 Hongyang Wang1*†
Hongyang Wang1*† Yun Gao1,2,3,4*†Jing Guan1,2,3,4Lan Lan1,2,3,4Ju Yang1,2,3,4Wenping Xiong1,2,3,4Cui Zhao1,2,3,4Linyi Xie1,2,3,4Lan Yu1,2,3,4Dayong Wang1,2,3,4Qiuju Wang1,2,3,4*
Yun Gao1,2,3,4*†Jing Guan1,2,3,4Lan Lan1,2,3,4Ju Yang1,2,3,4Wenping Xiong1,2,3,4Cui Zhao1,2,3,4Linyi Xie1,2,3,4Lan Yu1,2,3,4Dayong Wang1,2,3,4Qiuju Wang1,2,3,4*- 1College of Otolaryngology, Head and Neck Surgery, Chinese People’s Liberation Army (PLA) Institute of Otolaryngology, Chinese People’s Liberation Army (PLA) General Hospital, Beijing, China
- 2National Clinical Research Center for Otolaryngologic Diseases, Beijing, China
- 3Key Lab of Hearing Impairment Science of Ministry of Education, Beijing, China
- 4Key Lab of Hearing Impairment Prevention and Treatment of Beijing, Beijing, China
Objective: To report the phenotypic heterogeneity of GJB2 c.235delC homozygotes associated with post-lingual and/or milder hearing loss, and explore the possible mechanism of these unconditional phenotypes.
Methods: Mutation screening of GJB2 was performed on all ascertained members from Family 1006983 and three sporadic patients by polymerase chain reaction (PCR) amplification and Sanger sequencing. Next generation sequencing (NGS) was successively performed on some of the affected members and normal controls from Family 1006983 to explore additional possible genetic codes. Reverse transcriptase–quantitative PCR was conducted to test the expression of Connexin30.
Results: We identified a Chinese autosomal recessive hearing loss family with the GJB2 c.235delC homozygous mutation, affected members from which had post-lingual moderate to profound hearing impairment, and three sporadic patients with post-lingual moderate hearing impairment, instead of congenital profound hearing loss. NGS showed no other particular variants. Overexpression of Connexin30 in some of these cases was verified.
Conclusion: Post-lingual and/or moderate hearing impairment phenotypes of GJB2 c.235delC homozygotes are not the most common phenotype, revealing the heterogeneity of GJB2 pathogenic mutations. To determine the possible mechanism that rescues part of the hearing or postpones onset age of these cases, more cases are required to confirm both Connexin30 overexpression and the existence of modifier genes.
Introduction
Hearing loss is one of the major disabilities world-widely, which is often induced by loss of sensory hair cells (HCs) in the inner ear cochlea. HCs mainly function in transducing sound waves into the electric signals (Wang et al., 2017; Liu et al., 2019; Qi et al., 2019; He et al., 2020). Hearing loss could be caused by genetic factors, aging, chronic cochlear infections, infectious diseases, ototoxic drugs, and noise exposure (He et al., 2017; Cheng et al., 2019; Zhou et al., 2020); and genetic factors account for more than 60% of the hearing loss. GJB2, which encodes the gap junction protein Connexin 26, is the most common cause of genetic non-syndromic hearing loss (NSHL). Among the approximately 200 hearing loss associated GJB2 mutations (Stenson et al., 2017), the c.235delC mutation, a founder mutation (Yan et al., 2003; Shinagawa et al., 2020), is the most frequently known mutation in some East Asian groups, with a carrier frequency of approximately 1% (Yan et al., 2003). The mutation has a significant ethnic specificity, with increasing risk of NSHL in the East Asian and Southeast Asian populations (Dai et al., 2015), but no susceptibility in Oceania and European populations (Yao et al., 2012). Most patients with the GJB2 c.235delC homozygous mutation show pre-lingual severe to profound hearing impairment (Zhao et al., 2011). However, even cases with the GJB2 c.235delC homozygous mutation may be undetected by newborn hearing screening, suggesting that GJB2 c.235delC homozygous mutation-related hearing loss may not always be congenital at onset (Minami et al., 2013; Dai et al., 2019; Wang et al., 2019). Clinically, except for late-onset hearing impairment, some GJB2 c.235delC homozygous patients show post-lingual and/or mild to moderate hearing impairment instead of profound hearing loss with a highly heterogeneous phenotype.
In addition to GJB2, GJB6 is another gene located in DFNB1. GJB6 encodes the gap junction protein Connexin 30, which is co-assemble with Connexin 26 to form hybrid gap junctions (Ahmad et al., 2003). Connexin26 and Connexin 30 are predominant isoforms in the cochlea (Zhao et al., 2006). Deletions in GJB6 are also common genetic factors for NSHL in some populations, with ethnic specificity (del Castillo et al., 2002; Marlin et al., 2005; Falah et al., 2020). Cases with digenic heterozygous mutations in GJB2 and GJB6 are relatively rare in the NSHL population, with various phenotypes, such as prelingual or post-lingual, ranging from mild or moderate to severe or profound hearing loss (Bolz et al., 2004; Cama et al., 2009; Chan et al., 2010; del Castillo and del Castillo, 2011; Mei et al., 2017). In the GJB6 homozygous knockout mouse model, overexpression of Connexin26 restored hearing sensitivity and prevented hair cell death, suggesting that upregulation of Connexin might be a therapeutic strategy for patients with GJB6 mutations (Ahmad et al., 2007). However, there is no animal experiment to verify that the overexpression of Connexin30 can restore the hearing loss caused by GJB2 mutations, since Gjb2 knockout mice are embryonically lethal (Gabriel et al., 1998).
Modifier genes are a perspective for the study of hearing loss to interpret phenotypic variation. Linkage analysis (families) and association studies (unrelated patients) are two strategies can be adopted to identify modifier genes for human disorders (Bykhovskaya et al., 2004; Dunckley et al., 2007; Tao et al., 2019). However, association studies are not always effective, and a previous whole genome association (WGA) study on GJB2 c.35delG homozygous patients showed that phenotypic heterogeneity could not be explained by the effect of a single major modifier gene (Azaiez et al., 2004; Hilgert et al., 2009a).
In this study, we focused on some unconditional cases involving GJB2 c.235delC homozygotes, whose phenotypes were late-onset and/or moderate hearing loss. First, we found a Chinese family (Family 1006983) with the GJB2 c.235delC mutation, which was segregated with all of the recruited members. Unlike other patients with the GJB2 c.235delC homozygous mutation, whose phenotype were pre-lingual severe to profound hearing loss, patients in this family showed post-lingual moderate to profound sensorineural hearing loss. In addition, we further identified three sporadic cases with GJB2 c.235delC homozygotes, the phenotypes of which were post-lingual moderate hearing impairment. To decipher the mechanism of partial or late-onset hearing loss in GJB2 c.235delC homozygotes cases, we performed next generation sequencing (NGS), including targeted genes capture and genome sequencing on some of the affected members and controls from 1006983, but no pathogenic variations or modifier genes were identified. Since Connexin26 and Connexin 30 are co-assembled to form hybrid gap junctions and a previous mouse experiment suggested that up-regulation of Connexin or the slowing of its degradation might be a therapeutic strategy to prevent and treat deafness caused by Connexin 30 mutations (Ahmad et al., 2007), we hypothesised that for some of the patients with a more minor phenotype than other patients with the GJB2 c.235delC mutation, it is the overexpression of Connexin30 that plays a compensatory role. Thus, reverse transcriptase-quantitative polymerase chain reaction (RT-qPCR) was also conducted on some of these patients to test the expression of Connexin30, which is encoded by GJB6 (Supplementary Figure 1).
