 Qing Zhu
Qing Zhu Xue Rui
Xue Rui Ya Li
Ya Li Ya You
Ya You Xun-Lun Sheng
Xun-Lun Sheng Bo Lei
Bo Lei
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 01 March 2021
Sec. Molecular and Cellular Pathology
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.634843
This article is part of the Research Topic Genetic Mutations Associated with Ocular Diseases View all 19 articles
Purpose: The purpose of the study is to describe the genetic and clinical features of 17 patients with ABCA4-related inherited retinal degenerations (IRDs) and define the phenotype–genotype correlations.
Methods: In this multicenter retrospective study, 17 patients from 16 families were enrolled, and ABCA4 gene variants were detected using targeted next-generation sequencing using a custom designed panel for IRDs. Sanger sequencing and co-segregation analysis of the suspected pathogenic variants were performed with the family members. The pathogenicities of variants were evaluated according to the American College of Medical Genetics and Genomics guidelines (ACMG). Protein structure modifications mediated by the variants were studied using bioinformatic analyses.
Results: The probands were diagnosed with Stargardt disease 1 (7), cone-rod dystrophy type 3 (8), cone dystrophy (1), and retinitis pigmentosa 19 (1). Onset of symptoms occurred between 5 and 27 years of age (median age = 12.4 years). A total of 30 unique ABCA4 suspicious pathogenic variations were observed, including 18 missense mutations, seven frameshift mutations, two nonsense mutations, one canonical splice site mutation, one small in-frame deletion, and one insertion. Four novel ABCA4 variants were identified. Two novel frameshift variants, c.1290dupC (p.W431fs), and c.2967dupT (G990fs), were determined to be pathogenic. A novel missense variant c.G5761T (p.V1921L) was likely pathogenic, and another novel missense c.C170G (p.P57R) variant was of undetermined significance. All ABCA4 variants tested in this study inordinately changed the physico-chemical parameters and structure of protein based on in silico analysis.
Conclusion: ABCA4-related IRD is genetically and clinically highly heterogeneous. Four novel ABCA4 variants were identified. This study will expand the spectrum of disease-causing variants in ABCA4, which will further facilitate genetic counseling.
Inherited retinal degenerations (IRDs) are a group of blinding diseases that cause severe impairments of visual functions such as visual acuity and visual field. More than 270 disease-causing genes are associated with IRD (RetNet)1. Among them, ABCA4 is the most frequently identified gene (Kim et al., 2019; Pontikos et al., 2020). Mutations in the ABCA4 gene can cause various retinal diseases such as autosomal recessive (ar)-Stargardt disease (STGD1, OMIM # 248200), ar-cone-rod dystrophy (CORD3, OMIM # 604116), ar-cone dystrophy (COD), ar-retinitis pigmentosa (RP19, OMIM # 601718), and age-related macular degeneration-2 (ARMD2, OMIM # 153800) (van Driel et al., 1998; Baum et al., 2003; Burke and Tsang, 2011; Xin et al., 2015; Wang et al., 2018; Koyanagi et al., 2019).
ATP-binding cassette subfamily A member 4, denoted as ABCA4 (OMIM #601691), was first cloned by Allikmets et al. (Allikmets et al., 1997). The gene contains 50 exons, and it maps to position 22.1 (1p22.1) of the short (p) arm of chromosome 1 (Nasonkin et al., 1998). ABCA4 gene encodes a transmembrane protein that is exclusively expressed in photoreceptors and retina pigment epithelial cells (Lenis et al., 2018). The protein is composed of two non-equivalent tandem halves, and each half contains six transmembrane helices, a glycosylated extracytoplasmic domain (ECD) in the endolysosomes or disks, and a nucleotide binding domain in the cytoplasm. ATP-binding cassette transporter unidirectionally flips various compounds obtained from enzyme-catalyzed reactions of the visual cycle to the cytoplasm using the energy released from ATP hydrolysis (Kos and Ford, 2009; Quazi et al., 2012; Lenis et al., 2018). The misfolding and loss of functional activity of the transporter, caused by gene mutations, lead to accumulation of toxic substances such as all-trans-retinal and 11-cis-retinal in the outer segment of photoreceptor cells. Following diurnal phagocytosis of the distal outer segment of photoreceptors, excessive deposition of secondary toxic products, such as bisretinoid, eventually leads to the death of retinal pigment epithelium (Young and Bok, 1969; Weng et al., 1999; Quazi and Molday, 2014; Lenis et al., 2018). Different ABCA4 mutations lead to a broad range of IRD phenotypes (Rozet et al., 1998; van Driel et al., 1998; Xu et al., 2014; Garces et al., 2018). Phenotype severity mainly depends on the degree of influence of variations on protein functions (Fujinami et al., 2015).
Rapid advances in high-throughput next-generation sequencing technology have enabled an efficient and credible detection of gene mutations (Glöckle et al., 2014; Rong et al., 2018). Currently, 1,467 ABCA4 gene variants, containing a broad spectrum of IRD phenotypes, are present in the Human Gene Mutation Database2 (updated on April, 2019). However, the pathogenicities of numerous reported variants have not been elucidated yet, making their accurate clinical diagnoses difficult, let alone those of novel variants. Additionally, the analyses of genotype–phenotype correlations for the highly heterogeneous variants and clinical features are a challenge.
Therefore, we conducted this retrospective study to determine the pathogenicities of ABCA4 gene variants and novel genotype–phenotype correlations.
This multicenter retrospective study was conducted at the Henan Eye Hospital and the Ningxia Eye Hospital. The study was approved by the Medical Ethics Committees of both institutions, and it was conducted in accordance with the 1975 Declaration of Helsinki guidelines. Written informed consent was obtained from all included subjects or their guardians.
Seventeen patients with retinal degeneration, carrying the ABCA4 gene variants and belonging to 16 unrelated Chinese families, were recruited in the study from ophthalmic clinics. Of these, 11 patients were enrolled from Henan Eye Hospital (B1–11) and six patients from Ningxia Eye Hospital (A1–6). Family history, if available, was obtained from the patients and unaffected family members. Detailed clinical data, including age of onset, disease duration, best-corrected visual acuity (BCVA), and result of color vision testing, fundus photography, fundus autofluorescence imaging, fundus fluorescein angiography, optical coherence tomography, and full-field electroretinogram, were collected. Diagnostic criteria were adapted from previous studies (Fishman, 1976; Hamel, 2007; Sahel et al., 2014; Aboshiha et al., 2016; Verbakel et al., 2018). Retina specialists performed phenotype subgroup classification on all 17 included subjects.
Whole genomic DNA was extracted from the peripheral blood of the subjects using the TIANamp Blood DNA kit DP318 (TIANGEN, Beijing, China) according to the manufacturer’s protocol. A custom-designed posterior segment gene detection kit (PS400), containing 376 known causative IRD genes, their coding exons, flanking intronic sequences (50 bp), and all known intron mutations, was used for mutation screening. Sequence reads were aligned to the reference human genome (GRCh37/hg19) from the UCSC Genome Browser (Kent et al., 2002)3 using the XYGeneRanger2.0 software. Suspicious disease-relevant gene variants were routinely confirmed using Sanger sequencing, and co-segregation analyses were conducted for all affected families.
