 Nerea Deleyto-Seldas
Nerea Deleyto-Seldas Alejo Efeyan
Alejo Efeyan- Metabolism and Cell Signaling Laboratory, Spanish National Cancer Research Center (CNIO), Madrid, Spain
The mechanistic target of rapamycin (mTOR), master regulator of cellular metabolism, exists in two distinct complexes: mTOR complex 1 and mTOR complex 2 (mTORC1 and 2). MTORC1 is a master switch for most energetically onerous processes in the cell, driving cell growth and building cellular biomass in instances of nutrient sufficiency, and conversely, allowing autophagic recycling of cellular components upon nutrient limitation. The means by which the mTOR kinase blocks autophagy include direct inhibition of the early steps of the process, and the control of the lysosomal degradative capacity of the cell by inhibiting the transactivation of genes encoding structural, regulatory, and catalytic factors. Upon inhibition of mTOR, autophagic recycling of cellular components results in the reactivation of mTORC1; thus, autophagy lies both downstream and upstream of mTOR. The functional relationship between the mTOR pathway and autophagy involves complex regulatory loops that are significantly deciphered at the cellular level, but incompletely understood at the physiological level. Nevertheless, genetic evidence stemming from the use of engineered strains of mice has provided significant insight into the overlapping and complementary metabolic effects that physiological autophagy and the control of mTOR activity exert during fasting and nutrient overload.
The mTOR–Autophagy Axis
mTOR, Master Regulator of Metabolism
The mechanistic target of rapamycin (mTOR; also referred to as mammalian target of rapamycin) is an evolutionarily conserved kinase and the catalytic core of two distinct complexes: mTOR complex 1 and 2 (mTORC1 and mTORC2) defined by the presence of the key accessory proteins Raptor and Rictor, respectively. These two distinct complexes differ in substrate specificity, in their upstream regulatory cues, and in their subcellular localization. As part of mTORC1, mTOR drives most anabolic processes in the cell, including protein, lipid, cholesterol, and nucleotide synthesis, while it simultaneously boosts extracellular nutrient uptake and blocks autophagic catabolism (Buttgereit and Brand, 1995; Saxton and Sabatini, 2017; Valvezan and Manning, 2019). The coordination of such energetically onerous anabolic programs is coupled to (1) the availability of cellular nutrients and (2) the signals from the organismal nutritional state in the form of second messengers such as insulin (Efeyan et al., 2015; Shimobayashi and Hall, 2016).
Nutrients, Growth Factors, and mTORC1
The signal transduction cascade from hormones/growth factors that activates the mTOR kinase starts with the activation of a receptor tyrosine kinase at the plasma membrane that switches on the PI3K–Akt axis, which results in the inhibition of the tuberous sclerosis complex (TSC). TSC integrates inputs from cellular stress, such as hypoxia and limiting ATP levels, and is a GTPase-activating protein for the small GTPase Rheb (Garami et al., 2003; Inoki et al., 2003; Tee et al., 2003; Zhang et al., 2003). When bound to GTP, Rheb induces a conformational change in mTORC1 that results in kinase activation (Anandapadamanaban et al., 2019; Rogala et al., 2019). Importantly, Rheb is anchored at the outer surface of the lysosome, and it can only interact with and activate mTORC1 if mTORC1 is tethered to the outer lysosomal surface, a re-localization process that occurs in a cellular nutrient-dependent manner (Sancak et al., 2008; Figure 1).
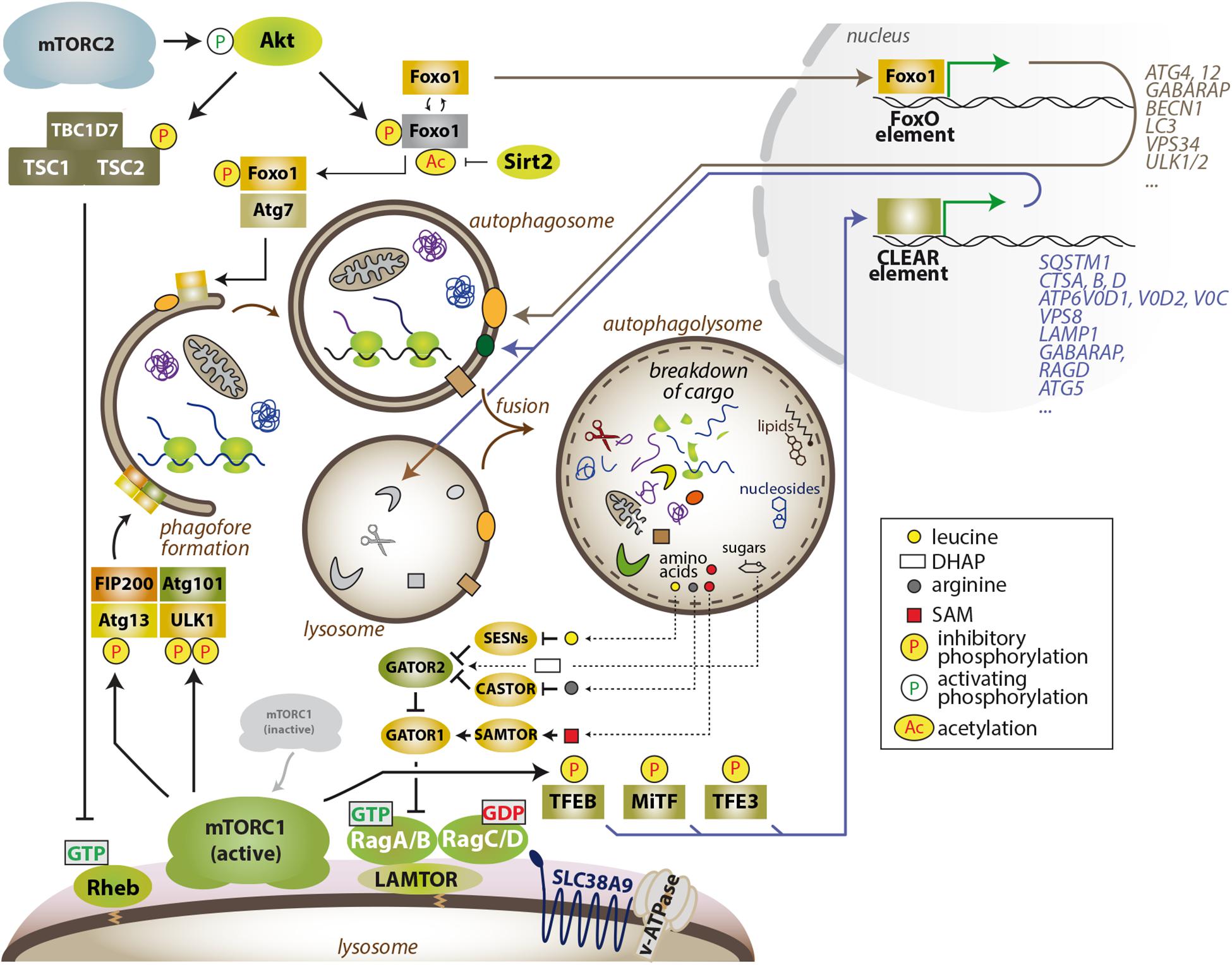
Figure 1. The mTOR–autophagy axis. MTORC1 blocks the early steps in autophagy by phosphorylation-dependent inhibition of Atg13 and ULK1 and also restrains the degradative capacity of the cell by inhibiting the activity of TFEB family members. Several autophagy-related proteins and other lysosomal factors are encoded by genes that harbor a CLEAR sequence in their promoter region, and are bound and transactivated by active TFEB family members. Thus, inhibition of mTORC1 enables autophagic degradation and degradative capacity of the cell, while also suppressing anabolism and, thus, lowering the demand for energy and nutrients. In turn, lysosome-derived amino acids (leucine and arginine) and also other nutrients (glucose-DHAP, cholesterol, SAM) can partially reactivate Rag GTPase – nutrient signaling upstream of mTORC1. MTORC2 and the AGC kinases Akt and SGK1 also limit the autophagic flux and lysosomal capacity via transcription-dependent and -independent processes. The cytoplasmic sequestration of the FoxO transcription factors indirectly limits autophagic degradation, as several FoxO targets encode proteins that directly participate in different steps of autophagy and lysosomal catabolism.
