
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 31 May 2021
Sec. Signaling
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.656604
This article is part of the Research Topic Emerging Mechanisms for Skeletal Muscle Mass Regulation View all 10 articles
 James P. White1,2,3*
James P. White1,2,3*Skeletal muscle protein synthesis is a highly complex process, influenced by nutritional status, mechanical stimuli, repair programs, hormones, and growth factors. The molecular aspects of protein synthesis are centered around the mTORC1 complex. However, the intricacies of mTORC1 regulation, both up and downstream, have expanded overtime. Moreover, the plastic nature of skeletal muscle makes it a unique tissue, having to coordinate between temporal changes in myofiber metabolism and hypertrophy/atrophy stimuli within a tissue with considerable protein content. Skeletal muscle manages the push and pull between anabolic and catabolic pathways through key regulatory proteins to promote energy production in times of nutrient deprivation or activate anabolic pathways in times of nutrient availability and anabolic stimuli. Branched-chain amino acids (BCAAs) can be used for both energy production and signaling to induce protein synthesis. The metabolism of BCAAs occur in tandem with energetic and anabolic processes, converging at several points along their respective pathways. The fate of intramuscular BCAAs adds another layer of regulation, which has consequences to promote or inhibit muscle fiber protein anabolism. This review will outline the general mechanisms of muscle protein synthesis and describe how metabolic pathways can regulate this process. Lastly, we will discuss how BCAA availability and demand coordinate with synthesis mechanisms and identify key factors involved in intramuscular BCAA trafficking.
Protein synthesis is regulated primarily at the initiation phase of protein translation. A series of signaling proteins, referred to as eukaryotic initiation factors (eIFs), ultimately control this process and depend on upstream signals to modulate their activity. The pathways involved in protein synthesis are extensive, however, two different events govern the translation process, described in Figure 1. The binding of the methionyl tRNA (met-tRNA) to the 40S ribosomal subunit is regulated by the eukaryotic initiation factor 2 eIF2 (Price and Proud, 1994). eIF2 binds GTP and the eIF2-GTP-met-tRNA binds to the 40S ribosomal complex forming the 43S preinitiation complex. Once the start codon of an mRNA binds to the complex, GTP is hydrolyzed back to GDP. The complex cannot form the 43S preinitiation complex until GTP is reformed (Panniers and Henshaw, 1983; Price and Proud, 1994). The enzyme guanine nucleotide exchange factor eIF-2B will return GDP back to GTP and allow the complex to be active again. The regulation of this process is at the phosphorylation state of the α subunit of eIF2. When eIF2 is phosphorylated, eIF2B is inhibited from recycling GDP back to GTP and translation is stopped (Rowlands et al., 1988). eIF2 is phosphorylated by several kinases including double stranded RNA-dependent protein kinase (PKR), heme-regulated inhibitor kinase (HRI), eukaryotic translation initiation factor 2-alpha kinase 3 (PERK), the yeast general control non-derepressible 2 (GCN2) (Proud, 2005), and more recently, glycogen synthase kinase-3 beta (GSK3β) (Welsh et al., 1998). Each kinase appears to target eIF2 under different cellular stresses. PKR is activated in the presence of double stranded RNA (dsRNA) commonly found from viral infections (Meurs et al., 1990). PKR inactivates eIF2 as a protective mechanism to shut down protein synthesis and stop viral replication. HRI was discovered in reticulocytes (Crosby et al., 2000). When heme is deprived from reticulocytes protein synthesis is shut off. This process is associated with the inactivation and subsequent phosphorylation of eIF2. Of these regulators, only GCN2, GSK3β, and PERK regulate eIF2 based on amino acid availability. Their respective actions are illustrated in Figure 1. GCN2 is extensively studied in yeast during inhibition of protein synthesis by amino acid deprivation (Marton et al., 1993). It inactivates eIF2 by phosphorylation at serine 51. GSK3β has been shown to be a key regulator in insulin-dependent protein synthesis in skeletal muscle by inactivating eIF2 through phosphorylation at serine 540 (Jefferson et al., 1999). GSK3β and its role in muscle protein synthesis will be discussed in more detail later in the review. PERK has been found to target and inhibit eIF2 by phosphorylation at serine 51 under various conditions, including iron or heme-deficiency, amino acid starvation, viral infection, and accumulation of unfolded proteins (Dever, 2002).
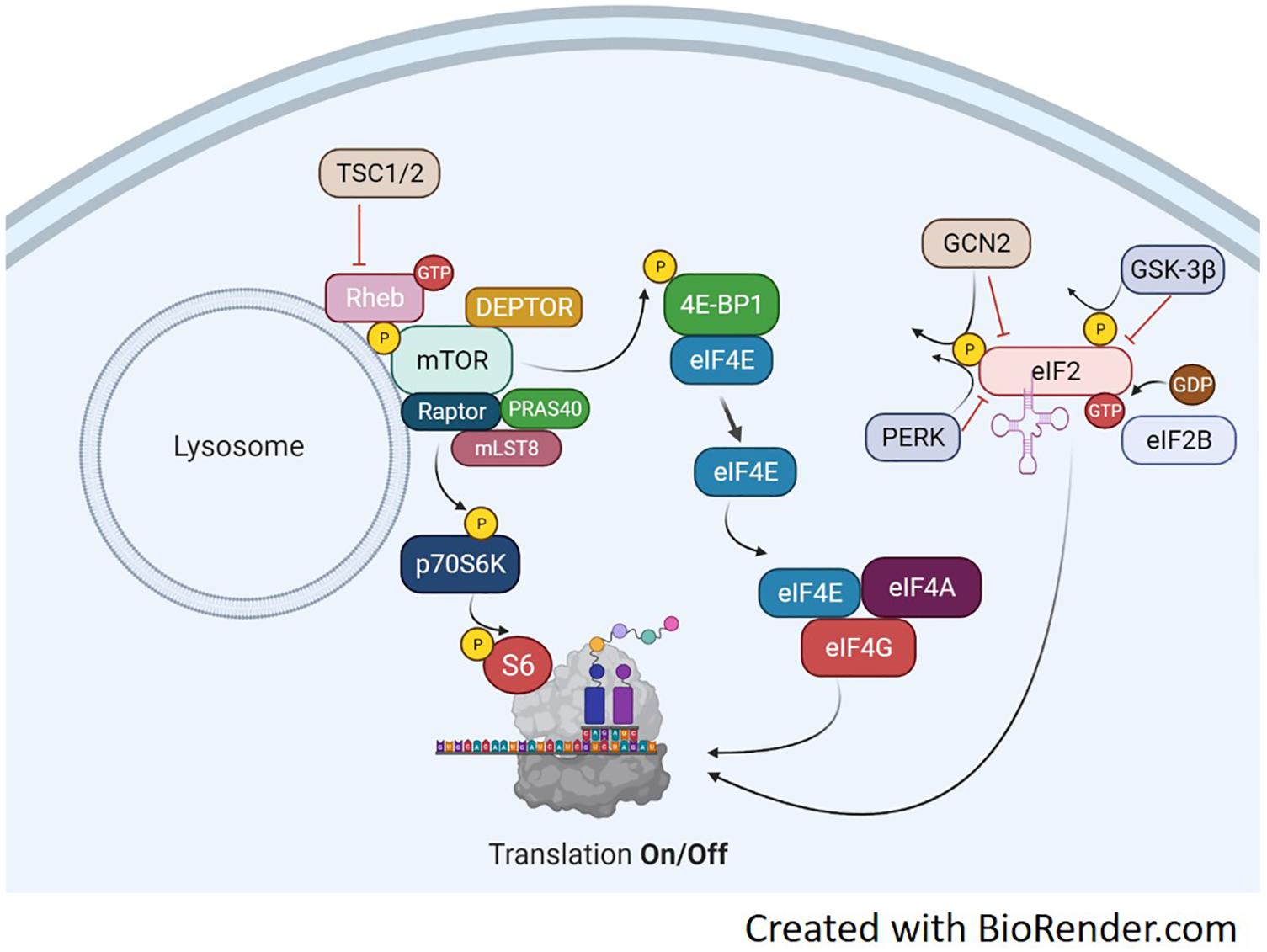
Figure 1. Translation Overview. Protein translation occurs, in part, by mTORC1-dependent regulation of p70S6K and the 4E-BP1/eIF4E complex and the mTORC1-independent eIF2 complex. mTORC1 activation is achieved through several points of regulation including, but not limited to phosphorylation at ser2448, inactivation of the TSC1/2 complex, binding of GTP-bound RHEB and translocation to the lysosome. Upon activation, mTORC1 will phosphorylate p70S6K at Thr389. This activates p70S6K to phosphorylate ribosomal protein S6 at ser236/236 and initiate RNA translation. mTORC1 can also regulate the eIF4E complex by phosphorylating 4E-BP1 which results in detachment of 4E-BP1 from eIF4E. This frees eIF4E to bind with eIF4A and eIF4G which will initiate binding of mRNA to the ribosome. A third point of translation initiation is the eIF2 complex. eIF2 forms a ternary complex with GTP and the Met-rRNA binds to the 40S ribosomal subunit to start the translation process. Its regulation is mediated through phosphorylation by various kinases including GSK-3β, PERK, and GCN2. Phosphorylation of eIF2 inhibits the guanine nucleotide exchange function of eIF2B and prevents translation.
The remaining regulatory mechanisms of protein translation are downstream of the mammalian target of rapamycin complex 1 (mTORC1). mTOR is a Ser/Thr kinase involved in a variety of processes including cell growth and differentiation, protein synthesis, and actin cytoskeletal organization. The mTORC1 complex can phosphorylate both 4E-BP1 and p70S6K to activate two downstream translation pathways, seen in Figure 1. Upon phosphorylation, the 4E-BP1 detaches from eIF4E and binds the eIF4F complex (Mader et al., 1995). The eIF4F complex recruits the 40S ribosomal subunit to mRNA through the 5′-cap structure (Proud, 2007). The eIF4F complex consists of three subunits each with distinct functions. eIF4E binds the 5′mRNA, eIF4A is an ATP-dependent RNA helicase and eIF4G serves as structural support for both eIF4E and 4A to form the eIF4F complex. eIF4E has been identified as a main regulatory protein for translation initiation through the eIF4F pathway. During instances of hypophosphorylation, 4E-BP1 remains bound to eIF4E and translation is turned off (Mader et al., 1995).
