 Elena Conte1†
Elena Conte1† Alessandra Pannunzio1†
Alessandra Pannunzio1† Paola Imbrici1
Paola Imbrici1 Giulia Maria Camerino1
Giulia Maria Camerino1 Lorenzo Maggi2
Lorenzo Maggi2 Marina Mora2
Marina Mora2 Sara Gibertini2
Sara Gibertini2 Ornella Cappellari1
Ornella Cappellari1 Annamaria De Luca1Mauro Coluccia1†
Annamaria De Luca1Mauro Coluccia1† Antonella Liantonio1*†
Antonella Liantonio1*†- 1Department of Pharmacy—Drug Sciences, University of Bari, Bari, Italy
- 2Neuromuscular Diseases and Neuroimmunology Unit, Foundation IRCCS Neurological Institute C. Besta, Milan, Italy
Tubular Aggregate Myopathy (TAM) is a hereditary ultra-rare muscle disorder characterized by muscle weakness and cramps or myasthenic features. Biopsies from TAM patients show the presence of tubular aggregates originated from sarcoplasmic reticulum due to altered Ca2+ homeostasis. TAM is caused by gain-of-function mutations in STIM1 or ORAI1, proteins responsible for Store-Operated-Calcium-Entry (SOCE), a pivotal mechanism in Ca2+ signaling. So far there is no cure for TAM and the mechanisms through which STIM1 or ORAI1 gene mutation lead to muscle dysfunction remain to be clarified. It has been established that post-natal myogenesis critically relies on Ca2+ influx through SOCE. To explore how Ca2+ homeostasis dysregulation associated with TAM impacts on muscle differentiation cascade, we here performed a functional characterization of myoblasts and myotubes deriving from patients carrying STIM1 L96V mutation by using fura-2 cytofluorimetry, high content imaging and real-time PCR. We demonstrated a higher resting Ca2+ concentration and an increased SOCE in STIM1 mutant compared with control, together with a compensatory down-regulation of genes involved in Ca2+ handling (RyR1, Atp2a1, Trpc1). Differentiating STIM1 L96V myoblasts persisted in a mononuclear state and the fewer multinucleated myotubes had distinct morphology and geometry of mitochondrial network compared to controls, indicating a defect in the late differentiation phase. The alteration in myogenic pathway was confirmed by gene expression analysis regarding early (Myf5, Mef2D) and late (DMD, Tnnt3) differentiation markers together with mitochondrial markers (IDH3A, OGDH). We provided evidences of mechanisms responsible for a defective myogenesis associated to TAM mutant and validated a reliable cellular model usefull for TAM preclinical studies.
Introduction
In its primary form, Tubular Aggregate Myopathy (TAM) is a clinically heterogeneous and very rare skeletal muscle disorder, in most cases inherited in an autosomal dominant pattern (Böhm and Laporte, 2018; Michelucci et al., 2018; Morin et al., 2020). Signs and symptoms typically begin in childhood and worsen over time. TAM patients can be characterized by asymptomatic elevated creatine kinase (CK) levels as well as by muscle weakness predominantly affecting the proximal muscles of lower limbs. Myalgia and cramps have also been described (Böhm et al., 2014; Hedberg et al., 2014; Walter et al., 2015) and in some cases the full picture of the multisystemic Stormorken syndrome develops (Morin et al., 2020). A consistent histopathological feature of TAM patients is represented by the presence of tubular aggregates (TAs) in skeletal muscle fibers. TAs are formed by regular arrays of densely packed membrane tubules, most likely originating from sarcoplasmic reticulum (SR) (Böhm et al., 2013, 2017; Endo et al., 2015; Harris et al., 2017; Böhm and Laporte, 2018). However, TAs represent a non-specific morphological alteration being present in several neuromuscular disorders associated to SR stress (Michelucci et al., 2018).
TAM can be caused by heterozygous mutations in STIM1 or ORAI1 gene (Morin et al., 2020), both encoding for Ca2+ homeostasis key regulators, and CASQ1 gene (Barone et al., 2017; Böhm et al., 2018), encoding for calsequestrin, the major Ca2+ buffering protein in skeletal muscle SR. Particularly, STIM1 and ORA1 are key components of the calcium release-activated calcium (CRAC) channels (Prakriya, 2009), which are activated following intracellular SR or endoplasmic reticulum (ER) Ca2+ store depletion and allow extracellular Ca2+ influx through a process called store operated Ca2+ entry (SOCE). SOCE is therefore fundamental in a variety of cellular functions, including secretion, transcription, motility, enzyme activity, Ca2+ store filling-state, and muscle contraction (Kiviluoto et al., 2011; Stiber and Rosenberg, 2011; Cho et al., 2017). SOCE-dependent Ca2+ signaling is also crucial for the onset of skeletal muscle development (Stiber et al., 2008; Darbellay et al., 2009, 2010; Li et al., 2012; Phuong et al., 2013; Wei-Lapierre et al., 2013; Tu et al., 2016). SOCE is indeed necessary for the activity of various Ca2+-dependent enzymes regulating myogenesis-associated transcription factors, as has been shown for Nuclear Factor of Activated T cells (NFAT) in murine models (Kegley et al., 2001; Armand et al., 2008), as well as for Myocyte Enhancer Factor-2 (MEF2) and Myogenin in myoblasts deriving from human biopsies (Louis et al., 2008; Darbellay et al., 2009, 2010).
STIM1 is an ER/SR transmembrane protein activated by a drop in ER/SR calcium levels. This is at the basis of the SOCE process. Indeed, following Ca2+ store depletion, the STIM1 Ca2+-sensing intraluminal EF-hands undergo a conformational switch leading to protein di- and oligomerization, to an extended active state, interacting and activating the plasma membrane Ca2+ channel ORAI1, thus finally leading to Ca2+ entry (Park et al., 2009; Cho et al., 2017; Gudlur et al., 2018).
To date, 18 distinct STIM1 mutations causing TAM have been identified, of which 15 cluster in the EF-hand domains (Morin et al., 2020). According to the resolved STIM1 protein structure (Yang et al., 2012; Zhu et al., 2017; Lopez et al., 2020), TAM-associated STIM1 EF-hand mutations appear to affect amino acids involved in Ca2+ coordination or forming a hydrophobic pocket which maintains STIM1 in a folded state (Stathopulos et al., 2008; Böhm et al., 2014). Functional impact of TAM-associated STIM1 EF-hand mutations has been investigated through their heterologous expression in murine C2C12 myoblasts (Böhm et al., 2013) as well as in other engineered cell lines (Hedberg et al., 2014; Nesin et al., 2014). All known EF-hand mutations were shown to induce STIM1 oligomerization and clustering independently from intraluminal SR/ER Ca2+ level, thus indicating a constitutive STIM1 and SOCE activation and a gain-of-function effect leading to intracellular Ca2+ accumulation (Böhm and Laporte, 2018). Importantly, only in selected cases, i.e., the STIM1 A84G (Böhm et al., 2013) and G81A (Walter et al., 2015) mutations, Ca2+ homeostasis alteration was directly confirmed on TAM patient-derived muscle cells.
So far, there is no cure for TAM and there is little information in literature regarding a therapy or management of this disorder. Considering that symptoms generally occur at a young age and significantly reduce the quality of life of the affected people, there is an urgent need to find new treatments. The resulting aberrant Ca2+ homeostasis associated to TAM mutants is likely the key cellular event causing serious damage on muscle development and integrity. Thus, the investigation of cellular processes dysfunction induced by Ca2+ homeostasis alteration associated with TAM mutants remains a pivotal strategy to identify novel druggable targets for this rare disease.
In this study, the STIM1 L96V-associated Ca2+ homeostasis dysregulation has been evaluated in patient-derived skeletal muscle cells. This mutation, located in the Ca2+ sensing canonical EF hand (cEF-hand) of STIM1 protein, was identified in a 13 year old girl showing CK elevation and lower limb weakness and myalgia, along with a muscle histology characterized by TAs, fiber size variability and internal nuclei (Böhm et al., 2014). Functional and morphological characterization of patient derived STIM1 L96V myoblasts and myotubes has been performed by using Ca2+ cytofluorimetry, molecular biology and high content imaging technologies. Our results confirm the STIM1 L96V-associated Ca2+ homeostasis dysfunction on patient-derived cells and demonstrate, for the first time, that the STIM1 L96V mutation alters the myogenic differentiation program, particularly regarding the terminal differentiation step, thereby providing new insights in the pathogenesis of STIM1-related TAM.
Materials and Methods
Cell Culture
Human muscle samples were provided by the Telethon biobank at Besta Neurological Institute in Milan. Written, informed consent was obtained from the subjects or their parents/legal guardians. Research was conducted according to protocols approved by the Institutional Review Board of the Besta Neurological Institute and University of Bari, and in compliance with the Helsinki Declaration and local legislation. Particularly, we used myoblasts and myotubes deriving from one TAM patient's biopsy carrying STIM1 L96V mutant. The patient's mutation was found by Böhm's laboratory in 2014 (Böhm et al., 2014). Particularly, patient' cells genetic characterization was performed on DNA extracted from blood cells. Sanger-sequencing has been used for all coding exons and the adjacent splice-relevant regions of STIM1 (NM_001277961). The mutation found in the proband was also analyzed in the parents (our patient belongs to Family 3 in Böhm et al., 2014). The patient carries a de novo heterozygous mutation. Mutations in DMD and lamin A/C (LMNA) were also exclude for the patient. A control muscle cell line was obtained from a patient not affected from TAM (who had no STIM, ORAI, or CASQ1 mutations) of the same age and sex of affected patients. To assess the myogenesis characteristics in control condition, gene expression analysis of genes involved in the differentiation of myoblasts to myotubes was performed both in cells deriving from a patient not affected from TAM (control) and for comparison in a control muscle cell line (HMb_2) (see Supplementary Material). Myoblast derived cells were isolated from patients' biopsy, cultured using the protocol described in Zanotti et al. (2007) and used under 2 different stages of differentiation. Primary myoblasts were derived directly from biopsied material by culturing in Dulbecco's modified Eagle's medium (DMEM; Lonza Group Ltd, Basel, Switzerland) containing 20% heat-inactivated fetal bovine serum (FBS) (Gibco Life Technologies), 1% penicillin-streptomycin (Lonza), L-glutamine (Lonza), 10 μg/ml insulin (Sigma Aldrich, St. Louis, MO), 2.5 ng/ml basic fibroblast growth factor (bFGF) (Gibco Life Technologies), and 10 ng/ml epidermal growth factor (EGF) (Gibco). Cells were grown on plastic. The medium was changed twice weekly and the cultures examined by inverted-phase microscopy. Once at 70% confluence, they were dissociated enzymatically with trypsin-EDTA (Sigma) and seeded for immediate propagation, or frozen in medium containing 10% DMSO (Sigma) for later propagation or other use. To obtain myotubes, the myoblasts were seeded into 35 mm dishes or in chamber slides in DMEM proliferating medium. At 70% confluence, proliferating medium was changed to differentiating medium (DMEM, 1% penicillin-streptomycin, L-glutamine and insulin, without FBS, or growth factors) and the myoblasts could differentiate into myotubes after 10 days (Zanotti et al., 2007). After the isolation, we analyzed phenotypic characteristic of the cell model carrying STIM1 L96V mutant, alongside with the non-mutated counterpart which represents control muscle cell. We evaluated various parameters on the proliferating and the differentiating myoblasts (T1, 5 days) and on the differentiated myotubes (T2, 10 days).
