 Marta Barbosa1
Marta Barbosa1 Cátia Gomes1
Cátia Gomes1 Catarina Sequeira1
Catarina Sequeira1 Joana Gonçalves-Ribeiro2,3
Joana Gonçalves-Ribeiro2,3 Carolina Campos Pina2,3
Carolina Campos Pina2,3 Luís A. Carvalho1
Luís A. Carvalho1 Rui Moreira1,4
Rui Moreira1,4 Sandra H. Vaz2,3
Sandra H. Vaz2,3 Ana Rita Vaz1,4
Ana Rita Vaz1,4 Dora Brites1,4*
Dora Brites1,4*- 1Instituto de Investigação do Medicamento (iMed.ULisboa), Faculdade de Farmácia, Universidade de Lisboa, Lisbon, Portugal
- 2Instituto de Medicina Molecular João Lobo Antunes, Faculdade de Medicina, Universidade de Lisboa, Lisbon, Portugal
- 3Instituto de Farmacologia e Neurociências, Faculdade de Medicina, Universidade de Lisboa, Lisbon, Portugal
- 4Departamento de Ciências Farmacêuticas e do Medicamento, Faculdade de Farmácia, Universidade de Lisboa, Lisbon, Portugal
Reactive astrocytes in Amyotrophic Lateral Sclerosis (ALS) change their molecular expression pattern and release toxic factors that contribute to neurodegeneration and microglial activation. We and others identified a dysregulated inflammatory miRNA profile in ALS patients and in mice models suggesting that they represent potential targets for therapeutic intervention. Such cellular miRNAs are known to be released into the secretome and to be carried by small extracellular vesicles (sEVs), which may be harmful to recipient cells. Thus, ALS astrocyte secretome may disrupt cell homeostasis and impact on ALS pathogenesis. Previously, we identified a specific aberrant signature in the cortical brain of symptomatic SOD1-G93A (mSOD1) mice, as well as in astrocytes isolated from the same region of 7-day-old mSOD1 mice, with upregulated S100B/HMGB1/Cx43/vimentin and downregulated GFAP. The presence of downregulated miR-146a on both cases suggests that it can be a promising target for modulation in ALS. Here, we upregulated miR-146a with pre-miR-146a, and tested glycoursodeoxycholic acid (GUDCA) and dipeptidyl vinyl sulfone (VS) for their immunoregulatory properties. VS was more effective in restoring astrocytic miR-146a, GFAP, S100B, HMGB1, Cx43, and vimentin levels than GUDCA, which only recovered Cx43 and vimentin mRNA. The miR-146a inhibitor generated typical ALS aberrancies in wild type astrocytes that were abolished by VS. Similarly, pre-miR-146a transfection into the mSOD1 astrocytes abrogated aberrant markers and intracellular Ca2+ overload. Such treatment counteracted miR-146a depletion in sEVs and led to secretome-mediated miR-146a enhancement in NSC-34-motor neurons (MNs) and N9-microglia. Secretome from mSOD1 astrocytes increased early/late apoptosis and FGFR3 mRNA in MNs and microglia, but not when derived from pre-miR-146a or VS-treated cells. These last strategies prevented the impairment of axonal transport and synaptic dynamics by the pathological secretome, while also averted microglia activation through either secretome, or their isolated sEVs. Proteomic analysis of the target cells indicated that pre-miR-146a regulates mitochondria and inflammation via paracrine signaling. We demonstrate that replenishment of miR-146a in mSOD1 cortical astrocytes with pre-miR-146a or by VS abrogates their phenotypic aberrancies and paracrine deleterious consequences to MNs and microglia. These results propose miR-146a as a new causal and emerging therapeutic target for astrocyte pathogenic processes in ALS.
Introduction
Amyotrophic Lateral Sclerosis (ALS) is a neurodegenerative disease with a life expectancy of only 3 years. It is unclear where the disease originates, or which are the specific targets, mediators, and the underlying ALS mechanisms involved, hindering the search for an effective therapeutic strategy. Though the majority of cases occurs in people with no prior family history, several mutations in different genes were already identified (Mejzini et al., 2019), and the mutant Cu/Zn superoxide dismutase 1 (mSOD1) mouse is the mostly widely used animal model in research. This model is well characterized in terms of behavior, histopathology, and molecular mechanisms, and recapitulates most of the pathological features of the human ALS (Al-Chalabi et al., 2017). Although motor neuron (MN) loss is believed to be the predominant feature, astrogliosis has a key role in MN degeneration due to the release of unknown toxic factors, either in the brain cortex or in the spinal cord (SC) (Robberecht and Philips, 2013; Gomes et al., 2020), and in both sporadic ALS (sALS) (Varga et al., 2018) and familiar ALS (fALS) (Meyer et al., 2014; van Rheenen et al., 2016).
Astrocytes isolated from the SC of symptomatic rats exhibit low levels of glial fibrillary acidic protein (GFAP) and glutamate transporter 1 (GLT-1), together with increased S100B and Cx43, and were designated as aberrant astrocytes (Díaz-Amarilla et al., 2011). We lately confirmed this same set of aberrant/reactive markers in astrocytes isolated from the SOD1G93A (mSOD1) mice at 7-day-old and cultured for 13 days in vitro (DIV) (Gomes et al., 2019; Gomes et al., 2020), pointing that such astrocytes acquire early deficits associated to a disease-specific phenotype, including low levels of GFAP. These low levels of GFAP were also found in other harmful conditions (Escartin et al., 2021).
The secretome from ALS aberrant astrocytes was shown to potentiate MN death (Díaz-Amarilla et al., 2011; Trias et al., 2017; Gomes et al., 2019), and to lead to activated microglia (Filipi et al., 2020), but the underlying neurotoxic processes and how the factors involved are disseminated are still to be clarified. One of the mechanisms may relate with the secretion of small extracellular vesicles (sEVs) from mSOD1 astrocytes with miRNAs (Jovicic and Gitler, 2017) and mSOD1 protein (Basso et al., 2013) that showed to cause the selective death of MNs. We previously found that the aberrant mSOD1 cortical astrocytes expressed reduced levels of miR-146a (Gomes et al., 2019). We also identified reduced levels of miR-146a in sEVs released from both cortical and spinal 13 DIV astrocytes that when cocultured with MNs showed to determine several dysfunctionalities (Gomes et al., 2020). We then wonder whether such depleted levels of miR-146a in mSOD1 astrocytes and their sEVs could relate with the cell pathological features and have harmful consequences to nearby MNs and microglia. In conformity, recent data with astrocytes directly converted from the C9orf72 patient fibroblasts has shown that their released sEVs with downregulated miR-494-3p caused neuronal network degeneration (Varcianna et al., 2019).
miRNAs are a class of small non-coding RNAs that play important roles in regulating gene expression. Among them some are more closely associated with inflammatory pathways, i.e., miR-155, miR-146a, miR-21 and miR-124, and are designed as inflamma-miRNAs (Brites, 2020). Their dysregulation was found in ALS (Butovsky et al., 2012; De Felice et al., 2014; Freischmidt et al., 2014), and they were suggested to be potential biomarkers (Ricci et al., 2018; Joilin et al., 2019). Upregulation of miR-155 was found in fALS and sALS patients (Koval et al., 2013; Butovsky et al., 2015), as well as in the SC of mSOD1 mice, in either pre-symptomatic or symptomatic stages (Cunha et al., 2018). When targeted in microglia, its reduction counteracted microglia activation and attenuated the disease in the mSOD1 mice (Butovsky et al., 2015). Our discovered depletion of miR-146a in aberrant cortical astrocytes and their sEVs may also determine a polarized pro-inflammatory microglia phenotype if we consider that it has a key role in repressing inflammation (Taganov et al., 2006; Iyer et al., 2012; Waters et al., 2019).
In this study we decided to use different strategies to recover the normal levels of miR-146a in the ALS aberrant cortical astrocytes, comprising either the pre-miR-146a or compounds recognized as immunoregulators. Our previous data evidenced that glycoursodeoxycholic acid (GUDCA) and dipeptidyl vinyl sulfone (VS) are effective in regulating inflammatory miRNAs and counteracting the release of pro-inflammatory cytokines (Fernandes et al., 2007; Vaz et al., 2015, 2019; Falcão et al., 2017). Thus, we hypothesized that replenishment of miR-146a in the cortical mSOD1 astrocytes toward normal levels could abrogate their phenotypic aberrancies. If that happens, we will avoid the release of neurotoxic factors leading to MN distress and microglia activation (Nagai et al., 2007; Ajmone-Cat et al., 2019; Gomes et al., 2019). Such polarized microglia release TNF-α what in turn further contributes to astrocyte reactivity and neurotoxicity (Liddelow and Barres, 2017). Therefore, restoration of the mSOD1 astrocyte steady-state phenotype will facilitate homeostatic balance recovery. This is important, once astrocyte-microglia crosstalk regulation or deregulation determines neuronal functions and dysfunctions (Jha et al., 2018). Actually, elimination of IL-1α, TNF-α, and C1q in the mSOD1 mice, extended their survival (Guttenplan et al., 2020), highlighting astrocyte reactivity and microglia activation as therapeutic targets to prevent neurodegeneration in ALS.
In this work we used the cortical astrocytes isolated from 7-day-old mSOD1 mice and cultured for 13 DIV, that were transfected with pre-miR-146a or treated with the immunomodulators GUDCA and VS, to test their ability to abolish miR-146a downregulation. Astrocyte reactivity markers usually characterizing their aberrant phenotype, together with the Ca2+ overload (Kawamata et al., 2014), were assessed with or without treatments. In addition, we evaluated the potential neuroprotective and anti-inflammatory properties of the secretome from treated astrocytes vs. the untreated ones, using the mouse NSC-34 MN-like cell line produced from the SC and the N9 microglial cell line from the brain. We explored the expression of synaptic and axonal genes in MNs, iNOS and TNF-α genes in microglia, as well as cell death by apoptosis, fibroblast growth factor receptors (FGFR) 1 and 3 mRNAs, miR-146a and proteomics in both cell types.
Our study is the first targeting the depressed levels of miR-146a in cortical ALS astrocytes toward its restoration, with ensuing data highlighting that both pre-miR-146a and VS may represent new therapeutic ALS strategies to reestablish astrocyte neuroprotection and microglia/MN homeostatic balance. Revival of disease-specific cortical astrocytes in ALS was found to be associated to the replenishment of miR-146a.
Materials and Methods
Ethics Statement
The present study was performed in accordance with the European Community guidelines (Directives 86/609/EU and 2010/63/EU, Recommendation 2007/526/CE, European Convention for the Protection of Vertebrate Animals used for Experimental or Other Scientific Purposes ETS 123/Appendix A) and Portuguese Laws on Animal Care (Decreto-Lei 129/92, Portaria 1005/92, Portaria 466/95, Decreto-Lei 197/96, Portaria 1131/97). All the protocols used in this study were approved by the Portuguese National Authority (General Direction of Veterinary) and the Ethics Committee of the Instituto de Medicina Molecular João Lobo Antunes (IMM) from Faculty of Medicine, University of Lisbon, Portugal. Every effort was made to minimize the number of animals used and their suffering, according with the 3R’s principle.
Transgenic SOD1-G93A Mouse Model
Transgenic B6SJL-TgN (SOD1-G93A)1Gur/J males (Jackson Laboratory, no. 002726) overexpressing the human(h)SOD1 gene carrying a glycine to alanine point mutation at residue 93 (mSOD1) (Gurney, 1994) and B6SJLF1/J non-transgenic wild type (WT) females were purchased from The Jackson Laboratory (Bar Harbor, ME, United States). Maintenance and handling took place at IMM animal house facilities, where a colony was established. Mice were maintained on a background B6SJL by breeding SOD1-G93A transgenic males with non-transgenic females. Both males and females were used as described by us (Gomes et al., 2020). Transgenic SOD1-G93A mice were compared to aged-matched WT mice. All animals were maintained on 12 h light/12 h dark cycle and received food and water ad libitum. Average number of animals per cage was 4 to 5.
Primary Culture of Astrocytes
Astrocytes were isolated from the cerebral cortices of WT and mSOD1 mice at postnatal day 7 (Gomes et al., 2019, 2020). Approximately, 2.0 × 105 cells/cm2 were plated on tissue culture plates in culture medium [DMEM (Biochrom AG, Berlin, Germany) supplemented with 11 mM sodium bicarbonate (Merck, Darmstadt, Germany), 38.9 mM glucose, 1% Antibiotic-Antimycotic solution (Sigma-Aldrich, St. Louis, MO, United States) and 10% Fetal Bovine Serum (FBS) (Biochrom AG)] and maintained at 37°C in a humidified atmosphere of 5% CO2 until 13 DIV, with changing of the culture medium at 7 DIV and 10 DIV. Then, cells were used for the evaluation of reactive and inflammatory mediators, with or without treatment strategies. These cells previously showed a set of specific-disease markers that were also found in symptomatic mice (Gomes et al., 2019), highlighting an early presentation of the mSOD1-associated disease phenotype in such neonatal astrocytes. Moreover, the culturing for 13 DIV guarantees the maturation of astrocytes, as we previously showed (Falcão et al., 2005, 2006). To assess astrocyte paracrine signaling, secretomes were collected from 13 DIV astrocytes, either modulated or not (as next explained), and sEVs were isolated.
Modulation of Cortical Astrocyte Aberrancies
At 12 DIV, cells were incubated with 50 μM GUDCA (Calbiochem; Darmstadt, Germany) or 10 μM VS (synthetized by the Medicinal Chemistry group from Instituto de Investigação do Medicamento) (Falcão et al., 2017), and maintained for further 24 h.
To downregulate miR-146a in WT astrocytes, we transfected cells with 15 nM Anti-miRTM 146a inhibitor (#AM10722, Ambion®, Thermo Fisher Scientific, Waltham, MA, United States) at 11 DIV. According with manufacturer’s description, they are chemically modified single-stranded oligonucleotides with a patented secondary structure that binds and inhibits the endogenous miR-146a.
To upregulate miR-146a in depleted mSOD1 astrocytes, the cells were transfected with 15 nM Pre-miRTM 146a Precursor (#PM10722, Ambion®), a chemically modified double-stranded RNA designed to mimic the endogenous precursor miR-146a and to ensure the uptake of the correct strand to the RISC complex. The mature miR-146a sequence was 5′UGAGAACUGAAUUC CAUGGGUU3′ (hsa-miR-146a-5p). Then, the oligonucleotides were mixed with X-tremeGENETM HP DNA Transfection Reagent (Sigma-Aldrich, St. Louis, MO, United States) in a proportion 2:1 and diluted in Opti-MEMTM (Thermo Scientific, Waltham, MA, United States). The controls used were the non-transfected WT and mSOD1 cells treated only with X-tremeGENETM HP DNA Transfection Reagent diluted in Opti-MEMTM (mock control). To make sure that the effect of miRNA-146a transfection in cells was due to its specificity, we also transfected WT cells with 15 nM of Anti-miR Negative Control and mSOD1 ones with 15 nM Pre-miR Negative Control (Ambion®). Results from mock controls, and respective Anti-miR Negative Control and Pre-miR Negative Control, were similar, validating the miR-146a specificity (Supplementary Table 1). After 12 h, medium was changed [DMEM supplemented with 11 mM sodium bicarbonate, 38.9 mM glucose, 1% FBS sEVs-depleted and 1% Antibiotic-Antimycotic solution] and maintained for 24 h. To evaluate which alterations in astrocyte phenotype treated with VS involved miR-146a regulation, we transfected WT astrocytes with 15 nM anti-miR-146a inhibitor at 11 DIV. After 12 h the medium was changed, and the VS compound was added. The cells were maintained in culture for further 24 h.
Culture of NSC-34 MN-Like Cells and Treatment With Astrocyte-Derived Secretome
In the present study we used the neuroblastoma hybrid cell line NSC-34 expressing the human SOD1 WT. These cells, produced by the fusion of MNs from mouse embryos with mouse neuroblastoma cells N18TG2 (Cashman et al., 1992), were shown to reproduce many features of MNs when differentiated and maturated, and have been used to assess neurotoxicity/neuroprotection (Maier et al., 2013; Vaz et al., 2015), including astrocyte-induced neurodegeneration (Benkler et al., 2013). As before, we decided to use these cells given our previous experience with them (Vaz et al., 2015; Pinto et al., 2017; Gomes et al., 2019) and considering the limitations of primary spinal MN cultures, and the immature phenotype of those generated from embryonic stem cells or from induced pluripotent stem cells (Ho et al., 2016; Bucchia et al., 2018).
NSC-34 cells were cultured in Poly-D-lysine coated culture plates at a density of 5 × 104 cells/ml and maintained for 48 h in the proliferation media [DMEM high glucose, w/o pyruvate, supplemented with 10% of FBS and 1% of penicillin/streptomycin, and geneticin sulfate (G418, 0.5 mg/ml) for selection], and then for additional 4 DIV in the differentiation media [DMEM-F12 plus FBS (1%), non-essential amino acids (1%) and penicillin/streptomycin (1%)], as previously described (Vaz et al., 2015). Media from the differentiated NSC-34 cells was removed and the cells incubated for 48 h with an equivalent volume of the secretome derived from untreated astrocytes and from pre-miR-146a and VS treated cells.
N9-Microglia Cell Culture and Treatment With Astrocyte-Derived Secretome and sEVs
Mouse microglial N9 cell line results from the immortalization of microglia isolated from the cortex of CD1 mouse embryos (Righi et al., 1989). This cell line shows properties that are analogous to primary cultures of microglia, such as migration, phagocytosis, and inflammation-related features (Fleisher-Berkovich et al., 2010), and respond similarly to lipopolysaccharide (LPS)-induced activation (Nikodemova and Watters, 2011; Cunha et al., 2016).
N9 microglial cells were plated on uncoated culture plates at a density of 1 × 105 cells/ml and maintained for 24 h in the culture medium [RPMI media (Sigma-Aldrich, St. Louis, MO, United States) supplemented with 10% FBS, 1% L-glutamine, and 1% antibiotic penicillin/streptomycin (1%), all from Biochrom AG, Berlin, Germany], as usual in our laboratory (Cunha et al., 2016; Pinto et al., 2017; Vaz et al., 2019). Then, culture media were removed, and the cells incubated for 24 h with an equivalent volume of the secretome derived from untreated astrocytes and from pre-miR-146a and VS treated cells. Secretome contains soluble factors and vesicular fractions as sEVs, essentially containing miRNAs (Pinho et al., 2020). We showed that sEVs were preferentially collected by microglia than by MNs, when in coculture (Pinto et al., 2017). To assess whether the treatment of astrocytes with pre-miR-146a or with VS would influence the number of sEVs engulfed by the N9 microglia, we incubated the isolated sEVs with microglia for 24 h. Subsequently, we assessed the influence of those sEVs in shifting the inflammatory state of microglia.
sEV Isolation, Characterization, and Labeling
sEVs were isolated from the secretome of WT and mSOD1 astrocytes, using the process implemented in our lab (Pinto et al., 2017). Briefly, the culture media was centrifuged at 1,000 g for 10 min to remove cell debris. After that, the supernatant was centrifuged at 16,000 g to separate large extracellular vesicles (lEVs, size 1,000 nm) and, the recovered supernatant was passed into a 0.22 μM filter and further centrifuged in the Ultra L-XP100 centrifuge (Beckman Coulter Inc., CA, United States) at 100,000 g to pellet sEVs, as we previously detailed (Pinto et al., 2017). Characterization of sEVs in terms of shape was performed using Negative-Staining Transmission Electron Microscopy (Jeol Jem 1400 TEM 120 kV, Tokyo, Japan). Concentration and size of sEVs were evaluated by Nanoparticle tracking analysis (NTA) using the Nanosight (model LM10-HSBF, Malvern, United Kingdom). Expression of alix and flotillin-1 was assessed by Western blot analysis, using 30 μg of total protein (antibodies are indicated in the Supplementary Table 2). To evaluate miRNA cargo, the final pellet containing sEVs was resuspended in lysis buffer, and the RNA extracted as explained below.
To monitor the incorporation of astrocyte-derived sEVs by N9 microglia, the sEVs were labeled with the PKH67 Fluorescent Linker Kit (Sigma Aldrich) in accordance with manufacturer specifications, resuspended in DMEM with 1% penicillin/streptomycin and added to N9 microglia cultures.
Immunocytochemistry
At 13 DIV, astrocytes were fixed with 4% paraformaldehyde and immunofluorescence staining performed as published (Gomes et al., 2019). Briefly, cells were incubated overnight at 4°C with GFAP (listed in the Supplementary Table 2). In the following day, cells were incubated with secondary antibody, also referred in the Supplementary Table 2. Cell nuclei were stained with Hoechst 33258 dye and then mounted onto uncoated slides using PBS-Glycerol (1:1). Fluorescence was visualized using an AxioCam HR camera adapted to an AxioScopeA1 microscope.
Fluorescence of ten random microscopic fields were acquired per sample using Zen (blue edition, 2012, Zeiss) software and the number of positive cells above a determined threshold or fluorescence per total number of cells was quantified. Area of sEVs labeled with PKH67 was determined per total number of PKH67-positive cells.
Western Blot Analysis
Cell lysis and protein isolation was made as usual in our lab (Gomes et al., 2019), and we used the protein assay kit (Bio-Rad, Hercules, CA, United States) to assess the protein concentration as manufacturer’s specifications. Then, equal amounts of protein were subjected to SDS-PAGE and transferred to a nitrocellulose membrane. After blocking with 5% (w/v) non-fat milk solution, nitrocellulose membranes were incubated overnight at 4°C with primary antibodies (depicted in the Supplementary Table 2). In the following day, secondary antibodies conjugated to horseradish peroxidase were used (Supplementary Table 2). The chemiluminescent detection was performed after membrane incubation with LumiGLO® (Cell Signaling). The relative intensities of protein bands were analyzed using the Image LabTM analysis software, after scanning with ChemiDocXRS, both from Bio-Rad Laboratories (Hercules, CA, United States). Results in cells were normalized to β-actin expression levels.
