIF1B mutations cause a post-natal neurodevelopmental syndrome associated with Golgi and primary cilium alterations. Brain 143, 2911–2928. doi: 10.1093/brain/awaa235
Duran, I., Taylor, S. P., Zhang, W., Martin, J., Forlenza, K. N., Spiro, R. P., et al. (2016). Destabilization of the IFT-B cilia core complex due to mutations in IFT81 causes a Spectrum of Short-Rib Polydactyly Syndrome. Sci. Rep. 6:34232.
Duran, I., Taylor, S. P., Zhang, W., Martin, J., Qureshi, F., Jacques, S. M., et al. (2017). Mutations in IFT-A satellite core component genes IFT43 and IFT121 produce short rib polydactyly syndrome with distinctive. Cilia 6:7.
Eggenschwiler, J. T., and Anderson, K. V. (2007). Cilia and developmental signaling. Annu Rev Cell Dev Biol 23, 345–373.
Eguether, T., Cordelieres, F. P., and Pazour, G. J. (2018). Intraflagellar transport is deeply integrated in hedgehog signaling. Mol. Biol. Cell 29, 1178–1189. doi: 10.1091/mbc.e17-10-0600
Eguether, T., San Agustin, J. T., Keady, B. T., Jonassen, J. A., Liang, Y., Francis, R., et al. (2014). IFT27 links the BBSome to IFT for maintenance of the ciliary signaling compartment. Dev. Cell. 31, 279–290. doi: 10.1016/j.devcel.2014.09.011
Ezratty, E. J., Stokes, N., Chai, S., Shah, A. S., Williams, S. E., and Fuchs, E. (2011). A role for the primary cilium in Notch signaling and epidermal differentiation during skin development. Cell 145, 1129–1141. doi: 10.1016/j.cell.2011.05.030
Fai, T. G., Mohapatra, L., Kar, P., Kondev, J., and Amir, A. (2019). Length regulation of multiple flagella that self-assemble from a shared pool of components. Elife 8:e42599.
Follit, J. A., Tuft, R. A., Fogarty, K. E., and Pazour, G. J. (2006). The intraflagellar transport protein IFT20 is associated with the Golgi complex and is required for cilia assembly. Mol. Biol. Cell 17, 3781–3792. doi: 10.1091/mbc.e06-02-0133
Forcioli-Conti, N., Lacas-Gervais, S., Dani, C., and Peraldi, P. (2015). The primary cilium undergoes dynamic size modifications during adipocyte differentiation of human adipose stem cells. Biochem. Biophys. Res. Commun. 458, 117–122. doi: 10.1016/j.bbrc.2015.01.078
Freedman, B. S., Lam, A. Q., Sundsbak, J. L., Iatrino, R., Su, X., Koon, S. J., et al. (2013). Reduced ciliary polycystin-2 in induced pluripotent stem cells from polycystic kidney disease patients with PKD1 mutations. J. Am. Soc. Nephrol. 24, 1571–1586. doi: 10.1681/asn.2012111089
Fu, W., Wang, L., Kim, S., Li, J., and Dynlacht, B. D. (2016). Role for the IFT-A complex in selective transport to the primary cilium. Cell Rep. 17, 1505–1517. doi: 10.1016/j.celrep.2016.10.018
Funabashi, T., Katoh, Y., Michisaka, S., Terada, M., Sugawa, M., and Nakayama, K. (2017). Ciliary entry of KIF17 is dependent on its binding to the IFT-B complex via IFT46-IFT56 as well as on its nuclear localization signal. Mol. Biol. Cell 28, 624–633. doi: 10.1091/mbc.e16-09-0648
Gadadhar, S., Dadi, H., Bodakuntla, S., Schnitzler, A., Bieche, I., Rusconi, F., et al. (2017). Tubulin glycylation controls primary cilia length. J. Cell Biol. 216, 2701–2713. doi: 10.1083/jcb.201612050
Garcia-Gonzalo, F. R., Corbit, K. C., Sirerol-Piquer, M. S., Ramaswami, G., Otto, E. A., Noriega, T. R., et al. (2011). A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat. Genet. 43, 776–784. doi: 10.1038/ng.891
Garcia-Gonzalo, F. R., and Reiter, J. F. (2017). Open sesame: how transition fibers and the transition zone control ciliary composition. Cold Spring Harb Perspect Biol. 9:a028134. doi: 10.1101/cshperspect.a028134
Gerakopoulos, V., Ngo, P., and Tsiokas, L. (2020). Loss of polycystins suppresses deciliation via the activation of the centrosomal integrity pathway. Life Sci. Alliance 3:e202000750. doi: 10.26508/lsa.202000750
Gonçalves, J., and Pelletier, L. (2017). The ciliary transition zone: finding the pieces and assembling the gate. Mol. Cells 40, 243–253. doi: 10.14348/molcells.2017.0054
Gorivodsky, M., Mukhopadhyay, M., Wilsch-Braeuninger, M., Phillips, M., Teufel, A., Kim, C., et al. (2009). Intraflagellar transport protein 172 is essential for primary cilia formation and plays a vital role in patterning the mammalian brain. Dev. Biol. 325, 24–32. doi: 10.1016/j.ydbio.2008.09.019
Halbritter, J., Bizet, A. A., Schmidts, M., Porath, J. D., Braun, D. A., Gee, H. Y., et al. (2013). Defects in the IFT-B component IFT172 cause Jeune and Mainzer-Saldino syndromes in humans. Am. J. Hum. Genet. 93, 915–925. doi: 10.1016/j.ajhg.2013.09.012
Han, S., Miyoshi, K., Shikada, S., Amano, G., Wang, Y., Yoshimura, T., et al. (2019). TULP3 is required for localization of membrane-associated proteins ARL13B and INPP5E to primary cilia. Biochem. Biophys. Res. Commun. 509, 227–234. doi: 10.1016/j.bbrc.2018.12.109
Han, S. J., Jang, H. S., Seu, S. Y., Cho, H. J., Hwang, Y. J., Kim, J. I., et al. (2017). Hepatic ischemia/reperfusion injury disrupts the homeostasis of kidney primary cilia via oxidative stress. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 1817–1828. doi: 10.1016/j.bbadis.2017.05.004
Han, Y. M., Kang, G. M., Byun, K., Ko, H. W., Kim, J., Shin, M. S., et al. (2014). Leptin-promoted cilia assembly is critical for normal energy balance. J. Clin. Invest. 124, 2193–2197. doi: 10.1172/jci69395
Harris, R. C., and Zhang, M. Z. (2012). Dopamine, the kidney, and hypertension. Curr. Hypertens. Rep. 14, 138–143. doi: 10.1007/s11906-012-0253-z
Hart, G. W., Slawson, C., Ramirez-Correa, G., and Lagerlof, O. (2011). Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 80, 825–858. doi: 10.1146/annurev-biochem-060608-102511
Haycraft, C. J., Zhang, Q., Song, B., Jackson, W. S., Detloff, P. J., Serra, R., et al. (2007). Intraflagellar transport is essential for endochondral bone formation. Development 134, 307–316. doi: 10.1242/dev.02732
He, K., Ma, X., Xu, T., Li, Y., Hodge, A., Zhang, Q., et al. (2018). Axoneme polyglutamylation regulated by Joubert syndrome protein ARL13B controls ciliary targeting of signaling molecules. Nat. Commun. 9:3310.
