 Dianne M. Perez
Dianne M. Perez- The Lerner Research Institute, The Cleveland Clinic Foundation, Cleveland, OH, United States
The α1-adrenergic receptors (ARs) are G-protein coupled receptors that bind the endogenous catecholamines, norepinephrine, and epinephrine. They play a key role in the regulation of the sympathetic nervous system along with β and α2-AR family members. While all of the adrenergic receptors bind with similar affinity to the catecholamines, they can regulate different physiologies and pathophysiologies in the body because they couple to different G-proteins and signal transduction pathways, commonly in opposition to one another. While α1-AR subtypes (α1A, α1B, α1C) have long been known to be primary regulators of vascular smooth muscle contraction, blood pressure, and cardiac hypertrophy, their role in neurotransmission, improving cognition, protecting the heart during ischemia and failure, and regulating whole body and organ metabolism are not well known and are more recent developments. These advancements have been made possible through the development of transgenic and knockout mouse models and more selective ligands to advance their research. Here, we will review the recent literature to provide new insights into these physiological functions and possible use as a therapeutic target.
Introduction
α1-Adrenergic receptors (ARs) regulate the sympathetic nervous system by binding and transducing the effects of the endogenous catecholamines, epinephrine, and norepinephrine (Graham and Lanier, 1986). ARs are members of the G-protein-coupled receptor (GPCR) superfamily and are composed of nine adrenergic receptor subtypes (α1A, α1B, α1D, α2A, α2B, α2C, β1, β2, and β3) from the three distinct families (α1, α2, β) which are activated by the same catecholamines and are related as paralogs.
The α1-AR subtype cDNAs were cloned in the late 1980s and early 1990s (Cotecchia et al., 1988; Schwinn et al., 1990; Lomasney et al., 1991; Perez et al., 1991, 1994; Laz et al., 1994). They have distinct pharmacological properties which helped to determine their classification and characterization. Before the cloning of the receptors, α1-ARs were already subdivided into the α1A- and α1B-AR subtypes based upon radioligand binding data in various tissues which showed two-site competition binding curves to the antagonists WB4101 and phentolamine. The α1A-AR subtype was defined as having a 10–100-fold higher binding affinity for these two antagonists while the α1B-AR subtype was defined as having the weaker binding affinity (Morrow and Creese, 1986). The α1C-AR designation is missing from the α1-AR subtype lineage because of a misclassification early on in the cloning of the receptors1.
α1-ARs are mainly coupled to the heterotrimeric Gq/11 (Gαq) family of G-proteins to activate phospholipase Cβ1 (PLCβ1), resulting in the hydrolysis of membrane-bound phosphatidylinositol 4,5-bisphosphate and the cytosolic release of inositol triphosphate (IP3) and diacylglycerol (DAG) (Piascik and Perez, 2001; Table 1). The IP3 plays a key role in calcium regulation by binding to IP3 receptors located on the endoplasmic reticulum resulting in calcium channel opening and the release of intracellular calcium. The DAG activates protein kinase C (PKC) which can phosphorylate many other types of proteins and signals downstream in the signaling cascade. There are also reports that α1-ARs can couple to Gi G-proteins under overexpressed conditions or in certain cell lines (Akhter et al., 1997; Melien et al., 2000; Snabaitis et al., 2005) but this has not been shown to occur in vivo. α1-ARs can also signal through G-protein-independent mechanisms involving β-arrestins which act as scaffolds to recruit and activate other second messengers such as ERK 1/2, p38, and Src (Perez-Aso et al., 2013; Segura et al., 2013). α1-ARs can also couple to phospholipase A2 and calcium channels though this may not be direct coupling (Perez et al., 1993).

Table 1. Properties of the α1-AR subtypes.
While the α1-AR subtypes display differences in internalization resulting in spatio-temporal changes in signaling (Stanasila et al., 2008; Perez-Aso et al., 2013; Segura et al., 2013), there is some evidence that the α1-AR subtypes differentially couple to different signaling proteins, such as Regulators of G-protein Signaling (RGS) (Hague et al., 2005). These G-protein modulators can interact with the alpha subunits of large G-proteins to increase the rate of GTP hydrolysis and to stop the receptor signaling process. RGS2 can directly bind to the third intracellular loop of the α1A-AR to inhibit its signaling process but does not bind at the α1B- or α1D-AR subtypes (Hague et al., 2005). As RGS2 plays a prominent role in regulating GPCR cardiovascular functions (Tang et al., 2003; Zou et al., 2006) and GPCR G11 signaling pathways (Cunningham et al., 2001), α1A-AR coupling to RGS2 may regulate many of its subtype-specific functions. Another way that α1-ARs create differential signaling pathways is through biased agonism (Wootten et al., 2018). Cirazoline or A61603, imidazolines which are α1A-AR selective agonists, can bias the receptor toward cAMP signaling rather than Ca+2 release or ERK phosphorylation (Evans et al., 2011; da Silva et al., 2017) or can enhance the α1A-AR desensitization and internalization process (Akinaga et al., 2013) leading to differential coupling to β-arrestin-mediated signaling.
Transgenic and Knockout Mouse Models
Due to the lack of sufficiently selective pharmacological agents to use in order to distinguish subtype-specific effects, a number of transgenic and knockout (KO) mouse models were developed that were used to determine long-term in vivo stimulatory or inhibitory effects of the α1-AR subtypes on physiology and pathophysiology (Table 2). KOs of the α1A-AR (Rokosh and Simpson, 2002; Zhang et al., 2020), α1B-AR (Cavalli et al., 1997), and α1D-AR (Tanoue et al., 2002) were developed using traditional insertion of the β-galactosidase or neomycin resistance gene in place of the first exon of the receptor. Recently, a cardiac-conditional KO of the α1A-AR was developed (Zhang et al., 2020). There is also a double KO model created by mating together the α1A and α1B-AR KO mice (O’Connell et al., 2003) and a triple KO of all three subtypes (Sanbe et al., 2007). Transgenic mice overexpressing α1-ARs were designed to either target to the myocyte using the α myosin heavy chain promoter to drive only cardiac expression of wild-type (WT) or constitutively active mutations (CAMs) in the receptor (Milano et al., 1994; Grupp et al., 1998; Eckhart et al., 2000; Lin et al., 2001) or used CAMs in the receptors that were driven by large fragments of the endogenous mouse promoters to generate systemic expression (Zuscik et al., 2000, 2001; Ross et al., 2003; Rorabaugh et al., 2005). The systemic expression of the CAMs also allows assessment of cardiovascular effects due to chronic α1-AR expression outside of the myocyte as well as in the brain or other organ systems. There is also only mild overexpression of the receptor in the heart and brain (2–3 fold) and throughout the body in using the endogenous promoters as compared to using the α myosin heavy chain promoter which caused very high amounts of receptor overexpression, often exceeding 100-fold. The use of CAMs instead of the WT receptor results in continuously activated receptors that do not need an agonist to be present and can be representative of a chronically stimulated condition, but this is still debated. In both overexpressed and KO mouse models, there is always the possibility of changes in the expression of other genes and receptors in compensation or as a result of additional insertion or deletion of genetic material, a widespread phenomenon that is hard to decipher and under reported (El-Brolosy and Stainier, 2017). Recognizing these limitations and seeing if general phenotypes repeat in the various mouse models of particular receptor subtypes is suggested. These different types of mouse models will be referred to throughout this review.

Table 2. Genetic animal models of the α1-AR subtypes.
Cognition
Localization in the Brain
The expression of the specific α1-AR subtypes in the brain was previously difficult to determine because of the lack of high avidity antibodies to the α1-ARs (Jensen et al., 2009c; Böhmer et al., 2014). Initial autoradiography studies used non-selective radiolabels that could not distinguish between the α1-ARs subtypes but did demonstrate high abundance throughout the rat brain (Unnerstall et al., 1985). Eventually, more specific and sensitive techniques were developed to determine the α1-AR subtype localization in the brain such as using the full-length cDNA sequence of the α1A-AR in hybridization studies (Domyancic and Morilak, 1997) or transgenic and knock-out (KO) mouse models of the α1-AR subtypes with the α1-ARs tagged with endogenous promoter-driven expression of EGFP or use of the β-galactosidase gene to KO the receptor (Papay et al., 2004, 2006). Using these approaches, the α1A- and α1B-ARs were shown to be expressed in similar areas of the brain, but the relative expression was different (Papay et al., 2004, 2006). The α1A-AR subtype was more noticeably expressed in the cognitive areas such as the hippocampus, amygdala, and particular cortical areas (Table 1; Papay et al., 2006), while the α1B-AR appeared more prominent throughout the cortex and thalamus (Drouin et al., 2002; Papay et al., 2004). The α1A-AR subtype was also more prominently expressed in neural progenitors and stem cells (Papay et al., 2006; Gupta et al., 2009). Using long sequences of antisense to the α1D-AR to assess brain localization, the α1D-AR although of low overall abundance, was present in the reticular thalamic nuclei, hippocampus, cortex and spinal cord (Harasawa et al., 2003). Using the α1-AR KO mice and comparing the total amount of α1-AR radioligand receptor binding to normal wild-type mice, it was concluded that the brain contains the highest amount of the α1A-AR subtype at ∼55% (Rokosh and Simpson, 2002), followed by the α1B-AR at 35% (Cavalli et al., 1997) but only 10% of the total α1-AR pool for the α1D-AR subtype (Tanoue et al., 2002; Sadalge et al., 2003).
The localization of the α1-ARs in the brain may have some species variation (Palacios et al., 1987; Zilles et al., 1991), but the cognitive areas appear similar in humans with high expression in the hippocampus and prefrontal cortex and the lowest expression in the caudate and putamen (Shimohama et al., 1986; Szot et al., 2005). The α1A-AR subtype appears to be prominent in expression in the hippocampus as assessed by RNA (Szot et al., 2005), single cell PCR (Hillman et al., 2005), protein localization using the EGFP-tagged transgenics (Papay et al., 2006) and functionally by regulating the CA1 hippocampal interneurons (Jurgens et al., 2009). In addition, the α1A-AR subtype regulated adult neurogenesis in the mouse subgranular and subventricular zones (Gupta et al., 2009; Jurgens et al., 2009; Collette et al., 2010) as assessed by increased BrdU incorporation and co-localization studies of EGFP-tagged α1A-ARs with stem cell and neural progenitor markers (Table 2). In addition, when normal WT mice were given the α1A-AR selective agonist, cirazoline, they also displayed increased neurogenesis (Gupta et al., 2009). The regulation of neurogenesis by the α1A-AR and its regulation of hippocampal function and translation to human brain domains may potentially play a therapeutic role to increase synaptic plasticity and cognition in diseases of dementia.
General Cognition
The α1-ARs have been previously associated with general roles in learning and memory functions (Sirviö and MacDonald, 1999) but these studies were not well characterized nor assigned to specific AR subtypes because of the lack of subtype-specific ligands. A few early studies suggested that α1-AR stimulation inhibits memory functions in monkeys (Arnsten and Jentsch, 1997; Mao et al., 1999) or in chickens (Gibbs and Summers, 2001) but used very low replicates, very high concentrations of ligands rendering them non-selective or attributed to species variation. However, as will be discussed, most of the recent studies indicate that α1-AR stimulation increases various types of memory in both formation and storage.
Long-Term Potentiation
Long-term potentiation (LTP) is a type of long-lasting synaptic plasticity that increases the strength of synaptic transmission over a long period of time (i.e., mins-hours) (Hopkins and Johnston, 1984; Kandel, 2001). LTP is considered a major mechanism of learning and memory, particularly in the hippocampus (Bliss and Collingridge, 1993). α1-AR stimulation can induce LTP in the hippocampus (Izumi and Zorumski, 1999; Sirviö and MacDonald, 1999; Lv et al., 2016) and there is one report in the neocortex (Pankratov and Lalo, 2015) which is also a center for neuronal spatial and recognition memory (Vann and Albasser, 2011). Interestingly, the α1-ARs can also stimulate ATP release on astrocytes to induce LTP via ATP receptors on the pyramidal neurons in the neocortex, suggesting that glial cell regulation by α1-ARs may also be involved in memory formation. Glia communicate through calcium signaling to neurons, causing the release of ATP and its subsequent increase in synaptic plasticity and LTP (Pascual et al., 2005). LTP stimulation by α1-ARs may be α1A-AR-specific as the CAM α1A-AR transgenic mice significantly increased LTP at hippocampal synapses (Doze et al., 2011; Table 2). The CAM α1A-AR mice also increased cognitive scores in a series of behavioral tests while the α1A-AR KO mice performed poorly compared to normal controls (Doze et al., 2011). The α1A-AR selective agonist, cirazoline also increased cognitive scores in normal mice when administered for 2 months. While the α1B-AR KO mice had impaired cognition in some behavior tests (Knauber and Müller, 2000a, b; Spreng et al., 2001), there was no assessment of effects of α1B- or α1D-AR KO on LTP.
Long-term depression (LTD) is also a form of long-term synaptic plasticity that can contribute to cognitive functions by increasing the flexibility of the synapse to store information (Heynen et al., 1996), such as remembering the exposure to novel objects (Manahan-Vaughan and Braunewell, 1999). Novelty exposure can reverse LTP in the hippocampus (Xu et al., 1998), suggesting a correlation between LTD and LTP that may impart different forms of synaptic information during spatial learning (Kemp and Manahan-Vaughan, 2004). There are reports that α1-AR mediated LTD required co-activation with a number of partners such as β-ARs (Katsuki et al., 1997), NMDA (Scheiderer et al., 2004) and the M1 muscarinic receptor (Scheiderer et al., 2008). α1-ARs have been shown to induce LTD at excitatory CA3–CA1 synapses in the rat hippocampus (Dyer-Reaves et al., 2019) through ERK signaling in the pyramidal neurons (Vanhoose et al., 2002; Scheiderer et al., 2008) and had characteristics of a novel form of synaptic plasticity (Hebb, 1949). However, there is no evidence of which α1-AR subtype(s) mediate LTD. This Hebbian LTD requires coincident presynaptic and postsynaptic NMDAR activity (Scheiderer et al., 2004) and is different and independent of the “classical” LTD which is induced by low frequency synaptic stimulation that is repetitive (Mulkey and Malenka, 1992). The mechanism of the Hebbian LTD also involves postsynaptic activation of the α1-AR as the paired pulse facilitation ratio did not change (Scheiderer et al., 2004). Paired pulse facilitation is a measurement of synaptic enhancement observed under a short period of time (i.e., milliseconds). For a pulse facilitation effect, a second evoked excitatory postsynaptic potential is increased when it follows immediately after a first evoked excitatory postsynaptic potential (Foster and McNaughton, 1991) and is used as evidence of an increase in the probability of neurotransmitter release. Increases in paired pulse facilitation that occur with LTP suggest a presynaptic mechanism (Schultz et al., 1994), because potentiated presynaptic neurons must increase neurotransmitter release.
Spatial Memory
The hippocampus also regulates spatial and associative learning functions (Mahmoodi et al., 2010) in addition to long-term memory functions. α1-AR blockage using the α1-AR antagonist prazosin in the hippocampus demonstrated impaired spatial learning (Petrasek et al., 2010) while stimulation of the α1-AR improved spatial memory (Puumala et al., 1998; Torkaman-Boutorabi et al., 2014). Transgenic mice overexpressing CAM α1A-ARs, or WT mice given the α1A-AR selective agonist cirazoline, displayed increased learning and memory using several spatial memory behavioral tests such as the Barnes, dry multi-T, and Morris water mazes (Doze et al., 2011), while α1A-AR KO mice showed decreased learning and memory compared to normal controls in the same cognitive tests (Doze et al., 2011; Collette et al., 2014; Table 2). The α1B-AR KO mice also had impaired spatial learning to novelty and exploration (Spreng et al., 2001) and a decrease in non-spatial memory functions such as memory consolidation, fear-motivated exploration (Knauber and Müller, 2000a), and short and long-term latency in a passive avoidance test (Knauber and Müller, 2000b). α1D-AR KO mice did not show changes in several different behavioral cognitive tests (Sadalge et al., 2003) but did show changes in locomotion and attention (Mishima et al., 2004). Together with enhancement of LTP and paired pulse facilitation (a type of short-term synaptic plasticity) in the CAM α1A-AR transgenic mice (Doze et al., 2011), these studies suggest that the α1A- and perhaps the α1B-AR to a lesser degree but not the α1D-AR are involved in spatial learning and memory processes.
Spatial Working Memory
Spatial working memory involves executive-type or motivational-related types of memory and relies more on the prefrontal cortex than the hippocampus as the task is more complex (Robbins, 1996). α1-AR stimulation increases while α1-AR blockade inhibits working memory (Pussinen et al., 1997; Puumala et al., 1998; Lapiz and Morilak, 2006; Hvoslef-Eide et al., 2015) by promoting both focused and flexible attention (Berridge et al., 2012; Berridge and Spencer, 2016). There is also an improvement in working memory with the cognitive-enhancing, wake-promoting neurochemical modafinil that is hypothesized to be mediated by α1-ARs since effects are blocked by prazosin (Duteil et al., 1990; Stone et al., 2002; Winder-Rhodes et al., 2010).
α1-ARs regulate spatial working memory through the release of glutamate in the prefrontal cortex due to a sustained excitatory effect on the pyramidal neurons increasing synaptic plasticity (Marek and Aghajanian, 1999; Zhang et al., 2013). When the ventral hippocampus was lesioned in vivo and α1-AR function was impaired, there was a decrease in glutamatergic synaptic plasticity within the prefrontal cortex which caused memory and learning dysfunction (Bhardwaj et al., 2014). Glutamatergic synaptic plasticity mediated through α1-ARs signals through PKC-dependent pathways in various cortical areas (Mouradian et al., 1991; Marek and Aghajanian, 1996; Chen et al., 2006; Kobayashi et al., 2008; Velásquez-Martinez et al., 2012; Luo et al., 2014, Luo et al., 2015a, b) and may require the co-signaling from both glutamate and the N-type Ca2+ channels (Luo et al., 2015a). PKC can increase synaptic plasticity and associated memory processes through the phosphorylation of synaptic proteins or enhancing the sensitivity to calcium which promotes the exocytosis of the synaptic vesicles, increasing neurotransmitter release (Shimazaki et al., 1996; Stevens and Sullivan, 1998; Hilfiker and Augustine, 1999; Wu and Wu, 2001).
Besides glutamatergic mechanisms, the disruption of GABAergic transmission in the prefrontal cortex can also cause a decrease in working memory (Enomoto et al., 2011; Bañuelos et al., 2014). α1-AR stimulation in the medial prefrontal cortex inhibits the inwardly rectifying potassium channels (Kirs) located on the interneuron, leading to depolarization and an increased calcium influx through calcium channels resulting in increased GABAergic transmission onto the pyramidal neurons (Luo et al., 2015b). The excitation can be enhanced when the α1-ARs stimulation is facilitated by postsynaptic α2-ARs decreasing the hyperpolarization of cyclic nucleotide-gated cation channels (Zhang et al., 2013). Therefore, α1-ARs may work to improve spatial working memory through both glutamatergic and GABAergic mechanisms which suggests that α1-AR agonists could be used to target enhancement of spatial working memory.
Memory Consolidation
α1-AR activation can enhance memory recall and consolidation. The process of memory consolidation changes recent and labile memories into long-lasting ones. The process starts in the hippocampus but as time passes and the memory is reorganized, the long-lasting memory is then distributed in the neocortex (Squire et al., 2015). The α1-AR antagonist, prazosin, blocked the norepinephrine-facilitated reconsolidation of memory during fear conditioning (Gazarini et al., 2013) and the consolidation of both short-term and intermediate-term memory in chickens (Gibbs and Bowser, 2010). The mechanism for α1-ARs to consolidate memories was suggested to be mediated through an increase in free cytosolic calcium in astrocytes as effects were blocked with glycolytic inhibitors (Gibbs and Bowser, 2010). Astrocytes, unlike neurons, mediate learning and memory utilizing glycogenolysis, which the astrocyte needs for the synthesis of glutamate (Gibbs et al., 2008; Newman et al., 2011).
The basolateral nucleus of the amygdala (BLA) can also be involved in the storage and consolidation of memory (Ferry and McGaugh, 2000). As cAMP signaling is mainly involved in mediating the effects of norepinephrine on memory consolidation, the β-ARs were previously considered the main AR to transduce those effects (Ikegaya et al., 1997; Ferry and McGaugh, 2000; Ferry and Quirarte, 2012). However, both β- and α1-ARs may be needed together to mediate memory storage in the BLA. The stimulation of cAMP through a β-AR agonist in the BLA can be blocked with an α1-AR antagonist and memory storage is increased with use of a synthetic cAMP analog (Ferry et al., 1999a, b). Similarly, stimulation of α1-ARs can potentiate β-AR-mediated cAMP formation in the BLA to enhance memory storage (Ferry et al., 1999a, b). α1B-AR KO mice had a decrease in latency in the passive avoidance test suggesting deficits in memory consolidation in vivo (Knauber and Müller, 2000b; Table 2). Research performed in amnesia patients developed the concept of memory consolidation as time was needed for this process to occur and greater memory deficits were seen in retrograde amnesia patients with loss of information from recent memory (Brown, 2002). α1-AR stimulation can reverse cannabinoid-induced (Moshfegh et al., 2011) and scopolamine-induced amnesia (Azami et al., 2010) and enhance recall when α1-AR agonists were administered before electroconvulsive shocks (Anand et al., 2001).
