 Ji-Hyun ParkKazuhide Hayakawa*
Ji-Hyun ParkKazuhide Hayakawa*- Neuroprotection Research Laboratory, Departments of Radiology and Neurology, Massachusetts General Hospital and Harvard Medical School, Charlestown, MA, United States
Mitochondria actively participate in the regulation of cell respiratory mechanisms, metabolic processes, and energy homeostasis in the central nervous system (CNS). Because of the requirement of high energy, neuronal functionality and viability are largely dependent on mitochondrial functionality. In the context of CNS disorders, disruptions of metabolic homeostasis caused by mitochondrial dysfunction lead to neuronal cell death and neuroinflammation. Therefore, restoring mitochondrial function becomes a primary therapeutic target. Recently, accumulating evidence suggests that active mitochondria are secreted into the extracellular fluid and potentially act as non-cell-autonomous signals in CNS pathophysiology. In this mini-review, we overview findings that implicate the presence of cell-free extracellular mitochondria and the critical role of intercellular mitochondrial transfer in various rodent models of CNS disorders. We also discuss isolated mitochondrial allograft as a novel therapeutic intervention for CNS disorders.
Introduction
Mitochondria are the powerhouse of cells and essential for maintaining cellular function in mammals (Devine and Kittler, 2018). The role of mitochondria is especially important in a high-metabolic-rate organ like the brain. Mitochondria produce adenosine triphosphate (ATP) (Jonckheere et al., 2012; Murphy et al., 2019), play a central role in fatty acid biosynthesis (Kastaniotis et al., 2017) and cellular calcium homeostasis (David and Barrett, 2003; Pivovarova and Andrews, 2010), and also regulate intracellular mechanisms that modulate viability, immune cell activation, and mitophagy (Tait and Green, 2012). Accumulating mitochondrial reactive oxygen species (ROS) and inflammasome along with imbalanced mitochondrial membrane permeability may cause progression of cell death and neuroinflammation (Green et al., 2011; Angelova and Abramov, 2018). Therefore, restoring mitochondrial perturbation within cells is a major therapeutic strategy in many CNS disorders including stroke (Rutkai et al., 2019), hemorrhagic stroke (Wang et al., 2018), spinal cord injury (Gollihue et al., 2018a), Alzheimer’s disease (AD) (Wang et al., 2020), and Parkinson’s disease (PD) (Requejo-Aguilar and Bolanos, 2016).
Recent studies in rodents and humans demonstrate that mitochondria may be secreted into the extracellular milieu and transported or exchanged between cells in the CNS (Spees et al., 2006; Islam et al., 2012; Hayakawa et al., 2016; Sinha et al., 2016; Yao et al., 2018; Gao J. et al., 2019; Gao L. et al., 2019; Liu et al., 2019; Marlein et al., 2019; Miliotis et al., 2019; Al Amir Dache et al., 2020; Eun Jung et al., 2020; Park et al., 2020b). Under pathophysiological conditions in the CNS, extracellular mitochondria and their components might contribute to signals between cells that could evoke detrimental inflammation or promote beneficial neuroprotection. In this regard, intact and active extracellular mitochondria may provide a novel therapeutic intervention to restore the bioenergetic needs of damaged or diseased recipient cells. In this mini-review, we collected studies of endogenous mitochondrial transfer as interdependent signals between cells, discuss extracellular mitochondria and its components as a novel class of mediators and biomarkers of injury or disease, and ultimately explore the clinical relevance of mitochondrial transplantation as a therapeutic approach for CNS disorders.
Mechanisms of Experimental Mitochondrial Transfer
Mitochondria support energy homeostasis in cells. But emerging evidence implicates that mitochondria are surprisingly transferred or exchanged between cells. The biological process was initially discovered in an in vitro cell culture system wherein mitochondrial DNA (mtDNA) from human mesenchymal progenitor cells (hMSCs) was delivered to A549 ρo cells (Spees et al., 2006). Similar observations have been subsequently reported by other research groups that showed evidence of mitochondrial transfer between progenitor cells and damaged cells via the formation of tunneling nanotubes (TNTs) (Onfelt et al., 2006; Kesner et al., 2016). The TNT-dependent mitochondrial transfer has been suggested in human-originated endothelial progenitors (Koyanagi et al., 2005) and MSCs but not in fibroblast (Yang et al., 2016) and rescued UV stress damage in PC12 cells, cigarette smoke-induced lung damage, and acute respiratory distress syndrome in rodents (Li et al., 2014; Wang and Gerdes, 2015; Jackson et al., 2016).
Extracellular vesicles (EVs) or exosomes can also deliver intracellular mitochondria or the components to other cells. It has been shown that glioblastoma cells and astrocytes produced exosome-enclosed mtDNA, supporting the idea that mtDNA may be delivered to cells through exosomal transfer (Guescini et al., 2010). In the retina, the vesicle-mediated transfer of mitochondria allows neurons to eliminate non-functional mitochondria within astrocytes via transmitophagy while these molecular mechanisms are still understudied (Davis et al., 2014). On contrary, astrocyte-to-neuron mitochondrial transfer has been reported as neuroprotection mechanism that reactive astrocytes after stroke produced extracellular mitochondria through CD38-cADPR signaling and these mitochondria were able to enter damaged neurons through an integrin-src/syk pathway to rescue damaged neurons (Babenko et al., 2015; Hayakawa et al., 2016).
It is important to note that the beneficiality of mitochondrial transfer may depend on context. For instance, activated astrocytes after focal cerebral ischemia may express regenerative genes (A2), whereas neurodegenerative diseases activate proinflammatory phenotype (A1) (Zamanian et al., 2012; Khakh et al., 2017; Liddelow et al., 2017; Trias et al., 2018; Yun et al., 2018). Inflammatory microenvironment where A1 astrocytes are enriched also increases proinflammatory microglia, and the crosstalk between inflammatory glia may exacerbate neurodegeneration through intercellular crosstalk via disrupted and fragmented mitochondria (Joshi et al., 2019). Collectively, extracellular mitochondria produced by glia can also be deleterious by expanding neuroinflammation and rigorous assessment of the functional property may provide a glimpse into the severity of neurodegeneration.
Although the molecular mechanisms underlying mitochondrial release and internalization remain to be fully elucidated, several pathways are being actively investigated. Recent proof-of-concept studies demonstrate that CD38 signaling may regulate mitochondrial transfer (Hayakawa et al., 2016; Huang et al., 2019; Lippert and Borlongan, 2019; Sun et al., 2019). Stimulating a CD38 downstream with cADPR or amplifying CD38 expression increased functional extracellular mitochondria secreted from astrocytes (Hayakawa et al., 2016). When astrocytes had a mutation R239C in GFAP gene, astrocyte-mediated mitochondrial transfer to neurons was disrupted accompanied by decreasing CD38 expression (Gao L. et al., 2019). What remains missing from the collective literature is the mechanism of how mitochondria are able to maintain their functionality. It has been proposed that mitochondrial protein posttranslational modification may support the functionality outside cells and be a key mechanism to support mitochondrial energy production, mitochondrial membrane potential, and motility in recipient cells postmitochondrial transfer (Yuzwa et al., 2012; Wang et al., 2016; Yang and Qian, 2017; Park et al., 2020a). A proof-of-concept study has been reported. When astrocytic CD38 was stimulated by NAD+, O-GlcNAc posttranslational modification in mitochondrial proteins was amplified, and these O-GlcNAcylated mitochondria maintained mtDNA content and membrane potential in extracellular space, thus improving neuroprotective effects after mitochondrial transfer (Park et al., 2020b).