Materials and Methods
Ethics Statement
The study was approved by the Committee of Medical Ethics of the Chinese PLA General Hospital. Written informed consents were obtained from all the participants in the family.
Family Recruitment and Clinical Evaluations
A five-generation family (Family 1006983) with 133 members presenting with autosomal recessive non-syndromic sensorineural hearing loss (ARNSHL) was ascertained from the Institute of Otolaryngology, Chinese PLA General Hospital (Figures 1, 2). Three independent sporadic patients with homozygous GJB2 c.235delC were also recruited in our study, who had late-onset moderate hearing impairment (Figure 3). Personal or family medical evidence of hearing loss, tinnitus, vestibular symptoms, and other clinical abnormalities of the participants were identified by a team of experienced physicians and audiologists. Audiometric evaluations included pure tone audiometry (PTA), which was calculated as the average hearing threshold at 0.5, 1, 2, and 4 kHz for the bilateral ears of the ascertained subjects. The severity of hearing impairment was defined as mild (26–40 dB HL), moderate (41–70 dB HL), severe (71–95 dB HL) and profound (>95 dB HL). The speech recognition score was tested and calculated for some patients. Caloric testing was performed on some patients and the normal controls to obtain data on semicircular canal function. Some patients who had tinnitus were estimated by tinnitus assessment. High resolution computed tomography (HRCT) of the temporal bone was conducted for some of the patients.
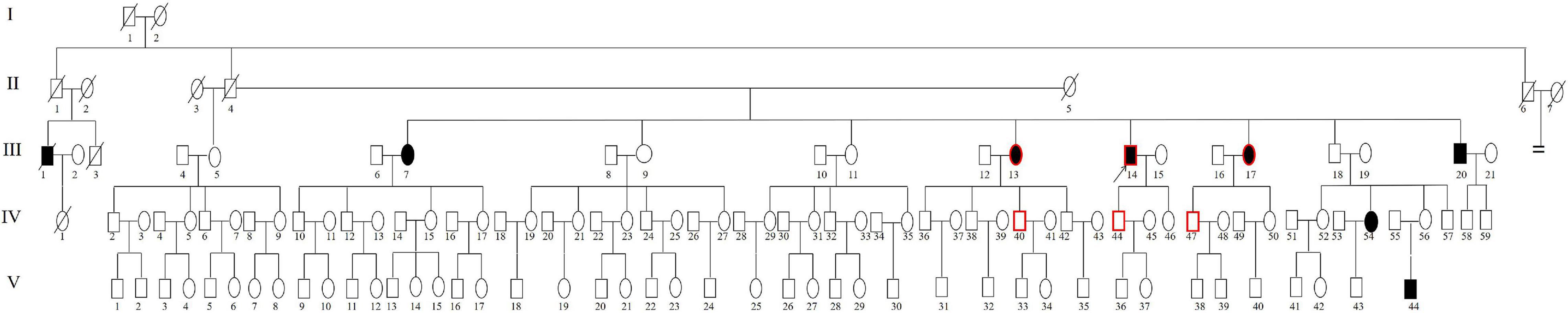
Figure 1. Pedigree of Family 1006983. Filled symbols for males (squares) and females (circles) represent affected individuals, and empty, unaffected individuals. An arrow denotes the proband. The symbols with a red border represent the cases for whom genome sequencing was performed.
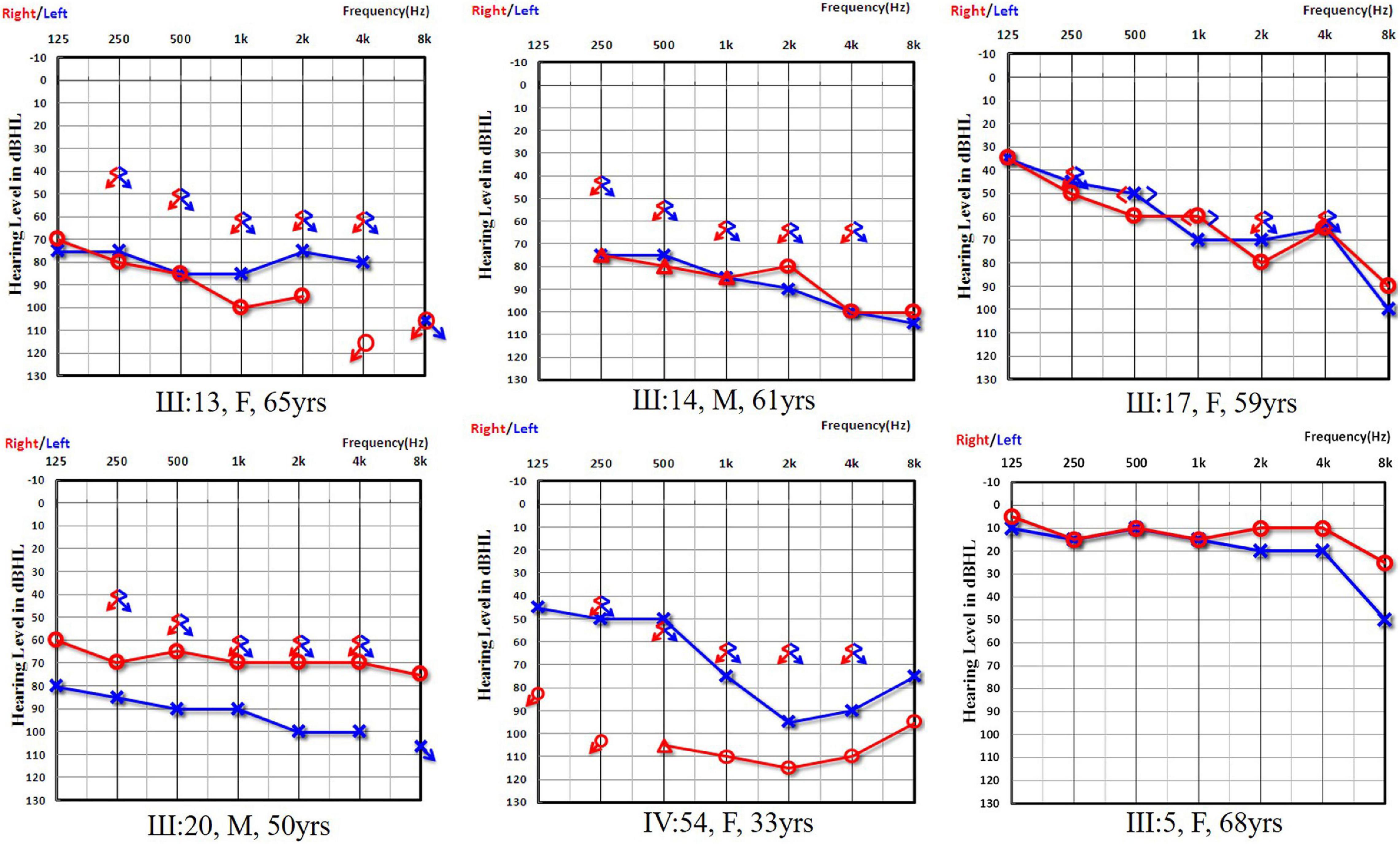
Figure 2. Audiograms of both ears from subjects in Family 1006983. Symbols “o” and “x” denote air conduction pure-tone thresholds at different frequencies in the right and left ear, respectively. dB HL, decibels hearing level; Hz, Hertz. III:13, III:14, III:17, III:20, and IV:54 are the affected cases from Family 1006983 with the GJB2 c.235delC homozygous mutation. III:5 is a normal control from the family with a GJB2 c.235delC heterozygous mutation.