RT-qPCR
Cellular RNA was isolated using TripleXtractor Reagent (GRISP, Porto, Portugal) (Gomes et al., 2019). For astrocyte-derived sEVs, RNA was extracted using miRNeasy Mini kit (Qiagen, Hilden, Germany). Quantification was performed using Nanodrop ND-100 Spectrophotometer (NanoDrop Technologies, Wilmington, DE, United States).
For gene expression, cellular RNA was converted to complementary DNA (cDNA) using GRS cDNA Synthesis Mastermix Kit (GRISP). Reverse transcriptase quantitative PCR (RT-qPCR) was accomplished using Xpert Fast SYBR Green Blue (GRISP) and the following optimized conditions: 94°C for 2 min followed by 40 cycles at 95°C for 0.05 min and 62°C for 0.3 min. To verify the specificity of the amplification, a melt-curve analysis was performed immediately after the amplification protocol. Non-specific products of PCR were not found in any case. β-actin was used as an endogenous control to normalize the expression levels. Primer sequences are listed in the Supplementary Table 3.
Expression of miRNA-146a (5′ UGAGAACUGAAUUCCAU GGGUU 3′) was assessed in cells and sEVs by RT-qPCR. After RNA quantification, cDNA conversion for miRNA quantification was performed with the miRCURY LNATM RT Kit (Qiagen), using 5 ng total RNA for cells and 10 ng for sEVs. The PowerUpTM SYBRTM Green Master Mix (Applied Biosystems, Life Technologies) was used in combination with pre-designed primers. The reaction conditions consisted of polymerase activation/denaturation and well-factor determination at 95°C for 10 min, followed by 50 amplification cycles at 95°C for 10 s and 60°C for 1 min (ramp-rate of 1.6°/s). SNORD110 (reference gene) was used as an endogenous control and to normalize the expression levels in cells. Both SNORD110 and spike-in were used as endogenous controls and geometric mean from both were calculated to normalize the expression levels in sEVs. RT-qPCR was accomplished on a QuantStudio 7 Flex Real-Time PCR System (Applied Biosystems, Life Technologies) and 384-well plates were used. Relative mRNA/miRNA concentrations were calculated using the 2–ΔΔCT method.
Cell Death Determination
Phycoerythrin-conjugated Annexin V (Annexin V-PE) and 7-amino-actinomycin D (7-AAD; Guava Nexin® Reagent, no. 4500-0450, Millipore) were used to determine the percentage of viable (Annexin V-PE and 7-AAD negative), early apoptotic (Annexin V-PE positive and 7-AAD negative), and late apoptotic/necrotic (Annexin V-PE and 7-AAD positive) cells by flow cytometry, as described (Gomes et al., 2020). After incubation, cells were trypsinized and added to the cells already detached in the culture medium. After centrifugation, the pellet of cells was resuspended in PBS containing 1% bovine serum albumin. Then, they were stained with Guava Nexin Reagent according to manufacturer’s instructions and analyzed on a Guava easyCyte 5HT flow cytometer (Guava Nexin® Software module, Millipore).
Ca2+ Imaging in Astrocytes and Analysis
Astrocyte Ca2+ imaging was performed as previously reported (Morais et al., 2017). Briefly, the suspension of astrocytes was plated on μ-slide 8 well chamber slide (Ibidi®, Gräfelfing, Germany) coated with 10 μg/ml of poly-D-lysine. At 13 DIV, cells were incubated at 37°C for 45 min with the Ca2+ sensitive fluorescent dye fura-2 acetoxymethyl ester (Fura-2AM; 5 mM; Calbiochem, Darmstadt, Germany). After three washes with artificial cerebrospinal fluid (aCSF: NaCl 125 mM, KCl 3 mM, NaH2PO4 1.25 mM, CaCl2 2 mM, MgSO4 2 mM, D(C)-glucose 10 mM and HEPES 10 mM; pH 7.4 adjusted with NaOH) cells were positioned on an inverted epifluorescent optics microscope (Axiovert 135TV, Zeiss, Germany) with a xenon lamp and band-pass filters of 340 and 380 nm wavelengths. Cells were continuously perfused with aCSF (with or without added compounds) at 1.5 ml/s and visualized with a 40x oil-immersion objective (Zeiss). The responses were recorded by a ratiometric method, in which image pairs were obtained every 250 ms by exciting the preparations at 340 and 380 nm, since Fura-2 has an absorbance at 340 nm if bound to calcium, and at 380 nm if not, while the emission wavelength was maintained at 510 nm. The magnitude of the changes in the emission fluorescence of Fura-2 was taken as a measure of the changes in intracellular Ca2+ concentration (Ca2+ T amplitude).
Excitation wavelengths were changed through a high-speed wavelength switcher, Lambda DG-4 (Sutter Instrument, Novato, CA, United States). An estimation of intracellular Ca2+ concentration was given by the ratio between the emissions derived from the two excitation wavelengths of Fura-2 (R340/380). All data were recorded by a cooled CCD camera (Photometrics CoolSNAP fx) and processed and analyzed by MetaFluor software (Universal Imaging, West Chester, PA, United States). During the first 5 min of the trial, baseline Ca2+ levels were established. Addition of glutamate, as the stimulating agent (100 μM), was performed after those first 5 min corresponding to the baseline, and the cell response recorded in the next 10 min, this completing 15 min of total time recording. Representative videos of intracellular Ca2+ changes were obtained with the MetaFluor Analyst and using AVI Creator and Movie Maker software programs.
The frequency of Ca2+ transients was determined with MATLAB and Statistics Toolbox Release 2016a, The MathWorks, Inc. (Natick, MA, United States), as previously described (Martins et al., 2020). In the first 5 min of the experiment, a baseline was obtained. Mean and standard error of the mean (SEM) of all data recorded by each cell were obtained.
Ca2+ transient was considered valid when fura-2 excitation wavelength at 340 nm and 380 nm (F340/380) ratio was higher than the mean of baseline values plus 5 times the SEM. In addition, the normalized ratio was considered when above this margin for more than 5 s. For every region of interest, the peak of each transient, as well as the occurrence of transients, were recorded.
Proteomics
Mass Spectrometric (MS) Analysis
The following cellular pellets were collected for proteomic analysis: (i) mSOD1 astrocytes, transfected or not with pre-miR-146a; (ii) naïve microglia; and (iii) WT motor neurons after incubation with mSOD1 astrocytic secretome (± pre-miR-146a). The cellular pellets were collected by trypsinization for 5 min, washed with PBS and immediately stored at −80°C. Each sample resulted from a pool of three biological replicates (cells isolated from different mice that were pooled down and sent for analysis at EMBL Proteomics Core Facility in Heidelberg, Germany. Samples were subjected to an in-solution tryptic digest using a modified version of the Single-Pot Solid-Phase-enhanced Sample Preparation (SP3) protocol (Moggridge et al., 2018). To this end, samples were added to Sera-Mag Beads (Thermo Scientific) in 10 μl 15% formic acid and 30 μl of ethanol. Binding of proteins was achieved by shaking for 15 min at room temperature. SDS was removed by 4 subsequent washes with 200 μl of 70% ethanol. Proteins were digested overnight at room temperature with 0.4 μg of sequencing grade modified trypsin (Promega) in 40 μl Hepes/NaOH, pH 8.4 in the presence of 1.25 mM TCEP and 5 mM chloroacetamide (Sigma-Aldrich). Beads were separated, washed with 10 μl of an aqueous solution of 2% DMSO and the combined eluates were dried down. Peptides were reconstituted in 10 μl of H2O and reacted for 1 h at room temperature with 80 μg of TMT10plex (Thermo Scientific) (Moggridge et al., 2018) label reagent dissolved in 4 μl of acetonitrile. Then, they were reacted with TMT-labeling reagent. 5% of each sample were mixed, purified by a reverse phase clean-up step (OASIS) and analyzed on a LUMOS system using a 1 h gradient. Calculated TMT ratios were used to adjust sample volumes to achieve a 1:1 ratio. The combined samples were subjected to a high pH offline fractionation yielding 12 fractions, each of those analyzed on a 2 h gradient on Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific). Peptides were separated using an UltiMate 3000 nano RSLC system (Dionex) equipped with a trapping cartridge (Precolumn; C18 PepMap 100, 5 mm, 300 μm i.d. ×5 μm, 100 A°) and an analytical column (Acclaim PepMap 100. 75 cm × 50 cm C18, 3 mm, 100 Å) connected to a nanospray-Flex ion source. The peptides were loaded onto the trap column at 30 μl per min using solvent A (0.1% formic acid) and eluted using a gradient from 2 to 40% Solvent B (0.1% formic acid in acetonitrile) over 2 h at 0.3 μl per min. The Orbitrap Fusion Lumos was operated in positive ion mode with a spray voltage of 2.4 kV and capillary temperature of 275°C to analyze the peptides. MS spectra with a mass range of 375–1.500 m/z were acquired in profile mode using a resolution of 120.000 [maximum fill time of 50 ms or a maximum of 4 × 105 ions (automatic gain control, AGC)]. Fragmentation was triggered for 3 s cycle time for peptide like features with charge states of 2–7 on the MS scan (data-dependent acquisition). Precursors were isolated using the quadrupole with a window of 0.7 m/z and fragmented with a normalized collision energy of 38. Fragment mass spectra were acquired in profile mode and a resolution of 30,000 in profile mode. Maximum fill time was set to 64 ms or an AGC target of 1e5 ions). The dynamic exclusion was set to 45 s.
Raw MS Data and Analysis
Acquired data were analyzed using IsobarQuant (Franken et al., 2015), Mascot V2.4 (Matrix Science) and a reverse UniProt FASTA Mus musculus (UP000000589) database, including common contaminants. The following modifications were considered: Carbamidomethyl (C, fixed), TMT10plex (K, fixed), Acetyl (N-term, variable), Oxidation (M, variable) and TMT10plex (N-term, variable). The mass error tolerance for full scan MS spectra was set to 10 ppm and for MS/MS spectra to 0.02 Da. A maximum of 2 missed cleavages were allowed. A minimum of 2 unique peptides with a peptide length of at least seven amino acids and a false discovery rate below 0.01 was required on the peptide and protein level (Savitski et al., 2015). 9083 proteins were identified, from which 6748 proteins were quantified. The raw output files of IsobarQuant were processed using the R programming language (R Development Core Team, 2020). As a quality filter, we only considered proteins that were quantified with at least two unique peptides. Raw TMT reporter ion intensities [Average expression (signal_sum)] were normalized using variance stabilization normalization (Huber et al., 2002). Differential expression was evaluated by computing the respective ratio of normalized TMT signals (additional parameters are indicted in Supplementary Tables 4–6). The proteins identified as hits were then classified by their link to biological processes using Gene Ontology (GO) annotation in Scaffold. Bioinformatic analysis of protein molecular function was done by using PANTHER Classification System (Version 16.0, released 2020-12-01) (Mi et al., 2021). Functional classification of gene lists for Mus musculus were examined using PANTHER Protein Class ontologies. MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE database1 (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD024294.
Statistical Analysis
Results of at least three independent experiments were expressed as mean values ± SEM. Results of untreated or treated mSOD1 astrocytes, astrocyte-derived secretome and astrocyte-derived sEVs were related to respective untreated WT samples. Since we always compared at least three groups, we determined the differences between them by using one-way ANOVA followed by Bonferroni post hoc test. Statistical analysis was performed using GraphPad PRISM 7.0 (GraphPad Software, San Diego, CA, United States) p < 0.05 was considered as statistically significant.
Results
VS Is More Competent Than GUDCA in Restoring miR-146a Expression and Counteracting mSOD1 Astrocytic Aberrancies
In the present study, we first assessed whether our promising immunomodulators GUDCA and VS were able to restore the normal levels of miR-146a in the cortical mSOD1 astrocytes isolated from the 7-day-old mSOD1 mice and cultured for 13 DIV. Such miR-146a was found downregulated in cells presenting decreased gene and protein levels of GFAP, and upregulated gene expression of vimentin, Cx43, S100B and HMGB1 (Gomes et al., 2019). This miR-146a was found to fine tune Toll-like receptors and cytokine signaling (Taganov et al., 2006) and to be a key regulator of astrocyte-mediated inflammatory response (Iyer et al., 2012). Though miR-146 has at least 488 predicted targets, as reviewed in Brites (2020), we decided to investigate whether miR-146a inhibited two of its recognized target genes, the interleukin-1 receptor-associated kinase (IRAK) 1 and TNF receptor-associated factor (TRAF) 6 (Iyer et al., 2012; Saba et al., 2014). Actually, we have found that both Irak1 and Traf6 transcripts were upregulated in mSOD1 cortical astrocytes (Gomes et al., 2019).
We confirmed that miR-146a was indeed downregulated in the mSOD1 cortical astrocytes (p < 0.05), while their target genes IRAK1 and TRAF6 were upregulated (Figures 1A–C, at least p < 0.05). We also validated that the expression of the aberrant-associated genes was deregulated (Figures 1D–J, at least p < 0.05), attesting the characteristic phenotype previously established (Gomes et al., 2019). From the treatment of cells with either GUDCA or VS, only the later showed to effectively upregulate miR-146a and downregulate IRAK1 and TRAF6 (Figures 1A–C, at least p < 0.01), as well as to restore GFAP protein levels (Figures 1E,F). Interestingly, both GUDCA and VS abrogated the increased levels of vimentin and Cx43 (Figures 1G,H, at least p < 0.05), and were not toxic to the cells through the induction of early or late apoptosis, as depicted in Supplementary Figure 1. VS was again the most efficient in downregulating S100B and HMGB1 gene expression toward the WT levels (Figures 1I,J at least p < 0.05). Thus, VS demonstrated to have a broader reparative ability than GUDCA in recovering miR-146a expression and in counteracting the mSOD1 astrocytes associated reactive markers.
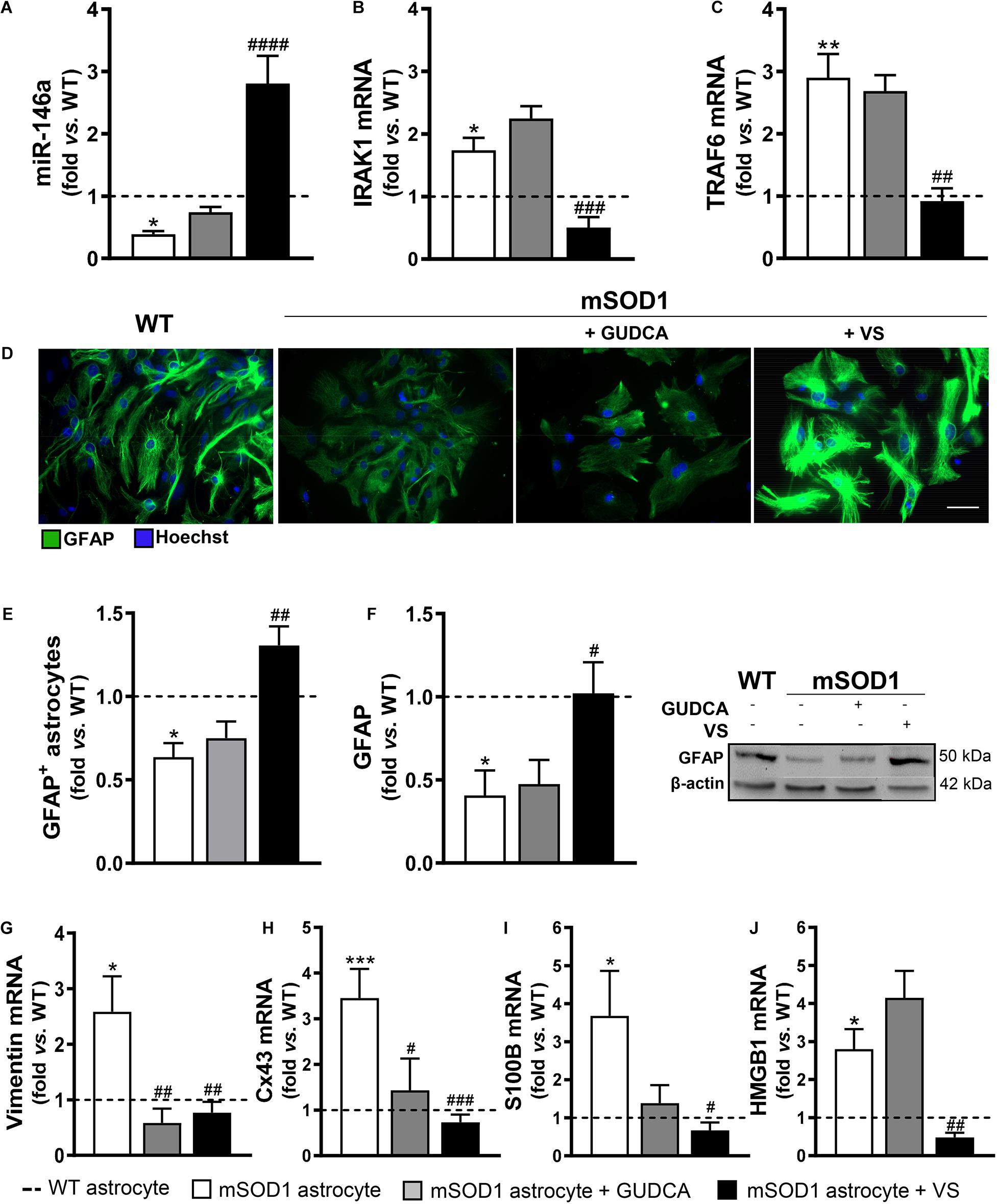
Figure 1. mSOD1 astrocytic aberrancies are more efficiently counteracted by VS than by GUDCA. Astrocytes were isolated from the cortex of SOD1-G93A (mSOD1) and wild type (WT) mice pups at 7-day-old and cultured for 13 days in vitro. Treatment with glycoursodeoxycholic acid (GUDCA) or dipeptidyl vinyl sulfone (VS) was performed in mSOD1 astrocytes. RT-qPCR analysis of (A) miRNA(miR)-146a, (B) interleukin-1 receptor associated kinase-1 (IRAK1) and (C) TNF receptor associated factor 6 (TRAF6), (G) vimentin, (H) connexin-43 (Cx43), (I) S100 calcium-binding protein B (S100B) and (J) high mobility group box 1 (HMGB1) was performed. (D) Representative images of astrocytes stained with glial fibrillary acidic protein (GFAP, in green) by immunocytochemistry and (E) quantification of GFAP-positive cells. Cell nuclei were stained with Hoechst dye (blue). (F) GFAP performed by Western blot analysis and the representative results from one blot are shown. Expression of β-actin was used as an endogenous control for Western Blot and RT-qPCR assays. SNORD110 was used as a reference gene for (A) analysis. Results are mean (±SEM) fold change vs. WT astrocytes from at least four independent experiments. *p < 0.05, **p < 0.01, and ***p < 0.001 vs. WT astrocytes; #p < 0.05, ##p < 0.01, ###p < 0.001, and ####p < 0.0001 vs. untreated mSOD1 astrocytes. One-way ANOVA followed by Bonferroni post hoc test was used. Scale bar represents 20 μm.
Treatment of mSOD1 Astrocytes With Pre-miR-146a Abolishes Their Phenotypic Aberrancies
To assess whether miR-146a downregulation in cortical astrocytes isolated from mSOD1 pups was a player in the deregulated astrocyte polarization, we decided to transfect the cells with pre-miR-146a, as indicated in Material and Methods section, and to evaluate the post-treatment signature of the cells.
We noticed that the mSOD1 astrocytes were more sensitive to the transfection process than the WT cells, as judged by the loss of viable cells (p < 0.05) due to an increase of cell late apoptosis (p < 0.01) (Supplementary Figure 2). Interestingly, such features disappeared in cells transfected with pre-miR-146a, reinforcing the regulatory role of miR-146a in stress/immune-associated stimuli. A 3-fold increase in the levels of miR-146a in the mSOD1 astrocytes was achieved by the transfection with the pre-miR-146a, with values close to those presented by WT astrocytes (Figure 2A, p < 0.001), and its target genes IRAK1 (Figure 2B, p < 0.05) and TRAF6 (Figure 2C, p < 0.001) were reduced near to normal values.
