 Amira Bouzidi1†
Amira Bouzidi1† Karim Labreche2†
Karim Labreche2† Marine Baron3‡
Marine Baron3‡ Marianne Veyri4‡
Marianne Veyri4‡ Jérôme Alexandre Denis5
Jérôme Alexandre Denis5 Mehdi Touat6
Mehdi Touat6 Marc Sanson6Frédéric Davi7Erell Guillerm8Stéphanie Jouannet9
Marc Sanson6Frédéric Davi7Erell Guillerm8Stéphanie Jouannet9 Frédéric Charlotte10Franck Bielle11
Frédéric Charlotte10Franck Bielle11 Sylvain Choquet3Pierre-Yves Boëlle2
Sylvain Choquet3Pierre-Yves Boëlle2 Jacques Cadranel12
Jacques Cadranel12 Véronique Leblond3
Véronique Leblond3 Brigitte Autran13
Brigitte Autran13 Jean-Marc Lacorte1§Jean-Philippe Spano4§
Jean-Marc Lacorte1§Jean-Philippe Spano4§ Florence Coulet8*§ and the IDEATION study group
Florence Coulet8*§ and the IDEATION study group- 1Sorbonne University, INSERM, Research Unit on Cardiovascular and Metabolic Disease UMR ICAN, Department of Endocrine Biochemistry and Oncology, AP-HP, Hôpital Pitié Salpêtrière, Paris, France
- 2Sorbonne University, INSERM, Pierre Louis Institute of Epidemiology and Public Health, Paris, France
- 3Sorbonne University, Center for Immunology and Infectious Diseases (CIMI-Paris), Department of Hematology, APHP, Hôpital Pitié Salpêtrière, Paris, France
- 4Sorbonne University, INSERM, Pierre Louis Institute of Epidemiology and Public Health, Theravir Team, Medical Oncology, AP-HP, Hôpital Pitié Salpêtrière, Paris, France
- 5Sorbonne University, INSERM, Saint-Antoine Research Center, Cancer Biology and Therapeutics, CRSA, Department of Endocrine Biochemistry and Oncology, AP-HP, Hôpital Pitié Salpêtrière, Paris, France
- 6Sorbonne University, INSERM, CNRS, Brain and Spine Institute, ICM, Department of Neurology 2-Mazarin, AP-HP, Hôpital Pitié Salpêtrière, Paris, France
- 7Sorbonne University, INSERM, Centre de Recherche des Cordeliers, Department of Biological Hematology, AP-HP, Hôpital Pitié Salpêtrière, Paris, France
- 8Sorbonne University, INSERM, Saint-Antoine Research Center, Microsatellites Instability and Cancer, CRSA, Genetics Department, AP-HP, Hôpital Pitié Salpêtrière, Paris, France
- 9Sorbonne University, Neurosurgery Department, AP-HP, Hôpital Pitié Salpêtrière, Paris, France
- 10Sorbonne University, Anatomy and Pathologic Cytology, AP-HP, Hôpital Pitié Salpêtrière, Paris, France
- 11Sorbonne University, Neuropathology Department, AP-HP, Hôpital Pitié Salpêtrière, Paris, France
- 12Sorbonne University, Chest Department and Thoracic Oncology, GRC 04, Theranoscan, AP-HP, Hôpital Tenon, Paris, France
- 13Sorbonne University, INSERM, CNRS, Center for Immunology and Infectious Diseases (CIMI-Paris), AP-HP, Pitié-Salpêtrière Hospital, Paris, France
Cell-free DNA (cfDNA) analysis is a minimally invasive method that can be used to detect genomic abnormalities by directly testing a blood sample. This method is particularly useful for immunosuppressed patients, who are at high risk of complications from tissue biopsy. The cfDNA tumor fraction (TF) varies greatly across cancer type and between patients. Thus, the detection of molecular alterations is highly dependent on the circulating TF. In our study, we aimed to calculate the TF and characterize the copy number aberration (CNA) profile of cfDNA from patients with rare malignancies occurring in immunosuppressed environments or immune-privileged sites. To accomplish this, we recruited 36 patients: 19 patients with non-Hodgkin lymphoma (NHL) who were either human immunodeficiency virus (HIV)-positive or organ transplant recipients, 5 HIV-positive lung cancer patients, and 12 patients with glioma. cfDNA was extracted from the patients’ plasma and sequenced using low-coverage whole genome sequencing (LC-WGS). The cfDNA TF was then calculated using the ichorCNA bioinformatic algorithm, based on the CNA profile. In parallel, we performed whole exome sequencing of patient tumor tissue and cfDNA samples with detectable TFs. We detected a cfDNA TF in 29% of immune-suppressed patients (one patient with lung cancer and six with systemic NHL), with a TF range from 8 to 70%. In these patients, the events detected in the CNA profile of cfDNA are well-known events associated with NHL and lung cancer. Moreover, cfDNA CNA profile correlated with the CNA profile of matched tumor tissue. No tumor-derived cfDNA was detected in the glioma patients. Our study shows that tumor genetic content is detectable in cfDNA from immunosuppressed patients with advanced NHL or lung cancer. LC-WGS is a time- and cost-effective method that can help select an appropriate strategy for performing extensive molecular analysis of cfDNA. This technique also enables characterization of CNAs in cfDNA when sufficient tumor content is available. Hence, this approach can be used to collect useful molecular information that is relevant to patient care.
Introduction
Patients infected with human immunodeficiency virus (HIV) or who are organ transplant recipients are at increased risk of developing several cancers compared to the general population (Hortlund et al., 2017; Yarchoan and Uldrick, 2018). Moreover, the risk of cancer-related mortality is increased in these patients (Sigel et al., 2011; Au et al., 2019; Horner et al., 2020). Tissue biopsies performed on immunosuppressed patients entails an elevated risk of complications and demonstrates lower diagnostic yields and increased mortality (Plattner et al., 2018; Cleveland et al., 2020). Compared with tissue biopsy, liquid biopsy such as cell-free DNA (cfDNA) has several benefits. Molecular profiling of cfDNA could allow patients to undergo close, longitudinal follow-up that can enable primary diagnosis of extensive disease, optimal choice of treatment, early diagnosis of clinical progression, and identification of secondary resistance mechanisms. In addition, cfDNA analysis offers comprehensive coverage that can help address issues of tumor heterogeneity (Ilie and Hofman, 2016). Collecting and analyzing cfDNA may be particularly advantageous for immunosuppressed patients, as this approach offers a minimally invasive and cost-effective alternative to more traditional molecular profiling methods.
The cfDNA tumor fraction (TF), which is the proportion of tumor molecules that are present in a cfDNA sample, varies greatly across cancer type and between patients, and represents a clinically relevant biomarker (Adalsteinsson et al., 2017; Van Roy et al., 2017; Chen et al., 2019). The detection of tumor-associated single nucleotide variants (SNVs) in the cfDNA can be achieved by using high-depth targeted sequencing methods (Newman et al., 2014; Ishii et al., 2020) or PCR-based methods (Denis et al., 2017). Broader analyses such as whole exome sequencing (WES) at standard depth (∼150 × coverage) are generally informative when the TF is ∼5% to 10% (Cibulskis et al., 2013). Therefore, the estimation of the TF could help guide selection of the appropriate molecular approach for subsequent detection of SNVs in cfDNA.
Recently, a cost-effective approach, ichorCNA11, was established to calculate the TF of cfDNA using low-coverage whole genome sequencing (LC-WGS). This method identifies segmental/arm-level copy number aberrations (CNAs), which enables the TF to be inferred (Adalsteinsson et al., 2017).
