 Junzhong Gu
Junzhong Gu Weiwei Zhang
Weiwei Zhang Lida Wu1*
Lida Wu1* Yuchun Gu
Yuchun Gu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol., 15 February 2021
Sec. Cellular Biochemistry
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.630654
This article is part of the Research TopicTransport of Nutrients, Metabolites and Ions linked to Bioenergetics: Relevance to Human PathologyView all 16 articles
Cystic Fibrosis (CF) is an autosomal recessive disorder caused by mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene. CF-related diabetes (CFRD) is one of the most prevalent comorbidities of CF. Altered glucose homeostasis has been reported in CF patients. The mechanism has not been fully elucidated. Besides the consequence of pancreatic endocrine dysfunction, we focus on insulin-responsive tissues and glucose transportation to explain glucose homeostasis alteration in CFRD. Herein, we found that CFTR knockout mice exhibited insulin resistance and glucose tolerance. Furthermore, we demonstrated insulin-induced glucose transporter 4 (GLUT4) translocation to the cell membrane was abnormal in the CFTR knockout mice muscle fibers, suggesting that defective intracellular GLUT4 transportation may be the cause of impaired insulin responses and glucose homeostasis. We further demonstrated that PI(4,5)P2 could rescue CFTR related defective intracellular GLUT4 transportation, and CFTR could regulate PI(4,5)P2 cellular level through PIP5KA, suggesting PI(4,5)P2 is a down-stream signal of CFTR. Our results revealed a new signal mechanism of CFTR in GLUT4 translocation regulation, which helps explain glucose homeostasis alteration in CF patients.
Cystic fibrosis (CF) is one of the most common autosomal recessive disorders in the Caucasian population. It is caused by mutations in the CFTR gene. CF-related diabetes (CFRD) affects 19% of adolescents with CF (Moran et al., 2010), 50% adults CF patients (Moran et al., 2009, 2010), indicating that CFRD is a progressive complication of CF. Despite advances in therapies designed to address the disease’s symptoms, the death rate is still high. As a result, it is crucial to study the mechanism of CFRD thoroughly.
One of the prevailing mechanistic beliefs is that CFRD results from defective insulin secretion of pancreatic β-cell, which is due to the combination of chronic pancreatitis and eventual loss of the islet cells (Cucinotta et al., 1994; Marshall et al., 2005; Costa et al., 2007; Mohan et al., 2009; Guo et al., 2014). Autopsy data confirmed the eventual loss of islet tissue and decreased β-cell numbers in aged CF patients (Iannucci et al., 1984; Lohr et al., 1989). However, most CF patients demonstrate abnormalities in glycaemic control regardless of the class and severity of the CFTR mutation (Wooldridge et al., 2015). Of the thousand CFTR mutations that have been identified, approximately 20 are understood to be disease-causing and are categorized into five classes of mutations of increasing disease severity (Manderson Koivula et al., 2017). CFTR in the regulation of insulin secretion and β cell function has also been reported as the mechanism of CFRD by several recent studies (Stalvey et al., 2006; Edlund et al., 2014; Guo et al., 2014; Fontes et al., 2015), which cannot fully explain the impaired insulin responses and glucose homeostasis reported in CF patients. In addition to the traditional view that CFRD is the consequence of pancreatic endocrine dysfunction, we speculated that CFRD might also affect insulin-responsive tissues’ function.
Several studies found that CF patients with overt diabetes exhibited peripheral and hepatic insulin resistance (Moran et al., 1994; Holl et al., 1995; Hardin et al., 2001; Fontes et al., 2015; Boudreau et al., 2016). However, little is known regarding the mechanism of CFTR loss-of-function in insulin resistance. Insulin facilitates the entry of glucose into muscle, adipose, and several other tissues. The typical mechanism by which cells can take up glucose is by facilitated diffusion through a family of hexose transporters. Skeletal muscle provides the most substantial contribution (∼70%) of insulin-dependent glucose disposal, and this process is mediated by the insulin-responsive glucose transporter 4(GLUT4) (Bouche et al., 2004; Leto and Saltiel, 2012). As a result, we speculated that CFTR deficiency might result in impaired insulin action and GLUT4 dysfunction in skeletal muscle. In this study, we found that CFTR knockout mice exhibited altered glucose homeostasis, and the reason was defective intracellular GLUT4 transportation.
Flexor Digitorum Brevis (FDB) muscle fibers isolation from mice skeletal muscle was previously described (Park et al., 2014). Mouse skeletal muscle cell lines, C2C12 myoblasts, were cultured in DMEM (GIBCO #11965) supplemented with 10% FBS (HyClone #30071.03). C2C12 myotubes differentiation was then induced by switching the medium to DMEM supplemented with 2% HS (GIBCO #26050070). The medium was changed every 24 h. The plasmid of myc–GLUT4–EGFP, bearing the c-Myc epitope tag in the first extracellular loop and GFP at the carboxyl terminus, was purchased from addgene (#52872). According to the manufacturer’s instructions, the plasmid was transfected into HEPG2 and Hela cells using Lipofectamine 2000 (Invitrogen). C2C12 myoblasts and myotubes were transfected with adenovirus (Vigenebio, China) to express myc–GLUT4–EGFP protein.
Cells that express myc–GLUT4–EGFP were cultured with media supplemented with 1% FBS overnight and then were treated with insulin (100 nM) for 30 min. Cells were then fixed with 4% paraformaldehyde, followed by immunocytochemistry using the anti-Myc antibody (Beijing TDY Biotech LTD, #TDY002) and Cy3 -conjugated anti-mouse IgG antibody (Beijing CWBIO, # CW0145S). The cells were imaged with a spin disk confocal system (Revolution XD; ColdSpring Science Corporation) with a CSU-X1 confocal head (Yokogawa) mounted on an inverted microscope (IX81ZDC2; Nikon) with Perfect Focus, using an EMCCD camera (iXon3 DU-897D-C00-#BV; Andor). MetaMorph controlled image acquisition and all other peripherals.
All methods were carried out following relevant guidelines and regulations. All experimental protocols were approved by the University Ethics Committee, Institute of Molecular Medicine, Peking University. All animals received humane care in compliance Guide for the Institutional Animal Care and Use Committee (IACUC). CFTR knockout (KO) and heterozygote (HET) mice were purchased from Jackson Lab(CFTR S489X-) and bred to generate homozygous mutants (KO), heterozygote (HET), and wild-type (WT) mice. CFTR KO mice were genotyped by standard PCR according to the genotyping protocol. Mice were housed under controlled temperature (21°C) on a 12 h light-dark cycle with unrestricted access to food and water.
FABP-hCFTR-CFTR transgenic mice harbor the FABP-hCFTR transgene and a targeted knock-out mutation of the CFTR. This model may be used to assess the effects of the null mutation and may be useful as a mouse model of severe phenotype cystic fibrosis.
For glucose tolerance tests (GTTs), the mice were starved overnight and were given 2.5 g/kg body weight of glucose by IP injection. Blood was then taken from the tail vein before and 15, 30, 60, and 120 min after injection to determine blood glucose by using an Accu-Chek blood glucose monitor (Roche). To determine insulin plasma levels, blood was collected, and the plasma was obtained, and analysis was performed with an insulin ELISA (Millipore). For ITT, 5 h fasted mice were given human insulin (0.75 U/kg, Novolin) by IP injection. The blood glucose concentration was monitored before and after 15, 30, 60, and 120 min after injection. During GTT and ITT, mice were caged with blinded identity and random orders.
The expression and phosphorylation of each protein were analyzed by Western blot analysis. The membrane protein was extracted by using a membrane protein extraction kit (Merck Millipore). The protein was quantified using the Bradford reagent (BioRad). Before loading into the 10% SDS-PAGE, the samples were mixed with a 5x loading buffer and boiled at 98°C for 5 min. After gel electrophoresis, the proteins were then transferred to a PVDF membrane (0.45 μm, Merck Millipore) under 250 mA for 90 min. The membranes were incubated with a blocking buffer (5% non-fat milk in Tris-buffered saline containing 0.5% Tween-20) for 1 h at room temperature and then incubated with the indicated primary antibodies at 4°C overnight. The membrane was incubated with horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. The target protein bands were visualized using Amersham Imager 600 (GE healthcare).
Rabbit polyclonal to GLUT4(ab654,1:2,000), Rabbit polyclonal to PIP5K1A(ab654,1:1,000), and Mouse monoclonal to alpha 1 Sodium Potassium ATPase(ab7671, 5 μg/ml) were purchased from Abcam (United Kingdom); Mouse Monoclonal Antibody to β-tubulin(TDY041, 1:1,000), Mouse Monoclonal Antibody to β-actin(TDY045, 1:1,000), Goat Polyclonal Antibody to Mouse IgG (H + L), HRP Conjugated(S001, 1:1,000), and Goat Polyclonal Antibody to Rabbit IgG (H + L), HRP Conjugated(S004, 1:1,000) were from TDY(Beijing, China).
PI(4,5)P2 were measured by dot-blot analysis. C2C12 myotubes were treated with CFTR (inh)-172(Chemegen) for 24 h. FDB muscle fibers isolated from CFTR KO, HET, WT mice. The whole-cell lysates were then extracted from C2C12 myotubes, and FDB muscle fibers were blotted onto nitrocellulose membranes (Bio-Rad Laboratories). These were probed with PI(4,5)P2 antibody (Abcam, # ab11039) at a 1:500 dilution and detected using a horseradish peroxidase-conjugated secondary antibody (Beijing TDY Biotech LTD, #E009) and were visualized using Amersham Imager 600 (GE healthcare). Experiments were repeated at least three times.
Uptake of [3H]2-deoxyglucose (Sigma, 73698-45-0) was measured in C2C12 myocytes differentiated in 24-well plates. After differentiation, cells were washed twice with DMEM (GIBCO #11965) and incubated in the DMEM for 2 h at 37°C. Cells were then washed twice in PBS and then incubated in KRBB (G-clone, RS1800) in the presence or absence of insulin for 30 min at 37°C. CFTR (inh)-172 was added 30 min before insulin. After insulin treatment, uptake of 10 μM [3H]2-deoxyglucose was measured for 15 min at 37°C. Reactions were terminated by rapidly washing the cells twice with cold KRBB. Cells were then were lysed in 0.1 N NaOH with SDS 0.1%, and radioactivity was determined by liquid scintillation counting and normalized according to the total protein content. Non-specific uptake was determined in the presence of 20 μM cytochalasin B.
The mice’s skeletal muscle was quickly dissected after killing the mice. Before the experiment, mice have fasted for 12 h. Do not disturb the mice and keep them in a resting state. The muscles were incubated in KRBB in the presence or absence of insulin for 30 min at 37°C, uptake of 10 μM [3H]2-deoxyglucose was measured for 15 min at 37°C, as described above.
All statistical calculations were carried out using Graphpad Prism 6. Data are expressed as average ± SEM of at least three independent experiments. The statistical significance was determined using a t-test when comparing two groups and ANOVA when comparing multiple groups. A value of P < 0.05 was considered statistically significant.
To investigate the connection between CFTR and glucose homeostasis, we performed insulin tolerance tests and glucose tolerance tests in 20-weeks-old male WT, heterozygous(HET), and CFTR KO mice (Figures 1A,B). During intraperitoneal insulin tolerance tests, the decrease in blood glucose was less marked in CFTR–/– mice than that of WT and HET mice following 10 mg/ml insulin injection (Figure 1A), suggesting insulin resistance. Moreover, the blood glucose level in CFTR–/– mice was also much higher than that of WT and HET mice 2 h after insulin injection (Figure 1A). It took CFTR–/– mice 60 min to reach the blood glucose summit in response to glucose tolerance tests, while it only took 15 min in WT and HET mice(Figure 1B). However, the glucose AUC was similar among WT and KO groups but slightly lower in the HE group (Figure 1C), indicating that the delay in attaining peak glucose levels during the OGTT suggests delayed glucose absorption. Reduced first, second, and amplifying phase secretion in CF islets have been reported by numerous researchers (Sun et al., 2017; Kelly et al., 2019); however, it was found that serum insulin level of CFTR–/– mice showed no significant difference than WT and HET mice by ELISA (Figure 1D), indicating that the amount of insulin secretion by CFTR–/– mice was normal before CFRD. Notably, maintained in a stable environment with standard chow and water, CFTR–/– mice’s average body weight was much lower than WT and HET mice (Figure 1E). We suspected that the lower body weight of CFTR–/– mice resulted from inadequate glucose intake, but other possibilities include CFTR-related glucosuria or increased metabolic rate, requiring further confirmation. In conclusion, CFTR dysfunction affects mice’s glucose homeostasis and body weight.
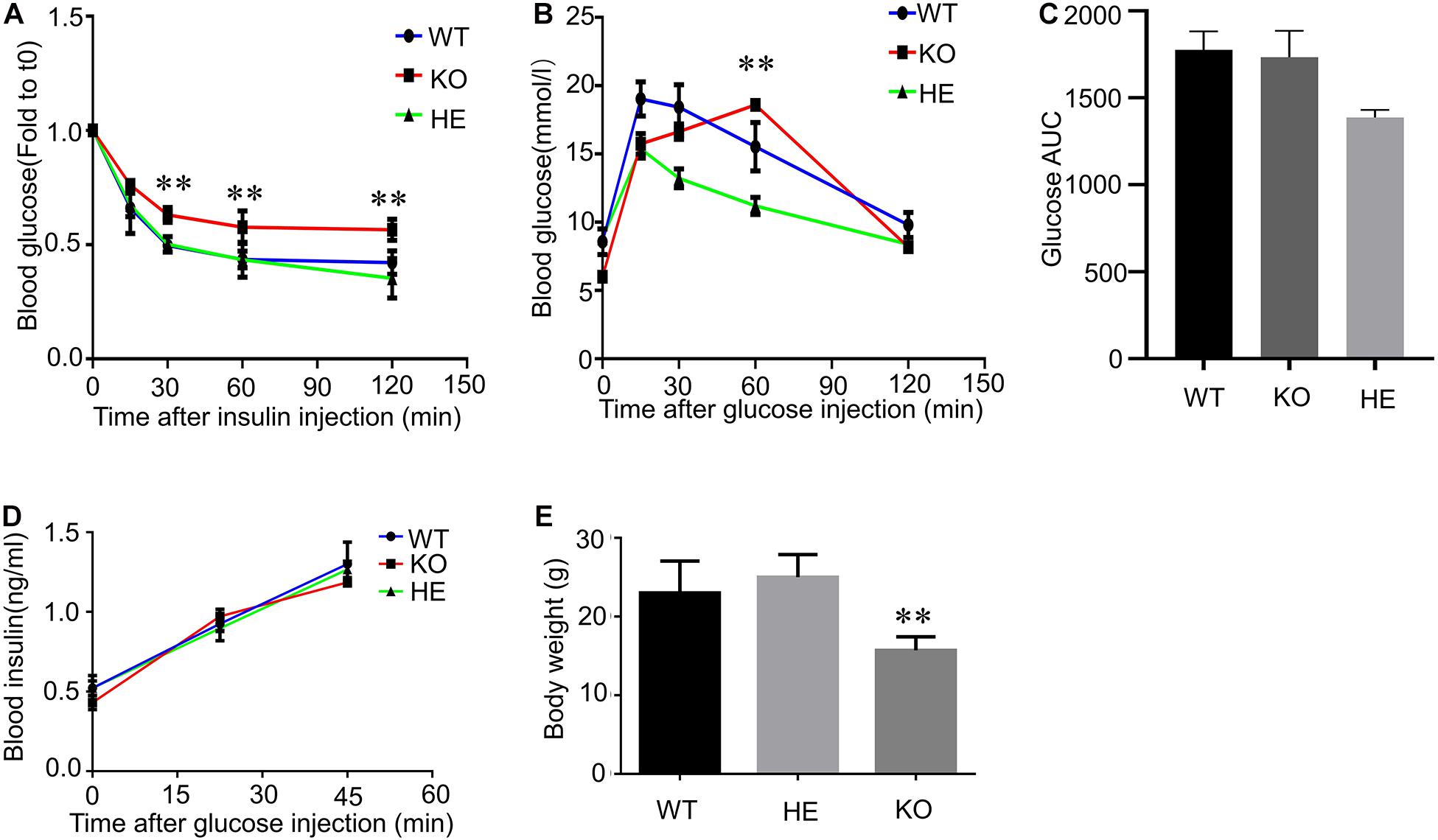
Figure 1. Insulin resistance and Glucose tolerance in CFTR knockout mice. (A) Intraperitoneal insulin tolerance tests were performed in 20-weeks-old male mice WT, HET, and CFTR KO mice. **P < 0.01; NS, not significant. (B) OGTTs were performed in 20-weeks-old male mice WT, HET, and CFTR KO mice. **P < 0.01; NS, not significant. (C) Area under the curve (AUC) of changes in blood glucose levels. (D) Following an injection of 250 mg/ml glucose, serum insulin concentration was measured by ELISA. NS, not significant. (E) Bodyweight was measured in 20-weeks-old male mice WT, HET, and KO mice. Data are mean ± SEM of 7–9 mice per group. **P < 0.01 vs. WT.
When GLUT4 glucose transporters are present in cytoplasmic vesicles, they are useless for transporting glucose. The binding of insulin to receptors on such cells leads rapidly to the fusion of those vesicles with the plasma membrane and the insertion of the glucose transporters, thereby giving the cell the ability to take up glucose efficiently. As a result, glucose uptake depends on insulin-stimulated membrane translocation of the GLUT4. To investigate the relation between CFTR and GLUT4, GLUT4 cell membrane expression was measured by western blot in mice skeletal muscle fibers and C2C12 myotubes. The Flexor Digitorum Brevis (FDB) muscle fibers total membrane protein were immediately isolated from WT, HET, and CFTR KO mice 30 min after insulin injection stimulation. Evident membrane translocation of GLUT4 was found in WT and HET mice muscle fibers with insulin stimulation (Figure 2A). However, there was no membrane expression of GLUT4 in CFTR–/– mice with or without insulin stimulation (Figure 2A). Consistent with this, the insulin-stimulated GLUT4 cell surface translocation was also blocked by a specific CFTR inhibitor [CFTR (inh)-172] in differentiated C2C12 myotubes (Figure 2B). To directly show the translocation of GLUT4, C2C12 myoblasts and myotubes were transiently transfected with myc–GLUT4–EGFP fusion plasmids. Myc epitope was inserted in the first exofacial loop of GLUT4 N terminus and presented outside of the cell membrane, which could be detected by the antibody without perforation. C2C12 Cells were then fixed (without perforation) and labeled with an anti-Myc antibody followed by Texas Red secondary to track the membrane translocation of GLUT4 (red). We found that GLUT4 was expressed in the cytoplasm (Figures 2C,D, Control), and insulin could induce translocation of GLUT4 to cell membranes both in C2C12 myoblasts and myotubes (Figures 2C,D). However, this process could be blocked by CFTR (inh)-172 (Figures 2C,D). The effects of CFTR (inh)-172 on insulin-stimulated glucose uptake activity were also tested in differentiated C2C12 myotubes and freshly isolated Flexor Digitorum Brevis (FDB) muscle fibers cells. We found that CFTR (inh)-172 significantly decreased 2-DG uptake in insulin-stimulated C2C12 myotubes (Figure 2E) and freshly isolated Flexor Digitorum Brevis (FDB) muscle fibers cells (Figure 2F).
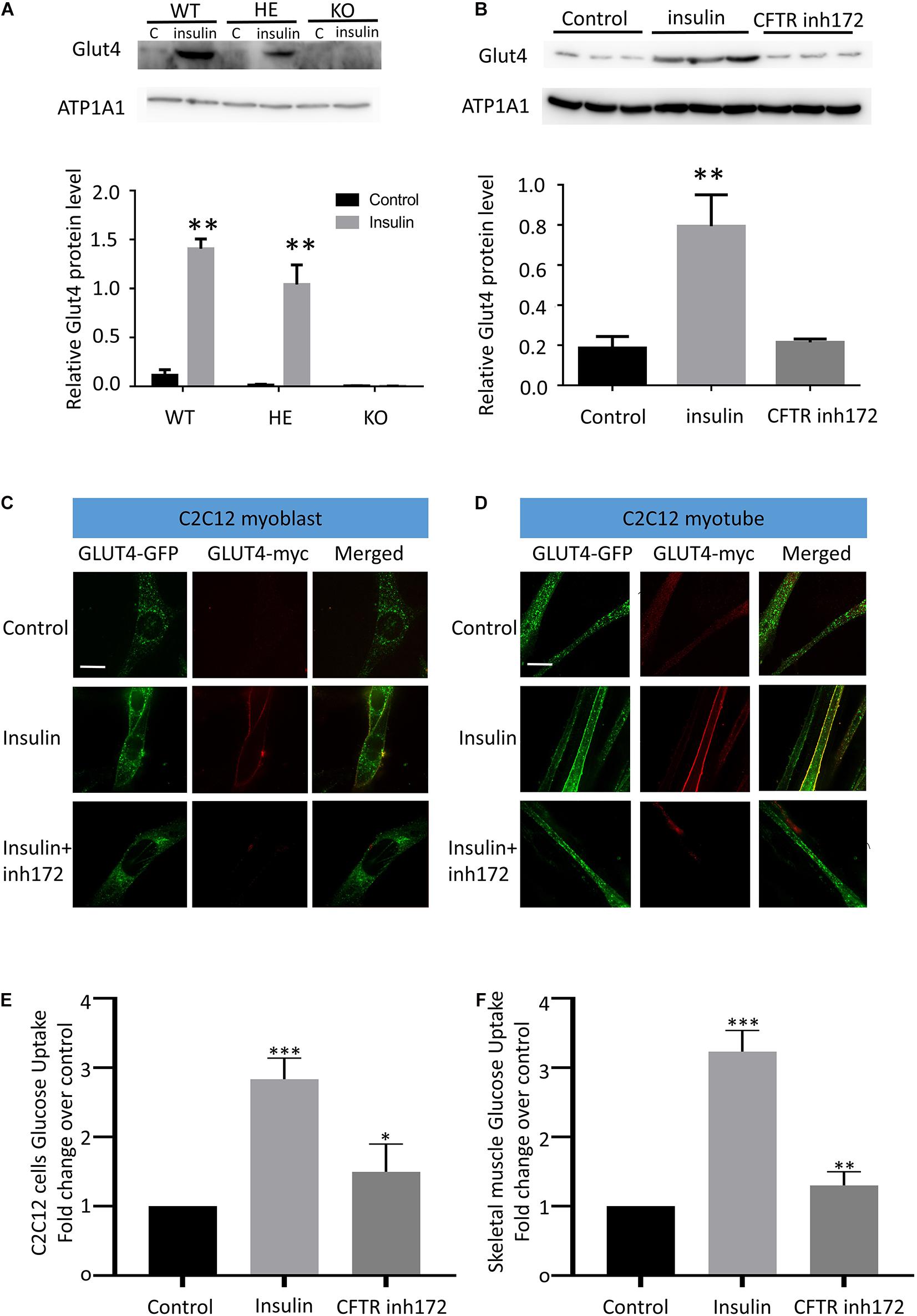
Figure 2. CFTR regulates GLUT4 membrane translocation and glucose homeostasis. (A) The membrane localization of GLUT4 in CFTR KO mice FDB muscle after injection of 10 mg/ml insulin was measured by Western blot. Images are representative of triplicate experiments with similar results. **P < 0.01 compared with control (n = 3). (B) The membrane expression of GLUT4 in C2C12 myotubes was measured by Western blot. **P < 0.01 compared with control (n = 3). (C) myc-GLUT4- GFP was transfected into C2C12 myoblasts by adenoviruses. The cells were stimulated with insulin (100 nM) for 30 min. GLUT4 translocation to the plasma membrane was detected by confocal microscopy. Glut4-GFP(green), Glut4-myc(red). Scale bar, 10 μm. (D) GLUT4 translocation to the plasma membrane of differentiated C2C12 myotubes was detected by confocal microscopy. Glut4-GFP(green), Glut4-myc(red). Scale bar, 10 μm. (E) The glucose uptake assay of C2C12 cells. Data are mean ± SD values for two independent assays performed in triplicate. ***P < 0.001, *P < 0.05. (F) The glucose uptake assay of Flexor Digitorum Brevis muscle fibers cells. Data are mean ± SD values for two independent assays performed in triplicate. ***P < 0.001, **P < 0.01.
Some tissues, such as the liver, do not require insulin for efficient uptake of glucose; this is because these cells do not use GLUT4 to import glucose, but rather, GLUT1 and GLUT2 are not insulin-dependent. We wondered if similar results could be observed in liver cells. HEPG2 cells were transiently transfected with myc–GLUT4–EGFP fusion plasmids. HEPG2 Cells were then fixed (without perforation) and labeled with an anti-Myc antibody followed by Texas Red secondary to track the membrane translocation of GLUT4 (red). It was found that CFTR (inh)-172 could also block insulin-induced GLUT4 membrane translocation in HEPG2 (Supplementary Figure S1A) and Hela cells (Supplementary Figure S1B). To further confirm CFTR’s role on GLUT4 translocation, as Hela cells endogenously express CFTR (Billet et al., 2017), we knocked out CFTR in Hela cells by CRISPR/Case9 (Supplementary Figure S1C). It was found that insulin could no longer induce GLUT4 translocation to the cell membrane in CFTR–/– cells (Supplementary Figure S1D). Taken together, these results suggested that CFTR was required in insulin-induced membrane translocation of GLUT4.
Several studies indicated that Rab8A and Rab13 could also contribute to the translocation of GLUT4 (Sano et al., 2007; Randhawa et al., 2008; Sun et al., 2010). Rab8A or Rab13 was co-transfected with myc-GLUT4-EGFP in Hela cells; we found that Rab8A or Rab13 could not rescue GLUT4 membrane translocation under CFTR (inh)-172 treatment (Figure 3A), suggesting Rab8A or Rab13 is not involved in this process. PI(4,5)P2 mediated insulin-induced actin remodeling is another crucial component in GLUT4 translocation (Kanzaki and Pessin, 2001; Kanzaki et al., 2004; Leto and Saltiel, 2012; Lim et al., 2015). To identify the role of PI(4,5)P2 in the process of GLUT4 translocation, C2C12 myotubes were transiently transfected with myc–GLUT4–EGFP fusion plasmids. Insulin-induced GLUT4 translocation in C2C12 myotubes was blocked after endogenous PI(4,5)P2 was depleted by application of both PI3K inhibitor (wortmanin) and PLC agonist (Figure 3B). The defective GLUT4 membrane translocation induced by CFTR (inh)-172 could also be rescued by exogenous PI(4,5)P2 (Figure 3B). As CFTR is a chloride channel, we suspected whether the chloride channel property of CFTR was also involved with GLUT4 translocation. By replacing the chloride with arginine in bath solution, we found that the translocation of GLUT4 was still blocked by CFTR (inh)-172 (Supplementary Figure S2A). In conclusion, these results indicated that CFTR regulated GLUT4 membrane translocation by PI(4,5)P2.
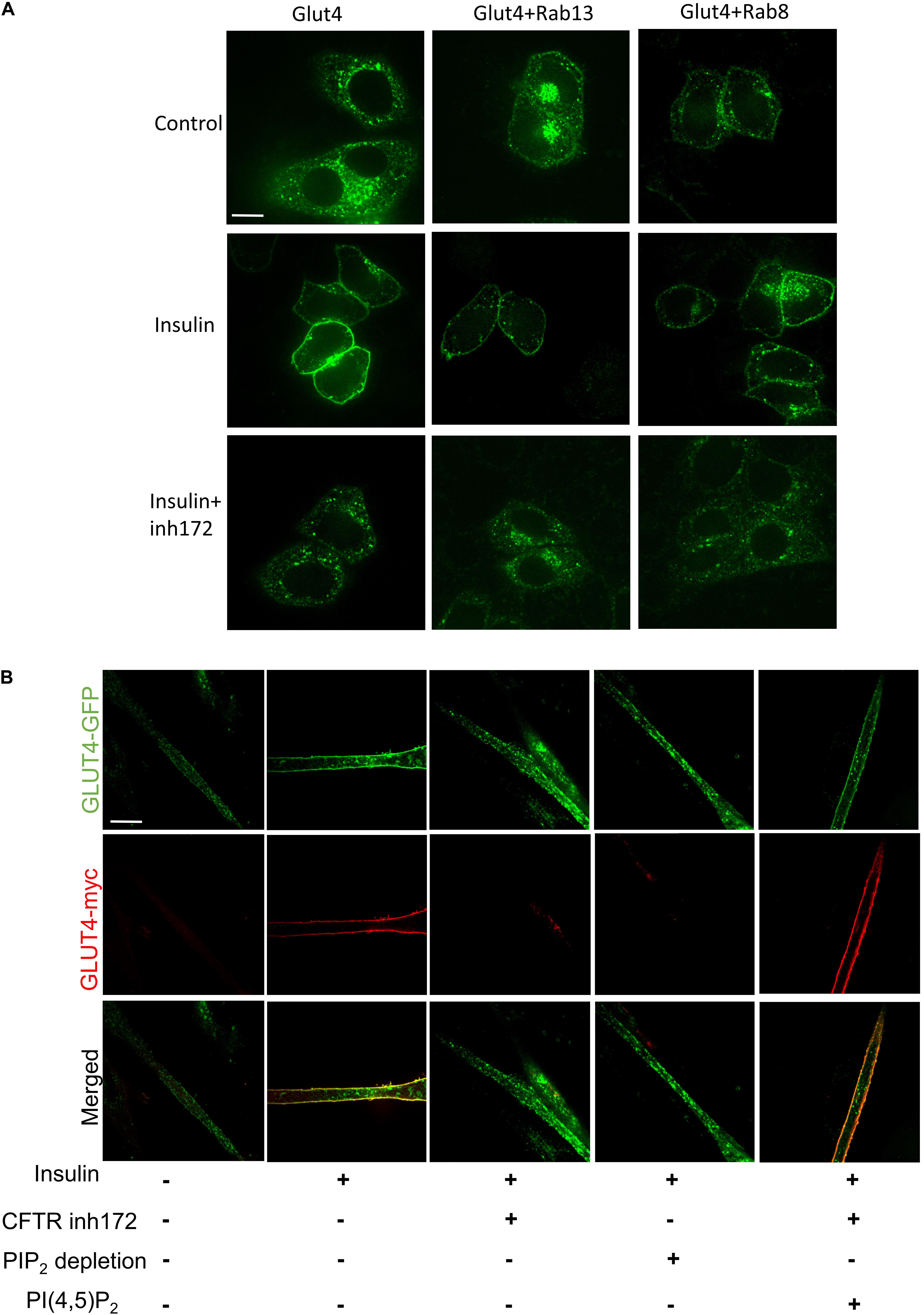
Figure 3. PI(4,5)P2 rescues CFTR related defective GLUT4 translocation. (A) The membrane localization of GLUT4 in Hela cells after overexpression of Rab8 and Rab13 was detected by confocal microscopy. Glut4 (green), Scale bar, 10 μm. (B) C2C12 myotubes were treated with insulin, CFTR inh172, PIP2 depletion agents, or PI(4,5)P2. PIP2 depletion agents are consist of PI3K inhibitor (1 μM wortmanin) and PLC agonist (50 μM m-3M3FBS). GLUT4 translocation to the plasma membrane was detected by confocal microscopy. Glut4-GFP (green), Glut4-myc (red). Scale bar, 10 μm.
To confirm the speculation that inhibition of CFTR might affect cellular PI(4,5)P2 concentration. PI(4,5)P2 concentration was detected by dot-blot analysis. C2C12 myotubes were treated with CFTR (inh)-172 for 24 h, and we found that CFTR (inh)-172 could significantly down-regulate PI(4,5)P2 concentration (Figure 4A). We also measured the concentration of PI(4,5)P2 in FDB muscle fibers isolated from CFTR WT, HET, and KO mice. The concentration of PI(4,5)P2 in CFTR–/– mice FDB muscle fibers was significantly lower than WT and HET mice (Figure 4B). PIP5K1A induces the phosphorylation of phosphatidylinositol 4-phosphate (PtdIns4P) to generation PI(4,5)P2. Studies indicated that that PI(4,5)P2 generation by PIP5K1A was associated with GLUT4 vesicle recycling (Kanzaki et al., 2004) and autophagic lysosome reformation (Rong et al., 2012). To understand how CFTR regulates PI(4,5)P2 expression, the PIP5K1A expression level was measured. We found that CFTR (inh)-172 induced a reduction of PIP5K1A expression in C2C12 myotubes (Figure 4C). The expression of PIP5K1A was also significantly decreased in CFTR–/– FDB muscle fibers (Figure 4D). These results suggest that the effect of CFTR on PI(4,5)P2 concentration is via the regulation of PIP5K1A expression.
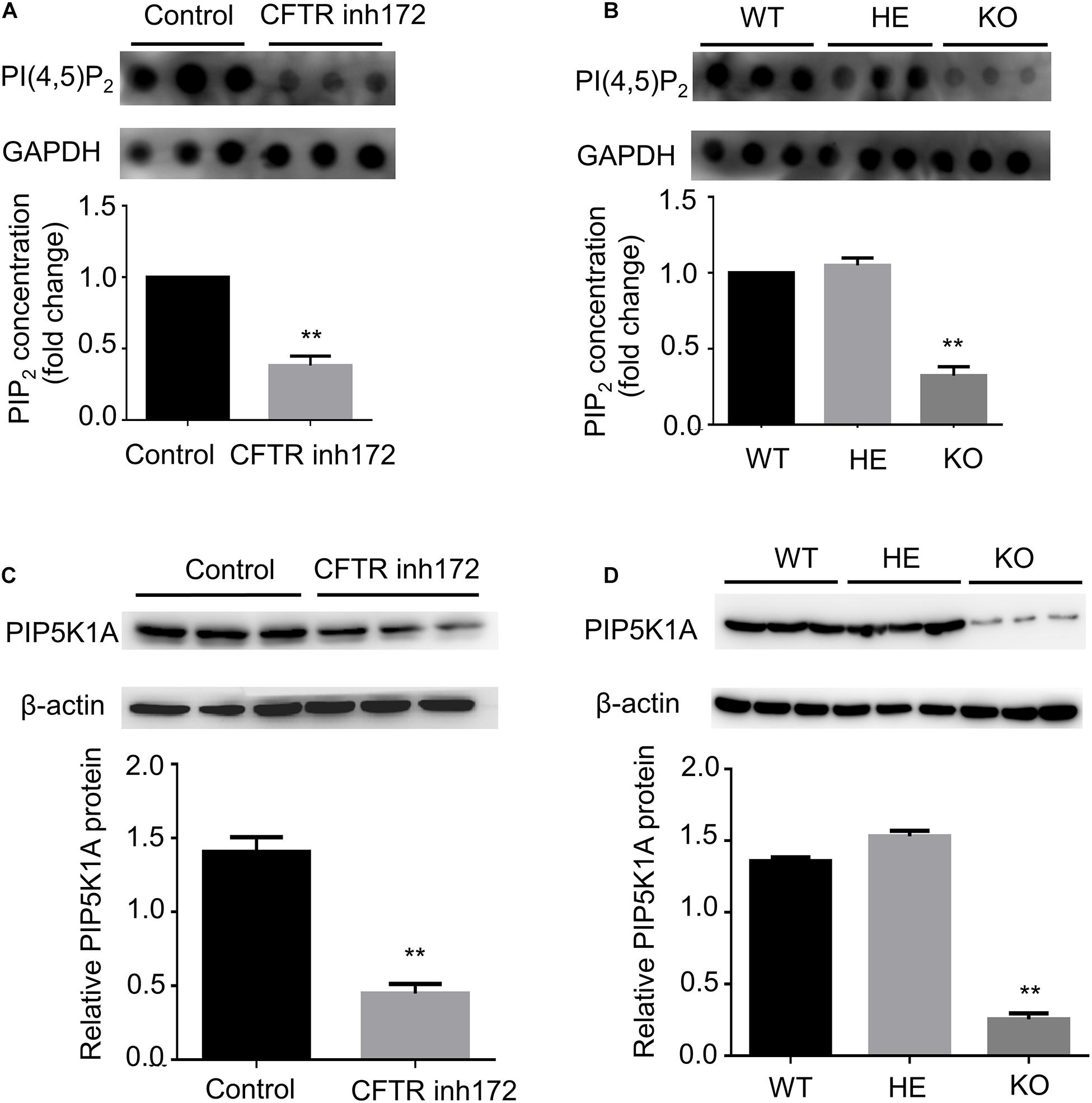
Figure 4. CFTR regulates PIP5KA expression and PI(4,5)P2 concentration. (A) C2C12 myotubes were treated with CFTR inh172, and the concentration of PI(4,5)P2 was measured by dot blot. **P < 0.01 compared with control (n = 3). (B) The concentration of PI(4,5)P2 in mice FDB muscle was measured by dot blot. **P < 0.01 compared with control (n = 3). (C) C2C12 myotubes were treated with CFTR inh172, and the protein level of PIP5KA was measured by western blot. **P < 0.01 compared with control (n = 3). (D) The protein level of PIP5KA in mice FDB muscle was measured by western blot. **P < 0.01 compared with control (n = 3).
In this study, we discovered a previously unrecognized regulation effect of CFTR to the GLUT4 cellular translocation, which provides a new perspective on the mechanism of CFRD. It was found that CFTR–/– mice exhibited normal insulin secretion but reduced insulin sensitivity and glucose tolerance. Insulin facilitates the entry of glucose into muscle through GLUT4. Meanwhile, Insulin-induced GLUT4 translocation to the cell membrane was also blocked in CFTR–/– mice, suggesting glucose homeostasis alteration from defective intracellular GLUT4 transportation. Unexpectedly, PI(4,5)P2 could rescue the effect of CFTR deficiency. Furthermore, it was found that CFTR deficiency leads to a significantly decreased expression of PIP5K1A, which reflected on PI(4,5)P2 concentration. Taken together, we discovered a new signal pathway of CFTR/GLUT4, which helps explain the glucose homeostasis disorder in CF patients.
To explain CFRD, several studies have shown that CFTR played a vital role in the regulation of insulin secretion and β cell function (Tofe et al., 2005; Noronha et al., 2011; Konrad et al., 2013; Edlund et al., 2014; Guo et al., 2014). Some studies suggested that insulin resistance was not an essential determinant in CFRD compared with insulin secretion (Cucinotta et al., 1994; Yung et al., 2002). Other studies proved that insulin resistance was highly involved in CFRD, and impaired insulin responses were also reported in CF patients (Hardin et al., 1997; Hardin et al., 2001; Fontes et al., 2015). Our data show that insulin secretion is normal in CFTR–/– mice (Figure 1C); the problem is insulin sensitivity (Figure 1A) and glucose absorption (Figure 1B). Consistent with previous studies (Paroni et al., 2013; Fontes et al., 2015), we also found that the body weight was lower in CFTR–/– mice than WT and HET controls (Figure 1D). Gradual pancreatic destruction in cystic fibrosis causes progressive insulin deficiency. Our results show that in the early stage of CFRD, insulin secretion is expected, so insulin deficiency may not be due to the destruction of pancreatic islets, leading to insufficient insulin secretion, but because CFTR dysfunction affects the insulin-responsive tissues, which manifests as insulin deficiency. In summary, we believe that CFTR affects the pancreas functionality and the insulin-responsive tissues; both contribute to cystic fibrosis-related diabetes (CFRD).
How does CFTR knock out affected insulin sensitivity and glucose tolerance? By binding and activating its cell-surface receptor, insulin triggers signaling cascades that regulate many cellular processes, including stimulating the muscle to dispose of dietary glucose (Richter and Hargreaves, 2013). Insulin stimulation of glucose uptake into muscle depends on the mobilization of GLUT4 to the plasma membrane (Lauritzen et al., 2010; Klip et al., 2014). As Akt2 is an essential kinase in insulin resistance (Cho et al., 2001; McCurdy and Cartee, 2005), we investigate the phosphorylation of Akt2 in CFTR–/– mice. Surprisingly, CFTR deficiency showed no connection with phosphorylation of Akt2 and its downstream protein AS160 (data not shown). Furthermore, other regulators, such as Rab8A and Rab13, also showed no significant impact on CFTR deficiency-induced GLUT4 translocation (Figure 3A). As a result, we focused on the “railway” of protein trafficking in the cell, and studies showed that actin remodeling also played an essential regulatory role in insulin-induced GLUT4 translocation (Kanzaki and Pessin, 2001; Tong et al., 2001). The role of actin dynamic regulated by PI(4,5)P2 in GLUT4 translocation has been proved by recent studies (Kanzaki and Pessin, 2001; Kanzaki et al., 2004; Leto and Saltiel, 2012; Lim et al., 2015). Our data show that the depletion of PI(4,5)P2 blocks GLUT4 trafficking to the cell membrane, and exogenous PI(4,5)P2 can rescue the effect of CFTR deficiency. Thus, we conclude that CFTR in GLUT4 translocation regulation is mediated by PI(4,5)P2. In addition to insulin-stimulation, the translocation of GLUT4 to the plasma membrane is also affected by skeletal muscle contraction. Whether CFTR has a role in skeletal muscle contraction needs further clarification.
As CFTR is an ion channel, which makes no sense in catalyzing PI(4,5)P2 generation. We proved that the chloride channel property of CFTR was not involved with GLUT4 translocation; only the protein itself is involved in translocation. Therefore, we speculated that CFTR allosteric affects some kinase proteins in the regulation of PI(4,5)P2 generation. The differential gene expression induced by CFTR deficiency has been reported by several studies (Ichikawa et al., 2000; Ogilvie et al., 2011; Chen et al., 2012). We suspected that inhibition or knockdown of CFTR might lead to decreased PIP5K1A expression, which regulates PI(4,5)P2 generation. We found that the expression of PIP5K1A in CFTR–/– mice and CFTR (inh)-172 treated C2C12 was significantly reduced, causing lower PI(4,5)P2 concentration. In conclusion, PIP5K1A is one of the regulators between CFTR and PI(4,5)P2, but further investigation is needed to assess how CFTR deficiency regulates PIP5K1A expression.
In summary, our results suggest a new mechanism of glucose homeostasis disorder in CF patients. These findings identify the role of GLUT4 translocation in CFTR related glucose homeostasis disorder in CF patients, which provides a new potential treatment strategy.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
The animal study was reviewed and approved by the University Ethics Committee, Institute of Molecular Medicine, Peking University.
YG and LW conceived and coordinated the study. JG and LW conducted molecular experiments and wrote the manuscript. WZ and JG performed the imaging-related experiments. All authors made critical contributions to data analysis, interpretation, discussion, and manuscript preparation and approved the final version.
This work was supported by the National Basic Research Program of China (973 Program) (2013CB531206 and 2012CB517803) and the National Natural Science Foundation of China (81170236 and 31127001).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.630654/full#supplementary-material
Supplementary Figure 1 | Insulin-induced GLUT4 translocation in HEPG2 and Hela cells.
Supplementary Figure 2 | Chloride channel property of CFTR and insulin-induced GLUT4 translocation.
CF, Cystic Fibrosis; CFTR, Cystic Fibrosis Transmembrane Conductance Regulator; CFRD, Cystic Fibrosis related diabetes; GLUT4, Glucose Transporter 4.
Billet, A., Froux, L., Hanrahan, J. W., and Becq, F. (2017). Development of automated patch clamp technique to investigate cftr chloride channel function. Front. Pharmacol. 8:195. doi: 10.3389/fphar.2017.00195
Bouche, C., Serdy, S., Kahn, C. R., and Goldfine, A. B. (2004). The cellular fate of glucose and its relevance in type 2 diabetes. Endocr. Rev. 25, 807–830. doi: 10.1210/er.2003-0026
Boudreau, V., Coriati, A., Hammana, I., Ziai, S., Desjardins, K., Berthiaume, Y., et al. (2016). Variation of glucose tolerance in adult patients with cystic fibrosis: what is the potential contribution of insulin sensitivity? J. Cystic Fibr. 15, 839–845. doi: 10.1016/j.jcf.2016.04.004
Chen, J., Fok, K. L., Chen, H., Zhang, X. H., Xu, W. M., and Chan, H. C. (2012). Cryptorchidism-induced CFTR down-regulation results in disruption of testicular tight junctions through up-regulation of NF-kappaB/COX-2/PGE2. Hum. Reprod. 27, 2585–2597. doi: 10.1093/humrep/des254
Cho, H., Mu, J., Kim, J. K., Thorvaldsen, J. L., Chu, Q., Crenshaw, E. B. III, et al. (2001). Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science 292, 1728–1731. doi: 10.1126/science.292.5522.1728
Costa, M., Potvin, S., Hammana, I., Malet, A., Berthiaume, Y., Jeanneret, A., et al. (2007). Increased glucose excursion in cystic fibrosis and its association with a worse clinical status. J. Cystic Fibr. 6, 376–383. doi: 10.1016/j.jcf.2007.02.005
Cucinotta, D., De Luca, F., Gigante, A., Arrigo, T., Di Benedetto, A., Tedeschi, A., et al. (1994). No changes of insulin sensitivity in cystic fibrosis patients with different degrees of glucose tolerance: an epidemiological and longitudinal study. Eur. J Endocrinol. 130, 253–258. doi: 10.1530/eje.0.1300253
Edlund, A., Esguerra, J. L., Wendt, A., Flodstrom-Tullberg, M., and Eliasson, L. (2014). CFTR and Anoctamin 1 (ANO1) contribute to cAMP amplified exocytosis and insulin secretion in human and murine pancreatic beta-cells. BMC Med. 12:87. doi: 10.1186/1741-7015-12-87
Fontes, G., Ghislain, J., Benterki, I., Zarrouki, B., Trudel, D., Berthiaume, Y., et al. (2015). The DeltaF508 mutation in the cystic fibrosis transmembrane conductance regulator is associated with progressive insulin resistance and decreased functional beta-cell mass in mice. Diabetes 64, 4112–4122. doi: 10.2337/db14-0810
Guo, J. H., Chen, H., Ruan, Y. C., Zhang, X. L., Zhang, X. H., Fok, K. L., et al. (2014). Glucose-induced electrical activities and insulin secretion in pancreatic islet beta-cells are modulated by CFTR. Nat. Commun. 5:4420.
Hardin, D. S., LeBlanc, A., Lukenbough, S., and Seilheimer, D. K. (1997). Insulin resistance is associated with decreased clinical status in cystic fibrosis. J. Pediatr. 130, 948–956. doi: 10.1016/s0022-3476(97)70282-8
Hardin, D. S., Leblanc, A., Marshall, G., and Seilheimer, D. K. (2001). Mechanisms of insulin resistance in cystic fibrosis. Am. J. Physiol. Endocrinol. Metab. 281, E1022–E1028.
Holl, R. W., Heinze, E., Wolf, A., Rank, M., and Teller, W. M. (1995). Reduced pancreatic insulin release and reduced peripheral insulin sensitivity contribute to hyperglycaemia in cystic fibrosis. Eur. J. Pediatr. 154, 356–361. doi: 10.1007/s004310050303
Iannucci, A., Mukai, K., Johnson, D., and Burke, B. (1984). Endocrine pancreas in cystic fibrosis: an immunohistochemical study. Hum. Pathol. 15, 278–284. doi: 10.1016/s0046-8177(84)80191-4
Ichikawa, J. K., Norris, A., Bangera, M. G., Geiss, G. K., van ‘t Wout, A. B., Bumgarner, R. E., et al. (2000). Interaction of Pseudomonas aeruginosa with epithelial cells: identification of differentially regulated genes by expression microarray analysis of human cDNAs. Proc. Natl. Acad. Sci. U.S.A. 97, 9659–9664. doi: 10.1073/pnas.160140297
Kanzaki, M., Furukawa, M., Raab, W., and Pessin, J. E. (2004). Phosphatidylinositol 4,5-bisphosphate regulates adipocyte actin dynamics and GLUT4 vesicle recycling. J. Biol. Chem. 279, 30622–30633. doi: 10.1074/jbc.m401443200
Kanzaki, M., and Pessin, J. E. (2001). Insulin-stimulated GLUT4 translocation in adipocytes is dependent upon cortical actin remodeling. J. Biol. Chem. 276, 42436–42444. doi: 10.1074/jbc.m108297200
Kelly, A., De Leon, D. D., Sheikh, S., Camburn, D., Kubrak, C., Peleckis, A. J., et al. (2019). Islet hormone and incretin secretion in cystic fibrosis after four months of ivacaftor therapy. Am. J. Respir. