 Leming Fu
Leming Fu Ya Li
Ya Li Shun Yao
Shun Yao Qingge Guo
Qingge Guo Ya You
Ya You Xianjun Zhu
Xianjun Zhu Bo Lei
Bo Lei
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol., 04 March 2021
Sec. Molecular and Cellular Pathology
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.635424
This article is part of the Research TopicGenetic Mutations Associated with Ocular DiseasesView all 19 articles
Purpose: ARL3 (ADP-ribosylation factor-like 3) variants cause autosomal dominant retinitis pigmentosa (RP) or autosomal recessive Joubert syndrome. We found a family with rod-cone dystrophy (RCD) and verified it was associated with compound heterozygous variants in ARL3 gene.
Methods: Ophthalmic examinations including optical coherence tomography and electroretinogram (ERG) were performed. Targeted next generation sequencing (NGS) was performed for the proband using a custom designed panel. Sanger sequencing and co-segregation were conducted in the family members. Changes of protein structure mediated by the variants were studied in vitro. ARL3 protein stability and its interaction with RP2 protein were assessed by cycloheximide chase assay and co-immunoprecipitation (Co-IP) assay.
Results: Visual acuity of the 18-year-old male proband was 0.25 in the right and 0.20 in the left eye, while his non-consanguineous parents and sister was normal. The proband showed signs of RCD, including nyctalopia, peripheral field loss, bone-spicule deposits in the retina, and reduced ERG responses. The father, aged 50 years old, showed visual acuity of 1.0 in both eyes. Unlike the proband, he presented late onset and mild cone-rod dystrophy (CRD), including macular atrophy, central scotomata, moderate reduction in photopic ERG responses. None of all the family members had hearing abnormality, mental dysplasia or gait instability. We identified two novel compound heterozygous variants (c.91A>G, p.T31A; c.353G>T, p.C118F) in ARL3 in the proband, while his father only had variant c.91A>G. Bioinformatics analysis indicated amino acid positions of the two variants are highly conserved among species. The in silico tools predicted the variants to be harmful. Protein structure analysis showed the two variants had potential to alter the protein structure. Based on the ACMG guidelines, the two variants were likely pathogenic. In addition, the ARL3 mutations destabilized ARL3 protein, and the mutation c.353G>T disrupted the interaction between ARL3 and RP2 in HEK293T cells.
Conclusions: We showed novel compound heterozygous variants in ARL3 were associated with early onset of autosomal recessive RCD, while c.91A>G along may be associated with a late onset of dominant CRD. The two variants in ARL3 could be causative by destabilizing ARL3 protein and impairing its interaction with RP2 protein.
Retinitis pigmentosa (RP) is a group of highly heterogeneous inherited retinal diseases, and it is one of the most important causes of blindness worldwide (Narayan et al., 2016). Almost 2.5 million individuals suffered with RP around the world (Hu et al., 2019). RP is typically characterized by night blindness (nyctalopia), progressive constriction of visual field, changes in the fundus and reduced electroretinogram (ERG), and ultimately resulting in complete blindness (Ali et al., 2017). Rod-cone dystrophy (RCD) is a common form of RP due to primary degeneration of rod cells followed by degeneration of cone cells (Pagon, 1988). RP manifests in a syndromic or a non-syndromic form. Non-syndromic RP can be in different traits, including autosomal dominant RP (adRP), autosomal recessive RP (arRP), X-linked recessive form and simplex/sporadic type. Nowadays, around 90 genes were identified to be associated with non-syndromic RP (https://sph.uth.tmc.edu/retnet/). The proteins encoded by RP-associated genes exert different roles in transcription, retina phototransduction, transport processes via the photoreceptor connecting cilium, cell growth, cellular structure, and metabolism of vitamin A (Collin et al., 2011; Bhatia et al., 2019).
Human ARL3 (ADP-ribosylation factor-like 3; MIM:604695) gene is mapped to chromosome 10q24.32, and contains 6 exons. The encoded protein ARL3 is a small molecule GTP-binding protein belonging to the ADP-ribosylation factor (ARF) family. In the mammalian retina, ARL3 is mainly localized to microtubule structures throughout the retina and is enriched in the connecting cilium of rod and cone photoreceptors (Grayson et al., 2002). ARL3 is crucial for ciliogenesis and axoneme formation, as well as cargo displacement of lipidated proteins in the cilium (Hanke-Gogokhia et al., 2016). Besides, ARL3 also acts as an allosteric factor for the release of lipidated proteins bound to PDE6D (delta subunit of phosphodiesterase) and UNC119A/B in an apparent GTP dependent manner (Hanke-Gogokhia et al., 2018). The activity of ARL3 is regulated by a GTP-exchange factor ARL13B (ADP-ribosylation factor-like protein 13B) (Gotthardt et al., 2015) and a GTPase-activating protein RP2 (Retinitis Pigmentosa 2) (Veltel et al., 2008).
A missense variant in ARL3 (NM_004311.3) (c.269A>G, p.Tyr90Cys) has previously been reported as a possible cause of non-syndromic autosomal dominant RP in two families (Strom et al., 2016; Holtan et al., 2019). In addition, homozygocity for two different ARL3 Arg149 missense variants (c.445C>T, p.Arg149Cys; c.446G>A, p.Arg149His) were reported to cause Joubert syndrome (Alkanderi et al., 2018). A most recent report indicated that a homozygous variant in ARL3 (c.296G>T, p. Arg99Ile) caused cone-rod dystrophy (CRD) in two consanguineous families (Sheikh et al., 2019). In this study, we found a Chinese family with typical rod-cone dystrophy (RCD), which was associated with novel compound heterozygous variants in ARL3. In addition, we presumed that one of the variants might be associated with a late onset of dominant CRD. This study expanded both the phenotype and genotype of ARL3 associated retinal dystrophy. Furthermore, our data suggested that the pathogenicity of the variants were related with destabilizing of ARL3 protein, as well as impairing its interaction with RP2, another protein that is associated with RP.
The study was performed in accordance with the Declaration of Helsinki and approved by the Ethics Committee of Henan Eye Hospital for the release of clinical information, family history, and blood extraction for genetic testing [IRB approval number: HNEECKY-2019 (15)]. Written informed consent was obtained from all participants after the study risks and benefits were thoroughly explained.
All the members were enquired about the family and medical history. Each member was accepted a complete ocular examination, including best corrected visual acuity (BCVA), intraocular pressure (IOP), slit-lamp biomicroscopy, color vision, fundus photography, visual field, swept-source optical coherence tomography (SS-OCT, VG200D SVision Imaging, China), and full-field electroretinogram (ERG).