All variants were classified according to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines (Richards et al., 2015). To exclude the possibility of non-pathogenic polymorphism, the frequency of variations in the healthy control population was determined using the 1,000 Genomes Project (1,000 Genomes)4 and the Genome Aggregation Database5. A variant was classified as benign if its minor allele frequency was ≥ 0.005. Prediction algorithms of the programs Polyphen-26, SIFT7, PROVEAN8, CADD9, FATHMM (Shihab et al., 2013)10, and MutationTaster11 were used to test variants for disease relevance. Clustal Omega12 was used to analyze the conservative loci. For the variants of uncertain significance. Human Splicing Finder (Desmet et al., 2009)13 and Project HOPE (Venselaar et al., 2010)14 were used to predict the splicing defects and structural effects of variants.
Clinical characteristics of patients are summarized in Table 1. Seven subjects were diagnosed with STGD1 (7/17, 41.2%), eight with CORD3 (8/17, 47%), one with RP19 (1/17, 5.9%), and one with COD (1/17, 5.9%). Hypopsia was the chief complaint in all 17 patients. The age of onset of diseases in patients ranged from 5 to 27 years with a median age of 12.4 years. The BCVA of the most recent evaluation ranged from 0.3 to 0.02 across the group. One RP19 patient (A1) and three patients with cone/cone–rod dystrophy had color vision disorders (A2, A4, and A5), all others possessed normal color vision.

Table 1. Clinical characteristics, including ABCA4 variations, of the 17 patients in the study.
Genotypic and phenotypic variations were observed in three patients, and others possessed a typical ABCA4-associated disease phenotype. Patient A1 was asymptomatic until 27 years of age. The patient was diagnosed with retinitis pigmentosa along with BCVA of 0.3 bilaterally. The patient carried heterozygous mutations for both ABCA4 (NM_000350: c.G2473A, p.G825R and c.G673A, p.V225M) and AHI1 (NM_001134830: c.G3267A, p.W1089X and c.T1979G, p.L660R), and color-vision testing revealed errors in the red–green axis. The cone–rod dystrophy patient (B1), who carried compound heterozygous variants (NM_000350: c.A2894G, p.N965S and c.2063dupA, p.N688fs) in the ABCA4 gene, complained of visual defects with strabismus for 25 years. Her latest BCVA was 0.05 bilaterally. Retinal degenerative features were observed in the fundus, and exposure of the underlying sclera was observed in the fovea of both eyes. Spectral domain optical coherence tomography scans through the fovea revealed retinal rupture with a subretinal cavitation in the fovea of the left eye (Figure 1). Patient B4, with two heterozygous missense mutations in ABCA4, had blurred vision in both eyes for 10 years, and fundus autofluorescence exhibited diffuse patches of macular atrophy with nascent fleck development in the mid-periphery (Figure 1), which is similar to the ABCA4 phenotype in the early stage of rapid-onset chorioretinopathy (Tanaka et al., 2018). Additionally, the STGD1 patients demonstrated typical fundus manifestations of macular atrophy with miscellaneous sizes (Figure 2).
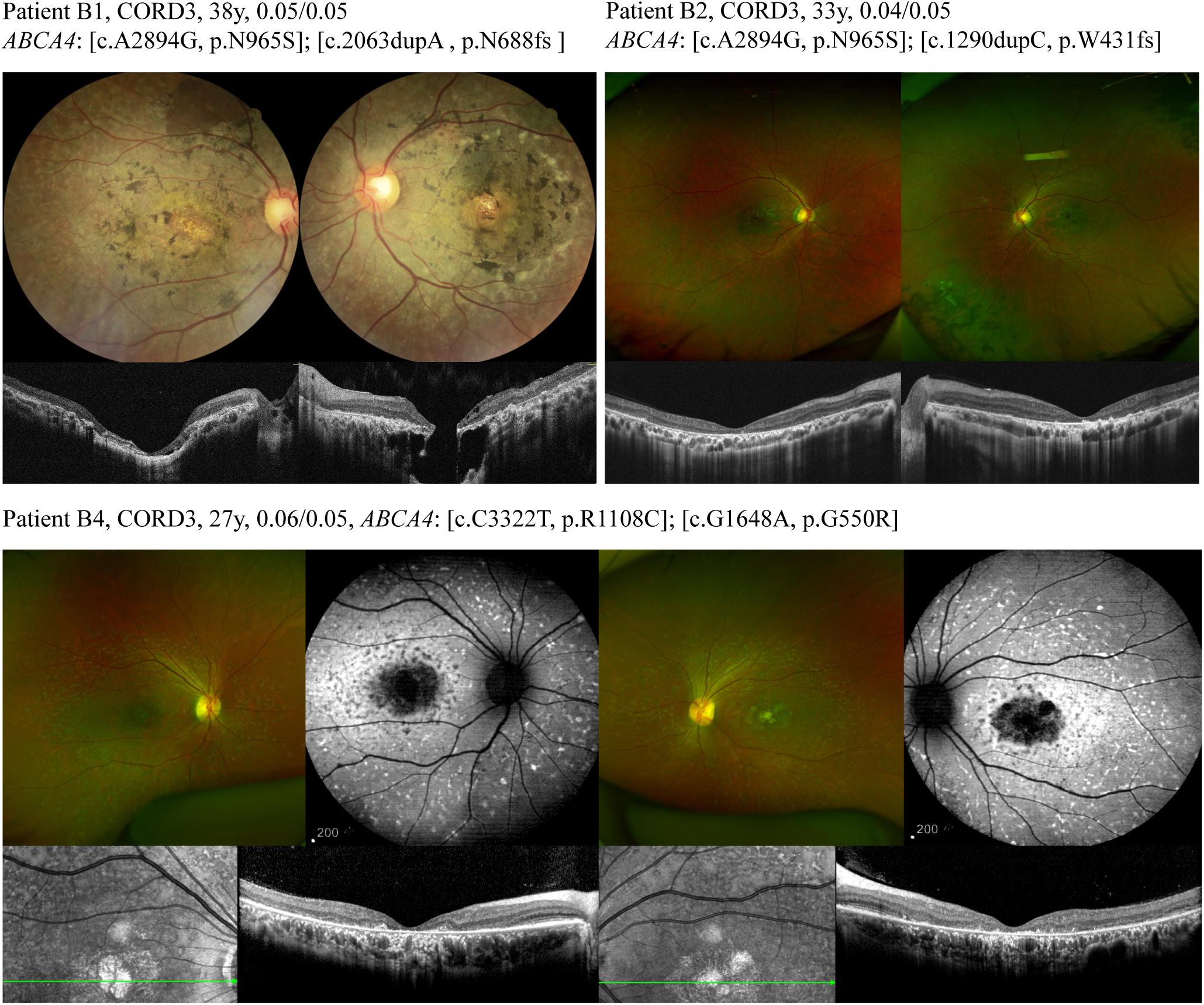
Figure 1. Different ABCA4 mutations lead to a broad range of phenotypes of cone-rod dystrophy. Fundus of patient B1 exhibited a relatively pale and blonde appearance due to exposure of the underlying sclera and nummular pigmentary deposition in the fovea of both eyes. The fundus retinal pigmentation in patient B2 was mainly distributed in the mid-periphery. Wide-field color fundus photograph of patient B4 demonstrated diffuse pisciform flecks throughout the posterior pole, macular atrophy, and no obvious retinal pigmentation.