Dementia-Related Diseases
α1-AR functions may change and contribute to the aging process in the loss of memory function. α1-AR protein is increased in the aging mouse brain and with improved learning, supporting a role for these receptors in age-related cognitive decline (Knauber and Müller, 2000b). In patients suffering from Alzheimer’s Disease (AD), α1-AR protein and mRNA is reduced in the prefrontal cortex (Shimohama et al., 1986; Kalaria, 1989; Szot et al., 2007). The mRNA levels of the α1A-AR were significantly decreased in the prefrontal cortex with AD with no changes in the mRNA of the α2-AR (Szot et al., 2007). There is also an α1A-AR polymorphism that associates with AD (Hong et al., 2001). Decreases in spatial memory that are due to the aging process were improved in rats when the α1-AR was stimulated (Riekkinen et al., 1997).
The 3xTG (Transgenic) is a widely used AD mouse model that contains three genetic mutations associated with familial AD (APP Swedish, MAPT P301L, and PSEN1 M146V) (Oddo et al., 2003). This AD mouse model displays β-amyloid deposits, tau immunoreactivity, cognitive impairment, and decreases in LTP and basal synaptic transmission (Oddo et al., 2003). When the 3xTG AD mouse model was given a selective α1A-AR positive allosteric modulator, spatial memory as assessed in the Barnes maze was improved along with LTP (Perez, 2021). These results suggest that selective agonists that increase α1A-AR functions may be able to improve cognitive decline in AD.
Another cognitive disease is vascular dementia which is the second-most frequent form of dementia after AD. α1-AR autoantibodies with agonistic function were found in 50% of people with dementia (Karczewski et al., 2010, 2012, 2018; Hempel et al., 2016; Thyrian et al., 2018). While these agonistic autoantibodies may also cause vascular damage, shown for several neurotransmitters (Wu and Li, 2016), one interpretation of the data consistent with the role of the α1A-AR in improving cognition, but also speculative, is that they may develop during dementia to compensate for the loss in receptor density as documented by Shimohama et al. (1986) and Szot et al. (2007).
Cardioprotection
The heart expresses both the α1A and α1B-AR subtypes with relative expression levels depending upon the species (Steinfath et al., 1992; Michel et al., 1994; Jensen et al., 2009a). The α1D-AR is weakly expressed if at all in the myocyte (Price et al., 1994; Scofield et al., 1995) but is present in vascular smooth muscle, particularly in the coronary arteries, mesenteric beds and the aorta (Table 1; Hrometz et al., 1999; Gisbert et al., 2002; Chalothorn et al., 2003; Turnbull et al., 2003; Hosoda et al., 2005; Jensen et al., 2009b; Methven et al., 2009; Martínez-Salas et al., 2011). A KO mouse model of the α1B-AR was created with a human placental alkaline phosphatase inserted into the first exon to facilitate reporting (Myagmar et al., 2017). Using this new KO model and the conventional α1A-KO which has the β-galactosidase reporter, the authors report a heterogenous population of the α1B and α1A-AR subtypes in the myocytes. The α1B was present in all of the myocytes but the α1A was present in only 60% of the myocytes and 20% of those had very high expression levels. This intermittent variable expression of the α1A-AR subtype was also observed in the mesenteric arteries in the α1B/D double KO and in the transgenic systemically expressing α1-AR WT mice that were tagged with the green fluorescent protein (Papay et al., 2004; McGrath, 2015). Since this intermittent expression is only present in genetically altered mouse models, this suggests that intermittent expression may be an artifact. However, the current lack of highly avid α1-AR antibodies that can be used for in vivo localization (Jensen et al., 2009c; Böhmer et al., 2014), precludes using immunoassays to determine if intermittent expression is an artifact. A potential experiment that may confirm intermittent expression in a WT mouse would be to perform autoradiography with and without selective α1-AR blockers such as niguldipine to block the α1A-AR subtype.
It is generally accepted that α1-AR stimulation can regulate a positive inotropic response in the heart, although the response can be variable and display negative inotropy depending upon the species and the region in the heart analyzed (Endoh et al., 1991; Nishimaru et al., 2001; Endoh, 2016). The α1A- and not the α1B-AR is suggested to play a role in positive inotropy (Lin et al., 2001; Ross et al., 2003; Luo et al., 2007; Janssen et al., 2018). The systemically over-expressed CAM α1B-AR mice had no changes in basal cardiac parameters but had autonomic failure (Zuscik et al., 2001). The autonomic failure in the CAM α1B-AR mice indicated reduced circulating catecholamine levels, bradycardia, reproductive problems and weight loss. Together with the widespread neurodegeneration and a phenotype that was consistent with a Parkinson Disease plus syndrome, the basal hypotension seen in these mice was likely due to the autonomic failure rather than a direct effect on the ability to contract vascular smooth muscle. The CAM α1B-AR mice also had a negative inotropic response to phenylephrine (Ross et al., 2003). Radioligand binding analysis revealed that there was decreased α1A-AR density which was likely causing the negative inotropic effect (Ross et al., 2003). This functional antagonism of the positive inotropy of the α1A-AR by the α1B-AR was also found in a mouse model of right ventricular failure (Cowley et al., 2015). The heart-targeted WT α1B-AR also displayed negative inotropy (Grupp et al., 1998). In contrast, both the cardiac-targeted WT and systemically expressed CAM α1A-AR mediated a positive inotropic response in the mouse heart (Lin et al., 2001; Rorabaugh et al., 2005; Table 2). In human myocardium, the α1A-AR selective agonist, A61603, had a strong positive inotropic response representing about 70% of the β-AR response (Janssen et al., 2018).
Heart Failure
In human heart failure, radioligand binding indicates that β1-ARs are downregulated (Bristow et al., 1982, 1986; Rockman et al., 2002) while α1-AR are either unchanged (Bristow et al., 1988; Jensen et al., 2009a) or decreased (Limas et al., 1989; Zhao et al., 1996; Fischer et al., 2008; Shi et al., 2013). MicroRNA-133 was found to be a key control in the downregulation of the β1-AR and several components of its signal transduction cascade in the heart (Castaldi et al., 2014), opening up new avenues of therapeutics in addition to β-blockers. Radioligand binding of human hearts with end-stage dilated cardiomyopathy versus non-failing controls revealed that while β1-ARs are downregulated as previously reported (Bristow et al., 1982, 1986), there was also a loss in the α1A-AR subtype receptor levels (Shi et al., 2013). The differences in these studies of the density of α1-ARs could be the severity of the heart failure (Limas et al., 1989), the level of sympathetic overdrive (Zhao et al., 1996) or the etiology of heart failure studied (ischemic versus non-ischemic) as α1-ARs are known to increase in density during ischemia (Corr et al., 1981; Maisel et al., 1987; Kurz et al., 1991) and could have masked the decrease in α1A-ARs during failure.
α1-ARs also can mediate cardiac hypertrophy, an increase in protein mass of the myocyte through an increase in protein synthesis which remodels the heart in response to various physiological and pathophysiological stimuli (Simpson, 1983; Fuller et al., 1990; Ikeda et al., 1991; Perez-Aso et al., 2013; Cotecchia et al., 2015). While both the α1A and α1B-ARs are involved in hypertrophy, the α1A-AR seems better coupled to enhance hypertrophic signaling pathways. The α1A-AR agonist, A-61603, increased the size of the myocyte by increasing the rate of protein synthesis (Autelitano and Woodcock, 1998). The various transgenic mouse models showed variable degrees of cardiac hypertrophy but have never been as robust as seen in cell cultures (Table 2). Cardiac hypertrophy can be a normal physiological response which is adaptive and improves function while hypertrophy that is associated with fibrosis or apoptosis is maladaptive and can lead to heart failure. Both the α1A- and α1B-AR subtypes are required for physiological cardiac hypertrophy (O’Connell et al., 2003) as single KO do not have decreased heart size (Vecchione et al., 2002; Table 2). The systemic-expressing CAM α1A displayed adaptive cardiac hypertrophy without increasing blood pressure (Papay et al., 2013). The heart-targeted CAM α1B mouse induced hypertrophy (Milano et al., 1994) but displayed maladaptive remodeling after pressure overload (Wang et al., 2000). The systemically expressing CAM α1B also induces cardiac hypertrophy (Zuscik et al., 2001) but was more pronounced when the mouse aged (Papay et al., 2013). A systemically expressing WT α1B-AR also displayed a lower degree of hypertrophy that only manifested in aged mice with fibrosis indicating a maladaptive cardiac hypertrophy (Zuscik et al., 2001). KO of the α1B-AR had a loss of NE-induced hypertrophy but not a decrease in heart size at birth (Vecchione et al., 2002). While a heart-targeted WT α1B with high overexpression did not induce hypertrophy, it did induce a maladaptive dilated cardiomyopathy (Akhter et al., 1997; Grupp et al., 1998; Lemire et al., 2001). The α1B-AR has been suggested to regulate cardiac hypertrophy differently than the α1A-AR and the two AR subtypes may need to be co-activated to regulate hypertrophy (Papay et al., 2013). The CAM α1A-AR mice selectively secreted interleukin-6 (IL-6) and atrial naturietic factor while the CAM α1B-AR mice activated nuclear factor-kB (Papay et al., 2013). The α1AB-AR double KO mice also failed to develop hypertrophy when stimulated with IL-6 but WT mice developed hypertrophy when given IL-6. These hypertrophic signals were blocked in each mouse model and no increase in heart weight observed when the other AR was coactivated or when the two transgenic mouse models were crossbred, resulting in a CAM α1A/B-AR double transgenic mouse model (Papay et al., 2013). Hypertrophy became apparent in the CAM α1AB-AR double transgenic when either the α1A-AR or α1B-AR were independently stimulated (Papay et al., 2013). These results suggest that both the AR subtypes can increase hypertrophy through different signaling pathways. Increased α1A-AR signaling can induce an adaptive hypertrophy consistent with its postulated role of cardiac protection while increased α1B-AR signaling induces a maladaptive hypertrophy in the heart. These differences between adaptive versus maladaptive hypertrophy may be due to differences in α1-AR mediation of IL-6, ANF, and NF-kB signaling pathways.
α1A-AR Mediated Protection in Heart Failure
It is postulated that selective α1A-AR stimulation may be a potential therapeutic in heart failure (Perez and Doze, 2011; Janssen et al., 2018) while α1B-AR stimulation, on the other hand, is maladaptive. This is evidenced by the heart-targeted WT α1B-AR mice induced dilated cardiomyopathy (Lemire et al., 2001) while heart-targeted CAM α1B-AR progressed to heart failure after pressure-overload (Wang et al., 2000; Table 2). In contrast, the heart-targeted WT α1A-AR mice were protected against pressure-overload induced heart failure (Du et al., 2004) or dysfunction due to myocardial infarction (Du et al., 2006) compared to non-transgenic controls. This mouse model also showed increased vascular endothelial growth factor-A expression which induced angiogenesis and resulted in increased capillary density and blood flow to the heart, postulated to be a contributing mechanism for cardioprotection (Zhao et al., 2015). This phenotype of induced angiogenesis could be reproduced when WT mice were given the α1A-AR agonist, A61603. A61603 or dabuzalgron also increased survival and prevented the damage due to the cardiotoxic agent, doxorubicin (Beak et al., 2017; Montgomery et al., 2017) and increased contraction in a mouse model of right heart failure (Cowley et al., 2015).
Preconditioning and Ischemia
The high metabolic rate of the heart can cause the heart to be sensitive to the lack of oxygen (i.e., ischemia) resulting in injury to the muscle. α1-AR have long been known to mediate protective effects against ischemia or preconditioning in ischemia in several species (Banerjee et al., 1993; Kitakaze et al., 1994; Tsuchida et al., 1994; Salvi, 2001; Rorabaugh et al., 2005; Zhao et al., 2012; Nazari et al., 2019; Papay and Perez, 2020). In preconditioning, short periods of ischemia can stimulate signaling in the heart that protects the cardiac muscle from subsequent ischemic injury. The mechanism has been multi-faceted and attributed to PKC (Tsuchida et al., 1994; Mitchell et al., 1995; Rehring et al., 1996; Rorabaugh et al., 2005), mitochondrial potassium channels (Nazari et al., 2019), mitochondrial permeability transition pore (Naderi et al., 2010), 5′-nucleotidase activity (Tsuchida et al., 1994) or angiogenesis (Zhao et al., 2012). In recent studies, the ischemic protective effect of the α1-AR observed in primary cardiomyocytes was also proposed to be through the metabolic effects of glucose (Papay and Perez, 2020). Most models of ischemic preconditioning and particularly those by α1-ARs converge first on PKC, then diverge to other downstream effectors (Downey and Cohen, 1997; Simkhovich et al., 2013) and are postulated to also do so in the human heart (Speechly-Dick et al., 1995).
α1A-AR Mediated Protection in Ischemia
The α1A-AR subtype has been shown to mediate the cardioprotective effects of α1-ARs in ischemic preconditioning. These studies have been performed in transgenic or KO mouse models as blocking one subtype is still not specific enough to perform with antagonists. The systemically expressed CAM α1A mice were inherently preconditioned against ischemia while the CAM α1B was not (Rorabaugh et al., 2005; Table 2). The heart-targeted CAM α1B-AR also did not show ischemic preconditioning (Gao et al., 2000). In corroboration, the heart-targeted WT α1A-AR transgenic rat exhibited preconditioning that appeared during the second window of protection that occurs days (and not minutes) after ischemia (Du et al., 2006; Zhao et al., 2012, 2015). There are also two reports that α1B-AR stimulation in WT mice can induce ischemic preconditioning involving PKC but used sensitivity to chloroethylclonidine as a criteria to block α1B-ARs selectively (Hu and Nattel, 1995; Gao et al., 2007). However, chloroethylclonidine was shown to not be selective against the α1B-AR but can block all the α1-AR subtypes (Xiao and Jeffries, 1998). Transgenic rats with myocyte-specific α1A-AR overexpression protected the heart from permanent coronary occlusion and during preconditioning (Zhao et al., 2012, 2015). The α1A-AR KO or conditional cardiac KO of the α1A-AR also had more pathological injury from myocardial infarction after left anterior descending ligation (Yeh et al., 2017; Zhang et al., 2020). Together, these results strongly suggest that the α1A-AR subtype mediates ischemic protection in the heart.
Hypertension
α1-ARs are highly expressed in vascular smooth muscle (Hussain and Marshall, 1997; Martí et al., 2005). The rise in calcium upon stimulation of α1-ARs in the vasculature activates myosin light chain kinase and actin/myosin cross-bridge formation to induce vascular muscle contraction and increased blood pressure (Somlyo and Somlyo, 2003). The smaller resistance arteries play a more important role in blood pressure regulation and are under stronger control from the sympathetic nervous system. Signals mediated through α1-AR activation have been shown to be involved in blood pressure regulation through their control of calcium release and sensitization and signaling through mechanisms involving PKC, PI3K, Rho Kinase, and MAPK (Woo and Lee, 1999; Wier and Morgan, 2003; Villalba et al., 2007; Gutiérrez et al., 2019).
While α1-AR antagonists are effective blockers to treat hypertension, they are used as a second line of defense (Chobanian et al., 2003) because of the side effects, poorer outcomes, and worsening or increased risk of heart failure (ALLHAT Collaborative Research Group, 2000). Using KO mice, the α1A was found to decrease blood pressure upon deletion, but only by 15% of the full phenylephrine effect (Rokosh and Simpson, 2002; Table 2). However, the α1B-AR KO mediated 45% of the phenylephrine response (Cavalli et al., 1997; Vecchione et al., 2002). Similar minor effects on blood pressure were observed in the α1D-AR KO compared to the α1A-AR or α1B-AR KOs (Cavalli et al., 1997; Hosoda et al., 2005). Only the α1D-AR KO decreased basal resting levels of blood pressure (Vecchione et al., 2002; Hosoda et al., 2005).
Since all of the α1-ARs appear to regulate blood pressure to a certain degree, specific blockage of the α1D-AR may provide better therapeutics to treat hypertension with less overall side effects on other organ systems. This is because the α1B-AR appears to have the strongest effect on blood pressure while α1D-AR blockage would still lower blood pressure but is not expressed or minimally expressed in the heart (Price et al., 1994; Scofield et al., 1995) or the brain (Tanoue et al., 2002; Sadalge et al., 2003), thereby reducing potential side effects. The α1D-AR is also expressed and regulates contraction in the small resistance mesenteric beds which is an important contributor to total peripheral resistance (Christensen and Mulvany, 1993; Hrometz et al., 1999; Gisbert et al., 2002; Methven et al., 2009). The α1B-AR subtype controls the neuroeffector junction and sympathetic regulation of the baroreflex response (Townsend et al., 2004) and both the α1A- and α1B-AR subtypes regulate physiological hypertrophy (O’Connell et al., 2003). The α1A-AR as reviewed above is a major regulator of neurotransmission and cognition; thus, blockage of α1A- or α1B-ARs would affect more off targets than vascular smooth muscle. Therefore, antagonists against the α1D-AR subtype might be more effective therapeutically against hypertension by avoiding negative side effects on the heart and brain but may focus effects better on blood pressure regulation.
Metabolism
The sympathetic nervous system is known to regulate many aspects of metabolism. α1-ARs stimulation has long been known to regulate gluconeogenesis in the liver (Chan and Exton, 1978; Hue et al., 1978; García-Sáinz and Hernández-Sotomayor, 1985; de Oliveira et al., 2013). α1-ARs also regulate somatostatin-induced gluconeogenesis in the kidney (Dileepan et al., 1982; Dileepan and Wagle, 1985). Gluconeogenesis generates the synthesis of glucose from non-carbohydrate sources while glycolysis breaks down glucose to yield energy (i.e., ATP). Gluconeogenesis becomes important during fasting or starvation when glucose is needed by the cell after glycogen is depleted. α1-AR agonists also stimulate glycogen phosphorylase activity, the rate limiting step in glycogen breakdown, which inhibits glycogen synthesis, and increases the breakdown of glycogen (Assimacopoulos-Jeannet et al., 1977; Aggerbeck et al., 1980; Thomas et al., 1985; Ballou et al., 2001; de Oliveira et al., 2013) and stimulates the release of glucagon from the pancreas (Ahrén and Lundquist, 1987; Skoglund et al., 1987; Vieira et al., 2004). However, recent studies have indicated that α1-ARs regulate metabolism at a much more systemic level as reviewed below.
α1-AR Stimulation Increases Glucose Tolerance
α1-AR stimulation is known to increase glucose uptake in the heart or in primary myocytes (Doenst and Taegtmeyer, 1999; Egert et al., 1999; Shi et al., 2016, 2017; Sato et al., 2018; Papay and Perez, 2020). The systemically expressing CAM α1A but not the CAM α1B-AR mice increased glucose uptake into the heart and only the α1A-AR KO mice displayed decreased glucose uptake into the heart (Shi et al., 2017). In corroboration, the α1A-selective agonist, A61603 increased glucose uptake into primary cardiomyocytes or human α1A-AR transfected Chinese hamster ovary (CHO) cells (Sato et al., 2018). While glucose uptake into the heart appears α1A-AR specific, both the α1A- and α1B-AR subtypes mediate glucose uptake into other tissues. The systemically expressing CAM α1A and α1B-AR mice both increased glucose uptake into adipose tissue and skeletal muscle while KO of the respective subtype decreased glucose uptake into those same tissues (Shi et al., 2017). The mechanism of α1A-AR mediated glucose uptake in the myocyte was through PKCδ signaling that resulted in GLUT 1/4 translocation which causes their activation to transport glucose into the cell (Shi et al., 2016).
The KO and CAM mice also displayed effects on glucose utilization and homeostasis. Both the systemically expressing CAM α1A- and α1B-AR mice had an increased tolerance for glucose, lower fasting glucose levels while KO mice had poor tolerance and high blood glucose after fasting (Shi et al., 2017). α1-AR stimulation also increased glucose absorption in the intestines (Mourad and Saadé, 2011). Hypothalamic central administration of prazosin increased plasma glucose levels (Murashita et al., 2007; Ikegami et al., 2013b) and glucose intolerance (Ikegami et al., 2013a). When fatty acid oxidation was suppressed centrally in the brain, α1-ARs stimulated the counter-regulatory increases in plasma glucose levels (Sajapitak et al., 2008). A metabolomic analysis in a neuronal cell culture also showed that α1-AR stimulation results in lower levels of carbohydrates (Wenner et al., 2016). These results are consistent with other studies in the α1B-AR KO mice which displayed insulin resistance and dysfunctional glucose homeostasis (Burcelin et al., 2004) and the use of prazosin treatment, an α1-AR antagonist, which increases risk of metabolic syndrome and high fasting plasma glucose levels in patients with benign prostatic hyperplasia (Lee et al., 2013). The mechanism of the increase in glucose tolerance and lowering of plasma glucose levels is likely due to the increased utilization of glucose through uptake and oxidation in various organs.