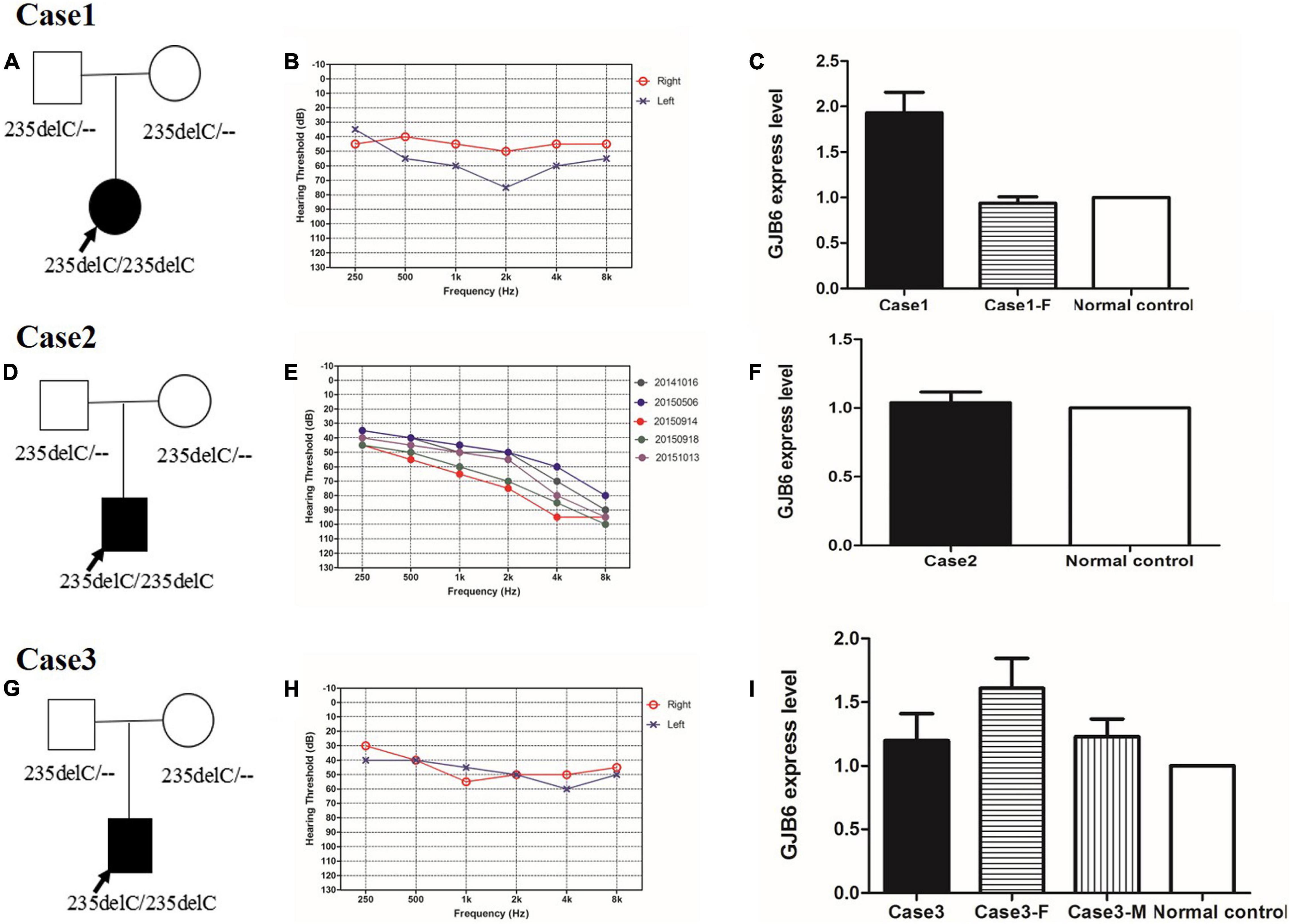
Figure 3. (A,D,G) Pedigree of case 1, case 2, and case 3, respectively. (B,E,H) Audiograms of three sporadic cases. (B) Audiograms of both ears from case 1. (E) Five audiograms of the left ear from case 2 in the last 2 years. Different color audiograms represent audiograms done at different times. (H) Audiograms of both ears from case 3. (B,E,H) Symbols “o” and “x” denote air conduction pure-tone thresholds at different frequencies in the right and left ear, respectively. dB, decibels; Hz, Hertz. (C,F,I) Connexin 30 expression in case 1, case 2, and case 3, respectively. Case 1-F is the father of case 1. Case3-F is the father of case 3, and Case 3-M is the mother of Case 3. Normal control is an independent subject with normal hearing from our institute.
A cohort of two hundred and ninety-five hearing loss cases who had the homozygous GJB2 c.235delC mutation in our institute were chosen as the controls to compare phenotypes (Zhao et al., 2011).
Sanger Sequencing
Genomic DNA was extracted from the whole blood samples using a Blood DNA kit according to the standard protocol (TIANGEN BIOTECH, Beijing, China). PCR and Sanger sequencing were performed on all available members from Family 1006983 and the three sporadic patients and their parents to determine whether the potential mutations in causative genes co-segregated with the disease phenotype in these families. Direct PCR products were sequenced using Bigdye Terminator v3.1 Cycle Sequencing Kits (Applied Biosystems, Foster City, CA) and analysed using an ABI 3700XL Genetic Analyser (primers were GJB2-Exon2-F: 5′-TTGGTGTTTGCTCAGGAAGA-3′ and GJB2-Exon2-R: 5′-GGCCTACAGGGGTTTCAAAT-3′).
Next Generation Sequencing
To examine whether other genetic defects or modifier genes were involved, targeted multi-genes capture and NGS were performed on case III:20, who had moderate hearing impairment in the better ear at the age of 50 years when tested. Targeted genes include 307 known and candidate hearing loss associated genes (Wang et al., 2015). And then genome sequencing was further performed on cases III:13, III:14, III:17 and controls IV;40, IV;44, IV:47. Sequencing was carried out on Illumina HiSeq2500 to generate paired end reads. Biologic information analysis was carried out according to a previous process (Wang et al., 2014), and specific analysis strategies to find the modifier genes were conducted on the genome sequencing data.
RT-qPCR of GJB6
Total RNA was extracted from the white cells of the whole blood samples using the cell RAN kit according to the standard protocol (TRIzol Reagent, Life Technologies). Synthesis of cDNA was carried out with a Revert Aid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). Primers were designed as follows by Primer5: GAPDH-H-F: GGAGCGAGATCCCTCCAAAAT; GAPDH-H-R: GGAGCGAGATCCCTCCAAAAT; GJB6-H-F: CAAGAGGACTTCGTCTGCAAC; GJB6-H-R: GTGGTTTCGTGCCTGTAGTAG. RT-qPCR was conducted according to the protocol of the “Power SYBR Green PCR Master Mix and RT-PCR” by Applied Biosystems. The quantitative analysis conditions were shown in Supplementary Figure 2. Expression stability analysis included geNorm analysis, NormFinder analysis and BestKeeper analysis. Statistical analysis was performed using Graphpad Prism version 5.01. All of the quantitative results were duplicated at least twice.