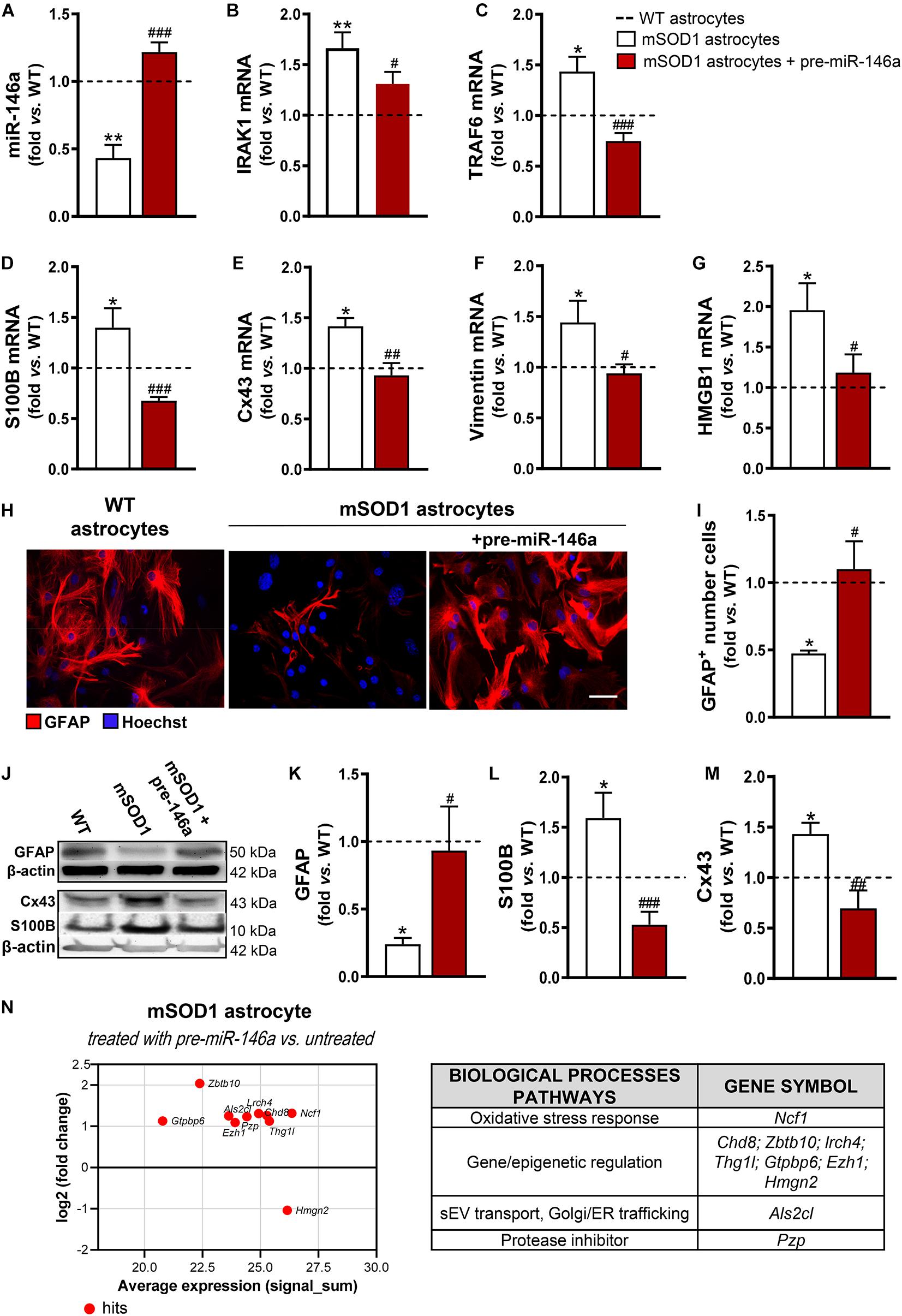
Figure 2. miR-146a upregulation successfully recovers GFAP expression and counteracts deregulated reactive markers in mSOD1 astrocytes. Astrocytes were isolated from the cortex of SOD1-G93A (mSOD1) and wild type (WT) mice with 7-day-old and cultured for 13 days in vitro. Transfection with pre-miR-146a was performed in mSOD1 astrocytes. RT-qPCR analysis of (A) miRNA(miR)-146a, (B) interleukin-1 receptor associated kinase-1 (IRAK1) and (C) TNF receptor associated factor 6 (TRAF6), (D) S100 calcium-binding protein B (S100B), (E) connexin-43 (Cx43), (F) vimentin, and (G) high mobility group box 1 (HMGB1) was performed. (H) Representative images of astrocytes stained with glial fibrillary acidic protein (GFAP, in red) by immunocytochemistry and (I) quantification of GFAP-positive cells. Cell nuclei were stained with Hoechst dye (blue). Protein expression of (K) GFAP, (L) S100B, and (M) Cx43 was performed by Western blot analysis and (J) representative results from one blot are shown. Expression of β-actin was used as an endogenous control for Western Blot and RT-qPCR assays. SNORD110 was used as reference gene for (A) analysis. Results are mean (±SEM) fold change vs. WT astrocytes from at least four independent experiments. *p < 0.05 and **p < 0.01 vs. WT astrocytes; #p < 0.05, ##p < 0.01, and ###p < 0.001 vs. untreated mSOD1 astrocytes. One-way ANOVA followed by Bonferroni post hoc test was used. Scale bar represents 20 μm. (N) MA plot (left) was obtained by comparison of the proteomic profiles of pre-miR-146a treated mSOD1 astrocytes and the untreated ones. Here, x axis is the mean log2 intensity (fold change) of each protein between two profiles (A-value) and y axis is the average expression (signal_sum) of protein intensities (M-value). Summary tables (right panels) show the hits and their classification according to biological processes using PANTHER Classification System, as indicated in methods.
Then, we investigated if the restoration of miR-146a levels was able to attenuate the aberrant phenotype of mSOD1 astrocytes. We verified that such treatment not only restored the gene control values of S100B (Figure 2D, p < 0.001), Cx43 (Figure 2E, p < 0.01), vimentin (Figure 2F, p < 0.05), and HMGB1 (Figure 2G, p < 0.05), but also recovered the GFAP protein expression levels (Figures 2H–K, p < 0.05). Besides, genetic upregulation of miR-146a restored the protein levels of S100B (Figures 2J,L, p < 0.001) and of Cx43 (Figures 2J,M, p < 0.01) toward the control values, attesting that the reduction of miR-146a expression may contribute to the cortical astrocyte aberrancies associated to the ALS disease and to MN degeneration.
To better understand the upregulation of miR-146a in the attenuation of ALS astrocyte-aberrancies and its potential targets we performed proteomic analysis. Data identified 10 hits after the treatment of mSOD1 astrocytes with pre-miR-146a vs. the untreated cells (Figure 2N and Supplementary Table 4). The increase of the neutrophil cytosol factor 1 encoded by the gene Ncf1 may prevent the immune response of mSOD1 challenged astrocytes (Holmdahl et al., 2016; Zhao et al., 2017). In the same way, the upregulated zinc finger and BTB domain-containing 10, encoded by Zbt10, and the downregulated non-histone chromosomal protein HMG-17, encoded by Hmgn2, may turn the neurotoxic into a neuroprotective astrocyte phenotype. Increased expression of proteins encoded by the genes Chd8, Lrch4, Thg1l, Gtpbp6, and Ezh1, involved in gene transcription and epigenetic modifications, suggest a role of miR-146a in the accurate control of stage-specific gene activities required for proper astrocyte function. Upregulation of the pregnancy zinc protein, encoded by Pzp and of the guanine nucleotide exchange factor for Rab5 (the homolog Als2cl) by pre-miR-146a may contribute to prevent mSOD1 pathology, since the first inhibits the aggregation of misfolded proteins (Cater et al., 2019) and the later controls endosome dynamics, thus favoring mSOD1 clearance by sEVs (Hessvik and Llorente, 2018) and autophagy (Otomo et al., 2012).
Overall, our data highlight miR-146a upregulation as a promising therapeutic strategy to restore the steady-state profile of ALS astrocytes and suggest a new mechanism of action of VS as upregulating miR-146a in defective mSOD1 cortical astrocytes.
Anti-miR-146a Transfection in WT Astrocytes Reproduces mSOD1 Cortical Astrocyte Aberrancies, Which Are Prevented by VS
To explore if the efficacy of VS in counteracting the aberrant profile of astrocytes was mediated through its direct action on miR-146a, we downregulated miR-146a expression in astrocytes isolated from the brain cortex of B6SJLF1/J non-transgenic WT mice. We confirmed that the transfection of cortical astrocytes with anti-miR-146a, after being isolated from 7-day-old WT mice and cultured for 13 days, led to a 2-fold decrease in the expression of miR-146a (Figure 3A, p < 0.01). In conformity, IRAK1 and TRAF6 gene expression levels were overexpressed (Figures 3B,C). Such immune deregulation was translated into the typical astrocyte ALS fingerprint, i.e., low GFAP protein and increased S100B, vimentin and Cx43 mRNAs (Figures 3D–H), as observed in mSOD1 cortical astrocytes.
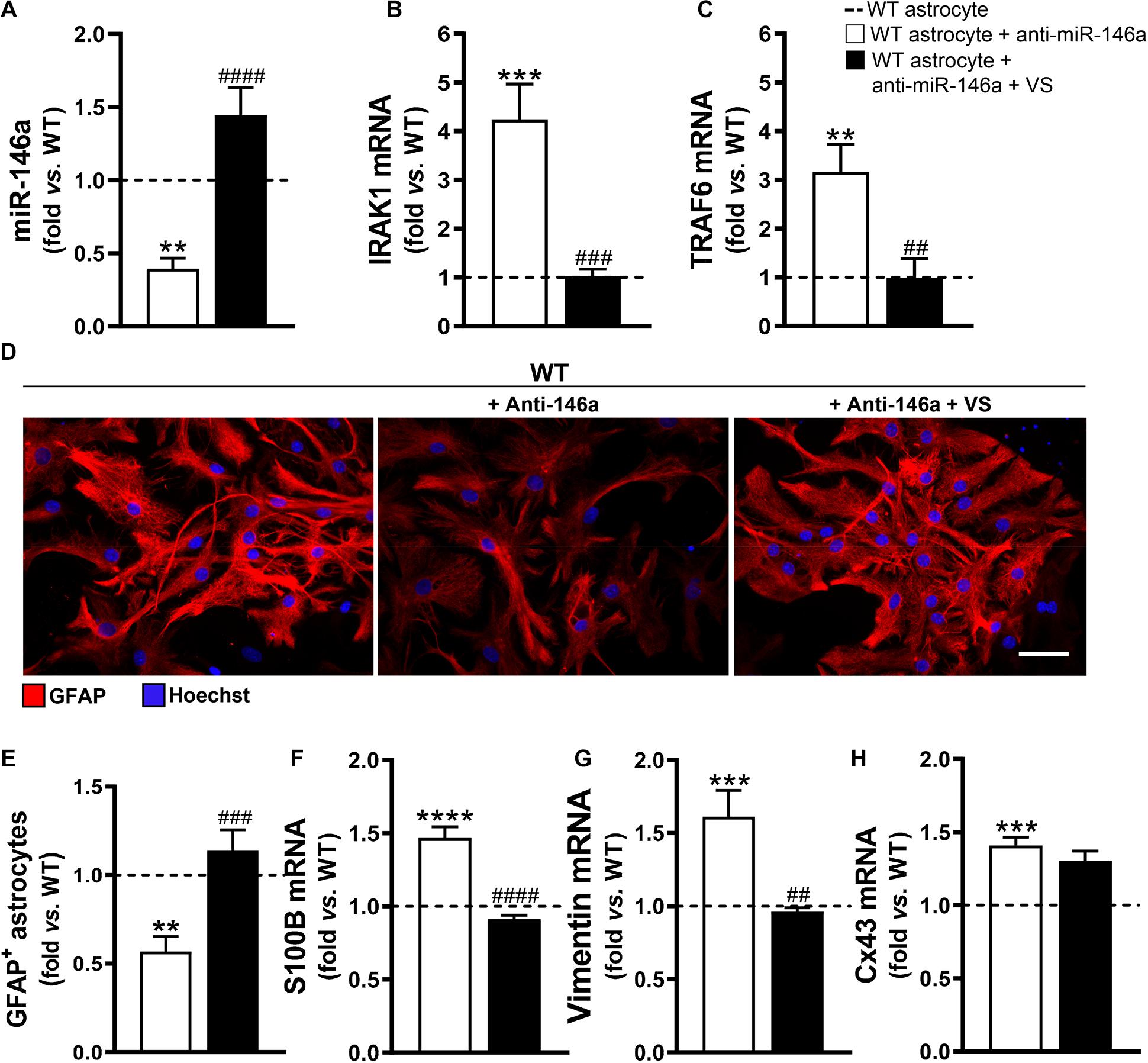
Figure 3. VS rescues GFAP levels and abolishes vimentin- and S100B-associated reactivity induced by the miR-146a inhibitor in WT astrocytes. Astrocytes were isolated from the cortex of wild type (WT) mice with 7-day-old and cultured for 13 days in vitro. Transfection with anti-miR-146a followed by treatment with dipeptidyl vinyl sulfone (VS) was performed in these cells. RT-qPCR analysis of (A) miRNA(miR)-146a, (B) interleukin-1 receptor associated kinase-1 (IRAK1), (C) TNF receptor associated factor 6 (TRAF6), (F) S100 calcium-binding protein B (S100B), (G) vimentin and (H) connexin-43 (Cx43) was performed. SNORD110 was used as reference gene for (A) analysis and β-actin for (B,C,F–H) analysis. (D) Representative images of astrocytes stained with glial fibrillary acidic protein (GFAP, red) in red by immunocytochemistry and (E) respective quantification of the GFAP-positive cells. Cell nuclei were stained with Hoechst dye (blue). Results are mean (±SEM) fold change vs. untreated WT astrocytes from at least three independent experiments. **p < 0.01, ***p < 0.001, and ****p < 0.0001 vs. untreated WT astrocytes; ##p < 0.01, ###p < 0.001, and ####p < 0.0001 vs. WT astrocytes treated with anti-miR-146a. One-way ANOVA followed by Bonferroni post hoc test was used. Scale bar represents 20 μm.
Then, we wonder whether VS would be able to invert the signature of ALS cells produced by miR-146a inhibition. Addition of VS was in fact able to restore the WT astrocyte profile even after the anti-miR-146a transfection. Indeed, WT astrocytes co-treated with VS and anti-miR-146a presented the steady-state signature of healthy cells for miR-146a and its targets IRAK1 and TRAF6 gene expression levels (Figures 3A–C), as well as for GFAP, S100B and vimentin (Figures 3D–G). Interestingly, VS was not as effective as pre-miR-146a in abrogating the elevated levels of Cx43 derived from the anti-miR-146a treatment in WT astrocytes.
Data suggest that miR-146a downregulation is associated with a pathological ALS astrocyte signature, which can be rescued through immunomodulation with pre-miR-146 or by addition of VS, reinforcing depleted miR-146a as a promising target for the restoration of cell homeostasis in the brain cortex.
VS Counteracts High Intracellular Ca2+ in mSOD1 Astrocytes, but Only Pre-miR-146a Prevents Enhanced Frequency and Amplitude of Glutamate-Induced Ca2+ Transients
Among the several toxic factors secreted by mSOD1 astrocytes is the excess of Ca2+ release that was found to contribute to the pathogenesis of ALS (Kawamata et al., 2014). Though that Ca2+ signals are fundamental for the regulation of intracellular signaling, as well as for vesicular secretion processes in astrocytes, their acute and chronic changes in response to brain injury were associated to dysregulated astrocyte-neuron communication (Vardjan et al., 2017). Evidence showed that aberrant Ca2+ signals in reactive astrocytes are implicated in the onset, progression and severity of neurodegenerative diseases (Shigetomi et al., 2019). Here, we intended to explore the intracellular calcium dysregulation in ALS astrocytes and the ability of pre-miR-146a and VS to prevent such abnormality and restore the neuroprotective profile of astrocytes.
We evaluated intracellular Ca2+ dynamics by live-cell imaging using the fluorescent dye Fura-2. Ca2+ signaling was measured without any stimulation (baseline) or in response to glutamate addition, which is a trigger of free cytoplasmic Ca2+ oscillations (Cornell-Bell et al., 1990) and known to be extracellularly elevated in ALS (Yamanaka and Komine, 2018).
Ca2+ signaling was assessed by the fluorescence intensity response of Fura-2 after consecutive stimulation at 340 and 380 nm and quantified in individual cells the ratio between the two responses (R340/380) (Figure 4 and Supplementary Videos 1–4). mSOD1 astrocytes revealed a significant increase of the basal Fura-2 340/380 ratio in (p < 0.01) when compared with matched vs. WT astrocytes (Figures 4A,B,E,F and Supplementary Videos 1, 2). Treatment with VS restored the WT values (p < 0.0001), but not pre-miR-146a upregulation (Figures 4A,B,G,H and Supplementary Videos 3, 4, respectively).
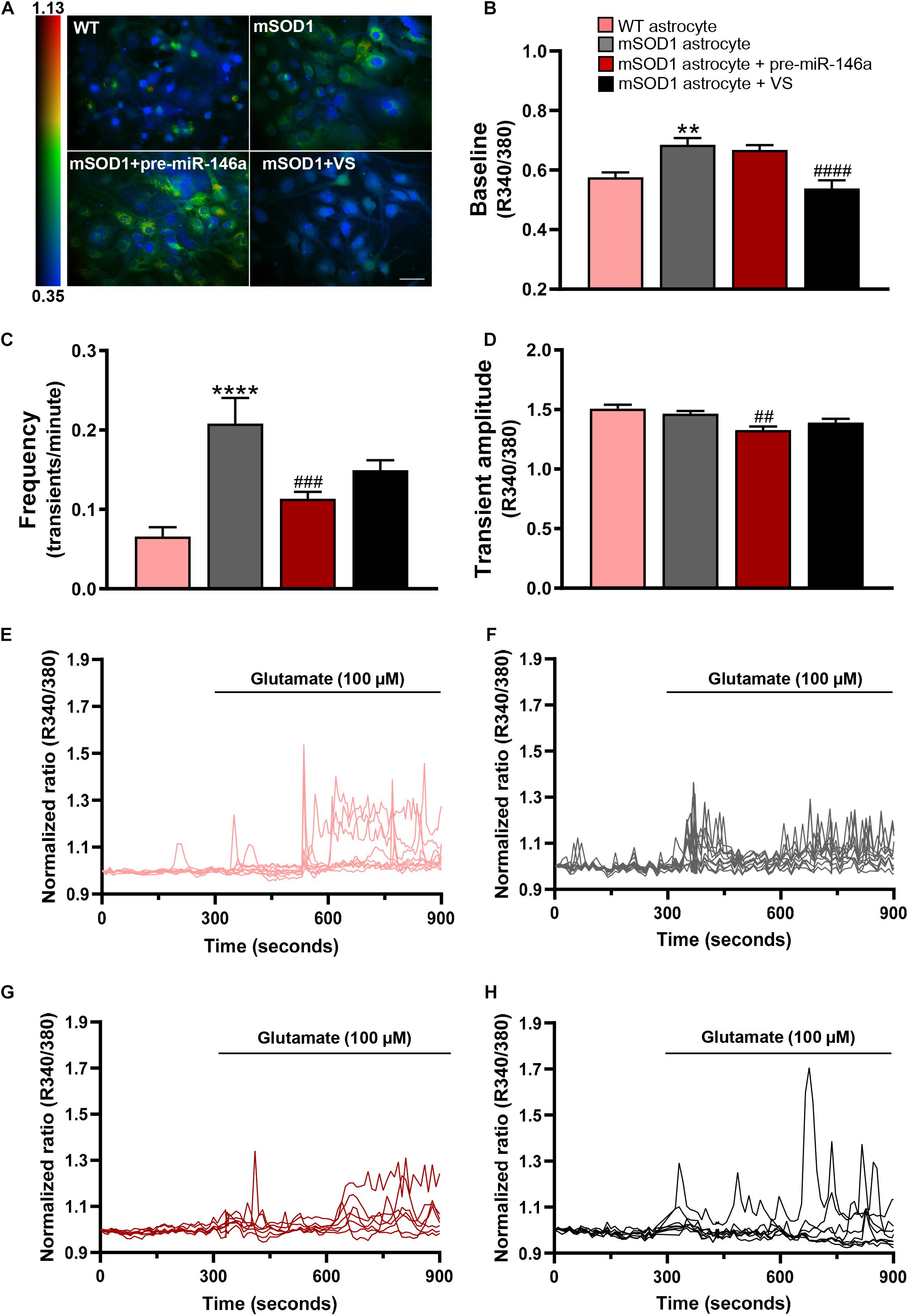
Figure 4. VS reduces the upregulated intracellular Ca2+, while pre-miR-146a normalizes the number and amplitude of glutamate-induced Ca2+ transients in mSOD1 astrocytes. Astrocytes were isolated from the cortex of SOD1-G93A (mSOD1) and wild type (WT) mice with 7-day-old and cultured for 13 days in vitro. Transfection with pre-miR-146a or treatment with dipeptidyl vinyl sulfone (VS) was performed in mSOD1 astrocytes. Cells were incubated at 37°C for 45 min with the calcium (Ca2+) sensitive fluorescent dye fura-2 acetoxymethyl ester (Fura-2), followed by glutamate addition (100 μM). (A) Pseudocolored Fluo4 fluorescence images in WT, untreated and treated-mSOD1 astrocytes show a prominent rise in intracellular Ca2+ and respective (B) changes in the baseline of Fura-2 fluorescence. The color code refers to the fluorescence ratio 340 nm/380 nm, with higher ratio reflecting higher intracellular Ca2+. (C) Summary plot of the frequency of transients per minute and (D) the amplitude of the Ca2+ responses. Results are mean (±SEM) from at least 40 responsive cells from six independent experiments. Representative profiles of normalized Ca2+ responses of at least 8 cells in (E) WT astrocytes and (F) mSOD1 astrocytes treated with (G) pre-miR-146a and (H) VS. The first 300 s represents the changes of Fura-2 fluorescence in the baseline. The remaining 600 s refers to changes after glutamate addition. **p < 0.01 and ****p < 0.0001 vs. WT astrocytes; ##p < 0.01, ###p < 0.001, and ####p < 0.0001 vs. untreated mSOD1 astrocytes. One-way ANOVA followed by Bonferroni post hoc test was used. Scale bar represents 20 μm.
Treatment of astrocytes with 100 μM glutamate produced a higher frequency of Ca2+ transients in mSOD1 astrocytes, as compared with the WT astrocytes (Figures 4C,E,F, p < 0.0001 and Supplementary Videos 5, 6). In the pre-miR-146a transfected mSOD1 astrocytes the addition of glutamate prevented the increased number of transient Ca2+ signaling events observed in the non-modulated cells (Figures 4C,G, p < 0.001 and Supplementary Video 7). A similar finding was observed in the amplitude of Ca2+ waves, where a small, but significant decrease occurred in glutamate-treated mSOD1 astrocytes previously transfected with pre-miR-146a, but not with VS (Figures 4D,H, p < 0.01 and Supplementary Video 8), supporting a delayed response of Ca2+ release in the mSOD1 astrocytes upregulated for miR-146a.
Results suggest that VS and pre-miR-146a differently regulate intracellular Ca2+ load. In the absence of increased extracellular glutamate, VS is a more promising strategy in sustaining intracellular Ca2+ homeostasis. However, if excitotoxity is predicted to occur, then pre-miR-146a treatment has advantage over the use of VS to control Ca2+ dynamic dysregulation.
