 Luca Pagliaroli
Luca Pagliaroli Marco Trizzino
Marco Trizzino
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol., 04 March 2021
Sec. Stem Cell Research
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.643361
This article is part of the Research TopicChromatin Regulation in Cell Fate DecisionsView all 15 articles
Organismal development is a process that requires a fine-tuned control of cell fate and identity, through timely regulation of lineage-specific genes. These processes are mediated by the concerted action of transcription factors and protein complexes that orchestrate the interaction between cis-regulatory elements (enhancers, promoters) and RNA Polymerase II to elicit transcription. A proper understanding of these dynamics is essential to elucidate the mechanisms underlying developmental diseases. Many developmental disorders, such as Coffin-Siris Syndrome, characterized by growth impairment and intellectual disability are associated with mutations in subunits of the SWI/SNF chromatin remodeler complex, which is an essential regulator of transcription. ARID1B and its paralog ARID1A encode for the two largest, mutually exclusive, subunits of the complex. Mutations in ARID1A and, especially, ARID1B are recurrently associated with a very wide array of developmental disorders, suggesting that these two SWI/SNF subunits play an important role in cell fate decision. In this mini-review we therefore discuss the available scientific literature linking ARID1A and ARID1B to cell fate determination, pluripotency maintenance, and organismal development.
The SWI/SNF (SWItch/Sucrose Non-Fermentable) chromatin remodeling complex leverages an ATP-dependent mechanism to modify the structure of the chromatin and modulate its accessibility to transcriptional regulators (Figure 1). It was first discovered in yeast (SWI/SNF) (Stern et al., 1984), later in Drosophila (Brm-associated protein, BAP) (Kennison and Tamkun, 1988; Tamkun et al., 1992) and finally in mammals (Brg/Brahma-associated factors, BAF) (Wang et al., 1996).
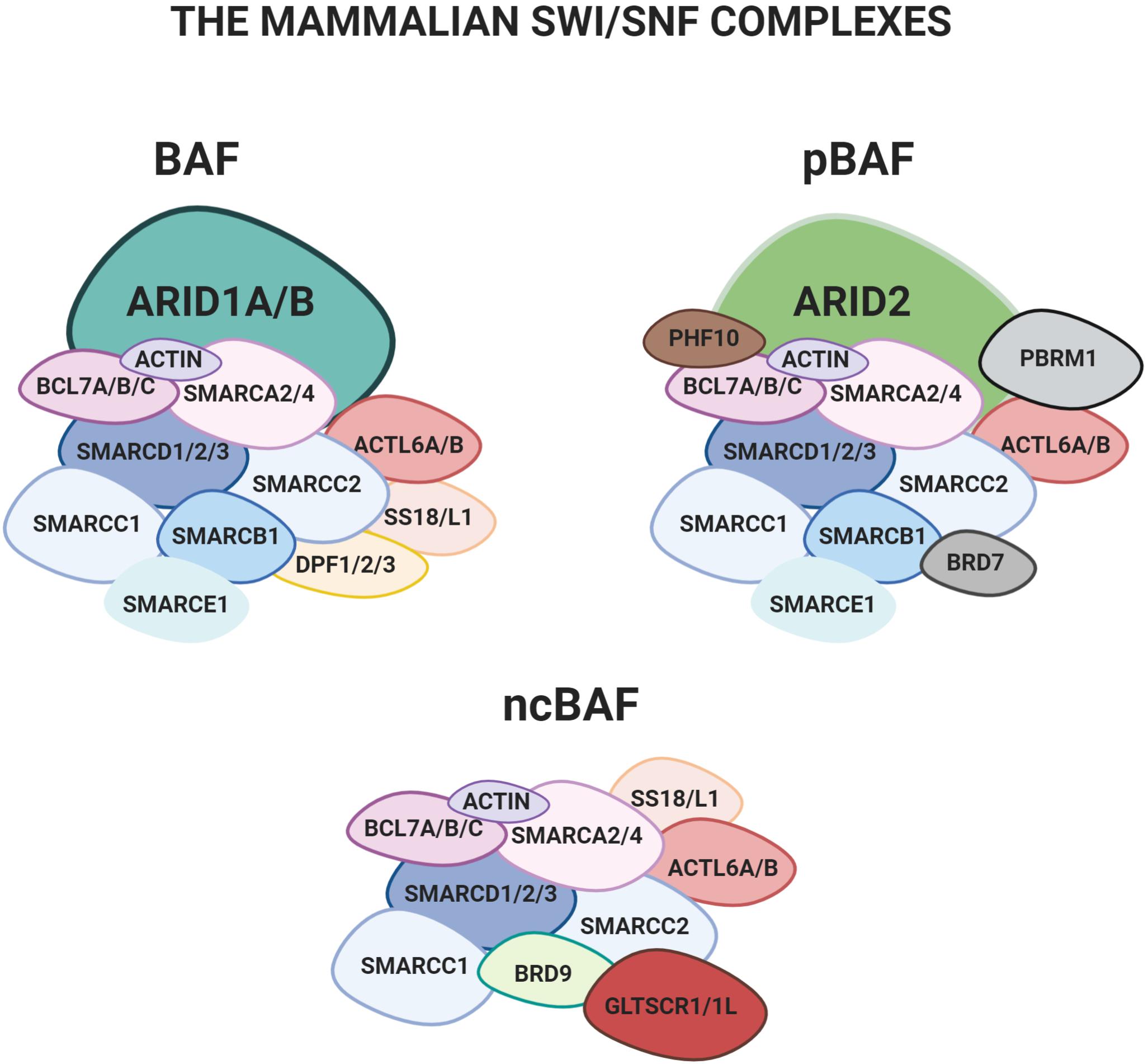
Figure 1. The three main configurations of the mSWI/SNF complex: BAF, pBAF e ncBAF. The two mutually exclusive subunits ARID1A and ARID1B, object of the present review, are only found in the BAF.
In mammals, the different subunits comprise eight different bromodomains, two PHD finger proteins, two chromodomain and multiple proteins with DNA binding domains (Wang et al., 1996a,b; Wang et al., 1998; Lessard et al., 2007). These various subunits are not always present at the same time.
Mammalian SWI/SNF (mSWI/SNF) complexes are assembled from subunits encoded by 29 genes, including multiple paralogs, which generate an extensive diversity in composition. Three versions of the mSWI/SNF were recently characterized in detail: 1) BRG1/BRM-associated factor complex (BAF), 2) polybromo containing complex (pBAF), and 3) a non-canonical version of the complex (ncBAF) (Figure 1) (Mashtalir et al., 2018).
The SWI/SNF complex is able to modify the structure of the chromatin leveraging the energy generated by the hydrolysis of ATP. SWI/SNF binds to the nucleosome in a central cavity where the DNA is exposed (Havas et al., 2000; Saha et al., 2005). Once bound, the complex uses the energy derived by the ATP hydrolysis to break the binding between histones and DNA, promoting the formation of a transient DNA loop that spreads around the nucleosome, ultimately orchestrating the changes in chromatin accessibility (Dechassa et al., 2008; Tang et al., 2010; Tyagi et al., 2016). As a consequence of this process, the chromatin becomes more accessible and permissive to the binding of transcription factors (Kadoch et al., 2017).