Results
Clinical Description
In Family 1006983, a total of 22 family members, composed of five clinically affected and 17 unaffected individuals were ascertained in this study. In this family, most of the affected members showed post-lingual, approximately symmetrical, and bilateral non-syndromic sensorineural hearing loss. The average onset age for all the affected cases was 6.2 years old. The hearing loss was presented at all frequencies (Table 1 and Figure 2). For the other three independent sporadic patients in this study, case 1 was a 5 years old girl who had post-lingual, bilateral symmetrical moderate sensorineural hearing impairment, with an onset age of 5 years old (Figures 3A,B); case 2 was a 37-year-old man suffering from sudden hearing loss of left ear accompanied by dizziness and tinnitus when he visited our hospital in 2015, whose hearing threshold had a fluctuation in the last 2 years, and whose onset age was not clear. His hearing threshold was first tested on 16th October, 2014, and the PTAs of the left and right ear were 52.5 and 56.25 dB HL, respectively (Figures 3D,E); Case 3 was a 6 year old boy with post-lingual, bilateral symmetrical moderate sensorineural hearing impairment (Figures 3G,H). All of these three cases had no family history of hearing loss, no symptoms in other organ systems and no other exposure to risk factors. All of the affected cases had associated tinnitus, but no vestibular symptoms or signs were reported except for case 2. HRCT of the temporal bone in the proband of Family 1006983 (III:14) and three sporadic cases showed normal inner ears structure. No exposure history that may account for the hearing impairment existed in any of the affected members.
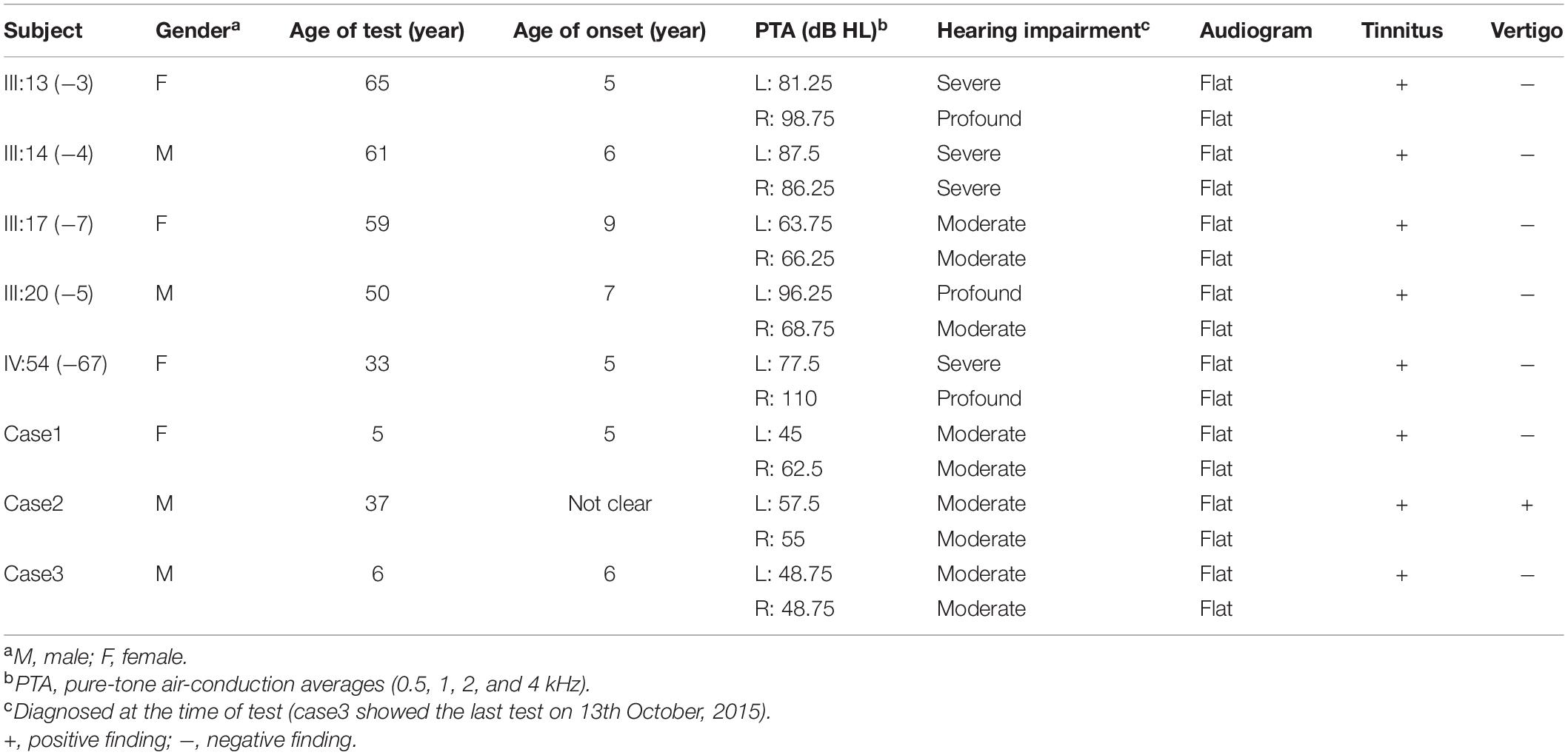
Table 1. Summary of clinical data for hearing impaired members in Family 1006983 and three sporadic cases.
Among the 295 cases with the homozygous GJB2 c.235delC mutation, four patients had late-onset hearing loss, an incidence of 1.36% (4/295) (Zhao et al., 2011). The onset ages of these four cases were 4, 4, 5, and 6 years old, with an average onset age of 4.75 years old. Among these four c.235delC homozygous cases, three children had severe hearing loss, and one had profound hearing impairment. There were also eight patients who had pre-lingual moderate hearing loss, an incidence of 2.71% (8/295).
Mutation Detection and Analysis
The c.235delC is a known pathogenic ARNSHL related mutation. Sanger sequencing confirmed the co-segregation of c.235delC with the disease phenotype in Family 1006983, the three sporadic patients and their parents (Figures 3A,D,G). All of the cases with hearing impairment had homozygous c.235delC mutation, and all of the normal controls recruited in this study had either no mutation or heterozygous c.235delC. The genotype frequency in dbSNP137, HapMap, 1,000 Genomes, and local dataset was less than 0.001. This mutation occurred at highly conserved amino acids, and was predicted to be deleterious by the PolyPhen 2, SIFT, and Mutationtaster programs.
A total of 307 hearing loss related genes were captured, and the targeted high-throughput sequencing data were analysed according to the standard process (Wang et al., 2014) and no pathogenic or likely pathogenic variations were identified according to the standards and guidelines for the interpretation of sequence variants (Richards et al., 2015).
To further analyse the modified genes that may exist in the 1006983 family, six samples from the family were selected for genome sequencing analysis. The total amount of data of the whole genome was shown in Supplementary Table 1. After screening, a total of 7,132,601 digits of variation were obtained. A total of 6.6% of them were novel variants. Sample sequencing depth and coverage are shown in Supplementary Table 2. Variation covers many different types. Four strategies were performed to decipher the possible effective genes. First, assuming that there was a modified gene in this family of patients, all of the heterozygous mutations on chromosome 13 shared by the three patients in Family 1006983, with a frequency of less than 0.0001 and functional mutations were selected (non-coding mutations may also be responsible for the possibility of modification, but due to the huge amount of data, so rare functional mutations were focused on first). Candidate mutation information was shown in Supplementary Table 3. Second, genes interacting with GJB2 were analysed emphatically. Third, copy number variants (CNVs) of genes interacting with GJB2 gene were analysed1 by selecting the 20M gene sequence before and after the target genes, with no related CNV were deciphered. Last, related genes located in the GJB2 signal pathway were determined (Supplementary Table 4), and TJP1 and TUBA3E were found to be most likely to be interacting with GJB2.