Other intracellular mechanisms have been studied. The interaction between mitochondria and the ER may facilitate mitochondrial transfer within the osteocyte dendritic network (Gao J. et al., 2019). Moreover, gap junction protein connexin 43 or mitochondrial transport coordinator Miro 1 may be involved in mitochondrial transfer mechanisms regulated by mesenchymal stem cells (Islam et al., 2012; Ahmad et al., 2014). Cellular stress or stimulus can also trigger mitochondrial secretion and transfer. It has been reported that stressed cells that had lost cytochrome c triggered mitochondrial transfer to prevent apoptosis in PC12 cells subjected to UV exposure (Wang and Gerdes, 2015). ROS produced during oxidative stress have been suggested to trigger the secretion of mitochondria (Islam et al., 2012; Torralba et al., 2016; Zhang et al., 2016). TNT was produced in induced pluripotent stem cells (iPSCs) by tumor necrosis factor alpha (TNF-α) and the microstructure interacted with cardiomyocytes through TNFαIP2 expression, which may promote the effective transfer of mitochondria (Zhang et al., 2016). Furthermore, when extracellular mitochondria were internalized into the cells, activation of several pathways including endocytosis (Wei et al., 2018), integrin-dependent pathways (Hayakawa et al., 2016), macro-pinocytosis (Kitani et al., 2014), TNT, or cell fusion (Spees et al., 2006; Torralba et al., 2016) have been observed. Future studies are warranted to identify the specific internalization pathway when extracellular mitochondria are either healthy or disrupted under pathophysiological conditions (Figure 1).
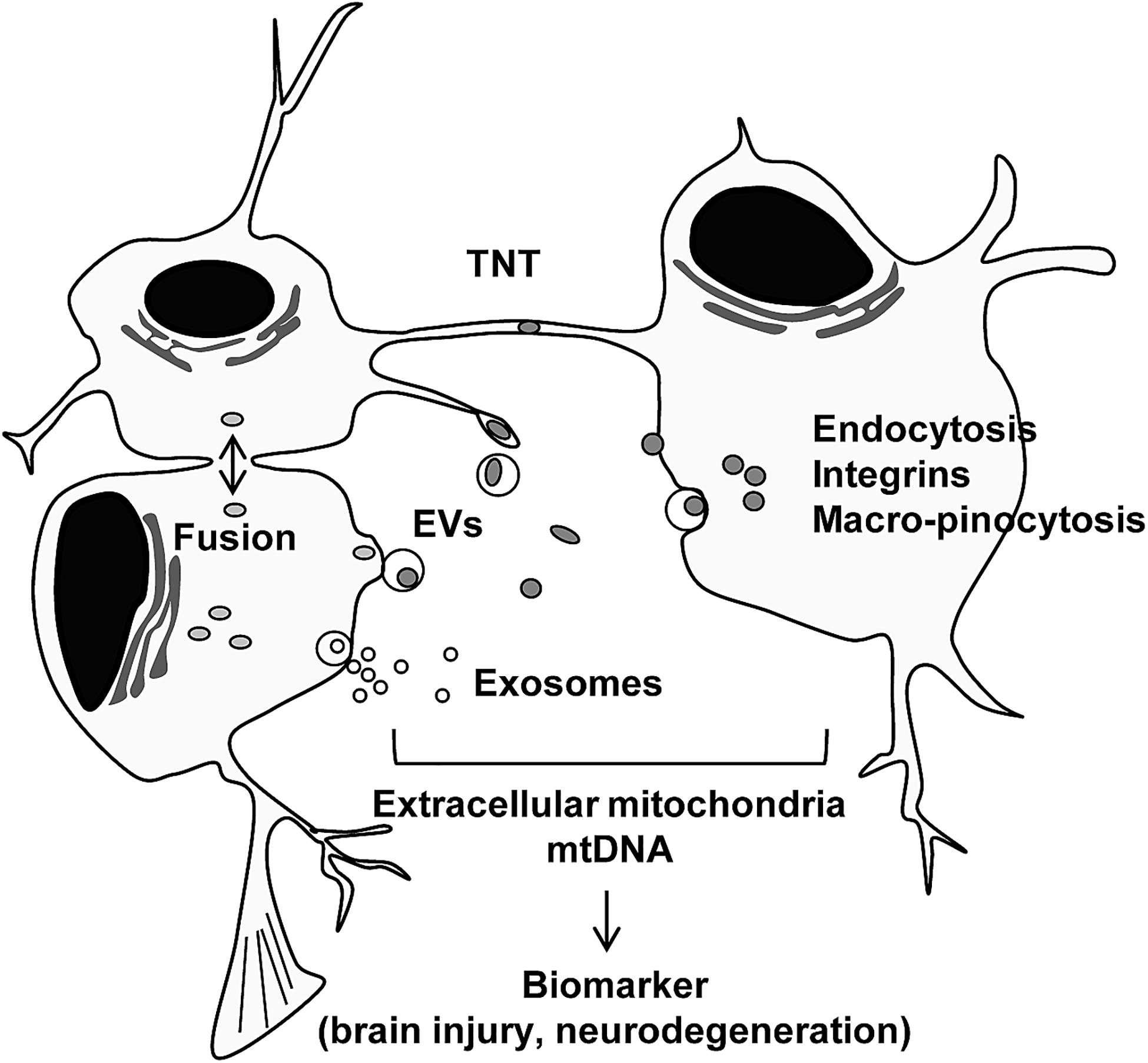
Figure 1. Intercellular mitochondrial transfer. Intercellular mitochondrial transfer can be regulated by tunneling nanotubles (TNT), extracellular vesicles (EVs), exosomes, and cell fusion. Extracellular mitochondria may be incorporated into cells through canonical endocytosis pathway, integrin-mediated pathway, and macro-pinocytosis. Extracellular mitochondria and mtDNA may provide a glimpse into the status of tissue metabolism, disease severity and recovery in the CNS.
Extracellular Mitochondria as a Biomarker for Mitochondrial Integrity in the CNS
Under normal conditions, healthy mitochondria maintain their functionality through continuous fission and fusion cycles. During injury or disease, the disruption of the process to integrate damaged mitochondria into the mitochondrial homeostatic system subsequently activates the process of mitochondrial elimination via mitophagy, mitochondria-specific autophagy that is a subtype of autophagy regulated by autophagosomes and lysosomes (Ashrafi and Schwarz, 2013), and recovered amino acids and fatty acids following the degradation process can be recycled to generate ATP (Twig et al., 2008; Nakatogawa et al., 2009), supporting that mitophagy is involved in maintaining mitochondrial quality and metabolic status.