For fluorescence imaging analysis, both control and Leu96Val STIM1 myoblasts were seeded in quadruplicate in 96-well culture plates (96-well CallCarrierTM, PerkinElmer) at 6,000/100 μL density and incubated at 37°C and 5% CO2 to allow cell growth and proliferation. After 48 h incubation and cell washing, the growth medium was replaced by differentiation medium (T0) and incubation continued for further five (T1) or ten (T2) days, taking care to change the medium every 2 days.
Cytosolic Calcium Measurements
Myoblasts or myotubes grown on coverslip were loaded for 30 min at room temperature with the cell permeant fluorescent Ca2+ indicator 5 μM Fura-2-AM (Molecular Probes-Invitrogen, Italy) mixed to 0.05% (v/v) Pluronic F-127 (Molecular Probes) in normal physiological solution. Ratiometric images of Fura-2 fluorescence were monitored using an inverted Eclipse TE300 microscope (Nikon, Japan) with a 40X Plan-Fluor objective (Nikon, Japan). Fluorescence measurements were made using a QuantiCell 900 integrated imaging system (Visitech International Ltd., Sunderland, UK) as previously described (Conte et al., 2017). During the experiments, pairs of background subtracted images of Fura-2 fluorescence (510 nm) emitted after excitation at 340 and 380 nm were acquired and ratiometric images (340/380 nm) were calculated for each cell using QC2000 software. Subsequently fluorescence ratio values were converted to the resting cytosolic calcium concentration, [Ca2+]i (nM), after a calibration procedure using the following equation: [Ca2+]i = (R-Rmin)/(Rmax-R)*β where R is the ratio of the fluorescence emitted after excitation at 340 nm to the fluorescence after excitation at 380 nm; KD is the affinity constant of fura-2 for calcium, which was taken as 145 nM (Molecular Probes); and β is a parameter according to Grynkiewicz et al. (1985) that was determined experimentally in situ in ionomycin-permeabilized muscle fibers as previously described (Conte et al., 2017).
To measure SOCE, 2 μM Thapsigargin was used to passively deplete the Ca2+ stores in the calcium free-solution and then extracellular Ca2+ was applied to myoblasts or myotubes (SOCE protocol). The normal physiological solution was composed of 148 mM NaCl, 4.5 mM KCl, 2.5 mM CaCl2, 1 mM MgCl2, 0.44 mM NaH2PO4, 12 mM NaHCO3, and 5.5 mM glucose. The pH of all solutions was adjusted to 7.3–7.4 by bubbling them with 95% O2/5% CO2. The calcium free-solution has the same composition of normal physiological solution except that CaCl2 was omitted and 10 mM ethylene glycol bis(β-aminoethyl ether)-N,N,N',N'-tetracetic acid (EGTA) was added. All chemicals cited above as well as ionomycin, caffeine and thapsigargin were purchased from Sigma (St. Louis, MO, USA).
Fluorescent Probes, Image Acquisition, and Analysis
Cells were stained with a fluorophore dye cocktail containing Hoechst 33,342 (Thermofisher) for nuclear staining and MitoTracker® Deep Red (Thermofisher), 1 μM and 50 nM final concentration, respectively. MitoTracker® Deep Red enables the detection of cell shape and is also used to evaluate cytoplasmic morphological parameters along with mitochondrial mass and network features (texture). After 30 min incubation with the dye cocktail (37°C, 5% CO2), live cell image acquisitions were performed from 16 distinct areas/well using a Perkin Elmer Operetta High Content Imaging system (40x WD objective). Cytoplasmic and nuclear morphological parameters of differentiating control and STIM1 L96V myoblasts at T0, T1, and T2 time points were analyzed at single-cell level using Harmony 3.1 software. Briefly, images were first segmented into nuclei and cytoplasm using “Find Nuclei” and “Find Cytoplasm” Building blocks on Hoechst and MitoTracker® Deep Red channels, respectively. After image segmentation, a set of basic intensity and morphological properties (e.g., area, roundness, etc.), and mitochondrial texture patterns of selected objects was calculated using “Calculate Intensity Properties,” “Calculate Morphology Properties” and “SER texture analysis” building blocks, respectively. Distinct mononuclear and multinuclear morphological phenotypes of skeletal muscle cells were manually identified and then used in the PhenoLOGICTM machine learning module of the Harmony software. The PhenoLOGIC module requires users to supervise training selecting about 100 representative cells/class, thus allowing the software to distinguish different phenotypes. The software performs a linear discriminant analysis to create a linear combination of the most relevant parameters that are then applied to untrained sample wells to classify cells. This approach is very helpful, considering the morphological variety of differentiating skeletal muscle cells. As far as the mitochondrial texture properties are concerned, the analysis was performed using the SER features method. Briefly, the image texture features usually described as smooth, rough, granular, homogeneous/inhomogeneous, linear etc. were quantified calculating the numerical properties which quantitatively describe the texture. The SER (Spot, Edge, Ridge) features method includes a set of eight properties (spot, hole, edge, ridge, valley, saddle, bright, and dark) sensitive to distinct intensity patterns according to the property geometry designation. All results are reported as mean ± SD from three independent experiments, each performed in quadruplicate. GraphPad Prism (GraphPad Software, Inc.) was used for calculating statistics and creation of graphs.
Real Time PCR
Total RNA was extracted from myoblasts and myotubes using RNeasy Micro Kit (Qiagen C.N. 74004, Valencia) according to the manufacturer's protocols and RNA quantity was assessed using a spectrophotometer (ND-1000 NanoDrop, Thermo Scientific, United States). Reverse transcription and real-time PCR analysis were performed as previously described (Conte et al., 2017). The mRNA expression of genes was normalized to the best housekeeping gene: Beta-actin (Actb) selected from beta-2-microglobulin (B2m) and ACTB by Normfinder software. Genes were analyzed by the use of TaqMan Hydrolysis primer and probe gene expression assays that are produced by Life-Technologies with the following assay IDs: ORAI1 assay ID: Hs03046013_m1; STIM1 assay ID: Hs00963373_m1; RYR1 assay ID: Hs00166991_m1; ATP2A1 (encoding SERCA1 protein) assay ID: HS01092295_m1; CACNA1S (encoding Cav1.1 protein) assay ID: Hs00163885_m1; ATP1A2 (encoding SERCA2 protein) assay ID: Hs00265131_m1; TRPC1 assay ID: Hs00608195_m1; TRPC4 assay ID: Hs01077392_m1; OGDH assay ID: Hs01081865_m1; IDH3A assay ID: Hs01051668_m1; PAX7 assay ID: Hs00242962_m1; MYF5 assay ID: Hs00271574_m1, MYOD1 assay ID: Hs00159528_m1; MEF2D assay ID: Hs00954735_m1; MYOG assay ID: Hs01072232_m1; TNNT3 (encoding Troponin protein) assay ID: Hs00952980_m1; DMD (encoding Dystrophin protein) assay ID: Hs00758098_m1; B2M assay ID: Hs00984230_m1 and Actb assay ID: Hs99999903_m1. For genes that were poorly expressed, such as Trpc1, Trpc4, Myf5, and Dmd, preamplification by TaqMan PreAmp Master Mix (Life Technologies C.N. 4391128) was made before real-time experiments with a set-up of pre-amplification detailed in Conte et al. (2017). The real-time PCR protocols were performed in line with the guidelines for qPCR (Bustin et al., 2009).
Statistics
Statistical analysis was performed using Student's t-test, with p < 0.05 or less considered as significant for calcium measurement and gene expression analysis. GraphPad Prism (GraphPad Software, Inc.) was used for calculating statistics (t-test, chi-square test, analysis of variance and Tukey HSD post-hoc test) and creation of graphs for high content fluorescence analysis.
Results
Calcium Homeostasis
In engineered murine myoblasts, all TAM-associated STIM1 EF-hand mutations so far identified, including the L96V one, have been shown to induce protein clustering and excessive Ca2+ entry independently from SR intraluminal Ca2+ level (Böhm and Laporte, 2018). Particularly, C2C12 myoblasts transfected with STIM1 L96V-YFP displayed statistically significant STIM1 clustering regardless of SR Ca2+ depletion (Böhm et al., 2014). Here, Ca2+ homeostasis alterations associated with STIM L96V mutation have been investigated on skeletal muscle cells deriving from TAM patient's biopsy. The clinical, histological and functional phenotype of this patient has been already defined (Böhm et al., 2014). Free intracellular Ca2+ was measured by using the ratiometric fluorescent dye Fura-2. Both STIM1 L96V myoblasts and myotubes, the latter obtained after 10 days in differentiation medium, showed higher basal cytoplasmic Ca2+ level with respect with control cells (Figures 1A,D).