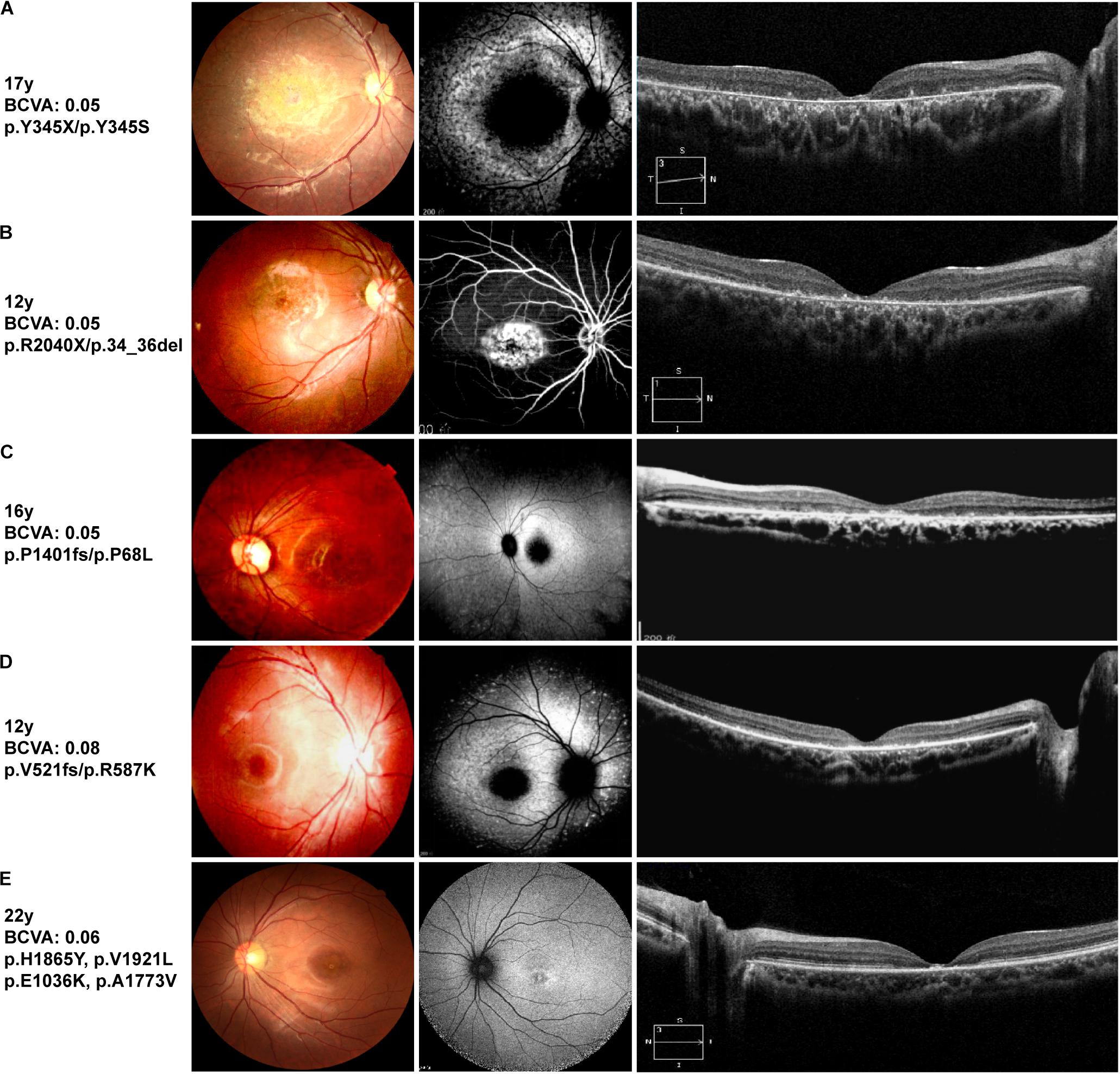
Figure 2. Representative clinical features of STGD1 patients. Left: color fundus images, center: retinal autofluorescence imaging or fundus photography, right: optical coherence tomography images. (A) Patient B6, (C) Patient B9, and (D) Patient B10 harboring a compound heterozygous truncating mutation and missense mutation. Fundus autofluorescence shows hypo-autofluorescence at the fovea with different sizes of macula atrophy. Optical coherence tomography shows atrophy around the fovea of STGD1 with the most severe degeneration for patient B6. (B) Center-fluoresce in angiography of the right eye (AV-transit phase) of patient B8 showing a window defect in the area of bull’s eye atrophy. The retinal deposits exhibit hyperfluorescence. (E) Fundus autofluorescence of patient B11, an individual with four ABCA4 missense mutations, showing a parafoveal hyperfluorescent ring.
We detected a total of 30 unique ABCA4 variants, including missense mutations (18), frameshift mutations (7), non-sense mutations (2), a splice site mutation (1), a small deletion (1), and an insertion (1). The 17 individuals, including one sibling pair, with at least two variants in the ABCA4 gene belonged to 16 families. Of the 30 rare ABCA4 variants, 24 were classified as pathogenic or likely pathogenic, and six were of uncertain significance. The pathogenicity of other mutated genes, except for the AHI1 in patient A1, was ruled out by phenotypic analysis. Frameshift mutations [c.1290dupC (p.W431fs) and c.2967dupT (G990fs)] and missense mutations [c.C170G (p.P57R) and c.G5761T (p.V1921L)] were novel unpublished variants (Table 2). The missense mutation c.A2894G (p.N965S) was detected four times without any exception in patients with cone/cone–rod dystrophy. The amino acid changes were primarily concentrated in ECD1 (47%, 14/30), and the amino acid variants were randomly distributed (Figure 3). Moreover, the HOPE online software revealed that all missense variants in this study inordinately changed the physico-chemical parameters or structure of ABCA4 (Appendix 1).