The similar mitochondria elimination mechanism may be present in neuron–glial network in retina (Davis et al., 2014). In this study, electron microscopy and confocal microscopy determined that retinal ganglion cells transferred mitochondria-contained vesicles to retinal astrocytes to eliminate damaged mitochondria through so called transmitophagy. In the context of PD, the mechanism of transmitophagy may be crucial to maintain mitochondrial function in dopaminergic neurons and prevent neuroinflammation mediated by disrupted mitochondrial secretion (Morales et al., 2020). In this study, Morales and colleagues observed that damaged dopaminergic neurons via 6-OHDA infusion generated spheroid-like structure containing mitochondria and transferred them to adjacent astrocytes for digesting damaged mitochondria through STX17/Lamp1/Lamp2/LC3+ autophagolysosomes (Morales et al., 2020). Collectively, the transmitophagy may be a mechanism of mitochondrial quality control through intercellular mitochondrial transfer in the CNS.
Secreted mitochondria and its components in blood and cerebrospinal fluid (CSF) can be considered promising biomarkers for injury or disease. The basic hypothesis is that extracellular mitochondrial functionality reflects intracellular metabolism and can be indirectly assessed the underlying tissue metabolic integrity (Hayakawa et al., 2018; Miliotis et al., 2019). In CSF analysis in subarachnoid hemorrhage (SAH), membrane potentials of extracellular mitochondria were decreased in SAH patients, and higher JC1 ratios were associated with better clinical recovery at 3 months after SAH (Chou et al., 2017; Youn et al., 2020). Cell-free mtDNA becomes a damage-associated molecular pattern (DAMP) acting as a “danger signal” (Galluzzi et al., 2012; Thurairajah et al., 2018). Therefore, the amount of mtDNA in CSF may also implicate inflammation status. In fact, higher mtDNA contents in CSF were correlated to the progression of AD (Cervera-Carles et al., 2017) or anti-NMDA receptor encephalitis (Peng et al., 2019).
Altogether, extracellular mitochondria and mtDNA may be a biomarker for mitochondrial integrity, disease progression, or recovery in CNS pathology including neuroinflammation, hemorrhagic stroke, and neurodegenerative disease (Figure 1). Additional studies are needed to address whether the concept of extracellular mitochondria as a biomarker is applicable in other CNS disorders such as spinal cord injury and PD.
Mitochondrial Transplantation as CNS Therapies
In the CNS, neuron-glial-vascular interaction is essential for a homeostatic network, the so-called neurovascular unit in the CNS (Iadecola, 2004; Lo et al., 2004; Hawkins and Davis, 2005; Zacchigna et al., 2008; Zlokovic, 2008; del Zoppo, 2009) and functional impairment of mitochondria within the unit can be one of the major reasons to cause CNS disorders. McCully and colleagues have studied mitochondrial transplantation to the heart as a promising therapy without inducing detrimental immune response (McCully et al., 2016; Shin et al., 2019). These studies have supported scientists to investigate therapeutic efficacy utilizing exogenous mitochondria for CNS injury or neurodegeneration. Accumulating evidence has shown the beneficial actions of mitochondrial transplantation and the limitation in various animal models of CNS disorders (Figure 2).
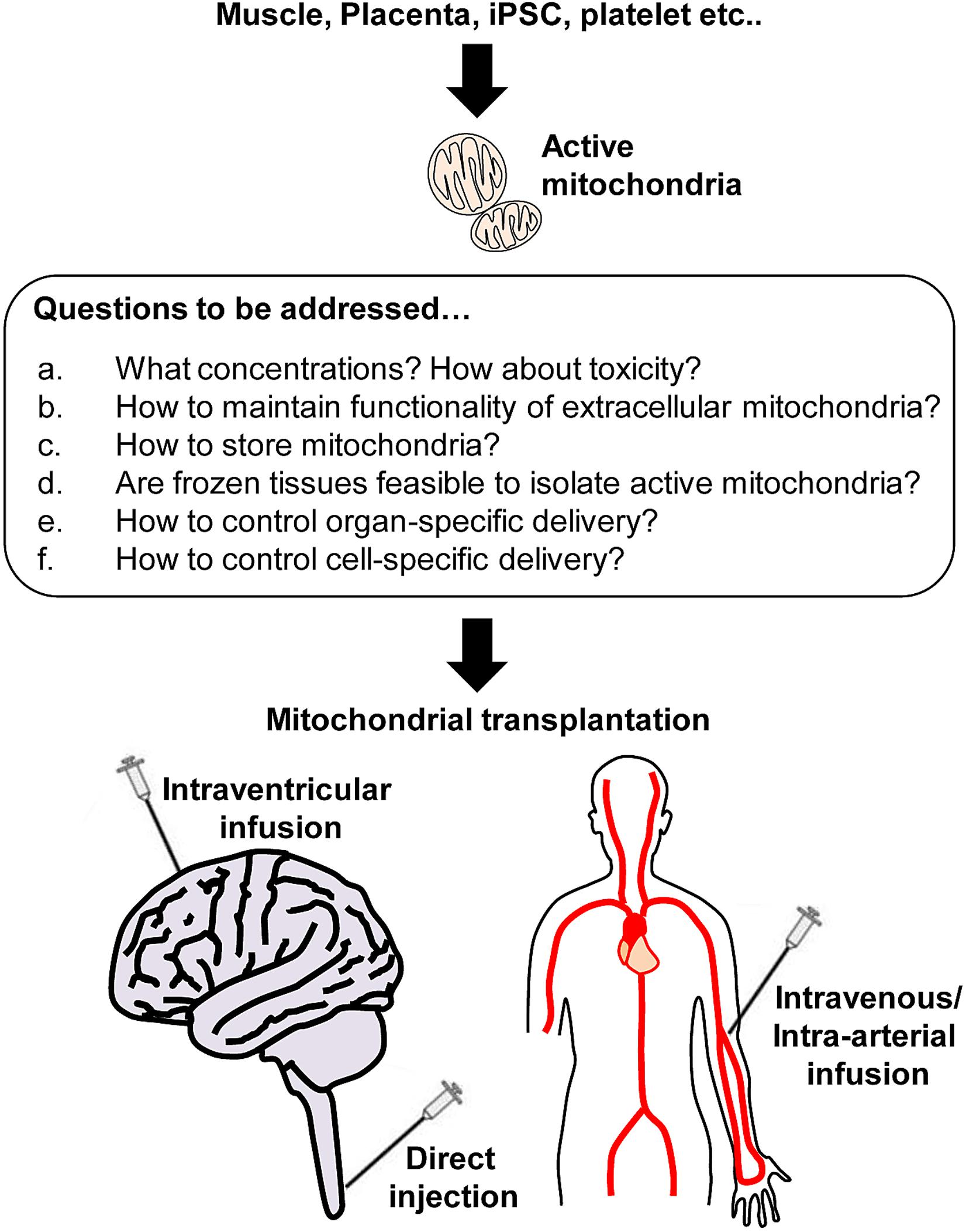
Figure 2. Active mitochondria isolation from various sources and beyond. Respiratory active mitochondria are able to isolate from various sources including skeletal muscle, placenta, iPSC, and platelets. However, many questions remain to be answered regarding mitochondrial transplantation. a: It is critical to identify the optimal concentration of mitochondria that do not induce toxicity. b: Protecting extracellular mitochondria against pathological environments (high calcium, ionic imbalance, high ROS, glycation etc.) would be challenging, but it is required to avoid disrupting mitochondria while maximize the benefit induced by mitochondrial transplantation. c-d: Storing mitochondria is the most challenging issue. Instead, it might be worth seeking if frozen tissues can be sources to isolate a larger amount of active mitochondria. e-f: Finally, it is critical to investigate how to deliver mitochondria to target tissues or cells to maximize the therapeutic efficacy.