The levels of cellular nutrients (amino acids, glucose, certain lipids, and likely other metabolites) are sensed and signaled by an expanding number of proteins (SESNs, CASTOR, KICSTOR, SAMTOR, GATOR1 and 2) (Saxton and Sabatini, 2017) that culminate in the control of the guanosine phosphate state of the members of the Rag family of GTPases, which bind mTORC1 in a nutrient-sensitive manner (Figure 1). In addition, Rag GTPase-independent amino acid control of mTORC1 exists (Efeyan et al., 2014; Jewell et al., 2015). The Rag GTPases operate as obligate heterodimeric partners composed of RagA or RagB plus RagC or RagD (Schurmann et al., 1995). In the presence of cellular nutrients, RagA or B are loaded with GTP, while RagC or D are loaded with GDP (Kim et al., 2008; Sancak et al., 2008). This specific nucleotide configuration allows the interaction with mTORC1 and its recruitment to the outer lysosomal surface, where the Rheb–mTORC1 interaction, and mTORC1 activation, occurs. Hence, the current paradigm of the regulation of mTORC1 states that maximal mTORC1 activity takes place when both signals are present: (1) cellular nutrients to recruit mTORC1 and (2) hormones/growth factors for kinase activation (Lawrence and Zoncu, 2019; Valvezan and Manning, 2019). This elegant mechanism of coincidence detection of inputs has been well illustrated by biochemical, cell biological, and structural approaches, but we still ignore how the convergent controls of mTORC1 activity result in the execution of a coherent, multifaceted metabolic program in real organs with physiological and pathological fluctuations in nutrients and hormones.
mTORC1, Catabolism, and Biosynthetic Capacity
At the cellular level, intense research in the last 15 years has shown that the anabolic program executed by mTORC1 is coupled to a block in catabolic processes. Otherwise, a disconnection would result in futile, cyclic synthesis and degradation of cellular components and biomass. Nonetheless, the increased demand for energy and building blocks for macromolecule synthesis upon mTORC1 activation, together with the block in autophagy, results in a biosynthetic burden that the cell alleviates with the execution of parallel programs that reinforce nutrient uptake (Park et al., 2017; Torrence et al., 2020) and synthesis (Robitaille et al., 2013; Ben-Sahra et al., 2016; Valvezan et al., 2017), and also enables the catabolic capacity of the cells through the proteasomal degradation of proteins (Zhang et al., 2014) to remove unwanted cellular material and to boost the pool of free amino acids available for protein synthesis. Reciprocally, an increased autophagic flux and the recycling of cellular macromolecules upon extended inhibition of mTORC1 is sufficient to partially replenish intracellular or intra-lysosomal amino acid pools and to partially reactivate mTORC1 (Yu et al., 2010; Figure 1).
mTORC1 Governs Autophagy at the Lysosomal Surface
It is not coincidental that the subcellular location where mTORC1 activation occurs is the cytoplasmic side of the organelle responsible for internal recycling of macromolecules: the lysosome. Indeed, such spatial association has remained fixed throughout eukaryotic and metazoan evolution. The cytoplasmic side of the lysosomal membrane provides a scaffold surface for controlling mTORC1 and also enables immediate control of specific mTORC1 targets associated to the lysosome (Rabanal-Ruiz and Korolchuk, 2018). Moreover, both abundance and positioning of lysosomes are important modulators of mTORC1 and mTORC2 (Korolchuk et al., 2011; Jia and Bonifacino, 2019; Mutvei et al., 2020). Targets of mTORC1 that are transiently or permanently associated to the lysosome include ULK1, ATG13, and TFEB and its family members, but exactly how and where mTORC1 reaches and phosphorylates its targets is not clear. The coordinated control of these targets results in complementary functions that, together, tightly restrict autophagy when mTORC1 is active. On one side, the inhibitory phosphorylation of the ULK1/ATG13/FIP200 complex by mTORC1 immediately blocks the initiation of the formation of the autophagosome (Ganley et al., 2009; Hosokawa et al., 2009; Kim et al., 2011). This early block is reinforced by the inhibitory phosphorylation of TFEB and MiTF-TFE family members (Settembre et al., 2011; Roczniak-Ferguson et al., 2012; Martina and Puertollano, 2013). TFEB and family members are themselves a signaling hub that computes information from nutrient sufficiency, calcium signaling (Medina et al., 2015), and overall lysosomal stress and health (Ballabio and Bonifacino, 2020), and ultimately execute a transcriptional program that includes the synthesis of proteins that boost the degradative capacity of the cell (Napolitano and Ballabio, 2016; Ballabio and Bonifacino, 2020). The lysosomal biogenesis program encompasses lysosomal membrane proteins, lysosomal lumen enzymes that execute the catalytic degradation of cargo, and factors directly involved in the execution of autophagy (Sardiello et al., 2009; Settembre et al., 2011; Figure 1). Thus, the coordinated actions of mTORC1 ensure a rapid block in autophagy and a slower inhibition of this transcriptional program that ultimately restricts lysosomal biogenesis and lysosomal function (Figure 1).
As mentioned, phosphorylation of mTORC1 targets results in induction and in inhibition of anabolic and catabolic functions, respectively. A consensus sequence that facilitates recruitment to, and phosphorylation by mTORC1 has been inceasingly refined (Hsu et al., 2011; Yu et al., 2011; Robitaille et al., 2013). While for years the paradigm has stated that all mTORC1 targets interact with mTORC1 by means of the TOR signaling (TOS) motif (Schalm and Blenis, 2002), recent compelling work has demonstrated that this is not universal. In particular, TFEB family members are orphan of a TOS motif and are instead recruited to mTORC1 through a direct interaction with the RagC GTPase (Napolitano et al., 2020). Importantly, inhibition of TFEB by mTORC1 is facilitated by RagC exclusively under cellular nutrient sufficiency, thus defining an input-dependent asymmetry in the activation of different targets downstream of mTORC1 (Napolitano et al., 2020). While targets with a TOS motif result phosphorylated only if the TSC-Rheb and the Rag GTPase arms are ON, TFEB is sensitive to any perturbation that would result in a change in the nucleotide to which RagC is bound, such as nutrient withdrawal/replenishment, but would be less affected by the inhibition and activation of the growth factor signaling cascade.