The aim of our study was to calculate the TF and determine the CNA profile of cfDNA in immunosuppressed patients or patients with tumors located in immune-privileged sites, such as the central nervous system (CNS) using LC-WGS and ichorCNA. This approach can guide the decision to subsequently perform a targeted PCR-based method, a targeted sequencing, or a WES, based on the circulating tumor content, to provide a more comprehensive molecular analysis of cfDNA in this specific, frail population.
For this purpose, we recruited patients with cancers known to be associated with variable amounts of cfDNA (from low to high) in immunocompetent patients (Mouliere et al., 2018). Specifically, we enrolled untreated immunodeficient patients with non-Hodgkin lymphoma (NHL) (either systemic or localized to the CNS), HIV-positive patients with non-small-cell lung cancer (NSCLC), and immunocompetent patients with glioma.
Materials and Methods
Patient Cohort
Patients were enrolled in the IDeATIon project (ClinicalTrials.gov identifier: NCT03706625), a multidisciplinary, multicentric, translational research program that focuses on severe tumors that appear in immunosuppressed patients or in immune-privileged sites such as the CNS. The protocol was approved by the French national IRB (no. 2018-A01099-46) and the “Commission Nationale de l’Informatique et des Libertés” (CNIL no. 918222).
Three types of rare and severe cancers were selected: (1) NHL (either systemic or localized to the CNS) occurring in patients with HIV or in transplant recipients, (2) NSCLC in patients with HIV, and (3) glioma. The inclusion criteria were as follows: (1) male or female adults (age ≥ 18 years old), (2) confirmed histopathological diagnosis of one of the three types of cancer listed above, (3) anti-tumoral treatment-naive (except for some glioma patients), (4) hemoglobin level > 9 g/dl for lung cancer patients and > 7 g/dl for NHL and glioma patients, and (5) written informed consent. From July 2018 to December 2020, 118 patients were enrolled in the IDeATIon project. This study includes 36 of these prospectively enrolled patients (Table 1).
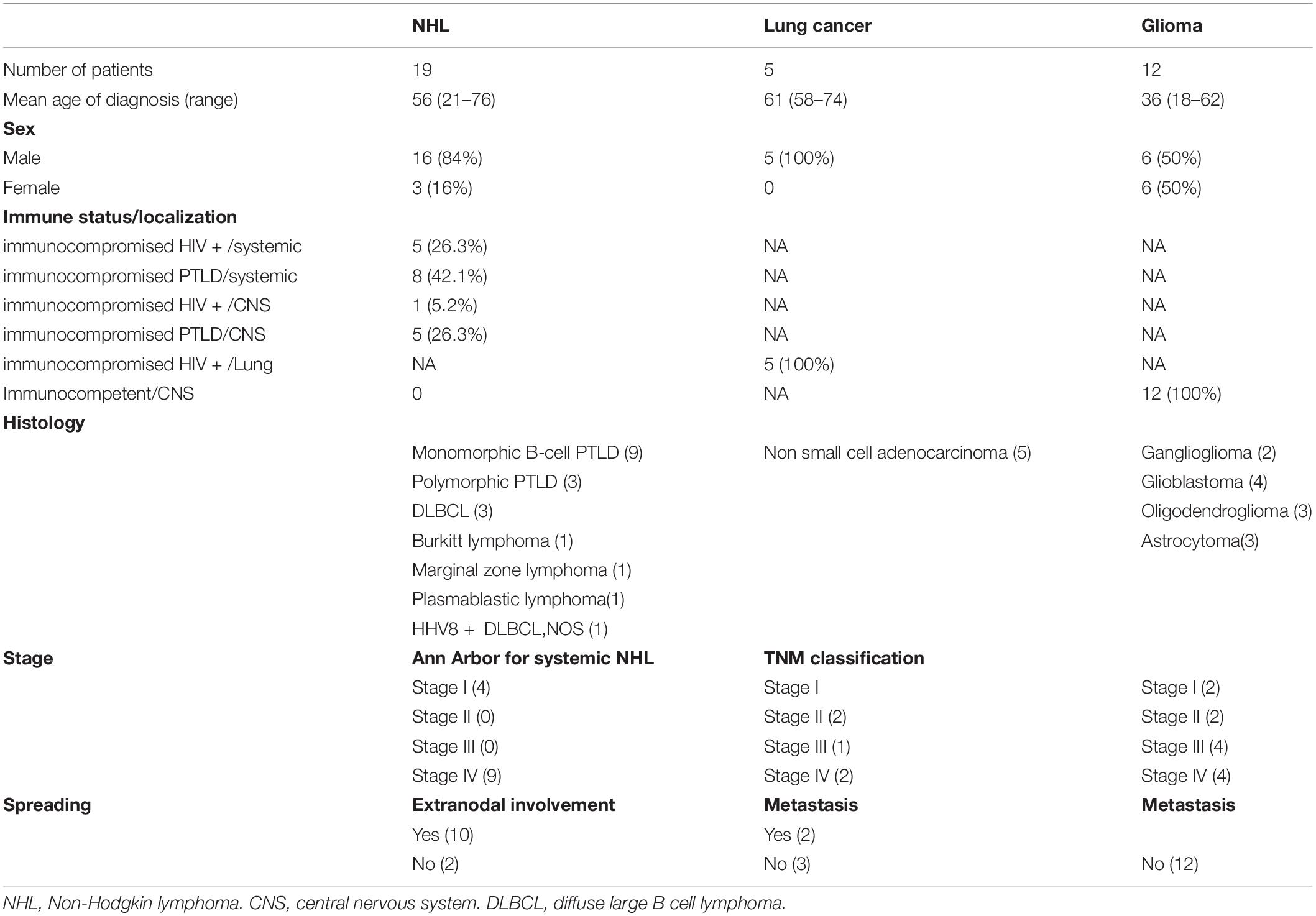
Table 1. Study cohort description.
Spike-In Experiment
Genomic DNA (gDNA) was isolated from two human NSCLC cell lines, H2030 (ATCC® CRL-5914Tm) and H1975 (ATCC® CRL-5908Tm), and healthy donor blood using a QIAamp DNA Blood Mini Kit (Qiagen). The DNA concentrations were quantified on a QuantusTm Fluorometer (Promega) using QuantiFluor® ONE dsDNA Dye (Promega). Genomic DNA was sheared to an average size of 150 bp using The Bioruptor® Pico (Diagenode). Fragment size was confirmed on a TapeStation 4200 electrophoresis system (Agilent). To create the spike-in mix, H2030 or H1975 DNA was mixed with healthy donor DNA to a final proportion of 5% or 15%, and the samples were analyzed by LC-WGS.
Plasma Preparation and cfDNA Extraction
Blood was collected in Cell-Free DNA Collection Tubes (ROCHE), which were then centrifuged for 10 min at 820 g. Next, the supernatant was transferred to a fresh tube, centrifuged at 20,000 g for a further 10 min, and stored in 1-ml aliquots at −80°C until later extraction. cfDNA was extracted from 5 ml of plasma using the QIAamp Circulating Nucleic Acid Kit (Qiagen) following the manufacturer’s instructions and eluted in a volume of 35 μl. cfDNA was quantified using a QuantusTm Fluorometer (Promega). The size distribution of the cfDNA was checked on a TapeStation 4200 electrophoresis system (Agilent) using Cell-free DNA ScreenTape.