Spinal Cord Injury
The therapeutic potential of extracellular mitochondria has been reported in a mouse model of spinal cord injury (SCI) (Gollihue et al.,2018a,b). In this study, GFP-labeled mitochondria (50–150 μg) were isolated from PC12 cells and then transplanted into the injury site after SCI onset. Following direct microinjection, macrophages, endothelial cells, and astrocytes incorporated injected mitochondria accompanied by improving acute mitochondrial bioenergetics and mitochondrial respiration (Gollihue et al., 2018a). Intriguingly, injected mitochondria were not found in neurons and did not support functional neuroprotection, implicating that mechanisms of mitochondrial internalization may be different among cell types. Ultimately, the study provides a very important aspect that mitochondrial incorporation efficiency in neurons may need to be improved to accelerate functional recovery after SCI.
Kuo and colleagues attempted to prevent axonal degeneration by mitochondrial transplantation in a rat model of sciatic nerve crush injury. In this study, BHK cell-derived mitochondria were utilized for therapeutic intervention. Dosages of mitochondria (0, 65, 130, 195, and 269 μg) were initially assessed in the cultured sciatic nerve. After the determination, extracellular mitochondria (195 μg) were injected directly into the injured nerve. Notably, mitochondria transplantation increased neuronal regeneration and decreased oxidative stress along with improving functional behaviors and physiological neuronal and muscle activities. Moreover, mitochondrial transplantation increased the expression of neurotrophic factors such as BDNF and CNTF in injured nerves along with restoring muscular integrity and increasing muscle progenitors and total muscle mass (Kuo et al., 2017).
Parkinson’s Disease
Many genes involved in familial PD have been identified and known to directly link to mitochondrial dysfunction (Lill, 2016). The impairment of the respiratory chain, redox homeostasis, mitophagy, and mitochondrial biogenesis in dopaminergic neurons may aggravate PD pathogenesis (Moon and Paek, 2015). Given the fact that mitochondria-targeting antioxidants such as coenzyme Q10 and creatine monohydrate did not effectively ameliorate PD pathology (Beal et al., 2014; Kieburtz et al., 2015), healthy mitochondrial transplants to the damaged brain area can be an alternative approach to recover mitochondria-mediated redox homeostasis within neurons if these mitochondria can successfully incorporate into disrupted cells.
A study has been conducted using a mouse model of MPTP-induced PD. Fluorescent-labeled mitochondria were delivered systemically via tail vein injections in this model. Intriguingly, infused mitochondria (IV route, 0.5 mg/kg body weight) were ubiquitously found in CNS tissues and peripheral organs. Most importantly, mitochondrial infusion significantly improved motor functions accompanied by recovering mitochondrial complex I activity and ATP production, thus inhibiting cell death pathways in the striatum in comparison with the vehicle group (Shi et al., 2017), suggesting that intravenous infusion of mitochondria may be feasible to target not only peripheral organs but also CNS tissues.
Another study has utilized a mitochondrial modification technique with Pep-1 to improve mitochondrial delivery into CNS tissues. Interestingly, mice treated with Pep-1-conjugated mitochondria into the substantia nigra pars compacta (1.05 μg/5 μl) showed better behavioral outcomes in locomotor activity, moving distance, and moving speed compared to ones treated with unmodified control mitochondria (Chang et al., 2016). Concomitantly, mitochondrial incorporation in TH+ dopaminergic neurons was improved, suggesting that mitochondrial modification can amplify the therapeutic potential of exogenous mitochondria and the beneficiality to enhance neuronal incorporation in the CNS.
Alzheimer’s Disease
Alzheimer’s disease is a neurodegenerative disease along with severe memory loss with impairment of episodic memory in the initial phases (Swerdlow et al., 2014). Accumulating evidence indicates that mitochondrial impairment may contribute to the pathology of AD. Indeed, brain mitochondria in AD have shown dysfunctional respiration chain complexes, decreased ATP generation, and an excessive amount of free radicals that may lead to neurodegeneration (Parker and Parks, 1995; Pohland et al., 2018). In addition to therapies attacking amyloid-beta, targeting mitochondrial dysfunction may indirectly ameliorate AD pathology. Nitzan and colleagues have attempted mitochondrial transplantation to restore mitochondrial function in a mouse AD model produced by Aβ 1–42 intracerebroventricular infusion (Nitzan et al., 2019). Mitochondria isolated from HeLa cells were injected through the tail vein. As a result, IV-injected mitochondria were observed in the liver, but there was no signal in the brain following IV injection. Intriguingly, mitochondrial infusion still improved cognitive functions accompanied by decreasing loss of neurons and suppressing hippocampal glial activation. Moreover, mitochondrial functional parameters in the liver and brain were also restored. Collectively, mitochondrial transplantation through IV route can be a new therapeutic strategy for AD (Nitzan et al., 2019).
Mitochondrial transplantation also improved diabetes-associated cognitive impairment (DACI) which is associated with elevated Aβ deposition (Ma et al., 2020). In this study, platelet-derived mitochondria were used as the donor source. After intracerebroventricular injection of mitochondria in db/db mice, mitochondrial internalization to hippocampal neurons was observed, and subsequently, DACI was alleviated accompanied by restored mitochondrial function as well as decreased accumulation of Aβ. Although further rigorous assessments are required, it may be promising that mitochondrial transplantation may ameliorate AD pathology via inhibiting Aβ generation and deposition.
Ischemic Stroke
Therapeutic interventions including recombinant tissue plasminogen activator or endovascular thrombectomy provide effective ways to achieve reperfusion for acute ischemic stroke (Vosler et al., 2009). While restoring cerebral blood flow is essential for decreasing infarction, reperfusion alone may not be sufficient to fully salvage penumbral tissue in some patients where cells are dying through active cell death pathways (Lo, 2008). Therefore, it is still important to continue the search for neuroprotective approaches that can be added to ischemia reperfusion (Vosler et al., 2009). The brain with ischemic stroke results in a lack of supplementations for glucose and oxygen and mitochondrial disruption to generate ATP, thus causing mitochondrial metabolic impairment and leading to neuronal apoptosis. Collectively, mitochondrial restoration therapy utilizing extracellular mitochondria may also be applicable in ischemic stroke (Watts et al., 2013; Baek et al., 2014; Russo et al., 2018).
Mitochondrial transplantation may indeed provide some beneficial effects to protect neurons in models of ischemic stroke. It has been shown that intact mitochondria isolated from BHK-21 cell lines successfully protected neurons against ischemic insult. In this study, mitochondria were treated directly into the striatum (75 μg) or infused through arteries (750 μg) after transient focal cerebral ischemia. Neurons and glia in the peri-infarct area appeared to incorporate mitochondria treated via both routes at 4 weeks after ischemic stroke. Moreover, treatment with isolated mitochondria significantly improved motor function accompanied by decreasing infarction and TUNEL-positive cells in acute stroke (Huang et al., 2016). Treatment with skeletal muscle-derived mitochondria may also provide beneficial neuroprotective effects (Zhang et al., 2019). Freshly isolated mitochondria from skeletal muscle (5 × 106/10 μl) were infused into the lateral ventricle immediately after reperfusion after focal cerebral ischemia. Consistent with prior findings, the peri-infarct neurons incorporated the treated mitochondria. Subsequently, the allograft remarkably attenuated infarct formation accompanied by restoring neurological impairments, reducing oxidative stress response and gliosis, as well as neuronal death at 28 days poststroke. Collectively, mitochondrial allograft may not only protect neurons against acute injury but also improve long-term outcomes after stroke.