α1-AR Mediated Glucose Oxidation in the Heart
α1-AR stimulation can also directly increase glucose oxidation in both normal and ischemic primary adult myocytes performed by measuring the rate of 14C-CO2 production using 14C-glucose as a substrate (Papay and Perez, 2020). This study confirmed that the glucose uptake into the heart also drives the oxidation of glucose for energy utilization to the heart. Stimulation of glucose oxidation in the heart improves the recovery from damage during ischemia (Dyck et al., 2006; Ussher et al., 2012; Masoud et al., 2014; Li Y. et al., 2017). Ischemia in the heart can increase glucose uptake by increasing the translocation of GLUT 1/4 (Egert et al., 1999), as this was also shown to be mediated by the α1A-AR (Shi et al., 2016). The α1-AR mediated glucose oxidation in primary myocytes was also blocked by PKC and AMPK inhibitors (Papay and Perez, 2020) consistent with the role of PKCδ in translocating the glucose transporters in the heart by the α1A-AR (Shi et al., 2016). α1-AR stimulation increased glucose uptake in the L6 skeletal muscle cell line also through an AMPK pathway (Hutchinson and Bengtsson, 2006). AMPK is an energy sensor that can regulate the rate of glucose and fatty acid uptake and oxidation according to the needs of the cell. AMPK signaling is cardioprotective during heart failure by switching the energy production in the heart from fatty acid oxidation to glucose oxidation (Kim et al., 2012). AMPK also can increase glucose uptake during ischemia to prevent post-ischemic cardiac damage and dysfunction (Russell et al., 2004; Kim et al., 2011). While α1A-AR mediated ischemic preconditioning was mediated through PKC (Rorabaugh et al., 2005), PKC was also shown to mediate its protection against ischemic damage through AMPK (Wang et al., 2011). These results suggest that glucose uptake and subsequent oxidation in the heart may be α1A-AR specific, signal through PKC/AMPK activation and may mediate α1A-AR’s cardioprotective effects during ischemia and heart failure.
α1-AR Mediated Glucose Metabolism in Other Tissues
α1-ARs are the main receptors that regulate the control of hepatic glucose metabolism in mice (Chu et al., 2000; Miyamoto et al., 2012; de Oliveira et al., 2013). α1-AR stimulation increased glucose uptake into L6 muscle cells (Hutchinson and Bengtsson, 2005, 2006) and C2C12 skeletal myoblasts (Liu et al., 2001). α1-AR stimulation also increases glucose uptake into brown and white adipocytes (Faintrenie and Géloën, 1998; Cheng et al., 2000; Boschmann et al., 2002; Flechtner-Mors et al., 2002, 2004; Chernogubova et al., 2005). The sympathetic nervous system enhances glucose uptake into human adipocytes independently of insulin action through the α1-AR (Flechtner-Mors et al., 2002, 2004; McCarty, 2004). In obese people that have insulin resistance, α1-AR stimulation may provide a critical alternative pathway for glucose uptake.
α1-ARs Mediated Fatty Acid Oxidation
The KO and transgenic mice of the α1-AR subtypes were used to discern effects of the specific subtypes on general whole-body metabolism. Systemically expressing CAM mice were assessed by indirect calorimetry and found that both CAM α1A- and α1B-AR mice decreased the respiratory exchange ratio (RER) (ratio of CO2 production and O2 consumption) which indicated an increase in whole body preference to metabolize fatty acids as a substrate (i.e., fatty acid oxidation) while the KO mice from both subtypes preferred to burn carbohydrates and increased the RER (Shi et al., 2017). It is likely that α1-AR stimulation increases fatty acid oxidation in the skeletal muscle as that muscle utilizes 40–50% of a body’s whole energy metabolism. While there is a report that prazosin can increase angiogenesis in skeletal muscle resulting in increased capillarization to improve the diffusion of glucose into the muscle and may increase glucose oxidation due to substrate availability (Akerstrom et al., 2014), prazosin’s effect was due to improved blood flow and not to GLUT 1/4 translocation.
Both systemically expressing CAM α1A- and α1B-AR mice displayed increased plasma levels of leptin while KO mice decreased leptin levels (Shi et al., 2017). In obese humans, α1-AR blockade reduces leptin levels (Ihara et al., 2006). While leptin can also directly increase glucose oxidation in the absence of insulin in skeletal muscle through a neural hypothalamic β-AR mechanism (Nevzorova et al., 2006; Glund et al., 2007; Shiuchi et al., 2009; Minokoshi et al., 2012; Cadaret et al., 2017), leptin mainly increases fatty acid oxidation in skeletal muscle and the liver through α1-AR stimulation of AMPK activity (Minokoshi et al., 2002, 2012; Miyamoto et al., 2012).
α1-ARs can also couple to peroxisome proliferator-activated receptor-delta (PPARs) to regulate fatty acid oxidation and utilization (Tanaka et al., 2003). PPAR subtypes β/δ are nuclear receptors and serve as sensors of fatty acid levels. They bind and are activated by fatty acids and their derivatives and activate transcription factors to regulate metabolism (Poulsen et al., 2012). Using midodrine to non-selectively stimulate α1-ARs, α1-ARs activated PPARs and AMPK to increase oxidative phosphorylation in rat skeletal muscle or in C2C12 skeletal muscle cells (Lee et al., 2020). PPARs are crucial to maintain normal cardiac function and its energy requirements. Cardiac-targeted KO of PPARδ decreases basal fatty acid oxidation leading to cardiac dysfunction, lipid accumulation and heart failure (Cheng et al., 2004). Overexpression of a CAM PPARβ/δ leads to increased levels of fatty acid oxidation (Barak et al., 2002).
Tissue transglutaminase (TG2) is an ubiquitous and multi-functional protein and enzyme with regulatory crosslinking functions in cell adhesion and the cytoskeleton but also has GTP hydrolyzing activities (Fesus and Piacentini, 2002; Eckert et al., 2014). Phenylephrine, an α1-AR non-selective agonist was injected into TG2 KO mice and resulted in a lowering of the RER indicating that the mice were burning more whole-body fatty acids than glucose when compared to normal mice with intact TG2 (Lénárt et al., 2020). α1-AR stimulation also resulted in lower organ damage particularly in the heart but also in the lung, liver, kidney, and skeletal muscle and a weaker vasoconstriction response compared to normal mice (Lénárt et al., 2020). When the same mice were given a β3-AR agonist, the RER was lowered and organ damage was changed to the same extent in both TG2 KO or normal mice (Lénárt et al., 2020). A β3 agonist lowers the RER because of its high density in adipose tissue (Ferrer-Lorente et al., 2005). These results concur with the whole-body indirect calorimetry studies that showed that the systemically expressing CAM α1-AR mice burned more fatty acids (Shi et al., 2017) and protected the heart from ischemic damage (Rorabaugh et al., 2005; Shi et al., 2016). TG2 is a protein ubiquitously found in cells and can function in both protein cross-linking and bind GTP to act as a G-protein transducer at α1-ARs (Nakaoka et al., 1994; Baek et al., 1996; Feng et al., 1996; Kang et al., 2004).
α1-AR stimulation can increase the rate of lipolysis in obese individuals (Flechtner-Mors et al., 2002) increasing the availability of fatty acids. α1-ARs stimulation also increase fatty acid oxidation in the liver or in hepatocytes (Sugden et al., 1980; Kosugi et al., 1983; Oberhaensli et al., 1985; de Oliveira et al., 2013) and during a high-fat diet can reduce hepatic steatosis (i.e., fatty liver disease) (Nakade et al., 2020). Using a metabolomic analysis, the α1A-AR selective agonist, A61603, produced a reduction in cardiac polyunsaturated fatty acids (Willis et al., 2016). The systemically expressed CAM α1A-AR mice displayed significantly decreased fasting plasma triglycerides while α1A-AR KO displayed increased levels of triglycerides (Shi et al., 2017). In contrast, α1-AR blockers such as prazosin or doxazosin have been reported to lower triglycerides and cholesterol but increase high density lipoproteins in humans (Ferrara et al., 1986; Weinberger, 1986; Trost et al., 1987). The reason for this discordance is unknown. However, α1-AR quinazoline-based antagonists and particularly prazosin and doxazosin have known non-α1-AR mediated off-target effects (Benning and Kyprianou, 2002; Lin et al., 2007; Isgor and Isgor, 2012).
Pharmacological Interventions
Development of α1-AR Subtype-Selective Ligands
Development of selective α1-AR subtype ligands has not been a focus in the pharmaceutical industry because of the ALLHAT Collaborative Research Group (2000) clinical trials and the major cardiovascular events that occur when α1-AR antagonists are used. There are still no selective blockers or agonists for the α1B-AR, and while BMY 7378 is somewhat selective for the α1D-AR (Goetz et al., 1995), there is no clear clinical target. α1A-AR antagonists have fared better in drug development because they target prostate and lower urinary tract problems which often affect men with increasing age and who also have high blood pressure; thus, tackling two problems with one therapeutic (Van Asseldonk et al., 2015). However, these therapeutics, as are all α1-AR antagonists, are contraindicated in people with heart problems (O’Connell et al., 2013). Recent studies also suggest that α1-AR antagonists increase mortality rates in hospitalized patients with Covid-19 (Rose et al., 2020).
The above review indicates that the α1A-AR subtype may be a target for drug development for cardioprotection and cognitive enhancement in dementia-type diseases. The potential for α1A-AR agonists to be used to treat these diseases has a major problematic side effect of increasing blood pressure (Woo and Lee, 1999; Wier and Morgan, 2003; Villalba et al., 2007; Gutiérrez et al., 2019). This drawback has limited the development of α1-AR-based therapeutics by pharmaceutical companies (Fordyce et al., 2015). However, there are two avenues of development that are recently being used to circumvent the blood pressure effect of α1A-AR agonists. The first one is the use of the imidazoline pharmacophore instead of the endogenous phenethylamine pharmacophore that is possessed by norepinephrine, epinephrine and several other α1-AR agonists (Figure 1).
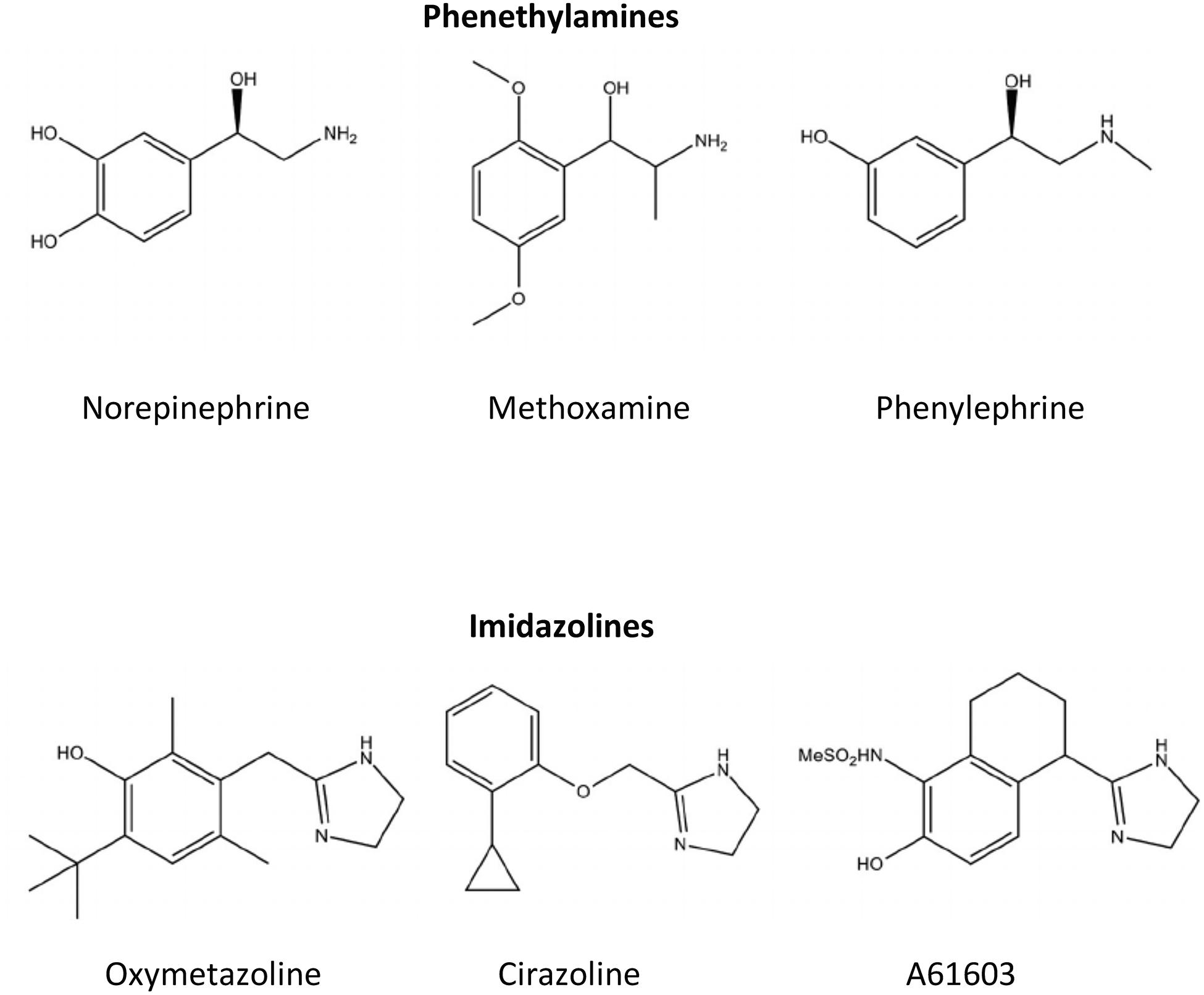
Figure 1. Chemical structures for common α1-AR agonists. Ligands such as norepinephrine that contain a phenyl, two-carbon ethyl, and amine group in their structural backbone are referred to as phenethylamines. Ligands that contain an imidazole ring in its structural backbone instead of the phenyl ring are referred to as imidazolines.
Imidazolines
In general, imidazolines have better binding and functional agonistic selectivity for α2-ARs and reduce blood pressure by decreasing norepinephrine release at the α2A-AR autoreceptor (Ruffolo et al., 1983). However, in the early days of α1-AR agonist drug development, it was noted imidazolines interacted with the α1-ARs in a different way structurally than with α2-ARs. The Easson-Stedman hypothesis states that adrenergic agonists that are chiral by possessing an asymmetric hydroxyl-substituted benzylic carbon atom will have higher binding affinity and potency for the R(–) (i.e., right hand) isomer when compared to the S(+) (i.e., desoxy) isomer (Easson and Stedman, 1933). Imidazoline binding to α1-ARs did not adhere to the Easson-Stedman hypothesis that held with phenethylamines, such as norepinephrine (Patil et al., 1974; Ruffolo et al., 1980, 1983; Hieble et al., 1986). While most imidazolines that selectively bind to the α2-AR are agonists, they become weak antagonists at the α1-AR (Ruffolo and Waddell, 1982). During drug development, specific substitutions off the imidazoline pharmacophore can convert imidazolines from α2-AR agonists to α1-AR agonists (Ruffolo et al., 1980; Hieble et al., 1986; Knepper et al., 1995). Furthermore, subsequent studies indicated that imidazolines that had higher affinity for the α1-AR than the α2-AR had agonist-selectivity for the α1A-AR subtype in both binding affinity and function compared to the other two α1-AR subtypes, the α1B- or α1D-AR (Minneman et al., 1994). Structure-function analysis revealed that imidazolines, while agonists at the α1A-AR, interact with amino acid residues closer to the cell surface in the α1A-AR binding pocket, similar to α1-AR antagonists, confirming the differences seen with the Easson-Stedman hypothesis (Waugh et al., 2001). These differences in binding also explained why most imidazolines are partial and not full agonists at the α1-ARs.
There are several commercially available imidazolines, such as cirazoline and A61603, that are selective for the α1A-AR versus the α1B- and α1D-AR subtypes and with lower affinity against the α2-AR. An analog of cirazoline and an α1A-AR partial agonist, RO 115–1240 and later by the commercial product dabuzalgron, was shown to reduce stress urinary incontinence without increasing blood pressure (Blue et al., 2004; Musselman et al., 2004). The therapeutic index is wide enough that R0 115–1240 can contract bladder smooth muscle at a much lower dose than required to contract vascular smooth muscle by the α1A-AR. This is possible because of the higher receptor density of the α1A-AR in the urinary tract compared with vascular smooth muscle and its partial agonist activity that allows reflex mechanisms to control changes in blood pressure (Ford et al., 1996; Walden et al., 1997; Kava et al., 1998; Musselman et al., 2004; Michel and Vrydag, 2006). While all of the above are indeed possible mechanisms for α1A-AR agonists to avoid increasing blood pressure, imidazolines were subsequently shown to have bias signaling or agonist trafficking which can lead to lower efficacy of the signaling pathways known to increase blood pressure. Imidazolines induce a more robust cAMP signaling response versus the inositol phosphate signal which increases calcium release to cause the vascular smooth muscle contraction (Evans et al., 2011; da Silva et al., 2017). Confirming the role of α1A-AR-selective imidazolines in cardioprotection, dabuzalgron was shown to protect against cardiac damage induced by doxorubicin (Beak et al., 2017; Montgomery et al., 2017) and A61603 increased inotropy in a mouse model of right heart failure (Cowley et al., 2015), but blood pressure was not assessed at the dosage used in these experiments. Confirming the role of α1A-AR-selective imidazolines in enhancing cognition, cirazoline, which crosses the blood brain barrier, was shown to increase cognition in normal mice (Doze et al., 2011).
Allosteric Modulators
A second avenue of drug development for α1A-AR agonists with a wide therapeutic index to avoid increases in blood pressure are allosteric modulators. Allosteric modulators offer greater selectivity in both binding and signaling than conventional ligands which bind to the natural endogenous site on the receptor (i.e., orthosteric) (Christopoulos, 2002). Besides greater selectivity because they bind in a different place than orthosteric agonists that is non-conserved between subtypes of the receptor, allosteric modulators offer many other benefits in therapeutics. These are the saturability of its binding site (i.e., ceiling effect) and conformational or probe bias that can alter the receptor to induce a bias in signaling and activation properties but only when the receptor is already occupied with a specific ligand or probe (Christopoulos, 2002).
Allosteric modulators are classified by their ability to modulate function. Positive allosteric modulators (PAMs) increase a receptor’s functional response while negative allosteric modulators (NAMs) decrease the functional response. There are also neutral or silent allosteric modulators (SAMs) that bind to the receptor and display no measurable changes in function (Lindsley et al., 2016) but can block the effects of PAMs or NAMS (Rodriguez et al., 2005). There are now many GPCR allosteric modulators that have been developed (Chen et al., 2008; Wold et al., 2019; Zhou and Cunningham, 2019; Fasciani et al., 2020) with several in clinical trials or with FDA approval (Wold et al., 2019). The HIV entry inhibitor maraviroc is the most known clinically used GPCR allosteric modulator against the CCR5 receptor (Maeda et al., 2012).
There are a few NAMs that have been characterized for the α1-AR but have not been developed for clinical use (Leppik et al., 2000; Sharpe et al., 2003; Chen et al., 2004; Lima et al., 2005; Ragnarsson et al., 2013; Campbell et al., 2017). We have developed the first PAM of the α1-ARs with selectivity at the α1A-AR subtype. It has the imidazoline pharmacophore and can cross the blood brain barrier in sufficient concentration to cause neurological effects without increased blood pressure (Perez, 2021). We have demonstrated its ability to significantly increase LTP in a mouse model of AD along with increases in cognitive behavior using the Barnes maze and fear-conditioning tests. This was achieved using a 10-month dosing scheme and studies are underway to test effects of this compound in a dose-efficacy preclinical trial for 3 months (Perez, 2021).
Therapeutic Autoantibodies and Vaccines Against α1-ARs
There has been recent work in therapeutic vaccines directed at the α1-AR subtypes and their roles in hypertension and cardiovascular disease. Autoantibodies against the α1-ARs were first found in patients over 20 years ago with severe hypertension (Fu et al., 1994; Luther et al., 1997; Wenzel et al., 2008). A vaccine made against the second extracellular loop of the α1D-AR was found to have antagonistic behavior (Li et al., 2019). The vaccine was injected into spontaneously hypertensive rats (SHR) with or without pre-treatment with NG-nitro-L-arginine methyl ester (L-NAME) to generate NO and to reduce blood pressure (Li et al., 2019). This α1D-AR vaccine reduced the systolic blood pressure up to 15 mmHg in the SHR group and up to 29 mmHG in the SHR + L-NAME group. This vaccine also prevented cardiac hypertrophy and fibrosis, vascular remodeling, and renal injury even better than compared to treatment with prazosin, suggesting that the antibody has blocking activity. There is one commercially available α1D-AR antagonist, BMY7378 (Goetz et al., 1995), but is not sufficiently selective to avoid blocking the other α1-AR subtypes for therapeutic use. Because of the unique amino acid sequence used in a non-conserved region of the second extracellular loop of the receptor, vaccines against the α1D-AR subtype would be highly selective and avid to regulate the blood pressure response and avoid blocking the other α1-AR subtypes.
However, the vast majority of autoantibodies are associated with agonistic activity resulting from a rise in intracellular calcium, and postulated to result in a vasoconstrictive effect (Bkaily et al., 2003; Karczewski et al., 2010; Yan et al., 2014). However, one controlled clinical study indicated that hypertensive patients with α1-AR autoantibodies displayed normal cardiovascular responses to α1-AR stimulation and removal of α1-AR autoantibodies by immunoadsorption did not alter that response (Schroeder et al., 2012).