The physiological implications of this asymmetric control of mTORC1 targets are enormous and have been already underlined in the context of brown adipose tissue function (Wada et al., 2016), and of the Birt–Hogg–Dubé syndrome (Napolitano et al., 2020), and may underlie the occurrence of activating mutations in RagC (but not in RagA) in B-cell lymphomas (Ortega-Molina et al., 2019).
The Regulation of Autophagy by mTORC2
In addition to the suppression of autophagy when part of mTORC1, the mTOR kinase restrains autophagy when part of mTORC2. Through mechanisms that are not entirely defined, growth factor signaling activates mTORC2, and upon its activation, mTORC2 phosphorylates and activates Akt and other members of the AGC kinase family (Hoxhaj and Manning, 2020). The FoxO transcription factors are phosphorylation targets of Akt (Brunet et al., 1999), and Akt-dependent phosphorylation of FoxO1/3a results innuclear exclusion, thus impairing the transactivation of autophagy-related factors and lysosome-tethered proteins (Mammucari et al., 2007; Zhao et al., 2007). In an interesting crosstalk between Akt and the NAD-dependent deacetylase family of Sirtuins, Sirt1 also modulates the transcriptional activity of FoxO1 (Frescas et al., 2005). Moreover, in an apparently counterintuitive manner, cytoplasmic FoxO1 can also induce the autophagic flux by its interaction with Atg7 (Zhao et al., 2010), and this cytoplasmic, transcription-independent function of FoxO1 requires its deacetylation by Sirt2, and is facilitated by nuclear exclusion of FoxO1 by Akt-dependent phosphorylation (Zhou et al., 2012). While this nucleo-cytoplasmic tug of war of FoxO1 shuttle in the control of autophagy deserves further investigation, the acetylation of FoxO1 in the cytoplasm ensures the rapid execution of autophagy even in conditions of high growth factor signaling (Figure 1). In addition, not all reported connections between mTORC2 and autophagy involve cytoplasmic and nuclear functions of FoxO, as Akt directly phosphorylates and inhibits ULK1 (Bach et al., 2011). Finally, transcriptional activity by FoxO includes the upregulation of Sestrins, negative regulators of the Rag-mTORC1 axis, in an additional loop of cross-regulation (Chen et al., 2010).
In addition to the functions of Akt, inhibition of another member of the AGC kinase family, SGK1, by either genetic and pharmacological means, induces autophagy in vitro and in vivo (Conza et al., 2017; Liu et al., 2018; Zuleger et al., 2018), and these effects may also be mediated by the phosphorylation of FoxO3a. In worms, suppression of the mTORC2–SGK1 axis induces autophagy in a DAF-16/FOXO-independent manner (Aspernig et al., 2019) downstream of altered mitochondrial permeability (Zhou et al., 2019), strongly suggesting that mTORC2 lies upstream of the control of autophagy by SGK1. Such control is prominent in mammalian skeletal muscle (Andres-Mateos et al., 2013; Zuleger et al., 2018). However, the fact that SGK1 knock-out mice (Wulff et al., 2002; Fejes-Tóth et al., 2008) show minimal phenotypic alterations, unlike autophagy-deficient mice (Kuma et al., 2004), may reflect the importance of SGK1 under specific stress conditions.
The enumerated multilayered, convergent, and complementary effects of mTORC1 and mTORC2 in the repression of autophagy deciphered at the molecular level point to (1) a direct, acute blockade of the phagophore–autophagosome formation, together with (2) a long-term, transcription-based limitation of the degradative machinery, but how is this complexity integrated to sustain metabolic homeostasis and how may its deregulation contribute to metabolic disease are far less understood. We next review the lessons that mouse genetic approaches have taught in the understanding of the physiology of the mTOR–autophagy axis.
The mTORC1–Autophagy Axis in Mammalian Metabolism
mTOR, Autophagy and Mammalian Adaptation to Fasting
Suppression of mTORC1 activity has proven essential to endure conditions of nutrient deprivation in mammals. Immediately after birth, glucose and amino acid levels in circulation drop dramatically due to the interruption of the trans-placental supply, and mTORC1 in mouse tissues is readily inhibited within 1 h after birth. This rapid and strong inhibition is critical to unleash autophagy, which in turn boosts free amino acid levels that feed gluconeogenesis to sustain glycaemia (Efeyan et al., 2013), and is key to endure the first hours of life, before significant maternal milk supply occurs. Neonatal mice carrying a point mutation in RagA (RagAQ66L, referred to as RagAGTP) that renders mTORC1 constitutively active regardless of nutrient levels are unable to trigger autophagy after birth, suffer a profound hypoglycemic state they do not recover from, and finally succumb within 1 day after birth (Efeyan et al., 2013). A strikingly similar phenomenon was observed in mice lacking Sestrin 1, 2 and 3, negative regulators of the Rag GTPases (Peng et al., 2014), and in Atg5–/– (Kuma et al., 2004) and Atg7–/– mice (Komatsu et al., 2005). The phenotypic resemblance of mice with constitutive nutrient signaling and autophagy-deficient mice provides strong genetic and physiological support for the importance of the regulation of autophagy by the nutrient–mTORC1 pathway in the maintenance of metabolic homeostasis (see Table 1).
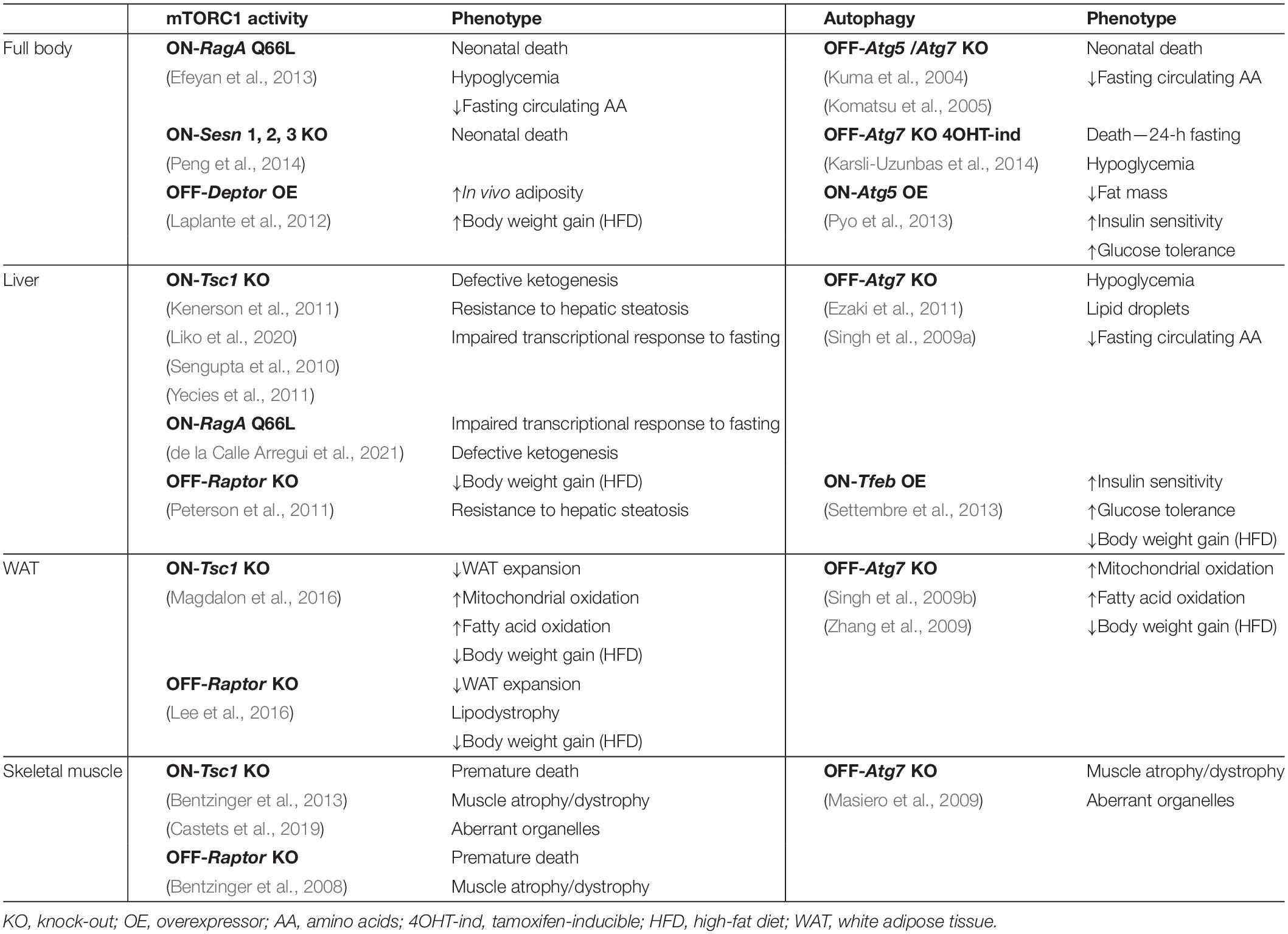
Table 1. Metabolic phenotypes of genetically-engineered mice with increased and decreased mTORC1 activity and enhanced or suppressed autophagy.