Genomic DNA was extracted from peripheral blood of all family members with the TIANGEN Blood DNA Kit (DP304, TIANGEN, China). Targeted next generation sequencing (NGS) was performed for the proband using a custom designed panel (PS400) containing 376 known genes associated with inherited retinal diseases (Zhu et al., 2021). The Nextseq 500 (Illumina, San Diego, CA, USA) platform was used for paired-end sequencing with read lengths of 150 bp and average sequencing depth of almost 300 X. Raw reads were mapped to the human genome reference (UCSC hg19) using three commercial software including XYGeneRanger 2.0 (Xunyin, Shanghai, China), TGex (LifeMap Sciences, Alameda, CA, USA) and Efficient Genosome Intepration System, EGIS (SierraVast Bio-Medical Technology Co., Ltd, Shanghai, China). Variant-filtering was based on public and in-house SNP databases, including 1000Genome project, HGMD, ExAC and ClinVar, as well as our internal database. The non-synonymous and splicing variants with MAF <2% were kept for further analysis. Sanger sequencing and co-segregation analysis were performed for the verification of suspicious disease-relevant gene variants in the available family members. Primer sequences and PCR conditions were shown in Supplementary Tables 1, 2, respectively.
To assess the possible pathogenicity of these mutations, and to predict whether a protein sequence variation affected protein function, the following web applications was used: LRT (Likelihood Ratio Test), PolyPhen-2 (Polymorphism Phenotyping v2), Mutation Taster (Schwarz et al., 2010), SIFT (Sorting Intolerant From Tolerant) (Kumar et al., 2009), FATHMM (Functional Analysis Through Hidden Markov Models) (Rogers et al., 2018), and CADD (Combined Annotation Dependent Depletion) (Kircher et al., 2014). Multiple protein sequence alignment among various species was carried out by Clustal Omega (Sievers and Higgins, 2018). The tertiary structure of protein was predicted by the Swiss-Model workspace (http://swissmodel.expasy.org) (Biasini et al., 2014). CDD online software (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) was used to conduct domains analysis. HOPE online software (http://www.cmbi.umcn.nl/hope) was used to analyze the structural effects of mutation. Superimposition of homology models of ARL3 was constructed using PyMOL software (https://pymol.org/2/). The protein stability of ARL3 mutations was predicted with the online tools MUpro (http://mupro.proteomics.ics.uci.edu/) (Cheng et al., 2006)and I-Mutant v2.0 (http://folding.biofold.org/i-mutant/i-mutant2.0) (Capriotti et al., 2005). All the sources were provided in the public domain.
HEK293T cell line was purchased from American Type Culture Collection. Cells were cultured in DMEM/HIGH GLUCOSE medium (SH30022.01, Hyclone, USA) containing 10% fetal bovine serum (35-081-CV, Corning, USA) with 100 units/mL penicillin and 100 μg/mL streptomycin at 37°C with 5% CO2.
The wild-type, c.91 A>G (p.T31A), and c.353G>T (p.C118F) CDS sequences of the ARL3 gene with the Flag tag at the C-terminal were synthesized and subcloned into the pcDNA3.1 (+) expression vector (Invitrogen, USA), respectively. The wild-type RP2 cDNA with the HA tag at the N-terminal was subcloned into the pcDNA3.1 (+) expression vector. All plasmids were confirmed by DNA sequencing and WB analysis. When cells grow to 80–90% confluence, cell transfection was performed using Lipofectamine 3000 (L3000015, Invitrogen, USA). After transfection, the cells were harvested at 24 h for protein extraction and further analysis.
The primary antibodies used were as follows: the flag antibody (147935, CST, USA), β-actin antibody (200068-8F10, ZENBIO, China), RP2 antibody (ab174840, Abcam, UK). Cells were lysed by RIPA lysis buffer (PC101, EpiZyme, China) with 1% protease inhibitor cocktail (GRF101, EpiZyme, China). The samples were resolved in 12% SDS-PAGE gel and transferred to a polyvinylidene difluoride (PVDF) membrane (IPVH00010, MILLIPORE, Ireland). The membranes were blocked with 5% non-fat dry milk for 1 h and then incubated with the primary antibody overnight at 4°C. After three washes with TBST (containing 1% Tween-20), the membranes were treated with horseradish peroxidase-conjugated secondary antibody at room temperature for 2 h. Then the membranes were exposed to X-ray film after washed for three times.
Protein stability was assessed by cycloheximide chase assay. Cycloheximide (CHX, 66-81-9, Selleck, USA), a protein synthesis inhibitor, was used. HEK293T cells were transfected with empty vector, wild-type, T31A and C118F mutation ARL3 vectors for 24 h. The cells were subsequently treated with 100 μg/ml CHX for 1, 3, and 6 h, respectively. The total protein was extracted for Western blot. Wild-type and mutant ARL3 protein levels were detected using flag antibody. The protein levels of β-actin were the endogenous control.
HEK293T cells were co-transfected with 6.25 μg of HA-RP2-WT plasmid and 6.25 μg of either wild-type or mutant Flag-ARL3 plasmids, and harvested for protein extraction after 24 h. Total protein lysate was extracted by immunoprecipitation buffer (BL509A, Biosharp, China), and the concentration of the supernatants was quantified with a BCA protein assay kit (P0011, Beyotime, China). Total proteins of 300 μg were added with 5 × SDS to prepare as the input sample. Total proteins of 500 μg were mixed with 10 μg anti-Flag magnetic beads (HY-K0207, MCE, USA) and shaken for 4 h at 4°C on rotor. The beads were collected by Magnetic Separator and washed three times by immunoprecipitation buffer with 1% protease inhibitor. The beads were mixed with sample loading buffer (1 ×) and boiled for 10 min. The supernatant was collected and used for western blot analysis.
We normalized the data from each group by dividing the measurement of the tested variant from that of the references. The value of the control was set to one and tested measurement was adjusted accordingly. GraphPad Prism 8.0 was used for the statistical analysis. Student's t-test and one-way analysis of variance (ANOVA) were performed with a 95% confidence level to evaluate the differences. P-value < 0.05 was considered statistically significant. All quantitative data were displayed as mean ± standard deviation (SD).
We found a Chinese family with RCD, the pedigree was presented in Figure 1A. In this family, the proband showed typical RCD but his father present with a late onset and mild CRD, while the mother and sister were unaffected. The proband, a 19-year-old male, had decrease of vision and nyctalopia for 4 years. The BCVA was 0.25 in the right and 0.20 in the left eye. The proband also had dyserythrochloropsia. Visual field testing revealed tunnel visual field (Figure 1B). Fundus photography showed peripheral bone spicule pigmentation (Figure 1C). SS-OCT presented atrophy of the retinal outer layers of the binocular macular area where the light reflection signal of ellipsoid zone and chimeric zone was weakened, coarse and interrupted. The reflection of RPE/Bruch membrane was rough (Figure 1D). ERG showed severe reduction in scotopic ERG responses and to a less extent in photopic ERG responses (Figure 1E).