The upstream pathways controlling mRNA translation are, in part, through the IGF-1/mTORC1 pathway (Glass, 2010), illustrated in Figure 2. The IGF-Akt (PKB) signaling pathway is well established for its role in regulation of skeletal muscle mass controlling both protein synthesis, degradation, and apoptotic pathways (Frost and Lang, 2007). The binding of IGF-1 activates the receptor tyrosine kinase IGF-1 receptor and recruits insulin receptor substrate (IRS), in particular IRS-1 (Sun et al., 1991; Yamauchi et al., 1998). This leads to the activation of phosphatidylinositol 3′-kinase (PI3K) and the eventual activation of the serine-threonine kinase Akt (PKB) via phosphorylation at serine 473 (Alessi et al., 1997; Andjelkovic et al., 1997; Moelling et al., 2002). Akt is a focal point in insulin and IGF-1 signaling in a variety of tissues. Akt is also phosphorylated by mTORC2 (Sarbassov et al., 2005), which will be discussed later in the review. Upon activation, Akt is involved in a multitude of downstream pathways that will promote muscle growth. During skeletal muscle hypertrophy, Akt activation is increased when examined in vivo (Bodine et al., 2001) and in cultured myotubes (Rommel et al., 2001). In addition, a genetically altered, constitutively active Akt was able to induce muscle hypertrophy independent of additional treatments (Bodine et al., 2001; Lai et al., 2004). Akt acts through mTORC1 pathways to initiate and enhance protein synthesis. In addition, Akt can enhance protein synthesis through inhibition of proteins that impede protein synthesis such as GSK-3β and PRAS40. As mentioned previously, GSK3β is an inhibitor of protein synthesis through phosphorylation and inhibition of eIF2. Akt phosphorylates GSK3β at Ser9 and inactivates its kinase activity, thus allowing the initiation of protein synthesis. Activation of the Akt/GSK3β pathway is observed in muscle in vitro, using anabolic stimulus on C2C12 myotubes. Administration of IGF-1 resulted in myotube hypertrophy, associated with hyperphosphorylation of both Akt and GSK3β (Rommel et al., 2001).
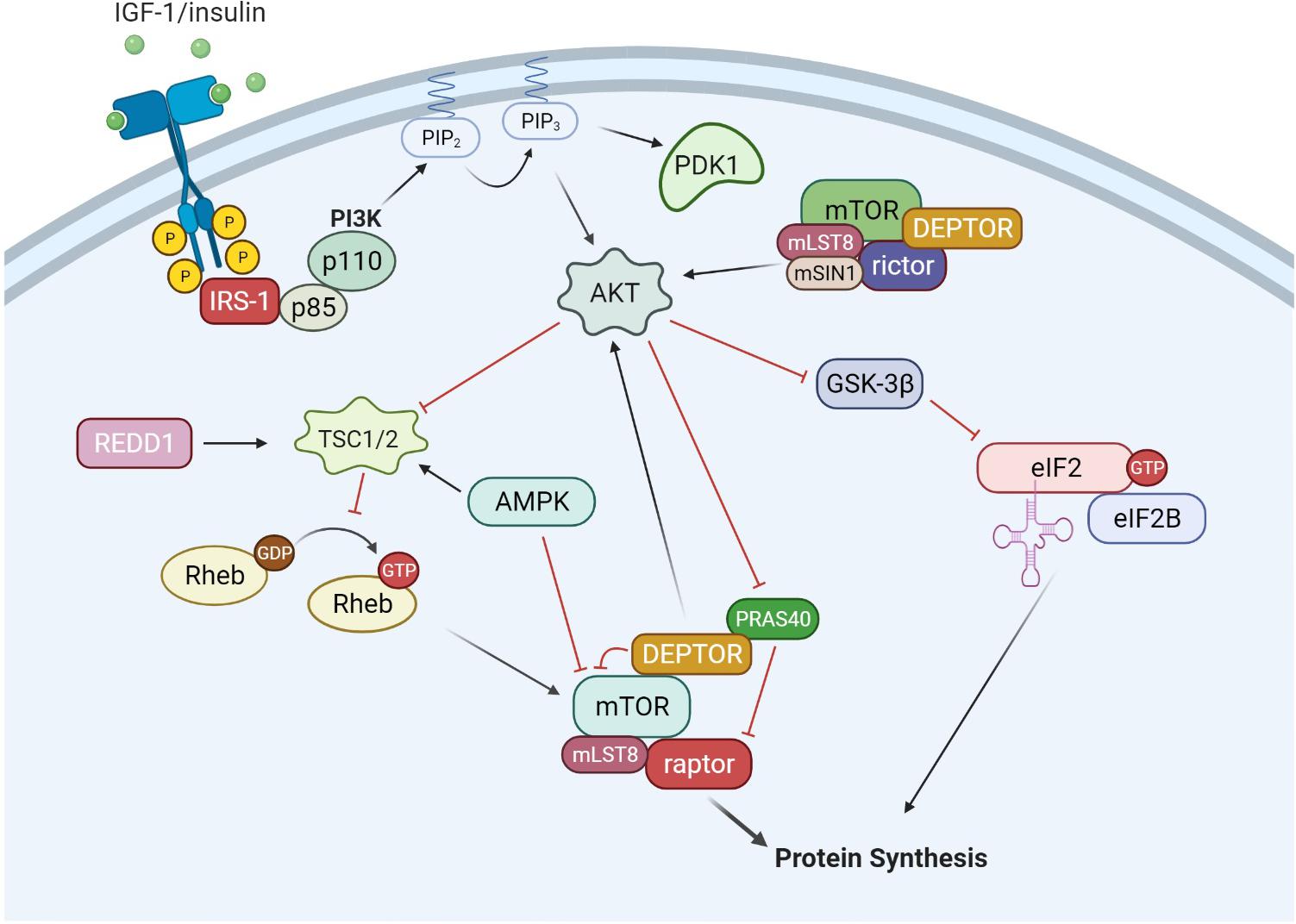
Figure 2. IGF-1/Akt signaling. The binding of growth factors such as IGF-1 and insulin activates receptor tyrosine kinases (RTKs), which recruits and activates IRS-1. IRS-1 then activates phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K), which consists of a regulator subunit (p85) and a catalytic subunit (p110). PI3K generates phosphoinositide 3,4, 5-phosphate (PIP3). PIP3 recruits and activates PDK1 and Akt. In addition to PIP3 signaling, mTORC2 can also phosphorylate and activate Akt. From there, Akt can signal through both mTORC1 and eIF2 pathways to increase protein synthesis. Akt can phosphorylate and inactivate GSK-3β, which is an inhibitor of the eIF2 complex. Akt can also activate the mTORC1 complex through inactivation of TSC1/2 and PRAS40. AMPK is a negative regulator of the IGF-1/Akt pathway, inhibiting mTORC1 activity through activation of the TSC1/2 complex and direct inhibition of mTORC1.
There are several regulatory points throughout the IGF-1/Akt/mTORC1 pathway, yet mTORC1 appears to be the gateway to downstream anabolic signaling. mTOR assembles into two distinct complexes, mTORC1 and mTORC2. mTORC1 consists of raptor (regulated associated protein of mTOR), mLST8, DEPTOR, PRAS40, and mTOR. mTORC2 consists of rictor (rapamycin insensitive companion of mTOR), mSIN1, mLST8, DEPTOR, and mTOR. mTORC1 activation is well described through PI3K/Akt signaling. Akt can phosphorylate several proteins that regulate mTORC1 activity including mTORC1 itself, PRAS40 and tuberous sclerosis complex 2 (TSC2) (Sancak et al., 2007; Vander Haar et al., 2007). Currently, the signaling mechanism for Akt through TSC2 is the most well described pathway. Akt phosphorylates TSC2 on multiple residues leading to its inactivation. TSC2 is a GTPase activating protein for Rheb. Therefore, inactivation of TSC2 by Akt increases the amount of GTP:Rheb complex bound to mTOR and leads to its activation. The second mechanism by which Akt can activate mTORC1 is through phosphorylation of the mTORC1 inhibitor PRAS40. Phosphorylated PRAS40 will disassociate from mTORC1, release its inhibition and increase mTOR kinase activity (Wang et al., 2012). In relation to mTORC1, there is a limited understanding of the role of mTORC2 in muscle protein synthesis and growth (Bentzinger et al., 2008). Initial studies have shown mTORC2 to be involved in organization of actin cytoskeleton and possibly phosphorylate Akt at Ser473 (Jacinto et al., 2004). In addition, there may be a coordinated effort for both mTOR complexes to work together for maximizing muscle protein synthesis under anabolic conditions (Ogasawara et al., 2020).
The mTORC1 complex has several related protein-protein complexes which regulate signaling activity. Each protein has a unique function in the complex. Raptor acts as a scaffolding protein to recruit downstream targets of mTOR, p70S6K, and 4E-BP1 (Hara et al., 2002; Kim et al., 2002). It is also the anchoring protein used by the Rag GTPases to recruit mTORC1 to the lysosome (Sancak et al., 2008). In skeletal muscle, raptor KO mice have a marked reduction in phosphorylation of both p70S6K and 4E-BP1 (Bentzinger et al., 2008). In addition, p70S6K and 4E-BP1 proteins have common mTORC1 signaling (TOS) motifs, which are essential for mTORC1-targeted phosphorylation (Dunlop et al., 2009). The raptor protein can also be modified at multiple phosphorylation sites. Phosphorylation of raptor appears to happen in a sequential manner (Foster et al., 2010). Once activated, mTOR will phosphorylate raptor at Ser863, which primes raptor for phosphorylation at several other sites including Ser859/855 (Foster et al., 2010). The phosphorylation events must be in the presence of adequate amino acid concentrations. In HEK293 cells, insulin-stimulated phosphorylation of raptor was not evident in amino acid depleted serum showing amino acid availability is critical for mTORC1 activity despite the availability of other growth factors (Foster et al., 2010). Raptor can also be phosphorylated by the 5′-adenosine monophosphate-activated protein kinase (AMPK) at Ser792, which inhibits raptor function and related mTORC1 activity (Gwinn et al., 2008). Skeletal muscle raptor phosphorylation at the AMPK targeted Ser792 is associated with body weight loss during cancer-associated muscle wasting (White et al., 2011).
The protein PRAS40 is another member of the TORC1 complex. PRAS40 has been shown to be an inhibitor of mTOR activity (Sancak et al., 2007; Wang et al., 2007). PRAS40 is bound to the inactive mTORC1 complex and directly inhibits substrate binding to raptor preventing downstream phosphorylation (Wang et al., 2007). Akt has been shown to phosphorylate and inhibit PRAS40 binding to raptor (Vander Haar et al., 2007), however, mTOR can phosphorylate PRAS40 as well (Foster et al., 2010). Upon activation from insulin or amino acids, activated mTOR can phosphorylate PRAS40 which facilitated its disassociation from the complex (Foster et al., 2010). Once PRAS40 is off the complex, raptor can bind p70S6K and 4E-BP1 for eventual phosphorylation. In male mice, muscle PRAS40 phosphorylation is responsive to circulating testosterone and muscle mass (White et al., 2012). Castration decreases phospho PRAS40, which is rescued with androgen add-back (White et al., 2012).