RT-qPCR of GJB6
The expression of Connexin30 in the affected members of Family 1006983 showed no significant difference from that in the control group (Figure 4). The expression levels in the three sporadic cases revealed no consistent results, with only case1 having double expression compared to normal control (Figure 3C). Case 2 and case 3 showed no significant increase in the expression level from that of the normal controls or the heterozygous controls (Figures 3F,I).
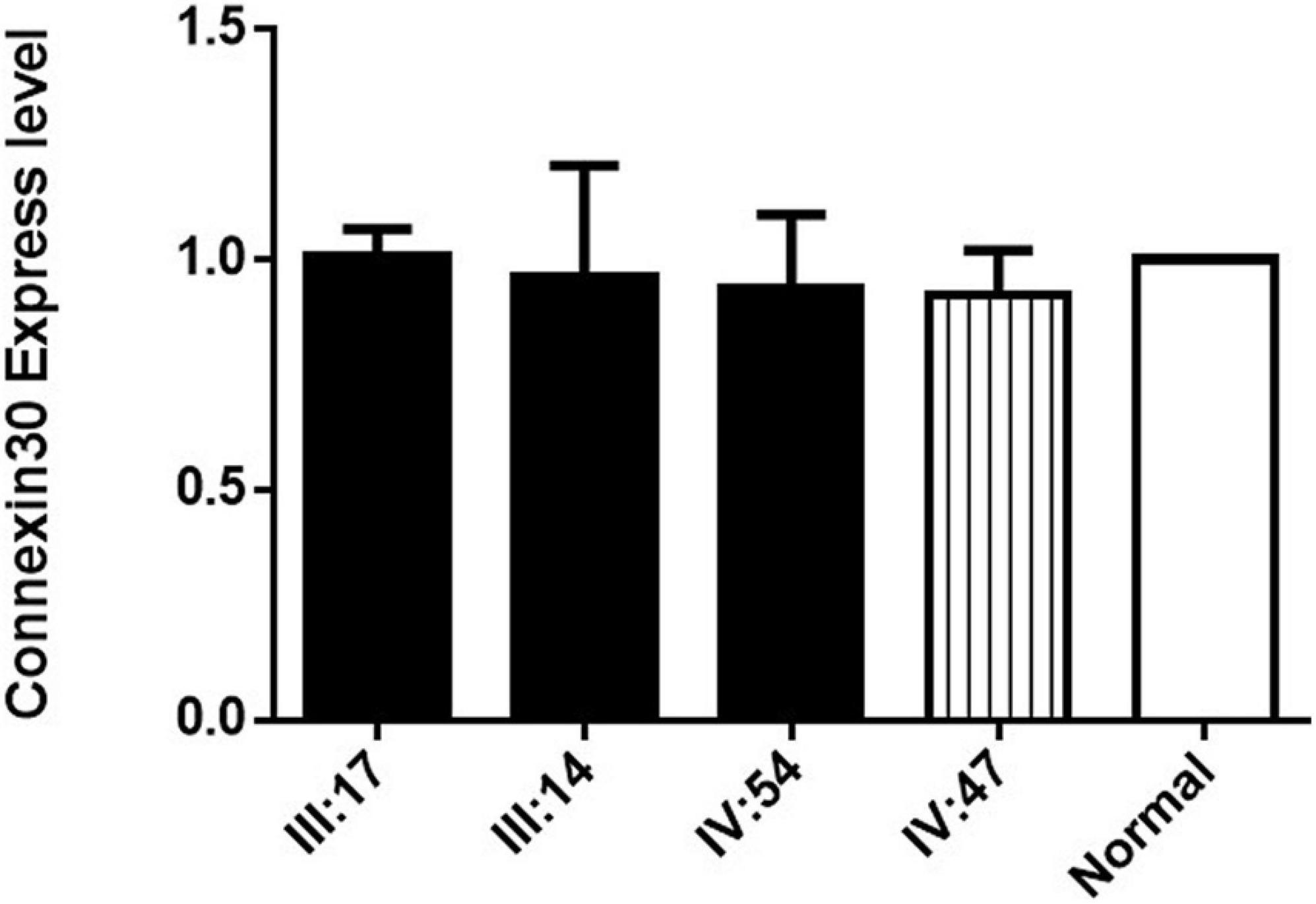
Figure 4. Connexin30 expression of some of the affected members and within-family normal controls from Family 1006983. III:17, III:14, and IV:54 are three hearing loss cases with the GJB2 235delC homozygous mutation, IV:47 is a normal control from Family 1006983. Normal represents the Connexin 30 expression of a person who is not a member of the family.
Discussion
Phenotype-Genotype Correlation Analysis
There is striking phenotypic heterogeneity in the onset age as well as degree of hearing loss caused by GJB2 mutations. For most genotypes, however, there is considerable variation. In clinic, cochlea developmental disorders are the mechanism of congenital hearing loss caused by Connexin26 deficiency, whereas the reduction of cochlea active amplification is the cause of late-onset hearing loss induced by Connexin26 deficiency (Mei et al., 2017). A multicenter genotype-phenotype correlation study showed that inactivating mutations cause a more severe phenotype than non-inactivating mutations (Snoeckx et al., 2005). Among c.235delC homozygotes, the majority do have pre-lingual profound hearing loss, while some c.235delC homozygotes have only post-lingual and/or moderate hearing loss. The phenotypic variances found among the cases recruited in this study are as follows.
Late-Onset Phenotype of Homozygous GJB2 c.235delC
All of the patients from Family 1006983 and the three sporadic cases had late-onset hearing impairment. Previous studies of 295 control cases with homozygous GJB2 c.235delC by our group reported that post-lingual cases accounted for 1.36% of the cohort, indicating a relatively rare variant (Zhao et al., 2011). Though relatively rare, the GJB2 c.35delG homozygous mutation, which is the most prevalent mutation in Caucasians, could also contribute to late postnatal onset hearing loss (Pagarkar et al., 2006). There are also some GJB2 pathogenic mutations associated with post-lingual hearing impairment, such as p.T55N, p.R75Q, p.D179N, and p.C202F (Morle et al., 2000; Primignani et al., 2003; Melchionda et al., 2005; Iossa et al., 2010), which are inherited as autosomal dominant patterns (Wang et al., 2017).
Milder Hearing Impairment Phenotype of Homozygous GJB2 c.235delC
One affected member from Family 1006983 and three sporadic cases had moderate hearing loss, accounting for 50% of the eight cases recruited in this study, and the other four cases from Family 1006983 had severe to profound hearing loss. In fact, among the four severe to profound cases in Family 1006983, the testing age was 33, 59, 61, and 65 years, showing a progressive hearing impairment tendency according to the complaints of these patients.
GJB2 Mutation and Sudden Hearing Loss
Case 2 complained of sudden hearing loss of the left ear when he came to our clinic on 14th September, 2015. In recent years, some studies have proposed that genetic deafness mutations may be associated with the pathogenesis of sudden hearing loss. Bora et al. once screened the GJB2, GJB3, and GJB6 genes in 40 sudden hearing loss patients and 40 normal controls, but the connection between Connexin and sudden hearing loss could not be verified (Bora et al., 2010). In the Chinese population, previous studies showed that the incidence of the GJB2 c.235delC mutation was low in sudden hearing loss patients (2.1%, 5/234) and had no significant difference from that of normal controls, suggesting that GJB2 c.235delC has no correlation with sudden hearing loss (Zhan et al., 2014). In our group, we once screened the GJB2 gene in 93 sudden hearing loss patients and 117 controls with normal hearing. Only two heterozygous c.235delC mutations were identified in the sudden hearing loss group, while five heterozygous c.235delC mutations were identified in the normal control group (data not published).