Tissue source for viable mitochondrial isolation is important for practical use of mitochondria in therapy. A proof-of-concept study using placenta-derived mitochondria was performed in a mouse stroke model. From the snap-frozen placentae obtained from E17 pregnant female mice, mitochondria were isolated and evaluated the functionality. Surprisingly, flow cytometry analysis demonstrated that up to 87% of placental mitochondria were viable and maintained JC1 membrane potentials after isolation. Placental mitochondrial fractions contained ATP equivalent to mitochondrial fractions isolated from skeletal muscle and brown fat tissue. Moreover, glutathione reductase, Mn-SOD, and HSP70 were highly preserved in placental mitochondrial fractions. Then, placental-derived mitochondrial fractions (100 μg/100 μl) were infused intravenously immediately after reperfusion with full blinding and randomization. Strikingly, treatment with placental mitochondrial fractions significantly decreased infarction after focal cerebral ischemia in mice. Collectively, cryopreserved placenta can be a feasible source for viable mitochondrial isolation and transplantation with placental mitochondria may amplify beneficial effects of reperfusion in stroke (Nakamura et al., 2020).
Conclusion
Status of mitochondrial function is a key for recovery after CNS injury or disease (Anne Stetler et al., 2013; Madsen et al., 2017). Accumulating evidences implicate that mitochondria can be surprisingly present outside cells and transported from cell to cell. Moreover, extracellular mitochondria are found not only in rodents but also in clinical samples, and assessments of mitochondrial functionality in extracellular fluids may provide us a biomarker-like glimpse into injury or repair/regeneration. Within the emerging paradigm of targeting mitochondria after CNS injury or disease (She et al., 2017; Vekaria et al., 2017; Simon et al., 2019), viable mitochondria may be feasible to isolate and utilized for CNS therapy. Nonetheless, more rigorous studies are required to validate the reliability, safety, and efficacy of extracellular mitochondria in a wide range of applications from diagnosis to therapeutic intervention in various CNS disorders.
Author Contributions
J-HP and KH collected literature and prepared the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by grants from NIH NINDS (R01NS094756).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Eng H. Lo for many helpful discussions.
References
Ahmad, T., Mukherjee, S., Pattnaik, B., Kumar, M., Singh, S., Rehman, R., et al. (2014). Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 33, 994–1010.
Al Amir Dache, Z., Otandault, A., Tanos, R., Pastor, B., Meddeb, R., Sanchez, C., et al. (2020). Blood contains circulating cell-free respiratory competent mitochondria. FASEB J. 34, 3616–3630. doi: 10.1096/fj.201901917rr
Angelova, P. R., and Abramov, A. Y. (2018). Role of mitochondrial ROS in the brain: from physiology to neurodegeneration. FEBS Lett. 592, 692–702. doi: 10.1002/1873-3468.12964
Anne Stetler, R., Leak, R. K., Gao, Y., and Chen, J. (2013). The dynamics of the mitochondrial organelle as a potential therapeutic target. J. Cereb. Blood Flow Metab. 33, 22–32. doi: 10.1038/jcbfm.2012.158
Ashrafi, G., and Schwarz, T. L. (2013). The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 20, 31–42. doi: 10.1038/cdd.2012.81
Babenko, V. A., Silachev, D. N., Zorova, L. D., Pevzner, I. B., Khutornenko, A. A., Plotnikov, E. Y., et al. (2015). Improving the post-stroke therapeutic potency of mesenchymal multipotent stromal cells by cocultivation with cortical neurons: the role of crosstalk between cells. Stem Cells Transl. Med. 4, 1011–1020. doi: 10.5966/sctm.2015-0010
Baek, S. H., Noh, A. R., Kim, K. A., Akram, M., Shin, Y. J., Kim, E. S., et al. (2014). Modulation of mitochondrial function and autophagy mediates carnosine neuroprotection against ischemic brain damage. Stroke 45, 2438–2443. doi: 10.1161/strokeaha.114.005183
Beal, M. F., Oakes, D., Shoulson, I., Henchcliffe, C., Galpern, W. R., Haas, R., et al. (2014). A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: no evidence of benefit. JAMA Neurol. 71, 543–552.
Cervera-Carles, L., Alcolea, D., Estanga, A., Ecay-Torres, M., Izagirre, A., Clerigue, M., et al. (2017). Cerebrospinal fluid mitochondrial DNA in the Alzheimer’s disease continuum. Neurobiol. Aging 53, 192.e1–192.e4.
Chang, J. C., Wu, S. L., Liu, K. H., Chen, Y. H., Chuang, C. S., Cheng, F. C., et al. (2016). Allogeneic/xenogeneic transplantation of peptide-labeled mitochondria in Parkinson’s disease: restoration of mitochondria functions and attenuation of 6-hydroxydopamine-induced neurotoxicity. Transl. Res. 170, 40.e–56.e.