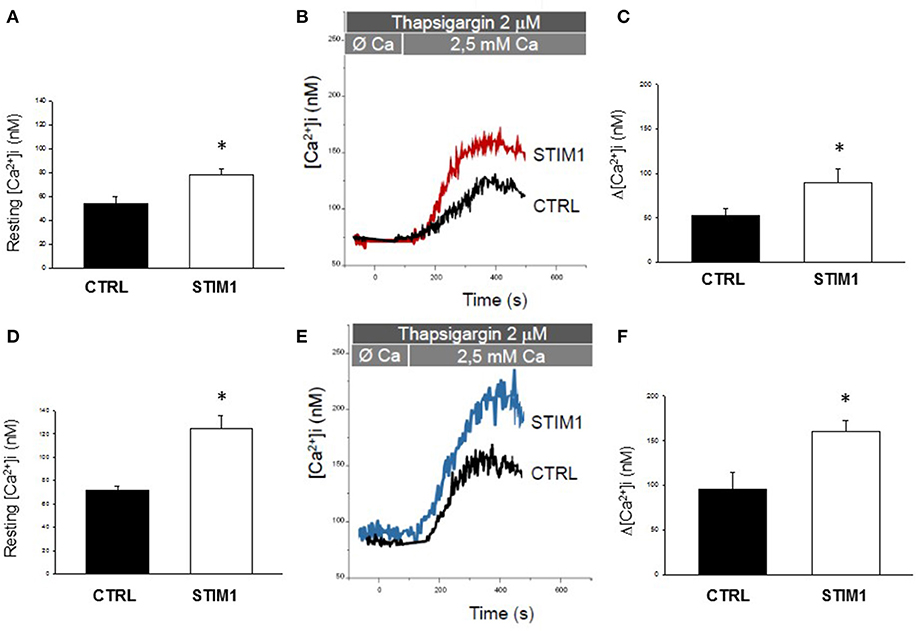
Figure 1. Calcium homeostasis characterization of myoblast and myotubes carrying STIM1 L96V mutation. (A–D) Resting intracellular calcium, resting [Ca2+]i measured in control and STIM1 L96V myoblasts and control and STIM1 L96V myotubes. Each bar represnts the mean ± SEM of resting [Ca2+]i measured in 35–40 cells; (B–E) Representative traces of increased Ca2+ entry in store depleted thapsigargin treated cells after addition of extracellular calcium (see SOCE protocol described in Material and Methods) in control and STIM1 L96V myoblasts and in control and STIM1 L96V multinucleated myotubes (C–F) Amplitude values of [Ca2+]i increase observed with SOCE protocol in control and STIM1 L96V myoblasts and in control and STIM1 L96V multinucleated myotubes. Each bar represents the mean ± SEM of [Ca2+]i increase measured in 25–30 cells. Statistical significance was determined by unpaired Student's t-test, with a value of P < 0.05 considered significant, *Significantly different.
To examine the effect of STIM L96V mutation on SOCE, Ca2+ was first depleted from the SR of myoblasts or myotubes with thapsigargin, in the absence of extracellular Ca2+, to avoid extracellular Ca2+ entry during depletion, and then extracellular Ca2+ was applied to myoblasts or myotubes to measure SOCE (Figures 1B,E, respectively). Both myoblasts and myotubes carrying STIM1 L96V mutation displayed a significant augmented SOCE compared to the respective control cells (Figures 1B,C,E,F).
Furthermore, to assess the Ca2+ amount in the SR, we treated STIM1 L96V myotubes with 40 mM caffeine to induce extensive store depletion. Importantly, more Ca2+ was released from the SR in STIM1 L96V myotubes than in control myotubes [Caffeine-induced Δ[Ca2+]i = 320 ± 27 nM and 214 ± 35 nM in mutant and control, respectively], thus demonstrating also an increase in the Ca2+ SR content of STIM1 L96V muscle cells.
Thus, our functional assay conducted for the first time on patient-derived myoblasts and myotubes revealed that STIM1 L96V mutation is consistent with SOCE constitutive activation, i.e., with a gain of function effect.
High Content Imaging of Differentiating Skeletal Muscle Cells
Different lines of evidence indicate that SOCE is a key factor controlling myoblasts fate (Louis et al., 2008; Darbellay et al., 2009, 2010; Michelucci et al., 2018) as well that mitochondria participate in the differentiating process of various cell types, including muscle cells (Noguchi and Kasahara, 2018). On this basis, cellular morphology properties and mitochondrial network features of control and STIM1 L96V differentiating cells have been investigated in vitro by automated fluorescence microscopy. The quantitative analysis of morphological changes associated with differentiation was performed on living myoblasts, at distinct time points after substitution of differentiation medium for growth medium (T0, T1, and T2, corresponding to 0, 5, and 10 days, respectively).
Differentiation-Associated Morphological Features
As shown in Figure 2A (panels A–C), the myogenic differentiation process of control myoblasts is characterized by the presence of mononuclear cells at T0, followed by the increase of cell size as well as the emergence of multinuclear cells after 5 days (T1) and, to a greater extent, after 10 days (T2) in differentiation medium. In parallel with the appearance of multinucleated elements, the number of control mononuclear cells progressively decreases (Figure 2B). In the case of STIM1 L96V cells, only rare multinucleated elements are present at T1, containing two-three nuclei [Figure 2A (panel E), arrowhead], whereas elements with more than 10 nuclei are already formed by control cells at this time [Figure 2A (panel B), arrow]. In parallel, mononuclear STIM1 L96V cells show a longer persistence, only at T2 their number being significantly reduced (Figure 2B).
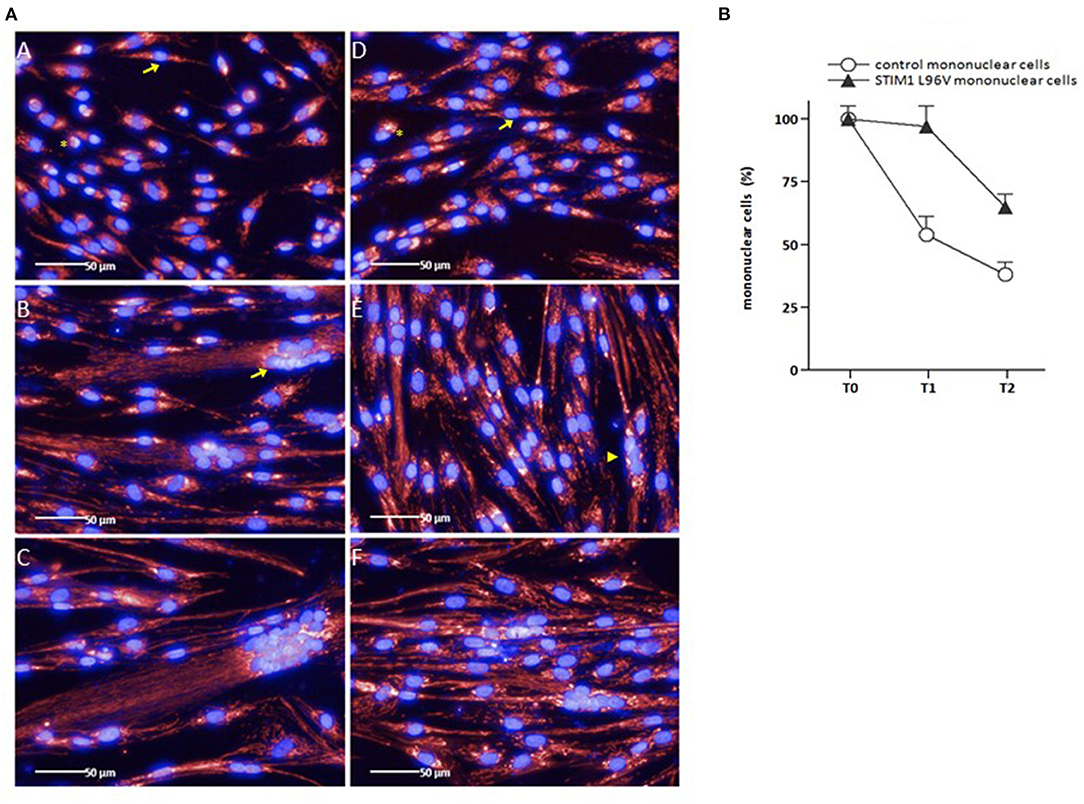
Figure 2. Representative images of control and STIM1 L96V skeletal muscle cells at different time points in differentiation medium (A), and differentiation-associated decrease of control and STIM1 L96V mononuclear cells (B). (A) Control and STIM1 L96V mononuclear mononuclear cells were seeded in 96-well cell culture plates (6,000/100 μL). Upon reaching sub-confluence (~60%), growth medium was replaced by differentiation medium to induce myoblast differentiation and myotube formation. For fluorescence staining of mitochondria (red) and nuclei (blue), MitoTracker Deep Red and Hoechst 33,342 were added into the media (final concentration 50 nM and 1 μM, respectively). After 30 min of incubation at 37°C, 5% CO2, live-cell images were recorded by automated fluorescence microscopy at day 0 (T0), 5 (T1), and 10 (T2) for control (panels A, B, C) and STIM1 L96V cells (panels D, E, F), respectively (Perkin Elmer Operetta High-Content Imaging System, 40x LWD objective). Asterisks and arrows in panels A and D indicate rounded and spindle-shaped control and STIM1 L96V myoblasts at T0, respectively. The arrow and arrowhead in panels B and E indicate a multinucleated control and STIM1 L96V cell at T1, respectively. (B) The percentage of mononuclear control and STIM1 L96V cells at distinct time points in differentiation medium is shown as mean ± SD.
Cell Morphometry and Mitochondrial Texture Properties of Mononuclear Control and STIM1 L96V Cells (T0)
Both control and STIM1 L96V myoblasts at T0 are mononuclear cells, characterized by variable size and rounded or spindle-shaped morphology (Figure 2, asterisks and arrows, respectively, in panels A and D). The relative amount of rounded and spindle-shaped myoblasts in the two cell populations was calculated by using the PhenoLOGIC module, finding that the percentage of spindle-shaped cells in STIM1 L96V myoblasts was significantly higher than that of normal cells (47 and 38%, respectively; p = 0.005, chi-square test; data not shown). The comparative analysis of cell morphometry parameters reveals the features of control and STIM1 L96V myoblasts at T0 (Table 1). STIM1 L96V myoblasts are about 20% bigger that normal myoblasts, as indicated by cell area values, and have a more pronounced spindle-shaped morphology (roundness index: 0.68 and 0.61, respectively). The mitochondrial mass mean concentration (mitotracker intensity mean) of control and STIM1 L96V myoblasts is similar, whereas the total mitochondrial mass (mitotracker intensity sum) of STIM1 L96V myoblasts is higher, according to their increased cell size.
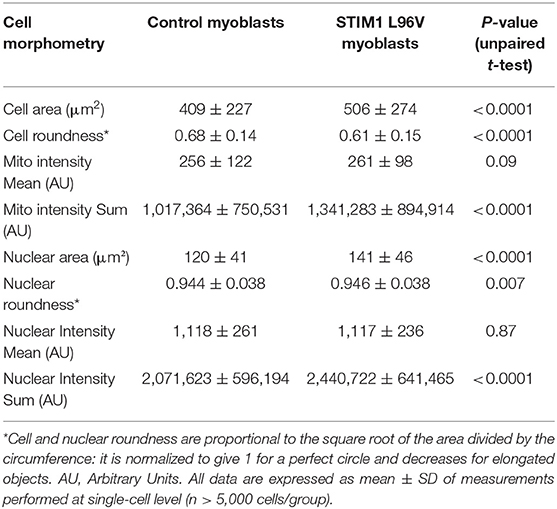
Table 1. Morphological features of control and STIM1 L96V myoblasts at T0.