LC-WGS and Analysis
Sequencing libraries were prepared from 4 to 10 ng of cfDNA using the SMARTer® ThruPLEX® Tag-seq Kit (Takara Bio). Libraries were quantified using a QubitTm dsDNA HS Assay Kit (Thermo Fisher), and the fragment sizes were confirmed using a TapeStation 4200 electrophoresis system (Agilent). Equal amounts of the sequencing libraries were pooled to generate a single sample for sequencing. WGS at an average coverage of 4 × was performed on a Novaseq (Illumina) system using 2 × 150-bp paired-end sequencing. BCL2FASTQ (Illumina) was used to convert bcl files to fastq files. The coverage statistics are summarized in Supplementary Figure 1. Paired-end fastq files were aligned to the hg38 human reference genome using the BWA-MEM algorithm (Li and Durbin, 2009). PCR duplicates were removed, and a Base Quality Score Recalibration (BQSR) was executed with the Genome Analysis Toolkit (GATK) version 4.1 (McKenna et al., 2010) following GATK best practice.
Based on the pre-processed aligned reads, the TF in the cfDNA was calculated using the ichorCNA workflow, as described previously (Adalsteinsson et al., 2017). Briefly, using the readCounter tools from the HMMcopy suite, we calculated the read count coverage of reads with a mapping quality greater than 20 in 1-Mb-window bins across all autosomal chromosomes. The read counts were then normalized to correct for GC content and mappability bias. The copy number analysis and TF prediction were performed using the ichorCNA R software package. LC-WGS sequencing of the healthy donor sample served as the normal WIG. Adjustments were made to the standard ichorCNA settings to account for ploidy (2 or 3) and an initial normal contamination range of 0.1 to 0.99. Genotype count at genomic position on LC-WGS data were performed using bcftools mpileup command (Supplementary Table 1).
DNA Extraction From Tissue Biopsy
Tumor gDNA was isolated from fresh or formalin-fixed paraffin-embedded (FFPE) diagnostic tissue biopsies using a QIAamp DNA Mini Kit (Qiagen) or a QIAamp DNA FFPE Tissue Kit (Qiagen), respectively, according to the manufacturer’s instructions.
WES and Tumor Tissue Analysis
Libraries were prepared and hybrid-captured using the SeqCap EZ MedExome Enrichment Kit (Roche) with 200 ng of DNA input. Sequencing was performed on an Illumina Novaseq system with 150-bp paired-end reads. Raw paired-end fastq files were pre-processed using fastp (Chen et al., 2018) for quality control, adapter trimming, and quality filtering. The filtered reads were aligned to the hg38 human reference genome using BWA-MEM (Adalsteinsson et al., 2017). Reads were pre-processed according to GATK best practice. Coverage summary statistics of the consensus coding sequence are shown in Supplementary Figure 2. Somatic SNVs and indels that differed between tumor–normal pairs were called using MuTecT (Cibulskis et al., 2013) and Strelka2 (Kim et al., 2018), respectively. We excluded potential oxidative damage-induced mutations as per Costello et al. (2013) using in-house scripts.
Copy number analysis for tumor biopsy WES was performed using the TitanCNA pipeline (Ha et al., 20141). Briefly, we first identified heterozygous single nucleotide polymorphisms (SNPs) from the matched normal germline blood sample using the Samtools mpileup command and the Exome captured regions bed file and computed read counts for the reference SNPs and alternative genotypes from the tumor biopsy samples. Next, we computed read counts for 50-kb bins using HMMcopy (Ha et al., 2012). Centromeres were filtered based on chromosome gap coordinates obtained from UCSC for the hg38 human reference genome. The read coverage for 50-kb bins across the genome was corrected for GC content and mappability. Finally, TitanCNA was used with default values (except for alphaK = 2500 and minDepth = 20) to generate solutions with one to five clonal clusters, with the initial ploidy set to 2 and 3. The solution with the optimal number of clonal clusters was selected using the minimum S_Dbw validity index (using log ratio and allele ratio) as recommended. Tumor purity estimation was performed using ABSOLUTE (Carter et al., 2012).
Comparing the LC-WGS Results for the cfDNA and Matched Tumor Biopsies
We performed a log2 ratio comparison for 23 patients for whom both LC-WGS cfDNA data and matched tumor biopsy WES data were available (Supplementary Figure 3). We excluded LC-WGS cfDNA segments that did not overlap the tumor biopsy sample. Additionally, we restricted the comparison to segments with cellular prevalence = 1, indicating a higher proportion of tumor cells containing that event. If more than one log2 ratio was assigned to the same segments, the median value of the log2 ratios in the LC-WGS segments was retained for comparison. Similarity between each cfDNA sample and the matching tumor biopsy was calculated by computing the Spearman correlation coefficient.
WES and cfDNA Analysis
WES was performed on three cfDNA samples with a detectable TF. The same libraries prepared for LC-WGS were hybrid-captured using a SeqCap EZ MedExome Enrichment Kit (Roche). The sequencing and bioinformatic analysis were performed as described above for tumor tissue, except that variant calling was performed on SNVs only.
RNA Sequencing and Expression Analysis
RNA was extracted from tumoral tissue using an RNeasy Micro Kit (Qiagen). Libraries were prepared from 500 ng of RNA. After end repair, A-tailing, ligation, and purification, sequencing was performed on an Illumina Novaseq with 150-bp paired-end reads. Reads were aligned to the human hg38 reference genome after an index was generated using STAR v2.7.2 (Dobin et al., 2013) by applying per-sample two-pass mapping. The generated BAM files were pre-processed according to GATK v4.1 RNA-seq best practice (Mark Duplicate, SplitNcigarReads and BQSR), followed by transcript quantification using Kallisto v0.45.0 (Bray et al., 2016). To summarize transcript-level abundance estimates for gene-level analysis, we used tximport R packages (Soneson et al., 2015). Gene expression comparison was achieved by simple extraction of the listed genes (ABC and GCB, Supplementary Figure 4C) from total gene expression level, followed by Wilcoxon test to compare the regularized-logarithm transformation (rlog) of the gene count between the two gene sets.
Statistical Analysis
We used R (version 4.0.0) to perform all statistical analyses and tests. The Wilcoxon test was used to compare ploidy across the exome and LC-WGS data. Copy number variation (CNV) segment tests and p-values were computed using Spearman’s test.
Results
Cohort Description
The clinical features of the 36 patients included in our cohort are shown in Table 1. Twenty-four patients were immunodeficient, namely, 19 who had NHL and 5 who had NSCLC. Among the patients with NHL, 6 were HIV-positive [1 with primary central nervous system lymphoma (PCNSL) and 5 with systemic NHL] and 13 were organ transplant recipients (5 with PCNSL and 8 with systemic NHL). All five patients with NSCLC were HIV-positive. The other 12 patients in the cohort were immunocompetent and had glioma. Biological material, including blood and tumor tissue samples, was collected at the time of diagnosis and prior to any anti-tumoral treatment (except for some glioma patients).
Estimation of the TF in the cfDNA
To establish the LC-WGS protocol and ensure reproducibility, we first performed a spike-in experiment by mixing gDNA from two lung cancer cell lines (H2030 and H1975) with known genomic profiles with DNA from a healthy donor at different ratios. We found that a detectable TF using our method was a TF > 5% (Supplementary Figure 1). Following LC-WGS and bioinformatics analysis, the TF in cfDNA from 24 immunodeficient patients with cancer (19 NHL and 5 NSCLC-HIV) and 12 immunocompetent patients with glioma was determined. We prioritized ichorCNA solutions with a ploidy of 2, as our cohort did not exhibit high-copy genome amplification, and subjected the CNV profiles to manual review to account for bias introduced to bin log2 ratio values after GC content correction.
The calculated TF values ranged from 8 to 70% (Figure 1A). Thirty-two percent of immunodeficient NHL patients (6/19) and 20% of HIV-positive NSCLC patients (1/5) had a TF greater than 5%. However, no tumor-derived cfDNA was detected in any of the glioma or PCNSL patients. The estimated TF for the HIV-positive patient with NSCLC was 23%; this patient had metastatic cancer (T4 N3 M1c, stage 4B disease).