All the existing mammalian configurations of the complex contain an ATPase subunit, either SMARCA4 (BRG1) or SMARCA2 (BRM), which catalyzes the hydrolysis of ATP. Several other “core-subunits” are shared by all the different configurations (e.g., SMARCD1/2/3, SMARCC1/2 and a few others; Mashtalir et al., 2018). Finally, some of the subunits are only present in specific configurations. Among these, the mutually exclusive AT-rich interactive domain proteins ARID1A or ARID1B are only found in the BAF (Wilsker et al., 2004; Patsialou et al., 2005; Raab et al., 2015; Mashtalir et al., 2018).
ARID1A and ARID1B are conserved throughout metazoans and expressed across most human cells and tissues. Mutations in the genes encoding for these two subunits are associated with a wide array of developmental disorders and cancers, suggesting that they are implicated in the maintenance of cell identity and in the determination of cell fate. Based on this premise, in the present review, we discuss the role of ARID1A and ARID1B in the maintenance of pluripotency, in the determination of cell fate, and, more broadly, in organismal development.
ARID1A encodes for the AT-Rich Interactive Domain-containing protein 1A (ARID1A/BAF250a). It is the most frequently mutated member of the SWI/SNF family in cancer. ARID1A mutations are associated with a wide range of cancers, including ovarian endometrioid/clear-cell carcinomas, pancreatic cancer, gastric carcinoma, esophageal adenocarcinoma, renal carcinoma and breast tumors (Biegel et al., 2014; Wu et al., 2014; Kadoch and Crabtree, 2015; Masliah-Planchon et al., 2015; Takeda et al., 2016). Despite this evidence, the role of ARID1A in cancer is still not fully understood, with some studies suggesting a tumor suppression role, while a few others indicate an oncogenic function (Fang et al., 2015; Gibson et al., 2016; Zhai et al., 2016; Zhao et al., 2016; Mathur et al., 2017). In the case of endometrial and ovarian cancers, these mutations might either hamper the nuclear import of ARID1A, or affect the ability of ARID1A to interact with the subunits of SWI/SNF complex (Guan et al., 2012). The most frequently dysregulated pathway is PI3K/AKT, along with the downstream signaling cascades PTEN and PIKC3A (Takeda et al., 2016).
Mutations in ARID1A also lead to Coffin-Siris Syndrome, a neurodevelopmental disorder which will be further discussed in this review (Santen et al., 2013; Wieczorek et al., 2013; Kosho et al., 2014; Tsurusaki et al., 2014; Lee and Ki, 2021). ARID1A mutations are typically frame-shift or nonsense, spread across the gene with no specific hotspots, resulting in loss of protein level.
ARID1B encodes for the AT-rich interactive domain-containing protein 1B (ARID1B/BAF250b). ARID1B mutations are commonly associated with neurodevelopmental disorders. Most frequently, ARID1B mutations are de novo haploinsufficient mutations, with no specific gene hotspots.
To date, the majority of the reported mutations are either nonsense or frameshift. These mutations result in non-functional truncated proteins, triggering ARID1B-haploinsufficiency and associated pathologies (Schweingruber et al., 2013; Sim et al., 2015). Mutations in ARID1B may disrupt the ability of the SWI/SNF complex to bind the chromatin (Sim et al., 2015).
Often, ARID1B mutations result in Coffin-Siris Syndrome, a relatively rare genetic disorder that manifests at birth and is characterized by both intellectual disabilities and physical phenotypes (Coffin and Siris, 1970; van der Sluijs et al., 2019). Coffin-Siris patients usually show coarse facial features, impaired craniofacial development, and hypoplastic fifth finger nails (Schrier et al., 2012). Further, most individuals present mild to severe intellectual disability, speech impairment and impaired motor skills (Vergano and Deardorff, 2014). Other characteristics of this disorder include respiratory infections, feeding issues, hearing loss, sparse scalp hair and hypermobility of joints (Vergano and Deardorff, 2014). Mutations in ARID1B have also been linked to Autism Spectrum Disorder (ASD), Intellectual Disabilities (ID), epilepsy and neuroblastoma (Vergano et al., 1993; Halgren et al., 2012; Hoyer et al., 2012; Santen et al., 2012; Vals et al., 2014; Yu et al., 2015; Ben-Salem et al., 2016; Sonmez et al., 2016; Jung et al., 2017; Lee et al., 2017; Shibutani et al., 2017; Yu et al., 2018; Demily et al., 2019; Filatova et al., 2019; Pranckeniene et al., 2019; Sekiguchi et al., 2019; van der Sluijs et al., 2019; Curcio et al., 2020; Fujita et al., 2020; Lian et al., 2020; Pascolini et al., 2020; Smith et al., 2020). ARID1B mutations can be associated with both syndromic and non-syndromic forms of ID (van der Sluijs et al., 2019). In this context, Coffin-Siris patients almost always show some degree of ID, and often present some characteristics that can be associated to Autism Spectrum Disorder. Recently, van der Sluijs et al. (2019) sought to determine genotypic and phenotypic differences between ARID1B-ID and ARID1B-CSS. They found only minor differences between ARID1B-ID and ARID1B-CSS patients, and suggested that ARID1B-related disorders seem to consist of a spectrum, and patients should be managed similarly.
Several studies tried to uncover genes and pathways most commonly dysregulated in ARID1B-ID and ARID1B-CSS. A recent paper looked at gene expression in monocytes of CSS patients. The study identified few differentially expressed genes (CRYZ, TRGV5, TSPAN33, TPPP3, SAMD9L, DDX60, FMN1, PER1, MIR3648, and GSTM1) (Kalmbach et al., 2019) and the pathway analysis did not reveal any statistically significant network. A previous study investigated gene expression in a single CSS patient carrying a novel microduplication of ARID1B, and identified EIF2 signaling and the regulation of eIF4 and p70S6K signaling as top canonical pathways (Seabra et al., 2017). Using an ARID1B-haploinsufficient mouse model, Celen et al. (2017) detected dysregulations in the Ephrin, nNOS, axonal guidance and glutamate receptor signaling pathways. Gene expression profile performed by Shibutani et al. (2017) suggested that ARID1B+/– mice exhibit a pattern very similar to autistic brains centered on immature fast spiking cells. Amongst the several differentially expressed genes, HOXB2, PRL, PODNL1, and PTH2 were the most downregulated, whereas AREG, GBP8, KLR2, and ZP2 were the most upregulated (Shibutani et al., 2017).
The contribution of ARID1A and ARID1B to cell pluripotency has been predominantly investigated in mouse embryos and in embryonic stem cells (ESCs). These cells are distinguished by their ability to differentiate into almost any cell lineage.
Gao et al. (2008) demonstrated that embryos carrying a homozygous ARID1A knockout are able to differentiate in primitive endoderm and epiblast layers but are unable to generate the mesodermal layer. Moreover, ARID1A–/– mouse ESCs fail to maintain a normal stem cell phenotype in culture and spontaneously differentiate (Gao et al., 2008). These pluripotency anomalies seem to be lineage specific, since the ESCs cannot differentiate into cardiomyocytes or adipocytes, but can differentiate into ectoderm-derived neurons (Gao et al., 2008). Consistent with this, Lei et al. (2015) observed dysregulated expression of key developmental and pluripotency genes in ARID1A–/– mouse ESCs. In particular, the most frequently affected genes were associated with the generation of the mesodermal and endodermal layers (Lei et al., 2015).