The effects of STIM1 L96V mutation on the mitochondrial architecture have been evaluated calculating the frequency of texture feature or combination of features using the “SER features” building block of the Harmony software (Supplementary Material). All SER texture indexes of STIM1 L96V myoblasts were significantly different from those of control myoblasts, except for spot and hole feature (Figure 3A). The major differences concern in the order saddle, valley, edge and ridge, indicating that STIM1 L96V myoblasts at T0 are characterized by a more elongated and networked mitochondrial architecture with respect to control myoblasts.
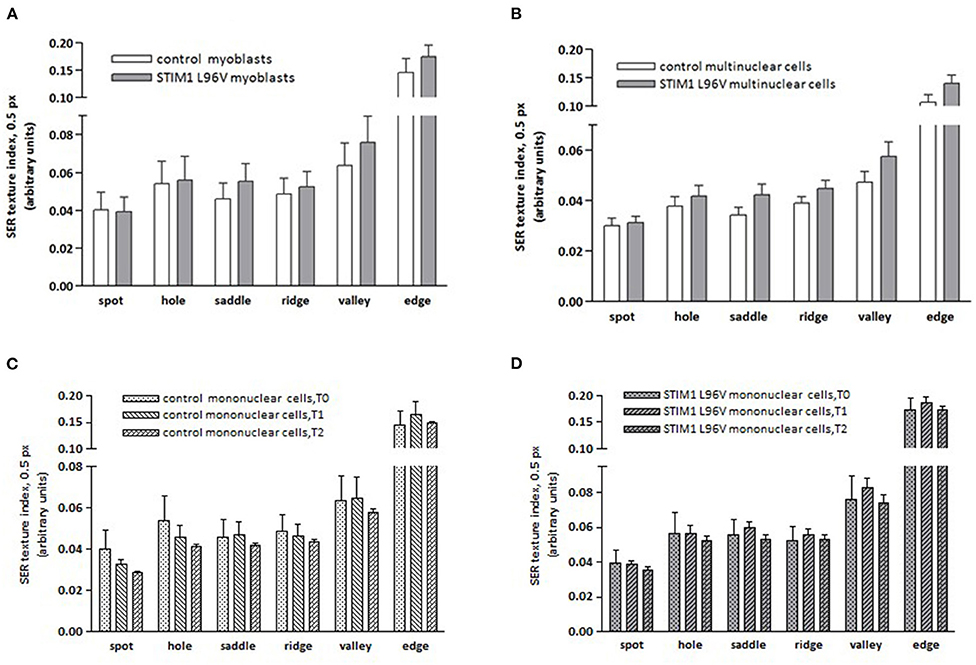
Figure 3. Mitochondrial SER texture indexes of control and STIM1 L96V skeletal muscle cells. (A) SER texture indexes of SER spot, hole, saddle, ridge, valley, and edge of normal and STIM1 L96V mononuclear cells at T0 are shown as mean ± SD (cell number > 5,000/group). All SER features of STIM1 L96V myoblasts are significantly different from those of control myoblasts (p < 0.0001, unpaired t-test), except for spot and hole. (B) SER texture indexes of SER spot, hole, saddle, ridge, valley, and edge of normal and STIM1 L96V multinuclear cells are shown as mean ± SD (cell number > 300). All variations of SER texture indexes are significantly different (p < 0.005 for spot; P < 0.0001 for the remaining filters, unpaired t-test). (C,D) Differentiation-associated variations of mitochondrial SER texture indexes of control (C) and STIM1 L96V (D) mononuclear cells; the SER texture indexes at distinct time points in differentiation medium are shown as mean ± SD (cell number>5,000 for T0 groups, and >3,000 for T1 and T2 groups). Time point-associated variations of SER texture indexes are significantly different for all SER features: P < 0.0001, ANOVA and Tukey HSD post-hoc test, in both control (C) and STIM1 L96V (D) mononuclear cells, except for hole in the T0–T1 comparison of STIM1 L96V cells.
Differentiation-Associated Modifications of Mononuclear Control and STIM1 L96V Cells
The differentiation-associated modifications of mononuclear cell morphometry of control and STIM1 L96V cells after 5 (T1) and 10 (T2) days in differentiation medium are reported in Tables 2, 3, respectively. The cell size of control cells progressively increases as well as the spindle-shaped morphology, the major increment occurring in the T0–T1 interval. Moreover, both mitotracker mean intensity and sum intensity values progressively increase, indicating a corresponding enhancement of mitochondrial mass and concentration (Table 2). As far as morphological modifications accompanying STIM1 L96V mononuclear cell differentiation are concerned, the results resemble those of control cells regarding size, spindle-shaped morphology and mitochondrial mass increase (Table 3). In contrast to control cells, however, the mitotracker mean intensity remains substantially unmodified from T0 to T2, suggesting that mitochondrial activity of mononuclear STIM1 L96V cells does not increase along with differentiation.
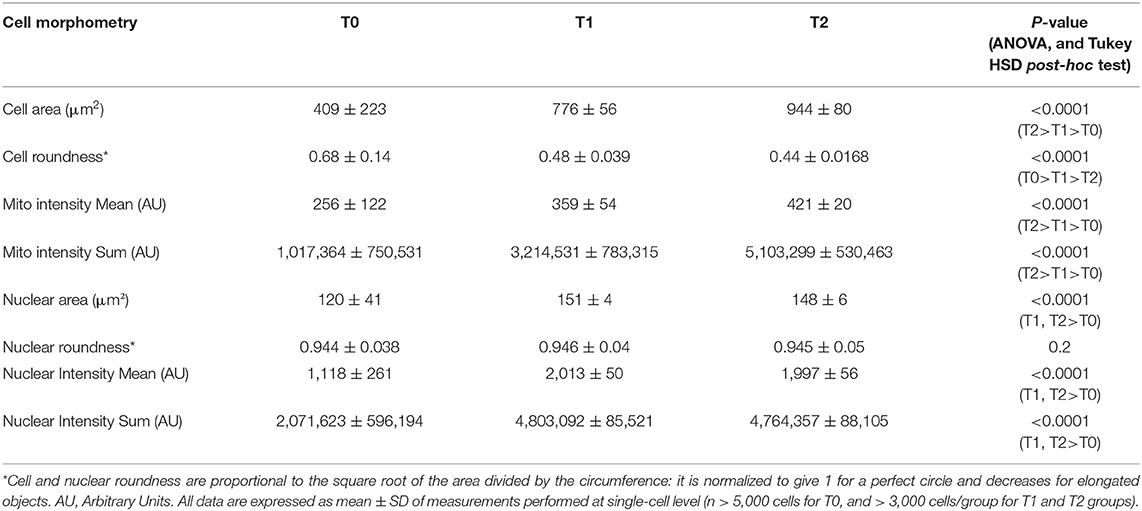
Table 2. Differentiation-associated modifications of control mononuclear cells.
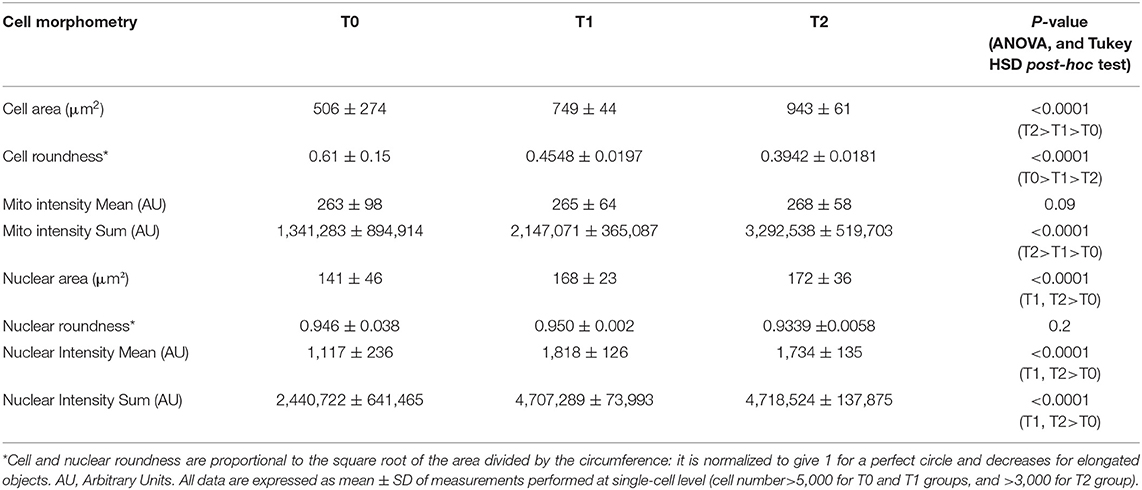
Table 3. Differentiation-associated modifications of STIM1 L96V mononuclear cells.
Differentiation-associated modifications of mitochondrial mass of control mononuclear cells were also accompanied by mitochondrial texture changes (Figure 3C). In the T0–T1 interval, the major variations of SER features concern the spot and hole filters, whose indexes reduce by around 18%, and the edge filter, which increases by 13%. This pattern suggests a relative increase of fragmentation over elongation in the mitochondrial architecture. Instead in the T1–T2 interval, SER feature variations suggestive of mitochondrial fragmentation, i.e., decrease of spot, hole, and saddle, are quantitatively similar to the elongation-associated features (ridge and edge), thus indicating a balance in the mixed morphology of mitochondrial network. Mitochondrial texture modifications accompanying STIM1 L96V mononuclear cell differentiation resemble those of control cells in both T0–T1 and T1–T2 intervals. However, the variations of SER indexes indicative of network fragmentation are less pronounced than those of control cells in the T0–T1 interval, and a parallel increase of the saddle and valley features indicates a mitochondrial elongation (Figure 3D).
Cell Morphometry and Mitochondrial Texture Properties of Control and STIM1 L96V Multinuclear Cells (T2)
A comparison of morphological features and mitochondrial architecture of multinuclear control and STIM1 L96V cells is reported in Table 4 and Figure 3B, respectively. Multinuclear STIM1 L96V cells are smaller than corresponding normal cells. Curiously, STIM1 L96V cells are also more spindle-shaped, this feature being present during the entire differentiation process (Tables 2, 3). The nuclear parameters of STIM1 L96V cells are all lower than those of control cells, according to the reduced number of nuclei in multinuclear STIM1 L96V cells. The mitotracker mean intensity of STIM1 multinuclear elements is slightly lower than that of control cells, suggesting a reduction of mitochondrial membrane potential. Interestingly, mitotracker mean intensity of STIM1 L96V multinuclear elements was higher than that of the corresponding mononuclear cells at T2, suggesting a fusion-associated increase of mitochondrial function. Mitochondrial texture features of multinuclear control and STIM1 L96V myotubes are reported in Figure 3B. The major variations concern saddle, valley, and hole features, indicating an elongated architecture, even though the increase of ridge, and edge indexes also denotes an increase of the fragmented phenotype.