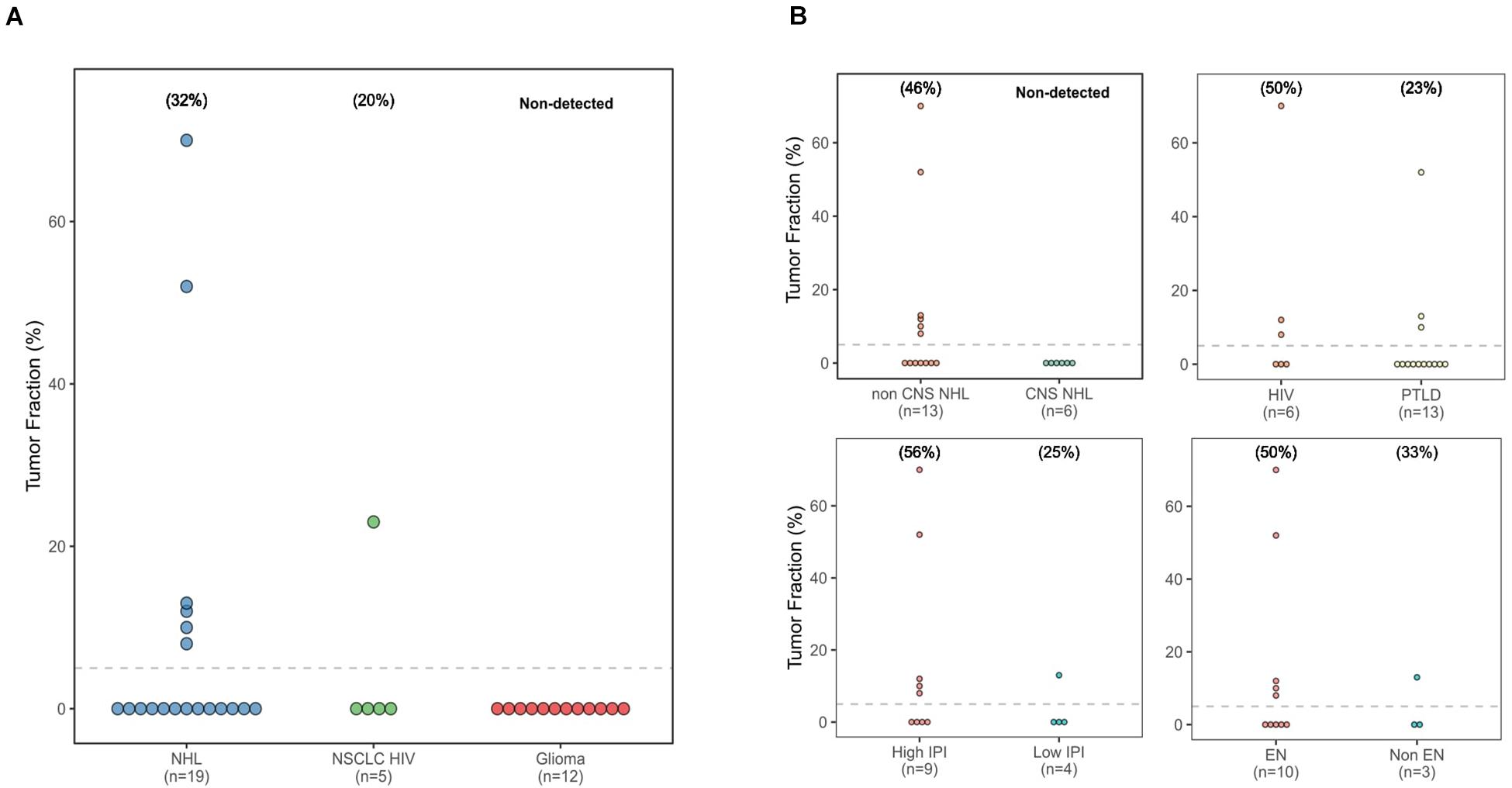
Figure 1. Tumor fraction dot plots. (A) All three cancer types studied. (B) Based on non-Hodgkin lymphoma location, immunodeficiency type, nodal location, and International Prognostic Index group.
Next, we compared TF values across different groups of patients with NHL, based on localization, International Prognostic Index (IPI) score, and type of immunodeficiency (Figure 1B). Six of the NHL patients with detectable TF (TF = 8%, 10%, 12%, 13%, 55%, and 70%) had exclusively systemic localization. In contrast, the TF was below the limit of detection of all the patients with PCNSL. Five out of the six patients with a TF value > 5% had extranodal manifestations of disease. No difference was observed in terms of type of immunodeficiency. Finally, five out of nine patients with a high IPI score (2 or 3) exhibited detectable levels of tumor cfDNA, whereas only one in four with a low IPI score (0 or 1) had a detectable TF.
Copy Number Alteration Profile Concordance Between Tumor Biopsies and cfDNA
Next, we compared the CNA profiles for the cfDNA samples as established by LC-WGS and ichorCNA with the CNA profiles deduced from WES of matched tumor biopsies (100× to 200×, n = 23). These analyses involve comparison of the normalized log2 copy ratios between the two sample types from same patients. We found that the Mb-scale copy numbers correlated well in samples with a TF > 5% (ranging from ρ = 0.34 to ρ = 0.81; p < 0.001, Figure 2A), while there was no significant correlation for patients with undetectable TFs (ρ = −0.31 to ρ = 0.08; Figure 2B, Supplementary Figure 3).