Overexpression of Pre-miR-146a in mSOD1 Astrocytes Is Recapitulated in Their Derived sEVs
Besides the paracrine influence of the Ca2+ signals derived from reactive astrocytes, it is currently accepted that other components of the astrocytic secretome contribute to neurotoxicity and microglia activation in ALS, mainly due to astrocytic-derived sEVs and their content in miRNAs and misfolded/mutated proteins (Silverman et al., 2016; Ferrara et al., 2018; Varcianna et al., 2019). Recently, we have shown that astrocytes isolated from the SC and the brain cortex of mSOD1 mouse pups are depleted in miR-146a, miR-155 and miR-21 (Gomes et al., 2020).
In this sense, we next assessed if the sEVs isolated from the mSOD1 astrocyte were depleted in miR-146a and whether the treatment of the cells with pre-miR-146a or VS was able to enrich the sEVs in such miRNA. We first characterized the sEVs for the presence of characteristic proteins such as Alix and Flotillin-1 (Figure 5A) and confirmed their cup-shape morphology by transmission electron microscopy (TEM) (Figure 5B), proving the efficiency of sEV isolation. Next, we attested that the transfection of the astrocytes with pre-miR-146 did not modified the number, size, or the concentration of sEVs (Figures 5C–E), relatively to those from WT and mSOD1 astrocytes. Interestingly, when assessed for miR-146a cargo, we observed a 22-fold increase of miR-146a in sEVs derived from the pre-miR-146a-treated astrocytes (Figure 5F, p < 0.01). In contrast, despite the ability of VS to increase the intracellular levels of miR-146a in the depleted mSOD1 astrocytes, it was unable to induce its integration in the secreted sEVs from VS-treated astrocytes, as compared with the untreated ones (Figure 5G).
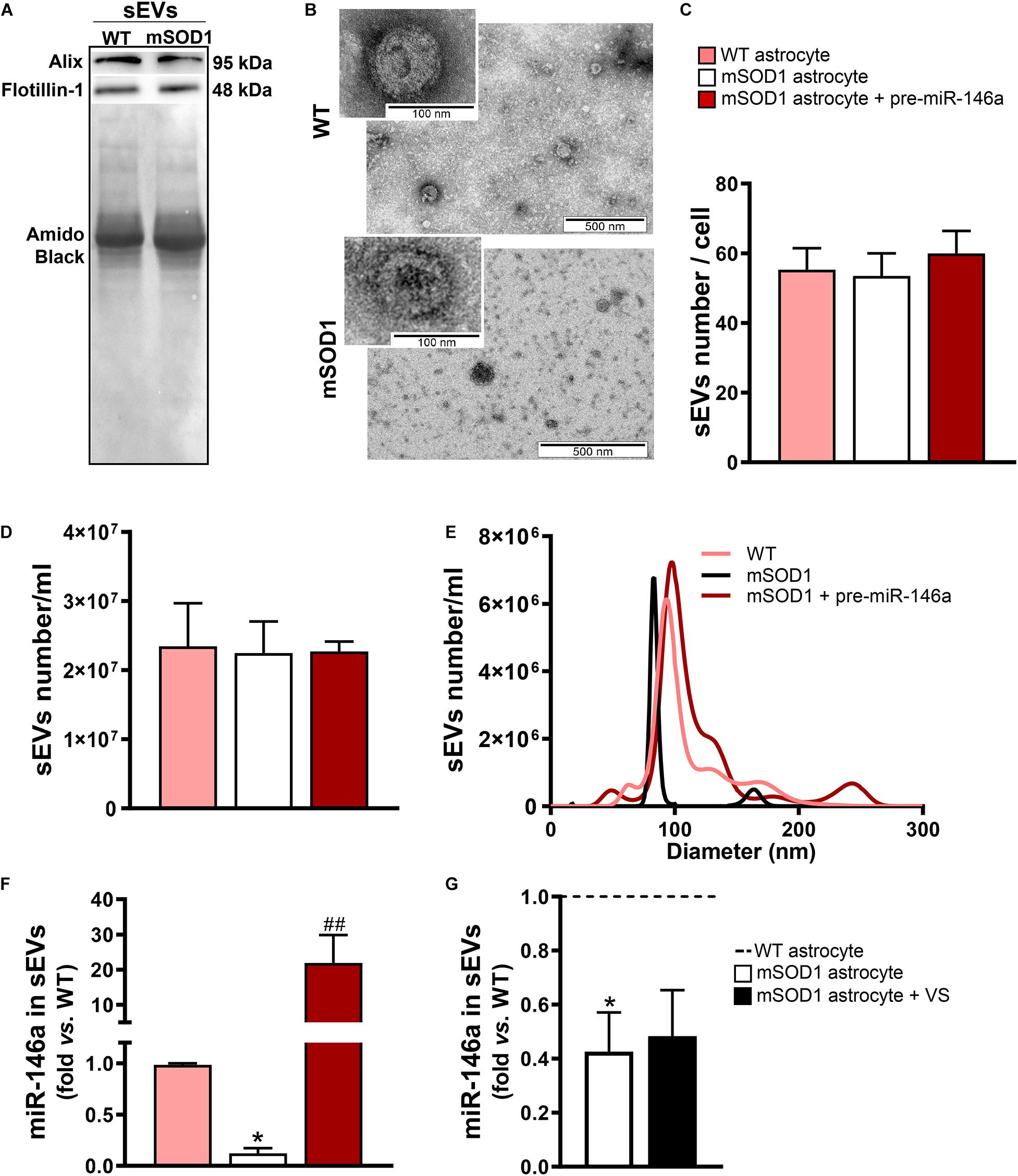
Figure 5. sEVs derived from pre-miR-146a-treated mSOD1 astrocytes show enriched content in miR-146a, but not those from VS-treated cells. Astrocytes were isolated from the cortex of SOD1-G93A (mSOD1) and wild type (WT) mice pups at 7-day-old and cultured for 13 days in vitro. Transfection with pre-miR-146a and treatment with dipeptidyl vinyl sulfone (VS) were performed in mSOD1 astrocytes. Small extracellular vesicles (sEVs) were isolated from the secretome of astrocytes by differential ultracentrifugation. (A) Results from one blot shows the expression of sEV markers (Alix and Flotillin-1). (B) Representative images obtained by transmission electron microscopy of sEVs show their cup shape morphology. Results from (C) number of sEVs per cell, (D) concentration (sEVs number/mL), and (E) size distribution derived from Nanoparticle Tracking Analysis using NanoSight. (F,G) RT-qPCR analysis of miRNA(miR)-146a expression in sEVs was performed. Spike and SNORD were used as endogenous controls. Results are mean (±SEM) fold change vs. sEVs-derived WT astrocytes from at least three independent experiments. *p < 0.05 vs. sEVs from WT astrocytes; ##p < 0.01 vs. sEVs from untreated mSOD1 astrocytes. One-way ANOVA followed by Bonferroni post hoc test was used.
From the data obtained we may conclude that only pre-miR-146a treatment of mSOD1 astrocytes has beneficial consequences in the active transfer of this miRNA into sEVs, thus re-establishing paracrine signaling to recipient cells, and effects in their target genes. We cannot, however, disregard that VS can also have positive influence on the neighboring cells through astrocyte-derived unidentified soluble factors.
Neuroprotective Strategies of mSOD1 Astrocytes by Pre-miR-146a or VS Immunomodulation Involve Distinct Signaling Pathways
Once mSOD1 astrocytes were shown to release neurotoxic factors that lead to MN demise (Nagai et al., 2007; Gomes et al., 2019, 2020), we next decided to explore whether the effects produced by pre-miR-146a or VS modulation in the cells would translate into a more neuroprotective secretome.
To explore this issue, we used WT MNs given the reasons explained in the Materials and Methods section, including our expertise with these cells and previous data (Vaz et al., 2015; Pinto et al., 2017; Gomes et al., 2019). We observed that the increase of miR-146a in the WT MNs was only achieved by the secretome from the mSOD1 astrocytes overexpressing miR-146a (Figure 6A, p < 0.001). Despite the upregulation of miR-146a in the mSOD1 astrocytes treated with VS (Figure 1A), miR-146a was not enriched in sEVs isolated from the secretome of VS-treated mSOD1 astrocytes (Figure 5G), or in WT MNs treated with the secretome from VS-treated mSOD1 astrocytes (Figure 6A). These findings suggest that soluble miR-146a may have accounted for its rise in MNs, an issue that deserves future studies for clarification.
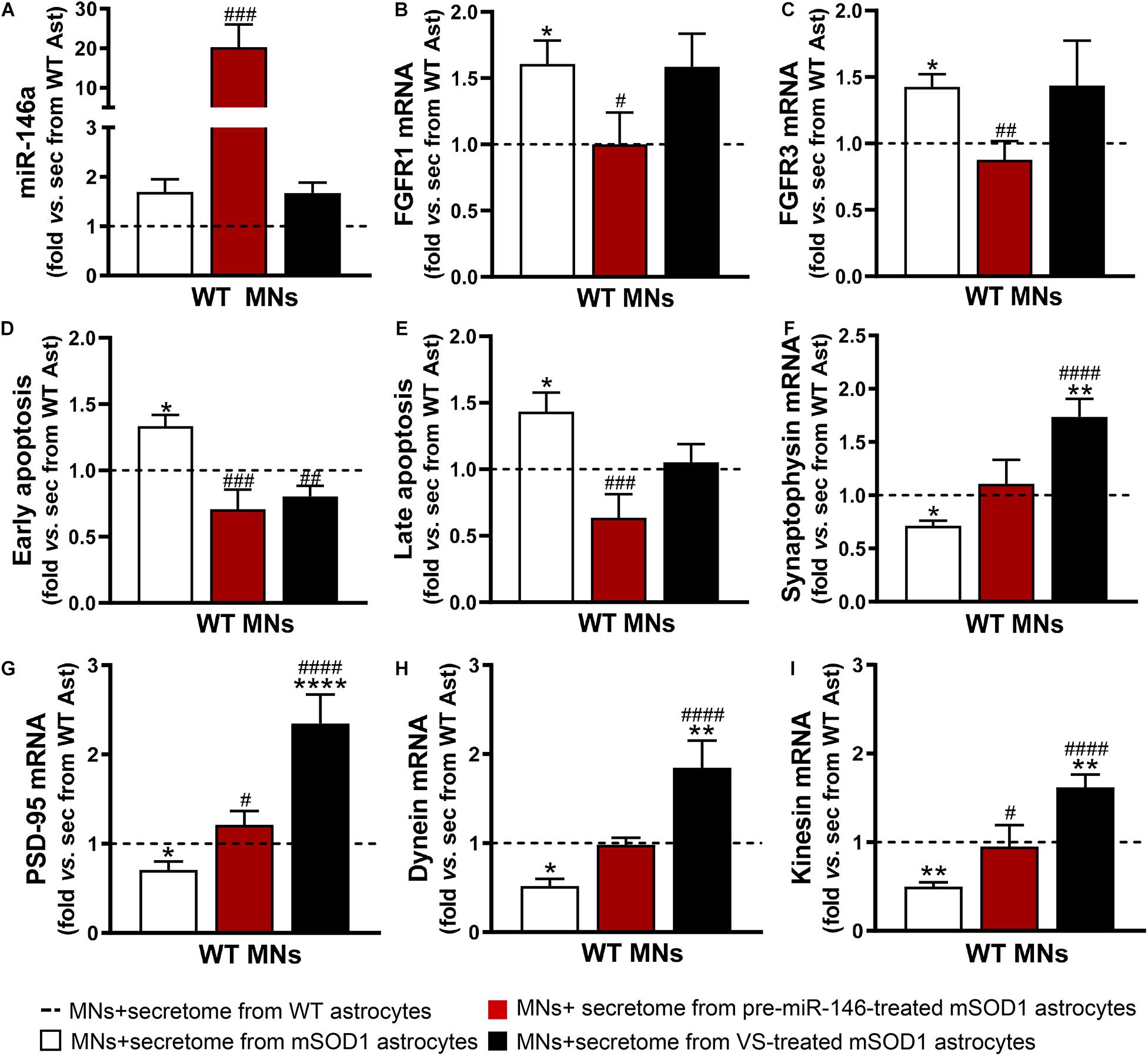
Figure 6. Both VS and pre-miR-146a treatment are effective in restoring the neuroprotective profile of mSOD1 astrocytes but reveal distinct benefits. Astrocytes (Ast) were isolated from the cortex of SOD1-G93A (mSOD1) and wild type (WT) mice with 7-day-old and cultured for 13 days in vitro. Transfection with pre-miR-146a or treatment with dipeptidyl vinyl sulfone (VS) was performed in mSOD1 astrocytes. Cell-derived secretome (sec) was incubated in WT NSC-34 motor neurons (MNs) for 48 h. Analysis of (A) miRNA(miR)-146a, (B) fibroblast growth factor receptor 1 (FGFR1), (C) FGFR3, (F) synaptophysin, (G) post-synaptic protein 95 (PSD95), (H) dynein, and (I) kinesin in WT MNs were assessed by RT-qPCR. SNORD110 was used as reference gene for (A) analysis and β-actin for (B,C,F–I) analysis. (D) Early apoptotic cells (annexin V-PE positive and 7-AA negative) and (E) late apoptotic/necrotic cells (annexin V-PE and 7-AA positive) were assessed by Guava Nexin® Reagent in the WT MNs after secretome interaction. Results are mean (±SEM) fold change vs. MNs + secretome from WT astrocytes from at least three independent experiments. ∗p < 0.05, ∗∗p < 0.01, and **** p < 0.0001 vs. MNs + secretome from WT astrocytes; #p < 0.05, ##p < 0.01, ###p < 0.001, and ####p < 0.0001 vs. MNs + secretome from mSOD1 astrocytes. One-way ANOVA followed by Bonferroni post hoc test was used.
Expression of the fibroblast growth factor receptor 1 (FGFR1) and 3 (FGFR3) were found in neurons and associated to major roles during neurodevelopment, namely in dendritogenesis (Huang et al., 2017; Okada et al., 2019). We found that the secretome from mSOD1 astrocytes induced the overexpression of both receptors in MNs (Figures 6B,C). When modulated, while that from VS-treated mSOD1 astrocytes did not cause any alteration, the secretome from pre-miR-146a-treated mSOD1 astrocytes sustained their levels close to WT ones, a finding not achieved with VS. Transfection of mSOD1 astrocytes with pre-miR-146a also prevented MN death by early and late apoptosis processes (Figures 6D,E, p < 0.05) induced by the secretome from the non-modulated pathological cells. Only the early apoptosis was prevented by the secretome from mSOD1 astrocytes previously treated with VS (p < 0.01).
As depicted in Figures 6F–I, secretome from mSOD1 astrocytes decreased pre- and post-synaptic proteins (synaptophysin and PSD-95, respectively), as well as proteins involved in anterograde and retrograde axonal transport (kinesin and dynein, respectively) (p < 0.05 for all). In this case, major benefits were obtained by the VS treatment that markedly upregulated the expression of all these genes (p < 0.001), though the secretome from pre-miR-146a-treated mSOD1 astrocytes reversed the deficient PSD-95 gene expression and that of the anterograde transport motor kinesin (p < 0.05). To be sure that the effects observed in MNs were due to the secretome from the VS-treated astrocytes and not to the presence of the own compound in the secretome, we evaluated the expression of PSD-95, kinesin and miR-146a in MNs exposed to the media from untreated and VS-treated WT astrocytes. Results showed no differences between such conditions (Supplementary Figure 3). Overall, our findings evidence that the treatment of mSOD1 astrocytes with VS prevents the release of toxic factors involved in the loss of MN synaptic and axonal dynamics.
Since we have observed an increase of miR-146a in the WT MNs treated with the secretome from the mSOD1 astrocytes transfected with pre-miR-46a, we next performed proteomic analysis to explore some of its potential targets in these cells and better understand related effects. We found one upregulated and 4 downregulated hits (Figure 7A and Supplementary Table 5). The first was associated to the induction of the pyruvate dehydrogenase complex encoded by Dlat (Goguet-Rubio et al., 2016) that enhances the production of ATP with benefits in preventing MN energetic stress that characterizes ALS (Vandoorne et al., 2018). To that it may have accounted the inhibition of the protein encoding for Atpif1 that also favors the ATP hydrolysis and ameliorates ALS MN mitochondrial dysfunctionalities (Li et al., 2010; Chen et al., 2014). These potential benefits may be increasingly sustained by the downregulation of FtsJ RNA 2′-O-Methyltransferase 1 (Ftsj1), known to reduce apotosis (He et al., 2020). Another important potential target was the inhibition of the Solute Carrier Family 2 (Facilitated Glucose Transporter) Member 13 protein encoded by Slc2a13, recently identified a risk factor in frontotemporal dementia (Sirkis et al., 2019). Its silencing was shown to diminish APP processing (Teranishi et al., 2015) and may impact on the crosstalk between APP and mSOD1, which seems to contribute to ALS pathology (Rabinovich-Toidman et al., 2015). Also, adding to a better MN functionality is the decrease of RNA-binding protein with multiple splicing encoded by the Rbpms gene that favors synaptogenesis and may be associated with the increased PSD-95 that we observed (Hornberg et al., 2013).
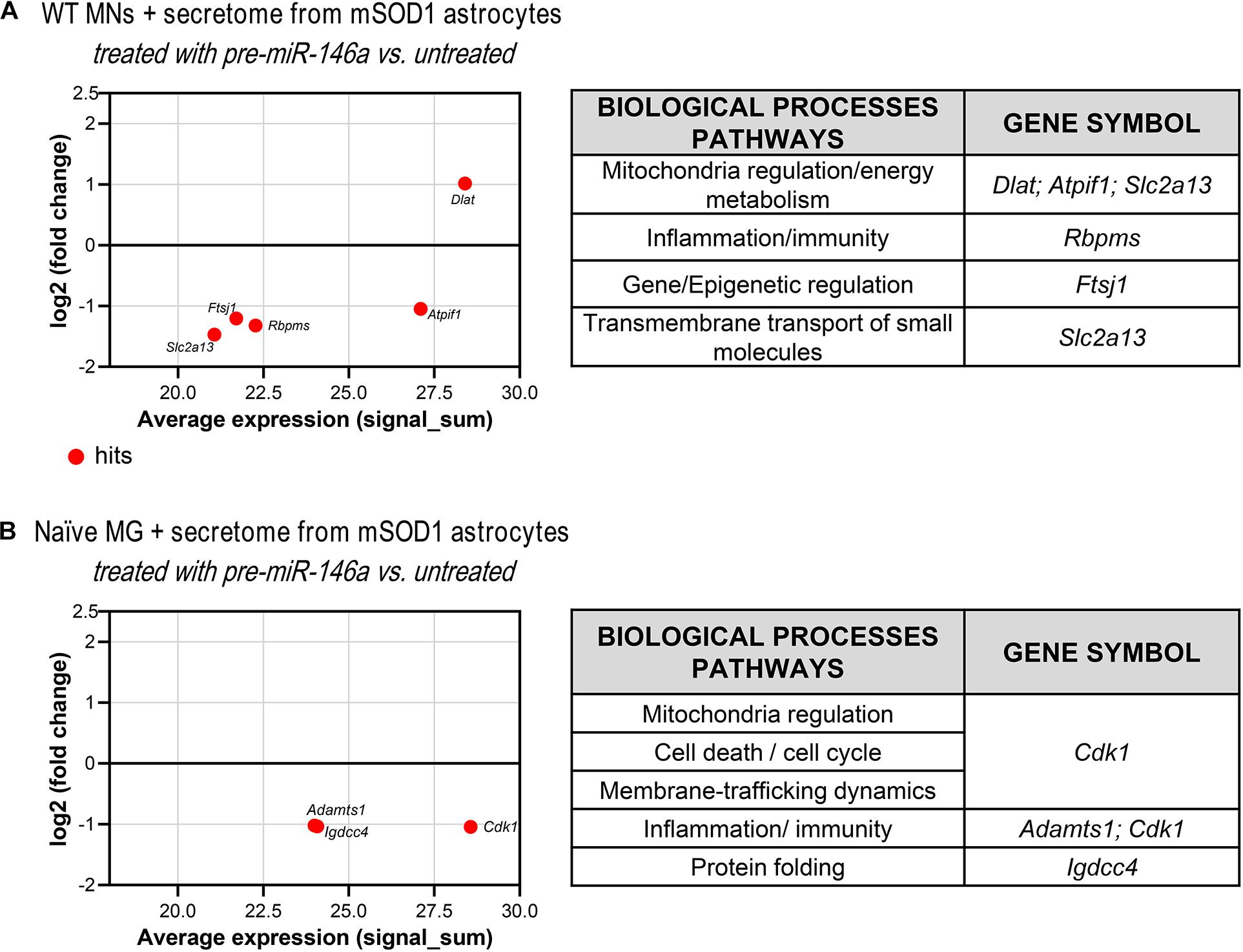
Figure 7. Proteomic analysis reveals that treatment of mSOD1 astrocytes with pre-miR-146a reverts MN dysfunction and microglia activation by paracrine mediators. Astrocytes were isolated from the cortex of SOD1-G93A (mSOD1) mice with 7-day-old and cultured for 13 days in vitro. Transfection with pre-miR-146a was performed. Cell-derived secretome was added to (A) WT NSC-34 motor neurons (MNs) for 48 h or (B) naïve N9 microglia for 24 h. MA plots (left panels) were obtained by comparison of the proteomic profiles of the treatment with the secretome of pre-miR-146a treated mSOD1 astrocytes and the untreated ones. Here, x axis is the mean log2 intensity (fold change) of each protein between two profiles (A-value) and y axis is the average expression (signal_sum) of protein intensities (M-value). Summary tables (right panels) show the obtained hits and their classification according to biological processes using PANTHER Classification System as indicated in methods.