Hildebrandt, F., Benzing, T., and Katsanis, N. (2011). Ciliopathies. N. Engl. J. Med. 364, 1533–1543.
Hilgendorf, K. I., Johnson, C. T., Mezger, A., Rice, S. L., Norris, A. M., Demeter, J., et al. (2019). Omega-3 fatty acids activate ciliary FFAR4 to control Adipogenesis. Cell 179:e1221.
Hirano, T., Katoh, Y., and Nakayama, K. (2017). Intraflagellar transport-A complex mediates ciliary entry and retrograde trafficking of ciliary G protein-coupled receptors. Mol. Biol. Cell 28, 429–439. doi: 10.1091/mbc.e16-11-0813
Hopp, K., Ward, C. J., Hommerding, C. J., Nasr, S. H., Tuan, H. F., Gainullin, V. G., et al. (2012). Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J. Clin. Invest. 122, 4257–4273. doi: 10.1172/jci64313
Huang, K., Diener, D. R., and Rosenbaum, J. L. (2009). The ubiquitin conjugation system is involved in the disassembly of cilia and flagella. J. Cell Biol. 186, 601–613. doi: 10.1083/jcb.200903066
Huangfu, D., Liu, A., Rakeman, A. S., Murcia, N. S., Niswander, L., and Anderson, K. V. (2003). Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 426, 83–87. doi: 10.1038/nature02061
Husson, H., Moreno, S., Smith, L. A., Smith, M. M., Russo, R. J., Pitstick, R., et al. (2016). Reduction of ciliary length through pharmacologic or genetic inhibition of CDK5 attenuates polycystic kidney disease in a model of nephronophthisis. Hum. Mol. Genet. 25, 2245–2255. doi: 10.1093/hmg/ddw093
Hwang, S. H., Somatilaka, B. N., Badgandi, H., Palicharla, V. R., Walker, R., Shelton, J. M., et al. (2019). Tulp3 regulates renal cystogenesis by trafficking of cystoproteins to cilia. Curr. Biol. 29:e795.
Ishikawa, H., Ide, T., Yagi, T., Jiang, X., Hirono, M., Sasaki, H., et al. (2014). TTC26/DYF13 is an intraflagellar transport protein required for transport of motility-related proteins into flagella. Elife 3:e01566.
Jacobs, D. T., Allard, B. A., Pottorf, T. S., Silva, L. M., Wang, W., Al-Naamani, A., et al. (2020). Intraflagellar-transport A dysfunction causes hyperphagia-induced systemic insulin resistance in a pre-obese state. FASEB J. 34, 148–160. doi: 10.1096/fj.201900751r
Jain, R., Javidan-Nejad, C., Alexander-Brett, J., Horani, A., Cabellon, M. C., Walter, M. J., et al. (2012). Sensory functions of motile cilia and implication for bronchiectasis. Front. Biosci. 4:1088–1098. doi: 10.2741/s320
Johnson, K. A., and Rosenbaum, J. L. (1992). Polarity of flagellar assembly in Chlamydomonas. J. Cell Biol. 119, 1605–1611. doi: 10.1083/jcb.119.6.1605
Jonassen, J. A., San Agustin, J., Follit, J. A., and Pazour, G. J. (2008). Deletion of IFT20 in the mouse kidney causes misorientation of the mitotic spindle and cystic kidney disease. J. Cell Biol. 183, 377–384. doi: 10.1083/jcb.200808137
Jonassen, J. A., Sanagustin, J., Baker, S. P., and Pazour, G. J. (2012). Disruption of IFT complex A causes cystic kidneys without mitotic spindle misorientation. J. Am. Soc. Nephrol. 23, 641–651. doi: 10.1681/asn.2011080829
Katoh, Y., Terada, M., Nishijima, Y., Takei, R., Nozaki, S., Hamada, H., et al. (2016). Overall architecture of the intraflagellar transport (IFT)-B complex containing cluap1/IFT38 as an essential component of the IFT-B peripheral subcomplex. J. Biol. Chem. 291, 10962–10975. doi: 10.1074/jbc.m116.713883
Keady, B. T., Samtani, R., Tobita, K., Tsuchya, M., San Agustin, J. T., Follit, J. A., et al. (2012). IFT25 links the signal-dependent movement of Hedgehog components to intraflagellar transport. Dev. Cell 22, 940–951. doi: 10.1016/j.devcel.2012.04.009
Kim, J. I., Kim, J., Jang, H. S., Noh, M. R., Lipschutz, J. H., and Park, K. M. (2013). Reduction of oxidative stress during recovery accelerates normalization of primary cilia length that is altered after ischemic injury in murine kidneys. Am. J. Physiol. Renal. Physio.l 304, F1283–F1294.