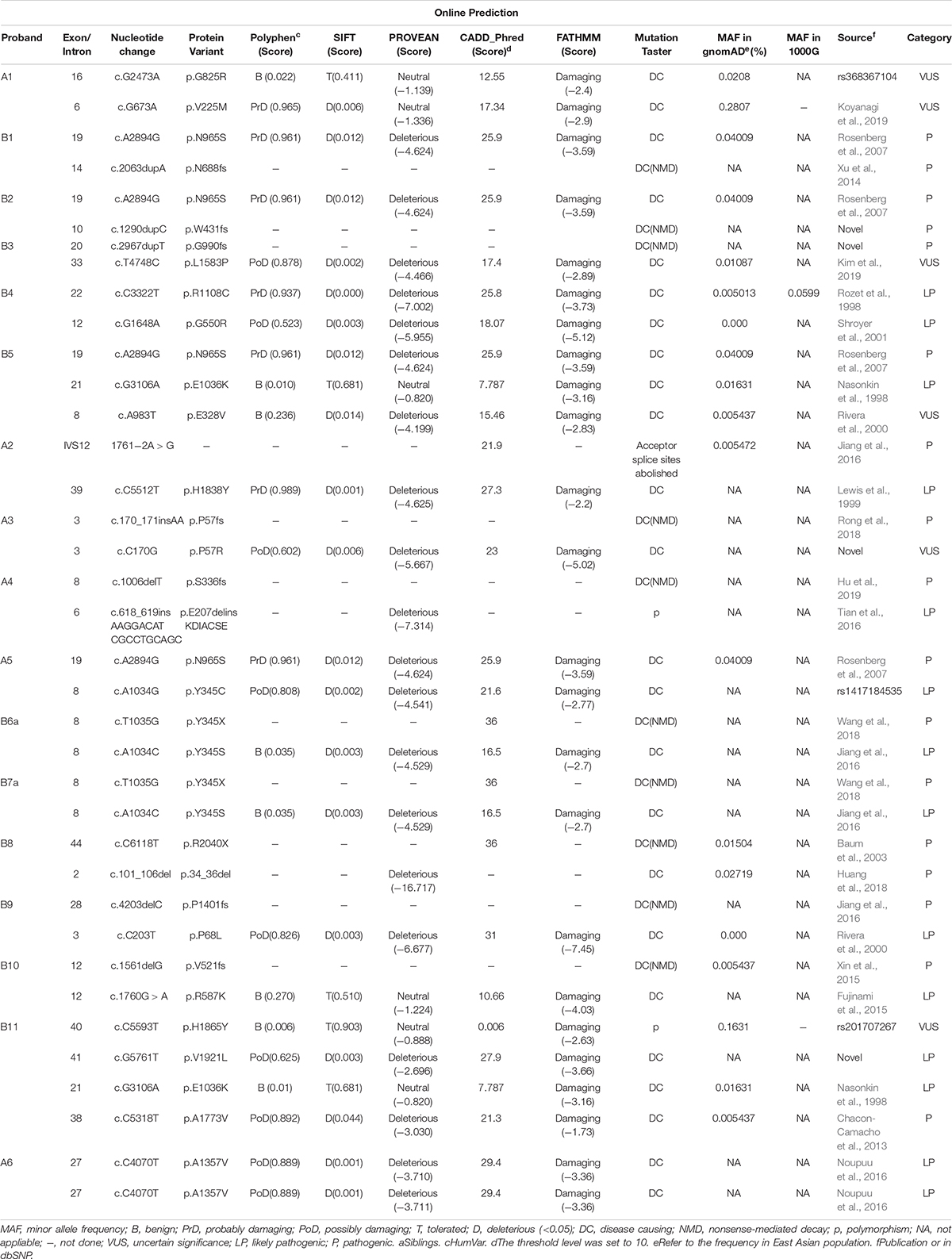
Table 2. Pathogenicity analyses of ABCA4 (NM_000350) variants in the 17 Chinese patients.
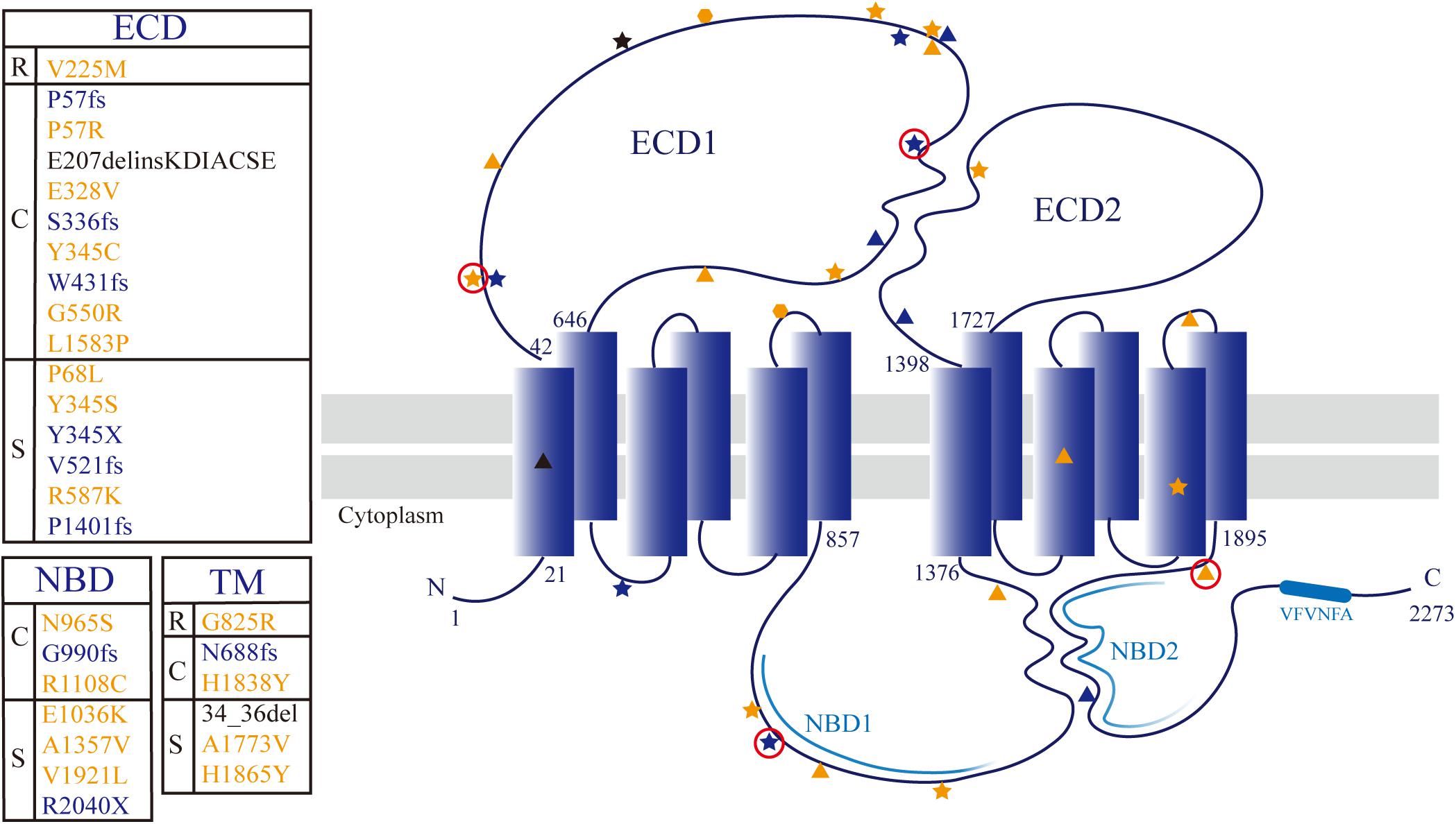
Figure 3. Topological model of ABCA4 protein showing the distribution of detected mutations. The textbox on the left of the model shows correspondence between the locus and phenotype (R: RP19; C: CORD3/COD; S: STGD1). The sites associated with RP19, CORD3/COD, and STGD1 phenotypes are denoted with hexagon, pentagram, and triangle, respectively. Missense mutation, protein truncating mutation, and non-frame-shift deletion are colored yellow, blue, and black, respectively. The novel variants are circled in red.
We identified four novel variants of the ABCA4 gene (Figure 4). According to ACMG standards and guidelines, two novel ABCA4 frameshift variants were pathogenic, one missense variant c.G5761T (p.V1921L) was likely pathogenic, and another missense variant c.C170G (p.P57R) was a variant of uncertain significance (Appendix 2).