DEP domain-containing mTOR-interacting protein is a relatively recent addition to the mTORC1 complex, having an inhibitory function on mTORC1 activity (Peterson et al., 2009). mTORC1 and DEPTOR negatively regulate each other, depending on nutrient availability. In a low nutrient state, the PZD domain of DEPTOR binds to the C-terminal portion of mTOR and inhibits downstream signaling to p70S6K and 4E-BP1. In addition, it also activates mTORC2/Akt signaling by relieving the mTORC1 negative feedback inhibition of PI3K (Peterson et al., 2009). During nutrient availably and subsequent mTORC1 activity, DEPTOR is phosphorylated and released from the mTORC1 complex. The reduction in protein expression is also accompanied with a suppression of DEPTOR mRNA expression (Peterson et al., 2009). In C2C12 myotubes, the knockdown of DEPTOR increased protein synthesis and associated mTORC1 signaling (Kazi et al., 2011). DEPTOR knockdown, in vivo, resulted in an attenuation of immobilization-induced muscle atrophy associated with increased muscle protein synthesis (Kazi et al., 2011). The sensitivity of DEPTOR to atrophy conditions has been replicated by others showing DEPTOR expression increases with limb immobilization (Shimkus et al., 2018) and hind limb unloading (Roberson et al., 2020). The regulation of DEPTOR from nutrient availability does not appear as strong as mechanical loading, as fasting/refeeding did not alter DEPTOR protein expression (Shimkus et al., 2018). However, further investigation is needed to determine how DEPTOR is regulated under different nutrients and availability of amino acids.
There are several negative regulators of mTORC1 activity existing outside the mTORC1 complex. Two well documented inhibitors are AMPK and the protein regulated in DNA damage and development 1 (REDD1, also referred to as Rtp801 and DDIT4). Their respective interactions with the Akt/mTOR pathways are shown in Figures 2, 3. AMPK and mTOR are key energy sensors in the cell, and function to regulate processes to either inhibit or enhance ATP production depending on nutrient availability. AMPK will be discussed in more detail later in this review. REDD1 is thought to inhibit mTORC1 signaling through activation of upstream TSC2 (Brugarolas et al., 2004; Reiling and Hafen, 2004; Sofer et al., 2005). In addition, REDD1 protein and mRNA expression are increased with cellular stress events including ATP depletion (Sofer et al., 2005), DNA damage (Lin et al., 2005), endoplasmic reticulum stress (Wang et al., 2003; Protiva et al., 2008), and hypoxia (Shoshani et al., 2002; Brugarolas et al., 2004; Reiling and Hafen, 2004; Schwarzer et al., 2005). Furthermore, treatment with the synthetic glucocorticoid dexamethasone has shown to increase REDD1 mRNA and protein in skeletal muscle as well as L6 myotubes (Wang et al., 2006). Glucocorticoids such as cortisone are elevated during fasting states which, in part through REDD1, may play a role in the inhibition of mTORC1 signaling and the subsequent reduction in protein synthesis. REDD1 protein and mRNA expression was increased with 18 h of starvation in rats which coincided with a reduction in mTORC1 signaling (McGhee et al., 2009). Upon refeeding, REDD1 protein and mRNA expression was returned to baseline and mTORC1 signaling was increased. Interestingly, fasting-induced glucocorticoid concentrations directly correlated with REDD1 expression showing evidence of cross talk between energy-sensitive hormones and energy-sensitive signaling within muscle (McGhee et al., 2009). Finally, the loss of REDD1 during a mechanical-overload hypertrophy stimuli enhanced the rate of muscle protein synthesis (Gordon et al., 2016), again, suggesting its role as a negative regulator of muscle protein anabolism.
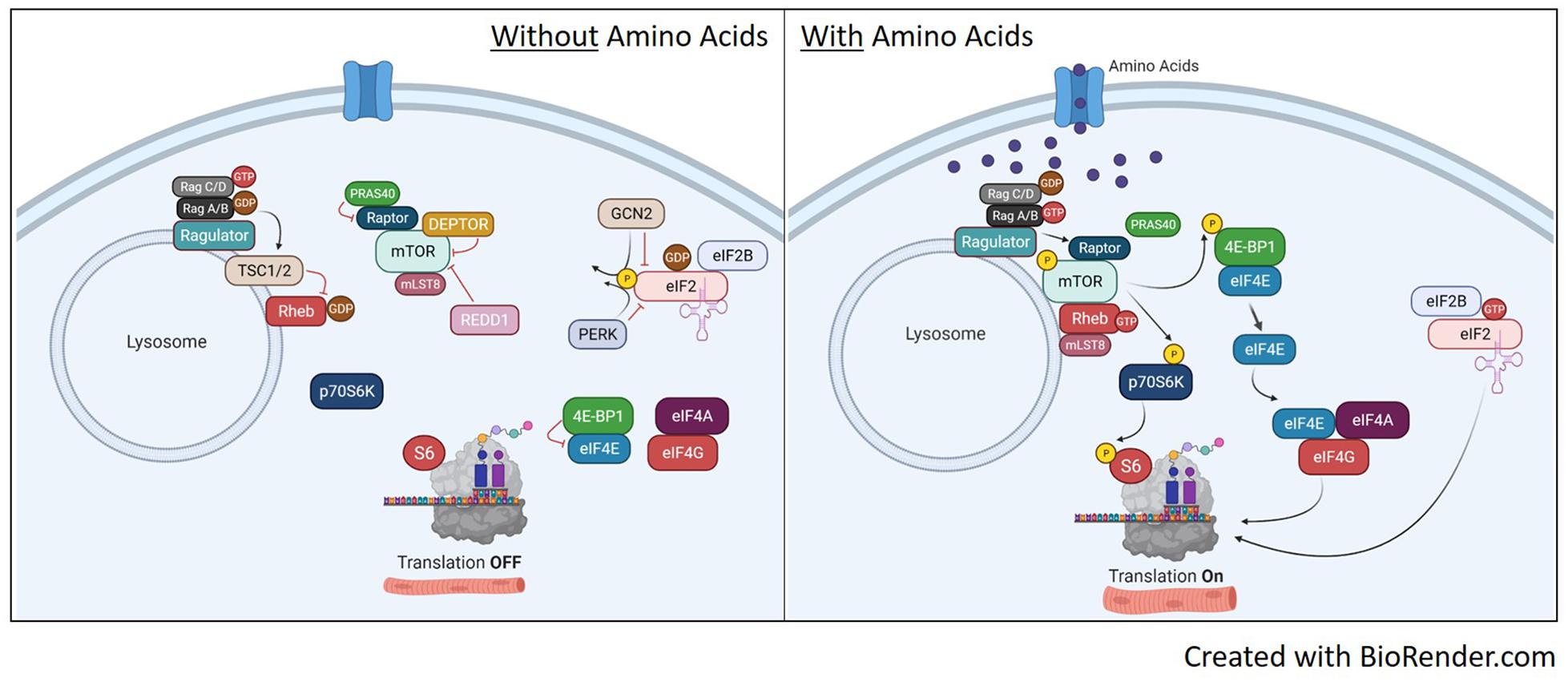
Figure 3. Translation pathways with or without amino acid availability. Without amino acid availability (left image), the heterodimeric Rag protein is loaded with GTP on Rag C/D while Rag A/B has the GDP. This conformation localizes the TSC1/2 complex to the lysosome to prevent GDP-Rheb conversion to GTP and prevents the recruitment of mTORC1 to the lysosome. This renders both S6 and 4E-BP1 unphosphorylated, shutting off translation. In addition, the inactive mTORC1 with be further suppressed by inhibitors DEPTOR, REDD1 and PRAS. On the eIF2 pathway, the absence of amino acids maintains GCN2 and PERK activity, which phosphorylates eIF2, inhibiting guanine nucleotide exchange of eIF2 by eIF2B. With eIF2 bound to GDP, it will release from the ribosome and stop translation. Upon availability of amino acids (right image), RagA/B is now loaded with GTP as it maintains binding to ragulator. This initiates removal of TSC1/2 from the lysosome and Rag-induced recruitment of mTORC1 to the lysosome via raptor. Once at the lysosome, mTOR kinase activity is activated by GTP-bound Rheb and will phosphorylate p70S6K and 4E-BP1 to initiate translation. mTORC1 activation will also disassociate inhibitors DEPTOR and PRAS40. Once dissociated, DEPTROR is quickly degraded. In relation to eIF2, the availability of amino acids will inhibit GCN2 and PERK, reversing phosphorylation on eIF2 and allowing guanine nucleotide exchange of eIF2 back to GTP. This will bind the eIF2 complex to the 40S ribosomal subunit and initiate protein translation.
AMPK is activated through the buildup of low energy phosphate group, AMP or by phosphorylation by one or more upstream kinases at a threonine residue within the activation loop of the α subunit (Hawley et al., 1996; Stein et al., 2000). The multiple targets that AMP can activate will induce a large activation in the activity of AMPK with relatively small changes in AMP. The energy state of the cell is not solely monitored by AMP concentrations. High ATP concentrations will oppose activation of AMP-induced pathways. Thus, the AMP:ATP ratio appears to the critical readout of cellular energy status and regulator of AMPK activity.
During physiological conditions, AMPK can be regulated by chemical mediators of metabolism in addition to ATP:AMP levels. Cellular levels of phosphocreatine can allosterically inhibit AMPK activity (Ponticos et al., 1998). In addition, glycogen content of the cell can affect AMPK activity (Hudson et al., 2003; Polekhina et al., 2003). The β-subunits of AMPK contain a glycogen binding domain. Reports in human and rodent muscle show high glycogen stores can inhibit AMPK activation (Wojtaszewski et al., 2002, 2003). Over expression of AMPK in culture has shown AMPK to localize in large glycogen granules (Hudson et al., 2003). Glycogen will not only bind AMPK, but also contain in close proximity glycogen synthase, a substrate of AMPK. Considering AMPK is allosterically regulated by phosphocreatine and glycogen stores, it has been speculated that AMPK is regulated by both short and long term energy stores (Hardie, 2003).