Kimura, Y., Tsutsumi, K., Konno, A., Ikegami, K., Hameed, S., Kaneko, T., et al. (2018). Environmental responsiveness of tubulin glutamylation in sensory cilia is regulated by the p38 MAPK pathway. Sci. Rep. 8:8392.
Kiseleva, A. A., Korobeynikov, V. A., Nikonova, A. S., Zhang, P., Makhov, P., Deneka, A. Y., et al. (2019). Unexpected activities in regulating ciliation contribute to off-target effects of targeted drugs. Clin. Cancer Res. 25, 4179–4193. doi: 10.1158/1078-0432.ccr-18-3535
Kobayashi, T., Ishida, Y., Hirano, T., Katoh, Y., and Nakayama, K. (2021). Cooperation of the IFT-A complex with the IFT-B complex is required for ciliary retrograde protein trafficking and GPCR import. Mol. Biol. Cell 32, 45–56. doi: 10.1091/mbc.e20-08-0556
Komarynets, O., Chassot, A., Bernabeu, E., Czogalla, J., Roth, I., Liaudet, N., et al. (2020). Aldosterone controls primary cilium length and cell size in renal collecting duct principal cells. FASEB J. 34, 2625–2640. doi: 10.1096/fj.201901947r
Kozminski, K. G., Johnson, K. A., Forscher, P., and Rosenbaum, J. L. (1993). A motility in the eukaryotic flagellum unrelated to flagellar beating. Proc. Natl. Acad. Sci. U.S.A. 90, 5519–5523. doi: 10.1073/pnas.90.12.5519
Kubo, T., Brown, J. M., Bellve, K., Craige, B., Craft, J. M., Fogarty, K., et al. (2016). Together, the IFT81 and IFT74 N-termini form the main module for intraflagellar transport of tubulin. J Cell Sci 129, 2106–2119. doi: 10.1242/jcs.187120
Kunova Bosakova, M., Varecha, M., Hampl, M., Duran, I., Nita, A., Buchtova, M., et al. (2018). Regulation of ciliary function by fibroblast growth factor signaling identifies FGFR3-related disorders achondroplasia and thanatophoric dysplasia as ciliopathies. Hum. Mol. Genet. 27, 1093–1105. doi: 10.1093/hmg/ddy031
Leaf, A., and Von Zastrow, M. (2015). Dopamine receptors reveal an essential role of IFT-B, KIF17, and Rab23 in delivering specific receptors to primary cilia. Elife 4:e06996.
Lee, C. H., Song, D. K., Park, C. B., Choi, J., Kang, G. M., Shin, S. H., et al. (2020). Primary cilia mediate early life programming of adiposity through lysosomal regulation in the developing mouse hypothalamus. Nat. Commun. 11:5772.
Lee, M. S., Hwang, K. S., Oh, H. W., Ji-Ae, K., Kim, H. T., Cho, H. S., et al. (2015). IFT46 plays an essential role in cilia development. Dev. Biol. 400, 248–257. doi: 10.1016/j.ydbio.2015.02.009
Legue, E., and Liem, K. F. Jr. (2019). Tulp3 Is a ciliary trafficking gene that regulates polycystic kidney disease. Curr. Biol. 29:e805.
Li, Y. H., Zhu, D., Cao, Z., Liu, Y., Sun, J., and Tan, L. (2020). Primary cilia respond to intermittent low-magnitude, high-frequency vibration and mediate vibration-induced effects in osteoblasts. Am. J. Physiol. Cell Physiol. 318, C73–C82.
Liem, K. F. Jr., Ashe, A., He, M., Satir, P., Moran, J., Beier, D., et al. (2012). The IFT-A complex regulates Shh signaling through cilia structure and membrane protein trafficking. J. Cell Biol. 197, 789–800. doi: 10.1083/jcb.201110049
Liew, G. M., Ye, F., Nager, A. R., Murphy, J. P., Lee, J. S., Aguiar, M., et al. (2014). The intraflagellar transport protein IFT27 promotes BBSome exit from cilia through the GTPase ARL6/BBS3. Dev. Cell 31, 265–278. doi: 10.1016/j.devcel.2014.09.004
Lim, J., Li, X., Yuan, X., Yang, S., Han, L., and Yang, S. (2020). Primary cilia control cell alignment and patterning in bone development via ceramide-PKCzeta-beta-catenin signaling. Commun. Bio.l 3:45.
Liu, M., Alharbi, M., Graves, D., and Yang, S. (2020). IFT80 is required for fracture healing through controlling the regulation of TGF-beta signaling in chondrocyte differentiation and function. J. Bone Miner Res. 35, 571–582. doi: 10.1002/jbmr.3902
Liu, X., Vien, T., Duan, J., Sheu, S. H., Decaen, P. G., and Clapham, D. E. (2018). Polycystin-2 is an essential ion channel subunit in the primary cilium of the renal collecting duct epithelium. Elife 7:e33183.
Long, H., and Huang, K. (2019). Transport of ciliary membrane proteins. Front. Cell Dev. Biol. 7:381.