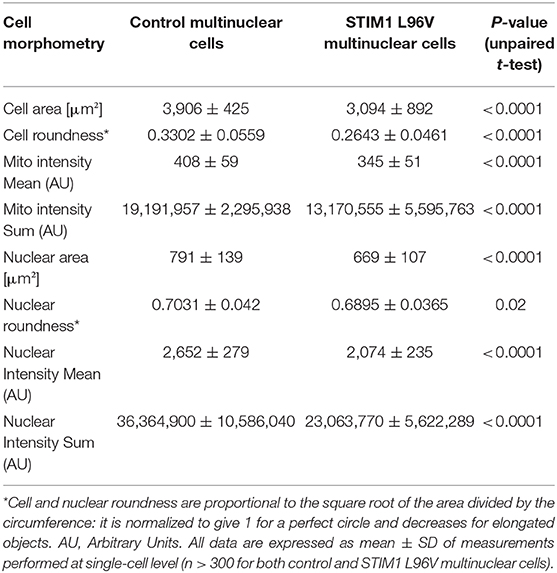
Table 4. Morphological features of control and STIM1 L96V multinuclear cells (T2).
Gene Expression Analysis
Genes Involved in Calcium Homeostasis and TA Formation
Tubular aggregates are displayed in patient derived muscle cells used in our study (Böhm et al., 2014). Different proteins involved in the uptake and Ca2+ storage such as STIM1, sarcoplasmic reticulum Ca2+-ATPase (SERCA1a) or ryanodine receptor 1 molecule (RyR1) were previously shown to be components of the aggregates (Chevessier et al., 2005; Böhm et al., 2013). In addition, STIM1 could directly interact with other proteins, such as the canonical-type transient receptor potential cationic channels (TRPCs) or the dihydropyridine receptor (DHPR) (Kiselyov and Patterson, 2009; Lee et al., 2013). By qPCR, we found a significant down-regulation of mRNA level of RyR1 and Atp2a1 (encoding for SERCA1a) in myoblasts and myotube carrying STIM1 L96V mutation compared to control myoblasts and myotubes, respectively (Figure 4). Any significant change was detected in the expression level of Stim1, Cacna1s (encoding for DHPR), Atp1a2 (encoding for Na/K ATPase) and Trpc4. Importantly, a trend of reduction of Trpc1 already observed in myoblasts carrying STIM1 L96V mutation, became significant in differentiated myotubes, while Orai1 expression, resulted unchanged in myoblasts, was significantly reduced in myotubes carrying STIM1 L96V (Figure 4).
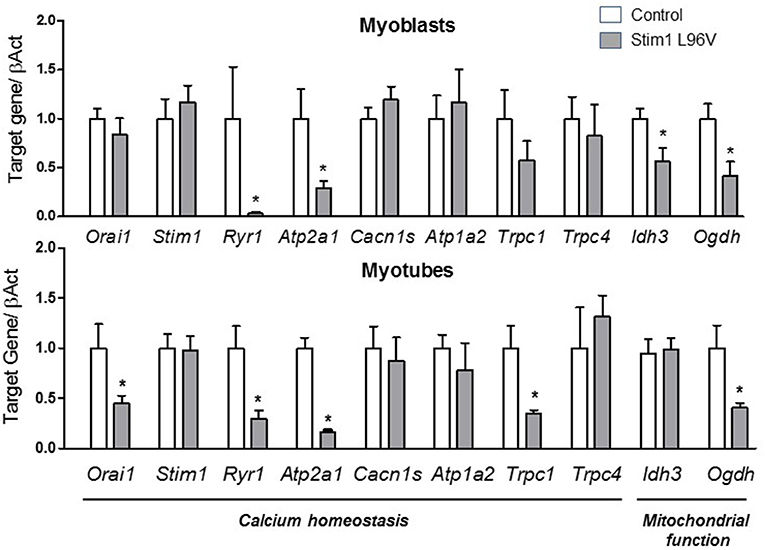
Figure 4. Expression levels of selected genes involved in calcium homeostasis and mitochondrial function. The histograms show the relative content of transcript levels for Orai1, Stim1, Ryr1, Atp2a1, Cacn1s, Atp1a2, Trpc1, Trpc4 genes involved in calcium homeostasis and Ogdh, and Idh3 involved in mitochondrial function normalized to β-actin gene in STIM1 L96V myoblasts (upper Panel) and myotubes (bottom Panel) with respect of control cells. Data are expressed as fold-difference compared with control cells; samples were analyzed in triplicate, and results are expressed as the means ± sem. Statistical significance was determined by unpaired Student's t-test, with a value of P < 0.05 considered significant, *Significantly different.
Genes Involved in Mitochondrial Function
To assess if STIM1 L96V mutation could affect mitochondrial function, we analyzed mRNA expression level of two mitochondrial Ca2+-sensitive dehydrogenases fundamental to generate NADH needed by the respiratory chain to generate ATP, such as an isoform of isocitrate dehydrogenases (IDH3A), which catalyzes the oxidative decarboxylation of the isocitrate in α-ketoglutarate, and 2-oxoglutarate dehydrogenase (OGDH), a component of the α-ketoglutarate dehydrogenase complex that converts α-ketoglutarate to succinate (Denton and McCormack, 1980). A significant reduction of the expression of IDH3A and OGDH was detected in myoblasts carrying STIM1 L96V mutation with respect to control myoblasts, which is however observed only for OGDH in mutated differentiated myotubes (Figure 4).
Genes Involved During the Differentiation of Myoblasts to Myotubes
High content imaging analysis revealed a defective myogenesis associated to STIM1 L96V mutant muscle cells with respect to control cells. On this basis, we gained insight into the mechanism underlying the myogenic pathway alteration by analyzing the mRNA levels in STIM1 L96V myoblasts and myotubes of the following genes: Pax7 (paired box 7), which is a member of the upstream regulators of myogenesis and a marker of satellite cells; Myf5 (Myogenic factor 5) and MyoD1 (Myogenic differentiation 1), which are members of the myogenic regulatory factor (MRF) family; Mef 2D (Myocyte enhancer factor 2D), which is a member of the myocyte enhancer factor 2 family (MEF2) and Myog (Myogenin), both markers of differentiation; finally, Tnnt3 (Troponin), and DMD (Dystrophin), were chosen as markers of late differentiation.
Interestingly, in STIM1 L96V myoblasts we found a significant reduction in DMD, Tnnt3 and an increase in Myf5 and Mef2D mRNA levels with respect to control myoblasts. A significant reduction in Pax7, DMD, and Tnnt3 was observed in mutated myotubes with respect to control ones (Figure 5). No significant alteration was observed for MyoD1 and Myog expression levels both in STIM1 L96V myoblasts and myotubes with respect to control cells.
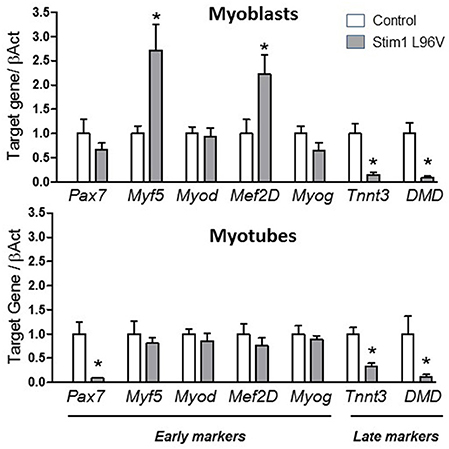
Figure 5. Expression levels of selected genes involved in muscle differentiation. The histograms show the relative content of transcript levels for early markers of differentiation Pax7, Myf5, Myod, Mef2D and Myog genes, and later markers of differentiation Tnnt3, DMD genes normalized to β-actin gene in STIM1 L96V myoblasts (upper Panel) and myotubes (bottom Panel) with respect of control cells. Data are expressed as fold-difference compared with control cells; samples were analyzed in triplicate, and results are expressed as the means ± sem. Statistical significance was determined by unpaired Student's t-test, with a value of P < 0.05 considered significant, *Significantly different.
All these findings indicate an altered myogenic pathway associated to STIM1 L96V mutation.
Discussion
TAM is a rare hereditary myopathy actually without a cure caused by mutations of genes involved in Ca2+ homeostasis. In more detail, gain-of-function mutations of STIM1 or ORAI1 genes inappropriately activate the SOCE process and induce an excessive extracellular Ca2+ entry despite repleted Ca2+ stores (Lacruz and Feske, 2015; Lee and Noguchi, 2016; Böhm and Laporte, 2018; Morin et al., 2020). Ca2+ homeostasis alterations affect a variety of cell functions, being Ca2+-dependent signaling involved in multiple cellular processes (Carafoli and Krebs, 2016). As far as skeletal muscle cells are concerned, the Ca2+-mediated coupling of excitation and contraction has long been well-established, but the recognition of Ca2+ relevance in muscle formation, growth and regeneration is also growing (Tu et al., 2016). Most of the STIM1 mutations responsible for TAM are located in the luminal Ca2+-sensing EF-hand domain, and affect amino acids thought to be involved in Ca2+ coordination or maintaining the protein in a folded and inactive conformation (Böhm and Laporte, 2018). The patients carrying EF-hand mutations are mainly characterized by a muscle phenotype, a proximal muscle weakness being generally reported. However, a precise genotype/phenotype correlation is still undefined, the onset and severity of muscle involvement being not uniform for the different STIM1 EF-hand mutations (Morin et al., 2020). Symptoms vary greatly from patient to patient, with a wide phenotypical spectrum ranging from childhood-onset muscle weakness to adult-onset myalgia. Functional effects of STIM1 mutations at cellular level have been investigated mainly in heterologous expression systems and, only in the case of STIM1 A84G (Böhm et al., 2013) and G81A (Walter et al., 2015) mutations, also in patient- derived myoblasts.
In this study, we have investigated the Ca2+ homeostasis alterations of skeletal muscle cells from a TAM patient carrying the STIM1 L96V mutation. This mutation, along with the more recently reported L92V (Morin et al., 2020), is located in the hydrophobic cleft which contributes to keeping the STIM1 inactive conformation, according to molecular modeling simulations (Schober et al., 2019). We confirm in human myoblasts the STIM1 L96V-dependent Ca2+ overload already reported in engineered murine myoblasts and detect the Ca2+ alteration persistence also in differentiated cells. The resting Ca2+ level is set by the balance between influx and efflux mechanisms at rest (Li et al., 2010; Ríos, 2010) and whether STIM1 L96V is the main source for the resting Ca2+ dysregulation remain to be defined. Indeed, this is an unsolved aspect also for other described STIM1 mutants (Morin et al., 2020). In this regards, the use of SOCE inhibitors at different times of myogenesis process would be useful to explore the observed differences in resting Ca2+ level between patient-derived muscle cells and control cells. Notably, in view of the non-specific effects mediated by SOCE inhibitors actually available (Le Guilcher et al., 2020; Meizoso-Huesca and Launikonis, 2021), focused studies will be required based on the use of several tools to unequivocally solve this issue.