AMPK has been shown to inhibit protein synthesis in skeletal muscle (Rolfe and Brown, 1997; Bolster et al., 2002; Deshmukh et al., 2008; Thomson et al., 2008; Mounier et al., 2009), liver (Reiter et al., 2005), and cultured myotubes (Williamson et al., 2006; Tong et al., 2009). The potency of AMPK signaling was described by Pruznak et al. (2008), showing AMPK activation can override the stimulatory effects of leucine on muscle protein synthesis (Pruznak et al., 2008). In contrast, deletion of the AMPKα1 gene in primary myotubes resulted in cell hypertrophy (Mounier et al., 2009). The mechanism by which AMPK inhibits muscle protein synthesis is through the inhibition of the mTORC1 complex (Bolster et al., 2002; Horman et al., 2002; Chan et al., 2004; Gwinn et al., 2008). There are currently three proposed mechanisms by which AMPK can inhibit mTORC1 signaling. The first is through phosphorylation of mTOR on Thr2446 (Cheng et al., 2004). This process does not directly inhibit mTOR activity, however, phosphorylation at Ser2446 prevents phosphorylation of Ser2448 which has been shown to increase mTOR activity (Bolster et al., 2002; Chiang and Abraham, 2005). The second method, and perhaps the best described mechanism, is through AMPK-mediated phosphorylation of the tuberous sclerosis complex 2 (TSC2) gene product Tuberin on Thr1227 and Ser1345 (Inoki et al., 2003). TSC2 combines with TSC1 to form a GTPase activator protein (GAP) for the Ras homolog enriched in brain (Rheb), causing an increase in GDP bound to Rheb (Zhang et al., 2003; Long et al., 2005a). The binding of the GDP:Rheb complex to mTORC1 inhibitors mTOR. The third mechanism, as discussed earlier in the review, is the phosphorylation of raptor (Gwinn et al., 2008). This promotes binding of the 14-3-3 protein and inhibition of raptor to signal downstream to p70S6K and 4E-BP1.
The AMPK pathways has been heavily investigated in muscle in regards to other aspects of mTORC1 signaling. In C2C12 cells, AICAR-induced AMPK activation showed a reduction in protein synthesis, polysome aggregation and downstream mTORC1 signaling proteins 4E-BP1, p70S6K and eEF2 (Williamson et al., 2006). Although Akt, upstream of mTORC1, remained unaffected with AICAR treatment, downstream AMPK targets raptor and TSC2 were effected with AMPK activation. In addition, AICAR increased the amount of TSC1 bound to TSC2 (Williamson et al., 2006), suggesting increased GTPase activity and inhibition of Rheb. A study by Du et al. (Tong et al., 2009) treated C2C12 cells with both AICAR and IGF-1 to determine if AMPK signaling can inhibit IGF-1 signaling, with the hypothesis that IGF-1 signaling originates upstream of mTORC1. AICAR treatment without IGF-1 resulted in cell atrophy caused by a reduction in signaling related to protein synthesis and an increase in markers of protein degradation. The addition of IGF-1 did not rescue the inhibition of AICAR treatment despite a significant increase in Akt, supporting the proposed mechanism of potent mTORC1 inhibition by AMPK.
AMPK has been examined in rodent models of muscle hypertrophy to determine its role in growth suppression. Overload-induced hypertrophy of the plantaris muscle was enhanced in AMPKα1−/− mice. This occurred in conjunction with an increase in phosphorylation of downstream targets of mTORC1 signaling p70S6K and 4E-BP1 (Mounier et al., 2009). In contrast, AICAR treatment resulted in a reduction in the percentage of plantaris muscle mass gained after 1 week of over load (Gordon et al., 2008). In the same study, there was a significant negative correlation between the percentage of plantaris hypertrophy and AMPK phosphorylation status in the plantaris muscle. In addition, there were also negative correlations between phosphorylation status of AMPK and p70S6K, eEF2 and 4E-BP1.
Catabolic signaling through AMPK can override amino acid-induced mTORC1 activation. AICAR treatment prevented leucine-stimulated protein synthesis in the mouse gastrocnemius muscle (Pruznak et al., 2008). The prevention of synthesis was accompanied with the prevention of mTOR activation. AMPK can phosphorylate and activate TSC-2, which subsequently inactivates mTOR. AICAR treatment did not increase the phosphorylation of TSC-2 or alter TSC-1/TSC-2 binding. However, AICAR treatment did increase phosphorylation of raptor independent of leucine treatment. The activation of downstream signaling proteins p70S6K1, 4E-BP1, and eIF4F were increased with leucine administration and prevented when leucine was given with AICAR treatment. This data is in agreement with the results from Du et al. (Du et al., 2007) who showed AICAR treatment prevents a leucine-induced increase in protein synthesis in C2C12 myoblasts. In support of these data, myoblasts expressing a dominant negative AMPKα subunit were administered AICAR. Without AMPK activation, leucine was able to increase protein synthesis even with AICAR treatment. Once again, suggesting AMPK-induced inhibition of protein synthesis was through the reduction in mTORC1. These results support the hypothesis that cellular energy demands can supersede the anabolic potential of amino acid availability.
Although mTORC1 is a key component of the IGF-1/insulin signaling pathway, mTORC1 can be activated independent of upstream signaling by amino acids (Potier et al., 2009). Branched-chain amino acids (BCAAs), especially leucine, are potent regulators of mTORC1 activity and increase rates of protein synthesis (Goberdhan et al., 2009). Infusion of an amino acid mixture into resting human subjects increased protein synthesis as early as 30 min after infusion and remained elevated for 90 min (Bohe et al., 2001). Amino acid infusion has been shown to increase phosphorylation of downstream targets of mTORC1, p70S6K, and 4E-BP1 (Greiwe et al., 2001; Liu et al., 2001) without effecting Akt (Greiwe et al., 2001) or its downstream target GSK3β (Liu et al., 2004). In the rodent, mTORC1 activity is necessary for BCAAs to induce anabolic signaling, as rapamycin prevented leucine-induced increased phosphorylation of p70S6K and 4E-BP1 (Anthony et al., 2000).
Despite strong evidence suggesting BCAAs activate mTORC1 signaling, the direct mechanism for mTORC1 activation remains unclear, especially in skeletal muscle. It has been proposed that BCAAs work through both a TSC1/2 dependent (Gao et al., 2002) and independent (Smith et al., 2005) manner to activate mTORC1 activity. The TSC1/2 complex can mediate amino acid signaling to mTORC1 (Gao et al., 2002), which could be regulated through relative localization of TSC1/2 and mTOR on the lysosome (Demetriades et al., 2014; Menon et al., 2014). Other reports have shown evidence for TSC1/2 independent activation of mTORC1 signaling through RAG and Rheb. In mammals, there are four RAG GTPases (A-D) shown to have a role in amino acid signaling to mTORC1 (Schurmann et al., 1995; Kim et al., 2008; Sancak et al., 2008). Amino acids can convert RAG to an active formation including Rag A/B loaded with GTP and Rag C/D loaded with a GDP. The active Rag A/B recruits the mTORC1 complex to the lysosome through raptor where it is activated by Rheb (Sancak et al., 2008). Rheb activates mTORC1 by direct associated and activation of the mTOR catalytic domain (Long et al., 2005a,b). The regulation of mTORC1 activation with and without amino acid availability is illustrated in Figure 3.
Amino acid metabolism has been well described, especially in the context of insulin resistant and obesity (White and Newgard, 2019). In skeletal muscle, the balance between amino acid catabolism and anabolism is complex, due to both metabolic and anabolic flux of the myofiber. BCAAs, in particular, are a widely used source to generate TCA/Krebs cycle intermediates for energy production. Although the majority of amino acid metabolism occurs in the liver, skeletal muscle has high expression of the branched-chain aminotransferase (BCAT). Interestingly, despite its metabolic nature, the liver does not express BCAT, rendering a unique pathway of BCAA metabolism to skeletal muscle (Hutson, 1989; White et al., 2020). BCAT-mediated transamination of leucine generates α-ketoisocaproate, the first step of BCAA catabolism in muscle. The second step of leucine catabolism is an irreversible oxidative decarboxylation of α-ketoisocaproate, which is catalyzed by the branched-chain α-keto acid dehydrogenase (BCKDH). This reaction is a rate-limiting step in leucine metabolism (Harris et al., 1990, 1994).
Branched-chain α-keto acid dehydrogenase is a highly regulated dehydrogenase enzyme responsible for metabolizing branched-chain keto acids (BCKA) into branched-chain acyl CoAs. The branched-chain CoAs are further metabolized into acetyl CoA or Succinyl CoA and used as TCA intermediates for energy (Walejko et al., 2021). The multi-subunit BCKDH complex consists of three components including the e1, e2, and e3 subunits. Each subunit carries out different reactions to convert BCKAs into branched-chain acyl CoAs. There are two opposing regulators of BCKDH activity, the BCKDH kinase (BDK) and the PPM1K phosphatase (also referred to as PP2Cm) (White et al., 2018). Both enzymes perform phosphorylation and dephosphorylation, respectively, of serine 293 of the e1a subunit. In the liver, increased phosphorylation of BCKDH on serine 293 occurs secondary to elevated expression of BDK, and decreased expression of PPM1K (She et al., 2007; Lian et al., 2015). Murine knockout models of either BDK or PPM1K (Joshi et al., 2006; Lu et al., 2009) show the significance of their respective role in BCAA metabolism, as each manipulation effects circulating BCAAs accordingly. Currently, there is limited understanding of this pathway in skeletal muscle, especially in regards to anabolic and catabolic conditions. BCAA fate and the antagonist relationship between BCKDH and PPM1K is shown in Figure 4.