Long, H., Zhang, F., Xu, N., Liu, G., Diener, D. R., Rosenbaum, J. L., et al. (2016). Comparative analysis of ciliary membranes and ectosomes. Curr. Biol. 26, 3327–3335. doi: 10.1016/j.cub.2016.09.055
Lu, H., Toh, M. T., Narasimhan, V., Thamilselvam, S. K., Choksi, S. P., and Roy, S. (2015). A function for the Joubert syndrome protein Arl13b in ciliary membrane extension and ciliary length regulation. Dev. Biol. 397, 225–236. doi: 10.1016/j.ydbio.2014.11.009
Ma, M., Tian, X., Igarashi, P., Pazour, G. J., and Somlo, S. (2013). Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat. Genet. 45, 1004–1012. doi: 10.1038/ng.2715
Malicki, J. J., and Johnson, C. A. (2017). The cilium: cellular antenna and central processing unit. Trends Cell Biol. 27, 126–140. doi: 10.1016/j.tcb.2016.08.002
Marshall, W. F., Qin, H., Rodrigo Brenni, M., and Rosenbaum, J. L. (2005). Flagellar length control system: testing a simple model based on intraflagellar transport and turnover. Mol. Biol. Cell 16, 270–278. doi: 10.1091/mbc.e04-07-0586
Marshall, W. F., and Rosenbaum, J. L. (2001). Intraflagellar transport balances continuous turnover of outer doublet microtubules: implications for flagellar length control. J. Cell Biol. 155, 405–414. doi: 10.1083/jcb.200106141
Martin, L., Kaci, N., Estibals, V., Goudin, N., Garfa-Traore, M., Benoist-Lasselin, C., et al. (2018). Constitutively-active FGFR3 disrupts primary cilium length and IFT20 trafficking in various chondrocyte models of achondroplasia. Hum. Mol. Genet. 27, 1–13. doi: 10.1093/hmg/ddx374
Miller, K. A., Ah-Cann, C. J., Welfare, M. F., Tan, T. Y., Pope, K., Caruana, G., et al. (2013). Cauli: a mouse strain with an Ift140 mutation that results in a skeletal ciliopathy modelling Jeune syndrome. PLoS Genet. 9:e1003746. doi: 10.1371/journal.pgen.1003746
Mirvis, M., Siemers, K. A., Nelson, W. J., and Stearns, T. P. (2019). Primary cilium loss in mammalian cells occurs predominantly by whole-cilium shedding. PLoS Biol. 17:e3000381. doi: 10.1371/journal.pbio.3000381
Mukherjee, M., Ratnayake, I., Janga, M., Fogarty, E., Scheidt, S., Grassmeyer, J., et al. (2020). Notch signaling regulates Akap12 expression and primary cilia length during renal tubule morphogenesis. FASEB J. 34, 9512–9530. doi: 10.1096/fj.201902358rr
Mukhopadhyay, S., Wen, X., Chih, B., Nelson, C. D., Lane, W. S., Scales, S. J., et al. (2010). TULP3 bridges the IFT-A complex and membrane phosphoinositides to promote trafficking of G protein-coupled receptors into primary cilia. Genes. Dev. 24, 2180–2193. doi: 10.1101/gad.1966210
Mukhopadhyay, S., Wen, X., Ratti, N., Loktev, A., Rangell, L., Scales, S. J., et al. (2013). The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell 152, 210–223. doi: 10.1016/j.cell.2012.12.026
Nachury, M. V. (2018). The molecular machines that traffic signaling receptors into and out of cilia. Curr. Opin. Cell Biol 51, 124–131. doi: 10.1016/j.ceb.2018.03.004
Nager, A. R., Goldstein, J. S., Herranz-Perez, V., Portran, D., Ye, F., Garcia-Verdugo, J. M., et al. (2017). An actin network dispatches ciliary gpcrs into extracellular vesicles to modulate signaling. Cell 168:e214.
Nakamura, K., Noguchi, T., Takahara, M., Omori, Y., Furukawa, T., Katoh, Y., et al. (2020). Anterograde trafficking of ciliary MAP kinase-like ICK/CILK1 by the intraflagellar transport machinery is required for intraciliary retrograde protein trafficking. J. Biol. Chem. 295, 13363–13376. doi: 10.1074/jbc.ra120.014142
Nakayama, K., and Katoh, Y. (2020). Architecture of the IFT ciliary trafficking machinery and interplay between its components. Crit. Rev. Biochem. Mol. Biol. 55, 179–196. doi: 10.1080/10409238.2020.1768206
Nandadasa, S., Kraft, C. M., Wang, L. W., O’donnell, A., Patel, R., Gee, H. Y., et al. (2019). Secreted metalloproteases ADAMTS9 and ADAMTS20 have a non-canonical role in ciliary vesicle growth during ciliogenesis. Nat. Commun. 10:953.
Nauli, S. M., Alenghat, F. J., Luo, Y., Williams, E., Vassilev, P., Li, X., et al. (2003). Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 33, 129–137. doi: 10.1038/ng1076
Nigg, E. A., and Stearns, T. (2011). The centrosome cycle: centriole biogenesis, duplication and inherent asymmetries. Nat. Cell Biol 13, 1154–1160. doi: 10.1038/ncb2345
Nikonova, A. S., Plotnikova, O. V., Serzhanova, V., Efimov, A., Bogush, I., Cai, K. Q., et al. (2014). Nedd9 restrains renal cystogenesis in Pkd1-/- mice. Proc. Natl. Acad. Sci. U.S.A. 111, 12859–12864. doi: 10.1073/pnas.1405362111
Ocbina, P. J., Eggenschwiler, J. T., Moskowitz, I., and Anderson, K. V. (2011). Complex interactions between genes controlling trafficking in primary cilia. Nat Genet 43, 547–553. doi: 10.1038/ng.832
Odabasi, E., Gul, S., Kavakli, I. H., and Firat-Karalar, E. N. (2019). Centriolar satellites are required for efficient ciliogenesis and ciliary content regulation. EMBO Rep. 20, e47723.