While the autoantibody against the α1D-AR appears antagonistic, several autoantibodies have been developed or discovered against the first or second extracellular loop of the α1-AR appear to be agonistic in behavior (Zhou et al., 2008; Karczewski et al., 2012; Hempel et al., 2016; Wallukat et al., 2020). While developing these autoantibodies for cardioprotective effects for the α1A-AR may be tempting, they may not be regulated by the normal desensitization and negative feedback mechanisms common in GPCRs to turn off or wane the signal, resulting in abnormal and non-physiological signaling and proliferation (Zhou et al., 2008; Karczewski et al., 2018; Becker et al., 2019; Wallukat et al., 2020). This abnormal signaling and proliferation may account for the vascular damage that many autoantibodies also impart (Zhou et al., 2008; Karczewski et al., 2012, 2018; Becker et al., 2019; Wallukat et al., 2020). Autoantibodies against the α1-AR have also been associated with coronary heart disease (Thyrian et al., 2018), cardiac remodeling and dysfunction (Zhou et al., 2005; Li T. et al., 2017), pre-eclampsia (Ma et al., 2013), thromboangiitis obliterans (Buerger’s Disease) (Klein-Weigel et al., 2014), AD and vascular dementia (Karczewski et al., 2012, 2018; Hempel et al., 2016), and prostate cancer (Wallukat et al., 2020). Therefore, both agonistic and antagonistic autoantibodies against the α1-AR subtypes would need to be thoroughly analyzed for off target effects.
Summary
α1-ARs are part of the adrenergic family of sympathetic control and have long been known to regulate blood pressure, smooth muscle contraction and cardiac hypertrophy. In recent work, α1A-AR stimulation also mediates adaptive effects and signals in the heart that lead to protective outcomes against ischemia and heart failure. They are also highly expressed in the cognitive centers of the brain and stimulation of α1-ARs, particularly the α1A-AR, can increase both short-term as well as LTP leading to increased learning and memory functions. With its ability to increase adult neurogenesis, there is a potential for α1A-AR agonists or positive allosteric modulators to treat AD and to protect the heart at the same time. α1-AR stimulation also mediates several aspects of whole-body and organ-specific metabolism to regulate glucose uptake, gluconeogenesis, glucose breakdown, lipolysis, and fatty acid oxidation for energy production. The regulation of cardiac metabolism by the α1A-AR is likely a contributing factor for its protective effects in the heart. For pharmacological interventions, it is suggested that therapeutics that focus on α1A-AR agonism be developed. To avoid the potential side effects on blood pressure, the imidazoline rather than the phenethylamine pharmacophore should be of primary focus for drug discovery. Several α1A-AR imidazoline-based agonists have been used in preclinical studies and allosteric agonists that will not increase blood pressure are now in development for heart failure and AD.
Author Contributions
DP performed all of the research, writing, and editing of this manuscript.
Funding
This manuscript was funded through grants from the National Institute on Aging (RO1-AG066627) and The Edward N. and Della L. Thome Memorial Foundation (award Program in Alzheimer’s Drug Discovery Research).
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
- ^ The α1C-AR was first designated novel but more detailed analysis revealed that it was a bovine analog of the α1A-AR (Schwinn et al., 1990; Laz et al., 1994; Perez et al., 1994). To avoid confusion, an actual novel α1-AR subtype discovered through molecular cloning was designated the α1D-AR (Perez et al., 1991) and the α1C designation was dropped from the classification scheme.
References
Aggerbeck, M., Guellaen, G., and Hanoune, J. (1980). Adrenergic receptor of the alpha1-subtype mediates the activation of the glycogen phosphorylase in normal rat liver. Biochem. Pharmacol. 29, 643–645. doi: 10.1016/0006-2952(80)90389-5
Ahrén, B., and Lundquist, I. (1987). Alpha-adrenoceptor blockade by phentolamine inhibits beta-adrenergically and cholinergically induced glucagon secretion in the mouse. Horm. Metab. Res. 19, 600–603. doi: 10.1055/s-2007-1011889
Akerstrom, T., Laub, L., Vedel, K., Brand, C. L., Pedersen, B. K., Lindqvist, A. K., et al. (2014). Increased skeletal muscle capillarization enhances insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 307, E1105–E1116.
Akhter, S. A., Milano, C. A., Shotwell, K. F., Cho, M. C., Rockman, H. A., Lefkowitz, R. J., et al. (1997). Transgenic mice with cardiac overexpression of α1B-adrenergic receptors. In vivo α1-adrenergic receptor-mediated regulation of β-adrenergic signaling. J. Biol. Chem. 272, 21253–21259.
Akinaga, J., Lima, V., Kiguti, L. R., Hebeler-Barbosa, F., Alcántara-Hernández, R., García-Sáinz, J. A., et al. (2013). Differential phosphorylation, desensitization, and internalization of α1A-adrenoceptors activated by norepinephrine and oxymetazoline. Mol. Pharmacol. 83, 870–881. doi: 10.1124/mol.112.082313
ALLHAT Collaborative Research Group (2000). Major cardiovascular events in hypertensive patients randomized to doxazosin vs chlorthalidone: the antihypertensive and lipid-lowering treatment to prevent heart attack trial (ALLHAT). ALLHAT collaborative research group. JAMA 283, 1967–1975. doi: 10.1001/jama.283.15.1967
Anand, A., Andrade, C., Sudha, S., Guido, S., and Venkataraman, B. V. (2001). Phenylephrine and ECS-induced retrograde amnesia. J. ECT 17, 166–169. doi: 10.1097/00124509-200109000-00003
Arnsten, A. F., and Jentsch, J. D. (1997). The α1-adrenergic agonist, cirazoline, impairs spatial working memory performance in aged monkeys. Pharmacol. Biochem. Behav. 58, 55–59. doi: 10.1016/s0091-3057(96)00477-7
Assimacopoulos-Jeannet, F. D., Blackmore, P. F., and Exton, J. H. (1977). Studies on alpha-adrenergic activation of hepatic glucose output. Studies on role of calcium in alpha-adrenergic activation of phosphorylase. J. Biol. Chem. 252, 2662–2669. doi: 10.1016/s0021-9258(17)40509-6
Auclair, A., Drouin, C., Cotecchia, S., Glowinski, J., and Tassin, J. P. (2004). 5-HT2A and alpha1b-adrenergic receptors entirely mediate dopamine release, locomotor response and behavioural sensitization to opiates and psychostimulants. Eur. J. Neurosci. 20, 3073–3084. doi: 10.1111/j.1460-9568.2004.03805.x
Autelitano, D. J., and Woodcock, E. A. (1998). Selective activation of alpha1A-adrenergic receptors in neonatal cardiac myocytes is sufficient to cause hypertrophy and differential regulation of alpha1-adrenergic receptor subtype mRNAs. J. Mol. Cell Cardiol. 30, 1515–1523. doi: 10.1006/jmcc.1998.0717
Azami, N. S., Piri, M., Oryan, S., Jahanshahi, M., Babapour, V., and Zarrindast, M. R. (2010). Involvement of dorsal hippocampal alpha-adrenergic receptors in the effect of scopolamine on memory retrieval in inhibitory avoidance task. Neurobiol. Learn. Mem. 93, 455–462. doi: 10.1016/j.nlm.2010.01.003
Baek, K. J., Das, T., Gray, C. D., Desai, S., Hwang, K. C., Gacchui, R., et al. (1996). A 50 KDa protein modulates guanine nucleotide binding of transglutaminase II. Biochemistry 35, 2651–2657. doi: 10.1021/bi9522965
Ballou, L. M., Tian, P. Y., Lin, H. Y., Jiang, Y. P., and Lin, R. Z. (2001). Dual regulation of glycogen synthase kinase-3beta by the alpha1A-adrenergic receptor. J. Biol. Chem. 276, 40910–40916.
Banerjee, A., Locke-Winter, C., Rogers, K. B., Mitchell, M. B., Brew, E. C., Cairnes, C. B., et al. (1993). Preconditioning against myocardial dysfunction after ischemia and reperfusion by an α1-adrenergic mechanism. Circ. Res. 73, 656–670. doi: 10.1161/01.res.73.4.656
Bañuelos, C., Beas, B. S., McQuail, J. A., Gilbert, R. J., Frazier, C. J., Setlow, B., et al. (2014). Prefrontal cortical GABAergic dysfunction contributes to age-related working memory impairment. J. Neurosci. 34, 3457–3466. doi: 10.1523/jneurosci.5192-13.2014
Barak, Y., Liao, D., He, W., Ong, E. S., Nelson, M. C., Olefsky, J. M., et al. (2002). Effects of peroxisome proliferator-activated receptor delta on placentation, adiposity, and colorectal cancer. Proc. Natl. Acad. Sci. U.S.A. 99, 303–308. doi: 10.1073/pnas.012610299
Beak, J., Huang, W., Parker, J. S., Hicks, S. T., Patterson, C., et al. (2017). An oral selective alpha-1A adrenergic receptor agonist prevents doxorubicin cardiotoxicity. JACC Basic Transl. Sci. 2, 39–53. doi: 10.1016/j.jacbts.2016.10.006
Becker, N. P., Goettel, P., Mueller, J., Wallukat, G., and Schimke, I. (2019). Functional autoantibody diseases: basics and treatment related to cardiomyopathies. Front. Biosci. 24:14.
Benning, C. M., and Kyprianou, N. (2002). Quinazoline-derived alpha1-adrenoceptor antagonists induce prostate cancer cell apoptosis via an alpha1-adrenoceptor-independent action. Cancer Res. 62, 597–602.
Berridge, C., Shumsky, J. S., Andrzejewski, M. E., McGaughy, J., Spencer, R. C., Devilbiss, D., et al. (2012). Differential sensitivity to psychostimulants across prefrontal cognitive tasks: differential involvement of Noradrenergic α1- vs. α2-Receptors. Biol. Psychiatry 71, 467–473. doi: 10.1016/j.biopsych.2011.07.022
Berridge, C. W., and Spencer, R. C. (2016). Differential cognitive actions of norepinephrine α2 and α1 receptor signaling in the prefrontal cortex. Brain Res. 1641(Pt B), 189–196. doi: 10.1016/j.brainres.2015.11.024
Bhardwaj, S. K., Tse, Y. C., Ryan, R., Wong, T. P., and Srivastava, L. K. (2014). Impaired adrenergic-mediated plasticity of prefrontal cortical glutamate synapses in rats with developmental disruption of the ventral hippocampus. Neuropsychopharmacology 39, 2963–2973. doi: 10.1038/npp.2014.142
Bkaily, G., El-Bizri, N., Bui, M., Sukarieh, R., Jacques, D., and Fu, M. L. (2003). Modulation of intracellular Ca2+ via L-type calcium channels in heart cells by the autoantibody directed against the second extracellular loop of the alpha1-adrenoceptors. Can. J. Physiol. Pharmacol. 81, 234–246. doi: 10.1139/y03-044
Bliss, T. V., and Collingridge, G. L. (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39. doi: 10.1038/361031a0
Blue, D. R., Daniels, D. V., Gever, J. R., Jett, M. F., O’Yang, C., Tang, H. M., et al. (2004). Pharmacological characteristics of Ro 115-1240, a selective α1A/L-adrenoceptor partial agonist: a potential therapy for stress urinary incontinence. BJU Int. 93, 162–170. doi: 10.1111/j.1464-410x.2004.04577.x
Böhmer, T., Pfeiffer, N., and Gericke, A. (2014). Three commercial antibodies against α1-adrenergic receptor subtypes lack specificity in paraffin-embedded sections of murine tissues. Naunyn. Schmiedebergs Arch. Pharmacol. 387, 703–706. doi: 10.1007/s00210-014-0992-2
Boschmann, M., Krupp, G., Luft, F. C., Klaus, S., and Jordan, J. (2002). In vivo response to alpha(1)-adrenoreceptor stimulation in human white adipose tissue. Obes. Res. 10, 555–558. doi: 10.1038/oby.2002.75
Bristow, M. R., Ginsburg, R., Minobe, W., Cubicciotti, R. S., Sageman, W. S., Lurie, K., et al. (1982). Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N. Engl. J. Med. 307, 205–211. doi: 10.1056/nejm198207223070401
Bristow, M. R., Ginsburg, R., Umans, V., Fowler, M., Minobe, W., Rasmussen, R., et al. (1986). β1- and β2-adrenergic-receptor subpopulations in nonfailing and failing human ventricular myocardium: coupling of both receptor subtypes to muscle contraction and selective β1-receptor down-regulation in heart failure. Circ. Res. 59, 297–309. doi: 10.1161/01.res.59.3.297
Bristow, M. R., Minobe, W., Rasmussen, R., Hershberger, R. E., and Hoffman, B. B. (1988). Alpha-1 adrenergic receptors in the nonfailing and failing human heart. J. Pharmacol. Exp. Ther. 247, 1039–1045.
Brown, A. S. (2002). Consolidation theory and retrograde amnesia in humans. Psychonom. Bull. Rev. 9, 403–425. doi: 10.3758/bf03196300
Burcelin, R., Uldry, M., Foretz, M., Perrin, C., Dacosta, A., Nenniger-Tosato, M., et al. (2004). Impaired glucose homeostasis in mice lacking the α1b-adrenergic receptor subtype. J. Biol. Chem. 279, 1108–1115. doi: 10.1074/jbc.m307788200
Cadaret, C. N., Beede, K. A., Riley, H. E., and Yates, D. T. (2017). Acute exposure of primary rat soleus muscle to zilpaterol HCl (β2 adrenergic agonist), TNFα, or IL-6 in culture increases glucose oxidation rates independent of the impact on insulin signaling or glucose uptake. Cytokine 96, 107–113. doi: 10.1016/j.cyto.2017.03.014
Campbell, A. P., Wakelin, L. P., Denny, W. A., and Finch, A. M. (2017). Homobivalent conjugation increases the allosteric effect of 9-aminoacridine at the α1-adrenergic receptors. Mol. Pharmacol. 91, 135–144. doi: 10.1124/mol.116.105874
Castaldi, A., Zaglia, T., Di Mauro, V., Carullo, P., Viggiani, G., Borile, G., et al. (2014). MicroRNA-133 modulates the β1-adrenergic receptor transduction cascade. Circ. Res. 115, 273–283. doi: 10.1161/circresaha.115.303252
Cavalli, A., Lattion, A. L., Hummler, E., Nenniger, M., Pedrazzini, T., Aubert, J. F., et al. (1997). Decreased blood pressure response in mice deficient of the alpha1b-adrenergic receptor. Proc. Natl. Acad. Sci. U.S.A. 94, 11589–11594. doi: 10.1073/pnas.94.21.11589
Chalothorn, D., McCune, D. F., Edelmann, S. E., Tobita, K., Keller, B. B., Lasley, R. D., et al. (2003). Differential cardiovascular regulatory activities of the α1B- and α1D-adrenoceptor subtypes. J. Pharmacol. Exp. Ther. 305, 1045–1053. doi: 10.1124/jpet.102.048553
Chan, T. M., and Exton, J. H. (1978). Studies on α-adrenergic activation of hepatic glucose output. Studies on α-adrenergic inhibition of hepatic pyruvate kinase and activation of gluconeogenesis. J. Biol. Chem. 253, 6393–6400. doi: 10.1016/s0021-9258(19)46946-9
Chaulet, H., Lin, F., Guo, J., Owens, W. A., Michalicek, J., Kesteven, S. H., et al. (2006). Sustained augmentation of cardiac alpha1A-adrenergic drive results in pathological remodeling with contractile dysfunction, progressive fibrosis and reactivation of matricellular protein genes. J. Mol. Cell Cardiol. 40, 540–552. doi: 10.1016/j.yjmcc.2006.01.015
Chen, Q., Li, D. P., and Pan, H. L. (2006). Presynaptic alpha1-adrenergic receptors differentially regulate synaptic glutamate and GABA release to hypothalamic presympathetic neurons. J. Pharmacol. Exp. Ther. 316, 733–742. doi: 10.1124/jpet.105.094797
Chen, Y., Goudet, C., Pin, J. P., and Conn, P. J. (2008). N-{4-Chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl) methyl]phenyl}-2-hydroxybenzamide (CPPHA) acts through a novel site as a positive allosteric modulator of group 1 metabotropic glutamate receptors. Mol. Pharmacol. 73, 909–918. doi: 10.1124/mol.107.040097
Chen, Z., Rogge, G., Hague, C., Alewood, D., Colless, B., Lewis, R. J., et al. (2004). Subtype-selective noncompetitive or competitive inhibition of human alpha1-adrenergic receptors by rho-TIA. J. Biol. Chem. 279, 35326–35333. doi: 10.1074/jbc.m403703200
Cheng, J. -T., Liu, I. -M., Yen, S. -T., and Chen, P. -C. (2000). Role of α1A-adrenoceptor in the regulation of glucose uptake into white adipocyte of rats in vitro. Auton. Neurosci. 84, 140–146. doi: 10.1016/s1566-0702(00)00197-1
Cheng, L., Ding, G., Qin, Q., Huang, Y., Lewis, W., He, N., et al. (2004). Cardiomyocyte-restricted peroxisome proliferator-activated receptor-δ deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat. Med. 10, 1245–1250. doi: 10.1038/nm1116
Chernogubova, E., Hutchinson, D. S., Nedergaard, J., and Bengtsson, T. (2005). Alpha1- and beta1-adrenoceptor signaling fully compensates for beta3-adrenoceptor deficiency in brown adipocyte norepinephrine-stimulated glucose uptake. Endocrinology 146, 2271–2284. doi: 10.1210/en.2004-1104
Chobanian, A. V., Bakris, G. L., Black, H. R., Cushman, W. C., Green, L. A., and Izzo, J. L. Jr., et al. (2003). The seventh report of the joint national committee on prevention, detection, evaluation, and treatment of high blood pressure: the JNC 7 report. JAMA 289, 2560–2571. doi: 10.1001/jama.289.19.2560
Christensen, K. L., and Mulvany, M. J. (1993). Mesenteric arcade arteries contribute substantially to vascular resistance in conscious rats. J. Vasc. Res. 30, 73–79. doi: 10.1159/000158978
Christopoulos, A. (2002). Allosteric binding sites on cell-surface receptors: novel targets for drug discovery. Nat. Rev. Drug Discov. 1, 198–210. doi: 10.1038/nrd746
Chu, C. A., Sindelar, D. K., Igawa, K., Sherck, S., Neal, D. W., Emshwiller, M., et al. (2000). The direct effects of catecholamines on hepatic glucose production occur via alpha(1)- and beta(2)-receptors in the dog. Am. J. Physiol. 279, E463–E473.
Collette, K., Fagerlie, R., Haselton, J., Perez, D. M., and Doze, V. (2010). Norepinephrine, through activation of α1A-ARs, stimulates production of new neurons, leading to an alleviation of depression and anxiety. FASEB J. 24, 1058–1057.
Collette, K. M., Zhou, X. D., Amoth, H. M., Lyons, M. J., Papay, R. S., Sens, D. A., et al. (2014). Long-term α1B-adrenergic receptor activation shortens lifespan, while α1A-adrenergic receptor stimulation prolongs lifespan in association with decreased cancer incidence. Age 36:9675.
Corr, P. B., Shayman, J. A., Kramer, J. B., and Kipnis, R. J. (1981). Increased α-adrenergic receptors in ischemic cat myocardium: a potential mediator of electrophysiological derangements. J. Clin. Invest. 67, 1232–1236. doi: 10.1172/jci110139
Cotecchia, S., Del Vescovo, C. D., Colella, M., Caso, S., and Diviani, D. (2015). The alpha1-adrenergic receptors in cardiac hypertrophy: signaling mechanisms and functional implications. Cell Signal. 27, 1984–1993. doi: 10.1016/j.cellsig.2015.06.009
Cotecchia, S., Schwinn, D. A., Randall, R. R., Lefkowitz, R. J., Caron, M. G., and Kobilka, B. K. (1988). Molecular cloning and expression of the cDNA for the hamster alpha 1-adrenergic receptor. Proc. Natl. Acad. Sci. U.S.A. 85, 7159–7163. doi: 10.1073/pnas.85.19.7159
Cowley, P. M., Wang, G., Chang, A. N., Makwana, O., Swigart, P. M., Lovett, D. H., et al. (2015). The alpha1A-adrenergic receptor subtype mediates increased contraction of failing right ventricular myocardium. Am. J. Physiol. Heart Circ. Physiol. 309, H888–H896.