The sequential steps in the transition from postprandial to early and long-term fasting metabolism in adult individuals encompass a shift from glucose to lipid and ketone body metabolism, through consistent and complementary responses in the liver, skeletal muscle, and white adipose tissue (WAT), coordinated systemically by hormonal signaling. An early fasting switch involves the release of liver glycogen stores as soon as insulin levels drop. When glycogen has been entirely mobilized, gluconeogenesis occurs together with increased lipolysis from the WAT to yield circulating glycerol, a substrate for gluconeogenesis, and free fatty acids (FFA). The ATP produced by β-oxidation of FFA in the liver sustains gluconeogenesis, while acetyl-CoA is shunted to the production of ketone bodies, an essential source of energy during fasting, and particularly important for the brain.
In addition to its essentiality in neonatal mice, autophagy is required by adult mice to endure periods of fasting. Acute, full-body ablation of autophagy by tamoxifen-mediated deletion of Atg7 impairs the survival of the mice following a 24-h fast due to severe hypoglycemia. Interestingly, amino acid levels in circulation are sustained by accelerated muscle wasting, but this increase fails to support gluconeogenesis, and decreased WAT and rapidly depleted hepatic glycogen stores compromise the endurance to long-term fasting (Karsli-Uzunbas et al., 2014).
As soon as insulin drops, autophagy is induced in the liver, and adult mice unable to trigger hepatic autophagy fail to release free amino acids and suffer from reduced blood glucose levels during fasting, an effect that can be rescued experimentally by exogenous administration of the gluconeogenic amino acid serine (Ezaki et al., 2011). It is noteworthy that in these mice, glycogen mobilization during fasting is not impaired, and neither is it in RagAGTP neonates (Efeyan et al., 2013) nor in adult Atg7-KO mice (Karsli-Uzunbas et al., 2014), in spite of the relevance of autophagic degradation of glycogen (glycophagy) during fasting (Schiaffino and Hanzlíková, 1972; Jiang et al., 2010). Surprisingly, mice with constitutive hepatic mTORC1 activity by means of genetic deletion of Tsc1 (Sengupta et al., 2010; Yecies et al., 2011; Liko et al., 2020), or liver-specific activation of RagA (de la Calle Arregui et al., 2021), do not phenocopy a deficiency in autophagy and instead have an impaired transcriptional adaptation to fasting that limits ketogenesis. Thus, in adult mice undergoing fasting, mTORC1 and autophagy control complementary adaptations to nutrient limitations (see Table 1).
mTOR, Autophagy, and Lipid Homeostasis
In addition to supporting fasting glucose homeostasis, autophagy contributes to the hydrolysis of lipid droplets upon nutrient deprivation by the delivery of lipid droplets to lysosomes, and pharmacologic and genetic inhibition of autophagy in the liver leads to the accumulation of lipid droplets and to lower rates of β-oxidation both in vitro and in vivo (Singh et al., 2009a). In vivo deletion of Atg7 specifically in WAT leads to a lean phenotype accompanied by morphological and functional changes of the WAT. Adipocytes from these animals are multilocular, with enriched content in mitochondria, and display increased rates of β-oxidation, and these mice also exhibit improved insulin sensitivity and resistance to high-fat-diet-induced obesity (Singh et al., 2009b; Zhang et al., 2009).
The phenotypes of mice with a hyper-active mTORC1 in the WAT have some features in common with those of defective autophagy. Deletion of Tsc1 in adipocytes compromises WAT expansion, results in a higher mitochondrial oxidative activity and fatty acid oxidation and to a leaner phenotype when mice were fed a high-fat diet (Magdalon et al., 2016). While deletion of Raptor in WAT also results in defective WAT expansion, lipodystrophy, and resistance to diet-induced obesity (Lee et al., 2016), the overexpression of Deptor, a negative regulator of mTOR, promotes adipogenesis and mice gain more weight upon high-fat feeding (Laplante et al., 2012). Deptor overexpression decreases the negative feedback loop to Akt, while not significantly affecting other mTORC1 targets, supporting the notion that both mTORC1 and Akt activities are required for adipogenesis.
The mTORC1 target TFEB controls lipid catabolism at the transcriptional level by promoting PGC1-α-dependent PPARα activation in response to fasting (Settembre et al., 2013). An analogous process occurs in C. elegans, where HLH-30 (ortholog of TFEB) regulates the expression of lysosomal triglyceride lipases (O’Rourke and Ruvkun, 2013). Overexpression of Tfeb in mice (TFEB-Tg mice) leads to an improvement of glucose tolerance, insulin sensitivity, and weight gain in mice fed with high-fat diet and, importantly, to a lean phenotype that is abolished in an Atg7-deficient background, genetically demonstrating the epistasis of TFEB and autophagy (Settembre et al., 2013). This work is consistent with a report showing that autophagy is impaired in obese (ob/ob) mice, and reconstitution of autophagy in the liver by expressing Atg7 using an adenoviral system improves their glucose tolerance and insulin sensitivity (Yang et al., 2010). Consistently, ubiquitous and moderate overexpression of Atg5 in mice, resulting in increased autophagy, leads to a leaner phenotype, an increased glucose tolerance and insulin sensitivity, and resistance to age-induced obesity (Pyo et al., 2013).