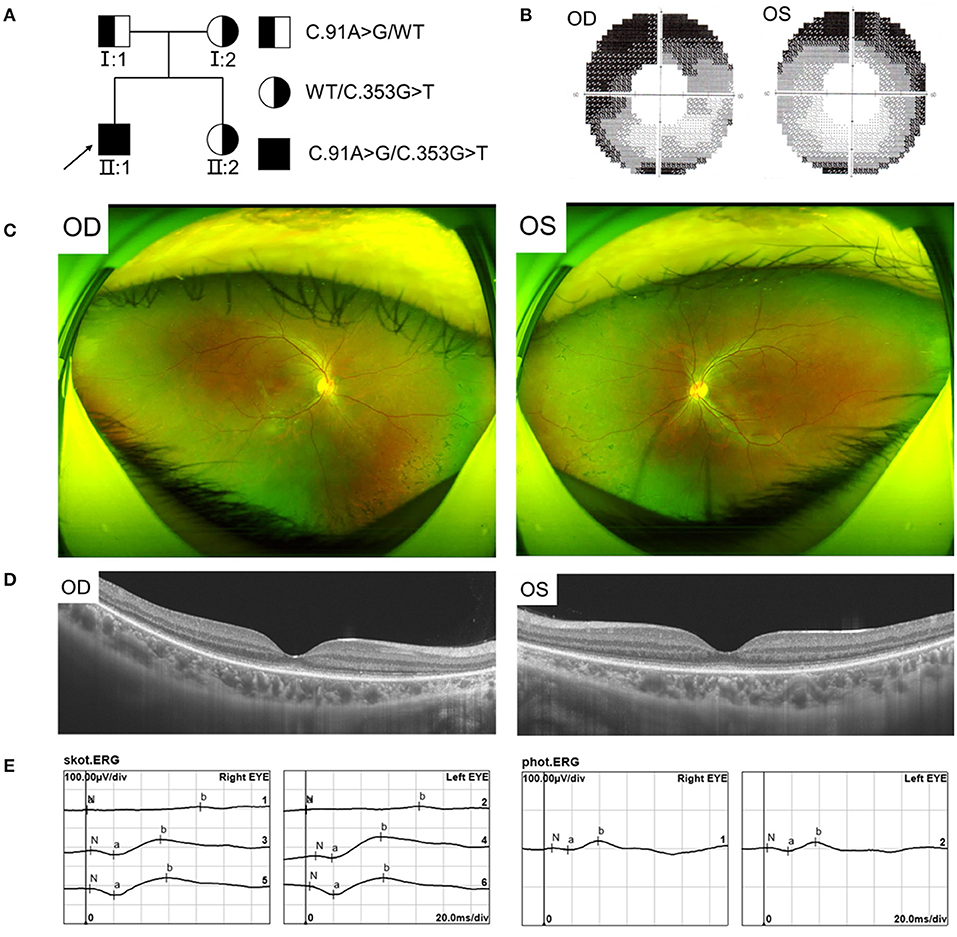
Figure 1. Pedigree and Clinical features of the proband with RCD. (A) Pedigree of the family with autosomal recessive RCD. (B) Visual field showed tunnel vision. (C) Fundus photographs showed peripheral bone spicule pigmentation. (D) SS-OCT showed atrophy of the retinal outer layers of the binocular macular area. (E) ERG showed severe reduced rod responses and to less extent cone responses.
The father, aged 50 years old, showed normal visual acuity of 1.0 in both eyes and had mild dyserythrochloropsia. Unlike the proband, the father's vision field appeared as central scotoma (Figure 2A). Fundus examination revealed circular degeneration around macular fovea without pigmentation of the peripheral retina (Figure 2B). SS-OCT revealed the binocular outer retinal layers of the macular area was thinned with macular sparing, and the light reflection signal of the ellipsoid zone in the parafovea was weakened and discontinuous (Figure 2C). The ERG showed a moderate decrease in the binocular cone and rod systems (Figure 2D).
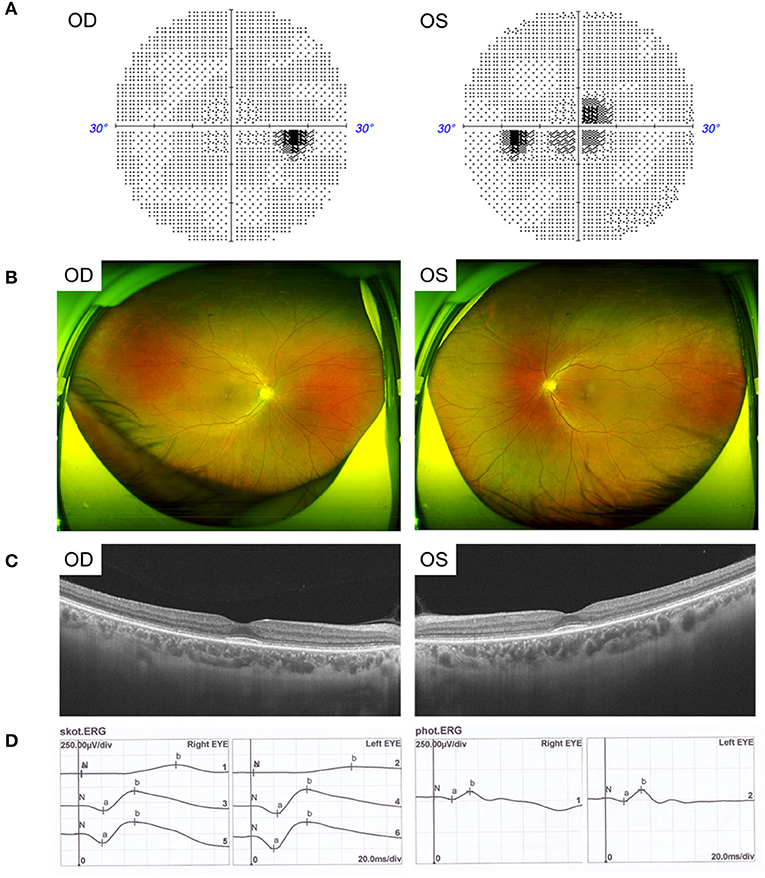
Figure 2. Clinical features of the father with CRD. (A) Visual field showed central scotoma. (B) Fundus photographs showed circular degeneration around macular fovea. (C) SS-OCT revealed the binocular macular area outer retinal was thinned by atrophy. (D) ERG showed a moderate decrease in the binocular cone and rod systems.