Cunningham, M. L., Waldo, G. L., Hollinger, S., Hepler, J. R., and Harden, T. K. (2001). Protein kinase C phosphorylates RGS2 and modulates its capacity for negative regulation of Galpha 11 signaling. J. Biol. Chem. 276, 5438–5444. doi: 10.1074/jbc.m007699200
da Silva, E. D., Sato, M., Merlin, J., Broxton, N., Hutchinson, D. S., Ventura, S., et al. (2017). Factors influencing biased agonism in recombinant cells expressing the human α1A-adrenoceptor. Br. J. Pharmacol. 174, 2318–2333. doi: 10.1111/bph.13837
de Oliveira, A. L., de Paula, M. N., Comar, J. F., Vilela, V. R., Peralta, R. M., and Bracht, A. (2013). Adrenergic metabolic and hemodynamic effects of octopamine in the liver. Int. J. Mol. Sci. 14, 21858–21872. doi: 10.3390/ijms141121858
Dileepan, K. N., Khawaja, A. M., and Wagle, S. R. (1982). Studies on the mechanism of action of somatostatin on renal gluconeogenesis: evidence for the involvement of alpha 1-adrenergic stimuli. Arch. Biochem. Biophys. 213, 169–176. doi: 10.1016/0003-9861(82)90452-0
Dileepan, K. N., and Wagle, S. R. (1985). Somatostatin: a metabolic regulator. Life Sci. 37, 2335–2343. doi: 10.1016/0024-3205(85)90100-6
Doenst, T., and Taegtmeyer, H. (1999). α-adrenergic stimulation mediates glucose uptake through phosphatidylinositol 3-kinase in rat heart. Circ. Res. 84, 467–474. doi: 10.1161/01.res.84.4.467
Domyancic, A. V., and Morilak, D. A. (1997). Distribution of alpha1A adrenergic receptor mRNA in the rat brain visualized by in situ hybridization. J. Comp. Neurol. 386, 358–378. doi: 10.1002/(sici)1096-9861(19970929)386:3<358::aid-cne3>3.0.co;2-0
Downey, J. M., and Cohen, M. V. (1997). Signal transduction in ischemic preconditioning. Adv. Exp. Med. Biol. 430, 39–55.
Doze, V. A., Papay, R. S., Goldenstein, B. L., Gupta, M. K., Collette, K. M., Nelson, B. W., et al. (2011). Long-term α1A-adrenergic receptor stimulation improves synaptic plasticity, cognitive function, mood, and longevity. Mol. Pharmacol. 80, 747–758. doi: 10.1124/mol.111.073734
Drouin, C., Darracq, L., Trovero, F., Blanc, G., Glowinski, J., Cotecchia, S., et al. (2002). Alpha1b-adrenergic receptors control locomotor and rewarding effects of psychostimulants and opiates. J. Neurosci. 22, 2873–2884. doi: 10.1523/jneurosci.22-07-02873.2002
Du, X. J., Fang, L., Gao, X. M., Kiriazis, H., Feng, X., Hotchkin, E., et al. (2004). Genetic enhancement of ventricular contractility protects against pressure-overload-induced cardiac dysfunction. J. Mol. Cell Cardiol. 37, 979–987. doi: 10.1016/j.yjmcc.2004.07.010
Du, X. J., Gao, X. M., Kiriazis, H., Moore, X. L., Ming, Z., Su, Y., et al. (2006). Transgenic alpha1A-adrenergic activation limits post-infarct ventricular remodeling and dysfunction and improves survival. Cardiovasc. Res. 71, 735–743. doi: 10.1016/j.cardiores.2006.06.015
Duteil, J., Rambert, F. A., Pessonnier, J., Hermant, J. F., Gombert, R., and Assous, E. (1990). Central alpha 1-adrenergic stimulation in relation to the behaviour stimulating effect of modafinil; studies with experimental animals. Eur. J. Pharmacol. 180, 49–58. doi: 10.1016/0014-2999(90)90591-s
Dyck, J. R., Hopkins, T. A., Bonnet, S., Michelakis, E. D., Young, M. E., Watanabe, M., et al. (2006). Absence of malonyl coenzyme A decarboxylase in mice increases cardiac glucose oxidation and protects the heart from ischemic injury. Circulation 114, 1721–1728. doi: 10.1161/circulationaha.106.642009
Dyer-Reaves, K., Goodman, A. M., Nelson, A. R., and McMahon, L. L. (2019). Alpha1-adrenergic receptor mediated long-term depression at CA3-CA1 synapses can be induced viaaccumulation of endogenous norepinephrine and is preserved following noradrenergic denervation. Front. Synaptic 11:27. doi: 10.3389/fnsyn.2019.00027
Easson, L. H., and Stedman, E. (1933). Studies on the relationship between chemical constitution and physiological action: molecular dissymmetry and physiological activity. Biochem. J. 27, 1257–1266. doi: 10.1042/bj0271257
Eckert, R. L., Kaartinen, M. T., Nurminskaya, M., Belkin, A. M., Colak, G., Johnson, G. V., et al. (2014). Transglutaminase regulation of cell function. Physiol. Rev. 94, 383–417. doi: 10.1152/physrev.00019.2013
Eckhart, A. D., Duncan, S. J., Penn, R. B., Benovic, J. L., Lefkowitz, R. J., and Koch, W. J. (2000). Hybrid transgenic mice reveal in vivo specificity of G protein-coupled receptor kinases in the heart. Circ. Res. 86, 43–50. doi: 10.1161/01.res.86.1.43
Egert, S., Nguyen, N., and Schwaiger, M. (1999). Contribution of alpha-adrenergic and beta-adrenergic stimulation to ischemia-induced glucose transporter (GLUT) 4 and GLUT1 translocation in the isolated perfused rat heart. Circ. Res. 84, 1407–1415. doi: 10.1161/01.res.84.12.1407
El-Brolosy, M. A., and Stainier, D. (2017). Genetic compensation: a phenomenon in search of mechanisms. PLoS Genet. 13:e1006780. doi: 10.1371/journal.pgen.1006780
Endoh, M. (2016). Cardiac α1-adrenoceptors and inotropy: myofilament Ca2+ sensitivity, intracellular Ca2+ mobilization, signaling pathway, and pathophysiological relevance. Circ. Res. 119, 587–590. doi: 10.1161/circresaha.116.309502
Endoh, M., Hiramoto, T., Ishihata, A., Takanashi, M., and Inui, J. (1991). Myocardial alpha 1-adrenoceptors mediate positive inotropic effect and changes in phosphatidylinositol metabolism. Species differences in receptor distribution and the intracellular coupling process in mammalian ventricular myocardium. Circ. Res. 68, 1179–1190. doi: 10.1161/01.res.68.5.1179
Enomoto, T., Tse, M. T., and Floresco, S. B. (2011). Reducing prefrontal gamma-aminobutyric acid activity induces cognitive, behavioral, and dopaminergic abnormalities that resemble schizophrenia. Biol. Psychiatry 69, 432–441. doi: 10.1016/j.biopsych.2010.09.038
Evans, B. A., Broxton, N., Merlin, J., Sato, M., Hutchinson, D. S., Christopoulos, A., et al. (2011). Quantification of functional selectivity at the human α1A-adrenoceptor. Mol. Pharmacol. 79, 298–307. doi: 10.1124/mol.110.067454
Faintrenie, G., and Géloën, A. (1998). Alpha-1 adrenergic stimulation of glucose uptake in rat white adipocytes. J. Pharmacol. Exp. Ther. 1286, 607–610.
Fasciani, I., Petragnano, F., Aloisi, G., Marampon, F., Carli, M., Scarselli, M., et al. (2020). Allosteric modulators of G protein-coupled dopamine and serotonin receptors: a new class of atypical antipsychotics. Pharmaceuticals. 13:388. doi: 10.3390/ph13110388
Feng, J. F., Rhee, S. G., and Im, M. J. (1996). Evidence that phospholipase delta-1 is the effector in the Gh (transglutaminase II)-mediated signalling. J. Biol. Chem. 271, 16451–16454. doi: 10.1074/jbc.271.28.16451
Ferrara, L. A., Marotta, T., Rubba, P., de Simone, B., Leccia, G., Son, S., et al. (1986). Effects of alpha-adrenergic and beta-adrenergic receptor blockade on lipid metabolism. Am. J. Med. 80(Suppl. 2A), 104–108. doi: 10.1016/0002-9343(86)90168-3
Ferrer-Lorente, R., Cabot, C., Fernández-López, J. A., and Alemany, M. (2005). Combined effects of oleoyl-estrone and a beta3-adrenergic agonist (CL316,243) on lipid stores of diet-induced overweight male Wistar rats. Life Sci. 77, 2051–2058. doi: 10.1016/j.lfs.2005.04.008
Ferry, B., and McGaugh, J. L. (2000). Role of amygdala norepinephrine in mediating stress hormone regulation of memory storage. Acta Pharmacol. Sin. 21, 481–493.
Ferry, B., and Quirarte, G. (2012). “Role of norepinephrine in mediating inhibitory avoidance memory storage: a critical involvement of the basolateral amygdala,” in The Amygdala: A Discrete Multitasking Manager, 980-953-307-188-1, ed. B. Ferry (London: In Tech – Open Science), 203–230.
Ferry, B., Roozendaal, B., and McGaugh, J. L. (1999a). Basolateral amygdala noradrenergic influences on memory storage are mediated by an interaction between beta- and alpha1-adrenoceptors. J. Neurosci. 19, 5119–5123. doi: 10.1523/jneurosci.19-12-05119.1999
Ferry, B., Roozendaal, B., and McGaugh, J. L. (1999b). Involvement of α1-adrenergic receptors in the basolateral amygdala in modulation of memory storage. Eur. J. Pharmacol. 372, 9–16. doi: 10.1016/s0014-2999(99)00169-7
Fesus, L., and Piacentini, M. (2002). Transglutaminase 2: an enigmatic enzyme with diverse functions. Trends Biochem. Sci. 27, 534–539. doi: 10.1016/s0968-0004(02)02182-5
Fischer, V., Gabauer, I., Tillinger, A., Novakova, M., Pechan, I., Krizanova, O., et al. (2008). Heart adrenoceptor gene expression and binding sites in the human failing heart. Ann. N. Y. Acad. Sci. 1148, 400–408. doi: 10.1196/annals.1410.013
Flechtner-Mors, M., Jenkinson, C. P., Alt, A., Biesalski, H. K., Adler, G., and Ditschuneit, H. H. (2004). Sympathetic regulation of glucose uptake by the alpha1-adrenoceptor in human obesity. Obes. Res. 12, 612–620. doi: 10.1038/oby.2004.70
Flechtner-Mors, M., Jenkinson, C. P., Alt, A., Adler, G., and Ditschuneit, H. H. (2002). In vivo alpha (1)-adrenergic lipolytic activity in subcutaneous adipose tissue of obese subjects. J. Pharmacol. Exp. Ther. 301, 229–233. doi: 10.1124/jpet.301.1.229
Ford, A. P., Arredondo, N. F., Blue, D. R. Jr., Bonhaus, D. W., Jasper, J., Kava, M. S., et al. (1996). RS-17053 (N-[2-(2-cyclopropylmethoxyphenoxy) ethyl]-5-chloro-alpha, alpha-dimethyl-1H-indole-3-ethanamine hydrochloride), a selective alpha 1A-adrenoceptor antagonist, displays low affinity for functional alpha 1-adrenoceptors in human prostate: implications for adrenoceptor classification. Mol. Pharmacol. 492, 209–215.
Fordyce, C. B., Roe, M. T., Ahmad, T., Libby, P., Borer, J. S., Hiatt, W. R., et al. (2015). Cardiovascular drug development: is it dead or just hibernating? J. Am. Coll. Cardiol. 65, 1567–1582.
Foster, T. C., and McNaughton, B. L. (1991). Long-term synaptic enhancement in CA1 is due to increased quanta1 size, not quanta1 content. Hippocampus 1, 79–79. doi: 10.1002/hipo.450010108
Fu, M. L., Herlitz, H., Wallukat, G., Hilme, E., Hedner, T., Hoebeke, J., et al. (1994). Functional autoimmune epitope on alpha 1-adrenergic receptors in patients with malignant hypertension. Lancet 344, 1660–1663. doi: 10.1016/s0140-6736(94)90456-1
Fuller, S. J., Gaitanaki, C. J., and Sugden, P. H. (1990). Effects of catecholamines on protein synthesis in cardiac myocytes and perfused hearts isolated from adult rats. Stimulation of translation is mediated through the alpha 1-adrenoceptor. Biochem. J. 266, 727–736. doi: 10.1042/bj2660727
Gao, H., Chen, L., and Yang, H.-T. (2007). Activation of α1B-adrenoceptors alleviates ischemia/reperfusion injury by limitation of mitochondrial Ca2+ overload in cardiomyocytes. Cardiovasc. Res. 75, 584–595. doi: 10.1016/j.cardiores.2007.04.008
Gao, X. M., Wang, B. H., Woodcock, E., and Du, X. J. (2000). Expression of active alpha(1B)-adrenergic receptors in the heart does not alleviate ischemic reperfusion injury. J. Mol. Cell Cardiol. 32, 1679–1686. doi: 10.1006/jmcc.2000.1201
García-Sáinz, J. A., and Hernández-Sotomayor, S. M. (1985). Adrenergic regulation of gluconeogenesis: possible involvement of two mechanisms of signal transduction in alpha 1-adrenergic action. Proc. Natl. Acad. Sci. U.S.A. 82, 6727–6730. doi: 10.1073/pnas.82.20.6727
Gazarini, L., Stern, C. A., Carobrez, A. P., and Bertoglio, L. J. (2013). Enhanced noradrenergic activity potentiates fear memory consolidation and reconsolidation by differentially recruiting α1- and β-adrenergic receptors. Learn. Mem. 20, 210–219. doi: 10.1101/lm.030007.112
Gibbs, M. E., and Bowser, D. N. (2010). Astrocytic adrenoceptors and learning: alpha1-adrenoceptors. Neurochem. Int. 57, 404–410. doi: 10.1016/j.neuint.2010.03.020
Gibbs, M. E., and Summers, R. J. (2001). Stimulation of α1-adrenoceptors inhibits memory consolidation in the chick. Eur. J. Neurosci. 14, 1369–1376. doi: 10.1046/j.0953-816x.2001.01742.x
Gibbs, M. E., Hutchinson, D., and Hertz, L. (2008). Astrocytic involvement in learning and memory consolidation. Neurosci. Biobehav. Rev. 32, 927–944.
Gisbert, R., Ziani, K., Miquel, R., Noguera, M. A., Ivorra, M. D., Anselmi, E., et al. (2002). Pathological role of a constitutively active population of alpha (1D)-adrenoceptors in arteries of spontaneously hypertensive rats. Br. J. Pharmacol. 135, 206–216. doi: 10.1038/sj.bjp.0704447
Glund, S., Deshmukh, A., Long, Y. C., Moller, T., Koistinen, H. A., Caidahl, K., et al. (2007). Interleukin-6 directly increases glucose metabolism in resting human skeletal muscle. Diabetes Metab. Res. Rev. 56, 1630–1637. doi: 10.2337/db06-1733
Goetz, A. S., King, H. K., Ward, S. D., True, T. A., Rimele, T. J., and Saussy, D. L. Jr. (1995). BMY 7378 is a selective antagonist of the D subtype of alpha 1-adrenoceptors. Eur. J. Pharmacol. 272, R5–R6.
Graham, R. M., and Lanier, S. M. (1986). “Identification and characterization of alpha- adrenergic receptors,” in The Heart, and Cardiovascular System, eds H. A. Fozzard et al. (New York, NY: Raven Press), 1059–1095.
Grupp, I. L., Lorenz, J. N., Walsh, R. A., Boivin, G. P., and Rindt, H. (1998). Overexpression of α1B-adrenergic receptor induces left ventricular dysfunction in the absence of hypertrophy. Am. J. Physiol. 275(Pt 2), H1338– H1350.
Gupta, M. K., Papay, R. S., Jurgens, C. W., Gaivin, R. J., Shi, T., Doze, V. A., et al. (2009). α1-Adrenergic receptors regulate neurogenesis and gliogenesis. Mol. Pharmacol. 76, 314–326. doi: 10.1124/mol.109.057307
Gutiérrez, A., Contreras, C., Sánchez, A., and Prieto, D. (2019). Role of phosphatidylinositol 3-kinase (PI3K), mitogen-activated protein kinase (MAPK), and protein kinase C (PKC) in calcium signaling pathways linked to the α1-adrenoceptor in resistance arteries. Front. Physiol. 10:55. doi: 10.3389/fphys.2019.00055
Hague, C., Bernstein, L. S., Ramineni, S., Chen, Z., Minneman, K. P., and Hepler, J. R. (2005). Selective inhibition of α1A-adrenergic receptor signaling by RGS2 association with the receptor third intracellular loop. J. Biol. Chem. 280, 27289–27295. doi: 10.1074/jbc.m502365200
Harasawa, I., Honda, K., Tanoue, A., Shinoura, H., Ishida, Y., Okamura, H., et al. (2003). Responses to noxious stimuli in mice lacking α1D-adrenergic receptors. Neuroreport 14, 1857–1860. doi: 10.1097/00001756-200310060-00020
Hempel, P., Heinig, B., Jerosch, C., Decius, I., Karczewski, P., Kassner, U., et al. (2016). Immunoadsorption of agonistic autoantibodies against α1-adrenergic receptors in patients with mild to moderate dementia. Ther. Apher. Dial. 20, 523–529. doi: 10.1111/1744-9987.12415
Heynen, A. J., Abraham, W. C., and Bear, M. F. (1996). Bidirectional modification of CA1 synapses in the adult hippocampus in vivo. Nature 381, 163–166. doi: 10.1038/381163a0
Hieble, J. P., DeMarinis, R. M., and Matthews, W. D. (1986). Evidence for and against heterogeneity of α1-adrenoceptors. Life Sci. 38, 1339–1350. doi: 10.1016/0024-3205(86)90466-2
Hilfiker, S., and Augustine, G. J. (1999). Regulation of synaptic vesicle fusion by protein kinase C. J. Physiol. 515:1. doi: 10.1111/j.1469-7793.1999.001ad.x
Hillman, K. L., Knudson, C. A., Carr, P. A., Doze, V. A., and Porter, J. E. (2005). Adrenergic receptor characterization of CA1 hippocampal neurons using real time single cell RT-PCR. Brain Res. Mol. Brain Res. 139, 267–276. doi: 10.1016/j.molbrainres.2005.05.033
Hong, C. J., Wang, Y. C., Liu, T. Y., Liu, H. C., and Tsai, S. J. (2001). A study of α-adrenoceptor gene polymorphisms and Alzheimer disease. J. Neural Transm. 108, 445–450. doi: 10.1007/s007020170065
Hopkins, W. F., and Johnston, D. (1984). Frequency-dependent noradrenergic modulation of long-term potentiation in the hippocampus. Science 226, 350–352. doi: 10.1126/science.6091272
Hosoda, C., Koshimizu, T. A., Tanoue, A., Nasa, Y., Oikawa, R., Tomabechi, T., et al. (2005). Two alpha1-adrenergic receptor subtypes regulating the vasopressor response have differential roles in blood pressure regulation. Mol. Pharmacol. 67, 912–922. doi: 10.1124/mol.104.007500
Hrometz, S. L., Edelmann, S. E., McCune, D. F., Olges, J. R., Hadley, R. W., Perez, D. M., et al. (1999). Expression of multiple α1-adrenoceptors on vascular smooth muscle: correlation with the regulation of contraction. J. Pharmacol. Exp. Ther. 290, 452–463.
Hu, K., and Nattel, S. (1995). Mechanisms of ischemic preconditioning in rat hearts. Involvement of alpha 1B-adrenoceptors, pertussis toxin-sensitive G proteins, and protein kinase C. Circulation 92, 2259–2265. doi: 10.1161/01.cir.92.8.2259
Hue, L., Felíu, J. E., and Hers, H. G. (1978). Control of gluconeogenesis and of enzymes of glycogen metabolism in isolated rat hepatocytes. A parallel study of the effect of phenylephrine and of glucagon. Biochem. J. 176, 791–797. doi: 10.1042/bj1760791
Hussain, M. B., and Marshall, I. (1997). Characterization of alpha1-adrenoceptor subtypes mediating contractions to phenylephrine in rat thoracic aorta, mesenteric artery and pulmonary artery. Br. J. Pharmacol. 122, 849–858. doi: 10.1038/sj.bjp.0701461
Hutchinson, D. S., and Bengtsson, T. (2005). α1A-adrenoceptors activate glucose uptake in L6 muscle cells through a phospholipase C-, phosphatidylinositol-3 kinase-, and atypical protein kinase C-dependent pathway. Endocrinology 146, 901–912. doi: 10.1210/en.2004-1083
Hutchinson, D. S., and Bengtsson, T. (2006). AMP-activated protein kinase activation by adrenoceptors in L6 skeletal muscle cells: mediation by alpha1-adrenoceptors causing glucose uptake. Diabetes Metab. Res. Rev. 55, 682–690. doi: 10.2337/diabetes.55.03.06.db05-0901
Hvoslef-Eide, M., Oomen, C. A., Fisher, B. M., Heath, C. J., Robbins, T. W., Saksida, L. M., et al. (2015). Facilitation of spatial working memory performance following intra-prefrontal cortical administration of the adrenergic alpha1 agonist phenylephrine. Psychopharmacology 232, 4005–4016. doi: 10.1007/s00213-015-4038-3
Ihara, S., Shimamoto, K., Watanabe, H., Sakai, R., and Kawana, M. (2006). An alpha1-receptor blocker reduces plasma leptin levels in hypertensive patients with obesity and hyperleptinemia. Hypertens. Res. 29, 805–811. doi: 10.1291/hypres.29.805
Ikeda, U., Tsuruya, Y., and Yaginuma, T. (1991). Alpha 1-adrenergic stimulation is coupled to cardiac myocyte hypertrophy. Am. J. Physiol. 260, H953–H956.