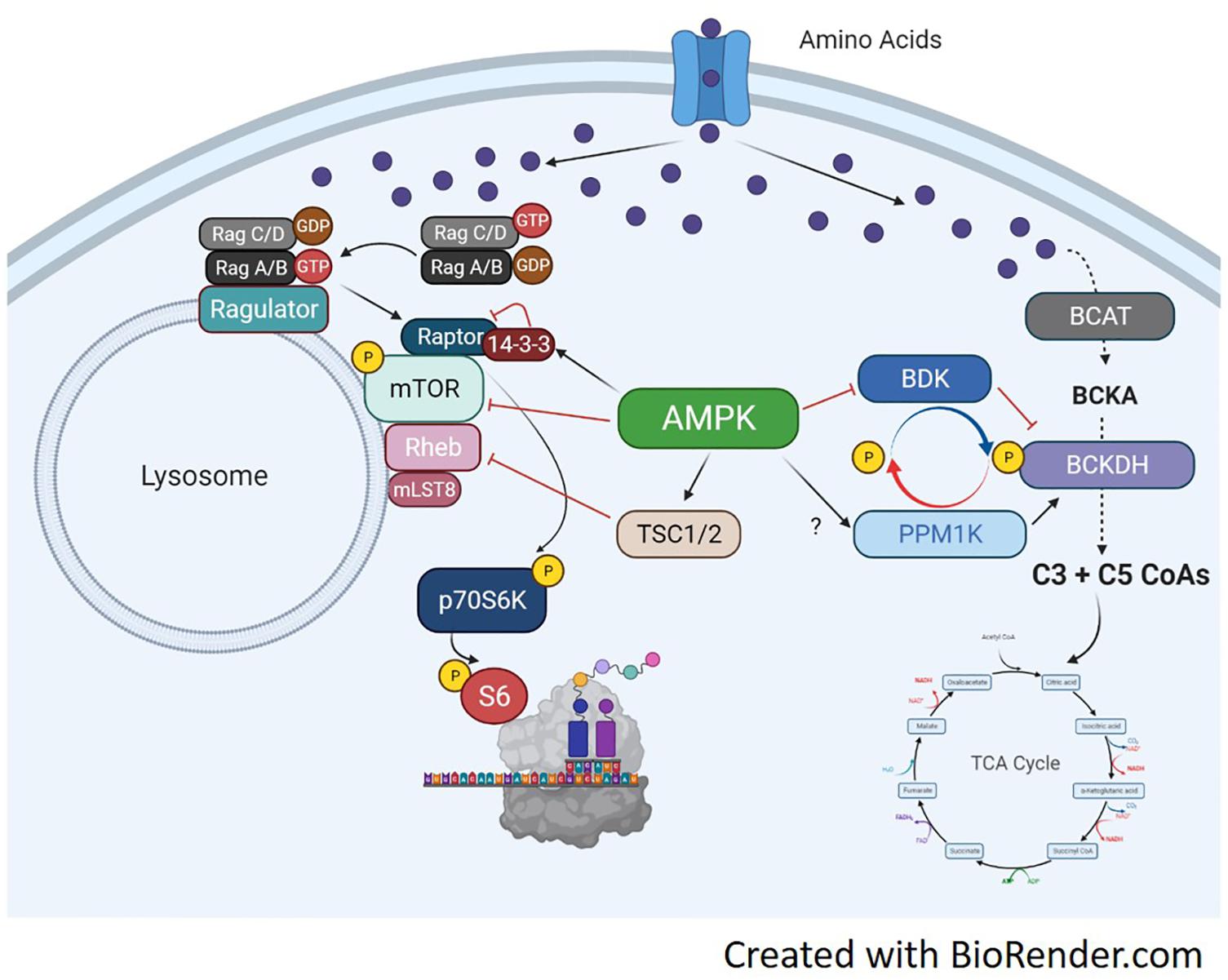
Figure 4. Molecular mechanisms of amino acid trafficking. Amino acids, especially BCAAs, enter the cell via their respective transporters. Once in the cell, depending on metabolic need, they can be metabolized for energy or used for other biochemical processes like protein synthesis. If needed for energy, the enzyme BCAT metabolizes the BCAA into branched-chain keto acids, which undergo a series of catabolic reactions to produce C3 and C5 CoAs by BCKDH. The C3/5 CoAs are then converted to either acetyl CoA or Succinyl CoA and can enter the TCA cycle. BCKDH is regulated by phosphorylation/dephosphorylation by the kinase BDK and the phosphatase PPM1K, respectively. Phosphorylation of BCKDH by BDK inhibits BCKDH activity while dephosphorylation by PPM1K activates BCKDH and increases generation of BCAA-derived CoAs for energy production. The activation of the BCAT/BCKDH pathway pulls amino acids away from the associated mTORC1 complexes and inhibits protein synthesis. If BDK is able to phosphorylate BCKDH, or PPM1K is inhibited, amino acids would be available to active mTORC1 and initiate protein translation. AMPK can regulate BCAA metabolism by increasing PPM1K and lowering BDK expression, which will activate BCKDH and increase BCAA flux to the TCA cycle. AMPK can simultaneously block mTORC1 signaling through activation of TSC1/2 or direct inhibition of mTOR and raptor.
As AMPK is a potent inhibitor of mTORC1 activity, it would also make sense that AMPK would regulate muscle amino acid metabolism. AICAR-induced AMPK activation can increase BCKDH activity in skeletal muscle through a reduction in BDK protein (Lian et al., 2015). Although AICAR increased PPM1K in liver and adipose tissue, muscle PPM1K was not effected by AICAR treatment (Lian et al., 2015). Skeletal muscle PGC-1α over-expression increases gene expression of branched-chain amino transferase (BCAT) 2 and BCKDH, while BDK was not changed. BCAAs levels in the PGC-1α mice were decreased in both muscle and blood (Hatazawa et al., 2014). These outcomes show the coupled relationship between two potent metabolic regulators, i.e., AMPK and PGC-1α, and the shift from anabolism to BCAA catabolism.
The extent of BCAA metabolism can have an impact on global muscle metabolism, as excess BCAAs or branched-chain ketoacids can inhibit insulin signaling in muscle in vitro (Moghei et al., 2016; Biswas et al., 2020) and in vivo (White et al., 2016, 2018; Wang et al., 2019). Interestingly, this result is dependent on certain BCAAs or a mixture of BCAAs as valine does seem to interfere with myotube insulin signaling in vitro (Rivera et al., 2020). Nutrient availability may also regulate metabolic fate of leucine. In C2C12 myotubes, leucine is used preferentially for protein synthesis rather than oxidation for energy production (Estrada-Alcalde et al., 2017). However, in the setting of high palmate, leucine oxidation increases and its incorporation into proteins decreases (Estrada-Alcalde et al., 2017). Moreover, high fat feeding increase BCKDH activity in muscle promoting amino acid catabolism (White et al., 2016). This again, points to the complexity of muscle metabolism and substrate availability altering BCAA trafficking. The proposed BCAA trafficking and related AMPK signaling pathways are shown in Figure 4.
In the context of muscle mass regulation, muscle BDK knockout mice have no overt muscle mass phenotype under a typical chow diet, despite a lower BCAA concentration in blood and muscle (Ishikawa et al., 2017). However, under a low protein diet, the lack of BDK magnifies myofibrillar protein loss associated with a reduction in mTORC1 signaling activity. Of note, protein restriction resulted in a reduction of myofibrillar protein synthesis, but not total (soluble) protein, indicating a preferential degradation of myofibrillar proteins to compensate for the low protein diet. A natural hypothesis would point to autophagy as a mechanism to provide amino acids during the restricted feeding. However, this study showed no difference in LC3I/II protein expression, and so the authors concluded autophagy was not playing a role and contributing to the myofibrillar degradation during the low protein diet (Ishikawa et al., 2017). This study highlights the interaction between BCAA metabolism and protein synthesis pathways, supporting an interactive crosstalk between the two processes. More work is needed to gain a better understanding of the molecular network between BCAA trafficking and mTORC1 signaling.
Together, muscle protein synthesis is an interactive process, taking input from numerous anabolic and catabolic pathways. The unique plasticity of skeletal muscle adds more layers of regulation, incorporating both metabolic demands and mechanical stimuli into these already intricate pathways. Moreover, there must be an adequate combination of mechanical stimuli and nutritional availability to maintain or hypertrophy myofiber size. In relation to other tissues, especially tumor biology, the molecular mechanisms involved in skeletal muscle protein synthesis are less developed. This concept is supported by the fact that the majority of citations in this review investigating amino acid metabolism and regulation of mTORC1 are not in muscle tissue. This is interesting, considering the extensive protein content of skeletal muscle and the potential utility of skeletal muscle as a model to investigate the complexities of protein synthesis. In regards to muscle signaling pathways, the IGF-1/insulin pathway dominates the literature since muscle is a major therapeutic target for diabetes drugs. However, the anabolic pathways in muscle have been relatively neglected since muscle atrophy/wasting is still considered a secondary symptom among various diseases and not targeted as commonly for pharmaceutical intervention.
A better understanding of the interface between muscle amino acid metabolism and synthesis pathways could uncover additional regulators of muscle protein synthesis. The high expression of BCAT in skeletal muscle supports the preference of branched-chain amino acids as a bioenergetic substrate. There is a gap in our understanding of fate decisions of BCAAs and anabolic signaling in muscle. This could be a result of the temporal nature of muscle energetics, having diverse metabolism with changes in nutrient availability and contractile activity. AMPK is an obvious regulator of BCAA fate, effecting both catabolic and anabolic signaling pathways, including direct/indirect effects on mTORC1 and BCKDH. However, it would not be surprising to identify additional regulators of BCAA metabolism having an impact on both catabolic and anabolic processes.
As for translational potential of these pathways to offset muscle atrophy/wasting, identification of anabolic pathways, unique to skeletal muscle, could open up therapeutic targets. The potency of these pathways to regulate muscle mass is supported by strong in vivo studies using preclinical models discussed throughout his review. Manipulation of key regulatory proteins within the mTORC1 signaling pathway can accelerate (Ishikawa et al., 2017) or preserve (Kazi et al., 2011) the loss in muscle mass and/or protein synthesis under atrophy-promoting conditions. Since we now have a general understanding of these pathways, why are there no available drugs to offset muscle wasting? The challenge is to identify key targets within these complex pathways and manipulate them in a muscle-specific manner. The mTORC1 pathway is tightly controlled and ubiquitous across many cell types. Promoting muscle anabolism by manipulating global mTORC1 activity will most likely alter the delicate balance of non-muscle cells and promote unchecked growth and malignancies. Finding key regulators within the mTORC1 pathway, specific to muscle would be ideal for drug development. This warrants continued investigation of anabolic pathways, especially within skeletal muscle.
The author confirms being the sole contributor of this work and has approved it for publication.
This work was supported by grants from the National Institutes of Health (grants K01AG056664 and R21AG065943).
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author would like to thank Phillip White for his valuable discussion and insight pertaining to this review.
4E-BP1, the eukaryotic initiation factor 4E binding protein 1; ADP, Adenine diphosphate; AICAR, 5-Aminoimidazole-4-carboxamide ribonucleotide; Akt, AKR thymoma; AMP, Adenine monophosphate; AMPK, AMP-activated kinase; ATP, Adenine triphosphate; BCAAs, Branched-chain amino acids; BCAT, Branched-chain amino acid aminotransferase; BCKDH, branched-chain α-keto acid dehydrogenase; BDK, BCKDH kinase; BKAs, branched-chain keto acids; DEPTOR, DEP domain-containing mTOR-interacting protein; eEF, Eukaryotic elongation factor; eIF, eukaryotic initiation factor; GAP, GTPase-activating protein; GCN2, general control non-derepressible 2; GDP, Guanosine diphosphate; GEF, guanine nucleotide exchange factor; GSK3β, glycogen synthase kinase-3; GTP, Guanosine-5 ′ -triphosphate; HRI, heme-regulated inhibitor kinase; IGF-1, Insulin-like growth factor; IRS, insulin receptor substrate; mLst8, mammalian LST8 homolog; mSin1, the mammalian stress-activated protein kinase-interacting 1; mTOR, mechanistic Target of Rapamycin; mTORC1, mammalian/mechanistic Target of Rapamycin Complex 1; mTORC2, mammalian/mechanistic Target of Rapamycin Complex 2; P70S6K, P70 Ribosomal protein S6 kinase; PDK-1, phosphoinositide-dependent kinase 1; PERK, eukaryotic translation initiation factor 2-alpha kinase 3; PGC-1α, Peroxisome proliferator-activated receptor gamma coactivator 1-alpha; PI3K, phosphatidylinositol 3-kinase; PI3P, phosphatidylinositol 3-phosphate; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; PKB, Protein kinase B; PKR, double stranded RNA-dependent protein kinase; PPM1K, protein phosphatase 1K; PRAS40, proline-rich Akt substrate, 40 kDa; Rag, Ras related GTP binding; Raptor, the regulatory-associated protein of mTOR (Raptor); REDD1, protein regulated in DNA damage and development 1; Rheb, Ras homolog enriched in brain; Rictor, the rapamycin-insensitive companion of mTOR; RTK, receptor tyrosine kinase; TCA, tricarboxylic acid; TOR, Target of Rapamycin; TSC, Tuberous sclerosis complex.