Other examinations including anterior segment and IOP of the proband and his father were normal. Neither the proband's mother nor sister showed any abnormalities in the eyes. None of all the family members, including the proband, presented hearing abnormality, mental dysplasia, or gait instability.
Targeted NGS identified two novel compound heterozygous variants (c.91A>G, p.T31A; c.353G>T, p.C118F) in ARL3 in the proband. Sanger sequencing of ARL3 revealed segregation of the two variants with the RCD phenotype in participating individuals (Figure 3A). Neither variant had been reported in the human gene mutation database (HGMD). The reported allele frequency for c.353G>T (p.C118F) was concentrated in the East Asian populations almost 0.0010 in the Genome Aggregation Database (GnomAD), while no record for the c.91A>G (p.T31A) mutation was found. Furthermore, c.353G>T (p.C118F) has not been reported homozygous in GnomAD. Multiple alignments of amino acid sequences of ARL3 protein from different species revealed that Thr31 is evolutionally conserved among species, while Cys118 is only conserved in mammals, but not in zebrafish and yeast (Figure 3B). Moreover, both of the variants T31A and C118F were considered “Deleterious” as predicted by LRT, Mutation Taster, SIFT, FATHMM, and CADD, but Polyphen-2 predicted the pathogenicity for the variant C118F benign (Table 1).
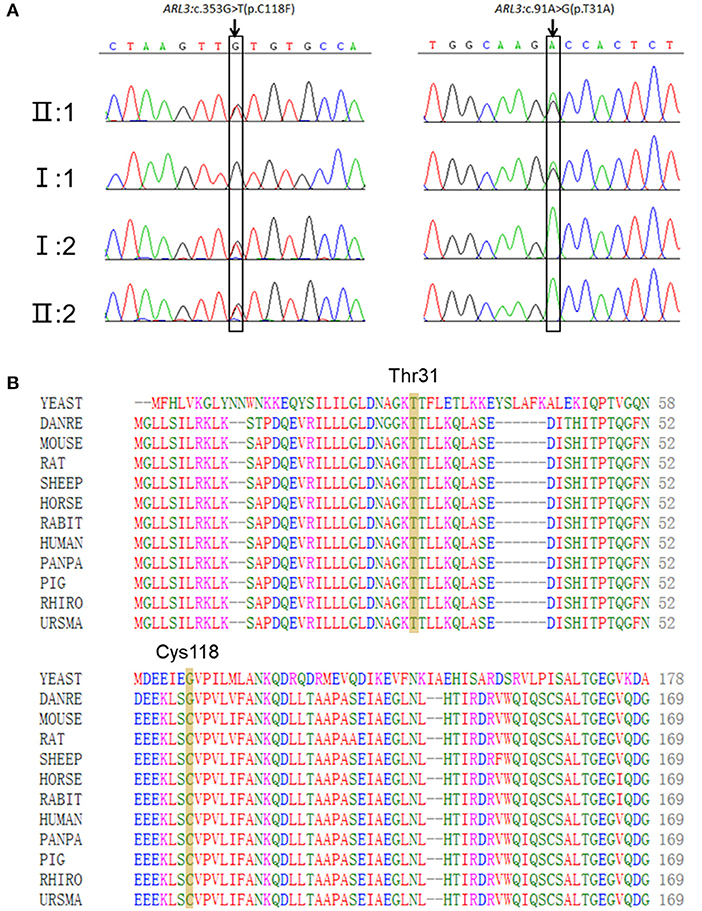
Figure 3. Two novel ARL3 variants (c.91A>G, p.T31A; c.353G>T, p.C118F) were identified in the RCD family. (A) Sanger sequencing in all family numbers. (B) Multiple alignments of Thr31 and Cys118 of ARL3 protein from different species. Both variants occurred on the conserved residues of the ARL3 protein.
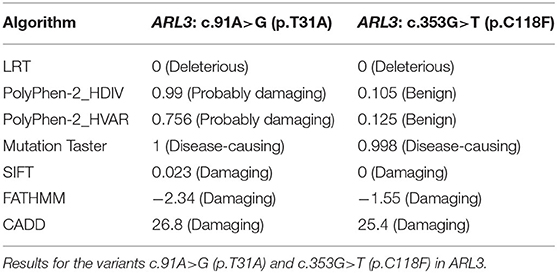
Table 1. In silico pathogenicity prediction.
In addition, other variants in the proband were screened out, including two heterozygous missense variants (c.13491T>A, p.F4497L; c.11197A>G, p.N3733D) in USH2A and a heterozygous missense variant (c.2432T>C, p.I811T) in FBN2 (Supplementary Table 3), all of which were inherited from his mother (Supplementary Figure 1). These two mutations in USH2A were cis-mutations, and the results predicted by in silico prediction such as SIFT, Polyphen-2, Mutation Taster, and CADD indicated that these two missense mutations might be benign. Moreover, neither the proband nor his mother showed sensorineural deafness. On the other hand, we noticed that the variant c.2432T>C in FBN2 was predicted to be possibly damaging by in silico prediction. Variants in FBN2 have been reported to associated with congenital contractural arachnodactyly (CCA) (Putnam et al., 1995) and early-onset macular degeneration (MD) (Ratnapriya et al., 2014), whereas the proband's mother and sister who carried the heterozygous missense variant (c.2432T>C, p.I811T) in FBN2 didn't have any phenotypes of CCA or MD.
To visualize the structure changes of the protein mediated by p.T31A and p.C118F mutations, we used HOPE online software. The schematic structures of the original and the mutant amino acid were shown in Figure 4A. The 3D-structure of wild-type and mutant proteins were shown in Figure 4B. Compared with the wild-type residue, the mutant residue at position 31 was smaller and more hydrophobic, while the mutant residue at position 118 was bigger and more hydrophobic. Among the interactors of ARL3 are RP2, ARL13B, UNC119A, who interact with ARL3 in a GTP dependent manner. In order to visualize the interactions, we superimposed the known structures of ARL3 in complex with the interactors (Figure 4C).