Chou, S. H., Lan, J., Esposito, E., Ning, M., Balaj, L., Ji, X., et al. (2017). Extracellular mitochondria in cerebrospinal fluid and neurological recovery after subarachnoid hemorrhage. Stroke 48, 2231–2237. doi: 10.1161/strokeaha.117.017758
David, G., and Barrett, E. F. (2003). Mitochondrial Ca2+ uptake prevents desynchronization of quantal release and minimizes depletion during repetitive stimulation of mouse motor nerve terminals. J. Physiol. 548(Pt 2), 425–438. doi: 10.1113/jphysiol.2002.035196
Davis, C. H., Kim, K. Y., Bushong, E. A., Mills, E. A., Boassa, D., Shih, T., et al. (2014). Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. U.S.A. 111, 9633–9638. doi: 10.1073/pnas.1404651111
del Zoppo, G. J. (2009). Inflammation and the neurovascular unit in the setting of focal cerebral ischemia. Neuroscience 158, 972–982. doi: 10.1016/j.neuroscience.2008.08.028
Devine, M. J., and Kittler, J. T. (2018). Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 19, 63–80. doi: 10.1038/nrn.2017.170
Eun Jung, J., Sun, G., Bautista Garrido, J., Obertas, L., Mobley, A. S., Ting, S. M., et al. (2020). The mitochondria-derived peptide humanin improves recovery from intracerebral hemorrhage: implication of mitochondria transfer and microglia phenotype change. J. Neurosci. 40, 2154–2165. doi: 10.1523/jneurosci.2212-19.2020
Galluzzi, L., Kepp, O., and Kroemer, G. (2012). Mitochondria: master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 13, 780–788. doi: 10.1038/nrm3479
Gao, J., Qin, A., Liu, D., Ruan, R., Wang, Q., Yuan, J., et al. (2019). Endoplasmic reticulum mediates mitochondrial transfer within the osteocyte dendritic network. Sci. Adv. 5:eaaw7215. doi: 10.1126/sciadv.aaw7215
Gao, L., Zhang, Z., Lu, J., and Pei, G. (2019). Mitochondria are dynamically transferring between human neural cells and alexander disease-associated GFAP mutations impair the astrocytic transfer. Front. Cell Neurosci. 13:316. doi: 10.3389/fncel.2019.00316
Gollihue, J. L., Patel, S. P., Eldahan, K. C., Cox, D. H., Donahue, R. R., Taylor, B. K., et al. (2018a). Effects of mitochondrial transplantation on bioenergetics, cellular incorporation, and functional recovery after spinal cord injury. J. Neurotrauma. 35, 1800–1818. doi: 10.1089/neu.2017.5605
Gollihue, J. L., Patel, S. P., and Rabchevsky, A. G. (2018b). Mitochondrial transplantation strategies as potential therapeutics for central nervous system trauma. Neural. Regen. Res. 13, 194–197. doi: 10.4103/1673-5374.226382
Green, D. R., Galluzzi, L., and Kroemer, G. (2011). Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 333, 1109–1112. doi: 10.1126/science.1201940
Guescini, M., Genedani, S., Stocchi, V., and Agnati, L. F. (2010). Astrocytes and Glioblastoma cells release exosomes carrying mtDNA. J. Neural Transm. (Vienna). 117, 1–4. doi: 10.1007/s00702-009-0288-8
Hawkins, B. T., and Davis, T. P. (2005). The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 57, 173–185. doi: 10.1124/pr.57.2.4
Hayakawa, K., Bruzzese, M., Chou, S. H., Ning, M., Ji, X., and Lo, E. H. (2018). Extracellular mitochondria for therapy and diagnosis in acute central nervous system injury. JAMA Neurol. 75, 119–122. doi: 10.1001/jamaneurol.2017.3475
Hayakawa, K., Esposito, E., Wang, X., Terasaki, Y., Liu, Y., Xing, C., et al. (2016). Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535, 551–555. doi: 10.1038/nature18928
Huang, L., Nakamura, Y., Lo, E. H., and Hayakawa, K. (2019). Astrocyte signaling in the neurovascular unit after central nervous system injury. Int. J. Mol. Sci. 20:282. doi: 10.3390/ijms20020282
Huang, P. J., Kuo, C. C., Lee, H. C., Shen, C. I., Cheng, F. C., Wu, S. F., et al. (2016). Transferring xenogenic mitochondria provides neural protection against ischemic stress in ischemic rat brains. Cell Transplant. 25, 913–927. doi: 10.3727/096368915x689785
Iadecola, C. (2004). Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 5, 347–360. doi: 10.1038/nrn1387
Islam, M. N., Das, S. R., Emin, M. T., Wei, M., Sun, L., Westphalen, K., et al. (2012). Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 18, 759–765. doi: 10.1038/nm.2736
Jackson, M. V., Morrison, T. J., Doherty, D. F., McAuley, D. F., Matthay, M. A., Kissenpfennig, A., et al. (2016). Mitochondrial transfer via tunneling nanotubes is an important mechanism by which mesenchymal stem cells enhance macrophage phagocytosis in the in Vitro and in Vivo models of ARDS. Stem Cells. 34, 2210–2223. doi: 10.1002/stem.2372
Jonckheere, A. I., Smeitink, J. A., and Rodenburg, R. J. (2012). Mitochondrial ATP synthase: architecture, function and pathology. J. Inherit. Metab. Dis. 35, 211–225. doi: 10.1007/s10545-011-9382-9
Joshi, A. U., Minhas, P. S., Liddelow, S. A., Haileselassie, B., Andreasson, K. I., and Dorn, G. W. 2nd et al., (2019). Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 22, 1635–1648. doi: 10.1038/s41593-019-0486-0
Kastaniotis, A. J., Autio, K. J., Keratar, J. M., Monteuuis, G., Makela, A. M., Nair, R. R., et al. (2017). Mitochondrial fatty acid synthesis, fatty acids and mitochondrial physiology. Biochim. Biophys. Acta Mol. Cell Biol. Lipids. 1862, 39–48.
Kesner, E. E., Saada-Reich, A., and Lorberboum-Galski, H. (2016). Characteristics of mitochondrial transformation into human cells. Sci. Rep. 6: 26057.
Khakh, B. S., Beaumont, V., Cachope, R., Munoz-Sanjuan, I., Goldman, S. A., and Grantyn, R. (2017). Unravelling and exploiting astrocyte dysfunction in huntington’s disease. Trends Neurosci. 40, 422–437. doi: 10.1016/j.tins.2017.05.002
Kieburtz, K., Tilley, B. C., Elm, J. J., Babcock, D., Hauser, R., Ross, G. W., et al. (2015). Effect of creatine monohydrate on clinical progression in patients with Parkinson disease: a randomized clinical trial. JAMA 313, 584–593.
Kitani, T., Kami, D., Matoba, S., and Gojo, S. (2014). Internalization of isolated functional mitochondria: involvement of macropinocytosis. J. Cell Mol. Med. 18, 1694–1703. doi: 10.1111/jcmm.12316
Koyanagi, M., Brandes, R. P., Haendeler, J., Zeiher, A. M., and Dimmeler, S. (2005). Cell-to-cell connection of endothelial progenitor cells with cardiac myocytes by nanotubes: a novel mechanism for cell fate changes? Circ. Res. 96, 1039–1041. doi: 10.1161/01.res.0000168650.23479.0c
Kuo, C. C., Su, H. L., Chang, T. L., Chiang, C. Y., Sheu, M. L., Cheng, F. C., et al. (2017). Prevention of axonal degeneration by perineurium injection of mitochondria in a sciatic nerve crush injury model. Neurosurgery 80, 475–488. doi: 10.1093/neuros/nyw090
Li, X., Zhang, Y., Yeung, S. C., Liang, Y., Liang, X., Ding, Y., et al. (2014). Mitochondrial transfer of induced pluripotent stem cell-derived mesenchymal stem cells to airway epithelial cells attenuates cigarette smoke-induced damage. Am. J. Respir. Cell Mol. Biol. 51, 455–465. doi: 10.1165/rcmb.2013-0529oc
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 541, 481–487.