In line with the phenotypes observed in TFEB-Tg mice and in Atg5-Tg mice, liver-specific Raptor KO mice are resistant to diet-induced obesity and to hepatic steatosis, and these effects are, at least in part, mediated by the mTORC1 target Lipin1 (Peterson et al., 2011). In an apparent contradiction, mice with hyperactive mTORC1 signaling in the liver (liver-specific Tsc1 KO) are also resistant to age- and diet-induced hepatic steatosis (Sengupta et al., 2010), but this apparently counterintuitive finding can be explained by an indirect attenuation of Akt-SREBP1 signaling (rather by suppression of autophagy) through the induction of a negative feedback loop to the insulin receptor substrate (IRS) (Kenerson et al., 2011; Yecies et al., 2011).
Mouse genetics for the study of WAT can produce phenotypic findings of difficult interpretation. This difficulty is probably related to the strength of perturbation caused by genetic deletion, in contrast to more physiological activation or inhibition of a signaling pathway, and to the expression patterns and leakiness of Cre recombinases. Nonetheless, the in vitro and in vivo lessons together have taught us that an appropriate, dynamic, and physiological regulation of both mTORC1 activity and autophagy is necessary for adipogenesis and for maintaining the correct accumulation and mobilization of lipids and the physiological functioning of WAT (Clemente-Postigo et al., 2020) (Table 1).
mTOR, Autophagy, and Skeletal Muscle Homeostasis
Another tissue that exhibits a remarkable sensitivity to changes in both autophagy and mTOR activity is skeletal muscle. The muscle has the intrinsic ability to hypertrophy and atrophy by a tight balance between protein synthesis and degradation (Fry and Rasmussen, 2011). Intense research has demonstrated that, although acute activation of the Akt–mTORC1 and the mTORC2–Akt axes drive hypertrophy, both chronically aberrant increase and decrease in mTOR activity yield muscle atrophy because dynamic synthesis–degradation oscillations are essential to maintain fiber homeostasis.
Muscle degradation occurs both in an autophagy-independent manner, by FoxO-dependent transactivation of the ubiquitin ligases Atrogin-1 and MuRF1 (Sandri et al., 2004; Stitt et al., 2004), and by autophagic degradation by FoxO-dependent transcription of many autophagy-related genes (Mammucari et al., 2007; Zhao et al., 2007). Thus, the Akt–FoxO signaling pathway coordinates the two main degradative pathways involved in atrophy: the ubiquitin–proteasome and the autophagy–lysosome.
In addition to a key role of the mTORC2–Akt–FoxO pathway in the control of muscle homeostasis, deletion of the mTORC1 component Raptor in the skeletal muscle results in profound atrophic and dystrophic muscles, with metabolic and structural changes incompatible with mouse survival (Bentzinger et al., 2008). While short-term genetic hyperactivation of mTORC1 in skeletal muscle by deletion of Tsc1 results in hypertrophy, sustained mTORC1 hyperactivation eventually precipitates atrophy in most muscles and premature death (Bentzinger et al., 2013; Castets et al., 2013). The features of this myopathy are very similar to the ones developed by mice with a specific deletion of Atg7 in the skeletal muscle, where aberrant membranous structures accumulate in muscle fibers (Masiero et al., 2009). The strong suppression of Akt through the mTORC1-mediated negative feedback loop allows the expression of Atrogin-1 and MuRF1 in young, but not in old, muscle-specific Tsc1-KO (sm-Tsc1 KO) mice. Instead, the late-onset myopathy in skeletal muscle of old sm-Tsc1 KO mice is caused by the inability to induce autophagy. Pharmacological inhibition of mTORC1 with rapamycin restores the autophagic flux and rescues the defects, pointing to pivotal and complex roles of both mTORC1 and TORC2 in the control of autophagy in the skeletal muscle from young and old individuals (Castets et al., 2013). Recent work has also shown that both the mTORC1–autophagy axis and mTORC1–Akt axis are critical for the coordination of recovery after denervation (Castets et al., 2019) (Table 1).
Concluding Remarks
The multilayered functional interactions of the mTOR signaling pathways and the process of autophagy are deciphered to a great extent at the cellular level, but we still have an incomplete understanding of their integration at the physiopathological level. To what extent does a blockade in autophagy explain the consequences of deregulated mTOR activity? Would the sole inhibition of mTOR unleash physiological autophagy and its benefits? Mouse genetics has contributed substantial snapshots of phenotypic similarities, epistasis, and complementary metabolic functions of mTOR inhibition and autophagy in different organs and metabolic states. A challenge for the future is to translate this body of knowledge into significant support for pharmacological manipulation of mTOR and autophagy to improve metabolism and control the pathologies associated with nutrient overload.
Author Contributions
ND-S and AE conceived, wrote, and edited the text and figures. Both authors contributed to the article and approved the submitted version.
Funding
This work was funded by RETOS projects Programme of Spanish Ministry of Science, Innovation and Universities, Spanish State Research Agency (AEI/10.13039/501100011033), co-funded by the European Regional Development Fund (Grant SAF2015-67538-R and PID2019-104012RB-I00), EU-H2020 Programme (ERC-2014-STG-638891), Excellence Network Grant from MICIU/AEI (SAF2016-81975-REDT), a Ramon y Cajal Award from MICIU/AEI (RYC-2013-13546), a Lab Grant from the Spanish Association Against Cancer Research Scientific Foundation (AECC), Beca de Investigación en Oncología Olivia Roddom, and FERO Grant for Research in Oncology (to AE). AE is an EMBO Young Investigator. ND-S is a recipient of Ayuda de contratos predoctorales para la formación de doctores from MICIU/AEI (BES-2016-077410).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
AE dedicates this work to the memory of Diego Armando Maradona.