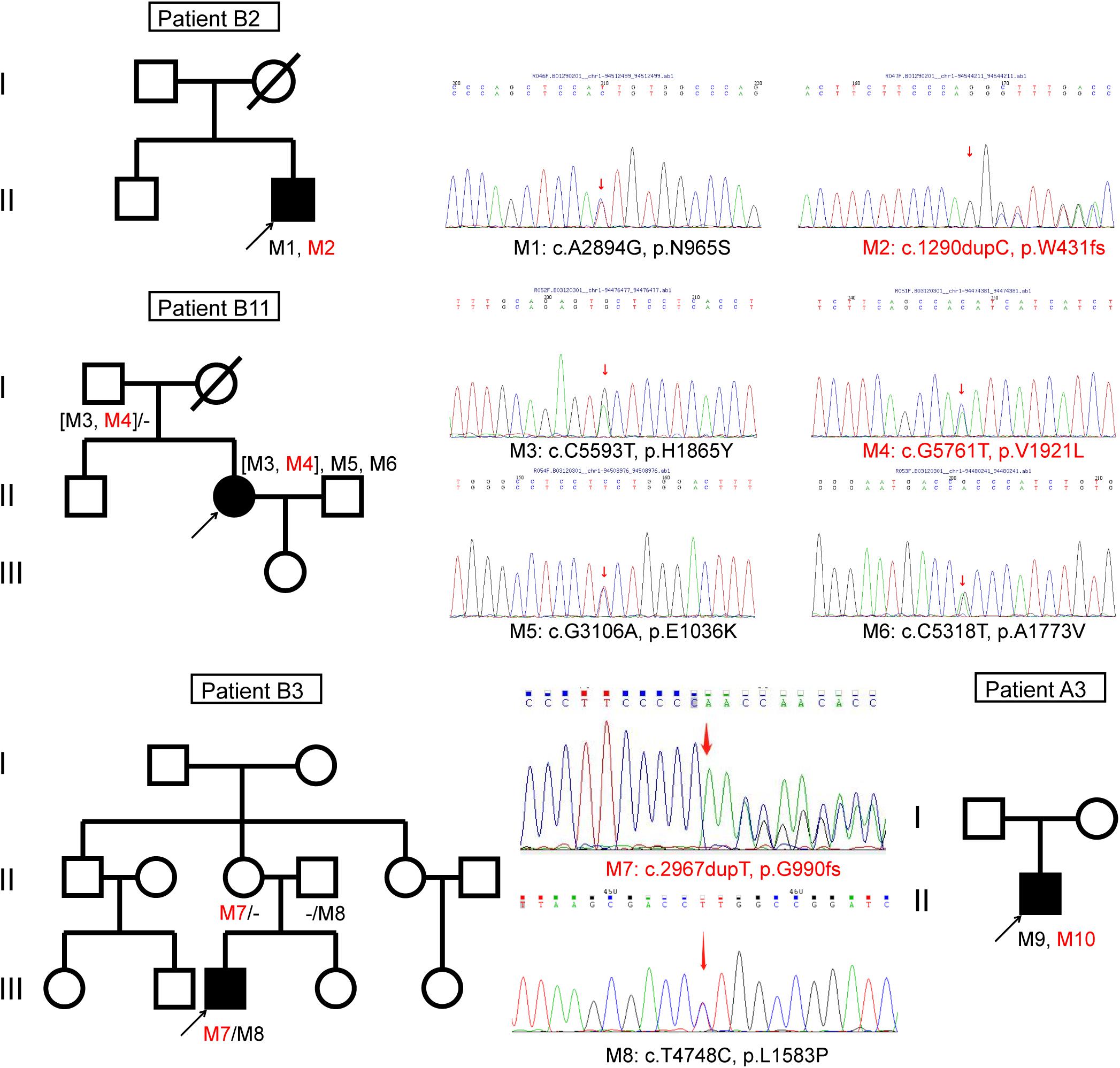
Figure 4. Pedigrees of four Chinese families with inherited retinal disorders harboring the ABCA4 novel variants (red font).
The well-conserved VFVNFA motif (amino acids 2244–2249) present within the C-terminal of ABCA4 enables folding of the polypeptide into a functionally active protein (Zhong et al., 2009). Deletion of the C-terminal domain, including the VFVNFA motif, leads to a loss of ABCA4 functions, such as N-retinylidene-phosphatidylethanolamine substrate binding, ATP photoaffinity labeling, retinal-stimulated ATPase activity, and nucleotide binding domain interactions (Zhong et al., 2009; Patel et al., 2019). All frameshift and nonsense mutations of ABCA4 caused a removal of the C-terminal conserved sequence VFVNFA in the polypeptide. The downstream sequence of the VFVNFA motif also plays a role in regulating the functions of ABCA4 (Zhong et al., 2009). Protein-truncating mutations in ABCA4 leads to truncated protein products or protein loss due to nonsense-mediated RNA decay (Khajavi et al., 2006; Makelainen et al., 2019). Hence, the frameshift and nonsense ABCA4 mutations, which are defined as pathogenic according to ACMG guidelines, may have a great impact on gene expression and protein functions (Table 2). The allele frequencies of the small deletion mutation c.101_106del (p.34_36del) in two Chinese patient cohorts were 3.1% (Jiang et al., 2016) and 10.5% (Hu et al., 2019). This mutation, located in the transmembrane domain, is predicted to break the transmembrane helical structure, leading to ABCA4 transport dysfunction. The variant c.618_619insAAGGACATCGCCTGCAGC (p.E207delinsKDIACSE) inserted six amino acids in ECD1 of ABCA4, which was predicted to make ECD1 more hydrophobic and alter the protein structure. The mutation is associated with STGD1 disease (Tian et al., 2016).
The majority of identified ABCA4 sequence variants are missense mutations. Therefore, analysis of ABCA4-associated retinal degeneration is difficult. Additionally, determination of the pathogenicity of a particular variant is difficult. Testing the phenotypic associations of all amino acid substitutions along with experimental characterizations of their effects on protein functions would be extremely expensive and time consuming. Therefore, an online study of their putative effects would be conducive to prioritize the most probable disease-causing variations associated with these diseases. To further study the pathogenesis and possibility of amino acid substitutions that damage the protein, we analyzed the effects of variants on the protein using an online software (Appendix 1). Biochemical studies on ABCA4 missense mutations have reported that insertion of charged amino acids in transmembrane domains leads to a decrease in protein expression (Sun et al., 2000). Apart from the variations located in nucleotide-binding domains (Sun et al., 2000; Ahn et al., 2003), the missense mutations of ABCA4 gene, in or close to the transmembrane region, causing protein conformational changes can impair azido-ATP labeling to the nucleotide binding domains (Sun et al., 2000; Garces et al., 2018). Three cone–rod dystrophy patients (B1, B2, and B5) and one cone dystrophy patient (A5) harbored the missense mutation c.A2894G (p.N965S). The prevalence of the c.A2894G (p.N965S) mutation was higher in STGD1-affected individuals than in controls (Rosenberg et al., 2007; Jiang et al., 2016; Hu et al., 2019). Therefore, we hypothesized that patients carrying c.A2894G (p.N965S) might more likely manifest cone–rod dystrophy as the disease progresses, although the early stage may be an STGD1-like phenotype. The mutation, which is a replacement of asparagine with serine, resides in the conserved WalkerA sequence of nucleotide binding domain 1, which participates in nucleotide binding (Tsybovsky et al., 2013). An in vivo experiment concluded that p.N965S mutation causes a loss of substrate-dependent ATPase activity of ABCA4 and protein misfolding (Molday et al., 2018).