Alessi, D. R., James, S. R., Downes, C. P., Holmes, A. B., Gaffney, P. R., Reese, C. B., et al. (1997). Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr. Biol. 7, 261–269. doi: 10.1016/s0960-9822(06)00122-9
Andjelkovic, M., Alessi, D. R., Meier, R., Fernandez, A., Lamb, N. J., Frech, M., et al. (1997). Role of translocation in the activation and function of protein kinase B. J. Biol. Chem. 272, 31515–31524.
Anthony, J. C., Yoshizawa, F., Anthony, T. G., Vary, T. C., Jefferson, L. S., and Kimball, S. R. (2000). Leucine stimulates translation initiation in skeletal muscle of postabsorptive rats via a rapamycin-sensitive pathway. J. Nutr. 130, 2413–2419. doi: 10.1093/jn/130.10.2413
Bentzinger, C. F., Romanino, K., Cloetta, D., Lin, S., Mascarenhas, J. B., Oliveri, F., et al. (2008). Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab. 8, 411–424. doi: 10.1016/j.cmet.2008.10.002
Biswas, D., Dao, K. T., Mercer, A., Cowie, A. M., Duffley, L., El Hiani, Y., et al. (2020). Branched-chain ketoacid overload inhibits insulin action in the muscle. J. Biol. Chem. 295, 15597–15621. doi: 10.1074/jbc.ra120.013121
Bodine, S. C., Stitt, T. N., Gonzalez, M., Kline, W. O., Stover, G. L., Bauerlein, R., et al. (2001). Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell Biol. 3, 1014–1019. doi: 10.1038/ncb1101-1014
Bohe, J., Low, J. F., Wolfe, R. R., and Rennie, M. J. (2001). Latency and duration of stimulation of human muscle protein synthesis during continuous infusion of amino acids. J. Physiol. 532, 575–579. doi: 10.1111/j.1469-7793.2001.0575f.x
Bolster, D. R., Crozier, S. J., Kimball, S. R., and Jefferson, L. S. (2002). AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J. Biol. Chem. 277, 23977–23980. doi: 10.1074/jbc.c200171200
Brugarolas, J., Lei, K., Hurley, R. L., Manning, B. D., Reiling, J. H., Hafen, E., et al. (2004). Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 18, 2893–2904. doi: 10.1101/gad.1256804
Chan, A. Y., Soltys, C. L., Young, M. E., Proud, C. G., and Dyck, J. R. (2004). Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. J. Biol. Chem. 279, 32771–32779. doi: 10.1074/jbc.m403528200
Cheng, S. W., Fryer, L. G., Carling, D., and Shepherd, P. R. (2004). Thr2446 is a novel mammalian target of rapamycin (mTOR) phosphorylation site regulated by nutrient status. J. Biol. Chem. 279, 15719–15722. doi: 10.1074/jbc.c300534200
Chiang, G. G., and Abraham, R. T. (2005). Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. J. Biol. Chem. 280, 25485–25490. doi: 10.1074/jbc.m501707200
Crosby, J. S., Chefalo, P. J., Yeh, I., Ying, S., London, I. M., Leboulch, P., et al. (2000). Regulation of hemoglobin synthesis and proliferation of differentiating erythroid cells by heme-regulated eIF-2alpha kinase. Blood 96, 3241–3248. doi: 10.1182/blood.v96.9.3241.h8003241_3241_3248
Demetriades, C., Doumpas, N., and Teleman, A. A. (2014). Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell 156, 786–799. doi: 10.1016/j.cell.2014.01.024
Deshmukh, A. S., Treebak, J. T., Long, Y. C., Viollet, B., Wojtaszewski, J. F., and Zierath, J. R. (2008). Role of adenosine 5’-monophosphate-activated protein kinase subunits in skeletal muscle mammalian target of rapamycin signaling. Mol. Endocrinol. 22, 1105–1112. doi: 10.1210/me.2007-0448
Dever, T. E. (2002). Gene-specific regulation by general translation factors. Cell 108, 545–556. doi: 10.1016/s0092-8674(02)00642-6
Du, M., Shen, Q. W., Zhu, M. J., and Ford, S. P. (2007). Leucine stimulates mammalian target of rapamycin signaling in C2C12 myoblasts in part through inhibition of adenosine monophosphate-activated protein kinase. J. Anim. Sci. 85, 919–927. doi: 10.2527/jas.2006-342
Dunlop, E. A., Dodd, K. M., Seymour, L. A., and Tee, A. R. (2009). Mammalian target of rapamycin complex 1-mediated phosphorylation of eukaryotic initiation factor 4E-binding protein 1 requires multiple protein-protein interactions for substrate recognition. Cell Signal. 21, 1073–1084. doi: 10.1016/j.cellsig.2009.02.024
Estrada-Alcalde, I., Tenorio-Guzman, M. R., Tovar, A. R., Salinas-Rubio, D., Torre-Villalvazo, I., Torres, N., et al. (2017). Metabolic fate of branched-chain amino acids during adipogenesis, in adipocytes from obese mice and C2C12 Myotubes. J. Cell Biochem. 118, 808–818. doi: 10.1002/jcb.25755
Foster, K. G., Acosta-Jaquez, H. A., Romeo, Y., Ekim, B., Soliman, G. A., Carriere, A., et al. (2010). Regulation of mTOR complex 1 (mTORC1) by raptor Ser863 and multisite phosphorylation. J. Biol. Chem. 285, 80–94. doi: 10.1074/jbc.m109.029637
Frost, R. A., and Lang, C. H. (2007). Protein Kinase B/Akt: a nexus of growth factor and cytokine signaling in determining muscle mass. J. Appl. Physiol. 103, 378–387. doi: 10.1152/japplphysiol.00089.2007
Gao, X., Zhang, Y., Arrazola, P., Hino, O., Kobayashi, T., Yeung, R. S., et al. (2002). Tsc tumour suppressor proteins antagonize amino-acid-TOR signalling. Nat. Cell Biol. 4, 699–704. doi: 10.1038/ncb847
Glass, D. J. (2010). PI3 kinase regulation of skeletal muscle hypertrophy and atrophy. Curr. Top. Microbiol. Immunol. 346, 267–278. doi: 10.1007/82_2010_78
Goberdhan, D. C., Ogmundsdottir, M. H., Kazi, S., Reynolds, B., Visvalingam, S. M., Wilson, C., et al. (2009). Amino acid sensing and mTOR regulation: inside or out? Biochem. Soc. Trans. 37, 248–252. doi: 10.1042/bst0370248
Gordon, B. S., Liu, C., Steiner, J. L., Nader, G. A., Jefferson, L. S., and Kimball, S. R. (2016). Loss of REDD1 augments the rate of the overload-induced increase in muscle mass. Am. J. Physiol. Regul. Integr. Comp. Physiol. 311, R545–R557.
Gordon, S. E., Lake, J. A., Westerkamp, C. M., and Thomson, D. M. (2008). Does AMP-activated protein kinase negatively mediate aged fast-twitch skeletal muscle mass? Exerc. Sport Sci. Rev. 36, 179–186. doi: 10.1097/jes.0b013e3181877e13
Greiwe, J. S., Kwon, G., McDaniel, M. L., and Semenkovich, C. F. (2001). Leucine and insulin activate p70 S6 kinase through different pathways in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 281, E466–E471.
Gwinn, D. M., Shackelford, D. B., Egan, D. F., Mihaylova, M. M., Mery, A., Vasquez, D. S., et al. (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell. 30, 214–226. doi: 10.1016/j.molcel.2008.03.003
Hara, K., Maruki, Y., Long, X., Yoshino, K., Oshiro, N., Hidayat, S., et al. (2002). Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110, 177–189. doi: 10.1016/s0092-8674(02)00833-4
Hardie, D. G. (2003). Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology 144, 5179–5183. doi: 10.1210/en.2003-0982
Harris, R. A., Popov, K. M., Zhao, Y., and Shimomura, Y. (1994). Regulation of branched-chain amino acid catabolism. J. Nutr. 124, 1499S–1502S.
Harris, R. A., Zhang, B., Goodwin, G. W., Kuntz, M. J., Shimomura, Y., Rougraff, P., et al. (1990). Regulation of the branched-chain alpha-ketoacid dehydrogenase and elucidation of a molecular basis for maple syrup urine disease. Adv. Enzyme Regul. 30, 245–263. doi: 10.1016/0065-2571(90)90021-s
Hatazawa, Y., Tadaishi, M., Nagaike, Y., Morita, A., Ogawa, Y., Ezaki, O., et al. (2014). PGC-1alpha-mediated branched-chain amino acid metabolism in the skeletal muscle. PLoS One 9:e91006. doi: 10.1371/journal.pone.0091006
Hawley, S. A., Davison, M., Woods, A., Davies, S. P., Beri, R. K., Carling, D., et al. (1996). Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J. Biol. Chem. 271, 27879–27887. doi: 10.1074/jbc.271.44.27879
Horman, S., Browne, G., Krause, U., Patel, J., Vertommen, D., Bertrand, L., et al. (2002). Activation of AMP-activated protein kinase leads to the phosphorylation of elongation factor 2 and an inhibition of protein synthesis. Curr. Biol. 12, 1419–1423. doi: 10.1016/s0960-9822(02)01077-1
Hudson, E. R., Pan, D. A., James, J., Lucocq, J. M., Hawley, S. A., Green, K. A., et al. (2003). A novel domain in AMP-activated protein kinase causes glycogen storage bodies similar to those seen in hereditary cardiac arrhythmias. Curr. Biol. 13, 861–866. doi: 10.1016/s0960-9822(03)00249-5
Hutson, S. M. (1989). Regulation of substrate availability for the branched-chain alpha-keto acid dehydrogenase enzyme complex. Ann. N. Y. Acad. Sci. 573, 230–239. doi: 10.1111/j.1749-6632.1989.tb15000.x
Inoki, K., Zhu, T., and Guan, K. L. (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590. doi: 10.1016/s0092-8674(03)00929-2
Ishikawa, T., Kitaura, Y., Kadota, Y., Morishita, Y., Ota, M., Yamanaka, F., et al. (2017). Muscle-specific deletion of BDK amplifies loss of myofibrillar protein during protein undernutrition. Sci. Rep. 7:39825.