These results clearly show distinct beneficial effects by the secretome from VS-treated astrocytes with protective properties on synaptic dynamics and axonal transport, while those depending on pre-miR-146a treated astrocytes were predominantly related with neuronal survival, miR-146a enrichment and regulation of FGFR1 and FGFR3. As far as we know, this is the first time that mSOD1 astrocytes are described as showing an elevation of such fibroblast growth factor receptors.
Secretome From mSOD1 Astrocytes Transfected With Pre-miR-146a Shows to Better Preserve Microglia Healthy State Than the Treatment With VS
Besides affecting MNs, astrocytes also release soluble factors that influence microglia activation, but such signaling events in the context of ALS disease are still unclarified.
It is known that the expression of FGFR3 in microglia has been linked to enhanced microglial migration and phagocytosis of neuronal debris (Noda et al., 2014). However, inappropriate phagocytosis of live neurons and excessive neuronal loss, termed phagoptosis (Brown and Neher, 2014), may have detrimental consequences on the patient outcome. Similarly, to our findings on the effects produced by the secretome from mSOD1 astrocytes on WT MNs, naïve N9 microglial cells showed increased gene expression of FGFR3 (Figure 8B). Both VS and pre-miR-146a immunomodulation of mSOD1 astrocytes were effective in reducing such expression (p < 0.001). Further results showed that the secretome of mSOD1 astrocytes induced early and late apoptosis (Figures 8C,D, at least p < 0.05) that were prevented when the cells were treated with pre-miR-146a or VS (only early apoptosis). As expected, the secretome from mSOD1 astrocytes also activated and polarized microglia to overexpress iNOS (Figure 8E, p < 0.01) and TNF-α mRNAs (Figure 8F, p < 0.01). Only the treatment of mSOD1 astrocytes with pre-miR-146a showed to be efficient in counteracting the expression of such inflammatory mediators and in switching microglia into their steady-state (p < 0.01).
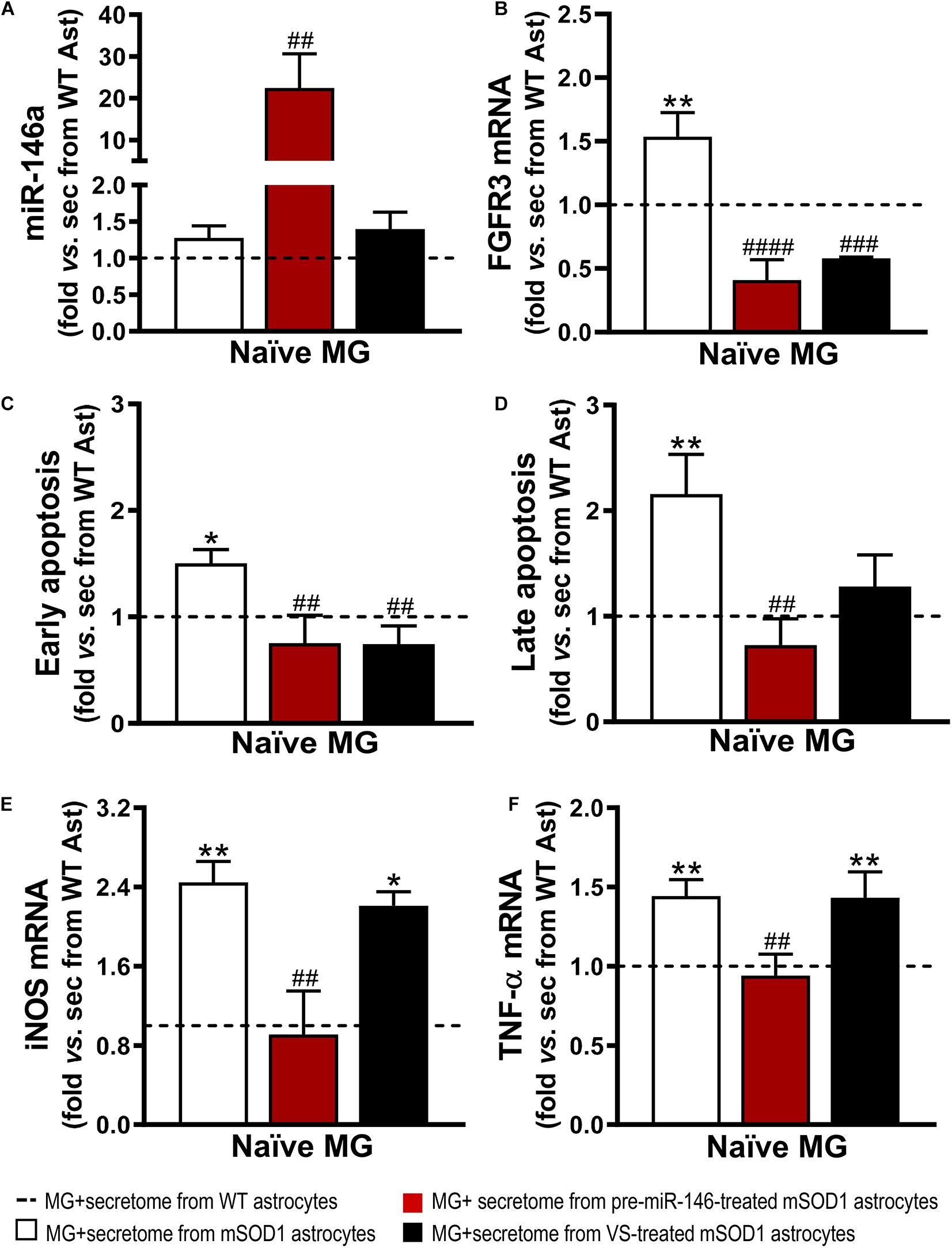
Figure 8. Secretome from pre-miR-146a-treated mSOD1 astrocytes translates into microglial miR-146 increase and regulation of cell activation by the untreated secretome, while also prevents cell demise similarly to VS modulation. Astrocytes (Ast) were isolated from the cortex of SOD1-G93A (mSOD1) and wild type (WT) mice with 7-day-old and cultured for 13 days in vitro. Transfection with pre-miR-146a or treatment with dipeptidyl vinyl sulfone (VS) was performed in mSOD1 astrocytes. Secretome was isolated and incubated in naïve N9 microglia for 24 h. Expression of (A) miRNA(miR)-146a, (B) fibroblast growth factor receptor 3 (FGFR3), (E) inducible nitric oxide synthase (iNOS) and (F) tumor necrosis alpha (TNF-α) in microglia was assessed by RT-qPCR. SNORD110 was used as reference gene for (A) analysis and β-actin for (B,E,F) analysis. (C) Early apoptotic (Annexin V-PE positive and 7-AAD negative), and (D) late apoptotic/necrotic cells (Annexin V-PE and 7-AAD positive) were assessed by Guava Nexin® Reagent in the microglia after secretome interaction. Results are mean (±SEM) fold change vs. MG + secretome from WT astrocytes from at least three independent experiments. *p < 0.05 and **p < 0.01 vs. MG + secretome from WT astrocytes; ##p < 0.01, ###p < 0.001, and ####p < 0.0001 vs. MG + secretome from mSOD1 astrocytes. One-way ANOVA followed by Bonferroni post hoc test was used.
As before, and since we found an overexpressed miR-146a in the microglia treated with the secretome from pre-miR-146a mSOD1 astrocytes, we proceeded to proteomic analysis to investigate how the potential targets related with the improved effects we observed. Downregulation of the cyclin-dependent kinase 1 levels (encoded by Cdk1) may have contributed to the regulation of iNOS and TNF-α in the microglia treated with the secretome from mSOD1 astrocytes relatively to the untreated one (Figure 7B and Supplementary Table 6). Actually, increase of iNOS and TNF-α is associated to microglia activation and neuroinflammation (Sheng et al., 2011) and CDK inhibitors were shown to reduce them in several conditions (Leitch et al., 2009; Skovira et al., 2016). As for the decrease in the metalloproteinase with thrombospondin motifs 1, encoded by the gene Adamts1, it was suggested to be a modulator of the immune cell response in tumors (Rodriguez-Baena et al., 2018), and found elevated in LPS-activated microglia (Chiu et al., 2013). In what concerns the immunoglobulin superfamily DCC subclass member 4, encoded by Igdcc4 and accepted as a biomarker of hepatocellular carcinoma (Zweerink et al., 2020) and of innate immune/inflammatory response (Sanz-Pamplona et al., 2014), it should be explored in the future as part of the recovering microglia steady-state profile by pre-miR-146a in ALS cortical astrocytes through paracrine signaling.
In sum, both pre-miR-146a and VS modulatory effects on ALS astrocytes prevented secretome-mediated microglial demise and FGFR3 increase. However, the transfection of the mSOD1 astrocytes with pre-miR-146a had a higher efficiency in preventing the microglial elevation of iNOS and TNF-α gene expression levels than the VS treatment. We next investigated if the effects observed on the microglia deactivation by the secretome from the mSOD1 astrocytes treated with pre-miR-146a were mediated by sEVs, previously shown to be enriched in this miRNA in the section “Overexpression of Pre-miR-146a in mSOD1 Astrocytes Is Recapitulated in Their Derived sEVs.”
sEVs From mSOD1 Astrocytes Treated With Pre-miR-146a Are Preferentially Captured by Microglia, Enhancing Its Intracellular Upregulation, and Together With VS Counteract Cell Activation by the Pathological sEVs
Misfolded and mutated SOD1 were shown to be propagated intercellularly by sEV-dependent and independent mechanisms (Grad et al., 2014), and to have impact on astrocyte protein secretion pathways, thus accounting to MN pathology and dissemination (Basso et al., 2013). We have previously demonstrated that sEVs from NSC-34 hSOD1G93A MN-like cells cause alterations on N9 microglia polarization (Pinto et al., 2017). However, the effects produced by the secretome of pathological astrocytes on microglia polarization have been scarcely investigated.
Similarly to what we have previously observed for WT MNs, the secretome from pre-miR-146a-treated mSOD1 astrocytes increased the expression of this miRNA on naïve N9 microglial cells (Figure 8A). However, while MNs are not able to collect entire sEVs, microglia easily engulf them, as we showed (Pinto et al., 2017). Therefore, we isolated sEVs from the secretome of mSOD1 astrocytes untreated or treated with either pre-miR-146a or VS, and we investigated the presence of labeled sEVs in the N9 microglial cells 24 h after incubation. As shown in Figures 9A,B no differences in the number of sEVs incorporated by microglia were obtained when isolated from mSOD1 astrocyte secretome relatively to the WT astrocyte one. However, sEVs were found more densely internalized by microglia (p < 0.01) in the case of the pre-miR-146a strategy, than by VS. This feature may have accounted for the miR-146a elevation in the cells (p < 0.001, Figure 9C), due to sEV enriched cargo in miR-146a (Figure 5F).
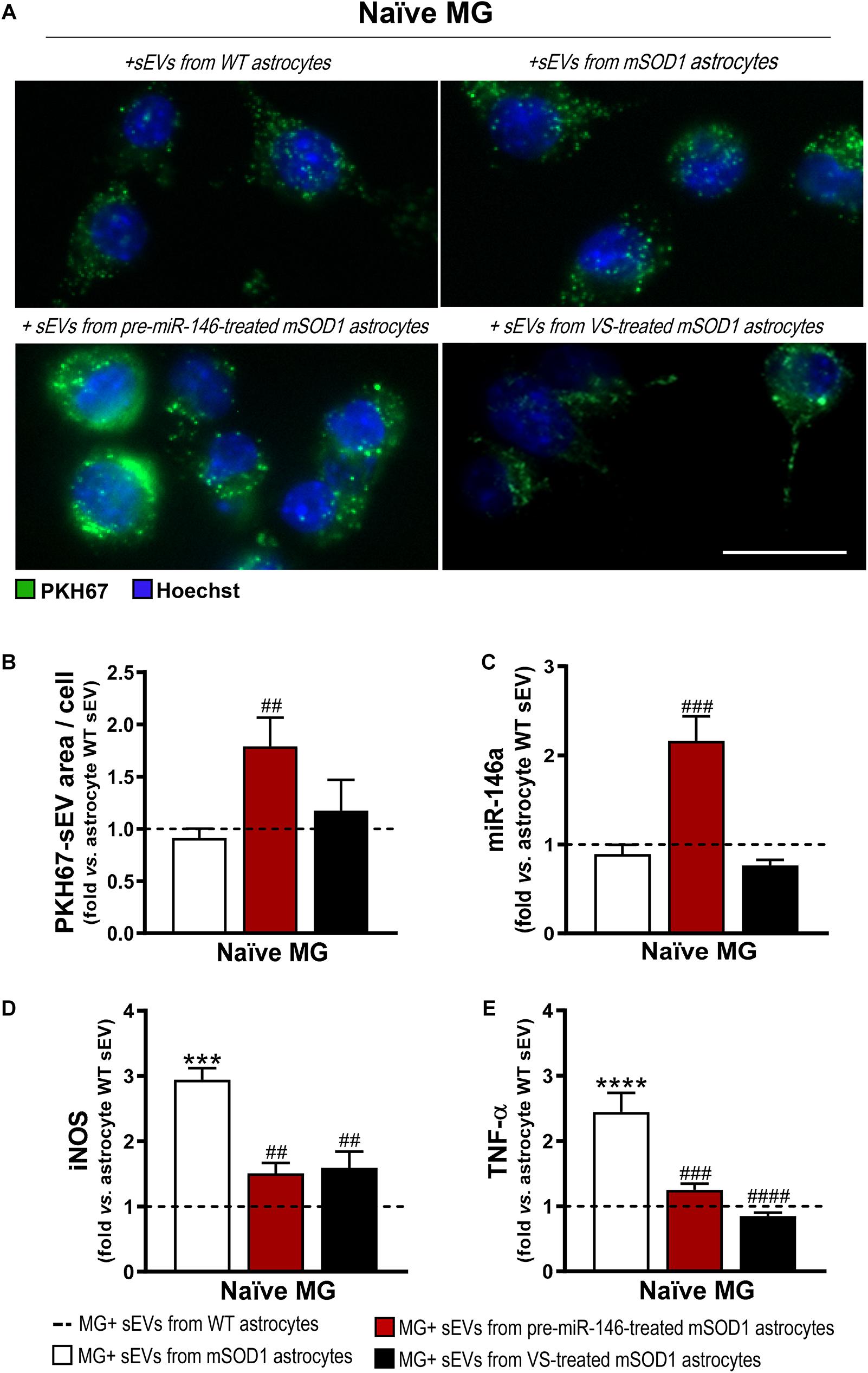
Figure 9. Microglia treated with sEVs from mSOD1 astrocytes modulated with pre-miR-146a or VS show different sEV internalization and miR-146a expression, but similar protection from activation by untreated-sEVs. Astrocytes were isolated from the cortex of SOD1-G93A (mSOD1) and wild type (WT) mice with 7-day-old and cultured for 13 days in vitro. Transfection with pre-miR-146a or VS treatment was performed in mSOD1 astrocytes. Small extracellular vesicles (sEVs) were isolated by differential ultracentrifugation and labeled with PKH67 cell linker, followed by incubation with naïve N9 microglia for 24 h. (A) Representative images of sEVs stained with PKH67 cell linker (green); (B) mean fluorescence area positive for PKH67-sEVs per cell. Cell nuclei were stained with Hoechst dye (blue). Expression of (C) miRNA(miR)-146a, (D) inducible nitric oxide synthase (iNOS) and (E) tumor necrosis alpha (TNF-α) in microglia was assessed by RT-qPCR. SNORD110 was used as reference gene for (C) analysis and β-actin for (D–E) analysis. Results are mean (±SEM) fold change vs. MG + sEVs from WT astrocytes from at least three independent experiments. ***p < 0.001 and ****p < 0.0001 vs. MG + sEVs from WT astrocytes; ##p < 0.01, ###p < 0.001, and ####p < 0.0001 vs. MG + sEVs from mSOD1 astrocytes. One-way ANOVA followed by Bonferroni post hoc test was used. Scale bar represents 20 μm.
Interestingly, both pre-miR-146a and VS treatment in mutated astrocytes produced sEVs able to prevent iNOS (p < 0.01, Figure 9D) and TNF-α (at least p < 0.001, Figure 9E) upregulation relatively to sEVs from untreated mSOD1 astrocytes. This is a very interesting result, if we consider that VS-derived secretome was unable to prevent microglia activation, while its isolated sEVs showed such property.
Data highlight the differential roles of the secretome (ineffective) and of the isolated sEVs (effective) from the VS-treated astrocytes in recovering microglia steady-state, while indicate to be independent of miR-146a content. However, in the case of astrocytes treated with pre-miR-146a, both the secretome and their isolated sEVs, suppressed microglia activation by a mechanism that is suggestive to depend on the paracrine influence of miR-146a expression.
Discussion
Currently, there are no effective therapies for ALS and the recent advances in this field converges in the modulation of the inflammatory and toxic environment by controlling glial activation (Liu and Wang, 2017). Indeed, increased evidences have shown that reactive astrocytes impair the survival of MNs (Díaz-Amarilla et al., 2011; Meyer et al., 2014; Qian et al., 2017; Gomes et al., 2019).
We have recently identified the presence of a deregulated set of reactive markers in mSOD1 cortical astrocytes isolated from 7-day-old mSOD1 mice cultured for 13 DIV, which were similarly present in the symptomatic stage of the same animals (Gomes et al., 2019). The ALS-disease characteristic phenotype included a low GFAP expression, but increased levels of S100B, Cx43, vimentin and HMGB1. By showing the same markers identified in the symptomatic stage of the mSOD1 mice their characteristic fingerprint with low GFAP levels seems to be disease specific. This profile is in agreement with the previous one found in the SC of adult symptomatic mSOD1 rats (Díaz-Amarilla et al., 2011). Astrocyte reactivity is usually associated to elevated levels of GFAP (Eng and Ghirnikar, 1994). Indeed, GFAP was found elevated in the cerebrospinal fluid and in the SC of patients with ALS (Fujita et al., 1998; Benninger et al., 2016), as well as in isolated adult astrocytes using supplementation with growth factors from the mSOD1 mice (Tripathi et al., 2017). However, our data and that of others (Rossi et al., 2008; Díaz-Amarilla et al., 2011; Cunha et al., 2018; Gomes et al., 2020) are consistent with a downregulation of GFAP in the presence of reactive markers that also include increased Ki-67, together contributing to denominate astrocytes in ALS as having an aberrant phenotype. In a recent consensus statement, it is referred that the increase in GFAP expression do not correlate with increased injury, reactive response to pathological stimuli or altered functions of reactive astrocytes (Escartin et al., 2021). In conformity, it should be used instead a combination of molecular markers to qualify astrocytes as reactive and as having a disease-specific signature. In this context, cortical homogenates from the symptomatic mSOD1 mice and isolated astrocytes from such animal pups showed increased reactive astrocyte markers, decreased GLT-1 and GFAP levels, and the pioneering downregulated expression of miR-146a (Gomes et al., 2019, 2020). Such reduction was even present in the presymptomatic stage and could not be found in astrocytes from the spinal cord (Cunha et al., 2018), suggesting regional astrocyte heterogeneity and their different contribution to bulbar and spinal forms of ALS disease. Such findings indicate that different therapeutic approaches may be required, depending on the local of disease onset, and that replenishment of miR-146a in cortical astrocytes may reveal a promising strategy.
Interestingly, miR-146a-enriched stem cell secretome was recently suggested to have proangiogenic and anti-inflammatory properties (Waters et al., 2019). For that reason and given the importance of miR-146a in controlling astrocyte inflammation (Iyer et al., 2012), we decided to test the efficacy of pre-miR-146a and immunomodulatory compounds, such as GUDCA and VS (Falcão et al., 2017; Vaz et al., 2019) in reverting astrocyte reactivity and miR-146a replenishment. Our results demonstrated that VS induced upregulation of miR-146a and downregulation of its targets, while GUDCA did not change such levels. This is in line with our previous studies that demonstrated the ability of VS in modulating miR-146a expression (Falcão et al., 2017; Vaz et al., 2019). Furthermore, GUDCA reduced the expression of vimentin and Cx43, exerting a modulatory role on astrocyte reactivity, in accordance with our previous data (Fernandes et al., 2007). However, VS showed more broader effects, not only by restoring the levels of GFAP and decreasing vimentin and Cx43 expression, but also by downregulating S100B and HMGB1 levels, corroborating its anti-inflammatory properties previously observed in microglial models of ALS (Vaz et al., 2019) and Alzheimer’s disease (Falcão et al., 2017). To note that neither GUDCA nor VS showed to compromise cell viability. Then, we hypothesized if the attenuation of such aberrancy by these compounds might be related with the modulation of miR-146a expression.
To verify such supposition, we restored the normal levels of miR-146a by transfecting mSOD1 astrocytes with pre-miR-146a. Re-establishment of GFAP, vimentin, Cx43, HMGB1 and S100B toward control WT levels revealed its therapeutic potential. Actually, vimentin is not only an intermediate filament protein involved in cellular processes, such as cell adhesion, migration and proliferation, promoting wound healing (Ivaska et al., 2007), but it is also a known marker of immature astrocytes, associated with a reactive gliosis and leading to thicker bundles in the astrocytic processes (Cheng and Eriksson, 2017). Usually, the expression of GFAP replaces vimentin in differentiated astrocytes (Roybon et al., 2013). In the case of the cortical ALS astrocytes data suggest that they sustain an immature-like reactive phenotype. The correction of the low levels of miR-146a seems to contribute to abolish such ALS-disease phenotype of cortical astrocytes. Regulation of vimentin and GFAP levels by pre-miR-146a are in accordance with a study wherein miR-146a overexpression increased GFAP expression and attenuated proliferation, migration and tumorigenic potential of glioma cells (Mei et al., 2011; Xiao et al., 2015). We also found decreased S100B and HMGB1 expression gene levels in pre-miR-146a-treated mSOD1 astrocytes. This is in line with a previous study that suggested the increased astrocytic S100B expression as an early occurring event in ALS and a contributor for neurodegeneration (Serrano et al., 2017). These Authors observed that its inhibition in mSOD1 astrocytes decreased the expression of proinflammatory/reactive genes. Besides, this attenuation of astrocyte-inflammatory potential supports the role of miR-146a as a negative regulator of the NF-κB inflammatory pathway in astrocytes (Taganov et al., 2006; Iyer et al., 2012). Concerning Cx43, it is known to modulate proliferation, migration and differentiation of astrocytes (Homkajorn et al., 2010). Its upregulation was detected in mSOD1 astrocytes, corroborating previous studies in rodent models (Díaz-Amarilla et al., 2011; Cui et al., 2014), and the restoration of Cx43 normal levels in pre-miR-146a-treated mSOD1 astrocytes may associate to its ability to regulate inflammatory response and attenuate gliosis (Mei et al., 2011). Like GUDCA and VS, pre-miR-146a transfection also did not activate early/late apoptosis.