Patient B11 harbored four unique ABCA4 gene missense variants. The variant c.C5593T (p.H1865Y) and a novel variant c.G5761T (p.V1921L) were inherited from his healthy father. The missense variant c.G5761T (p.V1921L), adjacent to the transmembrane region, was likely pathogenic according to ACMG standards (Table 2). The variant c.C5593T (p.H1865Y), which has not been reported to date, was predicted as “tolerated” using a five-function prediction software. The other two missense mutations c.G3106A (p.E1036K) and c.C5318T (p.A1773V) were classified as STGD1 disease-related variants (Rivera et al., 2000; Chacon-Camacho et al., 2013). The variant c.C5318T (p.A1773V) was conserved and located in the transmembrane domain of the ABCA4 protein. The allele frequency of this mutation in an STGD1 patient cohort is significantly higher than that in the control group (Chacon-Camacho et al., 2013). According to bioinformatic analysis, it was disease causing. Homozygous p.Ala1773Val mutation leads to a severe phenotype, which is similar to that of patients harboring null ABCA4 variants (Lopez-Rubio et al., 2018). However, the fundus autofluorescence of patient B11 did not reveal extensive retinal atrophy (Figure 2), which may be related to the heterozygous state of the c.C5318T (p.A1773V) mutation. The trans variant may be a “milder” mutation. Patient A6 harbored a highly conserved ABCA4 homozygous mutation c.C4070T (p.A1357V), which was revealed to be consistently pathogenic using the six-function software. A patient harboring compound heterozygous variants of p.A1357V and p.G1961E has been reported to possess a foveal sparing (Noupuu et al., 2016). The p.G1961E mutation is usually associated with a late onset and mild phenotype (Lewis et al., 1999; Burke et al., 2012). Patient A6 was asymptomatic until 17 years of age, and the phenotype was milder compared with that of other patients. Considering this, we hypothesized that p.A1357V is not a severe mutation.
Genetic studies revealed that the patient A1 harbored variations in both ABCA4 and AHI1 genes. The two missense mutations c.G2473A (p.G825R) and c.G673A (p.V225M), located in ABCA4, were two variants of uncertain significance according to ACMG standards. Based on the analyses of the ABCA4 protein structure, p.G825R was located in the torsion angles of the transmembrane domain, which will force the local backbone into an incorrect conformation to disturb the local structure. p.V225M was located in ECD1, which will slightly destabilize the local conformation. The c.G3267A (p.W1089X) and c.T1979G (p.L660R) variants were identified in the AHI1 gene. The nonsense mutation was located in the SH3 domain of AHI1, and the missense mutation was located in the WD40 domain. Missense mutations in the WD40 domain of the AHI1 gene can underlie non-syndromic retinitis pigmentosa (Nguyen et al., 2017). ABCA4-related retinitis pigmentosa in patients is frequently caused by combinations of ABCA4 null mutations such as frameshift and splicing site mutations (van Driel et al., 1998; Shroyer et al., 2001; Huang et al., 2018). Thus, we suspect that the phenotype of patient A1 with retinitis pigmentosa may be a result of variations in both ABCA4 and AHI1 genes, which highlights the genotypic variability associated with ABCA4-related retinitis pigmentosa.
The missense mutations c.C203T (p.P68L), c.A1034G (p.Y345C), c.A1034C (p.Y345S), c.C5512T (p.H1838Y), c.G1648A (p.G550R), c.C4070T(p.A1357V), and c.C3322T (p.R1108C) have been conserved during evolution. The minor allele frequencies of these mutations in 1,000 Genomes and gnomAD database were less than 0.005. They were predicted as “deleterious” using SIFT, PROVEAN, CADD, FATHMM, and MutationTaster. Considering the lack of functional experiments to verify the effect of mutant residues and based on the results of HOPE, we classified them as “likely pathogenic” in accordance with the ACMG guidelines (Table 2). The missense mutations c.A983T (p.E328V) and c.T4748C (p.L1583P), and novel mutation c.C170G (p.P57R) were all located in ECD1 of ABCA4. The biological functions of the ECDs of ABCA4 were unknown, and the results of computer prediction were contradictory. The clinical significance of the three missense mutations was unclear.
According to the genotype–phenotype correlation model proposed by van Driel et al., the severity of the phenotype of ABCA4-related diseases is inversely proportional to the residual function of the mutant protein (van Driel et al., 1998). Hence, we could evaluate the severity and prognosis of ABCA4-related diseases using the genotype. Because the protein-truncating mutations cause nearly a complete loss of protein function, the residual function of protein caused by trans mutations primarily determines the severity of the phenotype. In this study, all 11 patients (B1, B2, B3, A3, A4, B6, B7, B8, B9, and B10) carried a severe ABCA4 gene variant, including frameshift mutations, nonsense mutations, and the missense mutation c. C5318T (p. A1773V). Additionally, the severity of the phenotype was related to the course of the disease; therefore, follow-up was highly recommended.
ABCA4-related retinal degeneration is genetically and clinically heterogeneous. The high allelic heterogeneity makes molecular genetic analysis of ABCA4-associated retinal disease challenging. We described the findings of mutational profiling of the ABCA4 gene and related clinical phenotypes, and we predicted the pathogenicities of newly discovered variants, which will expand the spectrum of disease-causing variants in ABCA4, and further facilitate genetic counseling. For the variants of unknown significance due to limited data, further experimental verification is needed to provide new insights into the molecular mechanisms of the disease, which may help in the development of precision medicine.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number (s) can be found below: https://databases.lovd.nl/shared/individuals?search_owned_by_=%3D%22Qing%20Zhu%22.
The studies involving human participants were reviewed and approved by The Medical Ethics Committee of People’s Hospital of Ningxia Hui Autonomous Region and Henan Provincial Eye Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual (s), and minor (s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
BL and X-LS conceived and designed this study, directed the work, and finalized the manuscript. QZ and XR collected the clinical samples and clinical data. YL, YY, and QZ analyzed the sequencing data. QZ collected the information and drafted and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China grants (82071008, 81770949, and 81760180) and the Henan Key Laboratory of Ophthalmology and Vision Science.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank all patients and their families for participating in this study. We would like to thank Editage (www.editage.com) for English language editing.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.634843/full#supplementary-material
IRDs, inherited retinal degenerations; ar, autosomal recessive; ACMG, American College of Medical Genetics and Genomics; ECD, extracytoplasmic domain; BCVA, best-corrected visual acuity.