Ikegami, M., Ikeda, H., Ishikawa, Y., Ohsawa, M., Ohashi, T., Kai, M., et al. (2013a). Olanzapine induces glucose intolerance through the activation of AMPK in the mouse hypothalamus. Eur. J. Pharmacol. 718, 376–382. doi: 10.1016/j.ejphar.2013.08.006
Ikegami, M., Ikeda, H., Ohashi, T., Kai, M., Osada, M., Kamei, A., et al. (2013b). Olanzapine-induced hyperglycemia: possible involvement of histaminergic, dopaminergic and adrenergic functions in the central nervous system. Neuroendocrinology 98, 224–232. doi: 10.1159/000356119
Ikegaya, Y., Nakanishi, K., Saito, H., and Abe, K. (1997). Amygdala beta-noradrenergic influence on hippocampal long-term potentiation in vivo. Neuroreport 8, 3143–3146. doi: 10.1097/00001756-199709290-00027
Isgor, B. S., and Isgor, Y. G. (2012). Effect of alpha-1-adrenoceptor blocker on cytosolic enzyme targets for potential use in cancer chemotherapy. Int. J. Pharmacol. 8, 333–343. doi: 10.3923/ijp.2012.333.343
Izumi, Y., and Zorumski, C. F. (1999). Norepinephrine promotes long-term potentiation in the adult rat hippocampus in vitro. Synapse 31, 196–202. doi: 10.1002/(sici)1098-2396(19990301)31:3<196::aid-syn4>3.0.co;2-k
Janssen, P. M. L., Canan, B. D., Kilic, A., Whitson, B. A., and Baker, A. J. (2018). Human myocardium has a robust α1A-subtype adrenergic receptor inotropic response. J. Cardiovasc. Pharmacol. 72, 136–142. doi: 10.1097/fjc.0000000000000604
Jensen, B. C., Swigart, P. M., De Marco, T., Hoopes, C., and Simpson, P. C. (2009a). Alpha1-Adrenergic receptor subtypes in nonfailing and failing human myocardium. Circ. Heart Fail. 2, 654–663. doi: 10.1161/circheartfailure.108.846212
Jensen, B. C., Swigart, P. M., Laden, M. E., DeMarco, T., Hoopes, C., and Simpson, P. C. (2009b). The alpha-1D Is the predominant alpha-1-adrenergic receptor subtype in human epicardial coronary arteries. J. Am. Coll. Cardiol. 54, 1137–1145. doi: 10.1016/j.jacc.2009.05.056
Jensen, B. C., Swigart, P. M., and Simpson, P. C. (2009c). Ten commercial antibodies for alpha1-adrenergic receptor subtypes are nonspecific. Naunyn. Schmiedebergs. Arch. Pharmacol. 379, 409–412. doi: 10.1007/s00210-008-0368-6
Jurgens, C. W. D., Knudson, C. A., Carr, P. A., Perez, D. M., and Doze, V. A. (2009). α1-Adrenergic receptor regulation of interneuron function. FASEB J. 23:946.4.
Kalaria, R. N. (1989). Characterization of [125I]-HEAT binding to alpha 1-receptors in human brain: assessment in aging and Alzheimer’s disease. Brain Res. 501, 287–294. doi: 10.1016/0006-8993(89)90645-8
Kandel, E. R. (2001). The molecular biology of memory storage: a dialogue between genes and synapses. Science 294, 1030–1038. doi: 10.1126/science.1067020
Kang, S. K., Yi, K. S., Kwon, N. S., Park, K. H., Kim, U. H., Baek, K. J., et al. (2004). Alpha1B-adrenoceptor signaling and cell motility: GTPase function of Gh/transglutaminase 2 inhibits cell migration through interaction with cytoplasmic tail of integrin alpha subunits. J. Biol. Chem. 279, 36593–36600.
Karczewski, P., Haase, H., Hempel, P., and Bimmler, M. (2010). Agonistic antibody to the alpha1-adrenergic receptor mobilizes intracellular calcium and induces phosphorylation of a cardiac 15-kDa protein. Mol. Cell. Biochem. 333, 233–242. doi: 10.1007/s11010-009-0224-0
Karczewski, P., Hempel, P., and Bimmler, M. (2018). Role of alpha1-adrenergic receptor antibodies in Alzheimer’s disease. Front. Biosci. 23:2082–2089. doi: 10.2741/4691
Karczewski, P., Hempel, P., Kunze, R., and Bimmler, M. (2012). Agonistic autoantibodies to the α(1) -adrenergic receptor and the β(2) -adrenergic receptor in Alzheimer’s and vascular dementia. Scand. J. Immunol. 75, 524–530. doi: 10.1111/j.1365-3083.2012.02684.x
Katsuki, H., Izumi, Y., and Zorumski, C. F. (1997). Noradrenergic regulation of synaptic plasticity in the hippocampal CA1 region. J. Neurophysiol. 77, 3013–3020. doi: 10.1152/jn.1997.77.6.3013
Kava, M. S., Blue, D. R. Jr., Vimont, R., Clarke, D. E., and Ford, A. P. D. W. (1998). α1L-Adrenoceptor mediation of smooth muscle contraction in rabbit bladder neck: a model for lower urinary tract tissues of man. Br. J. Pharmacol. 123, 1359–1366. doi: 10.1038/sj.bjp.0701748
Kemp, A., and Manahan-Vaughan, D. (2004). Hippocampal long-term depression and long-term potentiation encode different aspects of novelty acquisition. Proc. Natl. Acad. Sci. U.S.A. 101, 8192–8197. doi: 10.1073/pnas.0402650101
Kim, A. S., Miller, E. J., Wright, T. M., Li, J., Qi, D., Atsina, K., et al. (2011). A small molecule AMPK activator protects the heart against ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 51, 24–32. doi: 10.1016/j.yjmcc.2011.03.003
Kim, M., Shen, M., Ngoy, S., Karamanlidis, G., Liao, R., and Tian, R. (2012). AMPK isoform expression in the normal and failing hearts. J. Mol. Cell. Cardiol. 52, 1066–1073. doi: 10.1016/j.yjmcc.2012.01.016
Kitakaze, M., Hori, M., Morioka, T., Minamino, T., Takashima, S., Sato, H., et al. (1994). Alpha 1-adrenoceptor activation mediates the infarct size-limiting effect of ischemic preconditioning through augmentation of 5’-nucleotidase activity. J. Clin. Invest. 93, 2197–2205. doi: 10.1172/jci117216
Klein-Weigel, P. F., Bimmler, M., Hempel, P., Schöpp, S., Dreusicke, S., Valerius, J., et al. (2014). G-protein coupled receptor auto-antibodies in thromboangiitis obliterans (Buerger’s disease) and their removal by immunoadsorption. Vasa 43, 347–352. doi: 10.1024/0301-1526/a000372
Knauber, J., and Müller, W. E. (2000a). Decreased exploratory activity and impaired passive avoidance behaviour in mice deficient for the α1b-adrenoceptor. Eur. Neuropsychopharmacol. 10, 423–427. doi: 10.1016/s0924-977x(00)00100-0
Knauber, J., and Müller, W. E. (2000b). Subchronic treatment with prazosin improves passive avoidance learning in aged mice: possible relationships to alpha1-receptor up-regulation. J. Neural. Transm. 107, 1413–1426. doi: 10.1007/s007020070005
Knepper, S. M., Buckner, S. A., Brune, M. E., DeBernardis, J. F., Meyer, M. D., and Hancock, A. A. (1995). A-61603, a potent α1-adrenergic receptor agonist, selective for the α1A receptor subtype. J. Pharmacol. Exp. Ther. 274, 97–10.
Kobayashi, M., Sasabe, T., Shiohama, Y., and Koshikawa, N. (2008). Activation of alpha1-adrenoceptors increases firing frequency through protein kinase C in pyramidal neurons of rat visual cortex. Neurosci. Lett. 430, 175–180. doi: 10.1016/j.neulet.2007.10.047
Kosugi, K., Harano, Y., Nakano, T., Suzuki, M., Kashiwagi, A., and Shigeta, Y. (1983). Mechanism of adrenergic stimulation of hepatic ketogenesis. Metabolism 32, 1081–1087. doi: 10.1016/0026-0495(83)90081-1
Kurz, T., Yamada, K. A., DaTorre, S. D., and Corr, P. B. (1991). Alpha 1-adrenergic system and arrhythmias in ischaemic heart disease. Eur. Heart J. 12(Suppl. F), 88–98. doi: 10.1093/eurheartj/12.suppl_f.88
Lapiz, M. D., and Morilak, D. A. (2006). Noradrenergic modulation of cognitive function in rat medial prefrontal cortex as measured by attentional set shifting capability. Neuroscience 137, 1039–1049. doi: 10.1016/j.neuroscience.2005.09.031
Laz, T. M., Forray, C., Smith, K. E., Bard, J. A., Vaysse, P. J., Branchek, T. A., et al. (1994). The rat homologue of the bovine alpha 1c-adrenergic receptor shows the pharmacological properties of the classical alpha 1A subtype. Mol. Pharmacol. 46, 414–422.
Lee, Y. C., Liu, C. C., Juan, Y. S., Wu, W. J., Li, W. M., Yeh, H. C., et al. (2013). The impact of metabolic syndrome on the responsiveness to α1-blocker in men with BPH/LUTS. Int. J. Clin. Pract. 67, 356–362. doi: 10.1111/ijcp.12086
Lee, Y.-J., Kim, H. S., Seo, H. S., Na, J. O., Jang, Y.-N., Han, Y.-M., et al. (2020). Stimulation of Alpha1-Adrenergic receptor ameliorates cellular functions of multiorgans beyond vasomotion through PPARδ. PPAR Res. 2020:3785137.
Lemire, I., Ducharme, A., Tardif, J. C., Poulin, F., Jones, L. R., Allen, B. G., et al. (2001). Cardiac-directed overexpression of wild-type alpha1B-adrenergic receptor induces dilated cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 281, H931–H938.
Lénárt, K., Pap, A., Pórszász, R., Oláh, V. A., Fésüs, L., and Mádi, A. (2020). Transglutaminase 2 has metabolic and vascular regulatory functions revealed by in vivo activation of alpha1-adrenergic receptor. Int. J. Mol. Sci. 21:3865. doi: 10.3390/ijms21113865
Leppik, R. A., Mynett, A., Lazareno, S., and Birdsall, N. J. (2000). Allosteric interactions between the antagonist prazosin and amiloride analogs at the human α1A-adrenergic receptor. Mol. Pharmacol. 57, 436–445. doi: 10.1124/mol.57.3.436
Li, C., Yan, X., Wu, D., Zhang, K., Liang, X., Pan, Y., et al. (2019). Vaccine targeted alpha 1D-adrenergic receptor for hypertension. Hypertension 74, 1551–1562. doi: 10.1161/hypertensionaha.119.13700
Li, T., Xu, J., Qin, X., Hou, Z., Guo, Y., Liu, Z., et al. (2017). Glucose oxidation positively regulates glucose uptake and improves cardiac function recovery after myocardial reperfusion. Am. J. Physiol. Endocrinol. Metab. 313, E577–E585.
Li, Y., Tian, J., Ma, X. R., Li, R. T., Zhang, S. L., Wang, P. L., et al. (2017). Increase in G protein-coupled receptor autoantibodies with decline of cardiac function in hypercholesterolemic rats. Eur. Rev. Med. Pharmacol. Sci. 21, 1065–1073.
Lima, V., Mueller, A., Kamikihara, S. Y., Raymundi, V., Alewood, D., Lewis, R. J., et al. (2005). Differential antagonism by conotoxin rho-TIA of contractions mediated by distinct alpha1-adrenoceptor subtypes in rat vas deferens, spleen and aorta. Eur. J. Pharmacol. 508, 183–192. doi: 10.1016/j.ejphar.2004.12.011
Limas, C. J., Limas, C., and Goldenberg, I. F. (1989). Intracellular distribution of adrenoceptors in the failing human myocardium. Am. Heart J. 117, 1310–1316. doi: 10.1016/0002-8703(89)90411-0
Lin, F., Owens, W. A., Chen, S., Stevens, M. E., Kesteven, S., Arthur, J. F., et al. (2001). Targeted alpha(1A)-adrenergic receptor overexpression induces enhanced cardiac contractility but not hypertrophy. Circ. Res. 89, 343–350. doi: 10.1161/hh1601.095912
Lin, S. C., Chueh, S. C., Hsiao, C. J., Li, T. K., Chen, T. H., Liao, C. H., et al. (2007). Prazosin displays anticancer activity against human prostate cancers: targeting DNA and cell cycle. Neoplasia 9, 830–839. doi: 10.1593/neo.07475
Lindsley, C. W., Emmitte, K. A., Hopkins, C. R., Bridges, T. M., Gregory, K. J., Niswender, C. M., et al. (2016). Practical strategies and concepts in GPCR allosteric modulator discovery: recent advances with metabotropic glutamate receptors. Chem. Rev. 116, 6707–6741. doi: 10.1021/acs.chemrev.5b00656
Liu, I. M., Tsai, C. C., Lai, T. Y., and Cheng, J. T. (2001). Stimulatory effect of isoferulic acid on α1A-adrenoceptor to increase glucose uptake into cultured myoblast C2C12 cells of mice. Auton. Neurosci. 88, 175–180. doi: 10.1016/s1566-0702(01)00241-7
Lomasney, J. W., Cotecchia, S., Lorenz, W., Leung, W. Y., Schwinn, D. A., Yang-Feng, T. L., et al. (1991). Molecular cloning and expression of the cDNA for the alpha 1Aadrenergic receptor. The gene for which is located on human chromosome 5. J. Biol. Chem. 266, 6365–6369. doi: 10.1016/s0021-9258(18)38126-2
Luo, D. L., Gao, J., Fan, L. L., Tang, Y., Zhang, Y. Y., and Han, Q. D. (2007). Receptor subtype involved in alpha 1-adrenergic receptor-mediated Ca2+ signaling in cardiomyocytes. Acta. Pharmacol. Sin. 28, 968–974. doi: 10.1111/j.1745-7254.2007.00605.x
Luo, F., Li, S. H., Tang, H., Deng, W. K., Zhang, Y., and Liu, Y. (2015a). Phenylephrine enhances glutamate release in the medial prefrontal cortex through interaction with N-type Ca2+ channels and release machinery. J. Neurochem. 132, 38–50. doi: 10.1111/jnc.12941
Luo, F., Tang, H., and Cheng, Z. Y. (2015b). Stimulation of α1-adrenoceptors facilitates GABAergic transmission onto pyramidal neurons in the medial prefrontal cortex. Neuroscience 300, 63–74. doi: 10.1016/j.neuroscience.2015.04.070
Luo, F., Tang, H., Li, B. M., and Li, S. H. (2014). Activation of alpha1-adrenoceptors enhances excitatory synaptic transmission via a pre- and postsynaptic protein kinase C-dependent mechanism in the medial prefrontal cortex of rats. Eur. J. Neurosci. 39, 1281–1293. doi: 10.1111/ejn.12495
Luther, H. P., Homuth, V., and Wallukat, G. (1997). Alpha 1-adrenergic receptor antibodies in patients with primary hypertension. Hypertension 29, 678–682. doi: 10.1161/01.hyp.29.2.678
Lv, J., Zhan, S. Y., Li, G. X., Wang, D., Li, Y. S., and Jin, Q. H. (2016). α1-Adrenoceptors in the hippocampal dentate gyrus involved in learning-dependent long-term potentiation during active-avoidance learning in rats. Neuroreport 27, 1211–1216. doi: 10.1097/wnr.0000000000000679
Ma, G., Li, Y., Zhang, J., Liu, H., Hou, D., Zhu, L., et al. (2013). Association between the presence of autoantibodies against adrenoreceptors and severe pre-eclampsia: a pilot study. PLoS One 8:e57983. doi: 10.1371/journal.pone.0057983
Maeda, K., Das, D., Nakata, H., and Mitsuya, H. (2012). CCR5 inhibitors: emergence, success, and challenges. Expert Opin. Emerg. Drugs 17, 135–145. doi: 10.1517/14728214.2012.673584
Mahmoodi, G., Ahmadi, S., Pourmotabbed, A., Oryan, S., and Zarrindast, M. R. (2010). Inhibitory avoidance memory deficit induced by scopolamine: Interaction of cholinergic and glutamatergic systems in the ventral tegmental area. Neurobiol. Learn. Mem. 94, 83–90. doi: 10.1016/j.nlm.2010.04.004
Maisel, A. S., Motulsky, H. J., Ziegler, M. G., and Insel, P. A. (1987). Ischemia- and agonist-induced changes in alpha- and beta-adrenergic receptor traffic in guinea pig hearts. Am. J. Physiol. 253(5 Pt 2), H1159–H1166.
Manahan-Vaughan, D., and Braunewell, K. H. (1999). Novelty acquisition is associated with induction of hippocampal long-term depression. Proc. Natl. Acad. Sci. U.S.A. 96, 8739–8744. doi: 10.1073/pnas.96.15.8739
Mao, Z. M., Arnsten, A. F., and Li, B. M. (1999). Local infusion of an α1-adrenergic agonist into the prefrontal cortex impairs spatial working memory performance in monkeys. Biol. Psychiatry 46, 1259–1265. doi: 10.1016/s0006-3223(99)00139-0
Marek, G. J., and Aghajanian, G. K. (1996). Alpha1B-adrenoceptor-mediated excitation of piriform cortical interneurons. Eur. J. Pharmacol. 305, 95–100. doi: 10.1016/0014-2999(96)00158-6
Marek, G. J., and Aghajanian, G. K. (1999). 5-HT2A receptor or α1-adrenoceptor activation induces excitatory postsynaptic currents in layer V pyramidal cells of the medial prefrontal cortex. Eur. J. Pharmacol. 367, 197–206. doi: 10.1016/s0014-2999(98)00945-5
Martí, D., Miquel, R., Ziani, K., Gisbert, R., Ivorra, M. D., Anselmi, E., et al. (2005). Correlation between mRNA levels and functional role of alpha1-adrenoceptor subtypes in arteries: evidence of alpha1L as a functional isoform of the alpha1A-adrenoceptor. Am. J. Physiol. Heart Circ. Physiol. 289, H1923–H1932.
Martínez-Salas, S. G., Campos-Peralta, J. M., Pardo, J. P., Hernández-Muñoz, R., Ibarra, M., Tanoue, A., et al. (2011). α(1D)-Adrenoceptor regulates the vasopressor action of α(1A)-adrenoceptor in mesenteric vascular bed of α(1D)-adrenoceptor knockout mice. Auton. Autacoid. Pharmacol. 31, 64–71. doi: 10.1111/j.1474-8673.2011.00468.x
Masoud, W. G., Ussher, J. R., Wang, W., Jaswal, J. S., Wagg, C. S., Dyck, J. R., et al. (2014). Failing mouse hearts utilize energy inefficiently and benefit from improved coupling of glycolysis and glucose oxidation. Cardiovasc. Res. 101, 30–38. doi: 10.1093/cvr/cvt216
McCarty, M. F. (2004). Elevated sympathetic activity may promote insulin resistance syndrome by activating alpha-1 adrenergic receptors on adipocytes. Med. Hypothes. 62, 830–838. doi: 10.1016/j.mehy.2003.11.007
McCloskey, D. T., Turnbull, L., Swigart, P., O’Connell, T. D., Simpson, P. C., and Baker, A. J. (2003). Abnormal myocardial contraction in α1A- and α1B-adrenoceptor double-knockout mice. J. Mol. Cell. Cardiol. 35, 1207–1216. doi: 10.1016/s0022-2828(03)00227-x
McGrath, J. C. (2015). Localization of α-adrenoceptors: JR vane medal lecture. Br. J. Pharmacol. 172, 1179–1194. doi: 10.1111/bph.13008
Melien, O., Sandnes, D., Johansen, E. J., and Christoffersen, T. (2000). Effects of pertussis toxin on extracellular signal-regulated kinase activation in hepatocytes by hormones and receptor-independent agents: evidence suggesting a stimulatory role of G(i) proteins at a level distal to receptor coupling. J. Cell Physiol. 184, 27–36. doi: 10.1002/(sici)1097-4652(200007)184:1<27::aid-jcp3>3.0.co;2-q
Methven, L., Simpson, P. C., and McGrath, J. C. (2009). Alpha1A/B-knockout mice explain the native alpha1D-adrenoceptor’s role in vasoconstriction and show that its location is independent of the other alpha1-subtypes. Br. J. Pharmacol. 158, 1663–1675. doi: 10.1111/j.1476-5381.2009.00462.x
Michel, M. C., Hanft, G., and Gross, G. (1994). Radioligand binding studies of alpha 1-adrenoceptor subtypes in rat heart. Br. J. Pharmacol. 111, 533–538. doi: 10.1111/j.1476-5381.1994.tb14770.x
Michel, M. C., and Vrydag, W. (2006). Alpha1-, alpha2- and beta-adrenoceptors in the urinary bladder, urethra and prostate. Br. J. Pharmacol. 147(Suppl. 2), S88–S119.
Milano, C. A., Dolber, P. C., Rockman, H. A., Bond, R. A., Venable, M. E., Allen, L. F., et al. (1994). Myocardial expression of a constitutively active alpha 1B-adrenergic receptor in transgenic mice induces cardiac hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 91, 10109–10113. doi: 10.1073/pnas.91.21.10109
Minneman, K. P., Theroux, T. L., Hollinger, S., Han, C., and Esbenshade, T. A. (1994). Selectivity of agonists for cloned α1-adrenergic receptor subtypes. Mol. Pharmacol. 46, 929–936.