Proteomic analysis showed that pre-miR-146a induced an upregulation of the NCF1 protein that we hypothesize to be an attempt to control the reactive/immune response in mSOD1 astrocytes, as demonstrated in chronic inflammatory disorders (Holmdahl et al., 2016; Zhao et al., 2017). ZBTB10 was found elevated during gliogenesis and linked to ZBTB20 for differentiation and maturation of neocortical astrocytes (Nagao et al., 2016; Araújo, 2019). Moreover, ZBTB10 was also suggested to be associated to DNA damage repair at telomeres (Bluhm et al., 2019). As for HMGN2, it belongs to a family of proteins associated to astrocyte differentiation during development (Nagao et al., 2014). Both ZBTB20 and HMGN2 may be relevant protein targets to be used toward the recovery of the neuroprotective phenotype by the ALS cortical astrocytes which, in our view, deserve to be explored. The observed upregulation of ALS2CL, by regulating the alsin-mediated endosome, might induces sEV formation (Hessvik and Llorente, 2018) and the activation of the autophagy-endolysosomal system (Otomo et al., 2012). Together with the PZP upregulation associated to a sustained extracellular proteostasis and inhibition of misfolded protein accumulation (Cater et al., 2019), they might act together to promote mSOD1 clearance from astrocytes by sEVs and autophagy. Indeed, PZP elevation was found in senile plaques and in glial cells in Alzheimer’s disease, possibly having a protective role (Ijsselstijn et al., 2011; Nijholt et al., 2015).
To assess whether reversal of mSOD1 astrocyte aberrancies by VS was mediated via miR-146a regulation, we depleted its expression in WT astrocytes with a miR-146a inhibitor, before using VS. Anti-miR-146a-transfected WT astrocytes acquired the aberrant phenotype that characterize ALS cortical astrocytes. Interestingly, VS was not only able to restore miR-146a levels in such condition, but also to revert GFAP, S100B and vimentin toward steady-state levels. In sum, our results indicate that targeting downregulated miR-146a, either with pre-miR-146a or VS, constitutes a good strategy to counteract the atypical reactivity of ALS cortical astrocytes associated to the miR-146a depletion.
Given the established contribution of ALS astrocytes on MN death and the possible involvement of astrocytic Ca2+ dysregulation in this process (Kawamata et al., 2014), we investigated its presence and if it was modulated by either pre-miR-146a or VS treatments. Our results revealed a baseline increase of intracellular Ca2+ in mSOD1 astrocytes that was prevented by VS co-incubation, together with elevated frequency of Ca2+ transients after glutamate stimulation, what may impact on synapse integrity (Guerra-Gomes et al., 2017). Astrocytes possess glutamate-sensitive ion channels and can respond to glutamate with a fast and oscillatory elevation of cytoplasmic free Ca2+ by autocrine and paracrine effects (Cornell-Bell et al., 1990). We hypothesize that the excess of Ca2+ in mSOD1 astrocytes may derive from ER stores, as observed in the SC of mSOD1 mice (Kawamata et al., 2014), or be caused by the aberrant Cx43 overexpression (Almad et al., 2016), inducing alterations in neuronal excitability, synaptic transmission and plasticity (Lu et al., 2010). As so, pre-miR-146a was able to reduce the number of [Ca+]i transients in mSOD1 astrocytes and to downregulate Cx43 expression. Previous studies observed the reduction in Ca2+ responses in the presence of a Cx43 blocker in ALS astrocytes (Almad et al., 2016, 2020).
Astrocytes are highly secretory cells, with their secretome containing hundreds of molecules. In ALS, it is accepted that astrocytes have a paracrine contribution to neurodegeneration and microglial activation, but the mechanisms associated are poorly explored (Pehar et al., 2017). As a component of secretome, sEVs mediate the interaction between neurons and glial cells and are responsible for the delivery of proteins, lipids, mRNAs and miRNAs in the recipient cell, being considered potential vehicles for drug delivery (Basso and Bonetto, 2016). Our observation that mSOD1 astrocytes release sEVs with low miR-146a content is in line with our previous findings (Gomes et al., 2020). In the same study, we established that mSOD1 astrocytes contribute to the neuronal degeneration when these cells are co-cultured with ALS MNs. Indeed, mSOD1 astrocytes selectively provoke toxicity to MNs (Di Giorgio et al., 2007; Nagai et al., 2007; Díaz-Amarilla et al., 2011; Haidet-Phillips et al., 2011; Gomes et al., 2019) and their secretome was found to play an important role (Nagai et al., 2007).
We observed that early and late apoptosis were induced by the secretome from the pathological ALS astrocytes, leading to a reduced expression of synaptophysin, PSD-95, dynein, and kinesin in MNs, thus compromising synaptic and axonal dynamics. Synaptophysin, a presynaptic membrane protein, was found reduced in MNs from mSOD1 mice and shown to contribute to motor deficits (Zang et al., 2005). The postsynaptic PSD-95, also important for synaptic plasticity (Zhang and Lisman, 2012), has been found reduced in neurodegenerative diseases, including ALS (Fogarty, 2019). Contribution of ALS astrocytes to synaptic failure is an established feature (Casas et al., 2016). In particular, these cortical mSOD1 astrocytes showed to reduce neurite length and synaptophysin expression in coculture with NSC-34 hSOD1 WT MN-like cells (Gomes et al., 2019). Defects in the anterograde and retrograde transports by kinesin and dynein, respectively, also occur in ALS neurodegeneration in patients and in mSOD1 mice (De Vos and Hafezparast, 2017). Inefficient anterograde transport was shown to reduce the transport of synaptic vesicle precursors, contributing to the decrease in synaptic vesicle density (Yonekawa et al., 1998) that is reflected by the reduced expression of the synaptic proteins, as we observed. Secretome from pre-miR-146a-treated mSOD1 astrocytes, but not from VS-treated ones, restored PSD-95 and kinesin levels found deregulated by the pathological paracrine signaling over WT MNs.
We identified for the first time that FGFR1 and FGFR3 gene expression levels, receptors of fibroblast growth factor-2 (FGF2), increased in MNs upon the secretome from mSOD1 astrocytes. FGF2 is highly produced in reactive astrocytes after injury and ischemia (Reilly and Kumari, 1996; Clarke et al., 2001) and their elevation was noticed in the CSF from ALS patients (Johansson et al., 2003; Ekestern, 2004). The upregulation of FGFR1 and FGFR3 identified in our study by the secretome from mSOD1 astrocytes may contribute to axonal damage involving the ERK/MAPK signaling (Huang et al., 2020). Instead, the secretome derived from pre-miR-146a-treated mSOD1 astrocyte was unique in decreasing the expression of these receptors and together with VS prevented the induced MN apoptosis upon treatment with the pathological astrocyte-derived secretome.
Motor neuron proteomic analysis revealed that pre-miR-146a increases the E2 component of the pyruvate dehydrogenase complex encoded by the Dlat gene (Goguet-Rubio et al., 2016), leading to the production of ATP that is required to continuously provide energy to maintain normal MN function with a high relevance in ALS (Vandoorne et al., 2018). By downregulating ATP1F1, pre-miR-146a may also ameliorate mitochondrial respiratory chain dysfunction because it inhibits ATPase activity (Chen et al., 2014), contributing to sustain ALS MN mitochondrial functionality (Li et al., 2010; Chen et al., 2014), and thus preventing the apoptosis induced by the secretome from pre-miR-146a-treated mSOD1 astrocytes. In conformity, the decreased expression of FTSJ1, as we found, was also associated to the inhibition of apoptosis in cancer (He et al., 2020). It was interesting to observe a reduction of SLCA13, since this transporter was identified as a risk factor for frontotemporal dementia (Sirkis et al., 2019). By being reduced, it prevents APP processing (Teranishi et al., 2015) and the interaction between SOD1 and APP, which are known to lead to synaptic dysfunctions and associated neuroinflammation and neurodegeneration events in ALS (Rabinovich-Toidman et al., 2015). We also found downregulation of RBPMS in MNs following the interaction with the secretome from pre-miR-146a-treated mSOD1 astrocytes. Its transcript was found downregulated in the axonal transcriptome from MNs overexpressing human mSOD1 (Nijssen et al., 2018) and showed to inversely affect synapse density (Hornberg et al., 2013).
VS treatment exceeded the beneficial effects of pre-miR-146a treatment by promoting MN functionality through upregulation of synaptophysin, PSD-95, dynein and kinesin transcripts and by avoiding early cell death by apoptosis. This is in line with a previous study describing vinyl sulfones as neuroprotective agents in the treatment of Parkinson’s disease (Woo et al., 2014; Choi et al., 2019). Increased levels of miR-146a in MNs were observed after treatment with the secretome from the pre-miR-146a-treated mSOD1 astrocytes, but not with that from VS treatment. We may hypothesize that the enriched content of the secretome or their isolated sEVs in miR-146a may have accounted for such MN upregulation and eventually the observed benefits. Indeed, previous studies demonstrated that miR-146a is important for neuronal survival and differentiation (Qu et al., 2019; Fregeac et al., 2020), as well as for axonal growth (Jia et al., 2016). This is not the case of VS, suggesting that its paracrine neuroprotective effect is not exclusively dependent on sEV-derived miR-146a. In contrast, their effectiveness might be related with soluble miR-146a or other neuroprotective factors, an interesting hypothesis to be explored in future studies.
The secretome from mSOD1 astrocytes also switched microglia to a pro-inflammatory subtype by increasing iNOS and TNF-α mRNA expression, in accordance with studies where mSOD1 astrocytes led to the activation of microglia, thus contributing to disease progression (Yamanaka et al., 2008). Moreover, we observed the activation of early/late apoptosis and an increase of FGFR3 expression. Injury was shown to upregulate the FGFR3 expression in both astrocytes and microglia and FGF2 appears to have a dual role, by firstly inducing gliosis and secondly neuroprotection (Goddard et al., 2002). Here, we may speculate that, as observed for damaged neurons, cortical ALS astrocytes release FGF2 and enhances the expression of FGFR3, which has been associated to neuroprotection against glutamate toxicity and increased microglial migration and phagocytosis (Noda et al., 2014). Nevertheless, excessive neuronal phagoptosis also contributes to a range of neurodegenerative diseases, including ALS (Brown and Neher, 2014). Therefore, if we consider that the secretome derived from both pre-miR-146a and VS-treated astrocytes reduces FGFR3 levels in microglia, it may then prevent reactivity and excessive phagoptosis. However, only the secretome derived from pre-miR-146a -treated astrocytes averted microglial inflammatory activation.
To better understand the effects produced by the secretome from mSOD1 astrocytes treated with pre-miR-146a on microglia we performed proteomic analysis. Among the effects produced we noticed a reduction in Cdk1 that may have contributed to the decrease of iNOS and TNF-α transcripts by the modulated secretome. Such proinflammatory signaling activation, characteristic of a polarized activated microglia (Sheng et al., 2011), was shown to be prevented by CDK inhibitors (Leitch et al., 2009; Skovira et al., 2016). Similarly contributing to counteract the activation of microglia is the reduction we found on Adamts1, once its upregulation was identified in LPS-activated microglia (Chiu et al., 2013) and associated to a tumor inflammatory response (Rodriguez-Baena et al., 2018). These findings indicate that pre-miR-146a contributes to a less polarized microglia phenotype. Regarding Igdcc4, also described as being involved in immune response (Sanz-Pamplona et al., 2014) and a biomarker in hepatocellular carcinoma (Zweerink et al., 2020), further studies should investigate its precise role in microglia.
Now going for the interaction of sEVs on microglia, though we did not observe changes in the concentration and size of sEVs derived from pre-miR-146a-treated astrocytes, an increased internalization of sEVs in microglia was noticed, when compared with sEVs derived from the pathological untreated mSOD1 astrocytes or VS-treated ones. This increased uptake by the microglial cells is not surprising since it was demonstrated that miR-146a leads to the upregulation of several microglia surface receptors (Saba et al., 2012) that most likely enable the uptake of sEVs. Also, previous studies demonstrated that microglia are one of the neural cells that show higher ability for sEV incorporation (Fitzner et al., 2011; Pinto et al., 2017; Xia et al., 2019).
In a previous study we evidenced that sEVs derived from the secretome of mSOD1 MNs with an enriched cargo in miR-124 were able to activate microglia (Pinto et al., 2017). Similarly, our sEVs depleted in miR-146a and isolated from the secretome of mSOD1 cortical astrocytes led to increased gene expression of iNOS and TNF-α. We further demonstrated that such effect was counteracted when we used pre-miR-146a or VS-treatment in mSOD1 cortical astrocytes. This is in line with studies evidencing that EVs containing miR-146a attenuate inflammation, supporting tissue repair and regeneration (Alexander et al., 2015; Waters et al., 2019; Wu et al., 2019). Moreover, miR-146a packaged into EVs and transferred into macrophages showed therapeutic efficacy against sepsis (Song et al., 2017), thus reinforcing its protective effects. Interestingly, despite not having increased cargo in miR-146a, sEVs isolated from the secretome of VS-treated mSOD1 astrocytes also demonstrated ability to switch the activated microglia into the steady-state phenotype, indicating that miR-146a is not the sole neuroprotective factor involved in the reestablishment of the healthy microglia state.
In sum, our study highlights that the downregulation of miR-146a in mSOD1 cortical astrocytes is responsible, at least in part, by the reactivity and aberrant features of these glial cells. We propose pre-miR-146a transfection and VS treatment as efficient strategies to induce miR-146a upregulation in cortical ALS astrocytes toward cell revival through the abrogation of their reactive/aberrant phenotype. These potential therapeutic approaches have additional advantages in re-establishing microglia and MN homeostatic states by paracrine signaling. Comparative effects of the miR-146a replenishing methods, i.e., pre-miR-146a, GUDCA and VS, in the ability to recovery of the neuroprotective phenotype of cortical astrocytes from mSOD1 mice, and the resulting paracrine signaling on WT MNs and N9 microglial cells are schematically represented in Figure 10. Overall, we consider sEV-enrichment in VS and pre-miR-146a cargoes as opportunities for neuro-glial restorative therapy in ALS disease.
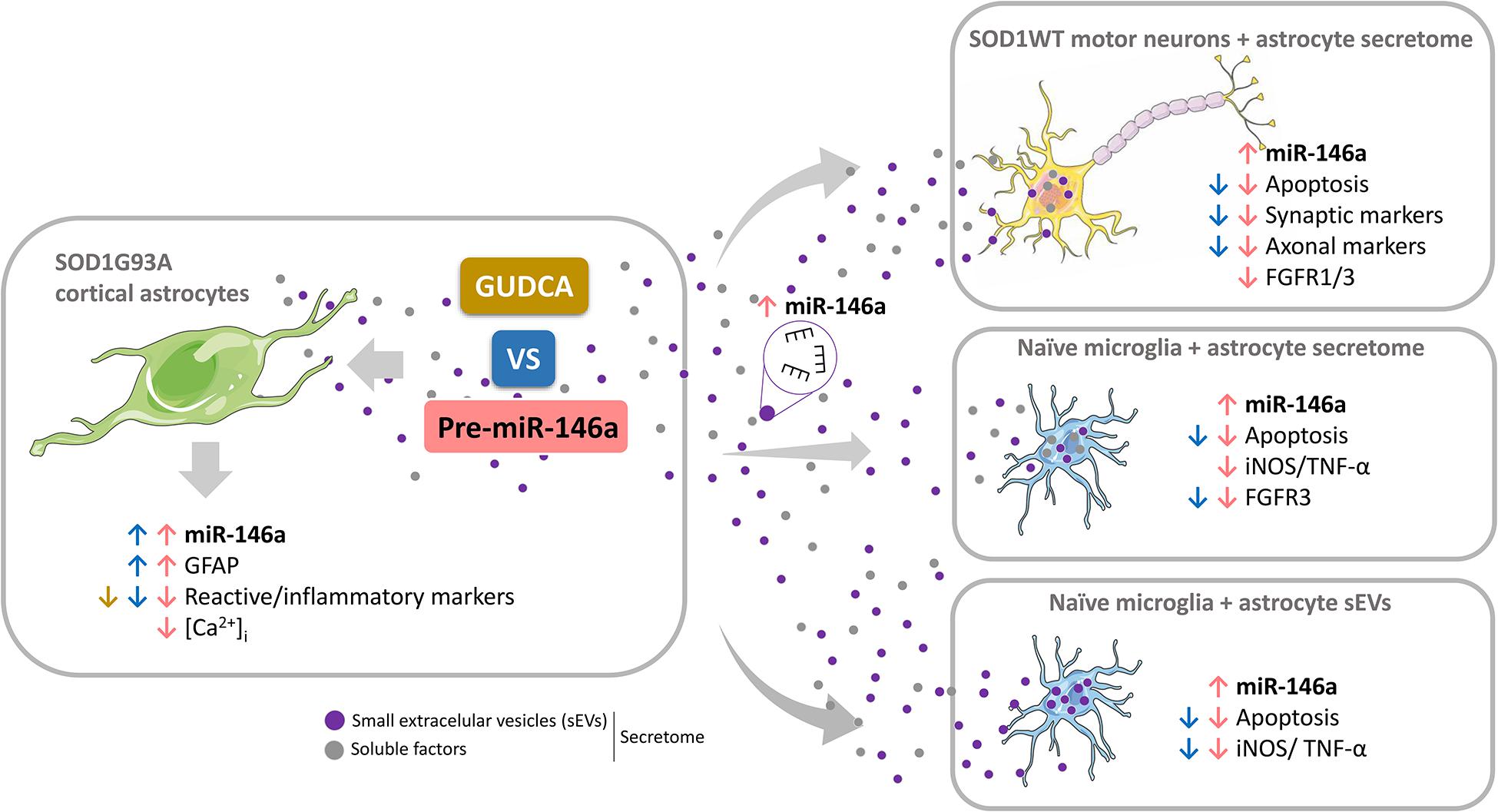
Figure 10. Schematic representation of the efficacy of miR-146a replenishing methods to recover the neuroprotective phenotype of aberrant mSOD1 cortical astrocytes, and in preventing toxic paracrine signaling toward motor neurons and microglia by the mediators released from the pathological astrocytes. Astrocytes were isolated from the cortex of SOD1-G93A (mSOD1) mice pups at 7-day-old and cultured for 13 days in vitro, showing an aberrant/reactive phenotype. Treatment with glycoursodeoxycholic acid (GUDCA) and dipeptidyl vinyl sulfone (VS) abrogated reactive markers, with additional re-establishment of glial fibrillary acidic protein (GFAP) and miR-146a by VS, evidencing their reparative ability. Transfection of mSOD1 astrocytes with pre-miR-146a also attenuated their phenotypic aberrancies and intracellular Ca2+ ([Ca2+]i) overload. Moreover, such treatment increased miR146a content in the cell-derived small extracellular vesicles (sEVs), and mediated miR-146a enrichment in SOD1-WT motor neurons (MNs) and naïve N9 microglial cell. Secretome from mSOD1 astrocytes increased early/late apoptosis and fibroblast growth factor receptor (FGFR) gene levels in MNs and microglia, effects that were prevented by pre-miR-146a or VS modulation. These strategies led to a secretome with preventable properties over the deregulation of synaptic dynamics and axonal transport upon the pathological extracellular milieu from mSOD1 astrocytes. The pre-miR-146a-treated cells also prevented microglia activation through their secretome or isolated sEVs, but in the case of VS only the isolated sEVs showed such property. Data reveal that both pre-miR-146a and VS-mediated miR-146a replenishment in mSOD1 cortical astrocytes are promising approaches to recover the neuroprotective phenotype of ALS cortical astrocytes and the microglia and MN homeostatic balance in the disease. [Ca2+]i, intracellular calcium; GFAP, glial fibrillary acidic protein; FGFR, fibroblast growth factor receptor; GUDCA, glycoursodeoxycholic acid; iNOS, inducible nitric oxide synthase; miR-146a, miRNA-146a; SOD1, superoxide dismutase 1; TNF-α, tumor necrosis alpha; VS, dipeptidyl vinyl sulfone.
Data Availability Statement
The datasets presented in this study are available in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/ Supplementary Material.
Ethics Statement
The animal study was reviewed and approved by Portuguese National Authority (General Direction of Veterinary) (Directives 86/609/EU and 2010/63/EU, Recommendation 2007/526/CE, European Convention for the Protection of Vertebrate Animals used for Experimental or Other Scientific Purposes ETS 123/Appendix A) and Portuguese Laws on Animal Care (Decreto-Lei 129/92, Portaria 1005/92, Portaria 466/95, Decreto-Lei 197/96, and Portaria 1131/97), Portugal; Ethics Committee of the Instituto de Medicina Molecular João Lobo Antunes (IMM), Faculdade de Medicina, Universidade de Lisboa, Portugal.