Aboshiha, J., Dubis, A. M., Carroll, J., Hardcastle, A. J., and Michaelides, M. (2016). The cone dysfunction syndromes. Br. J. Ophthalmol. 100, 115–121. doi: 10.1136/bjophthalmol-2014-306505
Ahn, J., Beharry, S., Molday, L. L., and Molday, R. S. (2003). Functional interaction between the two halves of the photoreceptor-specific ATP binding cassette protein ABCR (ABCA4). Evidence for a non-exchangeable ADP in the first nucleotide binding domain. J. Biol. Chem. 278, 39600–39608. doi: 10.1074/jbc.M304236200
Allikmets, R., Singh, N., Sun, H., Shroyer, N. F., Hutchinson, A., Chidambaram, A., et al. (1997). A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive stargardt macular dystrophy. Nat. Genet. 15, 236–246. doi: 10.1038/ng0397-236
Baum, L., Chan, W. M., Li, W. Y., Lam, D. S. C., Wang, P. B., and Pang, C. P. (2003). ABCA4 sequence variants in chinese patients with age-related macular degeneration or stargardt’s disease. Ophthalmologica. 217, 111–114. doi: 10.1159/000068553
Burke, T. R., Fishman, G. A., Zernant, J., Schubert, C., Tsang, S. H., Smith, R. T., et al. (2012). Retinal phenotypes in patients homozygous for the G1961E mutation in the ABCA4 gene. Invest. Ophthalmol. Vis. Sci. 53, 4458–4467. doi: 10.1167/iovs.11-9166
Burke, T. R., and Tsang, S. H. (2011). Allelic and phenotypic heterogeneity in ABCA4 mutations. Ophthalmic Genet. 32, 165–174. doi: 10.3109/13816810.2011.565397
Chacon-Camacho, O. F., Granillo-Alvarez, M., Ayala-Ramirez, R., and Zenteno, J. C. (2013). ABCA4 mutational spectrum in Mexican patients with Stargardt disease: identification of 12 novel mutations and evidence of a founder effect for the common p.A1773V mutation. Exp. Eye Res. 109, 77–82. doi: 10.1016/j.exer.2013.02.006
Desmet, F. O., Hamroun, D., Lalande, M., Collod-Béroud, G., Claustres, M., and Béroud, C. (2009). Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37:e67. doi: 10.1093/nar/gkp215
Fishman, G. A. (1976). Fundus flavimaculatus. a clinical classification. Arch. Ophthalmol. 94, 2061–2067. doi: 10.1001/archopht.1976.03910040721003
Fujinami, K., Zernant, J., Chana, R. K., Wright, G. A., Tsunoda, K., Ozawa, Y., et al. (2015). Clinical and molecular characteristics of childhood-onset stargardt disease. Ophthalmology 122, 326–334. doi: 10.1016/j.ophtha.2014.08.012
Garces, F., Jiang, K., Molday, L. L., Stohr, H., Weber, B. H., Lyons, C. J., et al. (2018). Correlating the expression and functional activity of ABCA4 disease variants with the phenotype of patients with stargardt disease. Invest. Ophthalmol. Vis. Sci. 59, 2305–2315. doi: 10.1167/iovs.17-23364
Glöckle, N., Kohl, S., Mohr, J., Scheurenbrand, T., Sprecher, A., Weisschuh, N., et al. (2014). Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur. J. Hum. Genet. 22, 99–104. doi: 10.1038/ejhg.2013.72
Hu, F. Y., Li, J. K., Gao, F. J., Qi, Y. H., Xu, P., Zhang, Y. J., et al. (2019). ABCA4 Gene screening in a chinese cohort with stargardt disease: identification of 37 novel variants. Front. Genet. 10:773. doi: 10.3389/fgene.2019.00773
Huang, X., Yuan, L., Xu, H., Zheng, W., Cao, Y., Yi, J., et al. (2018). Identification of a novel mutation in the ABCA4 gene in a chinese family with retinitis pigmentosa using exome sequencing. Biosci. Rep. 38. doi: 10.1042/BSR20171300
Jiang, F., Pan, Z., Xu, K., Tian, L., Xie, Y., Zhang, X., et al. (2016). Screening of ABCA4 gene in a chinese cohort with stargardt disease or cone-rod dystrophy with a report on 85 novel mutations. Invest. Ophthalmol. Vis. Sci. 57, 145–152. doi: 10.1167/iovs.15-18190
Kent, W. J., Sugnet, C. W., Furey, T. S., Roskin, K. M., Pringle, T. H., Zahler, A. M., et al. (2002). The human genome browser at UCSC. Genome Res. 12, 996–1006. doi: 10.1101/gr.229102
Khajavi, M., Inoue, K., and Lupski, J. R. (2006). Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur. J. Hum. Genet. 14, 1074–1081. doi: 10.1038/sj.ejhg.5201649
Kim, M. S., Joo, K., Seong, M. W., Kim, M. J., Park, K. H., Park, S. S., et al. (2019). Genetic mutation profiles in korean patients with inherited retinal diseases. J. Korean Med. Sci. 34:e161. doi: 10.3346/jkms.2019.34.e161
Kos, V., and Ford, R. C. (2009). The ATP-binding cassette family: a structural perspective. Cell Mol. Life. Sci. 66, 3111–3126. doi: 10.1007/s00018-009-0064-69
Koyanagi, Y., Akiyama, M., Nishiguchi, K. M., Momozawa, Y., Kamatani, Y., Takata, S., et al. (2019). Genetic characteristics of retinitis pigmentosa in 1204 japanese patients. J. Med. Genet. 56, 662–670. doi: 10.1136/jmedgenet-2018-105691
Lenis, T. L., Hu, J., Ng, S. Y., Jiang, Z., Sarfare, S., Lloyd, M. B., et al. (2018). Expression of ABCA4 in the retinal pigment epithelium and its implications for stargardt macular degeneration. Proc. Natl. Acad. Sci. U.S.A. 115, E11120–E11127. doi: 10.1073/pnas.1802519115
Lewis, R. A., Shroyer, N. F., Singh, N., Allikmets, R., Hutchinson, A., Li, Y., et al. (1999). Genotype/phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease. Am. J. Hum. Genet. 64, 422–434. doi: 10.1086/302251
Lopez-Rubio, S., Chacon-Camacho, O. F., Matsui, R., Guadarrama-Vallejo, D., Astiazaran, M. C., and Zenteno, J. C. (2018). Retinal phenotypic characterization of patients with ABCA4 retinopathydue to the homozygous p.Ala1773Val mutation. Mol. Vis. 24, 105–114.