References
Anandapadamanaban, M., Masson, G. R., Perisic, O., Berndt, A., Kaufman, J., Johnson, C. M., et al. (2019). Architecture of human Rag GTPase heterodimers and their complex with mTORC1. Science 366, 203–210. doi: 10.1126/science.aax3939
Andres-Mateos, E., Brinkmeier, H., Burks, T. N., Mejias, R., Files, D. C., Steinberger, M., et al. (2013). Activation of serum/glucocorticoid-induced kinase 1 (SGK1) is important to maintain skeletal muscle homeostasis and prevent atrophy. EMBO Mol. Med. 5, 80–91. doi: 10.1002/emmm.201201443
Aspernig, H., Heimbucher, T., Qi, W., Gangurde, D., Curic, S., Yan, Y., et al. (2019). Mitochondrial Perturbations Couple mTORC2 to Autophagy in C. elegans. Cell Rep. 29, 1399–1409.e5. doi: 10.1016/j.celrep.2019.09.072
Bach, M., Larance, M., James, D. E., and Ramm, G. (2011). The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem. J. 440, 283–291. doi: 10.1042/BJ20101894
Ballabio, A., and Bonifacino, J. S. (2020). Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 21, 101–118. doi: 10.1038/s41580-019-0185-4
Ben-Sahra, I., Hoxhaj, G., Ricoult, S. J. H., Asara, J. M., and Manning, B. D. (2016). mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 351, 728–733. doi: 10.1126/science.aad0489
Bentzinger, C. F., Lin, S., Romanino, K., Castets, P., Guridi, M., Summermatter, S., et al. (2013). Differential response of skeletal muscles to mTORC1 signaling during atrophy and hypertrophy. Skelet. Muscle 3:6. doi: 10.1186/2044-5040-3-6
Bentzinger, C. F., Romanino, K., Cloëtta, D., Lin, S., Mascarenhas, J. B., Oliveri, F., et al. (2008). Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab. 8, 411–424. doi: 10.1016/j.cmet.2008.10.002
Brunet, A., Bonni, A., Zigmond, M. J., Lin, M. Z., Juo, P., Hu, L. S., et al. (1999). Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell 96, 857–868. doi: 10.1016/S0092-8674(00)80595-4
Buttgereit, F., and Brand, M. D. (1995). A hierarchy of ATP-consuming processes in mammalian cells. Biochem. J. 312(Pt 1)(Pt 1), 163–167. doi: 10.1042/bj3120163
Castets, P., Lin, S., Rion, N., Di Fulvio, S., Romanino, K., Guridi, M., et al. (2013). Sustained activation of mTORC1 in skeletal muscle inhibits constitutive and starvation-induced autophagy and causes a severe, late-onset myopathy. Cell Metab. 17, 731–744. doi: 10.1016/j.cmet.2013.03.015
Castets, P., Rion, N., Théodore, M., Falcetta, D., Lin, S., Reischl, M., et al. (2019). mTORC1 and PKB/Akt control the muscle response to denervation by regulating autophagy and HDAC4. Nat. Commun. 10:3187. doi: 10.1038/s41467-019-11227-4
Chen, C. C., Jeon, S. M., Bhaskar, P. T., Nogueira, V., Sundararajan, D., Tonic, I., et al. (2010). FoxOs inhibit mTORC1 and activate Akt by inducing the expression of Sestrin3 and Rictor. Dev. Cell 18, 592–604. doi: 10.1016/j.devcel.2010.03.008
Clemente-Postigo, M., Tinahones, A., El Bekay, R., Malagón, M. M., and Tinahones, F. J. (2020). The role of autophagy in white adipose tissue function: implications for metabolic health. Metabolites 10:179. doi: 10.3390/metabo10050179
Conza, D., Mirra, P., Calì, G., Tortora, T., Insabato, L., Fiory, F., et al. (2017). The SGK1 inhibitor SI113 induces autophagy, apoptosis, and endoplasmic reticulum stress in endometrial cancer cells. J. Cell. Physiol. 232, 3735–3743. doi: 10.1002/jcp.25850
de la Calle Arregui, C., Plata-Gomez, A. B., Deleyto-Seldas, N., Garcia, F., Ortega-Molina, A., Abril-Garrido, J., et al. (2021). Limited survival and impaired hepatic fasting metabolism in mice with constitutive Rag GTPase signaling. Nat. Commun. doi: 10.1038/s41467-021-23857-8
Efeyan, A., Comb, W. C., and Sabatini, D. M. (2015). Nutrient-sensing mechanisms and pathways. Nature 517, 302–310. doi: 10.1038/nature14190
Efeyan, A., Schweitzer, L. D., Bilate, A. M., Chang, S., Kirak, O., Lamming, D. W., et al. (2014). RagA, but not RagB, is essential for embryonic development and adult mice. Dev. Cell 29, 321–329. doi: 10.1016/j.devcel.2014.03.017
Efeyan, A., Zoncu, R., Chang, S., Gumper, I., Snitkin, H., Wolfson, R. L., et al. (2013). Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature 493, 679–683. doi: 10.1038/nature11745
Ezaki, J., Matsumoto, N., Takeda-Ezaki, M., Komatsu, M., Takahashi, K., Hiraoka, Y., et al. (2011). Liver autophagy contributes to the maintenance of blood glucose and amino acid levels. Autophagy 7, 727–736. doi: 10.4161/auto.7.7.15371
Fejes-Tóth, G., Frindt, G., Náray-Fejes-Tóth, A., and Palmer, L. G. (2008). Epithelial Na+ channel activation and processing in mice lacking SGK1. Am. J. Physiol. Ren. Physiol. 294, F1298–F1305. doi: 10.1152/ajprenal.00579.2007
Frescas, D., Valenti, L., and Accili, D. (2005). Nuclear trapping of the forkhead transcription factor FoxO1 via sirt-dependent deacetylation promotes expression of glucogenetic genes. J. Biol. Chem. 280, 20589–20595. doi: 10.1074/jbc.M412357200
Fry, C. S., and Rasmussen, B. B. (2011). Skeletal muscle protein balance and metabolism in the elderly. Curr. Aging Sci. 4, 260–268. doi: 10.2174/1874609811104030260
Ganley, I. G., Lam, D. H., Wang, J., Ding, X., Chen, S., and Jiang, X. (2009). ULK1⋅ATG13⋅FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 284, 12297–12305. doi: 10.1074/jbc.M900573200
Garami, A., Zwartkruis, F. J., Nobukuni, T., Joaquin, M., Roccio, M., Stocker, H., et al. (2003). Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 11, 1457–1466. doi: 10.1016/s1097-2765(03)00220-x
Hosokawa, N., Hara, T., Kaizuka, T., Kishi, C., Takamura, A., Miura, Y., et al. (2009). Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 20, 1981–1991. doi: 10.1091/mbc.E08-12-1248
Hoxhaj, G., and Manning, B. D. (2020). The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 20, 74–88. doi: 10.1038/s41568-019-0216-7
Hsu, P. P., Kang, S. A., Rameseder, J., Zhang, Y., Ottina, K. A., Lim, D., et al. (2011). The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 332, 1317–1322. doi: 10.1126/science.1199498
Inoki, K., Li, Y., Xu, T., and Guan, K. L. (2003). Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 17, 1829–1834. doi: 10.1101/gad.1110003
Jewell, J. L., Kim, Y. C., Russell, R. C., Yu, F. X., Park, H. W., Plouffe, S. W., et al. (2015). Differential regulation of mTORC1 by leucine and glutamine. Science 347, 194–198. doi: 10.1126/science.1259472
Jia, R., and Bonifacino, J. S. (2019). Lysosome positioning influences mTORC2 and AKT signaling. Mol. Cell 75, 26–38.e3. doi: 10.1016/j.molcel.2019.05.009