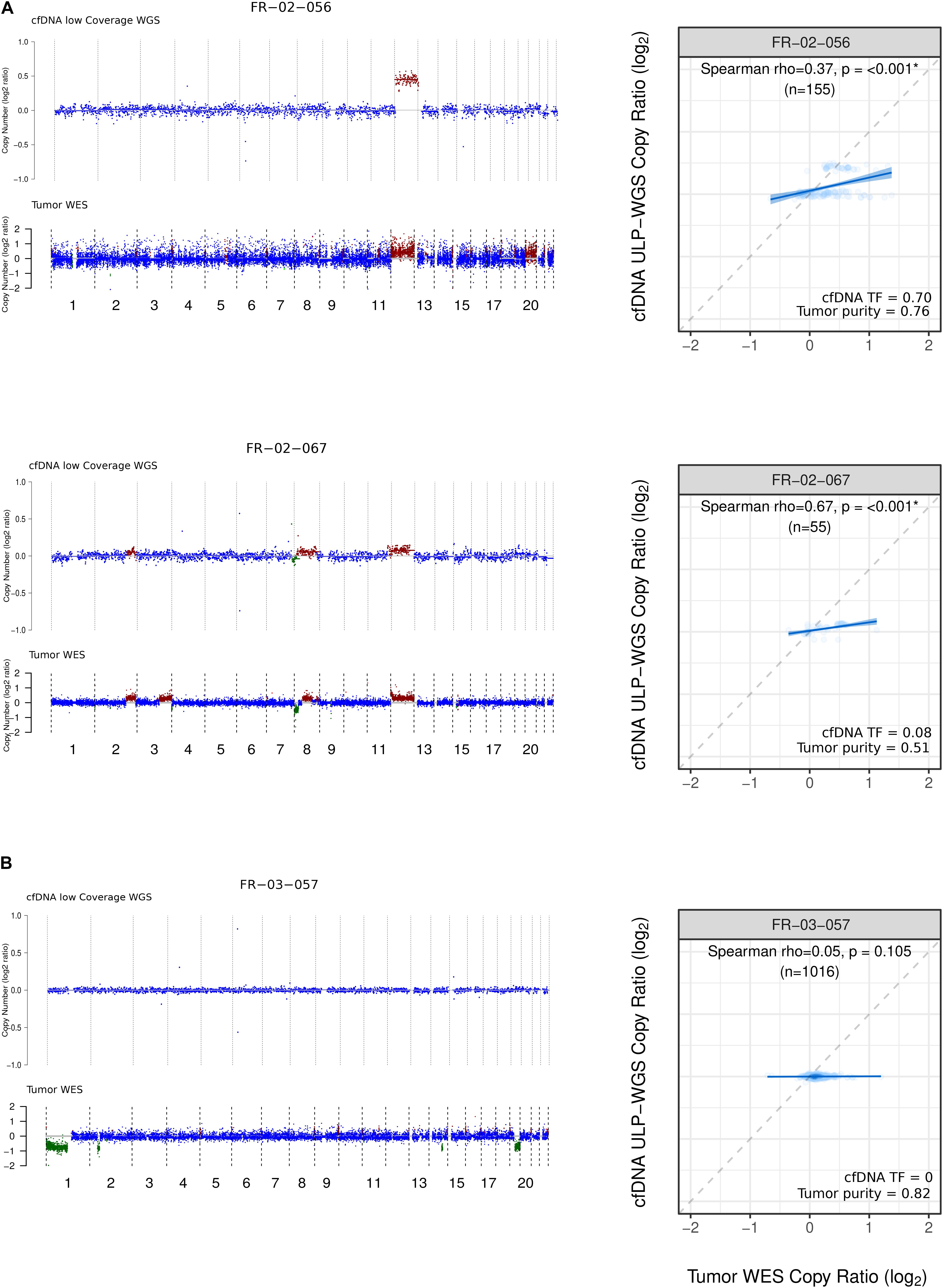
Figure 2. Copy number profile and comparison from LC-WGS cfDNA and WES tumor biopsy. (A) Left track shows genome-wide copy number from two ID-NHL patients (FR-02-056 and FR-02-067 with the following median coverage for cfDNA 4.24X, 3.49X and for WES tumor biopsy 216X, 211X, respectively). Right track shows cfDNA tumor fraction (TF) computed using ichorCNA and Tumor purity calculated using ABSOLUTE. (B) Left track shows genome-wide copy number from a 1p19q codeleted glioma patient FR-03-057 with the following median coverage for 4.30X for cfDNA and 166 X for WES tumor biopsy. Right track shows Spearman rank correlation between LC-WGS cfDNA and WES tumor biopsy (see Materials and Methods).
Copy Number Alteration Profile Analysis of cfDNA
To evaluate whether cfDNA can serve as a proxy for tumor biopsies in cancer analyses, we analyzed the CNA profiles of cfDNA from patients with a TF > 5%.
In the HIV-positive patient with NSCLC, chromosomal gains were identified for several genes known to be associated with NSCLC, such as SOX2 (3q), TERT (5p), and EGFR (7q). Furthermore, we found amplification of MYC and heterozygous deletion of the PTEN tumor suppressor, located at 8q and 10q, respectively (Figure 3B).
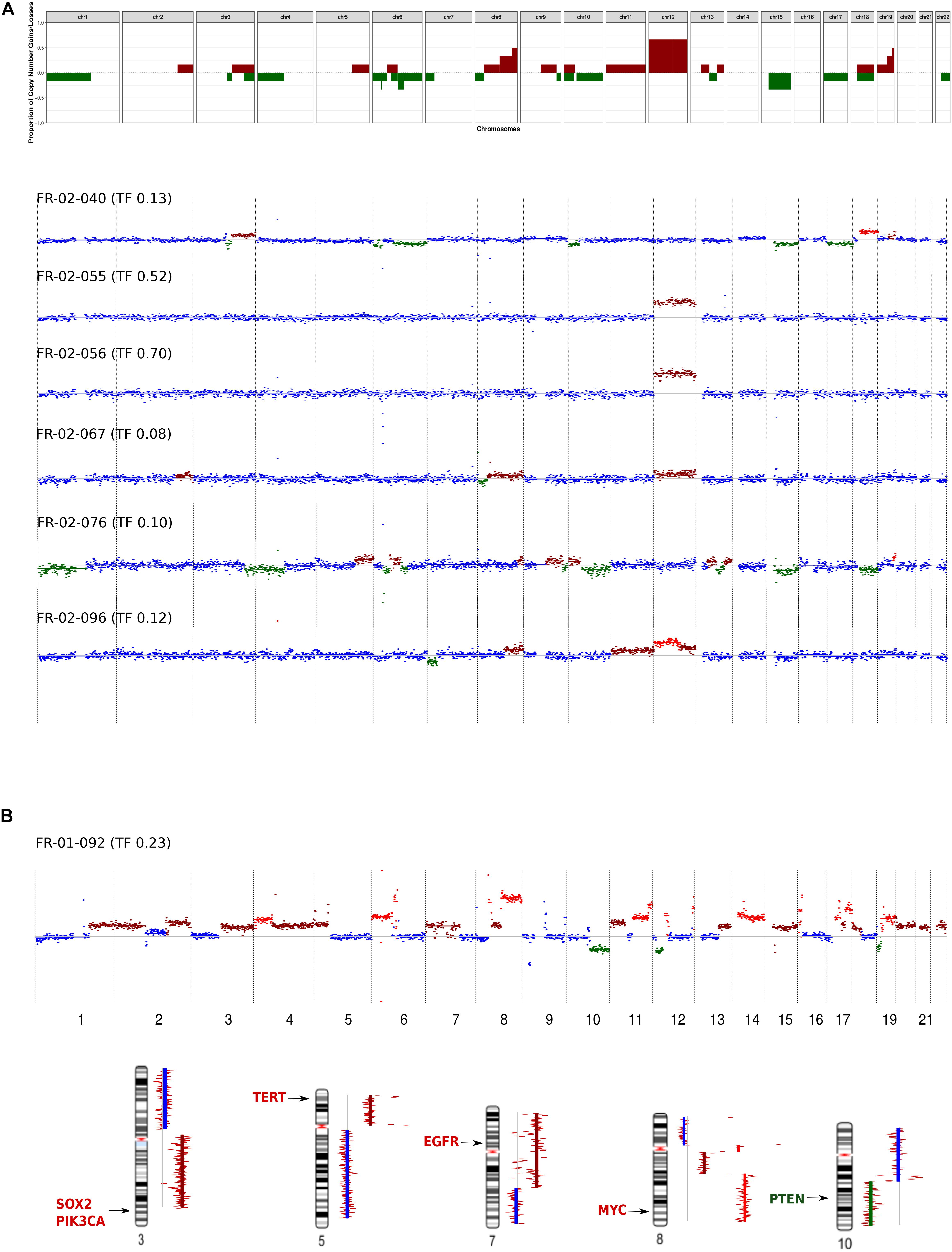
Figure 3. Genome-wide copy number alteration (CNA) landscape in patients with a tumor fraction >5%. (A) Cell-free DNA (cfDNA) results from six immunodeficient non-Hodgkin lymphoma patients. Top, frequency of copy number variation (CNV) regions detected by low-coverage whole-genome sequencing of cfDNA. Bottom, CNV pattern for each patient. The plots show the CNV log2 ratios between the cfDNA sample and the healthy donor (HD). Neutral copy numbers are shown in blue, copy number gains are shown in dark red, copy number amplifications are shown in red, and copy number losses are shown in green. (B) cfDNA results from the non-small-cell lung cancer (NSCLC) patients infected with human immunodeficiency virus. Top, CNV pattern. Bottom, genes affected by CNA known to be associated with NSCLC, and their chromosomal locations.