Jacinto, E., Loewith, R., Schmidt, A., Lin, S., Ruegg, M. A., Hall, A., et al. (2004). Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 6, 1122–1128. doi: 10.1038/ncb1183
Jefferson, L. S., Fabian, J. R., and Kimball, S. R. (1999). Glycogen synthase kinase-3 is the predominant insulin-regulated eukaryotic initiation factor 2B kinase in skeletal muscle. Int. J. Biochem. Cell Biol. 31, 191–200. doi: 10.1016/s1357-2725(98)00141-1
Joshi, M. A., Jeoung, N. H., Obayashi, M., Hattab, E. M., Brocken, E. G., Liechty, E. A., et al. (2006). Impaired growth and neurological abnormalities in branched-chain alpha-keto acid dehydrogenase kinase-deficient mice. Biochem. J. 400, 153–162. doi: 10.1042/bj20060869
Kazi, A. A., Hong-Brown, L., Lang, S. M., and Lang, C. H. (2011). Deptor knockdown enhances mTOR Activity and protein synthesis in myocytes and ameliorates disuse muscle atrophy. Mol. Med. 17, 925–936. doi: 10.2119/molmed.2011.00070
Kim, D. H., Sarbassov, D. D., Ali, S. M., King, J. E., Latek, R. R., Erdjument-Bromage, H., et al. (2002). mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175. doi: 10.1016/s0092-8674(02)00808-5
Kim, E., Goraksha-Hicks, P., Li, L., Neufeld, T. P., and Guan, K. L. (2008). Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 10, 935–945. doi: 10.1038/ncb1753
Lai, K. M., Gonzalez, M., Poueymirou, W. T., Kline, W. O., Na, E., Zlotchenko, E., et al. (2004). Conditional activation of akt in adult skeletal muscle induces rapid hypertrophy. Mol. Cell. Biol. 24, 9295–9304. doi: 10.1128/mcb.24.21.9295-9304.2004
Lian, K., Du, C., Liu, Y., Zhu, D., Yan, W., Zhang, H., et al. (2015). Impaired adiponectin signaling contributes to disturbed catabolism of branched-chain amino acids in diabetic mice. Diabetes 64, 49–59. doi: 10.2337/db14-0312
Lin, L., Qian, Y., Shi, X., and Chen, Y. (2005). Induction of a cell stress response gene RTP801 by DNA damaging agent methyl methanesulfonate through CCAAT/enhancer binding protein. Biochemistry 44, 3909–3914. doi: 10.1021/bi047574r
Liu, Z., Jahn, L. A., Long, W., Fryburg, D. A., Wei, L., and Barrett, E. J. (2001). Branched chain amino acids activate messenger ribonucleic acid translation regulatory proteins in human skeletal muscle, and glucocorticoids blunt this action. J. Clin. Endocrinol. Metab. 86, 2136–2143. doi: 10.1210/jc.86.5.2136
Liu, Z., Wu, Y., Nicklas, E. W., Jahn, L. A., Price, W. J., and Barrett, E. J. (2004). Unlike insulin, amino acids stimulate p70S6K but not GSK-3 or glycogen synthase in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 286, E523–E528.
Long, X., Lin, Y., Ortiz-Vega, S., Yonezawa, K., and Avruch, J. (2005a). Rheb binds and regulates the mTOR kinase. Curr. Biol. 15, 702–713. doi: 10.1016/j.cub.2005.02.053
Long, X., Ortiz-Vega, S., Lin, Y., and Avruch, J. (2005b). Rheb binding to mammalian target of rapamycin (mTOR) is regulated by amino acid sufficiency. J. Biol. Chem. 280, 23433–23436. doi: 10.1074/jbc.c500169200
Lu, G., Sun, H., She, P., Youn, J. Y., Warburton, S., Ping, P., et al. (2009). Protein phosphatase 2Cm is a critical regulator of branched-chain amino acid catabolism in mice and cultured cells. J. Clin. Invest. 119, 1678–1687. doi: 10.1172/jci38151
Mader, S., Lee, H., Pause, A., and Sonenberg, N. (1995). The translation initiation factor eIF-4E binds to a common motif shared by the translation factor eIF-4 gamma and the translational repressors 4E-binding proteins. Mol. Cell Biol. 15, 4990–4997. doi: 10.1128/mcb.15.9.4990
Marton, M. J., Crouch, D., and Hinnebusch, A. G. (1993). GCN1, a translational activator of GCN4 in Saccharomyces cerevisiae, is required for phosphorylation of eukaryotic translation initiation factor 2 by protein kinase GCN2. Mol. Cell Biol. 13, 3541–3556. doi: 10.1128/mcb.13.6.3541
McGhee, N. K., Jefferson, L. S., and Kimball, S. R. (2009). Elevated corticosterone associated with food deprivation upregulates expression in rat skeletal muscle of the mTORC1 repressor. REDD1. J Nutr. 139, 828–834. doi: 10.3945/jn.108.099846
Menon, S., Dibble, C. C., Talbott, G., Hoxhaj, G., Valvezan, A. J., Takahashi, H., et al. (2014). Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 156, 771–785. doi: 10.1016/j.cell.2013.11.049
Meurs, E., Chong, K., Galabru, J., Thomas, N. S., Kerr, I. M., Williams, B. R., et al. (1990). Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell 62, 379–390. doi: 10.1016/0092-8674(90)90374-n
Moelling, K., Schad, K., Bosse, M., Zimmermann, S., and Schweneker, M. (2002). Regulation of Raf-Akt Cross-talk. J. Biol. Chem. 277, 31099–31106. doi: 10.1074/jbc.m111974200
Moghei, M., Tavajohi-Fini, P., Beatty, B., and Adegoke, O. A. (2016). Ketoisocaproic acid, a metabolite of leucine, suppresses insulin-stimulated glucose transport in skeletal muscle cells in a BCAT2-dependent manner. Am. J. Physiol. Cell Physiol. 311, C518–C527.
Mounier, R., Lantier, L., Leclerc, J., Sotiropoulos, A., Pende, M., Daegelen, D., et al. (2009). Important role for AMPKalpha1 in limiting skeletal muscle cell hypertrophy. FASEB J. 23, 2264–2273. doi: 10.1096/fj.08-119057
Ogasawara, R., Knudsen, J. R., Li, J., Ato, S., and Jensen, T. E. (2020). Rapamycin and mTORC2 inhibition synergistically reduce contraction-stimulated muscle protein synthesis. J Physiol. 598, 5453–5466. doi: 10.1113/jp280528
Panniers, R., and Henshaw, E. C. (1983). A GDP/GTP exchange factor essential for eukaryotic initiation factor 2 cycling in Ehrlich ascites tumor cells and its regulation by eukaryotic initiation factor 2 phosphorylation. J. Biol. Chem. 258, 7928–7934. doi: 10.1016/s0021-9258(20)82007-9
Peterson, T. R., Laplante, M., Thoreen, C. C., Sancak, Y., Kang, S. A., Kuehl, W. M., et al. (2009). DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 137, 873–886. doi: 10.1016/j.cell.2009.03.046
Polekhina, G., Gupta, A., Michell, B. J., van Denderen, B., Murthy, S., Feil, S. C., et al. (2003). AMPK beta subunit targets metabolic stress sensing to glycogen. Curr. Biol. 13, 867–871. doi: 10.1016/s0960-9822(03)00292-6
Ponticos, M., Lu, Q. L., Morgan, J. E., Hardie, D. G., Partridge, T. A., and Carling, D. (1998). Dual regulation of the AMP-activated protein kinase provides a novel mechanism for the control of creatine kinase in skeletal muscle. Embo J. 17, 1688–1699. doi: 10.1093/emboj/17.6.1688
Potier, M., Darcel, N., and Tome, D. (2009). Protein, amino acids and the control of food intake. Curr. Opin. Clin. Nutr. Metab. Care 12, 54–58. doi: 10.1097/mco.0b013e32831b9e01
Price, N., and Proud, C. (1994). The guanine nucleotide-exchange factor, eIF-2B. Biochimie 76, 748–760. doi: 10.1016/0300-9084(94)90079-5
Protiva, P., Hopkins, M. E., Baggett, S., Yang, H., Lipkin, M., Holt, P. R., et al. (2008). Growth inhibition of colon cancer cells by polyisoprenylated benzophenones is associated with induction of the endoplasmic reticulum response. Int. J. Cancer 123, 687–694. doi: 10.1002/ijc.23515
Proud, C. G. (2005). eIF2 and the control of cell physiology. Semin. Cell Dev. Biol. 16, 3–12. doi: 10.1016/j.semcdb.2004.11.004
Proud, C. G. (2007). Signalling to translation: how signal transduction pathways control the protein synthetic machinery. Biochem. J. 403, 217–234. doi: 10.1042/bj20070024
Pruznak, A. M., Kazi, A. A., Frost, R. A., Vary, T. C., and Lang, C. H. (2008). Activation of AMP-activated protein kinase by 5-aminoimidazole-4-carboxamide-1-beta-D-ribonucleoside prevents leucine-stimulated protein synthesis in rat skeletal muscle. J. Nutr. 138, 1887–1894. doi: 10.1093/jn/138.10.1887
Reiling, J. H., and Hafen, E. (2004). The hypoxia-induced paralogs Scylla and Charybdis inhibit growth by down-regulating S6K activity upstream of TSC in Drosophila. Genes Dev. 18, 2879–2892. doi: 10.1101/gad.322704
Reiter, A. K., Bolster, D. R., Crozier, S. J., Kimball, S. R., and Jefferson, L. S. (2005). Repression of protein synthesis and mTOR signaling in rat liver mediated by the AMPK activator aminoimidazole carboxamide ribonucleoside. Am. J. Physiol. Endocrinol. Metab. 288, E980–E988.