Similar results were published by Liu J. et al. (2020), who investigated the role of ARID1A in early human cardiac development and neurogenesis. The study demonstrated that homozygous deletion of ARID1A in human ESCs results in spontaneous neuronal differentiation due to increased expression of several genes associated with neurodevelopment. Simultaneously, the same cells displayed downregulation of genes associated with cardiomyocyte differentiation (Liu J. et al., 2020).
Han et al. (2019) studied the function of ARID1A in hematopoietic stem cells (HSC), and uncovered that this SWI/SNF subunit is important for the generation of myeloid colonies, for normal T cell maturation, and for the differentiation of both myeloid and lymphoid lineages.
ARID1A loss/gain of function are thought to have context-dependent effects. For instance, ARID1A deletion is lethal in early embryonic mouse development (Gao et al., 2008). On the other hand, the depletion of this SWI/SNF subunit induces proliferation of ovarian clear cell carcinoma cells (Xiao et al., 2012; Yamamoto et al., 2012; Lakshminarasimhan et al., 2017). In contrast, another study leveraged a mouse ovarian cancer model and demonstrated that ARID1A loss enhances epithelial differentiation and prolongs survival (Chandler et al., 2015; Zhai et al., 2016). ARID1A is instead overexpressed in many hepatocellular carcinomas (Zhao et al., 2016), while the expression of this gene is reduced or lost in colorectal cancer (Mathur et al., 2017).
While there is extensive research investigating the role of ARID1A in cell differentiation, the work performed on ARID1B is thus far limited to a few studies. In this context, Yan et al. (2008) demonstrated that ARID1B–/– mouse ESCs are viable but exhibit a slower proliferation rate and tend to spontaneously differentiate. Consistent with this observation, ESCs with homozygous ARID1B deletion displayed reduced expression of several pluripotency markers, including OCT4 and NANOG. This suggests that ARID1B may be required to regulate stem cell pluripotency. Recently, Boerstler et al. (2020) leveraged CRISPR/Cas9 to generate a human ARID1B-haploinsufficient ESC line with an in-frame deletion of exons 5 and 6 of the gene. Future studies leveraging this cell line may help clarifying the role of ARID1B in pluripotency (Boerstler et al., 2020).
Shi et al. (2020) established ARID1A and ARID1B deletion mutant lines in zebrafish to investigate the effect of these subunits in neuroblastoma. The authors observed that depletion of ARID1A or ARID1B results in an increased rate of cell proliferation in the sympathoadrenal lineage, which ultimately leads to higher tumor penetrance (Shi et al., 2020).
A zebrafish model was also used to elucidate how ARID1B regulates organismal development (Liu X. et al., 2020). In this study, the authors demonstrated that ARID1B haploinsufficiency results in reduced body length due to dysregulated Wnt/β-catenin signaling pathway (Liu X. et al., 2020). An association between ARID1B and the Wnt/β-catenin signaling pathway had already been proposed by Vasileiou et al. (2015).
ARID1A was associated with neural crest differentiation and craniofacial development (Chandler and Magnuson, 2016). Neural crest cells are a transient, ectoderm-derived, cell population that can migrate throughout the embryo to give origin to craniofacial bone and cartilage, peripheral neurons and glia, melanocytes, and smooth muscle cells (Shakhova and Sommer, 2008). Chandler and Magnuson (2016) generated mice with a conditional, neural crest specific, heterozygous deletion of ARID1A. The ARID1A-depleted mice displayed craniofacial defects, including shortened snouts and low ears. Additionally, most of the bones involved in the ventral cranial skeleton were greatly reduced in size, leading to abnormal facial features (Chandler and Magnuson, 2016). The study also revealed that conditional haploinsufficiency of ARID1A results in defects in developing cardiac neural crest due to an incomplete colonization of the outflow tract and septation of the arterial trunk, ultimately producing defects in the pharyngeal arch arteries. Consistently, homozygous ARID1A mutants did not survive in utero (Chandler and Magnuson, 2016).
As mentioned, mutations in ARID1B often lead to a wide array of neurodevelopmental disorders, including Autism Spectrum Disorders, Coffin-Siris Syndrome, and other forms of Intellectual Disabilities (Vergano et al., 1993; Halgren et al., 2012; Hoyer et al., 2012; Santen et al., 2012; Vals et al., 2014; Yu et al., 2015; Ben-Salem et al., 2016; Sonmez et al., 2016; Jung et al., 2017; Lee et al., 2017; Shibutani et al., 2017; Yu et al., 2018; Demily et al., 2019; Filatova et al., 2019; Pranckeniene et al., 2019; Sekiguchi et al., 2019; van der Sluijs et al., 2019; Curcio et al., 2020; Fujita et al., 2020; Lian et al., 2020; Pascolini et al., 2020; Smith et al., 2020).
Based on these lines of evidence, several studies investigated the role of ARID1B in neurodevelopment (Ka et al., 2016; Celen et al., 2017; Jung et al., 2017; Shibutani et al., 2017; Smith et al., 2020). Ka et al. (2016) demonstrated that ARID1B is required for arborization and dendrite growth in cortical and hippocampal pyramidal neurons. ARID1B haploinsufficiency resulted in reduced dendritic innervation as well as diminished attachment of dendrites to the pial surface (Ka et al., 2016). In the same study, Ka et al. (2016) found that ARID1B mono-allelic loss impairs the formation and maturation of dendritic spines, generating malformations that morphologically resemble those reported in animal models of multiple neuropsychiatric disorders such as ID, ASD, Rett-Syndrome, Down-Syndrome and Fragile-X-Syndrome (Irwin et al., 2002; McKinney et al., 2005; Jentarra et al., 2010; Moffat et al., 2019).
Recently, Smith et al. (2020) leveraged a mouse model to elucidate the consequences of ARID1B-haploinsufficiency on the development and function of parvalbumin (PV) and somatostatin (SST) neurons, two of the most prevalent interneuron subtypes. Briefly, the authors discovered that ARID1B-haploinsufficiency in PV neurons leads to social and emotional impairments, which are key features of ASD, while ARID1B deficiency in the SST population results in learning and memory dysfunction (Smith et al., 2020).
In a similar study, Jung et al. (2017) demonstrated that ARID1B-haploinsufficient mice present a reduced number of cortical GABAergic interneurons and decreased proliferation of interneuron progenitors in the ganglionic eminence. These neurological phenotypes are often recovered in autism and schizophrenia patients (Benes and Berretta, 2001; Pizzarelli and Cherubini, 2011). Additionally, in a third mouse model study, Celen et al. (2017) showed that ARID1B-haploinsufficient mice are characterized by hydrocephalus, a condition frequently reported also in Coffin-Siris patients (Schrier Vergano et al., 1993). Brain abnormalities were detected in ARID1B-haploinsufficient mice also by Shibutani et al. (2017). The ARID1B-haploinsufficient mice also exhibited reduced size of the corpus callosum and dentate gyrus, along with impairment in social behavior, altered vocalization, presence of anxiety-like behavior, and growth deficit (Celen et al., 2017).
Recent studies conducted on mouse liver linked ARID1A to tissue regeneration (Sun et al., 2016; Li et al., 2019). Specifically, Li et al. (2019) demonstrated that ARID1A is required for the generation of liver-progenitor-like cells (LPLCs) in different types of periportal injuries. In detail, mice with conditional ARID1A knockout in the liver displayed impaired LPLCs formation and reduced regeneration of damaged liver tissue (Li et al., 2019). Moreover, ARID1A-knockout livers were characterized by significantly increased accumulation of fatty vacuoles and impaired liver function. Conversely, a prior study also performed in the mouse liver demonstrated that suppression of ARID1A is sufficient to promote liver regeneration (Sun et al., 2016). These two studies suggest a dual role for ARID1A in the hepatic context. Sun et al. (2016) proposed a mechanism focused on CYP-metabolism. On the other hand, Li et al. (2019) suggested the presence of a hepatocyte plasticity network where ARID1A promotes the formation of LPLC during injury, while hampering cell proliferation during the recovery stage.
The role of ARID1A and ARID1B in cell proliferation was also investigated using mouse derived preosteoblasts (Nagl Jr., Patsialou et al., 2005; Flores-Alcantar et al., 2011). Notably, Nagl Jr., Patsialou et al. (2005) showed that deletion of ARID1A leads to failure in cell cycle arrest. Conversely, the same study demonstrated that loss of ARID1B has not significant impact on the cell cycle (Nagl et al., 2005).
The SWI/SNF complex is mainly considered as a transcriptional activator, which antagonizes the Polycomb Repressor Complexes (PRC1 and PRC2) in the modulation of gene expression (Kadoch and Crabtree, 2015; Alfert et al., 2019). Nonetheless, repressing activity for the SWI/SNF has also been reported. For instance, a recent study performed on HepG2 cells (hepatocellular carcinoma line) uncovered that ARID1A-containing BAF activates and represses roughly equal numbers of genes (Raab et al., 2015). The same study also demonstrated that ARID1B-containing BAF is primarily a repressor of enhancer activity (Raab et al., 2015). More specifically, Raab et al. (2015) investigated the localization of ARID1A and ARID1B binding sites in HepG2 cells via chromatin immunoprecipitation followed by sequencing (ChIP-seq). The authors observed binding of ARID1A at most enhancers and promoters, while ARID1B was predominantly located at enhancers. Loss of ARID1A from HepG2 cells resulted in a roughly equal number of activated and repressed genes, whereas loss of ARID1B predominantly resulted in transcriptional activation (Raab et al., 2015).
Consistently, ARID1A and ARID1B have been recently associated with acetylation of histone tails at both enhancers and promoters (Chandler et al., 2013; Lei et al., 2015; Raab et al., 2015; Alver et al., 2017; Kelso et al., 2017; Trizzino et al., 2018). For example, Mathur et al. (2017) observed that human ARID1A–/– colorectal cancer cells display dampened acetylation levels at Histone H3 lysine 27 (H3K27ac), which is usually associated with transcriptional activity at enhancers and promoters. Correlation between ARID1A loss and attenuation of enhancer acetylation was also observed in zebrafish models (Shi et al., 2020).
Liu X. et al. (2020) profiled chromatin accessibility in wild-type and ARID1A-deleted human ES cells. With these experiments, the authors discovered that loss of ARID1A generated a loss in accessibility at cardiogenic genes, as well as an increase in accessibility at neurogenic genes (Liu X. et al., 2020). These data are thus consistent with a dual (activator/repressor) role of ARID1A in the transcriptional regulation of ESCs. An additional study on human ES cells also revealed that acute depletion of ARID1A increases nucleosome occupancy, and therefore repression, at a set of H3K4me3- and/or H3K27me3-associated promoters (Lei et al., 2015).
The consequences of ARID1A loss on chromatin accessibility were further investigated by Kelso et al. (2017) in colorectal carcinoma lines. The authors demonstrated that loss of ARID1A and ARID1B correlates with global dampening of chromatin accessibility, along with a significant decrease of histone modifications normally associated with transcriptional activation at enhancers (Kelso et al., 2017).
Recently, Wu et al. (2019) linked ARID1A-containing BAF to Condensin, a protein complex involved in the regulation of genomic organization and chromatin looping. The study demonstrated that ovarian cancer cell lines depleted of ARID1A exhibit decreased binding of Condensing-II at active enhancers. Further, they illustrated that ARID1A-loss leads to improper genome compartmentalization (Wu et al., 2019).
Finally, in a recent study conducted in ovarian cancer cell lines, Trizzino et al. (2018) demonstrated that ARID1A and ARID1B play a role in the regulation of RNA Polymerase II promoter-proximal pausing, a widespread mechanism that controls the timing of expression of developmental genes genome-wide.
In conclusion, multiple lines of evidence point toward a model in which the ARID1A- and ARID1B-containing configurations of the SWI/SNF complex (i.e., the BAF) play an important role in the regulation of pluripotency, as well as in cell fate determination and development (Table 1). Multiple molecular and genomic functions were ascribed to these two SWI/SNF subunits. However, the mechanisms by which ARID1A and ARID1B regulate pluripotency and cell fate are still not fully understood and are likely context-specific. The discovery of such mechanisms, along with the transcription factors and the molecular pathways involved, may open new roads for the diagnosis and the treatment of developmental disorders and cancer.
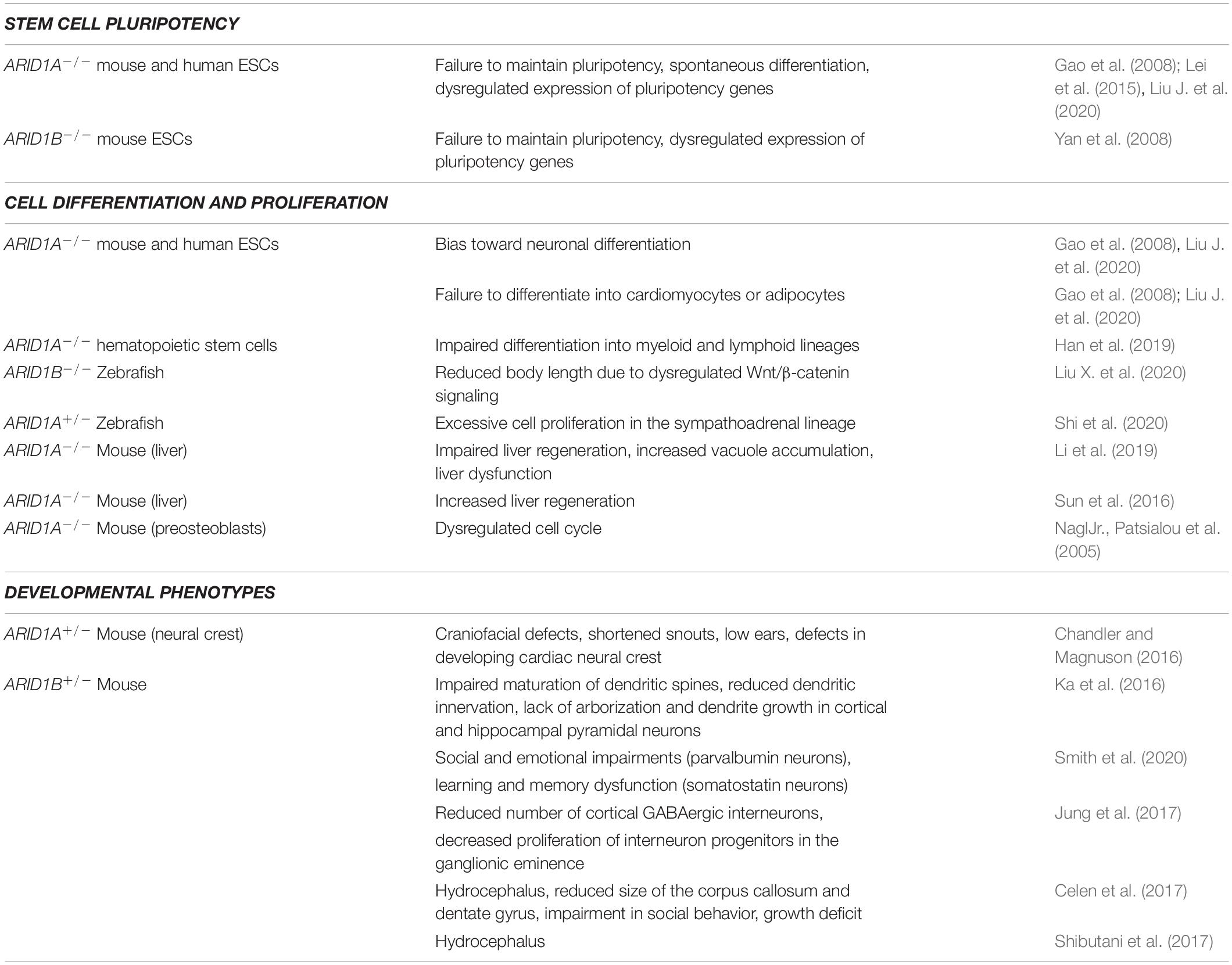
Table 1. Biological processes and phenotypes associated to ARID1A and ARID1B.
MT and LP designed and wrote the manuscript. Both authors contributed to the article and approved the submitted version.
Funding for this review were provided to MT by the G. Harold and Leila Y. Mathers Foundation. LP and MT were also supported by the Thomas Jefferson University Open Access Fund.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are thankful to Dr. Sruti Patoori (TJU) for providing insightful comments on the manuscript.
Alfert, A., Moreno, N., and Kerl, K. (2019). The BAF complex in development and disease. Epigenetics Chromatin 12:19.
Alver, B. H., Kim, K. H., Lu, P., Wang, X., Manchester, H. E., Wang, W., et al. (2017). The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat. Commun. 8:14648.
Benes, F. M., and Berretta, S. (2001). GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology 25, 1–27. doi: 10.1016/s0893-133x(01)00225-1
Ben-Salem, S., Sobreira, N., Akawi, N. A., Al-Shamsi, A. M., John, A., Pramathan, T., et al. (2016). Gonadal mosaicism in ARID1B gene causes intellectual disability and dysmorphic features in three siblings. Am. J. Med. Genet. A 170A, 156–161. doi: 10.1002/ajmg.a.37405
Biegel, J. A., Busse, T. M., and Weissman, B. E. (2014). SWI/SNF chromatin remodeling complexes and cancer. Am. J. Med. Genet. C Semin. Med. Genet. 166C, 350–366.
Boerstler, T., Wend, H., Krumbiegel, M., Kavyanifar, A., Reis, A., Lie, D. C., et al. (2020). CRISPR/Cas9 mediated generation of human ARID1B heterozygous knockout hESC lines to model Coffin-Siris syndrome. Stem. Cell Res. 47:101889. doi: 10.1016/j.scr.2020.101889
Celen, C., Chuang, J. C., Luo, X., Nijem, N., Walker, A. K., Chen, F., et al. (2017). Arid1b haploinsufficient mice reveal neuropsychiatric phenotypes and reversible causes of growth impairment. Elife 6:e25730.
Chandler, R. L., Brennan, J., Schisler, J. C., Serber, D., Patterson, C., and Magnuson, T. (2013). ARID1a-DNA interactions are required for promoter occupancy by SWI/SNF. Mol. Cell. Biol. 33, 265–280. doi: 10.1128/mcb.01008-12
Chandler, R. L., Damrauer, J. S., Raab, J. R., Schisler, J. C., Wilkerson, M. D., Didion, J. P., et al. (2015). Coexistent ARID1A-PIK3CA mutations promote ovarian clear-cell tumorigenesis through pro-tumorigenic inflammatory cytokine signalling. Nat. Commun. 6:6118.
Chandler, R. L., and Magnuson, T. (2016). The SWI/SNF BAF-A complex is essential for neural crest development. Dev. Biol. 411, 15–24. doi: 10.1016/j.ydbio.2016.01.015
Coffin, G. S., and Siris, E. (1970). Mental retardation with absent fifth fingernail and terminal phalanx. Am. J. Dis. Child. 119, 433–439. doi: 10.1001/archpedi.1970.02100050435009
Curcio, M. R., Ferranti, S., Lotti, F., and Grosso, S. (2020). Coffin-Siris syndrome and epilepsy. Neurol. Sci. 42, 727–729. doi: 10.1007/s10072-020-04782-y
Dechassa, M. L., Zhang, B., Horowitz-Scherer, R., Persinger, J., Woodcock, C. L., Peterson, C. L., et al. (2008). Architecture of the SWI/SNF-nucleosome complex. Mol. Cell. Biol. 28, 6010–6021. doi: 10.1128/mcb.00693-08
Demily, C., Duwime, C., Lopez, C., Hemimou, C., Poisson, A., Plasse, J., et al. (2019). Corpus callosum metrics predict severity of visuospatial and neuromotor dysfunctions in ARID1B mutations with Coffin-Siris syndrome. Psychiatr. Genet. 29, 237–242. doi: 10.1097/ypg.0000000000000225
Fang, J. Z., Li, C., Liu, X. Y., Hu, T. T., Fan, Z. S., and Han, Z. G. (2015). Hepatocyte-specific arid1a deficiency initiates mouse steatohepatitis and hepatocellular carcinoma. PLoS One 10:e0143042. doi: 10.1371/journal.pone.0143042
Filatova, A., Rey, L. K., Lechler, M. B., Schaper, J., Hempel, M., Posmyk, R., et al. (2019). Mutations in SMARCB1 and in other Coffin-Siris syndrome genes lead to various brain midline defects. Nat. Commun. 10:2966.
Flores-Alcantar, A., Gonzalez-Sandoval, A., Escalante-Alcalde, D., and Lomeli, H. (2011). Dynamics of expression of ARID1A and ARID1B subunits in mouse embryos and in cells during the cell cycle. Cell Tissue Res. 345, 137–148. doi: 10.1007/s00441-011-1182-x
Fujita, T., Ihara, Y., Hayashi, H., Ishii, A., Ideguchi, H., Inoue, T., et al. (2020). Coffin-Siris syndrome with bilateral macular dysplasia caused by a novel exonic deletion in ARID1B. Congenit. Anom. (Kyoto) 60, 189–193. doi: 10.1111/cga.12383
Gao, X., Tate, P., Hu, P., Tjian, R., Skarnes, W. C., and Wang, Z. (2008). ES cell pluripotency and germ-layer formation require the SWI/SNF chromatin remodeling component BAF250a. Proc. Natl. Acad. Sci. U.S.A. 105, 6656–6661. doi: 10.1073/pnas.0801802105
Gibson, W. J., Hoivik, E. A., Halle, M. K., Taylor-Weiner, A., Cherniack, A. D., Berg, A., et al. (2016). The genomic landscape and evolution of endometrial carcinoma progression and abdominopelvic metastasis. Nat. Genet. 48, 848–855. doi: 10.1038/ng.3602
Guan, B., Gao, M., Wu, C. H., Wang, T. L., and Shih Ie, M. (2012). Functional analysis of in-frame indel ARID1A mutations reveals new regulatory mechanisms of its tumor suppressor functions. Neoplasia 14, 986–993. doi: 10.1593/neo.121218
Halgren, C., Kjaergaard, S., Bak, M., Hansen, C., El-Schich, Z., Anderson, C. M., et al. (2012). Corpus callosum abnormalities, intellectual disability, speech impairment, and autism in patients with haploinsufficiency of ARID1B. Clin. Genet. 82, 248–255. doi: 10.1111/j.1399-0004.2011.01755.x
Han, L., Madan, V., Mayakonda, A., Dakle, P., Woon, T. W., Shyamsunder, P., et al. (2019). Chromatin remodeling mediated by ARID1A is indispensable for normal hematopoiesis in mice. Leukemia 33, 2291–2305. doi: 10.1038/s41375-019-0438-4
Havas, K., Flaus, A., Phelan, M., Kingston, R., Wade, P. A., Lilley, D. M., et al. (2000). Generation of superhelical torsion by ATP-dependent chromatin remodeling activities. Cell 103, 1133–1142. doi: 10.1016/s0092-8674(00)00215-4
Hoyer, J., Ekici, A. B., Endele, S., Popp, B., Zweier, C., Wiesener, A., et al. (2012). Haploinsufficiency of ARID1B, a member of the SWI/SNF-a chromatin-remodeling complex, is a frequent cause of intellectual disability. Am. J. Hum. Genet. 90, 565–572. doi: 10.1016/j.ajhg.2012.02.007
Irwin, S. A., Idupulapati, M., Gilbert, M. E., Harris, J. B., Chakravarti, A. B., Rogers, E. J., et al. (2002). Dendritic spine and dendritic field characteristics of layer V pyramidal neurons in the visual cortex of fragile-X knockout mice. Am. J. Med. Genet. 111, 140–146. doi: 10.1002/ajmg.10500
Jentarra, G. M., Olfers, S. L., Rice, S. G., Srivastava, N., Homanics, G. E., Blue, M., et al. (2010). Abnormalities of cell packing density and dendritic complexity in the MeCP2 A140V mouse model of Rett syndrome/X-linked mental retardation. BMC Neurosci. 11:19. doi: 10.1186/1471-2202-11-19
Jung, E. M., Moffat, J. J., Liu, J., Dravid, S. M., Gurumurthy, C. B., and Kim, W. Y. (2017). Arid1b haploinsufficiency disrupts cortical interneuron development and mouse behavior. Nat. Neurosci. 20, 1694–1707. doi: 10.1038/s41593-017-0013-0
Ka, M., Chopra, D. A., Dravid, S. M., and Kim, W. Y. (2016). Essential roles for ARID1B in dendritic arborization and spine morphology of developing pyramidal neurons. J. Neurosci. 36, 2723–2742. doi: 10.1523/jneurosci.2321-15.2016
Kadoch, C., and Crabtree, G. R. (2015). Mammalian SWI/SNF chromatin remodeling complexes and cancer: mechanistic insights gained from human genomics. Sci. Adv. 1:e1500447. doi: 10.1126/sciadv.1500447
Kadoch, C., Williams, R. T., Calarco, J. P., Miller, E. L., Weber, C. M., Braun, S. M., et al. (2017). Dynamics of BAF-Polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat. Genet. 49, 213–222. doi: 10.1038/ng.3734
Kalmbach, A., Schroder, C., Klein-Hitpass, L., Nordstrom, K., Ulz, P., Heitzer, E., et al. (2019). Genome-wide analysis of the nucleosome landscape in individuals with Coffin-Siris Syndrome. Cytogenet. Genome Res. 159, 1–11. doi: 10.1159/000503266
Kelso, T. W. R., Porter, D. K., Amaral, M. L., Shokhirev, M. N., Benner, C., and Hargreaves, D. C. (2017). Chromatin accessibility underlies synthetic lethality of SWI/SNF subunits in ARID1A-mutant cancers. Elife 6:e30506.
Kennison, J. A., and Tamkun, J. W. (1988). Dosage-dependent modifiers of polycomb and antennapedia mutations in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 85, 8136–8140. doi: 10.1073/pnas.85.21.8136
Kosho, T., Okamoto, N., and Coffin-Siris Syndrome International Collaborators. (2014). Genotype-phenotype correlation of Coffin-Siris syndrome caused by mutations in SMARCB1, SMARCA4, SMARCE1, and ARID1A. Am. J. Med. Genet. C Semin. Med. Genet. 166C, 262–275. doi: 10.1002/ajmg.c.31407
Lakshminarasimhan, R., Andreu-Vieyra, C., Lawrenson, K., Duymich, C. E., Gayther, S. A., Liang, G., et al. (2017). Down-regulation of ARID1A is sufficient to initiate neoplastic transformation along with epigenetic reprogramming in non-tumorigenic endometriotic cells. Cancer Lett. 401, 11–19. doi: 10.1016/j.canlet.2017.04.040
Lee, C. G., and Ki, C. S. (2021). A novel de novo heterozygous ARID1A missense variant cluster in cis c.[5954C>G;6314C>T;6334C>T;6843G>C] causes a Coffin-Siris syndrome. Ann. Lab. Med. 41, 350–353. doi: 10.3343/alm.2021.41.3.350
Lee, S. H., Kim, J. S., Zheng, S., Huse, J. T., Bae, J. S., Lee, J. W., et al. (2017). ARID1B alterations identify aggressive tumors in neuroblastoma. Oncotarget 8, 45943–45950. doi: 10.18632/oncotarget.17500
Lei, I., West, J., Yan, Z., Gao, X., Fang, P., Dennis, J. H., et al. (2015). BAF250a protein regulates nucleosome occupancy and histone modifications in priming embryonic stem cell differentiation. J. Biol. Chem. 290, 19343–19352. doi: 10.1074/jbc.m115.637389
Lessard, J., Wu, J. I., Ranish, J. A., Wan, M., Winslow, M. M., Staahl, B. T., et al. (2007). An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron 55, 201–215. doi: 10.1016/j.neuron.2007.06.019
Li, W., Yang, L., He, Q., Hu, C., Zhu, L., Ma, X., et al. (2019). A homeostatic arid1a-dependent permissive chromatin state licenses hepatocyte responsiveness to liver-injury-associated YAP signaling. Cell Stem Cell 25, 54–68.e5.
Lian, S., Ting, T. W., Lai, A. H. M., Tan, E. S., Wei, H., Cham, B., et al. (2020). Coffin-Siris syndrome-1: report of five cases from Asian populations with truncating mutations in the ARID1B gene. J. Neurol. Sci. 414, 116819. doi: 10.1016/j.jns.2020.116819
Liu, J., Liu, S., Gao, H., Han, L., Chu, X., Sheng, Y., et al. (2020). Genome-wide studies reveal the essential and opposite roles of ARID1A in controlling human cardiogenesis and neurogenesis from pluripotent stem cells. Genome Biol. 21:169.
Liu, X., Hu, G., Ye, J., Ye, B., Shen, N., Tao, Y., et al. (2020). De Novo ARID1B mutations cause growth delay associated with aberrant Wnt/beta-catenin signaling. Hum. Mutat. 41, 1012–1024. doi: 10.1002/humu.23990
Mashtalir, N., D’Avino, A. R., Michel, B. C., Luo, J., Pan, J., Otto, J. E., et al. (2018). Modular organization and assembly of SWI/SNF family chromatin remodeling complexes. Cell 175, 1272–1288e20.
Masliah-Planchon, J., Bieche, I., Guinebretiere, J. M., Bourdeaut, F., and Delattre, O. (2015). SWI/SNF chromatin remodeling and human malignancies. Annu. Rev. Pathol. 10, 145–171.
Mathur, R., Alver, B. H., San Roman, A. K., Wilson, B. G., Wang, X., Agoston, A. T., et al. (2017). ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat. Genet. 49, 296–302. doi: 10.1038/ng.3744
McKinney, B. C., Grossman, A. W., Elisseou, N. M., and Greenough, W. T. (2005). Dendritic spine abnormalities in the occipital cortex of C57BL/6 Fmr1 knockout mice. Am. J. Med. Genet. B Neuropsychiatr. Genet. 136B, 98–102. doi: 10.1002/ajmg.b.30183
Moffat, J. J., Jung, E. M., Ka, M., Smith, A. L., Jeon, B. T., Santen, G. W. E., et al. (2019). The role of ARID1B, a BAF chromatin remodeling complex subunit, in neural development and behavior. Prog. Neuropsychopharmacol. Biol. Psychiatry 89, 30–38. doi: 10.1016/j.pnpbp.2018.08.021
Nagl, N. G. Jr., Patsialou, A., Haines, D. S., Dallas, P. B., Beck, G. R. Jr., and Moran, E. (2005). The p270 (ARID1A/SMARCF1) subunit of mammalian SWI/SNF-related complexes is essential for normal cell cycle arrest. Cancer Res. 65, 9236–9244. doi: 10.1158/0008-5472.can-05-1225
Pascolini, G., Valiante, M., Bottillo, I., Laino, L., Fleischer, N., Ferraris, A., et al. (2020). Striking phenotypic overlap between Nicolaides-Baraitser and Coffin-Siris syndromes in monozygotic twins with ARID1B intragenic deletion. Eur. J. Med. Genet. 63:103739. doi: 10.1016/j.ejmg.2019.103739
Patsialou, A., Wilsker, D., and Moran, E. (2005). DNA-binding properties of ARID family proteins. Nucleic Acids Res. 33, 66–80. doi: 10.1093/nar/gki145
Pizzarelli, R., and Cherubini, E. (2011). Alterations of GABAergic signaling in autism spectrum disorders. Neural Plast. 2011:297153.
Pranckeniene, L., Siavriene, E., Gueneau, L., Preiksaitiene, E., Mikstiene, V., Reymond, A., et al. (2019). De novo splice site variant of ARID1B associated with pathogenesis of Coffin-Siris syndrome. Mol. Genet. Genomic. Med. 7:e1006.
Raab, J. R., Resnick, S., and Magnuson, T. (2015). Genome-wide transcriptional regulation mediated by biochemically distinct SWI/SNF complexes. PLoS Genet. 11:e1005748. doi: 10.1371/journal.pgen.1005748
Saha, A., Wittmeyer, J., and Cairns, B. R. (2005). Chromatin remodeling through directional DNA translocation from an internal nucleosomal site. Nat. Struct. Mol. Biol. 12, 747–755. doi: 10.1038/nsmb973
Santen, G. W., Aten, E., Sun, Y., Almomani, R., Gilissen, C., Nielsen, M., et al. (2012). Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat. Genet. 44, 379–380. doi: 10.1038/ng.2217
Santen, G. W., Aten, E., Vulto-van Silfhout, A. T., Pottinger, C., van Bon, B. W. I, and van Minderhout, J. (2013). Coffin-Siris syndrome and the BAF complex: genotype-phenotype study in 63 patients. Hum. Mutat. 34, 1519–1528.
Schrier, S. A., Bodurtha, J. N., Burton, B., Chudley, A. E., Chiong, M. A., D’Avanzo, G. M., et al. (2012). The Coffin-Siris syndrome: a proposed diagnostic approach and assessment of 15 overlapping cases. Am. J. Med. Genet. A 158A, 1865–1876. doi: 10.1002/ajmg.a.35415
Schrier Vergano, S., Santen, G., Wieczorek, D., Wollnik, B., Matsumoto, N., and Deardorff, M. A. (1993). “Coffin-Siris Syndrome,” in GeneReviews((R)), eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, et al. (Seattle, WA: University of Washington).
Schweingruber, C., Rufener, S. C., Zund, D., Yamashita, A., and Muhlemann, O. (2013). Nonsense-mediated mRNA decay – mechanisms of substrate mRNA recognition and degradation in mammalian cells. Biochim. Biophys. Acta 1829, 612–623. doi: 10.1016/j.bbagrm.2013.02.005
Seabra, C. M., Szoko, N., Erdin, S., Ragavendran, A., Stortchevoi, A., Maciel, P., et al. (2017). A novel microduplication of ARID1B: clinical, genetic, and proteomic findings. Am. J. Med. Genet A 173, 2478–2484. doi: 10.1002/ajmg.a.38327
Sekiguchi, F., Tsurusaki, Y., Okamoto, N., Teik, K. W., Mizuno, S., Suzumura, H., et al. (2019). Genetic abnormalities in a large cohort of Coffin-Siris syndrome patients. J. Hum. Genet. 64, 1173–1186.
Shakhova, O., and Sommer, L. (2008). “Neural crest-derived stem cells,” in StemBook [Internet], (Cambridge, MA: Harvard Stem Cell Institute).
Shi, H., Tao, T., Abraham, B. J., Durbin, A. D., Zimmerman, M. W., Kadoch, C., et al. (2020). ARID1A loss in neuroblastoma promotes the adrenergic-to-mesenchymal transition by regulating enhancer-mediated gene expression. Sci. Adv. 6:eaaz3440. doi: 10.1126/sciadv.aaz3440
Shibutani, M., Horii, T., Shoji, H., Morita, S., Kimura, M., Terawaki, N., et al. (2017). Arid1b haploinsufficiency causes abnormal brain gene expression and autism-related behaviors in mice. Int. J. Mol. Sci. 18:1872.
Sim, J. C., White, S. M., and Lockhart, P. J. (2015). ARID1B-mediated disorders: mutations and possible mechanisms. Intractable Rare Dis. Res. 4, 17–23.
Smith, A. L., Jung, E. M., Jeon, B. T., and Kim, W. Y. (2020). Arid1b haploinsufficiency in parvalbumin- or somatostatin-expressing interneurons leads to distinct ASD-like and ID-like behavior. Sci. Rep. 10:7834.
Sonmez, F. M., Uctepe, E., Gunduz, M., Gormez, Z., Erpolat, S., Oznur, M., et al. (2016). Coffin-Siris syndrome with cafe-au-lait spots, obesity and hyperinsulinism caused by a mutation in the ARID1B gene. Intractable Rare Dis. Res. 5, 222–226.
Stern, M., Jensen, R., and Herskowitz, I. (1984). Five SWI genes are required for expression of the HO gene in yeast. J. Mol Biol 178, 853–868.
Sun, X., Chuang, J. C., Kanchwala, M., Wu, L., Celen, C., Li, L., et al. (2016). Suppression of the SWI/SNF component arid1a promotes mammalian regeneration. Cell Stem Cell 18, 456–466.
Takeda, T., Banno, K., Okawa, R., Yanokura, M., Iijima, M., Irie-Kunitomi, H., et al. (2016). ARID1A gene mutation in ovarian and endometrial cancers (Review). Oncol. Rep. 35, 607–613.
Tamkun, J. W., Deuring, R., Scott, M. P., Kissinger, M., Pattatucci, A. M., Kaufman, T. C., et al. (1992). brahma: a regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2/SWI2. Cell 68, 561–572.
Tang, L., Nogales, E., and Ciferri, C. (2010). Structure and function of SWI/SNF chromatin remodeling complexes and mechanistic implications for transcription. Prog. Biophys. Mol. Biol. 102, 122–128.
Trizzino, M., Barbieri, E., Petracovici, A., Wu, S., Welsh, S. A., Owens, T. A., et al. (2018). The tumor suppressor ARID1A controls global transcription via pausing of RNA polymerase II. Cell Rep. 23, 3933–3945.
Tsurusaki, Y., Okamoto, N., Ohashi, H., Mizuno, S., Matsumoto, N., Makita, Y., et al. (2014). Coffin-Siris syndrome is a SWI/SNF complex disorder. Clin. Genet. 85, 548–554.
Tyagi, M., Imam, N., Verma, K., and Patel, A. K. (2016). Chromatin remodelers: we are the drivers!! Nucleus 7, 388–404.
Vals, M. A., Oiglane-Shlik, E., Noukas, M., Shor, R., Peet, A., Kals, M., et al. (2014). Coffin-Siris syndrome with obesity, macrocephaly, hepatomegaly and hyperinsulinism caused by a mutation in the ARID1B gene. Eur. J. Hum. Genet. 22, 1327–1329.
van der Sluijs, P. J., Jansen, S., Vergano, S. A., Adachi-Fukuda, M., Alanay, Y., AlKindy, A., et al. (2019). The ARID1B spectrum in 143 patients: from nonsyndromic intellectual disability to Coffin-Siris syndrome. Genet. Med. 21, 1295–1307.
Vasileiou, G., Ekici, A. B., Uebe, S., Zweier, C., Hoyer, J., Engels, H., et al. (2015). Chromatin-remodeling-factor ARID1B represses Wnt/beta-catenin signaling. Am. J. Hum. Genet. 97, 445–456.
Vergano, S. A., van der Sluijs, P. J., and Santen, G. (1993). “ARID1B-Related Disorder,” GeneReviews((R)), eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, G. Mirzaa, et al. (Seattle WA): University of Washington.
Vergano, S. S., and Deardorff, M. A. (2014). Clinical features, diagnostic criteria, and management of Coffin-Siris syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 166C, 252–256.
Wang, W., Chi, T., Xue, Y., Zhou, S., Kuo, A., and Crabtree, G. R. (1998). Architectural DNA binding by a high-mobility-group/kinesin-like subunit in mammalian SWI/SNF-related complexes. Proc. Natl. Acad. Sci. U.S.A. 95, 492–498.
Wang, W., Cote, J., Xue, Y., Zhou, S., Khavari, P. A., Biggar, S. R., et al. (1996a). Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J. 15, 5370–5382.
Wang, W., Xue, Y., Zhou, S., Kuo, A., Cairns, B. R., and Crabtree, G. R. (1996b). Diversity and specialization of mammalian SWI/SNF complexes. Genes Dev. 10, 2117–2130.
Wieczorek, D., Bogershausen, N., Beleggia, F., Steiner-Haldenstatt, S., Pohl, E., Li, Y., et al. (2013). A comprehensive molecular study on Coffin-Siris and Nicolaides-Baraitser syndromes identifies a broad molecular and clinical spectrum converging on altered chromatin remodeling. Hum. Mol. Genet. 22, 5121–5135.
Wilsker, D., Patsialou, A., Zumbrun, S. D., Kim, S., Chen, Y., Dallas, P. B., et al. (2004). The DNA-binding properties of the ARID-containing subunits of yeast and mammalian SWI/SNF complexes. Nucleic Acids Res. 32, 1345–1353.
Wu, R. C., Wang, T. L., and Shih, Ie.-M. (2014). The emerging roles of ARID1A in tumor suppression. Cancer Biol. Ther. 15, 655–664.
Wu, S., Fatkhutdinov, N., Rosin, L., Luppino, J. M., Iwasaki, O., Tanizawa, H., et al. (2019). ARID1A spatially partitions interphase chromosomes. Sci. Adv. 5:eaaw5294.
Xiao, W., Awadallah, A., and Xin, W. (2012). Loss of ARID1A/BAF250a expression in ovarian endometriosis and clear cell carcinoma. Int. J. Clin. Exp. Pathol. 5, 642–650.
Yamamoto, S., Tsuda, H., Takano, M., Tamai, S., and Matsubara, O. (2012). Loss of ARID1A protein expression occurs as an early event in ovarian clear-cell carcinoma development and frequently coexists with PIK3CA mutations. Mod. Pathol. 25, 615–624.
Yan, Z., Wang, Z., Sharova, L., Sharov, A. A., Ling, C., Piao, Y., et al. (2008). BAF250B-associated SWI/SNF chromatin-remodeling complex is required to maintain undifferentiated mouse embryonic stem cells. Stem Cells 26, 1155–1165.
Yu, D., Jiao, X., Cao, T., and Huang, F. (2018). Serum miRNA expression profiling reveals miR-486-3p may play a significant role in the development of autism by targeting ARID1B. Neuroreport 29, 1431–1436.
Yu, Y., Yao, R., Wang, L., Fan, Y., Huang, X., Hirschhorn, J., et al. (2015). De novo mutations in ARID1B associated with both syndromic and non-syndromic short stature. BMC Genomics 16:701. doi: 10.1186/s12864-015-1898-1
Zhai, Y., Kuick, R., Tipton, C., Wu, R., Sessine, M., Wang, Z., et al. (2016). Arid1a inactivation in an Apc- and Pten-defective mouse ovarian cancer model enhances epithelial differentiation and prolongs survival. J. Pathol. 238, 21–30.
Keywords: chromatin remodeling, SWI/SNF, ARID1A, ARID1B, development, pluripotency, cell identity
Citation: Pagliaroli L and Trizzino M (2021) The Evolutionary Conserved SWI/SNF Subunits ARID1A and ARID1B Are Key Modulators of Pluripotency and Cell-Fate Determination. Front. Cell Dev. Biol. 9:643361. doi: 10.3389/fcell.2021.643361
Received: 18 December 2020; Accepted: 15 February 2021;
Published: 04 March 2021.
Edited by:
Bruno Di Stefano, Baylor College of Medicine, United StatesReviewed by:
Michael Hoetker, Harvard Medical School, United StatesCopyright © 2021 Pagliaroli and Trizzino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marco Trizzino, bWFyY28udHJpenppbm9AamVmZmVyc29uLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.