In the NHL group, at the chromosomal level, chromosome 12 gain was the most frequent event (encountered in 4/6 patients), followed by chromosome 15 loss (detected in 2/6 patients). These CNAs were mutually exclusive in our cohort (Figure 3A). Focal chromosomal alterations were also detected, including 8q24 gain and 6p22, 6q21 loss, which were identified in 3/6 and 2/6 patients, respectively. These findings were compared to known cancer-associated somatic CNAs seen in patients with NHL (Lopez et al., 2019; Cascione et al., 2020) or NSCLC (Spoerke et al., 2012; Qiu et al., 2017).
The NHL patients in our cohort exhibited two distinct CNA profiles. The first group included patients [three patients with diffuse large B-cell lymphoma (DLBCL) and one patient with Burkitt lymphoma] who exhibited gain of chromosome 12, which is where STAT6, MDM2, and BCL7A are located. The second group (two patients with DLBCL) exhibited neutral chromosome 12 copy numbers but had gains in a region harboring the HLA locus, PRDM1, and POLG1 (Supplementary Figure 4A). Interestingly, patient FR-02-40 harbored alterations in genes associated with the activated B-cell (ABC) DLBCL subgroup (Testoni et al., 2015; Schmitz et al., 2018), such as PIK3CA, NFKBIZ, BCL2, and SPIB.
To confirm whether this patient belonged to the ABC molecular subgroup, we analyzed somatic mutations from WES analysis of a biopsy of the patient’s tumor. We identified mutations in the tumor suppressor gene TBL1XR1 and the proto-oncogene PIM1 (Supplementary Figure 4B). In addition, RNA-seq expression analysis of the matched tumor tissue revealed overexpression of ABC-associated genes (Alizadeh et al., 2000; Rosenwald et al., 2002) (Supplementary Figure 4C). Finally, immunohistochemistry analysis using the Hans algorithm classified patient FR-02-40 as non-GCB.
WES of cfDNA
The availability of sufficient tumor-derived cfDNA material allowed us to generate WES data (median coverage of 200×) on three of the cases with LC-WGS and tumor biopsy exome sequenced. We detected shared coding somatic SNV in cfDNA and tumor biopsy in FR-02-040-BP (16%), FR-02-056-LTT (28%), and FR-01-092-PA (83%). Across the common mutations, we identified mutations of known-driver genes in NSCLC as TP53 and MAP2K1 (Wislez et al., 2021) and recurrent mutated genes in NHL as MYC, TBL1XR1, IGLL5, GNA13, and PIM1. We found a strong correlation between the variant allele frequency (VAF) on cfDNA and the somatic VAF in tumor tissue biopsy (Pearson test, ρ = 0.7–0.78, all p-value < 0.001, Supplementary Figure 4). Notably, the median VAFs of shared SNV were larger as the estimated TFs were higher (Supplementary Table 2). In addition to somatic variant analysis, we estimated ploidy and purity in cfDNA WES data. Computed tumor purity on cfDNA was comparable to TFs calculated using LC-WGS (Supplementary Table 2).
Discussion
Studies have shown that LC-WGS and ichorCNA analysis can accurately detect low fractions of circulating tumor DNA in blood plasma (Adalsteinsson et al., 2017; Chen et al., 2019). Calculating the TF in the cfDNA can help select a method with appropriate sensitivity for performing a comprehensive molecular analysis. Using CNAs to calculate the TF is particularly relevant, as the vast majority of cancers harbor arm-level somatic CNAs (Beroukhim et al., 2010).
Our pilot study is the first to calculate TFs and characterize somatic CNA profiles for cfDNA using LC-WGS and ichorCNA in immunosuppressed patients (HIV-positive or transplant recipients) with NHL (systemic or CNS-localized) or lung cancer. Immunocompetent patients with glioma were also included, given the location of the tumor in an immunosuppressive environment, and, for the purposes of this pilot study, were expected to exhibit a low TF in the blood (Bettegowda et al., 2014).
A TF was detectable in 29% (7/24) of the immunodeficient patients in our cohort, with a TF range of 8% to 70%.
No TFs or CNAs were detected in PCNSL and glioma patients. The failure to detect a TF in these patients was presumably caused by insufficient ctDNA levels in the blood that were below the limit of detection for ichorCNA. Indeed, the lower limit of ichorCNA sensitivity for detecting the presence of tumor is 3% (Adalsteinsson et al., 2017). Glioma-associated mutations have been detected in liquid biopsies using highly sensitive droplet digital PCR (Muralidharan et al., 2021) or by analyzing cerebrospinal fluid (CSF) instead of plasma (Mouliere et al., 2018), suggesting that it could be valuable to test LC-WGS on CSF cfDNA in PCNSL patients. Our results support the use of targeted molecular analyses with high sensitivity when the TF is not detected or is very low.
In contrast, patients with a detectable TF should be selected for further larger-scale sequencing methods, such as WES, for which the calculated TF could help guide calibration of the sequencing depth.
One immunosuppressed lung cancer patient included in our study had a TF of 23%. This patient had non-small-cell adenocarcinoma, was HIV-positive, and had advanced metastatic disease (T4 N3 M1c, stage 4B disease). The CNA profile derived from the tumor tissue biopsy taken from this patient correlated well with the findings from the corresponding liquid biopsy (ρ = 0.81). Moreover, the CNA profile was consistent with patterns that are typically observed in patients with NSCLC (Chen et al., 2019; Raman et al., 2020). Determining the CNA profile from cfDNA could constitute an attractive method to support histological subtyping of lung cancer patients, as suggested by Raman et al.
In our study population, the immunodeficient NHL patient group had the highest proportion of patients with detectable aberrations (6/19, 32%). The proportion of patients with detectable cancer-derived cfDNA in this group was lower than that described in the literature for NHL patients. Bohers et al. identified tumor-associated mutations in the cfDNA in 19/30 patients (63%), and Rossi et al., in 20/30 patients (66.6%) (Rossi et al., 2017; Bohers et al., 2018). However, these two studies focused on targeted detection of somatic SNVs in recurrently mutated cancer genes using high-throughput targeted sequencing. It has been shown that a CNA is > 20 times more likely to be detected by LC-WGS than by arrays (Zhou et al., 2018).
Interestingly, 5/6 NHL patients with a TF > 5% had extranodal manifestations and a high IPI score. Given that our patients were untreated at the time of liquid biopsy, our results agree with a study concluding that ctDNA is a promising biomarker for identifying NHL patients at high risk of treatment failure and at risk of recurrence before clinical evidence of disease relapse (Roschewski et al., 2015).
The potential utility of cfDNA as a biomarker and as a way to molecularly characterize tumors is supported by the concordance between the cfDNA and tumor tissue CNA profiles. Despite the low number of patients with cfDNA WES data, VAF comparison and purity estimation collectively suggests an accurate sensitivity of TF estimation using LC-WGS/IchorCNA. Surprisingly, the strongest correlation was found in the patient with a TF of 23% and not in the patient with a TF of 70%. This may be attributable to the fact that several CNA events were detected in the patient with the lower TF, while only chromosome 12 gain was found in the patient with the higher TF. However, this patient harbors coding variants with high VAFs in cfDNA (median VAF = 0.28, Supplementary Table 2).
The CNA profiles derived from the cfDNA from the DLBCL subgroup are consistent with patterns that are usually observed in DLBCL and have been previously detected by analyzing tumor tissue (Testoni et al., 2015). However, based on events observed in the cfDNA CNA profiles, we were further able to distinguish two distinct groups of patients. These two groups (chromosome 12 gain or neutral copy number) correlate with the ABC and GCB gene expression subgroups (Schmitz et al., 2018). Furthermore, mutations in genes corresponding to one subgroup or the other were found in the matched tumor tissue. These results should be confirmed in a larger cohort.