Genetic Counselling and Clinical Management
Genetic diagnosis and counselling is increasingly affecting the clinical practice of genetic hearing loss, especially genotype-phenotype correlation analysis. GJB2 c.235delC is the most common deafness-related pathogenic variant in the Chinese population, and reporting the phenotypic heterogeneity of GJB2 c.235delC homozygous cases is helpful throughout the genetic counselling procedure, and would contribute greatly to clinical practice. Except for providing information about incidence, recurrent risk and prevention strategies, phenotype heterogeneity should be accounted both before and after gene testing. For example, information if provided on the possible gene testing results and relevant phenotype prognosis before gene testing, phenotype heterogeneity should be taken into consideration; for the management of an individual with GJB2 c.235delC homozygous mutation after gene testing, especially for a baby who has passed newborn hearing screening, audiology follow-up seems to be much more critical than performing cochlear implantation directly. Gene therapy as a potential clinical treatment method have been reported to be efficient in inherit hearing loss in animal models (Tan et al., 2019; Delmaghani and El-Amraoui, 2020; Niggemann et al., 2020). However, there are numerous challenges associated with in vivo gene therapy targeting the human inner ears to be addressed.
Phenotypic Heterogeneity Mechanisms
Cellular and deafness mechanisms underlying GJB2 induced hearing impairment are currently unclear, although numerous clinical reports indicate that Connexin26 mutations are associated with hearing impairment. Few studies have been performed on the pathogenesis of Connexin26 mutations. For example, in one clear functional study on a patient who had the Connexin26 c.35delG mutation and a p.E101G missense mutation, microscopic observation revealed nearly complete degeneration of hair cells and agenesis of the SV but no neural degeneration (Jun et al., 2000). In summary, previous studies on Connexin function showed that the cochlear development disorders lead to congenital deafness instead of hair cell degeneration and endocochlear potential reduction, while the reduction of active cochlear amplification leads to late-onset hearing loss, even though cochlear hair cells have no Connexin expression (Wingard and Zhao, 2015).
Most likely, the phenotypic heterogeneity is caused by modifier genes, but none has been identified to date. Modifier genes can be relatively easily identified in mice by crossing parental inbred strains that carry the disease-causing mutation and exhibit a difference in phenotype, such as the modifier gene Moth1 in the Tubby mouse (Ikeda et al., 2002). However, it is difficult to perform linkage or association analysis to determine the modifier gene in human cases with only one family and a limited number of mild/moderate sporadic patients, such as the situation in this study. In our study, the targeted multi-gene sequence of one of the affected members identified no other variation except for homozygous GJB2 c.235delC. Their identification could substantially contribute to a more accurate diagnosis and more appropriate genetic counselling (Hilgert et al., 2009b). Exome sequencing or genome sequencing may be better choice to find modifier genes in these unconditional cases. However, although we attempted different strategies, it is difficult to analyse the large amount of data of the genome sequencing, with numerous candidate genes being identified, but no further evidence was obtained to confirm the phenotypic correlation. Another family and many more sporadic cases with the GJB2 c.235delC homozygous mutation and late-onset milder hearing impairment may provide the possibility to decipher the molecular mechanism. A WGA study may also an effective way to decipher the major modifier genes of GJB2 c.235delC, as one had been previously performed on a separate set of GJB2 c.35delG cases (Hilgert et al., 2009a). The WGA study needs a second set of cases to replicate the possible SNPs, however, GJB2 c.235delC homozygote cases are limited.
From another point of view, we hypothesised that for some of the patients with more minor phenotypes than those with homozygous GJB2 c.235delC, the overexpression of Connexin30 plays a compensatory role. The mutual pathogenic mechanism of the biallelic GJB2 and GJB6 genes is unclear. These two genes have 77% sequence homology. Connexin26 and Connexin30, which are encoded by these two genes, assemble into a complete heterologous gap junction channel together (Liu et al., 2009), playing a key role in inner ear K+ regulation. GJB2 overexpression can effectively restore hearing impairment in GJB6 knockout mice (Ahmad et al., 2007), suggesting that genetic correction by overexpression may restore hearing function effectively. We found some of the cases overexpressed Connexin3, however, we could not find consistency between the family and sporadic patients in this study. In addition, the total RNA extracted from whole blood could not verify the located expression of Connexin30. Furthermore, biopsy of the human cochlear is impossible for now, and measurement of the gene transcription of Connexins by RT-qPCR and of the protein expression of Connexins by western blots analysis cannot be achieved. We will continue focus on the subjects with the same phenotype in this study, to find out the underlying mechanism of the phenotype heterogeneity. In addition, animal experiments are still needed to find out whether increasing the Connexin30 protein level in Gjb2 knock out background could restore hearing. To all knowledge, there is no data to confirm that up-regulation of Connexin6 expression in Gjb2 knockout mice could not rescue the hearing up to date.
Conclusion
In conclusion, we reported a family and three sporadic patients with homozygous c.235delC, who had an unconditional phenotype with post-lingual and/or moderate hearing impairment. Multi-genes sequencing showed no other pathogenic mutations or modifier genes. RT-qPCR of Connexin30 expression revealed that some of these cases overexpressed Connexin30, which may play a compensatory role in hearing impairment. However, the mechanism of the phenotypic heterogeneity and the specific mutation-induced pathological changes in vivo remain unclear, and there is little information is available for humans. Many more experiments and large series are needed to decipher the genetic code to extend this study in the future.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by the study was approved by the Committee of Medical Ethics of the Chinese PLA General Hospital. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
HW and YG conceived and designed the experiments. HW, YG, JG, LL, and JY performed the experiments. HW, JG, WX, LX, and LY analysed the data. JY and CZ contributed reagents, materials, and analysis tools. HW wrote the manuscript. DW and QW performed the critical reading and discussion of manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the grants of the National Natural Science Foundation of China (Major Project No. 81830028 and Youth Projects 81900950 and 81900951), Military Medical Technology Incubation Project for Youth (19QNP058), Military Family Planning Key Program (19JSZ14), and Beijing Municipal Natural Science Foundation Youth Projects (7204312). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.647240/full#supplementary-material
Supplementary Figure 1 | Flow chart to state the outline of the manuscript.
Supplementary Figure 2 | Quantitative condition of RT-qPCR.
Supplementary Table 1 | The total amount of the whole genome sequencing data of Family 1006983.
Supplementary Table 2 | The whole genome sequencing depth and coverage of Family 1006983.
Supplementary Table 3 | The candidate gene variants of whole genome sequencing from Family 1006983.
Supplementary Table 4 | related genes located in the GJB2 signal pathway (Whole genome sequencing data of Family 1006983).
Footnotes
References
Ahmad, S., Chen, S., Sun, J., and Lin, X. (2003). Connexins 26 and 30 are co-assembled to form gap junctions in the cochlea of mice. Biochem. Biophys. Res. Commun. 307, 362–368. doi: 10.1016/s0006-291x(03)01166-5
Ahmad, S., Tang, W., Chang, Q., Qu, Y., Hibshman, J., Li, Y., et al. (2007). Restoration of connexin26 protein level in the cochlea completely rescues hearing in a mouse model of human connexin30-linked deafness. Proc. Natl. Acad. Sci. U.S.A. 104, 1337–1341. doi: 10.1073/pnas.0606855104
Azaiez, H., Chamberlin, G. P., Fischer, S. M., Welp, C. L., Prasad, S. D., Taggart, R. T., et al. (2004). GJB2: the spectrum of deafness-causing allele variants and their phenotype. Hum. Mutat. 24, 305–311. doi: 10.1002/humu.20084
Bolz, H., Schade, G., Ehmer, S., Kothe, C., Hess, M., and Gal, A. (2004). Phenotypic variability of non-syndromic hearing loss in patients heterozygous for both c.35delG of GJB2 and the 342-kb deletion involving GJB6. Hear. Res. 188, 42–46. doi: 10.1016/s0378-5955(03)00346-0
Bora, A., Altuntas, E. E., Ozdemir, O., Uysal, I. O., and Muderris, S. (2010). [Genetic constitution analysis of idiopathic sudden hearing loss]. Kulak Burun Bogaz Ihtis. Derg. 20, 219–225.
Bykhovskaya, Y., Mengesha, E., Wang, D., Yang, H., Estivill, X., Shohat, M., et al. (2004). Phenotype of non-syndromic deafness associated with the mitochondrial A1555G mutation is modulated by mitochondrial RNA modifying enzymes MTO1 and GTPBP3. Mol. Genet. Metab. 83, 199–206. doi: 10.1016/j.ymgme.2004.07.009
Cama, E., Melchionda, S., Palladino, T., Carella, M., Santarelli, R., Genovese, E., et al. (2009). Hearing loss features in GJB2 biallelic mutations and GJB2/GJB6 digenic inheritance in a large Italian cohort. Int. J. Audiol. 48, 12–17. doi: 10.1080/14992020802400654
Chan, D. K., Schrijver, I., and Chang, K. W. (2010). Connexin-26-associated deafness: phenotypic variability and progression of hearing loss. Genet. Med. 12, 174–181. doi: 10.1097/gim.0b013e3181d0d42b
Cheng, C., Wang, Y., Guo, L., Lu, X., Zhu, W., Muhammad, W., et al. (2019). Age-related transcriptome changes in Sox2+ supporting cells in the mouse cochlea. Stem Cell Res. Ther. 10:365.
Dai, P., Huang, L. H., Wang, G. J., Gao, X., Qu, C. Y., Chen, X. W., et al. (2019). Concurrent hearing and genetic screening of 180,469 neonates with follow-up in Beijing, China. Am. J. Hum. Genet. 105, 803–812.
Dai, Z. Y., Sun, B. C., Huang, S. S., Yuan, Y. Y., Zhu, Y. H., Su, Y., et al. (2015). Correlation analysis of phenotype and genotype of GJB2 in patients with non-syndromic hearing loss in China. Gene 570, 272–276. doi: 10.1016/j.gene.2015.06.038
del Castillo, F. J., and del Castillo, I. (2011). The DFNB1 subtype of autosomal recessive non-syndromic hearing impairment. Front. Biosci. 16, 3252–3274. doi: 10.2741/3910
del Castillo, I., Villamar, M., Moreno-Pelayo, M. A., del Castillo, F. J., Alvarez, A., Tellería, D., et al. (2002). A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N. Engl. J. Med. 346, 243–249. doi: 10.1056/nejmoa012052
Delmaghani, S., and El-Amraoui, A. (2020). Inner ear gene therapies take off: current promises and future challenges. J. Clin. Med. 9:2309. doi: 10.3390/jcm9072309
Dunckley, T., Huentelman, M. J., Craig, D. W., Pearson, J. V., Szelinger, S., Joshipura, K., et al. (2007). Whole-genome analysis of sporadic amyotrophic lateral sclerosis. N. Engl. J. Med. 357, 775–788.
Falah, M., Houshmand, M., Balali, M., Asghari, A., Bagher, Z., Alizadeh, R., et al. (2020). Role of GJB2 and GJB6 in Iranian nonsyndromic hearing impairment: from molecular analysis to literature reviews. Fetal Pediatr. Pathol. 39, 1–12. doi: 10.1080/15513815.2019.1627625
Gabriel, H. D., Jung, D., Butzler, C., Temme, A., Traub, O., Winterhager, E., et al. (1998). Transplacental uptake of glucose is decreased in embryonic lethal connexin26-deficient mice. J. Cell Biol. 140, 1453–1461. doi: 10.1083/jcb.140.6.1453
He, Z., Guo, L., Shu, Y., Fang, Q., Zhou, H., Liu, Y., et al. (2017). Autophagy protects auditory hair cells against neomycin-induced damage. Autophagy 13, 1884–1904. doi: 10.1080/15548627.2017.1359449
He, Z. H., Zou, S. Y., Li, M., Liao, F. L., Wu, X., Sun, H. Y., et al. (2020). The nuclear transcription factor FoxG1 affects the sensitivity of mimetic aging hair cells to inflammation by regulating autophagy pathways. Redox Biol. 28:101364. doi: 10.1016/j.redox.2019.101364
Hilgert, N., Huentelman, M. J., Thorburn, A. Q., Fransen, E., Dieltjens, N., Mueller-Malesinska, M., et al. (2009a). Phenotypic variability of patients homozygous for the GJB2 mutation 35delG cannot be explained by the influence of one major modifier gene. Eur. J. H. Genet. 17, 517–524.
Hilgert, N., Smith, R. J. H., and Van Camp, G. (2009b). Forty-six genes causing nonsyndromic hearing impairment: which ones should be analyzed in DNA diagnostics? Mutat. Res. 681, 189–196. doi: 10.1016/j.mrrev.2008.08.002
Ikeda, A., Zheng, Q. Y., Zuberi, A. R., Johnson, K. R., Naggert, J. K., and Nishina, P. M. (2002). Microtubule-associated protein 1A is a modifier of tubby hearing (moth1). Nat. Genet. 30, 401–405. doi: 10.1038/ng838
Iossa, S., Chinetti, V., Corvino, V., Marciano, E., and Franze, A. (2010). R75Q dominant mutation in GJB2 gene silenced by the in Cis recessive mutation c.35delG. Am. J. Med. Genet. A 152A, 2658–2660. doi: 10.1002/ajmg.a.33630
Jun, A. I., McGuirt, W. T., Hinojosa, R., Green, G. E., Fischel-Ghodsian, N., and Smith, R. J. (2000). Temporal bone histopathology in connexin 26-related hearing loss. Laryngoscope 110(Pt 1), 269–275. doi: 10.1097/00005537-200002010-00016
Liu, X. Z., Yuan, Y., Yan, D., Ding, E. H., Ouyang, X. M., Fei, Y., et al. (2009). Digenic inheritance of non-syndromic deafness caused by mutations at the gap junction proteins Cx26 and Cx31. Hum. Genet. 125, 53–62. doi: 10.1007/s00439-008-0602-9
Liu, Y., Qi, J., Chen, X., Tang, M., Chu, C., Zhu, W., et al. (2019). Critical role of spectrin in hearing development and deafness. Sci. Adv. 5:eaav7803. doi: 10.1126/sciadv.aav7803
Marlin, S., Feldmann, D., Blons, H., Loundon, N., Rouillon, I., Albert, S., et al. (2005). GJB2 and GJB6 mutations: genotypic and phenotypic correlations in a large cohort of hearing-impaired patients. Arch. Otolaryngol. Head Neck Surg. 131, 481–487. doi: 10.1001/archotol.131.6.481
Mei, L., Chen, J., Zong, L., Zhu, Y., Liang, C., Jones, R. O., et al. (2017). A deafness mechanism of digenic Cx26 (GJB2) and Cx30 (GJB6) mutations: reduction of endocochlear potential by impairment of heterogeneous gap junctional function in the cochlear lateral wall. Neurobiol. Dis. 108, 195–203. doi: 10.1016/j.nbd.2017.08.002
Melchionda, S., Bicego, M., Marciano, E., Franzè, A., Morgutti, M., Bortone, G., et al. (2005). Functional characterization of a novel Cx26 (T55N) mutation associated to non-syndromic hearing loss. Biochem. Biophys. Res. Commun. 337, 799–805. doi: 10.1016/j.bbrc.2005.09.116
Minami, S. B., Mutai, H., Nakano, A., Arimoto, Y., Taiji, H., Morimoto, N., et al. (2013). GJB2-associated hearing loss undetected by hearing screening of newborns. Gene 532, 41–45. doi: 10.1016/j.gene.2013.08.094
Morle, L., Bozon, M., Alloisio, N., Latour, P., Vandenberghe, A., Plauchu, H., et al. (2000). A novel C202F mutation in the connexin26 gene (GJB2) associated with autosomal dominant isolated hearing loss. J. Med. Genet. 37, 368–370. doi: 10.1136/jmg.37.5.368
Niggemann, P., Gyorgy, B., and Chen, Z. Y. (2020). Genome and base editing for genetic hearing loss. Hear. Res. 394:107958. doi: 10.1016/j.heares.2020.107958
Pagarkar, W., Bitner-Glindzicz, M., Knight, J., and Sirimanna, T. (2006). Late postnatal onset of hearing loss due to GJB2 mutations. Int. J. Pediatr. Otorhinolaryngol. 70, 1119–1124. doi: 10.1016/j.ijporl.2005.10.026
Primignani, P., Castorina, P., Sironi, F., Curcio, C., Ambrosetti, U., and Coviello, D. A. (2003). A novel dominant missense mutation–D179N–in the GJB2 gene (Connexin 26) associated with non-syndromic hearing loss. Clin. Genet. 63, 516–521. doi: 10.1034/j.1399-0004.2003.00079.x
Qi, J., Liu, Y., Chu, C., Chen, X., Zhu, W., Shu, Y., et al. (2019). A cytoskeleton structure revealed by super-resolution fluorescence imaging in inner ear hair cells. Cell Discov. 5:12.