Jiang, S., Heller, B., Tagliabracci, V. S., Zhai, L., Irimia, J. M., DePaoli-Roach, A. A., et al. (2010). Starch binding domain-containing protein 1/genethonin 1 is a novel participant in glycogen metabolism. J. Biol. Chem. 285, 34960–34971. doi: 10.1074/jbc.M110.150839
Karsli-Uzunbas, G., Guo, J. Y., Price, S., Teng, X., Laddha, S. V., Khor, S., et al. (2014). Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 4, 914–927. doi: 10.1158/2159-8290.CD-14-0363
Kenerson, H. L., Yeh, M. M., and Yeung, R. S. (2011). Tuberous sclerosis complex-1 deficiency attenuates diet-induced hepatic lipid accumulation. PLoS One 6:e18075. doi: 10.1371/journal.pone.0018075
Kim, E., Goraksha-Hicks, P., Li, L., Neufeld, T. P., and Guan, K. L. (2008). Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 10, 935–945. doi: 10.1038/ncb1753
Kim, J., Kundu, M., Viollet, B., and Guan, K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. doi: 10.1038/ncb2152
Komatsu, M., Waguri, S., Ueno, T., Iwata, J., Murata, S., Tanida, I., et al. (2005). Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 169, 425–434. doi: 10.1083/jcb.200412022
Korolchuk, V. I., Saiki, S., Lichtenberg, M., Siddiqi, F. H., Roberts, E. A., Imarisio, S., et al. (2011). Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 13, 453–460. doi: 10.1038/ncb2204
Kuma, A., Hatano, M., Matsui, M., Yamamoto, A., Nakaya, H., Yoshimori, T., et al. (2004). The role of autophagy during the early neonatal starvation period. Nature 432, 1032–1036. doi: 10.1038/nature03029
Laplante, M., Horvat, S., Festuccia, W. T., Birsoy, K., Prevorsek, Z., Efeyan, A., et al. (2012). DEPTOR Cell-autonomously promotes adipogenesis, and its expression is associated with obesity. Cell Metab. 16, 202–212. doi: 10.1016/j.cmet.2012.07.008
Lawrence, R. E., and Zoncu, R. (2019). The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 21, 133–142. doi: 10.1038/s41556-018-0244-7
Lee, P. L., Tang, Y., Li, H., and Guertin, D. A. (2016). Raptor/mTORC1 loss in adipocytes causes progressive lipodystrophy and fatty liver disease. Mol. Metab. 5, 422–432. doi: 10.1016/j.molmet.2016.04.001
Liko, D., Rzepiela, A., Vukojevic, V., Zavolan, M., and Hall, M. N. (2020). Loss of TSC complex enhances gluconeogenesis via upregulation of Dlk1-Dio3 locus miRNAs. Proc. Natl. Acad. Sci. U.S.A. 117, 1524–1532. doi: 10.1073/pnas.1918931117
Liu, W., Wang, X., Wang, Y., Dai, Y., Xie, Y., Ping, Y., et al. (2018). SGK1 inhibition-induced autophagy impairs prostate cancer metastasis by reversing EMT. J. Exp. Clin. Cancer Res. 37:73. doi: 10.1186/s13046-018-0743-1
Magdalon, J., Chimin, P., Belchior, T., Neves, R. X., Vieira-Lara, M. A., Andrade, M. L., et al. (2016). Constitutive adipocyte mTORC1 activation enhances mitochondrial activity and reduces visceral adiposity in mice. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1861, 430–438. doi: 10.1016/j.bbalip.2016.02.023
Mammucari, C., Milan, G., Romanello, V., Masiero, E., Rudolf, R., Del Piccolo, P., et al. (2007). FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 6, 458–471. doi: 10.1016/j.cmet.2007.11.001
Martina, J. A., and Puertollano, R. (2013). Rag GTPases mediate amino acid-dependent recruitment of TFEB and MITF to lysosomes. J. Cell Biol. 200, 475–491. doi: 10.1083/jcb.201209135
Masiero, E., Agatea, L., Mammucari, C., Blaauw, B., Loro, E., Komatsu, M., et al. (2009). Autophagy is required to maintain muscle mass. Cell Metab. 10, 507–515. doi: 10.1016/j.cmet.2009.10.008
Medina, D. L., Di Paola, S., Peluso, I., Armani, A., De Stefani, D., Venditti, R., et al. (2015). Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 7, 288–299. doi: 10.1038/ncb3114
Mutvei, A. P., Nagiec, M. J., Hamann, J. C., Kim, S. G., Vincent, C. T., and Blenis, J. (2020). Rap1-GTPases control mTORC1 activity by coordinating lysosome organization with amino acid availability. Nat. Commun. 11:1416. doi: 10.1038/s41467-020-15156-5
Napolitano, G., and Ballabio, A. (2016). TFEB at a glance. J. Cell Sci. 129, 2475–2481. doi: 10.1242/jcs.146365
Napolitano, G., Di Malta, C., Esposito, A., de Araujo, M. E. G., Pece, S., Bertalot, G., et al. (2020). A substrate-specific mTORC1 pathway underlies Birt–Hogg–Dubé syndrome. Nature 585, 597–602. doi: 10.1038/s41586-020-2444-0
O’Rourke, E. J., and Ruvkun, G. (2013). MXL-3 and HLH-30 transcriptionally link lipolysis and autophagy to nutrient availability. Nat. Cell Biol. 15, 668–676. doi: 10.1038/ncb2741
Ortega-Molina, A., Deleyto-Seldas, N., Carreras, J., Sanz, A., Lebrero-Fernández, C., Menéndez, C., et al. (2019). Oncogenic Rag GTPase signalling enhances B cell activation and drives follicular lymphoma sensitive to pharmacological inhibition of mTOR. Nat. Metab. 1, 775–789. doi: 10.1038/s42255-019-0098-8
Park, Y., Reyna-Neyra, A., Philippe, L., and Thoreen, C. C. (2017). mTORC1 balances cellular amino acid supply with demand for protein synthesis through post-transcriptional control of ATF4. Cell Rep. 19, 775–789. doi: 10.1016/j.celrep.2017.04.042
Peng, M., Yin, N., and Li, M. O. (2014). Sestrins function as guanine nucleotide dissociation inhibitors for Rag GTPases to Control mTORC1 signaling. Cell 159, 122–133. doi: 10.1016/j.cell.2014.08.038
Peterson, T. R., Sengupta, S. S., Harris, T. E., Carmack, A. E., Kang, S. A., Balderas, E., et al. (2011). mTOR complex 1 regulates lipin 1 localization to control the srebp pathway. Cell 146, 408–420. doi: 10.1016/j.cell.2011.06.034
Pyo, J. O., Yoo, S. M., Ahn, H. H., Nah, J., Hong, S. H., Kam, T. I., et al. (2013). Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 4:2300. doi: 10.1038/ncomms3300
Rabanal-Ruiz, Y., and Korolchuk, V. I. (2018). mTORC1 and nutrient homeostasis: The central role of the lysosome. Int. J. Mol. Sci. 19, 818. doi: 10.3390/ijms19030818
Robitaille, A. M., Christen, S., Shimobayashi, M., Cornu, M., Fava, L. L., Moes, S., et al. (2013). Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 339, 1320–1323. doi: 10.1126/science.1228771
Roczniak-Ferguson, A., Petit, C. S., Froehlich, F., Qian, S., Ky, J., Angarola, B., et al. (2012). The Transcription Factor TFEB Links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 5:ra42. doi: 10.1126/scisignal.2002790
Rogala, K. B., Gu, X., Kedir, J. F., Abu-Remaileh, M., Bianchi, L. F., Bottino, A. M. S., et al. (2019). Structural basis for the docking of mTORC1 on the lysosomal surface. Science 366, 468–475. doi: 10.1126/science.aay0166
Sancak, Y., Peterson, T. R., Shaul, Y. D., Lindquist, R. A., Thoreen, C. C., Bar-Peled, L., et al. (2008). The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501. doi: 10.1126/science.1157535