Author Contributions
DB conceived the project. MB, AV, and DB planned and designed the experiments. MB performed most of the experimental work, analyzed data, and drafted the manuscript. AV, CG, and CS participated in the preparation and maintenance of primary astrocyte cultures and small extracellular vesicles characterization. SV planned and analyzed data of calcium experiments. CP and JG-R performed experimental work and analyzed data of calcium experiments. RM and LC synthesized the VS compound. MB, AV, and DB interpreted results and wrote the final version of the manuscript. DB supervised all aspects and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Research Grant of the Santa Casa Scientific Research Program on ALS, by Santa Casa da Misericórdia de Lisboa (SCML), Portugal, Project Ref. ELA-2015-002 (to DB). Fundação para a Ciência e a Tecnologia (FCT) also supported the project PTDC/MED-NEU/31395/2017 (to DB), PTDC/MED-QUI/30021/2017 (to RM) and iMed. ULisboa (UIDB/04138/2020 and UIDP/04138/2020), together with Programa Operacional Regional de Lisboa and the Programa Operacional Competitividade e Internacionalização (LISBOA-01-0145-FEDER-031395 to DB). Individual fellowships MB (SFRH/BD/129586/2017), CG (SFRH/BD/102718/2014), AV (SFRH/BPD/76590/2011), and JG-R PD/BD/150342/2019 were from FCT. CS was a research fellowship recipient from SCML. The funding organization had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Rui Fernandes and A. R. Malheiro from the Instituto de Investigação e Inovação em Saúde (i3S) and Histology and Electron Microscopy Service (HEMS/IBMC) at the Universidade do Porto, for acquisition and analysis of sEV TEM images. We also thank Drs. Bruno Costa-Silva and Cristian Bodo from Systems Oncology group, Fundação Champalimaud (Lisbon, Portugal) for their expertise and support in guiding Nanosight (model LM10-HSBF, Malvern, United Kingdom) operation and the application of NTA software version 3.4. Finally, we also thank Drs. Per Haberkant and Frank Stein from the EMBL Proteomics Core facility in Heidelberg for support with mass spectrometry and data analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.634355/full#supplementary-material
Footnote
References
Ajmone-Cat, M. A., Onori, A., Toselli, C., Stronati, E., Morlando, M., Bozzoni, I., et al. (2019). Increased FUS levels in astrocytes leads to astrocyte and microglia activation and neuronal death. Sci. Rep. 9:4572. doi: 10.1038/s41598-019-41040-4
Al-Chalabi, A., van den Berg, L. H., and Veldink, J. (2017). Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat. Rev. Neurol. 13, 96–104. doi: 10.1038/nrneurol.2016.182
Alexander, M., Hu, R., Runtsch, M. C., Kagele, D. A., Mosbruger, T. L., Tolmachova, T., et al. (2015). Exosome-delivered microRNAs modulate the inflammatory response to endotoxin. Nat. Commun. 6:7321. doi: 10.1038/ncomms8321
Almad, A. A., Doreswamy, A., Gross, S. K., Richard, J. P., Huo, Y., Haughey, N., et al. (2016). Connexin 43 in astrocytes contributes to motor neuron toxicity in amyotrophic lateral sclerosis. Glia 64, 1154–1169. doi: 10.1002/glia.22989
Almad, A. A., Taga, A., Joseph, J., Welsh, C., Patankar, A., Gross, S. K., et al. (2020). Connexin 43 hemichannels mediate spatial and temporal disease spread in ALS. BioRxiv [Preprint]. doi: 10.1101/2020.03.14.990747
Araújo, J. (2019). Roles of Zbtb20 in the Specification of Upper Layer Neurons and Astrocytes in the Neocortex. Ph.D. thesis. Natal: Universidade Federal do Rio Grande do Norte.
Basso, M., and Bonetto, V. (2016). Extracellular vesicles and a novel form of communication in the brain. Front. Neurosci. 10:127. doi: 10.3389/fnins.2016.00127
Basso, M., Pozzi, S., Tortarolo, M., Fiordaliso, F., Bisighini, C., Pasetto, L., et al. (2013). Mutant copper-zinc superoxide dismutase (SOD1) induces protein secretion pathway alterations and exosome release in astrocytes: implications for disease spreading and motor neuron pathology in amyotrophic lateral sclerosis. J. Biol. Chem. 288, 15699–15711. doi: 10.1074/jbc.M112.425066
Benkler, C., Ben-Zur, T., Barhum, Y., and Offen, D. (2013). Altered astrocytic response to activation in SOD1(G93A) mice and its implications on amyotrophic lateral sclerosis pathogenesis. Glia 61, 312–326. doi: 10.1002/glia.22428
Benninger, F., Glat, M. J., Offen, D., and Steiner, I. (2016). Glial fibrillary acidic protein as a marker of astrocytic activation in the cerebrospinal fluid of patients with amyotrophic lateral sclerosis. J. Clin. Neurosci. 26, 75–78. doi: 10.1016/j.jocn.2015.10.008
Bluhm, A., Viceconte, N., Li, F., Rane, G., Ritz, S., Wang, S., et al. (2019). ZBTB10 binds the telomeric variant repeat TTGGGG and interacts with TRF2. Nucleic Acids Res. 47, 1896–1907. doi: 10.1093/nar/gky1289
Brites, D. (2020). Regulatory function of microRNAs in microglia. Glia 68, 1631–1642. doi: 10.1002/glia.23846
Brown, G. C., and Neher, J. J. (2014). Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 15, 209–216. doi: 10.1038/nrn3710
Bucchia, M., Merwin, S. J., Re, D. B., and Kariya, S. (2018). Limitations and challenges in modeling diseases involving spinal motor neuron degeneration in vitro. Front. Cell. Neurosci. 12:61. doi: 10.3389/fncel.2018.00061
Butovsky, O., Jedrychowski, M. P., Cialic, R., Krasemann, S., Murugaiyan, G., Fanek, Z., et al. (2015). Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann. Neurol. 77, 75–99. doi: 10.1002/ana.24304
Butovsky, O., Siddiqui, S., Gabriely, G., Lanser, A. J., Dake, B., Murugaiyan, G., et al. (2012). Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J. Clin. Invest. 122, 3063–3087. doi: 10.1172/JCI62636
Casas, C., Manzano, R., Vaz, R., Osta, R., and Brites, D. (2016). Synaptic failure: focus in an integrative view of ALS. Brain Plast. 1, 159–175. doi: 10.3233/BPL-140001
Cashman, N. R., Durham, H. D., Blusztajn, J. K., Oda, K., Tabira, T., Shaw, I. T., et al. (1992). Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev. Dyn. 194, 209–221. doi: 10.1002/aja.1001940306
Cater, J. H., Kumita, J. R., Zeineddine Abdallah, R., Zhao, G., Bernardo-Gancedo, A., Henry, A., et al. (2019). Human pregnancy zone protein stabilizes misfolded proteins including preeclampsia- and Alzheimer’s-associated amyloid beta peptide. Proc. Natl. Acad. Sci. U.S.A. 116, 6101–6110. doi: 10.1073/pnas.1817298116
Chen, W. W., Birsoy, K., Mihaylova, M. M., Snitkin, H., Stasinski, I., Yucel, B., et al. (2014). Inhibition of ATPIF1 ameliorates severe mitochondrial respiratory chain dysfunction in mammalian cells. Cell Rep. 7, 27–34. doi: 10.1016/j.celrep.2014.02.046
Cheng, F., and Eriksson, J. E. (2017). Intermediate filaments and the regulation of cell motility during regeneration and wound healing. Cold Spring Harb. Perspect. Biol. 9:a022046. doi: 10.1101/cshperspect.a022046
Chiu, I. M., Morimoto, E. T., Goodarzi, H., Liao, J. T., O’Keeffe, S., Phatnani, H. P., et al. (2013). A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 4, 385–401. doi: 10.1016/j.celrep.2013.06.018
Choi, J. W., Kim, S., Park, J. H., Kim, H. J., Shin, S. J., Kim, J. W., et al. (2019). Optimization of vinyl sulfone derivatives as potent nuclear factor erythroid 2-related factor 2 (Nrf2) activators for Parkinson’s disease therapy. J. Med. Chem. 62, 811–830. doi: 10.1021/acs.jmedchem.8b01527
Clarke, W. E., Berry, M., Smith, C., Kent, A., and Logan, A. (2001). Coordination of fibroblast growth factor receptor 1 (FGFR1) and fibroblast growth factor-2 (FGF-2) trafficking to nuclei of reactive astrocytes around cerebral lesions in adult rats. Mol. Cell Neurosci. 17, 17–30. doi: 10.1006/mcne.2000.0920
Cornell-Bell, A. H., Finkbeiner, S. M., Cooper, M. S., and Smith, S. J. (1990). Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science 247, 470–473. doi: 10.1126/science.1967852
Cui, Y., Masaki, K., Yamasaki, R., Imamura, S., Suzuki, S. O., Hayashi, S., et al. (2014). Extensive dysregulations of oligodendrocytic and astrocytic connexins are associated with disease progression in an amyotrophic lateral sclerosis mouse model. J. Neuroinflammation 11:42. doi: 10.1186/1742-2094-11-42
Cunha, C., Gomes, C., Vaz, A. R., and Brites, D. (2016). Exploring new inflammatory biomarkers and pathways during LPS-induced M1 polarization. Mediators Inflamm. 2016:6986175. doi: 10.1155/2016/6986175
Cunha, C., Santos, C., Gomes, C., Fernandes, A., Correia, A. M., Sebastião, A. M., et al. (2018). Downregulated Glia interplay and INCREASED miRNA-155 as promising markers to track ALS at an early stage. Mol. Neurobiol. 55, 4207–4224. doi: 10.1007/s12035-017-0631-2
De Felice, B., Annunziata, A., Fiorentino, G., Borra, M., Biffali, E., Coppola, C., et al. (2014). miR-338-3p is over-expressed in blood, CFS, serum and spinal cord from sporadic amyotrophic lateral sclerosis patients. Neurogenetics 15, 243–253. doi: 10.1007/s10048-014-0420-2
De Vos, K. J., and Hafezparast, M. (2017). Neurobiology of axonal transport defects in motor neuron diseases: opportunities for translational research? Neurobiol. Dis. 105, 283–299. doi: 10.1016/j.nbd.2017.02.004
Di Giorgio, F. P., Carrasco, M. A., Siao, M. C., Maniatis, T., and Eggan, K. (2007). Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat. Neurosci. 10, 608–614. doi: 10.1038/nn1885
Díaz-Amarilla, P., Olivera-Bravo, S., Trias, E., Cragnolini, A., Martinez-Palma, L., Cassina, P., et al. (2011). Phenotypically aberrant astrocytes that promote motoneuron damage in a model of inherited amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 108, 18126–18131. doi: 10.1073/pnas.1110689108
Ekestern, E. (2004). Neurotrophic factors and amyotrophic lateral sclerosis. Neurodegener. Dis. 1, 88–100. doi: 10.1159/000080049
Eng, L. F., and Ghirnikar, R. S. (1994). GFAP and astrogliosis. Brain Pathol. 4, 229–237. doi: 10.1111/j.1750-3639.1994.tb00838.x
Escartin, C., Galea, E., Lakatos, A., O’Callaghan, J. P., Petzold, G. C., Serrano-Pozo, A., et al. (2021). Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 24, 312–325. doi: 10.1038/s41593-020-00783-4
Falcão, A. S., Carvalho, L. A., Lidónio, G., Vaz, A. R., Lucas, S. D., Moreira, R., et al. (2017). Dipeptidyl Vinyl sulfone as a novel chemical tool to inhibit HMGB1/NLRP3-inflammasome and inflamma-miRs in Abeta-mediated microglial inflammation. ACS Chem. Neurosci. 8, 89–99. doi: 10.1021/acschemneuro.6b00250
Falcão, A. S., Fernandes, A., Brito, M. A., Silva, R. F., and Brites, D. (2005). Bilirubin-induced inflammatory response, glutamate release, and cell death in rat cortical astrocytes are enhanced in younger cells. Neurobiol. Dis. 20, 199–206. doi: 10.1016/j.nbd.2005.03.001
Falcão, A. S., Fernandes, A., Brito, M. A., Silva, R. F., and Brites, D. (2006). Bilirubin-induced immunostimulant effects and toxicity vary with neural cell type and maturation state. Acta Neuropathol. 112, 95–105. doi: 10.1007/s00401-006-0078-4
Fernandes, A., Vaz, A. R., Falcão, A. S., Silva, R. F., Brito, M. A., and Brites, D. (2007). Glycoursodeoxycholic acid and interleukin-10 modulate the reactivity of rat cortical astrocytes to unconjugated bilirubin. J. Neuropathol. Exp. Neurol. 66, 789–798. doi: 10.1097/nen.0b013e3181461c74
Ferrara, D., Pasetto, L., Bonetto, V., and Basso, M. (2018). Role of extracellular vesicles in amyotrophic lateral sclerosis. Front. Neurosci. 12:574. doi: 10.3389/fnins.2018.00574
Filipi, T., Hermanova, Z., Tureckova, J., Vanatko, O., and Anderova, A. M. (2020). Glial cells-the strategic targets in amyotrophic lateral sclerosis treatment. J. Clin. Med. 9:261. doi: 10.3390/jcm9010261
Fitzner, D., Schnaars, M., van Rossum, D., Krishnamoorthy, G., Dibaj, P., Bakhti, M., et al. (2011). Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. J. Cell Sci. 124(Pt 3), 447–458. doi: 10.1242/jcs.074088
Fleisher-Berkovich, S., Filipovich-Rimon, T., Ben-Shmuel, S., Hulsmann, C., Kummer, M. P., and Heneka, M. T. (2010). Distinct modulation of microglial amyloid beta phagocytosis and migration by neuropeptides (i). J. Neuroinflammation 7:61. doi: 10.1186/1742-2094-7-61
Fogarty, M. J. (2019). Amyotrophic lateral sclerosis as a synaptopathy. Neural Regen. Res. 14, 189–192. doi: 10.4103/1673-5374.244782
Franken, H., Mathieson, T., Childs, D., Sweetman, G. M., Werner, T., Togel, I., et al. (2015). Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat. Protoc. 10, 1567–1593. doi: 10.1038/nprot.2015.101
Fregeac, J., Moriceau, S., Poli, A., Nguyen, L. S., Oury, F., and Colleaux, L. (2020). Loss of the neurodevelopmental disease-associated gene miR-146a impairs neural progenitor differentiation and causes learning and memory deficits. Mol. Autism 11:22. doi: 10.1186/s13229-020-00328-3
Freischmidt, A., Muller, K., Zondler, L., Weydt, P., Volk, A. E., Bozic, A. L., et al. (2014). Serum microRNAs in patients with genetic amyotrophic lateral sclerosis and pre-manifest mutation carriers. Brain 137(Pt 11), 2938–2950. doi: 10.1093/brain/awu249
Fujita, K., Kato, T., Yamauchi, M., Ando, M., Honda, M., and Nagata, Y. (1998). Increases in fragmented glial fibrillary acidic protein levels in the spinal cords of patients with amyotrophic lateral sclerosis. Neurochem. Res. 23, 169–174. doi: 10.1023/a:1022476724381
Goddard, D. R., Berry, M., Kirvell, S. L., and Butt, A. M. (2002). Fibroblast growth factor-2 induces astroglial and microglial reactivity in vivo. J. Anat. 200(Pt 1), 57–67. doi: 10.1046/j.0021-8782.2001.00002.x
Goguet-Rubio, P., Seyran, B., Gayte, L., Bernex, F., Sutter, A., Delpech, H., et al. (2016). E4F1-mediated control of pyruvate dehydrogenase activity is essential for skin homeostasis. Proc. Natl. Acad. Sci. U.S.A. 113, 11004–11009. doi: 10.1073/pnas.1602751113
Gomes, C., Cunha, C., Nascimento, F., Ribeiro, J. A., Vaz, A. R., and Brites, D. (2019). Cortical neurotoxic astrocytes with early ALS pathology and miR-146a deficit replicate gliosis markers of symptomatic SOD1G93A mouse model. Mol. Neurobiol. 56, 2137–2158. doi: 10.1007/s12035-018-1220-8
Gomes, C., Sequeira, C., Barbosa, M., Cunha, C., Vaz, A. R., and Brites, D. (2020). Astrocyte regional diversity in ALS includes distinct aberrant phenotypes with common and causal pathological processes. Exp. Cell Res. 395:112209. doi: 10.1016/j.yexcr.2020.112209
Grad, L. I., Yerbury, J. J., Turner, B. J., Guest, W. C., Pokrishevsky, E., O’Neill, M. A., et al. (2014). Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. U.S.A. 111, 3620–3625. doi: 10.1073/pnas.1312245111
Guerra-Gomes, S., Sousa, N., Pinto, L., and Oliveira, J. F. (2017). Functional roles of astrocyte calcium elevations: from synapses to behavior. Front. Cell. Neurosci. 11:427. doi: 10.3389/fncel.2017.00427
Gurney, M. E. (1994). Transgenic-mouse model of amyotrophic lateral sclerosis. N. Engl. J. Med. 331, 1721–1722. doi: 10.1056/NEJM199412223312516
Guttenplan, K. A., Weigel, M. K., Adler, D. I., Couthouis, J., Liddelow, S. A., Gitler, A. D., et al. (2020). Knockout of reactive astrocyte activating factors slows disease progression in an ALS mouse model. Nat. Commun. 11:3753. doi: 10.1038/s41467-020-17514-9
Haidet-Phillips, A. M., Hester, M. E., Miranda, C. J., Meyer, K., Braun, L., Frakes, A., et al. (2011). Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 29, 824–828. doi: 10.1038/nbt.1957
He, Q., Yang, L., Gao, K., Ding, P., Chen, Q., Xiong, J., et al. (2020). FTSJ1 regulates tRNA 2’-O-methyladenosine modification and suppresses the malignancy of NSCLC via inhibiting DRAM1 expression. Cell Death Dis. 11:348. doi: 10.1038/s41419-020-2525-x
Hessvik, N. P., and Llorente, A. (2018). Current knowledge on exosome biogenesis and release. Cell Mol. Life Sci. 75, 193–208. doi: 10.1007/s00018-017-2595-9
Ho, R., Sances, S., Gowing, G., Amoroso, M. W., O’Rourke, J. G., Sahabian, A., et al. (2016). ALS disrupts spinal motor neuron maturation and aging pathways within gene co-expression networks. Nat. Neurosci. 19, 1256–1267. doi: 10.1038/nn.4345
Holmdahl, R., Sareila, O., Olsson, L. M., Backdahl, L., and Wing, K. (2016). Ncf1 polymorphism reveals oxidative regulation of autoimmune chronic inflammation. Immunol. Rev. 269, 228–247. doi: 10.1111/imr.12378
Homkajorn, B., Sims, N. R., and Muyderman, H. (2010). Connexin 43 regulates astrocytic migration and proliferation in response to injury. Neurosci. Lett. 486, 197–201. doi: 10.1016/j.neulet.2010.09.051
Hornberg, H., Wollerton-van Horck, F., Maurus, D., Zwart, M., Svoboda, H., Harris, W. A., et al. (2013). RNA-binding protein Hermes/RBPMS inversely affects synapse density and axon arbor formation in retinal ganglion cells in vivo. J. Neurosci. 33, 10384–10395. doi: 10.1523/JNEUROSCI.5858-12.2013
Huang, J.-Y., Krebs, B. B., Miskus, M. L., Russell, M. L., Duffy, E. P., Graf, J. M., et al. (2020). Enhanced FGFR3 activity in post-mitotic principal neurons during brain development results in cortical dysplasia and axon miswiring. bioRxiv [Preprint]. doi: 10.1101/2020.05.02.073924
Huang, J. Y., Lynn Miskus, M., and Lu, H. C. (2017). FGF-FGFR mediates the activity-dependent dendritogenesis of Layer IV neurons during barrel formation. J. Neurosci. 37, 12094–12105. doi: 10.1523/JNEUROSCI.1174-17.2017
Huber, W., von Heydebreck, A., Sultmann, H., Poustka, A., and Vingron, M. (2002). Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 18(Suppl. 1), S96–S104. doi: 10.1093/bioinformatics/18.suppl_1.s96
Ijsselstijn, L., Dekker, L. J., Stingl, C., van der Weiden, M. M., Hofman, A., Kros, J. M., et al. (2011). Serum levels of pregnancy zone protein are elevated in presymptomatic Alzheimer’s disease. J Proteome Res. 10, 4902–4910. doi: 10.1021/pr200270z
Ivaska, J., Pallari, H. M., Nevo, J., and Eriksson, J. E. (2007). Novel functions of vimentin in cell adhesion, migration, and signaling. Exp. Cell Res. 313, 2050–2062. doi: 10.1016/j.yexcr.2007.03.040
Iyer, A., Zurolo, E., Prabowo, A., Fluiter, K., Spliet, W. G., van Rijen, P. C., et al. (2012). MicroRNA-146a: a key regulator of astrocyte-mediated inflammatory response. PLoS One 7:e44789. doi: 10.1371/journal.pone.0044789
Jha, M. K., Jo, M., Kim, J. H., and Suk, K. (2018). Microglia-astrocyte crosstalk: an intimate molecular conversation. Neuroscientist 25, 227–240. doi: 10.1177/1073858418783959
Jia, L., Wang, L., Chopp, M., Zhang, Y., Szalad, A., and Zhang, Z. G. (2016). MicroRNA 146a locally mediates distal axonal growth of dorsal root ganglia neurons under high glucose and sildenafil conditions. Neuroscience 329, 43–53. doi: 10.1016/j.neuroscience.2016.05.005