Lippert, T., and Borlongan, C. V. (2019). Prophylactic treatment of hyperbaric oxygen treatment mitigates inflammatory response via mitochondria transfer. CNS Neurosci. Ther. 25, 815–823. doi: 10.1111/cns.13124
Liu, K., Guo, L., Zhou, Z., Pan, M., and Yan, C. (2019). Mesenchymal stem cells transfer mitochondria into cerebral microvasculature and promote recovery from ischemic stroke. Microvasc Res. 123, 74–80. doi: 10.1016/j.mvr.2019.01.001
Lo, E. H. (2008). A new penumbra: transitioning from injury into repair after stroke. Nat. Med. 14, 497–500. doi: 10.1038/nm1735
Lo, E. H., Broderick, J. P., and Moskowitz, M. A. (2004). tPA and proteolysis in the neurovascular unit. Stroke 35, 354–356. doi: 10.1161/01.str.0000115164.80010.8a
Ma, H., Jiang, T., Tang, W., Ma, Z., Pu, K., Xu, F., et al. (2020). Transplantation of platelet-derived mitochondria alleviates cognitive impairment and mitochondrial dysfunction in db/db mice. Clin. Sci. (Lond.) 134, 2161–2175. doi: 10.1042/cs20200530
Madsen, P. M., Pinto, M., Patel, S., McCarthy, S., Gao, H., Taherian, M., et al. (2017). Mitochondrial DNA double-strand breaks in oligodendrocytes cause demyelination, axonal injury, and cns inflammation. J. Neurosci. 37, 10185–10199. doi: 10.1523/jneurosci.1378-17.2017
Marlein, C. R., Piddock, R. E., Mistry, J. J., Zaitseva, L., Hellmich, C., Horton, R. H., et al. (2019). CD38-Driven mitochondrial trafficking promotes bioenergetic plasticity in multiple myeloma. Cancer Res. 79, 2285–2297. doi: 10.1158/0008-5472.can-18-0773
McCully, J. D., Levitsky, S., Del Nido, P. J., and Cowan, D. B. (2016). Mitochondrial transplantation for therapeutic use. Clin. Transl. Med. 5:16.
Miliotis, S., Nicolalde, B., Ortega, M., Yepez, J., and Caicedo, A. (2019). Forms of extracellular mitochondria and their impact in health. Mitochondrion 48, 16–30. doi: 10.1016/j.mito.2019.02.002
Moon, H. E., and Paek, S. H. (2015). Mitochondrial dysfunction in Parkinson’s disease. Exp. Neurobiol. 24, 103–116.
Morales, I., Sanchez, A., Puertas-Avendano, R., Rodriguez-Sabate, C., Perez-Barreto, A., and Rodriguez, M. (2020). Neuroglial transmitophagy and Parkinson’s disease. Glia 68, 2277–2299.
Murphy, B. J., Klusch, N., Langer, J., Mills, D. J., Yildiz, O., and Kuhlbrandt, W. (2019). Rotary substates of mitochondrial ATP synthase reveal the basis of flexible F1-Fo coupling. Science. 364:eaaw9128. doi: 10.1126/science.aaw9128
Nakamura, Y., Lo, E. H., and Hayakawa, K. (2020). Placental mitochondria therapy for cerebral ischemia-reperfusion injury in mice. Stroke 51, 3142–3146. doi: 10.1161/strokeaha.120.030152
Nakatogawa, H., Suzuki, K., Kamada, Y., and Ohsumi, Y. (2009). Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat. Rev. Mol. Cell Biol. 10, 458–467. doi: 10.1038/nrm2708
Nitzan, K., Benhamron, S., Valitsky, M., Kesner, E. E., Lichtenstein, M., Ben-Zvi, A., et al. (2019). Mitochondrial transfer ameliorates cognitive deficits, neuronal loss, and gliosis in Alzheimer’s disease mice. J. Alzheimers Dis. 72, 587–604. doi: 10.3233/jad-190853
Onfelt, B., Nedvetzki, S., Benninger, R. K., Purbhoo, M. A., Sowinski, S., Hume, A. N., et al. (2006). Structurally distinct membrane nanotubes between human macrophages support long-distance vesicular traffic or surfing of bacteria. J. Immunol. 177, 8476–8483. doi: 10.4049/jimmunol.177.12.8476
Park, J., Lai, M. K. P., Arumugam, T. V., and Jo, D. G. O. (2020a). GlcNAcylation as a therapeutic target for alzheimer’s disease. Neuromolecular Med. 22, 171–193.
Park, J., Nakamura, Y., Li, W., Hamanaka, G., Arai, K., Lo, E., et al. (2020b). Effects of O-GlcNAcylation on functional mitochondrial transfer from astrocytes. J. Cereb. Blood Flow Metab. [Online ahead of print] doi: 10.1177/0271678X20969588
Parker, W. D. Jr., and Parks, J. K. (1995). Cytochrome c oxidase in Alzheimer’s disease brain: purification and characterization. Neurology 45(3 Pt 1), 482–486. doi: 10.1212/wnl.45.3.482
Peng, Y., Zheng, D., Zhang, X., Pan, S., Ji, T., Zhang, J., et al. (2019). Cell-Free mitochondrial DNA in the CSF: a potential prognostic biomarker of Anti-NMDAR encephalitis. Front. Immunol. 10:103. doi: 10.3389/fimmu.2019.00103
Pivovarova, N. B., and Andrews, S. B. (2010). Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 277, 3622–3636. doi: 10.1111/j.1742-4658.2010.07754.x
Pohland, M., Pellowska, M., Asseburg, H., Hagl, S., Reutzel, M., Joppe, A., et al. (2018). MH84 improves mitochondrial dysfunction in a mouse model of early Alzheimer’s disease. Alzheimers Res. Ther. 10:18.
Requejo-Aguilar, R., and Bolanos, J. P. (2016). Mitochondrial control of cell bioenergetics in Parkinson’s disease. Free Radic Biol. Med. 100, 123–137. doi: 10.1016/j.freeradbiomed.2016.04.012
Russo, E., Nguyen, H., Lippert, T., Tuazon, J., Borlongan, C. V., and Napoli, E. (2018). Mitochondrial targeting as a novel therapy for stroke. Brain Circ. 4, 84–94. doi: 10.4103/bc.bc_14_18
Rutkai, I., Merdzo, I., Wunnava, S. V., Curtin, G. T., Katakam, P. V., and Busija, D. W. (2019). Cerebrovascular function and mitochondrial bioenergetics after ischemia-reperfusion in male rats. J. Cereb. Blood Flow Metab. 39, 1056–1068. doi: 10.1177/0271678x17745028
She, D. T., Jo, D. G., and Arumugam, T. V. (2017). Emerging roles of sirtuins in ischemic stroke. Transl. Stroke Res. [Online ahead of print] doi: 10.1007/s12975-017-0544-4
Shi, X., Zhao, M., Fu, C., and Fu, A. (2017). Intravenous administration of mitochondria for treating experimental Parkinson’s disease. Mitochondrion 34, 91–100. doi: 10.1016/j.mito.2017.02.005
Shin, B., Saeed, M. Y., Esch, J. J., Guariento, A., Blitzer, D., Moskowitzova, K., et al. (2019). A novel biological strategy for myocardial protection by intracoronary delivery of mitochondria: safety and efficacy. JACC Basic Transl. Sci. 4, 871–888. doi: 10.1016/j.jacbts.2019.08.007
Simon, R., Meller, R., Yang, T., Pearson, A., and Wilson, G. (2019). Enhancing base excision repair of mitochondrial DNA to reduce ischemic injury following reperfusion. Transl. Stroke Res. 10, 664–671. doi: 10.1007/s12975-018-0680-5
Sinha, P., Islam, M. N., Bhattacharya, S., and Bhattacharya, J. (2016). Intercellular mitochondrial transfer: bioenergetic crosstalk between cells. Curr. Opin Genet. Dev. 38, 97–101. doi: 10.1016/j.gde.2016.05.002