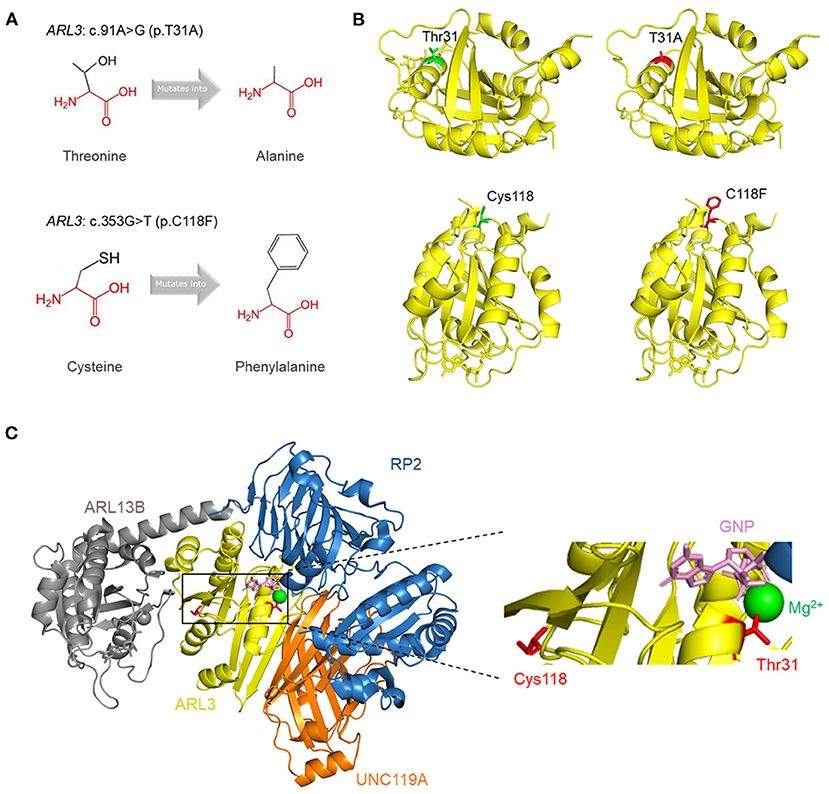
Figure 4. Tertiary structure prediction of mutant proteins. (A) The schematic structures of the original and the mutant amino acids. The backbone was colored in red; the side chain was colored in black. (B) The 3D-structure of the wild-type and mutant proteins. The main structure of the ARL3 protein was colored in yellow. The side chains of the wild-type and the mutant residues were colored in green and red, respectively. (C) Superimposition of the structures of ARL3 (yellow) in complex with its interactors: RP2 (blue; PDB: 3BH6) (Veltel et al., 2008), ARL13B (gray; PDB: 5DI3) (Gotthardt et al., 2015), and UNC119A (orange; PDB: 4GOJ) (Ismail et al., 2012). On the right side is a zoomed-in view of some structures, including GNP (pink), Mg2+ (green), and the side chains of Thr31 and Cys118 (red).
The conformational change of protein caused by the change of amino acid properties was closely related to the stability of protein. Thus, we wondered whether ARL3 T31A and C118F mutations affected the stability of the protein. The online tools MUpro and I-Mutant v2.0 were used to predict protein stability and results suggested that the two mutant ARL3 proteins showed decrease of stability (Table 2).
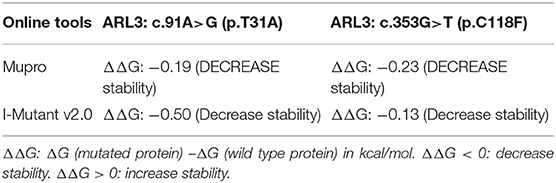
Table 2. Prediction of the protein stability with online tools.
To determine whether ARL3 T31A and C118F mutations destabilize ARL3 protein, CHX chase assays was performed. HEK293T cells were transfected with empty vector, wild-type and two mutant ARL3 vectors for 24 h, followed by treatment with cycloheximide (CHX) for 1, 3, and 6 h, respectively. Wild-type and two mutant ARL3 protein levels were analyzed using the flag antibody with Western blot. In Figure 5, both the half-lives of ARL3 T31A and ARL3 C118F protein showed a decreasing trend compared with the wild-type ARL3 protein. These results indicated that both T31A and C118F mutations decreased the stability of ARL3 protein.
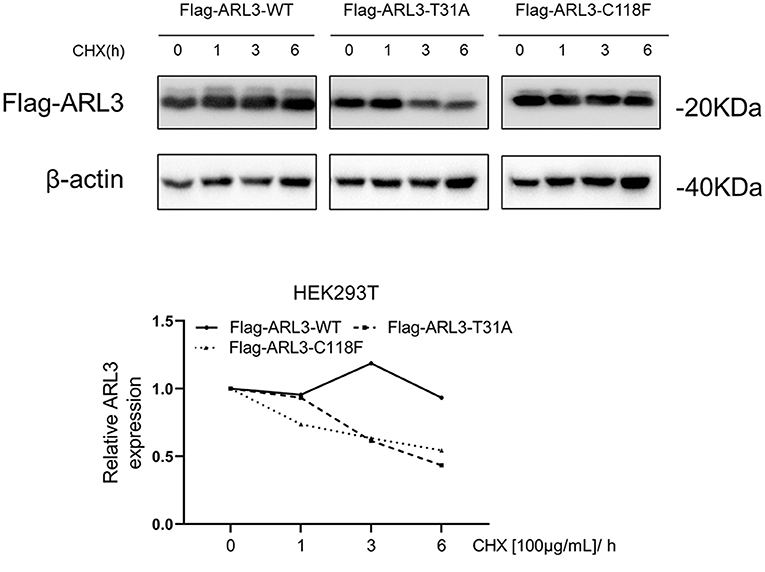
Figure 5. The ARL3 variants T31A and C118F impaired the stabilities of the encoded proteins in HEK293T cells. HEK293T cells were transfected with empty vector, wild-type, ARL3 T31A and C118F vectors for 24 h and then treated with 100 μg/ml cycloheximide (CHX) for 1, 3, and 6 h, respectively. ARL3 protein levels were detected using the flag antibody. The protein levels of β-actin were used as endogenous control (n = 1).
As a small molecule GTP-binding protein, ARL3 was activated when combined with GTP. But it depends on the presence of functional guanine nucleotide exchange factor, ARL13B (Gotthardt et al., 2015). Protein RP2 could interact with ARL3, and it was identified as a negative regulator by hydrolyzing GTP. In addition, it was reported that the impaired interaction between RP2 and ARL3 induced by RP2 mutation could cause RP (Kuhnel et al., 2006). To investigate whether the novel ARL3 variants affected the interaction between ARL3 and RP2, we conducted co-IP analysis. Plasmid of HA-RP2-WT was co-transfected with Flag-ARL3-WT, Flag-ARL3-T31A, or Flag-ARL3-C118F in HEK293T cells. As in Figure 6, RP2 was detected after Flag-ARL3 pull-down, suggesting interaction between ARL3 and RP2 in HEK293T cells. The relative level of RP2 protein was significantly reduced in the C118F mutant group compared with the control group, while there was no significant change in the T31A mutant group. The results demonstrated that ARL3 mutation C118F inhibited the interaction between ARL3 and RP2 proteins.