Conclusion
Our pilot study showed, for the first time, that ctDNA can be detected by LC-WGS/ichorCNA in the plasma of immunosuppressed patients with advanced NHL or lung cancer. This cost-effective method accurately calculated the TF and determined the CNA profile from the cfDNA and can be used to guide the choice of molecular method for further larger-scale investigations of cfDNA. Applying this method to a larger cohort of immunodeficient patients, as well as an immunocompetent cohort, may help shed light on cancer pathogenesis, the genomic mechanisms by which tumor cells acquire treatment resistance, and the response to treatment in this specific population. Liquid biopsy can be particularly useful in immunosuppressed patients, who are at higher risk for complications during tissue biopsy. Using this approach, molecular information from ctDNA can be collected before and during treatment to help improve patient care. Future analyses could help elucidate the direct impact that immune system impairment has on the circulating TF in these patients.
Members of the IDEATION Study Group
Ahmed Idbaih, Noureddine Balegroune, Amélie Guihot, Ioannis Theodorou, Agusti Alentorn, Isabelle Brocheriou, Anne-Geneviève, Damien Roos Weil, and Alberto Picca.
Data Availability Statement
The data presented in the study are deposited in the European Nucleotide Archive repository, accession number PRJEB43455, link: https://www.ebi.ac.uk/ena/browser/view/PRJEB43455.
Ethics Statement
The studies involving human participants were reviewed and approved by the IRB (no 2018-A01099-46) and “Commission Nationale de l’Informatique et des Libertés” (CNIL no 918222). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
AB and KL processed the experimental data, performed the analysis, drafted the manuscript, and designed the tures. AB and SJ carried out the experiments. KL and P-YB performed the bioinformatic analyses. FCo, JD, and J-ML were involved in planning and supervised the work and the manuscript draft. J-PS and BA conceived of the project IDEATION study. MV is the project manager of IDEATION study. EG helped for experimental process. FCh and FB are anatomopathologists involved for selection of tumor tissue. MB, MT, FD, SC, VL, and JC aided in interpreting the results and worked on the manuscript. All authors discussed the results and commented on the manuscript.
Funding
The IDeATIon project is financially supported by the MSDAvenir endowment fund. This work was supported by a grant from SIRIC CURAMUS (Cancer United Research Associating Medicine, University & Society integrated cancer research program).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the following individuals, belonging to the study group IDEATION, for their expertise and assistance throughout all aspects of our study: Ahmed Idbaih, Noureddine Balegroune, Am lie Guihot, Ioannis Theodorou, Agusti Alentorn, Isabelle Brocheriou, Anne-Genevi ve Marcelin, Damien Roos Weil, and Alberto Picca.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.661272/full#supplementary-material
Footnotes
References
Adalsteinsson, V. A., Ha, G., Freeman, S. S., Choudhury, A. D., Stover, D. G., Parsons, H. A., et al. (2017). Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 8:1324. doi: 10.1038/s41467-017-00965-y
Alizadeh, A. A., Eisen, M. B., Davis, R. E., Ma, C., Lossos, I. S., Rosenwald, A., et al. (2000). Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403, 503–511. doi: 10.1038/35000501
Au, E. H., Chapman, J. R., Craig, J. C., Lim, W. H., Teixeira-Pinto, A., Ullah, S., et al. (2019). Overall and site-specific cancer mortality in patients on dialysis and after kidney transplant. J. Am. Soc. Nephrol. 30, 471–480. doi: 10.1681/ASN.2018090906
Beroukhim, R., Mermel, C. H., Porter, D., Wei, G., Raychaudhuri, S., Donovan, J., et al. (2010). The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905. doi: 10.1038/nature08822
Bettegowda, C., Sausen, M., Leary, R. J., Kinde, I., Wang, Y., Agrawal, N., et al. (2014). Detection of circulating tumor DNA in early– and late-stage human malignancies. Sci. Transl. Med. 6:224ra24. doi: 10.1126/scitranslmed.3007094
Bohers, E., Viailly, P.-J., Becker, S., Marchand, V., Ruminy P., Maingonnat, C., et al. (2018). Non-invasive monitoring of diffuse large B-cell lymphoma by cell-free DNA high-throughput targeted sequencing: analysis of a prospective cohort. Blood Cancer J. 8:74. doi: 10.1038/s41408-018-0111-6
Bray, N. L., Pimentel, H., Melsted, P., and Pachter, L. (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527. doi: 10.1038/nbt.3519
Carter, S. L., Cibulskis, K., Helman, E., McKenna, A., Shen, H., Zack, T., et al. (2012). Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 30, 413–421. doi: 10.1038/nbt.2203
Cascione, L., Aresu, L., Baudis, M., and Bertoni, F. (2020). DNA copy number changes in diffuse large b cell lymphomas. Front. Oncol. 10:584095. doi: 10.3389/fonc.2020.584095
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Chen, X., Chang, C. W., Spoerke, J. M., Yoh, K. E., Kapoor, V., Baudo, C., et al. (2019). Low-pass whole-genome sequencing of circulating cell-free dna demonstrates dynamic changes in genomic copy number in a squamous lung cancer clinical cohort. Clin. Cancer Res. 25, 2254–2263. doi: 10.1158/1078-0432.CCR-18-1593
Cibulskis, K., Lawrence, M. S., Carter, S. L., Sivachenko, A., Jaffe, D., Sougnez, C., et al. (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219. doi: 10.1038/nbt.2514
Cleveland, H., Chau, A., Jeng, Z., Gardner, G., Yoo, R., Zhang, W., et al. (2020). Percutaneous lung biopsy in immunocompromised pediatric patients. J. Vasc. Interv. Radiol. 31, 93–98. doi: 10.1016/j.jvir.2019.07.016
Costello, M., Pugh, T. J., Fennell, T. J., Stewart, C., Lichtenstein, L., Meldrim, J. C., et al. (2013). Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 41:e67. doi: 10.1093/nar/gks1443
Denis, J. A., Guillerm, E., Coulet, F., Larsen, A. K., and Lacorte, J. M. (2017). The role of beaming and digital PCR for multiplexed analysis in molecular oncology in the era of next-generation sequencing. Mol. Diagn. Ther. 21, 587–600. doi: 10.1007/s40291-017-0287-7
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 29, 15–21. doi: 10.1093/bioinformatics/bts635
Ha, G., Roth, A., Khattra, J., Ho, J., Yap, D., Prentice, L. M., et al. (2014). TITAN: inference of copy number architectures in clonal cell populations from tumor whole-genome sequence data. Genome Res. 24, 1881–1893. doi: 10.1101/gr.180281.114
Ha, G., Roth, A., Lai, D., Bashashati, A., Ding, J., Goya, R., et al. (2012). Integrative analysis of genome-wide loss of heterozygosity and monoallelic expression at nucleotide resolution reveals disrupted pathways in triple-negative breast cancer. Genome Res. 22, 1995–2007. doi: 10.1101/gr.137570.112