O’hagan, R., Piasecki, B. P., Silva, M., Phirke, P., Nguyen, K. C., Hall, D. H., et al. (2011). The tubulin deglutamylase CCPP-1 regulates the function and stability of sensory cilia in C. elegans. Curr. Biol. 21, 1685–1694. doi: 10.1016/j.cub.2011.08.049
Olson, R. J., Hopp, K., Wells, H., Smith, J. M., Furtado, J., Constans, M. M., et al. (2019). Synergistic genetic interactions between Pkhd1 and Pkd1 result in an ARPKD-Like phenotype in murine models. J. Am. Soc. Nephrol. 30, 2113–2127. doi: 10.1681/ASN.2019020150
Orhon, I., Dupont, N., Zaidan, M., Boitez, V., Burtin, M., Schmitt, A., et al. (2016). Primary-cilium-dependent autophagy controls epithelial cell volume in response to fluid flow. Nat. Cell Biol. 18, 657–667. doi: 10.1038/ncb3360
Pala, R., Mohieldin, A. M., Shamloo, K., Sherpa, R. T., Kathem, S. H., Zhou, J., et al. (2019a). Personalized nanotherapy by specifically targeting cell organelles to improve vascular hypertension. Nano Lett. 19, 904–914. doi: 10.1021/acs.nanolett.8b04138
Pala, R., Mohieldin, A. M., Sherpa, R. T., Kathem, S. H., Shamloo, K., Luan, Z., et al. (2019b). Ciliotherapy: remote control of primary cilia movement and function by magnetic nanoparticles. ACS Nano 13, 3555–3572.
Pampliega, O., Orhon, I., Patel, B., Sridhar, S., Diaz-Carretero, A., Beau, I., et al. (2013). Functional interaction between autophagy and ciliogenesis. Nature 502, 194–200. doi: 10.1038/nature12639
Pan, J., Seeger-Nukpezah, T., and Golemis, E. A. (2013). The role of the cilium in normal and abnormal cell cycles: emphasis on renal cystic pathologies. Cell Mol. Life Sci. 70, 1849–1874. doi: 10.1007/s00018-012-1052-z
Paridaen, J. T., Wilsch-Brauninger, M., and Huttner, W. B. (2013). Asymmetric inheritance of centrosome-associated primary cilium membrane directs ciliogenesis after cell division. Cell 155, 333–344. doi: 10.1016/j.cell.2013.08.060
Park, K. M. (2018). Can tissue cilia lengths and urine cilia proteins be markers of kidney diseases? Chonnam. Med. J. 54, 83–89. doi: 10.4068/cmj.2018.54.2.83
Patel, V., Li, L., Cobo-Stark, P., Shao, X., Somlo, S., Lin, F., et al. (2008). Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Hum. Mol. Genet. 17, 1578–1590. doi: 10.1093/hmg/ddn045
Pazour, G. J., Dickert, B. L., Vucica, Y., Seeley, E. S., Rosenbaum, J. L., Witman, G. B., et al. (2000). Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J. Cell Biol. 151, 709–718. doi: 10.1083/jcb.151.3.709
Pazour, G. J., Dickert, B. L., and Witman, G. B. (1999). The DHC1b (DHC2) isoform of cytoplasmic dynein is required for flagellar assembly. J. Cell Biol. 144, 473–481. doi: 10.1083/jcb.144.3.473
Pazour, G. J., Wilkerson, C. G., and Witman, G. B. (1998). A dynein light chain is essential for the retrograde particle movement of intraflagellar transport (IFT). J. Cell Biol. 141, 979–992. doi: 10.1083/jcb.141.4.979
Perrault, I., Halbritter, J., Porath, J. D., Gerard, X., Braun, D. A., Gee, H. Y., et al. (2015). IFT81, encoding an IFT-B core protein, as a very rare cause of a ciliopathy phenotype. J. Med. Genet. 52, 657–665. doi: 10.1136/jmedgenet-2014-102838
Phua, S. C., Chiba, S., Suzuki, M., Su, E., Roberson, E. C., Pusapati, G. V., et al. (2017). Dynamic remodeling of membrane composition drives cell cycle through primary cilia excision. Cell 168:e215.
Picariello, T., Brown, J. M., Hou, Y., Swank, G., Cochran, D. A., King, O. D., et al. (2019). A global analysis of IFT-A function reveals specialization for transport of membrane-associated proteins into cilia. J. Cell Sci. 132, jcs220749.
Poole, C. A., Flint, M. H., and Beaumont, B. W. (1985). Analysis of the morphology and function of primary cilia in connective tissues: a cellular cybernetic probe? Cell Motil. 5, 175–193. doi: 10.1002/cm.970050302
Porter, M. E., Bower, R., Knott, J. A., Byrd, P., and Dentler, W. (1999). Cytoplasmic dynein heavy chain 1b is required for flagellar assembly in Chlamydomonas. Mol. Biol. Cell 10, 693–712. doi: 10.1091/mbc.10.3.693
Praetorius, H. A., and Spring, K. R. (2001). Bending the MDCK cell primary cilium increases intracellular calcium. J. Membr. Biol. 184, 71–79. doi: 10.1007/s00232-001-0075-4
Praetorius, H. A., and Spring, K. R. (2003). Removal of the MDCK cell primary cilium abolishes flow sensing. J. Membr. Biol. 191, 69–76. doi: 10.1007/s00232-002-1042-4
Pugacheva, E. N., Jablonski, S. A., Hartman, T. R., Henske, E. P., and Golemis, E. A. (2007). HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 129, 1351–1363. doi: 10.1016/j.cell.2007.04.035
Qin, J., Lin, Y., Norman, R. X., Ko, H. W., and Eggenschwiler, J. T. (2011). Intraflagellar transport protein 122 antagonizes Sonic Hedgehog signaling and controls ciliary localization of pathway components. Proc. Natl. Acad. Sci. U.S.A. 108, 1456–1461. doi: 10.1073/pnas.1011410108
R Ferreira, R., Fukui, H., Chow, R., Vilfan, A., and Vermot, J. (2019). The cilium as a force sensor-myth versus reality. J. Cell Sci. 132, jcs213496.