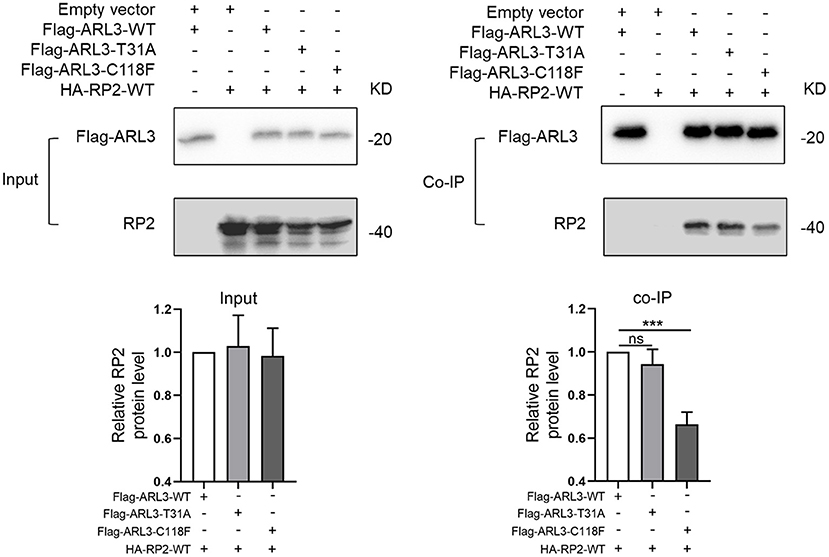
Figure 6. ARL3 variants T31A and C118F disrupted the combination between ARL3 and RP2. Plasmids of HA-RP2-WT were co-transfected with wild-type or mutant Flag-ARL3 plasmids into HEK293T cells. ARL3 was immunoprecipitated with anti-Flag antibody. Western blotting was performed to detect the specific proteins indicated on the left side of each panel (Error bars indicate means ± SD; n = 3, ***p < 0.001).
In this study, we identified two novel variants (c.91A>G, p.T31A; c.353G>T, p.C118F) in ARL3 gene in a Chinese RCD family. According to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines, the two variants were “likely pathogenic,” which was supported by the following evidences. The two variants, (1) were supported by functional tests in vitro (PS3); (2) located in an important domain that acts as GTP/Mg2+ binding site and GAP interaction site (PM1); (3) not found in the 200 unrelated health controls and absent from any databases (PM2); (4) The two variants were considered as “Deleterious” by function prediction software, and were highly conserved during evolution (PP3).
The ARL3 Thr31 and Cys118 residues are highly conserved among species, and in silico prediction tools suggested that either missense change was likely to be pathogenic. HOPE analysis showed that, compared with the wild-type residues (Threonine and Cysteine), the mutant residue (Alanine) of p.T31A was smaller and more hydrophobic, and the mutant residue (Phenylalanine) of p.C118F was bigger and more hydrophobic. The changes of size and hydrophobicity may cause the loss of hydrogen bonds in the core of the protein, and disturbing protein folding, and then destabilize ARL3 protein. In CHX chase assays, our results revealed that the T31A and C118F protein showed a rapid degradation while the wild-type ARL3 protein was stabilized, indicating the T31A and C118F mutation decreased the stability of ARL3 protein. On the other hand, we noticed that p. Thr31 was central to the P-loop NTPase domain in ARL3, and involved in direct binding of the beta and gamma phosphate moieties of GXP (GTP or GDP) and Mg2+ cation (Figure 4C). Another substitution at T31, T31N, has been widely used as a synthetic ARL3-GDP conformational mimic. In addition, the known interaction partners of ARL3 mainly include PDE6D, UNC119A/B, RP2 and ARL13B, whose interactions with ARL3 are GTP-dependent. Therefore, the replacement of Threonine with Arginine at residue 31 variant may impact GTP binding or exchange and thereby affected the normal interactions with these molecules or other parts of the protein, which is most likely a loss-of-function mechanism.
In the mammalian retina, ARL3 functions as a cargo displacement factor and plays an important role in the transport of lipidated protein to the outer segment of the photoreceptors. In addition, RP2 and ARL3 co-localize to the ciliary apparatus (Grayson et al., 2002), and RP2 could stimulate the exchange of GTP to GDP on ARL3, stimulating the release of the lipidated proteins bound to PDE6D and UNC119A/B (Hanke-Gogokhia et al., 2018). On the other hand, Arl3 and RP2 regulated the trafficking of specific ciliary tip kinesins, Kif7 and Kif17, independently of lipidated protein trafficking(Schwarz et al., 2017). The well-regulated protein trafficking in photoreceptors is crucial for normal visual function. The connecting cilium is a key player for the vectorial transport of proteins from the endoplasmic reticulum to the outer segment. Unsurprisingly, mutations in several connecting cilium-associated proteins have been shown to be associated with various types of retinal dystrophy like Joubert syndrome-associated RP, dominant RP, X-linked RP and dominant CRD (Liu et al., 2002; Cantagrel et al., 2008; Thomas et al., 2014; Zhang et al., 2019). It indicates that close cooperation of related proteins is necessary for normal operation of protein transport in photoreceptors.
Our co-IP results revealed that C118F mutant showed a reduced affinity for RP2. But it is unknown how C118F mutant contributes to the occurrence and development of RCD. Significantly, the reported allele frequency of c.353G>T (p.C118F) in GnomAD is concentrated in the East Asian population with almost 1:1,000. This allele frequency is borderline as being too common to cause a very rare disease like RCD. Furthermore, C118F is only conserved in mammals, but not in zebrafish and yeast, and not all the in silico prediction tools predicted the C118F variant would be pathogenic. Given that the mother and sister who carried the variant of C118F were unaffected, the mutation C118F may not cause disease when it exists alone as a heterozygote.
On the other hand, when T31A was carried by the father as a heterozygote, he showed a late onset and relatively mild phenotype of CRD. Significantly, the proband who carried the compound heterozygous variants of T31A and C118F typically presented with poor visual acuity, nyctalopia and peripheral field loss when he was young. ERG revealed severely reduced rod and cone system functions. We presumed these two variants may work together to promote the occurrence and development of RCD. Thus, compound heterozygous variants T31A and C118F in ARL3 caused earlier and severer RCD condition. However, considering the limitation of cell model, the in vivo tests of the two variants could not be inferred with confidence at this moment. And it could be helpful to identify whether his father had other gene mutation that caused this phenotype. Further functional studies are still necessary.