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and Genomics and the Association for molecular pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Shinagawa, J., Moteki, H., Nishio, S. Y., Noguchi, Y., and Usami, S. I. (2020). Haplotype analysis of GJB2 mutations: founder effect or mutational hot spot? Genes 11:250. doi: 10.3390/genes11030250
Snoeckx, R. L., Huygen, P. L. M., Feldmann, D., Marlin, S., Denoyelle, F., Waligora, J., et al. (2005). GJB2 mutations and degree of hearing loss: a multicenter study. Am. J. Hum. Genet. 77, 945–957.
Stenson, P. D., Mort, M., Ball, E. V., Evans, K., Hayden, M., Heywood, S., et al. (2017). The human gene mutation database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 136, 665–677. doi: 10.1007/s00439-017-1779-6
Tan, F., Chu, C., Qi, J., Li, W., You, D., Li, K., et al. (2019). AAV-ie enables safe and efficient gene transfer to inner ear cells. Nat. Commun. 10:3733.
Tao, F., Beecham, G. W., Rebelo, A. P., Blanton, S. H., Moran, J. J., Lopez-Anido, C., et al. (2019). Modifier gene candidates in Charcot-Marie-Tooth disease type 1a: a case-only genome-wide association study. J. Neuromuscul. Dis. 6, 201–211. doi: 10.3233/jnd-190377
Wang, H., Wu, K., Yu, L., Xie, L., Xiong, W., Wang, D., et al. (2017). A novel dominant GJB2 (DFNA3) mutation in a Chinese family. Sci. Rep. 7:34425.
Wang, H. Y., Zhao, Y. L., Liu, Q., Yuan, H., Gao, Y., Lan, L., et al. (2015). Identification of two disease-causing genes TJP2 and GJB2 in a Chinese family with unconditional autosomal dominant nonsyndromic hereditary hearing impairment. Chin. Med. J. 128, 3345–3351. doi: 10.4103/0366-6999.171440
Wang, H. Y., Zhao, Y. L., Yi, Y. T., Gao, Y., Liu, Q., Wang, D., et al. (2014). Targeted high-throughput sequencing identifies pathogenic mutations in KCNQ4 in two large chinese families with autosomal dominant hearing loss. PLoS One 9:e103133. doi: 10.1371/journal.pone.0103133
Wang, Q., Xiang, J., Sun, J., Yang, Y., Guan, J., Wang, D., et al. (2019). Nationwide population genetic screening improves outcomes of newborn screening for hearing loss in China. Genet. Med. 21, 2231–2238. doi: 10.1038/s41436-019-0481-6
Wang, Y., Li, J., Yao, X., Li, W., Du, H., Tang, M., et al. (2017). Loss of CIB2 causes profound hearing loss and abolishes mechanoelectrical transduction in mice. Front. Mol. Neurosci. 10:401. doi: 10.3389/fnmol.2017.00401
Wingard, J. C., and Zhao, H. B. (2015). Cellular and deafness mechanisms underlying connexin mutation-induced hearing loss – a common hereditary deafness. Front. Cell. Neurosci. 9:202. doi: 10.3389/fncel.2015.00202
Yan, D., Park, H. J., Ouyang, X. M., Pandya, A., Doi, K., Erdenetungalag, R., et al. (2003). Evidence of a founder effect for the 235delC mutation of GJB2 (connexin 26) in east Asians. Hum. Genet. 114, 44–50. doi: 10.1007/s00439-003-1018-1
Yao, J., Lu, Y., Wei, Q., Cao, X., and Xing, G. (2012). A systematic review and meta-analysis of 235delC mutation of GJB2 gene. J. Transl. Med. 10:136. doi: 10.1186/1479-5876-10-136
Zhan, Y., Hu, Y., Huang, X., Chen, H., Guo, C., Xiao, H., et al. (2014). [The study on 235delC mutation of GJB2 gene in patients with idiopathic sudden hearing loss]. Lin Chuang Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 28, 621–623, 634.
Zhao, F. F., Ji, Y. B., Wang, D. Y., Lan, L., Han, M. K., Li, Q., et al. (2011). Phenotype-genotype correlation in 295 chinese deaf subjects with biallelic causative mutations in the gjb2 gene. Genet. Test. Mol. Biomarkers 15, 619–625. doi: 10.1089/gtmb.2010.0192
Zhao, H. B., Kikuchi, T., Ngezahayo, A., and White, T. W. (2006). Gap junctions and cochlear homeostasis. J. Membr. Biol. 209, 177–186. doi: 10.1007/s00232-005-0832-x
Keywords: GJB2, Connexin30, modifier, heterogeneity, hearing loss
Citation: Wang H, Gao Y, Guan J, Lan L, Yang J, Xiong W, Zhao C, Xie L, Yu L, Wang D and Wang Q (2021) Phenotypic Heterogeneity of Post-lingual and/or Milder Hearing Loss for the Patients With the GJB2 c.235delC Homozygous Mutation. Front. Cell Dev. Biol. 9:647240. doi: 10.3389/fcell.2021.647240
Received: 29 December 2020; Accepted: 08 February 2021;
Published: 26 February 2021.
Edited by:
Renjie Chai, Southeast University, ChinaReviewed by:
Lei Xu, Shandong Provincial Hospital, ChinaYong Feng, University of South China Affiliated Changsha Central Hospital, China
Chufeng He, Central South University, China
Copyright © 2021 Wang, Gao, Guan, Lan, Yang, Xiong, Zhao, Xie, Yu, Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongyang Wang, d2h5eDMwMUBmb3htYWlsLmNvbQ==; Qiuju Wang, d3FjcjMwMUB2aXAuc2luYS5jb20=
†These authors have contributed equally to this work