Johansson, A., Larsson, A., Nygren, I., Blennow, K., and Askmark, H. (2003). Increased serum and cerebrospinal fluid FGF-2 levels in amyotrophic lateral sclerosis. Neuroreport 14, 1867–1869. doi: 10.1097/00001756-200310060-00022
Joilin, G., Leigh, P. N., Newbury, S. F., and Hafezparast, M. (2019). An Overview of MicroRNAs as Biomarkers of ALS. Front. Neurol. 10:186. doi: 10.3389/fneur.2019.00186
Jovicic, A., and Gitler, A. D. (2017). Distinct repertoires of microRNAs present in mouse astrocytes compared to astrocyte-secreted exosomes. PLoS One 12:e0171418. doi: 10.1371/journal.pone.0171418
Kawamata, H., Ng, S. K., Diaz, N., Burstein, S., Morel, L., Osgood, A., et al. (2014). Abnormal intracellular calcium signaling and SNARE-dependent exocytosis contributes to SOD1G93A astrocyte-mediated toxicity in amyotrophic lateral sclerosis. J. Neurosci. 34, 2331–2348. doi: 10.1523/JNEUROSCI.2689-13.2014
Koval, E. D., Shaner, C., Zhang, P., du Maine, X., Fischer, K., Tay, J., et al. (2013). Method for widespread microRNA-155 inhibition prolongs survival in ALS-model mice. Hum. Mol. Genet. 22, 4127–4135. doi: 10.1093/hmg/ddt261
Leitch, A. E., Haslett, C., and Rossi, A. G. (2009). Cyclin-dependent kinase inhibitor drugs as potential novel anti-inflammatory and pro-resolution agents. Br. J. Pharmacol. 158, 1004–1016. doi: 10.1111/j.1476-5381.2009.00402.x
Li, Q., Vande Velde, C., Israelson, A., Xie, J., Bailey, A. O., Dong, M. Q., et al. (2010). ALS-linked mutant superoxide dismutase 1 (SOD1) alters mitochondrial protein composition and decreases protein import. Proc. Natl. Acad. Sci. U.S.A. 107, 21146–21151. doi: 10.1073/pnas.1014862107
Liddelow, S. A., and Barres, B. A. (2017). Reactive astrocytes: production, function, and therapeutic potential. Immunity 46, 957–967. doi: 10.1016/j.immuni.2017.06.006
Liu, J., and Wang, F. (2017). Role of neuroinflammation in amyotrophic lateral sclerosis: cellular mechanisms and therapeutic implications. Front. Immunol. 8:1005. doi: 10.3389/fimmu.2017.01005
Lu, B., Zhang, Q., Wang, H., Wang, Y., Nakayama, M., and Ren, D. (2010). Extracellular calcium controls background current and neuronal excitability via an UNC79-UNC80-NALCN cation channel complex. Neuron 68, 488–499. doi: 10.1016/j.neuron.2010.09.014
Maier, O., Bohm, J., Dahm, M., Bruck, S., Beyer, C., and Johann, S. (2013). Differentiated NSC-34 motoneuron-like cells as experimental model for cholinergic neurodegeneration. Neurochem. Int. 62, 1029–1038. doi: 10.1016/j.neuint.2013.03.008
Martins, R. S., Rombo, D. M., Gonçalves-Ribeiro, J., Meneses, C., Borges-Martins, V. P. P., Ribeiro, J. A., et al. (2020). Caffeine has a dual influence on NMDA receptor-mediated glutamatergic transmission at the hippocampus. Purinergic Signal. 16, 503–518. doi: 10.1007/s11302-020-09724-z
Mei, J., Bachoo, R., and Zhang, C. L. (2011). MicroRNA-146a inhibits glioma development by targeting Notch1. Mol. Cell Biol. 31, 3584–3592. doi: 10.1128/MCB.05821-11
Mejzini, R., Flynn, L. L., Pitout, I. L., Fletcher, S., Wilton, S. D., and Akkari, P. A. (2019). ALS genetics, mechanisms, and therapeutics: where are we now? Front. Neurosci. 13:1310. doi: 10.3389/fnins.2019.01310
Meyer, K., Ferraiuolo, L., Miranda, C. J., Likhite, S., McElroy, S., Renusch, S., et al. (2014). Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc. Natl. Acad. Sci. U.S.A. 111, 829–832. doi: 10.1073/pnas.1314085111
Mi, H., Ebert, D., Muruganujan, A., Mills, C., Albou, L. P., Mushayamaha, T., et al. (2021). PANTHER version 16: a revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 49, D394–D403. doi: 10.1093/nar/gkaa1106
Moggridge, S., Sorensen, P. H., Morin, G. B., and Hughes, C. S. (2018). Extending the compatibility of the SP3 paramagnetic bead processing approach for proteomics. J. Proteome Res. 17, 1730–1740. doi: 10.1021/acs.jproteome.7b00913
Morais, T. P., Coelho, D., Vaz, S. H., Sebastião, A. M., and Valente, C. A. (2017). Glycine receptor activation impairs ATP-induced calcium transients in cultured cortical astrocytes. Front. Mol. Neurosci. 10:444. doi: 10.3389/fnmol.2017.00444
Nagai, M., Re, D. B., Nagata, T., Chalazonitis, A., Jessell, T. M., Wichterle, H., et al. (2007). Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10, 615–622. doi: 10.1038/nn1876
Nagao, M., Lanjakornsiripan, D., Itoh, Y., Kishi, Y., Ogata, T., and Gotoh, Y. (2014). High mobility group nucleosome-binding family proteins promote astrocyte differentiation of neural precursor cells. Stem Cells 32, 2983–2997. doi: 10.1002/stem.1787
Nagao, M., Ogata, T., Sawada, Y., and Gotoh, Y. (2016). Zbtb20 promotes astrocytogenesis during neocortical development. Nat. Commun. 7:11102. doi: 10.1038/ncomms11102
Nijholt, D. A., Ijsselstijn, L., van der Weiden, M. M., Zheng, P. P., Sillevis Smitt, P. A., Koudstaal, P. J., et al. (2015). Pregnancy zone protein is increased in the Alzheimer’s disease brain and associates with senile plaques. J. Alzheimers Dis. 46, 227–238. doi: 10.3233/JAD-131628
Nijssen, J., Aguila, J., Hoogstraaten, R., Kee, N., and Hedlund, E. (2018). Axon-Seq Decodes the motor axon transcriptome and its modulation in response to ALS. Stem Cell Rep. 11, 1565–1578. doi: 10.1016/j.stemcr.2018.11.005
Nikodemova, M., and Watters, J. J. (2011). Outbred ICR/CD1 mice display more severe neuroinflammation mediated by microglial TLR4/CD14 activation than inbred C57Bl/6 mice. Neuroscience 190, 67–74. doi: 10.1016/j.neuroscience.2011.06.006
Noda, M., Takii, K., Parajuli, B., Kawanokuchi, J., Sonobe, Y., Takeuchi, H., et al. (2014). FGF-2 released from degenerating neurons exerts microglial-induced neuroprotection via FGFR3-ERK signaling pathway. J. Neuroinflammation 11:76. doi: 10.1186/1742-2094-11-76
Okada, T., Enkhjargal, B., Travis, Z. D., Ocak, U., Tang, J., Suzuki, H., et al. (2019). FGF-2 attenuates neuronal Apoptosis via FGFR3/PI3k/Akt signaling pathway after subarachnoid hemorrhage. Mol. Neurobiol. 56, 8203–8219. doi: 10.1007/s12035-019-01668-9
Otomo, A., Pan, L., and Hadano, S. (2012). Dysregulation of the autophagy-endolysosomal system in amyotrophic lateral sclerosis and related motor neuron diseases. Neurol. Res. Int. 2012:498428. doi: 10.1155/2012/498428
Pehar, M., Harlan, B. A., Killoy, K. M., and Vargas, M. R. (2017). Role and therapeutic potential of astrocytes in amyotrophic lateral sclerosis. Curr. Pharm. Des. 23, 5010–5021. doi: 10.2174/1381612823666170622095802
Perez-Riverol, Y., Csordas, A., Bai, J., Bernal-Llinares, M., Hewapathirana, S., Kundu, D. J., et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450. doi: 10.1093/nar/gky1106
Pinho, A. G., Cibrão, J. R., Silva, N. A., Monteiro, S., and Salgado, A. J. (2020). Cell secretome: basic insights and therapeutic opportunities for CNS disorders. Pharmaceuticals (Basel) 13:31. doi: 10.3390/ph13020031
Pinto, S., Cunha, C., Barbosa, M., Vaz, A. R., and Brites, D. (2017). Exosomes from NSC-34 cells transfected with hSOD1-G93A are enriched in miR-124 and drive alterations in microglia phenotype. Front. Neurosci. 11:273. doi: 10.3389/fnins.2017.00273
Qian, K., Huang, H., Peterson, A., Hu, B., Maragakis, N. J., Ming, G. L., et al. (2017). Sporadic ALS astrocytes induce neuronal degeneration in vivo. Stem Cell Reports 8, 843–855. doi: 10.1016/j.stemcr.2017.03.003
Qu, X., Wang, N., Cheng, W., Xue, Y., Chen, W., and Qi, M. (2019). MicroRNA-146a protects against intracerebral hemorrhage by inhibiting inflammation and oxidative stress. Exp. Ther. Med. 18, 3920–3928. doi: 10.3892/etm.2019.8060
R Development Core Team (2020). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rabinovich-Toidman, P., Rabinovich-Nikitin, I., Ezra, A., Barbiro, B., Fogel, H., Slutsky, I., et al. (2015). Mutant SOD1 Increases APP expression and phosphorylation in cellular and animal models of ALS. PLoS One 10:e0143420. doi: 10.1371/journal.pone.0143420
Reilly, J. F., and Kumari, V. G. (1996). Alterations in fibroblast growth factor receptor expression following brain injury. Exp. Neurol. 140, 139–150. doi: 10.1006/exnr.1996.0124
Ricci, C., Marzocchi, C., and Battistini, S. (2018). MicroRNAs as biomarkers in amyotrophic lateral sclerosis. Cells 7:219. doi: 10.3390/cells7110219
Righi, M., Mori, L., De Libero, G., Sironi, M., Biondi, A., Mantovani, A., et al. (1989). Monokine production by microglial cell clones. Eur. J. Immunol. 19, 1443–1448. doi: 10.1002/eji.1830190815
Robberecht, W., and Philips, T. (2013). The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 14, 248–264. doi: 10.1038/nrn3430
Rodriguez-Baena, F. J., Redondo-Garcia, S., Peris-Torres, C., Martino-Echarri, E., Fernandez-Rodriguez, R., Plaza-Calonge, M. D. C., et al. (2018). ADAMTS1 protease is required for a balanced immune cell repertoire and tumour inflammatory response. Sci. Rep. 8:13103. doi: 10.1038/s41598-018-31288-7
Rossi, D., Brambilla, L., Valori, C. F., Roncoroni, C., Crugnola, A., Yokota, T., et al. (2008). Focal degeneration of astrocytes in amyotrophic lateral sclerosis. Cell Death Differ. 15, 1691–1700. doi: 10.1038/cdd.2008.99
Roybon, L., Lamas, N. J., Garcia, A. D., Yang, E. J., Sattler, R., Lewis, V. J., et al. (2013). Human stem cell-derived spinal cord astrocytes with defined mature or reactive phenotypes. Cell Rep. 4, 1035–1048. doi: 10.1016/j.celrep.2013.06.021
Saba, R., Gushue, S., Huzarewich, R. L., Manguiat, K., Medina, S., Robertson, C., et al. (2012). MicroRNA 146a (miR-146a) is over-expressed during prion disease and modulates the innate immune response and the microglial activation state. PLoS One 7:e30832. doi: 10.1371/journal.pone.0030832
Saba, R., Sorensen, D. L., and Booth, S. A. (2014). MicroRNA-146a: a dominant, negative regulator of the innate immune response. Front. Immunol. 5:578. doi: 10.3389/fimmu.2014.00578
Sanz-Pamplona, R., Berenguer, A., Cordero, D., Mollevi, D. G., Crous-Bou, M., Sole, X., et al. (2014). Aberrant gene expression in mucosa adjacent to tumor reveals a molecular crosstalk in colon cancer. Mol. Cancer 13:46. doi: 10.1186/1476-4598-13-46
Savitski, M. M., Wilhelm, M., Hahne, H., Kuster, B., and Bantscheff, M. (2015). A scalable approach for protein false discovery rate estimation in large proteomic data sets. Mol. Cell Proteomics 14, 2394–2404. doi: 10.1074/mcp.M114.046995
Serrano, A., Donno, C., Giannetti, S., Peric, M., Andjus, P., D’Ambrosi, N., et al. (2017). The astrocytic S100B protein with its receptor RAGE is aberrantly expressed in SOD1(G93A) models, and its inhibition decreases the expression of proinflammatory genes. Mediators Inflamm. 2017:1626204. doi: 10.1155/2017/1626204
Sheng, W., Zong, Y., Mohammad, A., Ajit, D., Cui, J., Han, D., et al. (2011). Pro-inflammatory cytokines and lipopolysaccharide induce changes in cell morphology, and upregulation of ERK1/2, iNOS and sPLA(2)-IIA expression in astrocytes and microglia. J. Neuroinflammation 8:121. doi: 10.1186/1742-2094-8-121
Shigetomi, E., Saito, K., Sano, F., and Koizumi, S. (2019). Aberrant calcium signals in reactive astrocytes: a key process in neurological disorders. Int. J. Mol. Sci. 20:996. doi: 10.3390/ijms20040996
Silverman, J. M., Fernando, S. M., Grad, L. I., Hill, A. F., Turner, B. J., Yerbury, J. J., et al. (2016). Disease mechanisms in ALS: misfolded SOD1 transferred through exosome-dependent and exosome-independent pathways. Cell Mol. Neurobiol. 36, 377–381. doi: 10.1007/s10571-015-0294-3
Sirkis, D. W., Geier, E. G., Bonham, L. W., Karch, C. M., and Yokoyama, J. S. (2019). Recent advances in the genetics of frontotemporal dementia. Curr. Genet. Med. Rep. 7, 41–52. doi: 10.1007/s40142-019-0160-6
Skovira, J. W., Wu, J., Matyas, J. J., Kumar, A., Hanscom, M., Kabadi, S. V., et al. (2016). Cell cycle inhibition reduces inflammatory responses, neuronal loss, and cognitive deficits induced by hypobaria exposure following traumatic brain injury. J. Neuroinflammation 13:299. doi: 10.1186/s12974-016-0769-2
Song, Y., Dou, H., Li, X., Zhao, X., Li, Y., Liu, D., et al. (2017). Exosomal miR-146a contributes to the enhanced therapeutic efficacy of interleukin-1beta-primed mesenchymal stem cells against sepsis. Stem Cells 35, 1208–1221. doi: 10.1002/stem.2564
Taganov, K. D., Boldin, M. P., Chang, K. J., and Baltimore, D. (2006). NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. U.S.A. 103, 12481–12486. doi: 10.1073/pnas.0605298103
Teranishi, Y., Inoue, M., Yamamoto, N. G., Kihara, T., Wiehager, B., Ishikawa, T., et al. (2015). Proton myo-inositol cotransporter is a novel gamma-secretase associated protein that regulates Abeta production without affecting Notch cleavage. FEBS J. 282, 3438–3451. doi: 10.1111/febs.13353
Trias, E., Ibarburu, S., Barreto-Nunez, R., and Barbeito, L. (2017). Significance of aberrant glial cell phenotypes in pathophysiology of amyotrophic lateral sclerosis. Neurosci. Lett. 636, 27–31. doi: 10.1016/j.neulet.2016.07.052
Tripathi, P., Rodriguez-Muela, N., Klim, J. R., de Boer, A. S., Agrawal, S., Sandoe, J., et al. (2017). Reactive astrocytes promote als-like degeneration and intracellular protein aggregation in human motor neurons by disrupting autophagy through TGF-beta1. Stem Cell Reports 9, 667–680. doi: 10.1016/j.stemcr.2017.06.008
van Rheenen, W., Shatunov, A., Dekker, A. M., McLaughlin, R. L., Diekstra, F. P., Pulit, S. L., et al. (2016). Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 48, 1043–1048. doi: 10.1038/ng.3622
Vandoorne, T., De Bock, K., and Van Den Bosch, L. (2018). Energy metabolism in ALS: an underappreciated opportunity? Acta Neuropathol. 135, 489–509. doi: 10.1007/s00401-018-1835-x
Varcianna, A., Myszczynska, M. A., Castelli, L. M., O’Neill, B., Kim, Y., Talbot, J., et al. (2019). Micro-RNAs secreted through astrocyte-derived extracellular vesicles cause neuronal network degeneration in C9orf72 ALS. EBioMedicine 40, 626–635. doi: 10.1016/j.ebiom.2018.11.067
Vardjan, N., Verkhratsky, A., and Zorec, R. (2017). Astrocytic pathological calcium homeostasis and impaired vesicle trafficking in neurodegeneration. Int. J. Mol. Sci. 18:358. doi: 10.3390/ijms18020358
Varga, B., Martin-Fernandez, M., Hilaire, C., Sanchez-Vicente, A., Areias, J., Salsac, C., et al. (2018). Myotube elasticity of an amyotrophic lateral sclerosis mouse model. Sci. Rep. 8:5917. doi: 10.1038/s41598-018-24027-5
Vaz, A. R., Cunha, C., Gomes, C., Schmucki, N., Barbosa, M., and Brites, D. (2015). Glycoursodeoxycholic acid reduces matrix metalloproteinase-9 and caspase-9 activation in a cellular model of superoxide dismutase-1 neurodegeneration. Mol. Neurobiol. 51, 864–877. doi: 10.1007/s12035-014-8731-8
Vaz, A. R., Pinto, S., Ezequiel, C., Cunha, C., Carvalho, L. A., Moreira, R., et al. (2019). Phenotypic effects of wild-type and mutant SOD1 expression in N9 murine microglia at steady state, inflammatory and immunomodulatory conditions. Front. Cell Neurosci. 13:109. doi: 10.3389/fncel.2019.00109
Waters, R., Subham, S., Pacelli, S., Modaresi, S., Chakravarti, A. R., and Paul, A. (2019). Development of MicroRNA-146a-enriched stem cell secretome for wound-healing applications. Mol. Pharm. 16, 4302–4312. doi: 10.1021/acs.molpharmaceut.9b00639
Woo, S. Y., Kim, J. H., Moon, M. K., Han, S. H., Yeon, S. K., Choi, J. W., et al. (2014). Discovery of vinyl sulfones as a novel class of neuroprotective agents toward Parkinson’s disease therapy. J. Med. Chem. 57, 1473–1487. doi: 10.1021/jm401788m
Wu, H., Fan, H., Shou, Z., Xu, M., Chen, Q., Ai, C., et al. (2019). Extracellular vesicles containing miR-146a attenuate experimental colitis by targeting TRAF6 and IRAK1. Int. Immunopharmacol. 68, 204–212. doi: 10.1016/j.intimp.2018.12.043
Xia, Y., Zhang, G., Han, C., Ma, K., Guo, X., Wan, F., et al. (2019). Microglia as modulators of exosomal alpha-synuclein transmission. Cell Death Dis. 10:174. doi: 10.1038/s41419-019-1404-9
Xiao, W. Z., Lu, A. Q., Liu, X. W., Li, Z., Zi, Y., and Wang, Z. W. (2015). Role of miRNA-146 in proliferation and differentiation of mouse neural stem cells. Biosci. Rep. 35:e00245. doi: 10.1042/BSR20150088
Yamanaka, K., Chun, S. J., Boillee, S., Fujimori-Tonou, N., Yamashita, H., Gutmann, D. H., et al. (2008). Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 11, 251–253. doi: 10.1038/nn2047
Yamanaka, K., and Komine, O. (2018). The multi-dimensional roles of astrocytes in ALS. Neurosci. Res. 126, 31–38. doi: 10.1016/j.neures.2017.09.011
Yonekawa, Y., Harada, A., Okada, Y., Funakoshi, T., Kanai, Y., Takei, Y., et al. (1998). Defect in synaptic vesicle precursor transport and neuronal cell death in KIF1A motor protein-deficient mice. J. Cell Biol. 141, 431–441. doi: 10.1083/jcb.141.2.431
Zang, D. W., Lopes, E. C., and Cheema, S. S. (2005). Loss of synaptophysin-positive boutons on lumbar motor neurons innervating the medial gastrocnemius muscle of the SOD1G93A G1H transgenic mouse model of ALS. J. Neurosci. Res. 79, 694–699. doi: 10.1002/jnr.20379
Zhang, P., and Lisman, J. E. (2012). Activity-dependent regulation of synaptic strength by PSD-95 in CA1 neurons. J. Neurophysiol. 107, 1058–1066. doi: 10.1152/jn.00526.2011
Zhao, J., Ma, J., Deng, Y., Kelly, J. A., Kim, K., Bang, S. Y., et al. (2017). A missense variant in NCF1 is associated with susceptibility to multiple autoimmune diseases. Nat. Genet. 49, 433–437. doi: 10.1038/ng.3782
Keywords: amyotrophic lateral sclerosis, astrocyte-microglia communication, astrocyte-motor neuron crosstalk, calcium signaling aberrancies, glycoursodeoxycholic acid, reactive astrocytes, small extracellular vesicles, vinyl sulfone
Citation: Barbosa M, Gomes C, Sequeira C, Gonçalves-Ribeiro J, Pina CC, Carvalho LA, Moreira R, Vaz SH, Vaz AR and Brites D (2021) Recovery of Depleted miR-146a in ALS Cortical Astrocytes Reverts Cell Aberrancies and Prevents Paracrine Pathogenicity on Microglia and Motor Neurons. Front. Cell Dev. Biol. 9:634355. doi: 10.3389/fcell.2021.634355
Received: 27 November 2020; Accepted: 26 March 2021;
Published: 23 April 2021.
Edited by:
Oleg Shupliakov, Karolinska Institutet (KI), SwedenReviewed by:
Emiliano Trias, Institut Pasteur de Montevideo, UruguayPavle R. Andjus, University of Belgrade, Serbia
Copyright © 2021 Barbosa, Gomes, Sequeira, Gonçalves-Ribeiro, Pina, Carvalho, Moreira, Vaz, Vaz and Brites. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dora Brites, ZGJyaXRlc0BmZi51bGlzYm9hLnB0