So far, four ARL3 mutations have been identified (Figure 7), most of which locate at the important domain, affecting the stability of encoded protein or the affinity with interactors. Previously, two different ARL3 Arg149 missense variants (c.445C>T, p.Arg149Cys; c.446G>A, p.Arg149His) were reported to cause recessive Joubert syndrome, characterized by hypoplasia of the cerebellar vermis, developmental delay, renal anomalies, and RCD in two families (Alkanderi et al., 2018). The study showed that substitution of arginine at position 149 disrupted the known interaction between ARL3 and ARL13B and thus prevented the correct release of intra-ciliary cargos. Patients with heterozygous missense variant (c.269A>G, p.Tyr90Cys) in ARL3 have recently been found to cause non-syndromic autosomal dominant RP, confirming earlier reports of this missense variant causing retinal disease. The missense variant is predicted to disrupt protein folding and compromise GTP binding or exchange. A most recent report indicated that a homozygous variant in ARL3 (c.296G>T, p. Arg99Ile) caused CRD in two consanguineous families. The study suggested that the mutation p.Arg99Ile may alter the affinity of encoded protein for guanine nucleotides, resulting in a much less stable protein. Thus, although ARL3 is not a common cause of retinal degeneration in humans, it is a strong prior candidate gene.
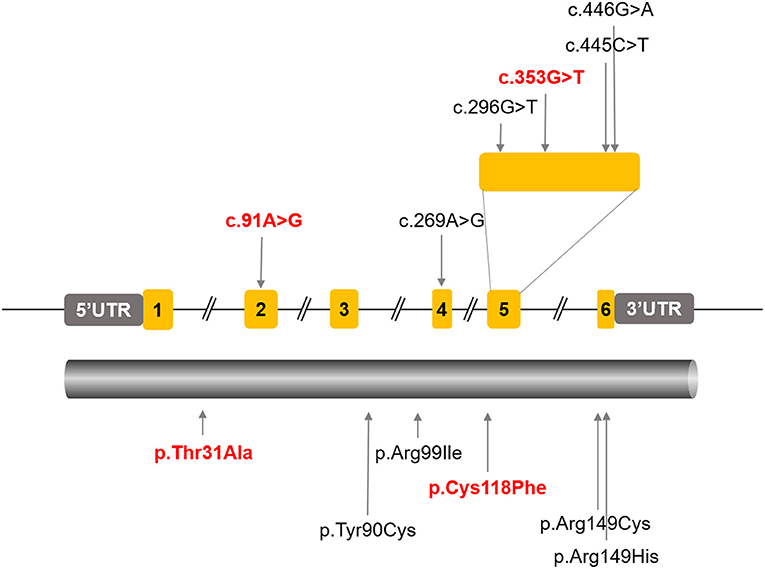
Figure 7. All the reported ARL3 pathogenic mutations were labeled in the schematic diagram of ARL3 gene and protein. The ARL3 mutations c.91A>G (p.T31A) and c.353G>T (p.C118F) were indicated in red.
In conclusion, our results extended both genotype and phenotype of ARL3 associated retinal dystrophy. We defined two novel pathogenic variants (c.91A>G, p.T31A; c.353G>T, p.C118F) of ARL3 in a Chinese family with typical RCD. We presumed that these compound heterozygous variants were associated with the early onset of recessive RCD, while c.91A>G along might be associated with a late onset of dominant CRD. We further found the ARL3 variants T31A and C118F destabilized ARL3 protein, and the C118F disrupted the interaction with RP2 in HEK293T cells. On the other hand, considering the limitation of cell model and number of patients, the detailed mechanisms that the two ARL3 variants causing RCD deserve further studies.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number (s) can be found below: https://databases.lovd.nl/shared/individuals?search_owned_by_=%3D%22Leming%20Fu%22.
The studies involving human participants were reviewed and approved by The Ethics Committee of Henan Eye Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
BL conceptualized and designed the study. YL, QG, and YY collected the clinical samples and clinical data. LF, YL, and YY performed the genetic analysis and bioinformatics evaluations. LF conducted the molecular biology experiments and drafted the manuscript. BL, SY, and XZ interpreted the results. BL and XZ reviewed and edited the draft. All authors agreed to be accountable for the content of the work and approved the final manuscript.
This work was supported by National Natural Science Foundation of China grants (82071008, 81770949), Henan Science and Technology Bureau (212102310308), and the Henan Key Laboratory of Ophthalmology and Vision Science.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank all the patients and their families for participating in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.635424/full#supplementary-material
Ali, M. U., Rahman, M. S. U., Cao, J., and Yuan, P. X. (2017). Genetic characterization and disease mechanism of retinitis pigmentosa; current scenario. Biotech 7:251. doi: 10.1007/s13205-017-0878-3
Alkanderi, S., Molinari, E., Shaheen, R., Elmaghloob, Y., Stephen, L. A., Sammut, V., et al. (2018). ARL3 mutations cause Joubert syndrome by disrupting ciliary protein composition. Am. J. Hum. Genet. 103, 612–620. doi: 10.1016/j.ajhg.2018.08.015
Bhatia, S., Kaur, N., Singh, I. R., and Vanita, V. (2019). A novel mutation in MERTK for rod-cone dystrophy in a North Indian family. Can. J. Ophthalmol. 54, 40–50. doi: 10.1016/j.jcjo.2018.02.008
Biasini, M., Bienert, S., Waterhouse, A., Arnold, K., Studer, G., Schmidt, T., et al. (2014). SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–258. doi: 10.1093/nar/gku340
Cantagrel, V., Silhavy, J. L., Bielas, S. L., Swistun, D., Marsh, S. E., Bertrand, J. Y., et al. (2008). Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am. J. Hum. Genet. 83, 170–179. doi: 10.1016/j.ajhg.2008.06.023
Capriotti, E., Fariselli, P., and Casadio, R. (2005). I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic. Acids Res. 33, W306–310. doi: 10.1093/nar/gki375
Cheng, J., Randall, A., and Baldi, P. (2006). Prediction of protein stability changes for single-site mutations using support vector machines. Proteins 62, 1125–1132. doi: 10.1002/prot.20810
Collin, R. W., van den Born, L. I., Klevering, B. J., de Castro-Miro, M., Littink, K. W., Arimadyo, K., et al. (2011). High-resolution homozygosity mapping is a powerful tool to detect novel mutations causative of autosomal recessive RP in the Dutch population. Invest. Ophthalmol. Vis. Sci. 52, 2227–2239. doi: 10.1167/iovs.10-6185
Gotthardt, K., Lokaj, M., Koerner, C., Falk, N., Giessl, A., and Wittinghofer, A. (2015). A G-protein activation cascade from Arl13B to Arl3 and implications for ciliary targeting of lipidated proteins. Elife 4:15. doi: 10.7554/eLife.11859.015