Ritter, A., Friemel, A., Kreis, N. N., Hoock, S. C., Roth, S., Kielland-Kaisen, U., et al. (2018). Primary cilia are dysfunctional in obese adipose-derived mesenchymal stem cells. Stem Cell Rep. 10, 583–599. doi: 10.1016/j.stemcr.2017.12.022
Rix, S., Calmont, A., Scambler, P. J., and Beales, P. L. (2011). An Ift80 mouse model of short rib polydactyly syndromes shows defects in hedgehog signalling without loss or malformation of cilia. Hum. Mol. Genet. 20, 1306–1314. doi: 10.1093/hmg/ddr013
Rosenbaum, J. L., and Witman, G. B. (2002). Intraflagellar transport. Nat. Rev. Mol. Cell Biol. 3, 813–825.
Saraga-Babic, M., Vukojevic, K., Bocina, I., Drnasin, K., and Saraga, M. (2012). Ciliogenesis in normal human kidney development and post-natal life. Pediatr. Nephrol. 27, 55–63. doi: 10.1007/s00467-011-1941-7
Satir, P. (2005). Tour of organelles through the electron microscope: a reprinting of Keith R. Porter’s classic harvey lecture with a new introduction. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 287, 1184–1185. doi: 10.1002/ar.a.20222
Schneider, L., Clement, C. A., Teilmann, S. C., Pazour, G. J., Hoffmann, E. K., Satir, P., et al. (2005). PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr. Biol. 15, 1861–1866. doi: 10.1016/j.cub.2005.09.012
Shaheen, R., Alsahli, S., Ewida, N., Alzahrani, F., Shamseldin, H. E., Patel, N., et al. (2020). Biallelic mutations in tetratricopeptide repeat domain 26 (Intraflagellar Transport 56) cause severe biliary ciliopathy in humans. Hepatology 71, 2067–2079. doi: 10.1002/hep.30982
Shamloo, K., Chen, J., Sardar, J., Sherpa, R. T., Pala, R., Atkinson, K. F., et al. (2017). Chronic hypobaric hypoxia modulates primary cilia differently in adult and fetal ovine kidneys. Front. Physiol. 8:677.
Shao, L., El-Jouni, W., Kong, F., Ramesh, J., Kumar, R. S., Shen, X., et al. (2020). Genetic reduction of cilium-length by targeting intraflagellar transport 88 protein impedes kidney and liver cysts formation in mouse models of autosomal polycystic kidney disease. Kidney Int. 98, 1225–1241. doi: 10.1016/j.kint.2020.05.049
Shida, T., Cueva, J. G., Xu, Z., Goodman, M. B., and Nachury, M. V. (2010). The major alpha-tubulin K40 acetyltransferase alphaTAT1 promotes rapid ciliogenesis and efficient mechanosensation. Proc. Natl. Acad. Sci. U.S.A. 107, 21517–21522. doi: 10.1073/pnas.1013728107
Shillingford, J. M., Leamon, C. P., Vlahov, I. R., and Weimbs, T. (2012). Folate-conjugated rapamycin slows progression of polycystic kidney disease. J. Am. Soc. Nephrol. 23, 1674–1681. doi: 10.1681/asn.2012040367
Signor, D., Wedaman, K. P., Orozco, J. T., Dwyer, N. D., Bargmann, C. I., Rose, L. S., et al. (1999). Role of a class DHC1b dynein in retrograde transport of IFT motors and IFT raft particles along cilia, but not dendrites, in chemosensory neurons of living Caenorhabditis elegans. J. Cell Biol. 147, 519–530. doi: 10.1083/jcb.147.3.519
Smith, L. A., Bukanov, N. O., Husson, H., Russo, R. J., Barry, T. C., Taylor, A. L., et al. (2006). Development of polycystic kidney disease in juvenile cystic kidney mice: insights into pathogenesis, ciliary abnormalities, and common features with human disease. J. Am. Soc. Nephrol. 17, 2821–2831. doi: 10.1681/asn.2006020136
Sorokin, S. (1962). Centrioles and the formation of rudimentary cilia by fibroblasts and smooth muscle cells. J. Cell Biol. 15, 363–377. doi: 10.1083/jcb.15.2.363
Spasic, M., and Jacobs, C. R. (2017). Lengthening primary cilia enhances cellular mechanosensitivity. Eur. Cell Mater. 33, 158–168. doi: 10.22203/ecm.v033a12
Srivastava, S., Ramsbottom, S. A., Molinari, E., Alkanderi, S., Filby, A., White, K., et al. (2017). A human patient-derived cellular model of Joubert syndrome reveals ciliary defects which can be rescued with targeted therapies. Hum. Mol. Genet. 26, 4657–4667. doi: 10.1093/hmg/ddx347
Stephan, A., Vaughan, S., Shaw, M. K., Gull, K., and Mckean, P. G. (2007). An essential quality control mechanism at the eukaryotic basal body prior to intraflagellar transport. Traffic 8, 1323–1330. doi: 10.1111/j.1600-0854.2007.00611.x
Streets, A. J., Prosseda, P. P., and Ong, A. C. (2020). Polycystin-1 regulates ARHGAP35-dependent centrosomal RhoA activation and ROCK signaling. JCI Insight 5:e135385.
Stroope, A., Radtke, B., Huang, B., Masyuk, T., Torres, V., Ritman, E., et al. (2010). Hepato-renal pathology in pkd2ws25/- mice, an animal model of autosomal dominant polycystic kidney disease. Am. J. Pathol. 176, 1282–1291. doi: 10.2353/ajpath.2010.090658
Szempruch, A. J., Sykes, S. E., Kieft, R., Dennison, L., Becker, A. C., Gartrell, A., et al. (2016). Extracellular vesicles from trypanosoma brucei mediate virulence factor transfer and cause Host Anemia. Cell 164, 246–257. doi: 10.1016/j.cell.2015.11.051
Takahara, M., Katoh, Y., Nakamura, K., Hirano, T., Sugawa, M., Tsurumi, Y., et al. (2018). Ciliopathy-associated mutations of IFT122 impair ciliary protein trafficking but not ciliogenesis. Hum. Mol. Genet. 27, 516–528. doi: 10.1093/hmg/ddx421
Takei, R., Katoh, Y., and Nakayama, K. (2018). Robust interaction of IFT70 with IFT52-IFT88 in the IFT-B complex is required for ciliogenesis. Biol. Open 7:bio033241.
Taschner, M., Weber, K., Mourao, A., Vetter, M., Awasthi, M., Stiegler, M., et al. (2016). Intraflagellar transport proteins 172, 80, 57, 54, 38, and 20 form a stable tubulin-binding IFT-B2 complex. EMBO J. 35, 773–790. doi: 10.15252/embj.201593164
Teves, M. E., Sundaresan, G., Cohen, D. J., Hyzy, S. L., Kajan, I., Maczis, M., et al. (2015). Spag17 deficiency results in skeletal malformations and bone abnormalities. PLoS One 10:e0125936. doi: 10.1371/journal.pone.0125936
Thevenon, J., Duplomb, L., Phadke, S., Eguether, T., Saunier, A., Avila, M., et al. (2016). Autosomal recessive IFT57 hypomorphic mutation cause ciliary transport defect in unclassified oral-facial-digital syndrome with short stature and brachymesophalangia. Clin. Genet. 90, 509–517. doi: 10.1111/cge.12785
Tian, J. L., and Qin, H. (2019). O-GlcNAcylation regulates primary ciliary length by promoting microtubule disassembly. iScience 12, 379–391. doi: 10.1016/j.isci.2019.01.031
Tran, P. V., Haycraft, C. J., Besschetnova, T. Y., Turbe-Doan, A., Stottmann, R. W., Herron, B. J., et al. (2008). THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia. Nat. Genet. 40, 403–410. doi: 10.1038/ng.105
Tran, P. V., Talbott, G. C., Turbe-Doan, A., Jacobs, D. T., Schonfeld, M. P., Silva, L. M., et al. (2014). Downregulating hedgehog signaling reduces renal cystogenic potential of mouse models. J. Am. Soc. Nephrol. 25, 2201–2212. doi: 10.1681/asn.2013070735
Upadhyay, V. S., Muntean, B. S., Kathem, S. H., Hwang, J. J., Aboualaiwi, W. A., and Nauli, S. M. (2014). Roles of dopamine receptor on chemosensory and mechanosensory primary cilia in renal epithelial cells. Front. Physiol. 5:72.
Verghese, E., Martelotto, L. G., Cain, J. E., Williams, T. M., Wise, A. F., Hill, P. A., et al. (2019). Renal epithelial cells retain primary cilia during human acute renal allograft rejection injury. BMC Res. Notes 12:718.
Verghese, E., Ricardo, S. D., Weidenfeld, R., Zhuang, J., Hill, P. A., Langham, R. G., et al. (2009). Renal primary cilia lengthen after acute tubular necrosis. J. Am. Soc. Nephrol. 20, 2147–2153. doi: 10.1681/asn.2008101105
Verghese, E., Weidenfeld, R., Bertram, J. F., Ricardo, S. D., and Deane, J. A. (2008). Renal cilia display length alterations following tubular injury and are present early in epithelial repair. Nephrol. Dial. Transplant 23, 834–841. doi: 10.1093/ndt/gfm743
Verghese, E., Zhuang, J., Saiti, D., Ricardo, S. D., and Deane, J. A. (2011). In vitro investigation of renal epithelial injury suggests that primary cilium length is regulated by hypoxia-inducible mechanisms. Cell Biol. Int 35, 909–913. doi: 10.1042/cbi20090154
Viau, A., Bienaime, F., Lukas, K., Todkar, A. P., Knoll, M., Yakulov, T. A., et al. (2018). Cilia-localized LKB1 regulates chemokine signaling, macrophage recruitment, and tissue homeostasis in the kidney. EMBO J. 37:e98615.
Walker, R. V., Keynton, J. L., Grimes, D. T., Sreekumar, V., Williams, D. J., Esapa, C., et al. (2019). Ciliary exclusion of Polycystin-2 promotes kidney cystogenesis in an autosomal dominant polycystic kidney disease model. Nat. Commun. 10:4072.
Wang, J., Silva, M., Haas, L. A., Morsci, N. S., Nguyen, K. C., Hall, D. H., et al. (2014). C. elegans ciliated sensory neurons release extracellular vesicles that function in animal communication. Curr. Biol. 24, 519–525. doi: 10.1016/j.cub.2014.01.002
Wang, L., and Dynlacht, B. D. (2018). The regulation of cilium assembly and disassembly in development and disease. Development 145:dev151407.
Wang, Q., Peng, Z., Long, H., Deng, X., and Huang, K. (2019). Polyubiquitylation of alpha-tubulin at K304 is required for flagellar disassembly in Chlamydomonas. J. Cell Sci. 132:jcs229047.
Wang, Q., Taschner, M., Ganzinger, K. A., Kelley, C., Villasenor, A., Heymann, M., et al. (2018). Membrane association and remodeling by intraflagellar transport protein IFT172. Nat. Commun. 9:4684.
Wang, W., Allard, B. A., Pottorf, T. S., Wang, H. H., Vivian, J. L., and Tran, P. V. (2020). Genetic interaction of mammalian IFT-A paralogs regulates cilia disassembly, ciliary entry of membrane protein, Hedgehog signaling, and embryogenesis. FASEB J. 34, 6369–6381. doi: 10.1096/fj.201902611r
Wemmer, K., Ludington, W., and Marshall, W. F. (2020). Testing the role of intraflagellar transport in flagellar length control using length-altering mutants of Chlamydomonas. Philos. Trans. R. Soc. Lond B Biol. Sci. 375:20190159. doi: 10.1098/rstb.2019.0159
Westlake, C. J., Baye, L. M., Nachury, M. V., Wright, K. J., Ervin, K. E., Phu, L., et al. (2011). Primary cilia membrane assembly is initiated by Rab11 and transport protein particle II (TRAPPII) complex-dependent trafficking of Rabin8 to the centrosome. Proc. Natl. Acad. Sci. U.S.A. 108, 2759–2764. doi: 10.1073/pnas.1018823108
Wheway, G., Nazlamova, L., and Hancock, J. T. (2018). Signaling through the primary cilium. Front. Cell Dev. Biol. 6:8.
Williams, C. L., Li, C., Kida, K., Inglis, P. N., Mohan, S., Semenec, L., et al. (2011). MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. J. Cell Biol. 192, 1023–1041. doi: 10.1083/jcb.201012116
Witman, G. B. (1975). The site of in vivo assembly of flagellar microtubules. Ann. N. Y. Acad. Sci. 253, 178–191. doi: 10.1111/j.1749-6632.1975.tb19199.x
Wood, C. R., Huang, K., Diener, D. R., and Rosenbaum, J. L. (2013). The cilium secretes bioactive ectosomes. Curr. Biol. 23, 906–911. doi: 10.1016/j.cub.2013.04.019
Wren, K. N., Craft, J. M., Tritschler, D., Schauer, A., Patel, D. K., Smith, E. F., et al. (2013). A differential cargo-loading model of ciliary length regulation by IFT. Curr. Biol. 23, 2463–2471. doi: 10.1016/j.cub.2013.10.044
Xin, D., Christopher, K. J., Zeng, L., Kong, Y., and Weatherbee, S. D. (2017). IFT56 regulates vertebrate developmental patterning by maintaining IFTB complex integrity and ciliary microtubule architecture. Development 144, 1544–1553. doi: 10.1242/dev.143255
Yadav, S. P., Sharma, N. K., Liu, C., Dong, L., Li, T., and Swaroop, A. (2016). Centrosomal protein CP110 controls maturation of the mother centriole during cilia biogenesis. Development 143, 1491–1501. doi: 10.1242/dev.130120
Yamaguchi, T., Wallace, D. P., Magenheimer, B. S., Hempson, S. J., Grantham, J. J., and Calvet, J. P. (2004). Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J. Biol. Chem. 279, 40419–40430. doi: 10.1074/jbc.m405079200
Yang, N., Li, L., Eguether, T., Sundberg, J. P., Pazour, G. J., and Chen, J. (2015). Intraflagellar transport 27 is essential for hedgehog signaling but dispensable for ciliogenesis during hair follicle morphogenesis. Development 142, 2194–2202. doi: 10.1242/dev.115261
Yang, S., and Wang, C. (2012). The intraflagellar transport protein IFT80 is required for cilia formation and osteogenesis. Bone 51, 407–417. doi: 10.1016/j.bone.2012.06.021
Ye, F., Nager, A. R., and Nachury, M. V. (2018). BBSome trains remove activated GPCRs from cilia by enabling passage through the transition zone. J. Cell Biol. 217, 1847–1868. doi: 10.1083/jcb.201709041
Yoder, B. K., Richards, W. G., Sweeney, W. E., Wilkinson, J. E., Avener, E. D., and Woychik, R. P. (1995). Insertional mutagenesis and molecular analysis of a new gene associated with polycystic kidney disease. Proc. Assoc. Am. Physicians 107, 314–323.
Yoder, B. K., Tousson, A., Millican, L., Wu, J. H., Bugg, C. E. Jr., Schafer, J. A., et al. (2002). Polaris, a protein disrupted in orpk mutant mice, is required for assembly of renal cilium. Am. J. Physiol. Renal. Physiol. 282, F541–F552.
Yu, F., Guo, S., Li, T., Ran, J., Zhao, W., Li, D., et al. (2019). Ciliary defects caused by dysregulation of O-GlcNAc modification are associated with diabetic complications. Cell Res. 29, 171–173. doi: 10.1038/s41422-018-0114-7
Yu, F., Li, T., Sui, Y., Chen, Q., Yang, S., Yang, J., et al. (2020). O-GlcNAc transferase regulates centriole behavior and intraflagellar transport to promote ciliogenesis. Protein Cell 11, 852–857. doi: 10.1007/s13238-020-00746-2
Zhang, W., Taylor, S. P., Nevarez, L., Lachman, R. S., Nickerson, D. A., Bamshad, M., et al. (2016). IFT52 mutations destabilize anterograde complex assembly, disrupt ciliogenesis and result in short rib polydactyly syndrome. Hum. Mol. Genet. 25, 4012–4020. doi: 10.1093/hmg/ddw241
Zhu, B., Zhu, X., Wang, L., Liang, Y., Feng, Q., and Pan, J. (2017a). Functional exploration of the IFT-A complex in intraflagellar transport and ciliogenesis. PLoS Genet. 13:e1006627. doi: 10.1371/journal.pgen.1006627
Zhu, X., Liang, Y., Gao, F., and Pan, J. (2017b). IFT54 regulates IFT20 stability but is not essential for tubulin transport during ciliogenesis. Cell Mol. Life Sci. 74, 3425–3437. doi: 10.1007/s00018-017-2525-x
Keywords: IFT-B, IFT-A, ciliogenesis, cilia disassembly, ectocytosis, posttranslational modification, kidney
Citation: Wang W, Jack BM, Wang HH, Kavanaugh MA, Maser RL and Tran PV (2021) Intraflagellar Transport Proteins as Regulators of Primary Cilia Length. Front. Cell Dev. Biol. 9:661350. doi: 10.3389/fcell.2021.661350
Received: 30 January 2021; Accepted: 06 April 2021;
Published: 19 May 2021.
Edited by:
Susana Santos Lopes, New University of Lisbon, PortugalReviewed by:
Bipul R. Acharya, University of Virginia, United StatesCopyright © 2021 Wang, Jack, Wang, Kavanaugh, Maser and Tran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pamela V. Tran, cHRyYW5Aa3VtYy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.