Makelainen, S., Godia, M., Hellsand, M., Viluma, A., Hahn, D., Makdoumi, K., et al. (2019). An ABCA4 loss-of-function mutation causes a canine form of Stargardt disease. PLoS Genet. 15:e1007873. doi: 10.1371/journal.pgen.1007873
Molday, L. L., Wahl, D., Sarunic, M. V., and Molday, R. S. (2018). Localization and functional characterization of the p.Asn965Ser (N965S) ABCA4 variant in mice reveal pathogenic mechanisms underlying Stargardt macular degeneration. Hum. Mol. Genet. 27, 295–306. doi: 10.1093/hmg/ddx400
Nasonkin, I., Illing, M., Koehler, M. R., Schmid, M., Molday, R. S., and Weber, B. H. (1998). Mapping of the rod photoreceptor ABC transporter (ABCR) to 1p21–p22.1 and identification of novel mutations in stargardt’s disease. Hum. Genet. 102, 21–26. doi: 10.1007/s004390050649
Nguyen, T. T., Hull, S., Roepman, R., van den Born, L. I., Oud, M. M., de Vrieze, E., et al. (2017). Missense mutations in the WD40 domain of AHI1 cause non-syndromic retinitis pigmentosa. J. Med. Genet. 54, 624–632. doi: 10.1136/jmedgenet-2016-104200
Noupuu, K., Lee, W., Zernant, J., Greenstein, V. C., Tsang, S., and Allikmets, R. (2016). Recessive Stargardt disease phenocopying hydroxychloroquine retinopathy. Graefes Arch. Clin. Exp. Ophthalmol. 254, 865–872. doi: 10.1007/s00417-015-3142-3148
Patel, M. J., Biswas, S. B., and Biswas-Fiss, E. E. (2019). Functional significance of the conserved C-Terminal VFVNFA motif in the retina-specific ABC transporter, ABCA4, and its role in inherited visual disease. Biochem. Biophys. Res. Commun. 519, 46–52. doi: 10.1016/j.bbrc.2019.08.121
Pontikos, N., Arno, G., Jurkute, N., Schiff, E., Ba-Abbad, R., Malka, S., et al. (2020). Genetic basis of inherited retinal disease in a molecularly characterized cohort of more than 3000 families from the United Kingdom. Ophthalmology 127, 1384–1394. doi: 10.1016/j.ophtha.2020.04.008
Quazi, F., Lenevich, S., and Molday, R. S. (2012). ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat. Commun. 3:925. doi: 10.1038/ncomms1927
Quazi, F., and Molday, R. S. (2014). ATP-binding cassette transporter ABCA4 and chemical isomerization protect photoreceptor cells from the toxic accumulation of excess 11-cis-retinal. Proc. Natl. Acad. Sci. U S A. 111, 5024–5029. doi: 10.1073/pnas.1400780111
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Rivera, A., White, K., Stohr, H., Steiner, K., Hemmrich, N., Grimm, T., et al. (2000). A comprehensive survey of sequence variation in the ABCA4 (ABCR) gene in Stargardt disease and age-related macular degeneration. Am. J. Hum. Genet. 67, 800–813. doi: 10.1086/303090
Rong, W. N., Wang, X. G., and Sheng, X. L. (2018). ABCA4 mutations and phenotype of different hereditary retinopathies in 3 pedigrees. Chin. J. Ophthalmol. 54, 775–781. doi: 10.3760/cma.j.issn.0412-4081.2018.10.011
Rosenberg, T., Klie, F., Garred, P., and Schwartz, M. (2007). N965S is a common ABCA4 variant in stargardt-related retinopathies in the danish population. Mol. Vis. 13, 1962–1969.
Rozet, J. M., Gerber, S., Souied, E., Perrault, I., Châtelin, S., Ghazi, I., et al. (1998). Spectrum of ABCR gene mutations in autosomal recessive macular dystrophies. Eur. J Hum. Geneti. 6, 291–295. doi: 10.1038/sj.ejhg.5200221
Sahel, J.-A., Marazova, K., and Audo, I. (2014). Clinical characteristics and current therapies for inherited retinal degenerations. Cold Spring Harb. Perspect. Med. 5:a017111. doi: 10.1101/cshperspect.a017111
Shihab, H. A., Gough, J., Cooper, D. N., Stenson, P. D., Barker, G. L. A., Edwards, K. J., et al. (2013). Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 34, 57–65. doi: 10.1002/humu.22225
Shroyer, N. F., Lewis, R. A., Yatsenko, A. N., and Lupski, J. R. (2001). Null missense ABCR (ABCA4) mutations in a family with stargardt disease and retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 42, 2757–2761.
Sun, H., Smallwood, P. M., and Nathans, J. (2000). Biochemical defects in ABCR protein variants associated with human retinopathies. Nat. Genet. 26, 242–246. doi: 10.1038/79994
Tanaka, K., Lee, W., Zernant, J., Schuerch, K., Ciccone, L., Tsang, S. H., et al. (2018). The rapid-onset chorioretinopathy phenotype of ABCA4 disease. Ophthalmology 125, 89–99. doi: 10.1016/j.ophtha.2017.07.019
Tian, L., Jiang, F., Xu, K., Zhang, X. H., Sun, T. Y., Lu, N., et al. (2016). Characteristics of ABCA4 genotype in Chinese patients with Stargardt disease. Ophthalmol CHN 25, 219–224.
Tsybovsky, Y., Orban, T., Molday, R. S., Taylor, D., and Palczewski, K. (2013). Molecular organization and ATP-induced conformational changes of ABCA4, the photoreceptor-specific ABC transporter. Structure 21, 854–860. doi: 10.1016/j.str.2013.03.001
van Driel, M. A., Maugeri, A., Klevering, B. J., Hoyng, C. B., and Cremers, F. P. (1998). ABCR unites what ophthalmologists divide(s). Ophthalmic Genet. 19, 117–122. doi: 10.1076/opge.19.3.117.2187
Venselaar, H., Te Beek, T. A., Kuipers, R. K., Hekkelman, M. L., and Vriend, G. (2010). Protein structure analysis of mutations causing inheritable diseases. an e-science approach with life scientist friendly interfaces. BMC Bioinformatics 11:548. doi: 10.1186/1471-2105-11-548
Verbakel, S. K., van Huet, R. A. C., Boon, C. J. F., den Hollander, A. I., Collin, R. W. J., Klaver, C. C. W., et al. (2018). Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 66, 157–186. doi: 10.1016/j.preteyeres.2018.03.005
Wang, X. G., Liu, H. J., Zhang, S. C., Qi, X. L., Pan, B., Zhuang, W. J., et al. (2018). Genotype and clinical phenotype analysis in patients with retinitis pigmentosa and cone rod dystrophy. Chin. J. Ocul. Fundus. Dis. 34, 526–535. doi: 10.3760/cma.j.issn.1005-1015.2018.06.002
Weng, J., Mata, N. L., Azarian, S. M., Tzekov, R. T., Birch, D. G., and Travis, G. H. (1999). Insights into the function of Rim protein in photoreceptors and etiology of Stargardt’s disease from the phenotype in abcr knockout mice. Cell 98, 13–23. doi: 10.1016/s0092-8674(00)80602-80609
Xin, W., Xiao, X., Li, S., Jia, X., Guo, X., and Zhang, Q. (2015). Identification of genetic defects in 33 probands with stargardt disease by WES-based bioinformatics gene panel analysis. PloS One 10:e0132635. doi: 10.1371/journal.pone.0132635
Xu, Y., Guan, L., Shen, T., Zhang, J., Xiao, X., Jiang, H., et al. (2014). Mutations of 60 known causative genes in 157 families with retinitis pigmentosa based on exome sequencing. Hum. Genet. 133, 1255–1271. doi: 10.1007/s00439-014-1460-2
Young, R. W., and Bok, D. (1969). Participation of the retinal pigment epithelium in the rod outer segment renewal process. J. Cell Biol. 42, 392–403. doi: 10.1083/jcb.42.2.392
Keywords: ABCA4, inherited retinal degeneration, Stargardt diseases, cone-rod dystrophy, photoreceptor degeneration
Citation: Zhu Q, Rui X, Li Y, You Y, Sheng X-L and Lei B (2021) Identification of Four Novel Variants and Determination of Genotype–Phenotype Correlations for ABCA4 Variants Associated With Inherited Retinal Degenerations. Front. Cell Dev. Biol. 9:634843. doi: 10.3389/fcell.2021.634843
Received: 29 November 2020; Accepted: 26 January 2021;
Published: 01 March 2021.
Edited by:
Minzhong Yu, Case Western Reserve University, United StatesReviewed by:
Renan Paulo Martin, Johns Hopkins University, United StatesCopyright © 2021 Zhu, Rui, Li, You, Sheng and Lei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xun-Lun Sheng, c2hlbmd4dW5sdW5AMTYzLmNvbQ==; Bo Lei, Ym9sZWk5OUAxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.