Horner, M. J., Shiels, M. S., Pfeiffer, R. M., and Engels, E. A. (2020). Deaths attributable to cancer in the United States HIV population during 2001-2015. Clin. Infect. Dis. 72, e224–e231. doi: 10.1093/cid/ciaa1016
Hortlund, M., Arroyo Muhr, L. S., Storm, H., Engholm, G., Dillner, J., and Bzhalava, D. (2017). Cancer risks after solid organ transplantation and after long-term dialysis. Int. J. Cancer 140, 1091–1101. doi: 10.1002/ijc.30531
Ilie, M., and Hofman, P. (2016). Pros: Can tissue biopsy be replaced by liquid biopsy? Transl. Lung Cancer Res. 5, 420–423. doi: 10.21037/tlcr.2016.08.06
Ishii, H., Azuma, K., Sakai, K., Naito, Y., Matsuo, N., Tokito, T., et al. (2020). Determination of somatic mutations and tumor mutation burden in plasma by capp-seq during afatinib treatment in nsclc patients resistance to osimertinib. Sci. Rep. 10:691. doi: 10.1038/s41598-020-57624-4
Kim, S., Scheffler, K., Halpern, A. L., Bekritsky, M. A., Noh, E., Kallberg, M., et al. (2018). Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods 15, 591–594. doi: 10.1038/s41592-018-0051-x
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Lopez, C., Kleinheinz, K., Aukema, S. M., Rohde, M., Bernhart, S. H., Hubschmann, D., et al. (2019). Genomic and transcriptomic changes complement each other in the pathogenesis of sporadic Burkitt lymphoma. Nat. Commun. 10:1459. doi: 10.1038/s41467-019-08578-3
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Mouliere, F., Mair, R., Chandrananda, D., Marass, F., Smith, C. G., Su, J., et al. (2018). Detection of cell-free DNA fragmentation and copy number alterations in cerebrospinal fluid from glioma patients. EMBO Mol. Med. 10:e9323. doi: 10.15252/emmm.201809323
Muralidharan, K., Yekula, A., Small, J. L., Rosh, Z. S., Kang, K. M., Wang, L., et al. (2021). TERT promoter mutation analysis for blood-based diagnosis and monitoring of gliomas. Clin. Cancer Res. 27, 169–178. doi: 10.1158/1078-0432.CCR-20-3083
Newman, A. M., Bratman, S. V., To, J., Wynne, J. F., Eclov, N. C., Modlin, L. A., et al. (2014). An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 20, 548–554. doi: 10.1038/nm.3519
Plattner, B. W., Chen, P., Cross, R., Leavitt, M. A., Killen, P. D., and Heung, M. (2018). Complications and adequacy of transplant kidney biopsies: a comparison of techniques. J. Vasc. Access 19, 291–296. doi: 10.1177/1129729817747543
Qiu, Z. W., Bi, J. H., Gazdar, A. F., and Song, K. (2017). Genome-wide copy number variation pattern analysis and a classification signature for non-small cell lung cancer. Genes Chromosomes Cancer 56, 559–569. doi: 10.1002/gcc.22460
Raman, L., Van der Linden, M., Van der Eecken, K., Vermaelen, K., Demedts, I., Surmont, V., et al. (2020). Shallow whole-genome sequencing of plasma cell-free DNA accurately differentiates small from non-small cell lung carcinoma. Genome Med. 12:35. doi: 10.1186/s13073-020-00735-4
Roschewski, M., Dunleavy, K., Pittaluga, S., Moorhead, M., Pepin, F., Kong, K., et al. (2015). Circulating tumour DNA and CT monitoring in patients with untreated diffuse large B-cell lymphoma: a correlative biomarker study. Lancet Oncol. 16, 541–549. doi: 10.1016/S1470-2045(15)70106-3
Rosenwald, A., Wright, G., Chan, W. C., Connors, J. M., Campo, E., Fisher, R. I., et al. (2002). The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N. Engl. J. Med. 346, 1937–1947. doi: 10.1056/NEJMoa012914
Rossi, D., Diop, F., Spaccarotella, E., Monti, S., Zanni, M., Rasi, S., et al. (2017). Diffuse large B-cell lymphoma genotyping on the liquid biopsy. Blood 129, 1947–1957. doi: 10.1182/blood-2016-05-719641
Schmitz, R., Wright, G. W., Huang, D. W., Johnson, C. A., Phelan, J. D., Wang, J. Q., et al. (2018). Genetics and Pathogenesis of diffuse large b-cell lymphoma. N. Engl. J. Med. 378, 1396–1407. doi: 10.1056/NEJMoa1801445
Sigel, K., Dubrow, R., Silverberg, M., Crothers, K., Braithwaite, S., and Justice, A. (2011). Cancer screening in patients infected with HIV. Curr. HIV/AIDS Rep. 8, 142–152. doi: 10.1007/s11904-011-0085-5
Soneson, C., Love, M. I., and Robinson, M. D. (2015). Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Research 4:1521. doi: 10.12688/f1000research.7563.2
Spoerke, J. M., O’Brien, C., Huw, L., Koeppen, H., Fridlyand, J., Brachmann, R. K., et al. (2012). Phosphoinositide 3-kinase (PI3K) pathway alterations are associated with histologic subtypes and are predictive of sensitivity to PI3K inhibitors in lung cancer preclinical models. Clin. Cancer Res. 18, 6771–6783. doi: 10.1158/1078-0432.CCR-12-2347
Testoni, M., Zucca, E., Young, K. H., and Bertoni, F. (2015). Genetic lesions in diffuse large B-cell lymphomas. Ann. Oncol. 26, 1069–1080. doi: 10.1093/annonc/mdv019
Van Roy, N., Van Der Linden, M., Menten, B., Dheedene, A., Vandeputte, C., Van Dorpe, J., et al. (2017). Shallow whole genome sequencing on circulating cell-free dna allows reliable noninvasive copy-number profiling in neuroblastoma patients. Clin. Cancer Res. 23, 6305–6314. doi: 10.1158/1078-0432.CCR-17-0675
Wislez, M., Domblides, C., Greillier, L., Mazières, J., Monnet, I., Kiakouama-Maleka, L., et al. (2021). Circulating tumor DNA in advanced non-small-cell lung cancer patients with HIV is associated with shorter overall survival: Results from a Phase II trial (IFCT-1001 CHIVA). Lung Cancer 157, 124–130. doi: 10.1016/j.lungcan.2021.05.013
Yarchoan, R., and Uldrick, T. S. (2018). HIV-associated cancers and related diseases. N. Engl. J. Med. 378, 1029–1041. doi: 10.1056/NEJMra1615896
Keywords: liquid biopsy, cfDNA, CNA profile, cancer, immunodeficiency
Citation: Bouzidi A, Labreche K, Baron M, Veyri M, Denis JA, Touat M, Sanson M, Davi F, Guillerm E, Jouannet S, Charlotte F, Bielle F, Choquet S, Boëlle P-Y, Cadranel J, Leblond V, Autran B, Lacorte J-M, Spano J-P, Coulet F and the IDEATION study group (2021) Low-Coverage Whole Genome Sequencing of Cell-Free DNA From Immunosuppressed Cancer Patients Enables Tumor Fraction Determination and Reveals Relevant Copy Number Alterations. Front. Cell Dev. Biol. 9:661272. doi: 10.3389/fcell.2021.661272
Received: 30 January 2021; Accepted: 18 June 2021;
Published: 03 August 2021.
Edited by:
Catherine Alix-Panabieres, Centre Hospitalier Universitaire de Montpellier, FranceReviewed by:
Florent Mouliere, Amsterdam University Medical Center, NetherlandsSimon Heeke, University of Texas MD Anderson Cancer Center, United States
Copyright © 2021 Bouzidi, Labreche, Baron, Veyri, Denis, Touat, Sanson, Davi, Guillerm, Jouannet, Charlotte, Bielle, Choquet, Boëlle, Cadranel, Leblond, Autran, Lacorte, Spano, Coulet and the IDEATION study group. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Florence Coulet, ZmxvcmVuY2UuY291bGV0QGFwaHAuZnI=
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work
§These authors have contributed equally to this work and share last authorship