Rivera, M. E., Lyon, E. S., Johnson, M. A., Sunderland, K. L., and Vaughan, R. A. (2020). Effect of valine on myotube insulin sensitivity and metabolism with and without insulin resistance. Mol. Cell Biochem. 468, 169–183. doi: 10.1007/s11010-020-03720-y
Roberson, P. A., Shimkus, K. L., Welles, J. E., Xu, D., Whitsell, A. L., Kimball, E. M., et al. (2020). A time course for markers of protein synthesis and degradation with hindlimb unloading and the accompanying anabolic resistance to refeeding. J. Appl. Physiol. 129, 36–46. doi: 10.1152/japplphysiol.00155.2020
Rolfe, D. F., and Brown, G. C. (1997). Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol. Rev. 77, 731–758. doi: 10.1152/physrev.1997.77.3.731
Rommel, C., Bodine, S. C., Clarke, B. A., Rossman, R., Nunez, L., Stitt, T. N., et al. (2001). Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat. Cell Biol. 3, 1009–1013. doi: 10.1038/ncb1101-1009
Rowlands, A. G., Panniers, R., and Henshaw, E. C. (1988). The catalytic mechanism of guanine nucleotide exchange factor action and competitive inhibition by phosphorylated eukaryotic initiation factor 2. J. Biol. Chem. 263, 5526–5533. doi: 10.1016/s0021-9258(18)60596-4
Sancak, Y., Peterson, T. R., Shaul, Y. D., Lindquist, R. A., Thoreen, C. C., Bar-Peled, L., et al. (2008). The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501. doi: 10.1126/science.1157535
Sancak, Y., Thoreen, C. C., Peterson, T. R., Lindquist, R. A., Kang, S. A., Spooner, E., et al. (2007). PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 25, 903–915. doi: 10.1016/j.molcel.2007.03.003
Sarbassov, D. D., Guertin, D. A., Ali, S. M., and Sabatini, D. M. (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101. doi: 10.1126/science.1106148
Schurmann, A., Brauers, A., Massmann, S., Becker, W., and Joost, H. G. (1995). Cloning of a novel family of mammalian GTP-binding proteins (RagA, RagBs, RagB1) with remote similarity to the Ras-related GTPases. J. Biol. Chem. 270, 28982–28988. doi: 10.1074/jbc.270.48.28982
Schwarzer, R., Tondera, D., Arnold, W., Giese, K., Klippel, A., and Kaufmann, J. (2005). REDD1 integrates hypoxia-mediated survival signaling downstream of phosphatidylinositol 3-kinase. Oncogene 24, 1138–1149. doi: 10.1038/sj.onc.1208236
She, P., Van Horn, C., Reid, T., Hutson, S. M., Cooney, R. N., and Lynch, C. J. (2007). Obesity-related elevations in plasma leucine are associated with alterations in enzymes involved in branched-chain amino acid metabolism. Am. J. Physiol. Endocrinol. Metab. 293, E1552–E1563.
Shimkus, K. L., Jefferson, L. S., Gordon, B. S., and Kimball, S. R. (2018). Repressors of mTORC1 act to blunt the anabolic response to feeding in the soleus muscle of a cast-immobilized mouse hindlimb. Physiol. Rep. 6:e13891. doi: 10.14814/phy2.13891
Shoshani, T., Faerman, A., Mett, I., Zelin, E., Tenne, T., Gorodin, S., et al. (2002). Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol. Cell Biol. 22, 2283–2293. doi: 10.1128/mcb.22.7.2283-2293.2002
Smith, E. M., Finn, S. G., Tee, A. R., Browne, G. J., and Proud, C. G. (2005). The tuberous sclerosis protein TSC2 is not required for the regulation of the mammalian target of rapamycin by amino acids and certain cellular stresses. J. Biol. Chem. 280, 18717–18727. doi: 10.1074/jbc.m414499200
Sofer, A., Lei, K., Johannessen, C. M., and Ellisen, L. W. (2005). Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol. Cell Biol. 25, 5834–5845. doi: 10.1128/mcb.25.14.5834-5845.2005
Stein, S. C., Woods, A., Jones, N. A., Davison, M. D., and Carling, D. (2000). The regulation of AMP-activated protein kinase by phosphorylation. Biochem. J. 345(Pt 3), 437–443.
Sun, X. J., Rothenberg, P., Kahn, C. R., Backer, J. M., Araki, E., Wilden, P. A., et al. (1991). Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature 352, 73–77. doi: 10.1038/352073a0
Thomson, D. M., Fick, C. A., and Gordon, S. E. (2008). AMPK activation attenuates S6K1, 4E-BP1, and eEF2 signaling responses to high-frequency electrically stimulated skeletal muscle contractions. J. Appl. Physiol. 104, 625–632. doi: 10.1152/japplphysiol.00915.2007
Tong, J. F., Yan, X., Zhu, M. J., and Du, M. (2009). AMP-activated protein kinase enhances the expression of muscle-specific ubiquitin ligases despite its activation of IGF-1/Akt signaling in C2C12 myotubes. J. Cell Biochem. 108, 458–468. doi: 10.1002/jcb.22272
Vander Haar, E., Lee, S. I., Bandhakavi, S., Griffin, T. J., and Kim, D. H. (2007). Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 9, 316–323. doi: 10.1038/ncb1547
Walejko, J. M., Christopher, B. A., Crown, S. B., Zhang, G. F., Pickar-Oliver, A., Yoneshiro, T., et al. (2021). Branched-chain alpha-ketoacids are preferentially reaminated and activate protein synthesis in the heart. Nat. Commun. 12:1680.
Wang, H., Kubica, N., Ellisen, L. W., Jefferson, L. S., and Kimball, S. R. (2006). Dexamethasone represses signaling through the mammalian target of rapamycin in muscle cells by enhancing expression of REDD1. J. Biol. Chem. 281, 39128–39134. doi: 10.1074/jbc.m610023200
Wang, H., Zhang, Q., Wen, Q., Zheng, Y., Lazarovici, P., Jiang, H., et al. (2012). Proline-rich Akt substrate of 40kDa (PRAS40): a novel downstream target of PI3k/Akt signaling pathway. Cell Signal. 24, 17–24. doi: 10.1016/j.cellsig.2011.08.010
Wang, J., Liu, Y., Lian, K., Shentu, X., Fang, J., Shao, J., et al. (2019). BCAA catabolic defect alters glucose metabolism in lean mice. Front. Physiol. 10:1140. doi: 10.3389/fphys.2019.01140
Wang, L., Harris, T. E., Roth, R. A., and Lawrence, J. C. Jr. (2007). PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J. Biol. Chem. 282, 20036–20044. doi: 10.1074/jbc.m702376200
Wang, Z., Malone, M. H., Thomenius, M. J., Zhong, F., Xu, F., and Distelhorst, C. W. (2003). Dexamethasone-induced gene 2 (dig2) is a novel pro-survival stress gene induced rapidly by diverse apoptotic signals. J. Biol. Chem. 278, 27053–27058. doi: 10.1074/jbc.m303723200
Welsh, G. I., Miller, C. M., Loughlin, A. J., Price, N. T., and Proud, C. G. (1998). Regulation of eukaryotic initiation factor eIF2B: glycogen synthase kinase-3 phosphorylates a conserved serine which undergoes dephosphorylation in response to insulin. FEBS Lett. 421, 125–130. doi: 10.1016/s0014-5793(97)01548-2
White, J. P., Baynes, J. W., Welle, S. L., Kostek, M. C., Matesic, L. E., Sato, S., et al. (2011). The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the Apc(Min/+) mouse. PLoS One 6:e24650. doi: 10.1371/journal.pone.0024650
White, J. P., Gao, S., Puppa, M. J., Sato, S., Welle, S. L., and Carson, J. A. (2012). Testosterone regulation of Akt/mTORC1/FoxO3a signaling in skeletal muscle. Mol. Cell Endocrinol. 365, 174–186. doi: 10.1016/j.mce.2012.10.019
White, P. J., Lapworth, A. L., An, J., Wang, L., McGarrah, R. W., Stevens, R. D., et al. (2016). Branched-chain amino acid restriction in Zucker-fatty rats improves muscle insulin sensitivity by enhancing efficiency of fatty acid oxidation and acyl-glycine export. Mol. Metab. 5, 538–551. doi: 10.1016/j.molmet.2016.04.006
White, P. J., Lapworth, A. L., McGarrah, R. W., Kwee, L. C., Crown, S. B., Ilkayeva, O., et al. (2020). Muscle-liver trafficking of BCAA-derived nitrogen underlies obesity-related glycine depletion. Cell Rep. 33:108375. doi: 10.1016/j.celrep.2020.108375
White, P. J., McGarrah, R. W., Grimsrud, P. A., Tso, S. C., Yang, W. H., Haldeman, J. M., et al. (2018). The BCKDH kinase and phosphatase integrate BCAA and lipid metabolism via regulation of ATP-citrate lyase. Cell Metab. 27, 1281.e7–1293.e7.
White, P. J., and Newgard, C. B. (2019). Branched-chain amino acids in disease. Science 363, 582–583.
Williamson, D. L., Bolster, D. R., Kimball, S. R., and Jefferson, L. S. (2006). Time course changes in signaling pathways and protein synthesis in C2C12 myotubes following AMPK activation by AICAR. Am. J. Physiol. Endocrinol. Metab. 291, E80–E89.
Wojtaszewski, J. F., Jorgensen, S. B., Hellsten, Y., Hardie, D. G., and Richter, E. A. (2002). Glycogen-dependent effects of 5-aminoimidazole-4-carboxamide (AICA)-riboside on AMP-activated protein kinase and glycogen synthase activities in rat skeletal muscle. Diabetes 51, 284–292. doi: 10.2337/diabetes.51.2.284
Wojtaszewski, J. F., MacDonald, C., Nielsen, J. N., Hellsten, Y., Hardie, D. G., Kemp, B. E., et al. (2003). Regulation of 5’AMP-activated protein kinase activity and substrate utilization in exercising human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 284, E813–E822.
Yamauchi, T., Kaburagi, Y., Ueki, K., Tsuji, Y., Stark, G. R., Kerr, I. M., et al. (1998). Growth hormone and prolactin stimulate tyrosine phosphorylation of insulin receptor substrate-1, -2, and -3, their association with p85 phosphatidylinositol 3-kinase (PI3-kinase), and concomitantly PI3-kinase activation via JAK2 kinase. J. Biol. Chem. 273, 15719–15726. doi: 10.1074/jbc.273.25.15719
Keywords: skeletal muscle, branch chain amino acids, protein synthesis, BCKD, branched-chain α-ketoacid dehydrogenase, AMPK (5′-AMP activated kinase), mammalian target of rapamycin
Citation: White JP (2021) Amino Acid Trafficking and Skeletal Muscle Protein Synthesis: A Case of Supply and Demand. Front. Cell Dev. Biol. 9:656604. doi: 10.3389/fcell.2021.656604
Received: 21 January 2021; Accepted: 28 April 2021;
Published: 31 May 2021.
Edited by:
Shuichi Sato, University of Louisiana at Lafayette, United StatesReviewed by:
Olasunkanmi Adegoke, York University, CanadaCopyright © 2021 White. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James P. White, SmFtZXMud2hpdGVAZHVrZS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.