Spees, J. L., Olson, S. D., Whitney, M. J., and Prockop, D. J. (2006). Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. U.S.A. 103, 1283–1288. doi: 10.1073/pnas.0510511103
Sun, C., Liu, X., Wang, B., Wang, Z., Liu, Y., Di, C., et al. (2019). Endocytosis-mediated mitochondrial transplantation: transferring normal human astrocytic mitochondria into glioma cells rescues aerobic respiration and enhances radiosensitivity. Theranostics 9, 3595–3607. doi: 10.7150/thno.33100
Swerdlow, R. H., Burns, J. M., and Khan, S. M. (2014). The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochim. Biophys. Acta 1842, 1219–1231. doi: 10.1016/j.bbadis.2013.09.010
Tait, S. W., and Green, D. R. (2012). Mitochondria and cell signalling. J. Cell Sci. 125(Pt 4), 807–815. doi: 10.1242/jcs.099234
Thurairajah, K., Briggs, G. D., and Balogh, Z. J. (2018). The source of cell-free mitochondrial DNA in trauma and potential therapeutic strategies. Eur. J. Trauma Emerg. Surg. 44, 325–334. doi: 10.1007/s00068-018-0954-3
Torralba, D., Baixauli, F., and Sanchez-Madrid, F. (2016). Mitochondria know no boundaries: mechanisms and functions of intercellular mitochondrial transfer. Front. Cell Dev. Biol. 4:107. doi: 10.3389/fcell.2016.00107
Trias, E., Barbeito, L., and Yamanaka, K. (2018). Phenotypic heterogeneity of astrocytes in motor neuron disease. Clin. Exp. Neuroimmunol. 9, 225–234. doi: 10.1111/cen3.12476
Twig, G., Elorza, A., Molina, A. J., Mohamed, H., Wikstrom, J. D., Walzer, G., et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446. doi: 10.1038/sj.emboj.7601963
Vekaria, H. J., Talley Watts, L., Lin, A. L., and Sullivan, P. G. (2017). Targeting mitochondrial dysfunction in CNS injury using Methylene Blue; still a magic bullet? Neurochem. Int. 109, 117–125. doi: 10.1016/j.neuint.2017.04.004
Vosler, P. S., Graham, S. H., Wechsler, L. R., and Chen, J. (2009). Mitochondrial targets for stroke: focusing basic science research toward development of clinically translatable therapeutics. Stroke 40, 3149–3155. doi: 10.1161/strokeaha.108.543769
Wang, A. C., Jensen, E. H., Rexach, J. E., Vinters, H. V., and Hsieh-Wilson, L. C. (2016). Loss of O-GlcNAc glycosylation in forebrain excitatory neurons induces neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 113, 15120–15125. doi: 10.1073/pnas.1606899113
Wang, W., Zhao, F., Ma, X., Perry, G., and Zhu, X. (2020). Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol. Neurodegener. 15:30.
Wang, X., and Gerdes, H. H. (2015). Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ. 22, 1181–1191. doi: 10.1038/cdd.2014.211
Wang, Z., Zhou, F., Dou, Y., Tian, X., Liu, C., Li, H., et al. (2018). Melatonin alleviates intracerebral hemorrhage-induced secondary brain injury in rats via suppressing apoptosis, inflammation, oxidative stress, DNA damage, and mitochondria injury. Transl. Stroke Res. 9, 74–91. doi: 10.1007/s12975-017-0559-x
Watts, L. T., Lloyd, R., Garling, R. J., and Duong, T. (2013). Stroke neuroprotection: targeting mitochondria. Brain Sci. 3, 540–560. doi: 10.3390/brainsci3020540
Wei, Z., Su, W., Lou, H., Duan, S., and Chen, G. (2018). Trafficking pathway between plasma membrane and mitochondria via clathrin-mediated endocytosis. J. Mol. Cell Biol. 10, 539–548. doi: 10.1093/jmcb/mjy060
Yang, H., Borg, T. K., Ma, Z., Xu, M., Wetzel, G., Saraf, L. V., et al. (2016). Biochip-based study of unidirectional mitochondrial transfer from stem cells to myocytes via tunneling nanotubes. Biofabrication 8:015012. doi: 10.1088/1758-5090/8/1/015012
Yang, X., and Qian, K. (2017). Protein O-GlcNAcylation: emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol. 18, 452–465. doi: 10.1038/nrm.2017.22
Yao, Y., Fan, X. L., Jiang, D., Zhang, Y., Li, X., Xu, Z. B., et al. (2018). Connexin 43-mediated mitochondrial transfer of iPSC-MSCs alleviates asthma inflammation. Stem Cell Rep. 11, 1120–1135. doi: 10.1016/j.stemcr.2018.09.012
Youn, D. H., Kim, B. J., Kim, Y., and Jeon, J. P. (2020). Extracellular mitochondrial dysfunction in cerebrospinal fluid of patients with delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Neurocrit. Care 33, 422–428. doi: 10.1007/s12028-019-00895-1
Yun, S. P., Kam, T. I., Panicker, N., Kim, S., Oh, Y., Park, J. S., et al. (2018). Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 24, 931–938. doi: 10.1038/s41591-018-0051-5
Yuzwa, S. A., Shan, X., Macauley, M. S., Clark, T., Skorobogatko, Y., Vosseller, K., et al. (2012). Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat. Chem. Biol. 8, 393–399. doi: 10.1038/nchembio.797
Zacchigna, S., Lambrechts, D., and Carmeliet, P. (2008). Neurovascular signalling defects in neurodegeneration. Nat. Rev. Neurosci. 9, 169–181. doi: 10.1038/nrn2336
Zamanian, J. L., Xu, L., Foo, L. C., Nouri, N., Zhou, L., Giffard, R. G., et al. (2012). Genomic analysis of reactive astrogliosis. J. Neurosci. 32, 6391–6410. doi: 10.1523/jneurosci.6221-11.2012
Zhang, Y., Yu, Z., Jiang, D., Liang, X., Liao, S., Zhang, Z., et al. (2016). iPSC-MSCs with high intrinsic MIRO1 and sensitivity to TNF-alpha yield efficacious mitochondrial transfer to rescue anthracycline-induced cardiomyopathy. Stem Cell Rep. 7, 749–763. doi: 10.1016/j.stemcr.2016.08.009
Zhang, Z., Ma, Z., Yan, C., Pu, K., Wu, M., Bai, J., et al. (2019). Muscle-derived autologous mitochondrial transplantation: a novel strategy for treating cerebral ischemic injury. Behav. Brain Res. 356, 322–331. doi: 10.1016/j.bbr.2018.09.005
Keywords: extracellular mitochondria, central nervous system, mitochondrial transfer, biomarker, neurovascular unit
Citation: Park J-H and Hayakawa K (2021) Extracellular Mitochondria Signals in CNS Disorders. Front. Cell Dev. Biol. 9:642853. doi: 10.3389/fcell.2021.642853
Received: 16 December 2020; Accepted: 26 January 2021;
Published: 05 March 2021.
Edited by:
Fabricio Simao, Harvard Medical School, United StatesReviewed by:
Prasad V. Katakam, Tulane University, United StatesPatrick G. Sullivan, University of Kentucky, United States
Copyright © 2021 Park and Hayakawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kazuhide Hayakawa, khayakawa1@mgh.harvard.edu; KHAYAKAWA1@partners.org