Grayson, C., Bartolini, F., Chapple, J. P., Willison, K. R., Bhamidipati, A., Lewis, S. A., et al. (2002). Localization in the human retina of the X-linked retinitis pigmentosa protein RP2, its homologue cofactor C and the RP2 interacting protein Arl3. Hum. Mol. Genet. 11, 3065–3074. doi: 10.1093/hmg/11.24.3065
Hanke-Gogokhia, C., Frederick, J. M., Zhang, H., and Baehr, W. (2018). Binary function of ARL3-GTP revealed by gene knockouts. Adv. Exp. Med. Biol. 1074, 317–325. doi: 10.1007/978-3-319-75402-4_39
Hanke-Gogokhia, C., Wu, Z., Gerstner, C. D., Frederick, J. M., Zhang, H., and Baehr, W. (2016). Arf-like protein 3 (ARL3) regulates protein trafficking and ciliogenesis in mouse photoreceptors. J. Biol. Chem. 291, 7142–7155. doi: 10.1074/jbc.M115.710954
Holtan, J. P., Teigen, K., Aukrust, I., Bragadottir, R., and Houge, G. (2019). Dominant ARL3-related retinitis pigmentosa. Ophthalmic Genet. 40, 124–128. doi: 10.1080/13816810.2019.1586965
Hu, Y. S., Song, H., Li, Y., Xiao, Z. Y., and Li, T. (2019). Whole-exome sequencing identifies novel mutations in genes responsible for retinitis pigmentosa in 2 nonconsanguineous Chinese families. Int. J. Ophthalmol. 12, 915–923. doi: 10.18240/ijo.2019.06.06
Ismail, S. A., Chen, Y. X., Miertzschke, M., Vetter, I. R., Koerner, C., and Wittinghofer, A. (2012). Structural basis for Arl3-specific release of myristoylated ciliary cargo from UNC119. EMBO J. 31, 4085–4094. doi: 10.1038/emboj.2012.257
Kircher, M., Witten, D. M., Jain, P., O'Roak, B. J., Cooper, G. M., and Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315. doi: 10.1038/ng.2892
Kuhnel, K., Veltel, S., Schlichting, I., and Wittinghofer, A. (2006). Crystal structure of the human retinitis pigmentosa 2 protein and its interaction with Arl3. Structure 14, 367–378. doi: 10.1016/j.str.2005.11.008
Kumar, P., Henikoff, S., and Ng, P. C. (2009). Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081. doi: 10.1038/nprot.2009.86
Liu, Q., Zhou, J., Daiger, S. P., Farber, D. B., Heckenlively, J. R., Smith, J. E., et al. (2002). Identification and subcellular localization of the RP1 protein in human and mouse photoreceptors. Invest. Ophthalmol. Vis. Sci. 43, 22–32.
Narayan, D. S., Wood, J. P., Chidlow, G., and Casson, R. J. (2016). A review of the mechanisms of cone degeneration in retinitis pigmentosa. Acta Ophthalmol. 94, 748–754. doi: 10.1111/aos.13141
Pagon, R. A. (1988). Retinitis pigmentosa. Surv. Ophthalmol. 33, 137–177. doi: 10.1016/0039-6257(88)90085-9
Putnam, E. A., Zhang, H., Ramirez, F., and Milewicz, D. M. (1995). Fibrillin-2 (FBN2) mutations result in the Marfan-like disorder, congenital contractural arachnodactyly. Nat. Genet. 11, 456–458. doi: 10.1038/ng1295-456
Ratnapriya, R., Zhan, X., Fariss, R. N., Branham, K. E., Zipprer, D., Chakarova, C. F., et al. (2014). Rare and common variants in extracellular matrix gene Fibrillin 2 (FBN2) are associated with macular degeneration. Hum. Mol. Genet. 23, 5827–5837. doi: 10.1093/hmg/ddu276
Rogers, M. F., Shihab, H. A., Mort, M., Cooper, D. N., Gaunt, T. R., and Campbell, C. (2018). FATHMM-XF: accurate prediction of pathogenic point mutations via extended features. Bioinformatics 34, 511–513. doi: 10.1093/bioinformatics/btx536
Schwarz, J. M., Rodelsperger, C., Schuelke, M., and Seelow, D. (2010). MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7, 575–576. doi: 10.1038/nmeth0810-575
Schwarz, N., Lane, A., Jovanovic, K., Parfitt, D. A., Aguila, M., Thompson, C. L., et al. (2017). Arl3 and RP2 regulate the trafficking of ciliary tip kinesins. Hum. Mol. Genet. 26:3451. doi: 10.1093/hmg/ddx245
Sheikh, S. A., Sisk, R. A., Schiavon, C. R., Waryah, Y. M., Usmani, M. A., Steel, D. H., et al. (2019). Homozygous variant in ARL3 causes autosomal recessive cone rod dystrophy. Invest. Ophthalmol. Vis. Sci. 60, 4811–4819. doi: 10.1167/iovs.19-27263
Sievers, F., and Higgins, D. G. (2018). Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 27, 135–145. doi: 10.1002/pro.3290
Strom, S. P., Clark, M. J., Martinez, A., Garcia, S., Abelazeem, A. A., Matynia, A., et al. (2016). De novo occurrence of a variant in ARL3 and apparent autosomal dominant transmission of retinitis pigmentosa. PLoS ONE 11:e0150944. doi: 10.1371/journal.pone.0150944
Thomas, S., Wright, K. J., Le Corre, S., Micalizzi, A., Romani, M., Abhyankar, A., et al. (2014). A homozygous PDE6D mutation in Joubert syndrome impairs targeting of farnesylated INPP5E protein to the primary cilium. Hum. Mutat. 35, 137–146. doi: 10.1002/humu.22470
Veltel, S., Gasper, R., Eisenacher, E., and Wittinghofer, A. (2008). The retinitis pigmentosa 2 gene product is a GTPase-activating protein for Arf-like 3. Nat. Struct. Mol. Biol. 15, 373–380. doi: 10.1038/nsmb.1396
Zhang, J., Gao, F., Du, C., Wang, J., Pi, X., Guo, W., et al. (2019). A novel RP2 missense mutation Q158P identified in an X-linked retinitis pigmentosa family impaired RP2 protein stability. Gene 707, 86–92. doi: 10.1016/j.gene.2019.05.006
Keywords: ARL3, compound heterozygous variants, rod-cone dystrophy, cone-rod dystrophy, RP2
Citation: Fu L, Li Y, Yao S, Guo Q, You Y, Zhu X and Lei B (2021) Autosomal Recessive Rod-Cone Dystrophy Associated With Compound Heterozygous Variants in ARL3 Gene. Front. Cell Dev. Biol. 9:635424. doi: 10.3389/fcell.2021.635424
Received: 30 November 2020; Accepted: 15 February 2021;
Published: 04 March 2021.
Edited by:
Minzhong Yu, Case Western Reserve University, United StatesReviewed by:
Said El Shamieh, Beirut Arab University, LebanonCopyright © 2021 Fu, Li, Yao, Guo, You, Zhu and Lei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bo Lei, Ym9sZWk5OUAxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.