The measured excess of intraluminal and cytosolic Ca2+ is accompanied by expression profile modifications of genes involved in Ca2+ homeostasis. Our findings of expression profile regarding genes encoding proteins involved in Ca2+ homeostasis indicate an adaptive or compensatory response of the cells to the increased SOCE, which was already observable in myoblast and became clear in differentiated myotubes. Indeed, the significant reduction of Orai1, RyR1, and Atp2a1 expression could be considered an attempt of muscle cells to counteract the Ca2+ homeostasis dysfunction. The parallel reduction of Trpc1 could corroborate the involvement of these cationic channels into the STIM1-induced aggregate composition, as reported in other studies (Kiselyov and Patterson, 2009; Lee et al., 2013). Futhermore, RyR1 expression reduction could be also correlate with the expression of the other RyR isoform expressed in skeletal muscle, i.e., RyR3. Indeed, RyR3 isoform has a significant effect on resting Ca2+ levels and a precise balance between RyR1 and RyR3 expression physiologically tightly regulate the diversity of cellular responses that muscle cells undergo during their early development (Protasi et al., 2000; Perez et al., 2005). Particularly, in myotubes RyR3 has virtually no role in initiating or maintaining EC coupling. Thus, it may be postulated that the reduction of RyR1 gene expression we observed in Stim1 mutant myotubes is likely related with a consequent RyR1 and RyR3 expression imbalance. This could be supported by the increased level of resting Ca2+ as well as by the significantly increased responsiveness to caffeine in STIM1 mutant muscle cells with respect to control cells. Indeed, it has been reported that altered expression of RyR3 resulted in myotubes with significantly higher resting Ca2+ level as well as with a different caffeine sensitivity than myotubes expressing RyR1 (Perez et al., 2005).
The STIM1/ORAI1-mediated SOCE is emerging as a critical process in regulating long-term muscle functions and a growing evidence supports its relevance in muscle differentiation, development, and growth (Louis et al., 2008; Darbellay et al., 2009, 2010; Michelucci et al., 2018). To evaluate the cellular response to Ca2+ dyshomeostasis, in this study, we compared the morphological features of STIM1 L96V myoblasts and control myoblasts, and investigated the effects of STIM1 L96V mutation on in vitro myogenic differentiation process through high content imaging and gene expression analysis. Cells morphology and mitochondrial network are critical for several biological processess that control nuclear programs and are strictley related to Ca2+ handeling in skeletal muscle (Favaro et al., 2019). Thus, we particularly focused on these cellular features. First of all, high content imaging was employed for evaluating similarities and differences between control and STIM1 L96V myoblasts at T0, i.e., when differentiation medium was substituted for growth medium. The variability of cellular shape and dimension of both control and STIM1 L96V myoblasts at T0 indicates that cells are in various growth stages and that some of them probably already started the differentiation process. This is most likely related to the use of non-synchronized cell cultures, an ex-ante choice undertaken with the aim of investigating differentiating muscle cells with minimal manipulation. Interestingly, STIM1 L96V myoblasts are larger and more splindle-shaped than control cells. More generally, the morphological features of STIM1 L96V myoblasts at T0 reflect the cellular adaptive response to the STIM1 mutation. In this respect, mitochondria might also be involved in cellular response to Ca2+ dyshomeostasis (Bagur and Hajnóczky, 2017). Mitochondria are dynamic organelles characterized by high mobility as well as shape changes, therefore they exhibit a mixed morphology, with small particles and tubular and highly networked structures (Karbowski and Youle, 2003). Organizational changes of mitochondrial morphology are controlled by multiple processes, including biogenesis and fusion and fission events, which adapt mitochondrial shape to the cell physiological needs. Mitochondrial fusion and fission could be related to STIM1-mediated intracellular Ca2+ movements and SOCE and alterations of STIM1 expression or activity is associated with mitochondrial abnormalities in skeletal muscle cells (Goonasekera et al., 2014; Choi et al., 2019). SER texture analysis of mitochondrial network indicate that STIM1 L96V myoblasts at T0 are characterized by a more elongated and networked mitochondrial architecture with respect to control myoblasts. The Ca2+ overload associated with STIM1 L96V mutation could prematurely activate and/or upregulate the Ca2+-dependent pathways, thus inducing an earlier onset of the differentiation process. This possibility is supported by the early appearance of spindle-shaped STIM1 L96V cells as well as by their more marked spindle-shaped morphology. If that was the case, it should be hypothesized that the mitochondrial system develops alongside the SR and myofibrils, as suggested by the elongated mitochondrial architecture observed in STIM1 L96V myoblasts in the early phase of differentiation.
High content imaging analysis of the differentiation process of control myoblasts shows a progressive increase of cell size and splindle-cell morphology. The parallel increase of mitochondrial mass and concentration also suggests an enhancement of mitochondrial activity. Mitotraker deep red accumulation is indeed dependent upon mitochondrial membrane potential (Poot et al., 1996), and an increase of mitotracker signal intensity associated with myogenic differentiation has already been reported (Miyake et al., 2011). Differentiation-associated modifications of mitochondrial mass of control myoblasts are also accompanied by mitochondrial network modifications, the SER texture analysis likely indicating an initial prevalence of mitochondrial fragmentation followed by a balance condition of fragmented/elongated architecture. Indeed, the mitochondrial network SER modifications of differentiating control myoblasts are in agreement with results reported by Sin et al. (2016), who showed a mitochondrial dynamic remodeling in differentiating C2C12 myoblasts, characterized by a network fragmentation in the early phases of the process. Interestingly, the mitochondrial architecture modifications here reported could represent the morphological counterpart of the mitochondrial function modifications associated to human muscle cell differentiation (Hoffmann et al., 2018). In view of the well-established dysfunction of SR structure in TAM (Chevessier et al., 2005; Böhm et al., 2013, 2017; Morin et al., 2020), we cannot not rule out that mitochondrial alterations we observed herein are a secondary phenomenon to the altered SR functionality.
The STIM1 L96V mutation affects the myoblast differentiation process, the most striking differences with respect to control myoblasts being the longer persistence of mononuclear cells along with the formation of multinuclear elements with reduced size, mitochondrial mass and concentration, and nuclei number. Overall, these findings indicate that STIM1 L96V mutation and the associated Ca2+ overload induce a complex cell response affecting the myoblast differentiation program. In particular, the alterations of mitochondrial mass and texture along with the reduced expression of IDH3A and OGDH, two key genes of mitochondrial metabolism, strongly suggest a mitochondrial dysfunction associated with STIM1 L96V mutation. Interestingly, a recent reported mouse model bearing a gain-of-function mutation in STIM1 displays histological and muscle alteration associated with mitochondria dysfunction evidenced by the presence of enlarged mitochondria with abnormal morphology (Cordero-Sanchez et al., 2019).
A major phenotypic effect of STIM1 L96V mutation upon myoblast differentiation is represented by the reduced formation of multinuclear elements, this suggesting a STIM1 L96V-dependent delay in the fusion process. A critical event in muscle formation during both embryonic development and regeneration upon injury, is the fusion of myoblasts into multinucleated myotubes. Molecular and cellular mechanisms of myoblast fusion are less known than those of preceding events, even though significant advances have been recently achieved. Cell fusion is a complex event that requires the coordination of various processes culminating in the activation of dedicated proteins, named fusogens, responsible for mediating membrane fusion (Hernández and Podbilewicz, 2017; Sampath et al., 2018). It is generally acknowledged that the fusion of myoblasts into multinucleated myotubes is regulated by calcium-dependent signaling. In particular, the increase of intracellular Ca2+ determines the calcineurin-mediated activation of NFAT transcription factor and myoblast fusion (Hindi et al., 2013). However, coordination and crosstalk of many signaling pathways are involved in myoblast fusion, and a complete picture of underlying cellular events is still lacking. In this context, it cannot be excluded that also a reduction of intracellular Ca2+ could be at some point required for cell fusion, as in the case of choriocarcinoma BeWo cells (Vatish et al., 2012), and that this event could be compromised by the STIM1 L96V-dependent Ca2+ overload.
By assessing the gene expression of some key myogenic factors leading to cell differentiation and fusion into multinucleated myotubes, we confirm the finding of our morphological imaging analysis highlighting an alteration of the myogenic pathway associated to TAM mutant. Particularly the gene expression increase of early differentiation markers such as Myf5 and Mef2D together with the reduction of late differentiation markers such as DMD and Tnnt3, encoding for dystrophin and troponin, in mutant myoblasts and myotubes strongly supported an altered myogenesis associated to TAM mutant, mainly regarding the late differentiation phase. In comparison with respect to control cells, STIM1 Leu96Val myoblasts differentiation into myotubes is early started but is not concluded. Future focused studies aimed to detect the protein expression levels of the genes of interest will certainly contribute to gain further insight into the role of these differentiation biomarkers.
Cellular Models for Personalized Therapeutics
To date, there is no specific treatment recommended for TAM patients. At least in principle, TAM could potentially benefit from treatment with SOCE/CRAC channel inhibitors, a group of putative immunomodulatory agents proposed for some chronic immune-related disorders (Riva et al., 2018; Stauderman, 2018). As far as preclinical investigations are concerned, there are two reported murine models bearing STIM1 gain-of function mutations and exhibiting a muscular phenotype (Gamage et al., 2018; Silva-Rojas et al., 2019), and only one is characterized by the luminal EF-hand mutation I115F (Cordero-Sanchez et al., 2019). In this respect, in our study, besides highilighting new etiopathological mechanisms underlying TAM disease, we validated a preclinical cellular model for TAM. We believe that preclinical investigations could benefit also from a preliminary phenotypic in vitro screening based upon the findings here reported, i.e., upon the ability of candidate SOCE/CRAC inhibitors to counteract the differentiation alterations associated with Ca2+ overload. As it is usually desired for neuromusculuar disorders (Silva-Rojas et al., 2020; van Putten et al., 2020), a such experimental approach could finally allow a reliable translation in the clinical management of TAM patients.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by human muscle samples were provided by the Telethon biobank at Besta Neurological Institute in Milan. Research was conducted according to protocols approved by the Institutional Review Board of the Besta Neurological Institute and University of Bari, and in compliance with the Helsinki Declaration and local legislation. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
EC and AL conceived and coordinated the study. LM, MM, and SG provided the muscle biopsy. PI and SG performed cell culture. EC and AL performed calcium cytofluorimetry experiments. AP and MC performed high content imaging experiments. EC and GC performed quantitative PCR experiments. PI, OC, and AD contributed ideas and critically interpreted results. EC, AP, MC, and AL wrote the paper. All authors approved the final version of the manuscript.
Funding
This work was in part supported by the research grants from Brain to the South (Fondazione con il Sud) with Project number 2018-PDR-00351 to OC.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Prof. Lorenzo Puri for kindly providing the additional human control muscle cell line (HMb_2).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.635063/full#supplementary-material
References
Armand, A. S., Bourajjaj, M., Martínez-Martínez, S., El Azzouzi, H., da Costa Martins, P. A., Hatzis, P., et al. (2008). Cooperative synergy between NFAT and MyoD regulates myogenin expression and myogenesis. J. Biol. Chem. 283, 29004–29010. doi: 10.1074/jbc.M801297200
Bagur, R., and Hajnóczky, G. (2017). Intracellular Ca2+ sensing: its role in calcium homeostasis and signaling. Mol. Cell. 66, 780–788. doi: 10.1016/j.molcel.2017.05.028
Barone, V., Del Re, V., Gamberucci, A., Polverino, V., Galli, L., Rossi, D., et al. (2017). Identification and characterization of three novel mutations in the CASQ1 gene in four patients with tubular aggregate myopathy. Hum. Mutat. 38, 1761–1773. doi: 10.1002/humu.23338
Böhm, J., Bulla, M., Urquhart, J. E., Malfatti, E., Williams, S. G., O'Sullivan, J., et al. (2017). ORAI1 Mutations with Distinct Channel Gating Defects in Tubular Aggregate Myopathy. Hum. Mutat. 38, 426–438. doi: 10.1002/humu.23172
Böhm, J., Chevessier, F., De Paula, A. M., Koch, C., Attarian, S., Feger, C., et al. (2013). Constitutive activation of the calcium sensor STIM1 causes tubular-aggregate myopathy. Am. J. Hum. Genet. 92, 271–278. doi: 10.1016/j.ajhg.2012.12.007
Böhm, J., Chevessier, F., Koch, C., Peche, A., Mora, M., Morandi, L., et al. (2014). Clinical, histological and genetic characterisation of patients with tubular aggregate myopathy caused by mutations in STIM1. J. Med. Genet. 51, 824–833. doi: 10.1136/jmedgenet-2014-102623
Böhm, J., and Laporte, J. (2018). Gain-of-function mutations in STIM1 and ORAI1 causing tubular aggregate myopathy and Stormorken syndrome. Cell Calcium 76, 1–9. doi: 10.1016/j.ceca.2018.07.008
Böhm, J., Lornage, X., Chevessier, F., Birck, C., Zanotti, S., Cudia, P., et al. (2018). CASQ1 mutations impair calsequestrin polymerization and cause tubular aggregate myopathy. Acta Neuropathol. 135, 149–151. doi: 10.1007/s00401-017-1775-x
Bustin, S. A., Benes, V., Garson, J. A., Hellemans, J., Huggett, J., Kubista, M., et al. (2009). The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622. doi: 10.1373/clinchem.2008.112797
Carafoli, E., and Krebs, J. (2016). Why calcium? How calcium became the best communicator. J. Biol. Chem. 291, 20849–20857. doi: 10.1074/jbc.R116.735894
Chevessier, F., Bauché-Godard, S., Leroy, J. P., Koenig, J., Paturneau-Jouas, M., Eymard, B., et al. (2005). The origin of tubular aggregates in human myopathies. J. Pathol. 207, 313–323. doi: 10.1002/path.1832
Cho, C. H., Woo, J. S., Perez, C. F., and Lee, E. H. (2017). A focus on extracellular Ca2+ entry into skeletal muscle. Exp. Mol. Med. 49:e378. doi: 10.1038/emm.2017.208
Choi, J. H., Huang, M., Hyun, C., Oh, M. R., Lee, K. J., Cho, C. H., et al. (2019). A muscular hypotonia-associated STIM1 mutant at R429 induces abnormalities in intracellular Ca2+ movement and extracellular Ca2+ entry in skeletal muscle. Sci. Rep. 9:19140. doi: 10.1038/s41598-019-55745-z
Conte, E., Camerino, G. M., Mele, A., De Bellis, M., Pierno, S., Rana, F., et al. (2017). Growth hormone secretagogues prevent dysregulation of skeletal muscle calcium homeostasis in a rat model of cisplatin-induced cachexia. J. Cachexia Sarcopenia Muscle 8, 386–404. doi: 10.1002/jcsm.12185
Cordero-Sanchez, C., Riva, B., Reano, S., Clemente, N., Zaggia, I., Ruffinatti, F. A., et al. (2019). A luminal EF-hand mutation in STIM1 in mice causes the clinical hallmarks of tubular aggregate myopathy. Dis. Model. Mech. 13:dmm041111. doi: 10.1242/dmm.041111
Darbellay, B., Arnaudeau, S., Ceroni, D., Bader, C. R., Konig, S., and Bernheim, L. (2010). Human muscle economy myoblast differentiation and excitation-contraction coupling use the same molecular partners, STIM1 and STIM2. J. Biol. Chem. 285, 22437–22447. doi: 10.1074/jbc.M110.118984
Darbellay, B., Arnaudeau, S., König, S., Jousset, H., Bader, C., Demaurex, N., et al. (2009). STIM1- and Orai1-dependent store-operated calcium entry regulates human myoblast differentiation. J. Biol. Chem. 284, 5370–5380. doi: 10.1074/jbc.M806726200
Denton, R. M., and McCormack, J. G. (1980). The role of calcium in the regulation of mitochondrial metabolism. Biochem. Soc. Trans. 8, 266–268. doi: 10.1042/bst0080266
Endo, Y., Noguchi, S., Hara, Y., Hayashi, Y. K., Motomura, K., Miyatake, S., et al. (2015). Dominant mutations in ORAI1 cause tubular aggregate myopathy with hypocalcemia via constitutive activation of store-operated Ca2? channels. Hum. Mol. Genet. 24, 637–648. doi: 10.1093/hmg/ddu477
Favaro, G., Romanello, V., Varanita, T., Andrea Desbats, M., Morbidoni, V., Tezze, C., et al. (2019). DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat. Commun. 10:2576. doi: 10.1038/s41467-019-10226-9
Gamage, T. H., Gunnes, G., Lee, R. H., Louch, W. E., Holmgren, A., Bruton, J. D., et al. (2018). STIM1 R304W causes muscle degeneration and impaired platelet activation in mice. Cell Calcium 76, 87–100. doi: 10.1016/j.ceca.2018.10.001
Goonasekera, S. A., Davis, J., Kwong, J. Q., Accornero, F., Wei-LaPierre, L., Sargent, M. A., et al. (2014). Enhanced Ca2? influx from STIM1-Orai1 induces muscle pathology in mouse models of muscular dystrophy. Hum. Mol. Genet. 23, 3706–3715. doi: 10.1093/hmg/ddu079
Grynkiewicz, G., Poenie, M., and Tsien, R. Y. (1985). A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260, 3440–3450. doi: 10.1016/S0021-9258(19)83641-4
Gudlur, A., Zeraik, A. E., Hirve, N., Rajanikanth, V., Bobkov, A. A., Ma, G., et al. (2018). Calcium sensing by the STIM1 ER-luminal domain. Nat. Commun. 9:4536. doi: 10.1038/s41467-018-06816-8
Harris, E., Burki, U., Marini-Bettolo, C., Neri, M., Scotton, C., Hudson, J., et al. (2017). Complex phenotypes associated with STIM1 mutations in both coiled coil and EF-hand domains. Neuromuscul. Disord. 27, 861–872. doi: 10.1016/j.nmd.2017.05.002
Hedberg, C., Niceta, M., Fattori, F., Lindvall, B., Ciolfi, A., D'Amico, A., et al. (2014). Childhood onset tubular aggregate myopathy associated with de novo STIM1 mutations. J. Neurol. 261, 870–876. doi: 10.1007/s00415-014-7287-x
Hernández, J. M., and Podbilewicz, B. (2017). The hallmarks of cell-cell fusion. Development 144, 4481–4495. doi: 10.1242/dev.155523
Hindi, S. M., Tajrishi, M. M., and Kumar, A. (2013). Signaling mechanisms in mammalian myoblast fusion. Sci. Signal. 6:re2. doi: 10.1126/scisignal.2003832
Hoffmann, C., Höckele, S., Kappler, L., Hrabe de Angelis, M., Häring, H. U., and Weigert, C. (2018). The effect of differentiation and TGFβ on mitochondrial respiration and mitochondrial enzyme abundance in cultured primary human skeletal muscle cells. Sci. Rep. 8:737. doi: 10.1038/s41598-017-18658-3
Karbowski, M., and Youle, R. (2003). Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell. Death Differ. 10, 870–880. doi: 10.1038/sj.cdd.4401260
Kegley, K. M., Gephart, J., Warren, G. L., and Pavlath, G. K. (2001). Altered Primary Myogenesis in NFATC3−/− mice leads to decreased muscle size in the adult. Dev. Biol. 232, 115–126. doi: 10.1006/dbio.2001.0179
Kiselyov, K., and Patterson, R. L. (2009). The integrative function of TRPC channels. Front. Biosci. 14, 45–58. doi: 10.2741/3230
Kiviluoto, S., Decuypere, J. P., De Smedt, H., Missiaen, L., Parys, J. B., and Bultynck, G. (2011). STIM1 as a key regulator for Ca2+ homeostasis in skeletal-muscle development and function. Skelet. Muscle 1:16. doi: 10.1186/2044-5040-1-16
Lacruz, R. S., and Feske, S. (2015). Diseases caused by mutations in ORAI1 and STIM1. Ann N Y Acad Sci. 1356, 45–79. doi: 10.1111/nyas.12938
Le Guilcher, C., Luyten, T., Parys, J. B., Pucheault, M., and Dellis, O. (2020). Synthesis and characterization of store-operated calcium entry inhibitors active in the submicromolar range. Int. J. Mol. Sci. 21:9777. doi: 10.3390/ijms21249777
Lee, J. M., and Noguchi, S. (2016). Calcium dyshomeostasis in tubular aggregate myopathy. Int. J. Mol. Sci. 17:1952. doi: 10.3390/ijms17111952
Lee, K. J., Woo, J. S., Hwang, J. H., Hyun, C., Cho, C. H., Kim, D. H., et al. (2013). STIM1 negatively regulates Ca2? release from the sarcoplasmic reticulum in skeletal myotubes. Biochem. J. 453, 187–200. doi: 10.1042/BJ20130178
Li, H., Ding, X., Lopez, J. R., Takeshima, H., Ma, J., Allen, P. D., et al. (2010). Impaired Orai1-mediated resting Ca2+ entry reduces the cytosolic [Ca2+] and sarcoplasmic reticulum Ca2+ loading in quiescent junctophilin 1 knock-out myotubes. J. Biol. Chem. 285, 39171–39179. doi: 10.1074/jbc.M110.149690
Li, T., Finch, E. A., Graham, V., Zhang, Z. S., Ding, J. D., Burch, J., et al. (2012). STIM1-Ca(2+) signaling is required for the hypertrophic growth of skeletal muscle in mice. Mol. Cell. Biol. 32, 3009–3017. doi: 10.1128/MCB.06599-11
Lopez, J. J., Jardin, I., Albarrán, L., Sanchez-Collado, J., Cantonero, C., Salido, G. M., et al. (2020). Molecular basis and regulation of store-operated calcium entry. Adv. Exp. Med. Biol. 1131, 445–469. doi: 10.1007/978-3-030-12457-1_17
Louis, M., Zanou, N., Van Schoor, M., and Gailly, P. (2008). TRPC1 regulates skeletal myoblast migration and differentiation. J. Cell. Sci. 121(Pt 23), 3951–3959. doi: 10.1242/jcs.037218
Meizoso-Huesca, A., and Launikonis, B. S. (2021). The Orai1 inhibitor BTP2 has multiple effects on Ca2+ handling in skeletal muscle. J Gen Physiol. 153:e202012747. doi: 10.1085/jgp.202012747
Michelucci, A., García-Castañeda, M., Boncompagni, S., and Dirksen, R. T. (2018). Role of STIM1/ORAI1-mediated store-operated Ca2+ entry in skeletal muscle physiology and disease. Cell Calcium. 76, 101–115. doi: 10.1016/j.ceca.2018.10.004
Miyake, T., McDermott, J. C., and Gramolini, A. O. (2011). A method for the direct identification of differentiating muscle cells by a fluorescent mitochondrial dye. PLoS ONE 6:e28628. doi: 10.1371/journal.pone.0028628
Morin, G., Biancalana, V., Echaniz-Laguna, A., Noury, J. B., Lornage, X., Moggio, M., et al. (2020). Tubular aggregate myopathy and stormorken syndrome: mutation spectrum and genotype/phenotype correlation. Hum. Mutat. 41, 17–37. doi: 10.1002/humu.23899
Nesin, V., Wiley, G., Kousi, M., Ong, E. C., Lehmann, T., Nicholl, D. J., et al. (2014). Activating mutations in STIM1 and ORAI1 cause overlapping syndromes of tubular myopathy and congenital miosis. Proc. Natl. Acad. Sci. U.S.A. 111, 4197–4202. doi: 10.1073/pnas.1312520111
Noguchi, M., and Kasahara, A. (2018). Mitochondrial dynamics coordinate cell differentiation. Biochem. Biophys. Res. Commun. 500, 59–64. doi: 10.1016/j.bbrc.2017.06.094
Park, C. Y., Hoover, P. J., Mullins, F. M., Bachhawat, P., Covington, E. D., Raunser, S., et al. (2009). STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 136, 876–890. doi: 10.1016/j.cell.2009.02.014
Perez, C. F., López, J. R., and Allen, P. D. (2005). Expression levels of RyR1 and RyR3 control resting free Ca2+ in skeletal muscle. Am. J. Physiol. Cell. Physiol. 288, C640–C649. doi: 10.1152/ajpcell.00407.2004
Phuong, T. T., Yun, Y. H., Kim, S. J., and Kang, T. M. (2013). Positive feedback control between STIM1 and NFATc3 is required for C2C12 myoblast differentiation. Biochem. Biophys. Res. Commun. 430, 722–728. doi: 10.1016/j.bbrc.2012.11.082
Poot, M., Zhang, Y. Z., Krämer, J. A., Wells, K. S., Jones, L. J., Hanzel, D. K., et al. (1996). Analysis of mitochondrial morphology and function with novel fixable fluorescent stains. J. Histochem. Cytochem. 44, 1363–1372. doi: 10.1177/44.12.8985128
Prakriya, M. (2009). The molecular physiology of CRAC channels. Immunol. Rev. 231, 88–98. doi: 10.1111/j.1600-065X.2009.00820.x
Protasi, F., Takekura, H., Wang, Y., Chen, S. R., Meissner, G., Allen, P. D., et al. (2000). RYR1 and RYR3 have different roles in the assembly of calcium release units of skeletal muscle. Biophys. J. 79, 2494–2508. doi: 10.1016/S0006-3495(00)76491-5
Ríos, E. (2010). The cell boundary theorem: a simple law of the control of cytosolic calcium concentration. J. Physiol. Sci. 60, 81–84. doi: 10.1007/s12576-009-0069-z
Riva, B., Griglio, A., Serafini, M., Cordero-Sanchez, C., Aprile, S., Di Paola, R., et al. (2018). Pyrtriazoles, a novel class of store-operated calcium entry modulators: discovery, biological profiling, and in vivo proof-of-concept efficacy in acute pancreatitis. J. Med. Chem. 61, 9756–9783. doi: 10.1021/acs.jmedchem.8b01512
Sampath, S. C., Sampath, S. C., and Millay, D. P. (2018). Myoblast fusion confusion: the resolution begins. Skelet. Muscle. 8:3. doi: 10.1186/s13395-017-0149-3
Schober, R., Bonhenry, D., Lunz, V., Zhu, J., Krizova, A., Frischauf, I., et al. (2019). Sequential activation of STIM1 links Ca2+ with luminal domain unfolding. Sci Signal. 12:eaax3194. doi: 10.1126/scisignal.aax3194
Silva-Rojas, R., Laporte, J., and Böhm, J. (2020). STIM1/ORAI1 loss-of-function and gain-of-function mutations inversely impact on SOCE and calcium homeostasis and cause multi-systemic mirror diseases. Front. Physiol. 11:604941. doi: 10.3389/fphys.2020.604941
Silva-Rojas, R., Treves, S., Jacobs, H., Kessler, P., Messaddeq, N., Laporte, J., et al. (2019). STIM1 over-activation generates a multi-systemic phenotype affecting the skeletal muscle, spleen, eye, skin, bones and immune system in mice. Hum. Mol. Genet. 28, 1579–1593. doi: 10.1093/hmg/ddy446
Sin, J., Andres, A. M., Taylor, D. J., Weston, T., Hiraumi, Y., Stotland, A., et al. (2016). Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy 12, 369–380. doi: 10.1080/15548627.2015.1115172
Stathopulos, P. B., Zheng, L., Li, G. Y., Plevin, M. J., and Ikura, M. (2008). Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell 135, 110–122. doi: 10.1016/j.cell.2008.08.006
Stauderman, K. A. (2018). CRAC channels as targets for drug discovery and development. Cell Calcium 74, 147–159. doi: 10.1016/j.ceca.2018.07.005
Stiber, J., Hawkins, A., Zhang, Z. S., Wang, S., Burch, J., Graham, V., et al. (2008). STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat. Cell. Biol. 10, 688–697. doi: 10.1038/ncb1731
Stiber, J. A., and Rosenberg, P. B. (2011). The role of store-operated calcium influx in skeletal muscle signaling. Cell Calcium. 49, 341–349. doi: 10.1016/j.ceca.2010.11.012
Tu, M. K., Levin, J. B., Hamilton, A. M., and Borodinsky, L. N. (2016). Calcium signaling in skeletal muscle development, maintenance and regeneration. Cell Calcium 59, 91–97. doi: 10.1016/j.ceca.2016.02.005
van Putten, M., Hmeljak, J., Aartsma-Rus, A., and Dowling, J. J. (2020). Moving neuromuscular disorders research forward: from novel models to clinical studies. Dis. Model. Mech. 13:dmm044370. doi: 10.1242/dmm.044370
Vatish, M., Tesfa, L., Grammatopoulos, D., Yamada, E., Bastie, C. C., and Pessin, J. E. (2012). Inhibition of Akt activity and calcium channel function coordinately drive cell-cell fusion in the BeWO choriocarcinoma placental cell line. PLoS ONE 7:e29353. doi: 10.1371/journal.pone.0029353
Walter, M. C., Rossius, M., Zitzelsberger, M., Vorgerd, M., Muller-Felber, W., Ertl-Wagner, B., et al. (2015). 50 years to diagnosis: Autosomal dominant tubular aggregate myopathy caused by a novel STIM1 mutation. Neuromuscul. Disord. 25, 577–584. doi: 10.1016/j.nmd.2015.04.005
Wei-Lapierre, L., Carrell, E. M., Boncompagni, S., Protasi, F., and Dirksen, R. T. (2013). Orai1-dependent calcium entry promotes skeletal muscle growth and limits fatigue. Nat. Commun. 4:2805. doi: 10.1038/ncomms3805
Yang, X., Jin, H., Cai, X., Li, S., and Shen, Y. (2012). Structural and mechanistic insights into the activation of stromal interaction molecule 1 (STIM1). Proc. Natl. Acad. Sci. U.S.A. 109, 5657–5662. doi: 10.1073/pnas.1118947109
Zanotti, S., Saredi, S., Ruggieri, A., Fabbri, M., Blasevich, F., Romaggi, S., et al. (2007). Altered extracellular matrix transcript expression and protein modulation in primary Duchenne muscular dystrophy myotubes. Matrix Biol. 26, 615–624. doi: 10.1016/j.matbio.2007.06.004
Keywords: STIM1, tubular aggregate myopathy, myogenesis (in vitro), high content imaging, calcium homeostasis
Citation: Conte E, Pannunzio A, Imbrici P, Camerino GM, Maggi L, Mora M, Gibertini S, Cappellari O, De Luca A, Coluccia M and Liantonio A (2021) Gain-of-Function STIM1 L96V Mutation Causes Myogenesis Alteration in Muscle Cells From a Patient Affected by Tubular Aggregate Myopathy. Front. Cell Dev. Biol. 9:635063. doi: 10.3389/fcell.2021.635063
Received: 29 November 2020; Accepted: 02 February 2021;
Published: 26 February 2021.
Edited by:
Agnese Secondo, University of Naples Federico II, ItalyReviewed by:
Jose M. Eltit, Virginia Commonwealth University, United StatesEdoardo Malfatti, INSERM U1179 Handicap neuromusculaire: Physiopathologie, Biothérapie et Pharmacologie appliquées (END-ICAP), France
Copyright © 2021 Conte, Pannunzio, Imbrici, Camerino, Maggi, Mora, Gibertini, Cappellari, De Luca, Coluccia and Liantonio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonella Liantonio, YW50b25lbGxhLmxpYW50b25pb0B1bmliYS5pdA==
†These authors have contributed equally to this work