Sandri, M., Sandri, C., Gilbert, A., Skurk, C., Calabria, E., Picard, A., et al. (2004). Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117, 399–412. doi: 10.1016/S0092-8674(04)00400-3
Sardiello, M., Palmieri, M., di Ronza, A., Medina, D. L., Valenza, M., Gennarino, V. A., et al. (2009). A gene network regulating lysosomal biogenesis and function. Science 325, 473–477. doi: 10.1126/science.1174447
Saxton, R. A., and Sabatini, D. M. (2017). mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976. doi: 10.1016/j.cell.2017.02.004
Schalm, S. S., and Blenis, J. (2002). Identification of a conserved motif required for mTOR signaling. Curr. Biol. 12, 632–639. doi: 10.1016/S0960-9822(02)00762-5
Schiaffino, S., and Hanzlíková, V. (1972). Autophagic degradation of glycogen in skeletal muscles of the Newborn rat. J. Cell Biol. 52, 41–51. doi: 10.1083/jcb.52.1.41
Schurmann, A., Brauers, A., Massmann, S., Becker, W., and Joost, H. G. (1995). Cloning of a novel family of mammalian GTP-binding proteins (RagA, RagBs, RagB1) with remote similarity to the Ras-related GTPases. J. Biol. Chem. 270, 28982–28988. doi: 10.1074/jbc.270.48.28982
Sengupta, S., Peterson, T. R., Laplante, M., Oh, S., and Sabatini, D. M. (2010). mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature 468, 1100–1104. doi: 10.1038/nature09584
Settembre, C., De Cegli, R., Mansueto, G., Saha, P. K., Vetrini, F., Visvikis, O., et al. (2013). TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 15, 647–658. doi: 10.1038/ncb2718
Settembre, C., Di Malta, C., Polito, V. A., Garcia Arencibia, M., Vetrini, F., Erdin, S., et al. (2011). TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433. doi: 10.1126/science.1204592
Shimobayashi, M., and Hall, M. N. (2016). Multiple amino acid sensing inputs to mTORC1. Cell Res. 26, 7–20. doi: 10.1038/cr.2015.146
Singh, R., Kaushik, S., Wang, Y., Xiang, Y., Novak, I., Komatsu, M., et al. (2009a). Autophagy regulates lipid metabolism. Nature 458, 1131–1135. doi: 10.1038/nature07976
Singh, R., Xiang, Y., Wang, Y., Baikati, K., Cuervo, A. M., Luu, Y. K., et al. (2009b). Autophagy regulates adipose mass and differentiation in mice. J. Clin. Invest. 119, 3329–3339. doi: 10.1172/JCI39228
Stitt, T. N., Drujan, D., Clarke, B. A., Panaro, F., Timofeyva, Y., Kline, W. O., et al. (2004). The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol. Cell 14, 395–403. doi: 10.1016/S1097-2765(04)00211-4
Tee, A. R., Manning, B. D., Roux, P. P., Cantley, L. C., and Blenis, J. (2003). Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 13, 1259–1268. doi: 10.1016/s0960-9822(03)00506-2
Torrence, M. E., MacArthur, M. R., Hosios, A. M., Asara, J. M., Mitchell, J. R., and Manning, B. D. (2020). The mTORC1-mediated activation of ATF4 promotes protein and glutathione 1 synthesis downstream of growth signals 2. bioRxiv [Preprint]. doi: 10.1101/2020.10.03.324186
Valvezan, A. J., and Manning, B. D. (2019). Molecular logic of mTORC1 signalling as a metabolic rheostat. Nat. Metab. 1, 321–333. doi: 10.1038/s42255-019-0038-7
Valvezan, A. J., Turner, M., Belaid, A., Lam, H. C., Miller, S. K., McNamara, M. C., et al. (2017). mTORC1 couples nucleotide synthesis to nucleotide demand resulting in a targetable metabolic vulnerability. Cancer Cell 32, 624–638.e5. doi: 10.1016/j.ccell.2017.09.013
Wada, S., Neinast, M., Jang, C., Ibrahim, Y. H., Lee, G., Babu, A., et al. (2016). The tumor suppressor FLCN mediates an alternate mTOR pathway to regulate browning of adipose tissue. Genes Dev. 30, 2551–2564. doi: 10.1101/gad.287953.116
Wulff, P., Vallon, V., Huang, D. Y., Völkl, H., Yu, F., Richter, K., et al. (2002). Impaired renal Na+ retention in the sgk1-knockout mouse. J. Clin. Invest. 110, 1263–1268. doi: 10.1172/jci15696
Yang, L., Li, P., Fu, S., Calay, E. S., and Hotamisligil, G. S. (2010). Defective hepatic autophagy in obesity promotes er stress and causes insulin resistance. Cell Metab. 11, 467–478. doi: 10.1016/j.cmet.2010.04.005
Yecies, J. L., Zhang, H. H., Menon, S., Liu, S., Yecies, D., Lipovsky, A. I., et al. (2011). Akt stimulates hepatic SREBP1c and lipogenesis through Parallel mTORC1-dependent and independent pathways. Cell Metab. 14, 21–32. doi: 10.1016/j.cmet.2011.06.002
Yu, L., McPhee, C. K., Zheng, L., Mardones, G. A., Rong, Y., Peng, J., et al. (2010). Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465, 942–946. doi: 10.1038/nature09076
Yu, Y., Yoon, S.-O., Poulogiannis, G., Yang, Q., Ma, X. M., Villen, J., et al. (2011). Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 332, 1322–1326. doi: 10.1126/science.1199484
Zhang, Y., Gao, X., Saucedo, L. J., Ru, B., Edgar, B. A., and Pan, D. (2003). Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat. Cell Biol. 5, 578–581. doi: 10.1038/ncb999
Zhang, Y., Goldman, S., Baerga, R., Zhao, Y., Komatsu, M., and Jin, S. (2009). Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc. Natl. Acad. Sci. U.S.A. 106, 19860–19865. doi: 10.1073/pnas.0906048106
Zhang, Y., Nicholatos, J., Dreier, J. R., Ricoult, S. J. H., Widenmaier, S. B., Hotamisligil, G. S., et al. (2014). Coordinated regulation of protein synthesis and degradation by mTORC1. Nature 513, 440–443. doi: 10.1038/nature13492
Zhao, J., Brault, J. J., Schild, A., Cao, P., Sandri, M., Schiaffino, S., et al. (2007). FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 6, 472–483. doi: 10.1016/j.cmet.2007.11.004
Zhao, Y., Yang, J., Liao, W., Liu, X., Zhang, H., Wang, S., et al. (2010). Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol. 12, 665–675. doi: 10.1038/ncb2069
Zhou, B., Kreuzer, J., Kumsta, C., Wu, L., Kamer, K. J., Cedillo, L., et al. (2019). Mitochondrial permeability uncouples elevated autophagy and lifespan extension. Cell 177, 299–314.e16. doi: 10.1016/j.cell.2019.02.013
Zhou, J., Liao, W., Yang, J., Ma, K., Li, X., Wang, Y., et al. (2012). FOXO3 induces FOXO1-dependent autophagy by activating the AKT1 signaling pathway. Autophagy 8, 1712–1723. doi: 10.4161/auto.21830
Keywords: autophagy, mechanistic target of rapamycin, lysosome, metabolism, nutrients
Citation: Deleyto-Seldas N and Efeyan A (2021) The mTOR–Autophagy Axis and the Control of Metabolism. Front. Cell Dev. Biol. 9:655731. doi: 10.3389/fcell.2021.655731
Received: 19 January 2021; Accepted: 19 May 2021;
Published: 01 July 2021.
Edited by:
William T. Festuccia, University of São Paulo, BrazilReviewed by:
Yoana Rabanal, University of Castilla-La Mancha, SpainGustavo J. S. Pereira, Federal University of São Paulo, Brazil
Copyright © 2021 Deleyto-Seldas and Efeyan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alejo Efeyan, YWVmZXlhbkBjbmlvLmVz