Minokoshi, Y., Kim, Y. B., Peroni, O. D., Fryer, L. G., Müller, C., Carling, D., et al. (2002). Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 415, 339–343. doi: 10.1038/415339a
Minokoshi, Y., Toda, C., and Okamoto, S. (2012). Regulatory role of leptin in glucose and lipid metabolism in skeletal muscle. Indian J. Endocrinol. Metab. 16(Suppl. 3), S562–S568.
Mishima, K., Tanoue, A., Tsuda, M., Hasebe, N., Fukue, Y., Egashira, N., et al. (2004). Characteristics of behavioral abnormalities in alpha1d-adrenoceptors deficient mice. Behav. Brain Res. 152, 365–373. doi: 10.1016/j.bbr.2003.10.038
Mitchell, M. B., Meng, X., Ao, L., Brown, J. M., Harken, A. H., and Banerjee, A. (1995). Preconditioning of isolated rat heart is mediated by protein kinase C. Circ. Res. 76, 73–81. doi: 10.1161/01.res.76.1.73
Miyamoto, L., Ebihara, K., Kusakabe, T., Aotani, D., Yamamoto-Kataoka, S., Sakai, T., et al. (2012). Leptin activates hepatic 5’-AMP-activated protein kinase through sympathetic nervous system and alpha1-adrenergic receptor: a potential mechanism for improvement of fatty liver in lipodystrophy by leptin. J. Biol. Chem. 287, 40441–40447. doi: 10.1074/jbc.m112.384545
Montgomery, M. D., Chan, T., Swigart, P. M., Myagmar, B. E., Dash, R., and Simpson, P. C. (2017). An alpha-1A adrenergic receptor agonist prevents acute doxorubicin cardiomyopathy in male mice. PLoS ONE 12:e0168409. doi: 10.1371/journal.pone.0168409
Morrow, A. L., and Creese, I. (1986). Characterization of alpha 1-adrenergic receptor subtypes in rat brain: a reevaluation of [3H]-WB4104 and [3H]-prazosin binding. Mol. Pharmacol. 29, 321–330.
Moshfegh, A., Babaei, P., Oryan, S., Soltani, B., Zarrindast, M. R., Moshfegh, A., et al. (2011). Involvement of dorsal hippocampal α1-adrenergic receptors in the effect of WIN55,212-2 on memory retrieval in inhibitory avoidance task. Neurosci. Lett. 489, 69–73. doi: 10.1016/j.neulet.2010.07.079
Mourad, F. H., and Saadé, N. E. (2011). Neural regulation of intestinal nutrient absorption. Prog. Neurobiol. 95, 149–162. doi: 10.1016/j.pneurobio.2011.07.010
Mouradian, R. D., Sessler, F. M., and Waterhouse, B. D. (1991). Noradrenergic potentiation of excitatory transmitter action in cerebrocortical slices: evidence for mediation by an alpha 1 receptor-linked second messenger pathway. Brain Res. 546, 83–95. doi: 10.1016/0006-8993(91)91162-t
Mulkey, R. M., and Malenka, R. C. (1992). Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron 9, 967–975. doi: 10.1016/0896-6273(92)90248-c
Murashita, M., Kusumi, I., Hosoda, H., Kangawa, K., and Koyama, T. (2007). Acute administration of clozapine concurrently increases blood glucose and circulating plasma ghrelin levels in rats. Psychoneuroendocrinology 32, 777–784. doi: 10.1016/j.psyneuen.2007.05.007
Musselman, D. M., Ford, A. P., Gennevois, D. J., Harbison, M. L., Laurent, A. L., et al. (2004). A randomized crossover study to evaluate Ro 115-1240, a selective α1A/L -adrenoceptor partial agonist in women with stress urinary incontinence. BJU Int. 93, 78–83. doi: 10.1111/j.1464-410x.2004.04560.x
Myagmar, B. E., Flynn, J. M., Cowley, P. M., Swigart, P. M., Montgomery, M. D., Thai, K., et al. (2017). Adrenergic receptors in individual ventricular myocytes: the beta-1 and alpha-1B are in all cells, the alpha-1A is in a subpopulation, and the beta-2 and beta-3 are mostly absent. Circ. Res. 120, 1103–1115. doi: 10.1161/circresaha.117.310520
Naderi, R., Imani, A., Faghihi, M., and Moghimian, M. (2010). Phenylephrine induces early and late cardioprotection through mitochondrial permeability transition pore in the isolated rat heart. J. Surg. Res. 164, e37–e42.
Nakade, Y., Kitano, R., Yamauchi, T., Kimoto, S., Sakamoto, K., Inoue, T., et al. (2020). Effect of adrenergic agonists on high-fat diet-induced hepatic steatosis in mice. Int. J. Mol. Sci. 21:9392. doi: 10.3390/ijms21249392
Nakaoka, H., Perez, D. M., Baek, K. J., Das, T., Husain, A., Misono, K., et al. (1994). Gh: A GTP binding protein with transglutaminase activity and receptor signaling function. Science 254, 1593–1596. doi: 10.1126/science.7911253
Nazari, A., Sedighi, M., Dalvand, P., Azizi, Y., Moghimian, M., and Boroujeni, S. N. (2019). Late cardiac perconditioning by phenylephrine in an isolated rat heart model is mediated by mitochondrial potassium channels. Braz. J. Pharmac. Sci. 55:e18075.
Nevzorova, J., Evans, B. A., Bengtsson, T., and Summers, R. J. (2006). Multiple signalling pathways involved in β2-adrenoceptor-mediated glucose uptake in rat skeletal muscle cells. Br. J. Pharmacol. 147, 446–454. doi: 10.1038/sj.bjp.0706626
Newman, L. A., Korol, D. L., and Gold, P. E. (2011). Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS One 6:e28427. doi: 10.1371/journal.pone.0028427
Nishimaru, K., Kobayashi, M., Matsuda, T., Tanaka, Y., Tanaka, T., and Shigenobu, K. (2001). alpha-Adrenoceptor stimulation-mediated negative inotropism and enhanced Na(+)/Ca(2+) exchange in mouse ventricle. Am. J. Physiol. Heart Circ. Physiol. 280, H132–H141.
Oberhaensli, R. D., Schwendimann, R., and Keller, U. (1985). Effect of norepinephrine on ketogenesis, fatty acid oxidation, and esterification in isolated rat hepatocytes. Diabetes Metab. Res. Rev. 34, 774–779. doi: 10.2337/diabetes.34.8.774
O’Connell, T. D., Ishizaka, S., Nakamura, A., Swigart, P. M., Rodrigo, M. C., Simpson, G. L., et al. (2003). The α(1a/c)- and α(1b)-adrenergic receptors are required for physiological cardiac hypertrophy in the double-knockout mouse. J. Clin. Invest. 111, 1783–1791. doi: 10.1172/jci200316100
O’Connell, T. D., Jensen, B. C., Baker, A. J., and Simpson, P. C. (2013). Cardiac alpha1-adrenergic receptors: novel aspects of expression, signaling mechanisms, physiologic function, and clinical importance. Pharmacol. Rev. 66, 308–333. doi: 10.1124/pr.112.007203
Oddo, S., Caccamo, A., Shepherd, J. D., Murphy, M. P., Golde, T. E., Kayed, R., et al. (2003). Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421.
Palacios, J. M., Hoyer, D., and Cortés, R. (1987). Alpha 1-Adrenoceptors in the mammalian brain: similar pharmacology but different distribution in rodents and primates. Brain Res. 419, 65–75. doi: 10.1016/0006-8993(87)90569-5
Pankratov, Y., and Lalo, U. (2015). Role for astroglial α1-adrenoreceptors in gliotransmission and control of synaptic plasticity in the neocortex. Front. Cell Neurosci. 9:230. doi: 10.3389/fncel.2015.00230
Papay, R., Gaivin, R., Jha, A., McCune, D. F., McGrath, J. C., Rodrigo, M. C., et al. (2006). Localization of the mouse alpha1A-adrenergic receptor (AR) in the brain: alpha1AAR is expressed in neurons, GABAergic interneurons, and NG2 oligodendrocyte progenitors. J. Comp. Neurol. 497, 209–222. doi: 10.1002/cne.20992
Papay, R., Gaivin, R., McCune, D. F., Rorabaugh, B. R., Macklin, W. B., McGrath, J. C., et al. (2004). Mouse alpha1B-adrenergic receptor is expressed in neurons and NG2 oligodendrocytes. J. Comp. Neurol. 478, 1–10. doi: 10.1002/cne.20215
Papay, R. S., and Perez, D. M. (2020). α1-Adrenergic receptors increase glucose oxidation under normal and ischemic conditions in adult mouse cardiomyocytes. J. Recept. Signal Transduct. Res. 5, 1–7.
Papay, R. S., Shi, T., Piascik, M. T., Naga Prasad, S. V., and Perez, D. M. (2013). α1A-adrenergic receptors regulate cardiac hypertrophy in vivo through interleukin-6 secretion. Mol. Pharmacol. 83, 939–948. doi: 10.1124/mol.112.084483
Pascual, O., Casper, K. B., Kubera, C., Zhang, J., Revilla-Sanchez, R., et al. (2005). Astrocytic purinergic signaling coordinates synaptic networks. Science 310, 113–116. doi: 10.1126/science.1116916
Patil, P. N., Miller, D. D., and Trendelenburg, U. (1974). Molecular geometry and adrenergic drug activity. Pharmacol. Rev. 26, 323–392.
Perez, D. M. (2021). Novel positive allosteric modulators of the α1A-adrenergic receptor to treat Alzheimer’s disease. Brain Connect.. 11, A1–A8.
Perez, D. M., DeYoung, M. B., and Graham, R. M. (1993). Coupling of expressed alpha 1B- and alpha 1D-adrenergic receptor to multiple signaling pathways is both G protein and cell type specific. Mol. Pharmacol. 44, 784–795.
Perez, D. M., and Doze, V. A. (2011). Cardiac and neuroprotection regulated by α(1)-adrenergic receptor subtypes. J. Recept. Signal Transduct. Res. 31, 98–110. doi: 10.3109/10799893.2010.550008
Perez, D. M., Piascik, M. T., and Graham, R. M. (1991). Solution-phase library screening for the identification of rare clones: isolation of an alpha 1D-adrenergic receptor cDNA. Mol. Pharmacol. 40, 876–883.
Perez, D. M., Piascik, M. T., Malik, N., Gaivin, R., and Graham, R. M. (1994). Cloning, expression, and tissue distribution of the rat homolog of the bovine alpha 1C-adrenergic receptor provide evidence for its classification as the alpha 1A subtype. Mol. Pharmacol. 46, 823–831.
Perez-Aso, M., Segura, V., Monto, F., Barettino, D., Noguera, M. A., Milligan, G., et al. (2013). The three α1-adrenoceptor subtypes show different spatio-temporal mechanisms of internalization and ERK1/2 phosphorylation. Biochim.. Biophys. Acta 1833, 2322–2333. doi: 10.1016/j.bbamcr.2013.06.013
Petrasek, T., Doulames, V., Prokopova, I., Vales, K., Stuchlik, A., Petrasek, T., et al. (2010). Combined administration of alpha1-adrenoceptor antagonist prazosin and beta-blocker propranolol impairs spatial avoidance learning on a dry arena. Behav. Brain Res. 208, 402–407. doi: 10.1016/j.bbr.2009.12.025
Piascik, M. T., and Perez, D. M. (2001). Alpha1-adrenergic receptors: new insights and directions. J. Pharmacol. Exp. Ther. 298, 403–410.
Poulsen, L. L., Siersbæk, M., and Mandrup, S. (2012). PPARs: fatty acid sensors controlling metabolism. Semin. Cell Dev. Biol. 23, 631–639. doi: 10.1016/j.semcdb.2012.01.003
Price, D. T., Lefkowitz, R. J., Caron, M. G., Berkowitz, D., and Schwinn, D. A. (1994). Localization of mRNA for three distinct alpha 1-adrenergic receptor subtypes in human tissues: implications for human alpha-adrenergic physiology. Mol. Pharmacol. 45, 171–175.
Pussinen, R., Nieminen, S., Koivisto, E., Haapalinna, A., Riekkinen, P., and Sirvio, J. (1997). Enhancement of intermediate-term memory by an α-1 agonist or a partial agonist at the glycine site of the NMDA receptor. Neurobiol. Learn. Mem. 67, 69–74. doi: 10.1006/nlme.1996.3738
Puumala, T., Greijus, S., Narinen, K., Haapalinna, A., Riekkinen, P., and Sirviö, J. (1998). Stimulation of α-1 adrenergic receptors facilitates spatial learning in rats. Eur. Neuropsychopharmacol. 8, 17–26. doi: 10.1016/s0924-977x(97)00040-0
Ragnarsson, L., Wang, C. I., Andersson, Å, Fajarningsih, D., Monks, T., Brust, A., et al. (2013). Conopeptide ρ-TIA defines a new allosteric site on the extracellular surface of the α1B-adrenoceptor. J. Biol. Chem. 288, 1814–1827. doi: 10.1074/jbc.m112.430785
Rehring, T. F., Friese, R. S., Cleveland, J. C., Meng, X., Robertson, F. G., Harken, A. H., et al. (1996). Alpha-adrenergic preservation of myocardial pH during ischemia is PKC isoform dependent. J. Surg. Res. 63, 324–327. doi: 10.1006/jsre.1996.0269
Riekkinen, M., Kemppainen, S., and Riekkinen, P. Jr. (1997). Effects of stimulation of alpha 1-adrenergic and NMDA/glycine-B receptors on learning defects in aged rats. Psychopharmacology 131, 49–56. doi: 10.1007/s002130050264
Robbins, T. W. (1996). Dissociating executive functions of the prefrontal cortex. Philos. Trans. R. Soc. Lond. B Biol. Sci. 351, 1463–1470. doi: 10.1098/rstb.1996.0131
Rockman, H., Koch, W., and Lefkowitz, R. (2002). Seven-transmembrane-spanning receptors and heart function. Nature 415, 206–212. doi: 10.1038/415206a
Rodriguez, A. L., Nong, Y., Sekaran, N. K., Alagille, D., Tamagnan, G. D., and Conn, P. J. (2005). A close structural analog of 2-methyl-6-(phenylethynyl)-pyridine acts as a neutral allosteric site ligand on metabotropic glutamate receptor subtype 5 and blocks the effects of multiple allosteric modulators. Mol. Pharmacol. 68, 1793–1802. doi: 10.1124/mol.105.016139
Rokosh, D. G., and Simpson, P. C. (2002). Knockout of the alpha1A/C-adrenergic receptor subtype: the alpha1A/C is expressed in resistance arteries and is required to maintain arterial blood pressure. Proc. Natl. Acad. Sci. U.S.A. 99, 9474–9479. doi: 10.1073/pnas.132552699
Rorabaugh, B. R., Ross, S. A., Gaivin, R. J., Papay, R. S., McCune, D. F., Simpson, P. C., et al. (2005). alpha1A- but not alpha1B-adrenergic receptors precondition the ischemic heart by a staurosporine-sensitive, chelerythrinein-sensitive mechanism. Cardiovasc. Res. 65, 436–445. doi: 10.1016/j.cardiores.2004.10.009
Rose, L., Graham, L., Koenecke, A., Powell, M., Xiong, R., Shen, Z., et al. (2020). The association between Alpha-1 adrenergic receptor antagonists and in-hospital mortality from COVID-19. medRxiv doi: 10.1101/2020.12.18.20248346
Ross, S. A., Rorabaugh, B. R., Chalothorn, D., Yun, J., Gonzalez-Cabrera, P. J., McCune, D. F., et al. (2003). The alpha(1B)-adrenergic receptor decreases the inotropic response in the mouse Langendorff heart model. Cardiovasc. Res. 60, 598–607. doi: 10.1016/j.cardiores.2003.09.020
Ruffolo, R. R. Jr., Rice, P. J., Patil, P. N., Hamada, A., and Miller, D. D. (1983). Differences in the applicability of the Easson-Stedman hypothesis to the α1- and α2-adrenergic effects of phenethylamines and imidazolines. Eur. J. Pharmacol. 86, 471–475. doi: 10.1016/0014-2999(83)90199-1
Ruffolo, R. R. Jr., and Waddell, J. E. (1982). Receptor interactions of imidazolines. IX. Cirazoline is an alpha-1 adrenergic agonist and an alpha-2 adrenergic antagonist. J. Pharmacol. Exp. Ther. 222, 29–36.
Ruffolo, R. R. Jr., Yaden, E. L., Waddell, J. E., and Dillard, R. D. (1980). Receptor interactions of imidazolines. VI. Significance of carbon bridge separating phenyl and imidazoline rings of tolazoline-like α-adrenergic imidazolines. J. Pharmacol. Exp. Ther. 214, 535–540.
Russell, R. R. III, Li, J., Coven, D. L., Pypaert, M., Zechner, C., Palmeri, M., et al. (2004). AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J. Clin. Invest. 114, 495–503. doi: 10.1172/jci19297
Sadalge, A., Coughlin, L., Fu, H., Wang, B., Valladares, O., Valentino, R., et al. (2003). α1d Adrenoceptor signaling is required for stimulus induced locomotor activity. Mol. Psychiatry. 8, 664–672. doi: 10.1038/sj.mp.4001351
Sajapitak, S., Uenoyama, Y., Yamada, S., Kinoshita, M., Iwata, K., Bari, F. Y., et al. (2008). Paraventricular alpha1- and alpha2-adrenergic receptors mediate hindbrain lipoprivation-induced suppression of luteinizing hormone pulses in female rats. J. Reprod. Dev. 54, 198–202. doi: 10.1262/jrd.20024
Salvi, S. (2001). Protecting the myocardium from ischemic injury: a critical role for alpha(1)-adrenoreceptors? Chest 119, 1242–1249. doi: 10.1378/chest.119.4.1242
Sanbe, A., Tanaka, Y., Fujiwara, Y., Tsumura, H., Yamauchi, J., Cotecchia, S., et al. (2007). Alpha1-adrenoceptors are required for normal male sexual function. Br. J. Pharmacol. 152, 332–340. doi: 10.1038/sj.bjp.0707366
Sato, M., Evans, B. A., Sandström, A. L., Chia, L. Y., Mukaida, S., Thai, B. S., et al. (2018). α1A-Adrenoceptors activate mTOR signalling and glucose uptake in cardiomyocytes. Biochem. Pharmacol. 148, 27–40. doi: 10.1016/j.bcp.2017.11.016
Scheiderer, C. L., Dobrunz, L. E., and McMahon, L. L. (2004). Novel form of long-term synaptic depression in rat hippocampus induced by activation of α1-adrenergic receptors. J. Neurophysiol. 91, 1071–1077. doi: 10.1152/jn.00420.2003
Scheiderer, C. L., Smith, C. C., McCutchen, E., McCoy, P. A., Thacker, E. E., Kolasa, K., et al. (2008). Coactivation of M(1) muscarinic and alpha1 adrenergic receptors stimulates extracellular signal-regulated protein kinase and induces long-term depression at CA3-CA1 synapses in rat hippocampus. J. Neurosci. 14, 5350–5358. doi: 10.1523/jneurosci.5058-06.2008
Schroeder, C., Stabroth, C., Luft, F. C., and Jordan, J. (2012). Adrenergic cardiovascular control before and after removal of stimulatory α1-adrenoreceptor antibodies. Hypertension 59, e6–e7.
Schultz, P. E., Cook, E. P., and Johnston, D. (1994). Changes in paired-pulse facilitation suggest presynaptic involvement in long-term potentiation. J. Neurosci. 14, 5325–5337. doi: 10.1523/jneurosci.14-09-05325.1994
Schwinn, D. A., Lomasney, J. W., Lorenz, W., Szklut, P. J., Fremeau, R. T. Jr., Yang-Feng, T. L., et al. (1990). Molecular cloning and expression of the cDNA for a novel α1- adrenergic receptor subtype. J. Biol. Chem. 265, 8183–8189. doi: 10.1016/s0021-9258(19)39055-6
Scofield, M. A., Liu, F., Abel, P. W., and Jeffries, W. B. (1995). Quantification of steady state expression of mRNA for α1-adrenergic receptor subtypes using reverse transcription and a competitive polymerase chain reaction. J. Pharmacol. Exp. Ther. 275, 1035–1042.
Segura, V., Perez-Aso, M., Monto, F., Carceller, E., Noguera, M. A., Pediani, J., et al. (2013). Differences in the signaling pathways of α1A- and α1B-adrenoceptors are related to different endosomal targeting. PLoS One 8, e64996. doi: 10.1371/journal.pone.0064996
Sharpe, I. A., Thomas, L., Loughnan, M., Motin, L., Palant, E., Croker, D. E., et al. (2003). Allosteric α1-adrenoreceptor antagonism by the conopeptide ρ-TIA. J. Biol. Chem. 278, 34451–34457. doi: 10.1074/jbc.m305410200
Shi, T., Moravec, C. S., and Perez, D. M. (2013). Novel proteins associated with human dilated cardiomyopathy: selective reduction in α(1A)-adrenergic receptors and increased desensitization proteins. J. Recept. Signal Transduct. Res. 33, 96–106. doi: 10.3109/10799893.2013.764897
Shi, T., Papay, R. S., and Perez, D. M. (2016). α1A-Adrenergic receptor prevents cardiac ischemic damage through PKCδ/GLUT1/4-mediated glucose uptake. J. Recept. Signal Transduct. Res. 36, 261–270. doi: 10.3109/10799893.2015.1091475
Shi, T., Papay, R. S., and Perez, D. M. (2017). The role of α1-adrenergic receptors in regulating metabolism: increased glucose tolerance, leptin secretion and lipid oxidation. J. Recept. Signal Transduct. Res. 37, 124–132. doi: 10.1080/10799893.2016.1193522
Shimazaki, Y., Nishiki, T., Omori, A., Sekiguchi, M., Kamata, Y., Kozaki, S., et al. (1996). Phosphorylation of 25-kDa synaptosome-associated protein. Possible involvement in protein kinase C-mediated regulation of neurotransmitter release. J. Biol. Chem. 271, 14548–14553. doi: 1074/jbc.271.24.14548
Shimohama, S., Taniguchi, T., Fujiwara, M., and Kameyama, M. (1986). Biochemical characterization of α1-adrenergic receptors in human brain and changes in Alzheimer-type dementia. J. Neurochem. 47, 1295–1301.
Shiuchi, T., Haque, M. S., Okamoto, S., Inoue, T., Kageyama, H., Lee, S., et al. (2009). Hypothalamic orexin stimulates feeding-associated glucose utilization in skeletal muscle via sympathetic nervous system. Cell Metab. 10, 466–480. doi: 10.1016/j.cmet.2009.09.013
Simkhovich, B. Z., Przyklenk, K., and Kloner, R. A. (2013). Role of protein kinase C in ischemic “conditioning”: from first evidence to current perspectives. J. Cardiovasc. Pharmacol. Ther. 18, 525–532. doi: 10.1177/1074248413494814
Simpson, P. (1983). Norepinephrine-stimulated hypertrophy of cultured rat myocardial cells is an alpha1-adrenergic response. J. Clin. Invest. 72, 732–738. doi: 10.1172/jci111023
Sirviö, J., and MacDonald, E. (1999). Central alpha1-adrenoceptors: their role in the modulation of attention and memory formation. Pharmacol. Ther. 83, 49–65.
Skoglund, G., Lundquist, I., and Ahren, B. (1987). Alpha 1- and alpha 2-adrenoceptor activation increases plasma glucagon levels in the mouse. Eur. J. Pharmacol. 143, 83–88. doi: 10.1016/0014-2999(87)90737-0
Snabaitis, A. K., Muntendorf, A., Wieland, T., and Avkiran, M. (2005). Regulation of the extracellular signal-regulated kinase pathway in adult myocardium: differential roles of G(q/11), Gi and G(12/13) proteins in signalling by alpha1-adrenergic, endothelin-1 and thrombin-sensitive protease-activated receptors. Cell Signal. 17, 655–664. doi: 10.1016/j.cellsig.2004.10.008
Somlyo, A. P., and Somlyo, A. V. (2003). Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 83, 1325–1358. doi: 10.1152/physrev.00023.2003
Speechly-Dick, M. E., Grover, G. J., and Yellon, D. M. (1995). Does ischemic preconditioning in the human involve protein kinase C and the ATP-dependent K+ channel? Studies of contractile function after simulated ischemia in an atrial in vitro model. Circ. Res. 77, 1030–1035. doi: 10.1161/01.res.77.5.1030
Spreng, M., Cotecchia, S., and Schenk, F. (2001). A behavioral study of alpha-1b adrenergic receptor knockout mice: increased reaction to novelty and selectively reduced learning capacities. Neurobiol. Learn Mem. 75, 214–229. doi: 10.1006/nlme.2000.3965
Squire, L. R., Genzel, L., Wixted, J. T., and Morris, R. G. (2015). Memory consolidation. Cold Spring Harb. Perspect. Biol. 7:a021766.
Stanasila, L., Abuin, L., Dey, J., and Cotecchia, S. (2008). Different internalization properties of the alpha1a- and alpha1b-adrenergic receptor subtypes: the potential role of receptor interaction with beta-arrestins and AP50. Mol. Pharmacol. 74, 562–573. doi: 10.1124/mol.107.043422
Steinfath, M., Chen, Y. Y., Lavicky, J., Magnussen, O., Nose, M., Rosswag, S., et al. (1992). Cardiac alpha 1-adrenoceptor densities in different mammalian species. Br. J. Pharmacol. 107, 185–188. doi: 10.1111/j.1476-5381.1992.tb14484.x
Stone, E. A., Cotecchia, S., Lin, Y., and Quartermain, D. (2002). Role of brain alpha 1B-adrenoceptors in modafinil-induced behavioral activity. Synapse 46, 269–270. doi: 10.1002/syn.10127
Sugden, M. C., Tordoff, A. F., Ilic, V., and Williamson, D. H. (1980). α-adrenergic stimulation of [1-14C]-oleate oxidation to 14CO2 in isolated rat hepatocytes. FEBS Lett. 120, 80–84. doi: 10.1016/0014-5793(80)81051-9
Szot, P., White, S. S., Greenup, J. L., Leverenz, J. B., Peskind, E. R., and Raskind, M. A. (2005). Alpha1-adrenoreceptor in human hippocampus: binding and receptor subtype mRNA expression. Brain Res. Mol. Brain Res. 139, 367–371. doi: 10.1016/j.molbrainres.2005.06.013
Szot, P., White, S. S., Greenup, J. L., Leverenz, J. B., Peskind, E. R., and Raskind, M. A. (2007). Changes in adrenoreceptors in the prefrontal cortex of subjects with dementia: evidence of compensatory changes. Neuroscience 146, 471–480. doi: 10.1016/j.neuroscience.2007.01.031
Tanaka, T., Yamamoto, J., Iwasaki, S., Asaba, H., Hamura, H., Ikeda, Y., et al. (2003). Activation of peroxisome proliferator-activated receptor δ induces fatty acid β-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl. Acad. Sci. U.S.A. 100, 15924–15929. doi: 10.1073/pnas.0306981100
Tang, K. M., Wang, G. R., Lu, P., Karas, R. H., Aronovitz, M., Heximer, S. P., et al. (2003). Regulator of G-protein signaling-2 mediates vascular smooth muscle relaxation and blood pressure. Nat. Med. 9, 1506–1512. doi: 10.1038/nm958
Tanoue, A., Nasa, Y., Koshimizu, T., Shinoura, H., Oshikawa, S., Kawai, T., et al. (2002). The alpha(1D)-adrenergic receptor directly regulates arterial blood pressure via vasoconstriction. J. Clin. Invest. 109, 765–775. doi: 10.1172/jci200214001
Thomas, A. P., Martin-Requero, A., and Williamson, J. R. (1985). Interactions between insulin and alpha 1-adrenergic agents in the regulation of glycogen metabolism in isolated hepatocytes. J. Biol. Chem. 260, 5963–5973. doi: 10.1016/s0021-9258(18)88923-2
Thyrian, J. R., Hertel, J., Schulze, L. N., Dörr, M., Prüss, H., Hempel, P., et al. (2018). Prevalence and determinants of agonistic autoantibodies against α1-adrenergic receptors in patients screened positive for dementia: results from the population-based DelpHi-study. J. Alzheimers Dis. 64, 1091–1097. doi: 10.3233/jad-171096
Torkaman-Boutorabi, A., Danyali, F., Oryan, S., Ebrahimi-Ghiri, M., and Zarrindast, M. R. (2014). Hippocampal α-adrenoceptors involved in the effect of histamine on spatial learning. Physiol. Behav. 129, 17–24. doi: 10.1016/j.physbeh.2014.02.009
Townsend, S. A., Jung, A. S., Hoe, Y. S., Lefkowitz, R. Y., Khan, S. A., Lemmon, C. A., et al. (2004). Critical role for the alpha-1B adrenergic receptor at the sympathetic neuroeffector junction. Hypertension 44, 776–782. doi: 10.1161/01.hyp.0000145405.01113.0e
Trost, B. N., Weidmann, P., Riesen, W., Claessens, J., Streulens, Y., and Nelemans, F. (1987). Comparative effects of doxazosin and hydrochlorothiazide on serum lipids and blood pressure in essential hypertension. Am. J. Cardiol. 59, 99–104.
Tsuchida, A., Liu, Y., Liu, G. S., Cohen, M. V., and Downey, J. M. (1994). Alpha 1-adrenergic agonists precondition rabbit ischemic myocardium independent of adenosine by direct activation of protein kinase C. Circ. Res. 75, 576–585. doi: 10.1161/01.res.75.3.576
Turnbull, L., McCloskey, D. T., O’Connell, T. D., Simpson, P. C., and Baker, A. J. (2003). Alpha 1-adrenergic receptor responses in alpha 1AB-AR knockout mouse hearts suggest the presence of alpha 1D-AR. Am. J. Physiol. Heart Circ. Physiol. 284, H1104–H1109.
Unnerstall, J. R., Fernandez, I., and Orensanz, L. M. (1985). The alpha-adrenergic receptor: radiohistochemical analysis of functional characteristics and biochemical differences. Pharmacol. Biochem. Behav. 22, 859–874. doi: 10.1016/0091-3057(85)90538-6
Ussher, J. R., Wang, W., Gandhi, M., Keung, W., Samokhvalov, V., Oka, T., et al. (2012). Stimulation of glucose oxidation protects against acute myocardial infarction and reperfusion injury. Cardiovasc. Res. 94, 359–369. doi: 10.1093/cvr/cvs129
Van Asseldonk, B., Barkin, J., and Elterman, D. S. (2015). Medical therapy for benign prostatic hyperplasia: a review. Can. J. Urol. 22(Suppl. 1), 7–17.
Vanhoose, A. M., Emery, M., Jimenez, L., and Winder, D. G. (2002). ERK activation by G-protein-coupled receptors in mouse brain is receptor identity-specific. J. Biol. Chem. 277, 9049–9053. doi: 10.1074/jbc.m108309200
Vann, S. D., and Albasser, M. M. (2011). Hippocampus and neocortex: recognition and spatial memory. Curr. Opin. Neurobiol. 21, 1–6.
Vecchione, C., Fratta, L., Rizzoni, D., Notte, A., Poulet, R., Porteri, E., et al. (2002). Cardiovascular influences of alpha1b-adrenergic receptor defect in mice. Circulation 105, 1700–1707. doi: 10.1161/01.cir.0000012750.08480.55
Velásquez-Martinez, M. C., Vázquez-Torres, R., and Jiménez-Rivera, C. A. (2012). Activation of alpha1-adrenoceptors enhances glutamate release onto ventral tegmental area dopamine cells. Neuroscience 216, 18–30. doi: 10.1016/j.neuroscience.2012.03.056
Vieira, E., Liu, Y. J., and Gylfe, E. (2004). Involvement of alpha1 and beta-adrenoceptors in adrenaline stimulation of the glucagon-secreting mouse alpha-cell. Naunyn. Schmiedebergs Arch. Pharmacol. 369, 179–183. doi: 10.1007/s00210-003-0858-5
Villalba, N., Stankevicius, E., Garcia-Sacristán, A., Simonsen, U., and Prieto, D. (2007). Contribution of both Ca2+ entry and Ca2+ sensitization to the alpha1-adrenergic vasoconstriction of rat penile small arteries. Am. J. Physiol. Heart Circ. Physiol. 292, H1157–H1169.
Walden, P. D., Durkin, M. M., Lepor, H., Wetzel, J. M., Gluchowski, C., and Gustafson, E. L. (1997). Localization of mRNA and receptor binding sites for the alpha 1a-adrenoceptor subtype in the rat, monkey and human urinary bladder and prostate. J. Urol. 157, 1032–1038. doi: 10.1016/s0022-5347(01)65136-x
Wallukat, G., Jandrig, B., Becker, N. P., Wendler, J. J., Göttel, P., Müller, J., et al. (2020). Autoantibodies directed against α1-adrenergic receptor and endothelin receptor A in patients with prostate cancer. Auto. Immun. Highlights 11:13. doi: 10.1186/s13317-020-00136-y
Wang, B. H., Du, X. J., Autelitano, D. J., Milano, C. A., and Woodcock, E. A. (2000). Adverse effects of constitutively active alpha(1B)-adrenergic receptors after pressure overload in mouse hearts. Am. J. Physiol. Heart Circ. Physiol. 279, H1079–H1086.
Wang, J., Yang, L., Rezaie, A. R., and Li, J. (2011). Activated protein C protects against myocardial ischemic/reperfusion injury through AMP-activated protein kinase signaling. J. Thromb. Haemost. 9, 1308–1317. doi: 10.1111/j.1538-7836.2011.04331.x
Waugh, D. J. J., Gaivin, R. J., Zuscik, M. J., Gonzalez-Cabrera, P., Ross, S. A., Yun, J., et al. (2001). Phe308 and Phe312 in TM VII are major sites of α1-adrenergic receptor antagonist binding: imidazoline agonists bind like antagonists. J. Biol. Chem. 276, 25366–25371.
Weinberger, M. H. (1986). Antihypertensive therapy and lipids: Paradoxical influences on cardiovascular disease risk. Am. J. Med. 80(Suppl. 2A), 64–70. doi: 10.1016/0002-9343(86)90162-2
Wenner, M. I., Maker, G. L., Dawson, L. F., Drummond, P. D., and Mullaney, I. (2016). The potential of metabolomic analysis techniques for the characterisation of α1-adrenergic receptors in cultured N1E-115 mouse neuroblastoma cells. Cytotechnology 68, 1561–1575. doi: 10.1007/s10616-015-9915-4
Wenzel, K., Haase, H., Wallukat, G., Derer, W., Bartel, S., Homuth, V., et al. (2008). Potential relevance of a1-adrenergic receptor autoantibodies in refractory hypertension. PLoS One 3:e3742. doi: 10.1371/journal.pone.0003742
Wier, W. G., and Morgan, K. G. (2003). Alpha1-adrenergic signaling mechanisms in contraction of resistance arteries. Rev. Physiol. Biochem. Pharmacol. 150, 91–139. doi: 10.1007/s10254-003-0019-8
Willis, M. S., Ilaiwy, A., Montgomery, M. D., Simpson, P. C., and Jensen, B. C. (2016). The alpha-1A adrenergic receptor agonist A61603 reduces cardiac polyunsaturated fatty acid and endocannabinoid metabolites associated with inflammation in vivo. Metabolomics 12:155.
Winder-Rhodes, S. E., Chamberlain, S. R., Idris, M. I., Robbins, T. W., Sahakian, B. J., and Müller, U. (2010). Effects of modafinil and prazosin on cognitive and physiological functions in healthy volunteers. J. Psychopharmacol. Oxf. Engl. 24, 1649–1657. doi: 10.1177/0269881109105899
Wold, E. A., Chen, J., Cunningham, K. A., and Zhou, J. (2019). Allosteric modulation of class A GPCRs: targets, agents, and emerging concepts. J. Med. Chem. 62, 88–127. doi: 10.1021/acs.jmedchem.8b00875
Woo, S. H., and Lee, C. O. (1999). Role of PKC in the effects of alpha1-adrenergic stimulation on Ca2+ transients, contraction and Ca2+ current in guinea-pig ventricular myocytes. Pflugers Arch. 437, 335–344. doi: 10.1007/s004240050787
Wootten, D., Christopoulos, A., Marti-Solano, M., Babu, M. M., and Sexton, M. (2018). Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 19, 638–653. doi: 10.1038/s41580-018-0049-3
Wu, J., and Li, L. (2016). Autoantibodies in Alzheimer’s disease: potential biomarkers, pathogenic roles, and therapeutic implications. J. Biomed. Res. 30, 361–372.
Wu, X. S., and Wu, L. G. (2001). Protein kinase C increases the apparent affinity of the release machinery to Ca2 + by enhancing the release machinery downstream of the Ca2 + sensor. J. Neurosci. 21, 7928–7936. doi: 10.1523/JNEUROSCI.21-20-07928.2001
Xiao, L., and Jeffries, W. B. (1998). Kinetics of alkylation of cloned rat α1-adrenoceptor subtypes by chloroethylclonidine. Eur. J. Pharmacol 347, 319–327. doi: 10.1016/s0014-2999(98)00109-5
Xu, L., Anwyl, R., and Rowan, M. J. (1998). Spatial exploration induces a persistent reversal of long-term potentiation in rat hippocampus. Nature 394, 891–894. doi: 10.1038/29783
Yan, L., Tan, X., Chen, W., Zhu, H., Cao, J., and Liu, H. (2014). Enhanced vasoconstriction to α1 adrenoceptor autoantibody in spontaneously hypertensive rats. Sci. China Life Sci. 57, 681–689. doi: 10.1007/s11427-014-4672-8
Yeh, C. C., Fan, Y., Xu, Y., Yang, Y. L., Simpson, P. C., and Mann, M. J. (2017). Shift toward greater pathologic post-myocardial infarction remodeling with loss of the adaptive hypertrophic signaling of alpha1 adrenergic receptors in mice. PLoS One 12:e0188471. doi: 10.1371/journal.pone.0188471
Zhang, J., Ash, T., Huang, W., Smith, A., Huang, H., and Jensen, B. (2020). An essential protective role for cardiomyocyte alpha1A-adrenergic receptors in a mouse model of myocardial infarction. Circ. Res. 127:A408.
Zhang, Z., Cordeiro Matos, S., Jego, S., Adamantidis, A., and Seguela, P. (2013). Norepinephrine drives persistent activity in prefrontal cortex via synergistic alpha1 and alpha2 adrenoceptors. PLoS One 8:e66122. doi: 10.1371/journal.pone.0066122
Zhao, M., Hagler, H. K., and Muntz, K. H. (1996). Regulation of alpha 1-, beta 1-, and beta 2-adrenergic receptors in rat heart by norepinephrine. Am. J. Physiol. 271(5 Pt 2), H1762–H1768.
Zhao, X., Balaji, P., Pachon, R., Beniamen, D. M., Vatner, D. E., Graham, R. M., et al. (2015). Overexpression of cardiomyocyte α1A-adrenergic receptors attenuates postinfarct remodeling by inducing angiogenesis through heterocellular signaling. Arterioscler. Thromb. Vasc. Biol. 35, 2451–2459. doi: 10.1161/atvbaha.115.305919
Zhao, X., Park, J., Ho, D., Gao, S., Yan, L., Ge, H., et al. (2012). Cardiomyocyte overexpression of the α1A-adrenergic receptor in the rat phenocopies second but not first window preconditioning. Am. J. Physiol. Heart Circ. Physiol. 302, H1614–H1624.
Zhou, J., and Cunningham, K. A. (2019). Positive-allosteric modulation of the 5-HT2C receptor: implications for neuropsychopharmacology and neurotherapeutics. Neuropsychopharmacology 44, 230–231. doi: 10.1038/s41386-018-0190-x
Zhou, Z., Liao, Y., Li, L., Wei, F., Wang, B., Wei, Y., et al. (2008). Vascular damages in rats immunized by alpha1-adrenoceptor peptides. Cell. Mol. Immunol. 5, 349–356. doi: 10.1038/cmi.2008.43
Zhou, Z., Liao, Y. H., Wei, Y., Wei, F., Wang, B., Li, L., et al. (2005). Cardiac remodeling after long-term stimulation by antibodies against the alpha1-adrenergic receptor in rats. Clin. Immunol. 114, 164–173. doi: 10.1016/j.clim.2004.09.011
Zilles, K., Gross, G., Schleicher, A., Schildgen, S., Bauer, A., Bahro, M., et al. (1991). Regional and laminar distributions of alpha1-adrenoceptors and their subtypes in human and rat hippocampus. Neuroscience 40, 307–320. doi: 10.1016/0306-4522(91)90122-5
Zou, M. X., Roy, A. A., Zhao, Q., Kirshenbaum, L. A., Karmazyn, M., and Chidiac, P. (2006). RGS2 is upregulated by and attenuates the hypertrophic effect of alpha1-adrenergic activation in cultured ventricular myocytes. Cell Signal. 18, 1655–1663. doi: 10.1016/j.cellsig.2006.01.012
Zuscik, M. J., Chalothorn, D., Hellard, D., Deighan, C., McGee, A., Daly, C. J., et al. (2001). Hypotension, autonomic failure, and cardiac hypertrophy in transgenic mice overexpressing the alpha 1B-adrenergic receptor. J. Biol. Chem. 276, 13738–13743. doi: 10.1074/jbc.m008693200
Keywords: adrenergic receptor, G-protein coupled receptor, cognition, cardioprotection, metabolism
Citation: Perez DM (2021) Current Developments on the Role of α1-Adrenergic Receptors in Cognition, Cardioprotection, and Metabolism. Front. Cell Dev. Biol. 9:652152. doi: 10.3389/fcell.2021.652152
Received: 11 January 2021; Accepted: 29 April 2021;
Published: 25 May 2021.
Edited by:
Muheeb Beg, University of Gothenburg, SwedenReviewed by:
Alan Bush, Massachusetts General Hospital and Harvard Medical School, United StatesGianluigi Pironti, Karolinska Institutet (KI), Sweden
Van Doze, University of North Dakota, United States
Copyright © 2021 Perez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dianne M. Perez, UGVyZXpkQGNjZi5vcmc=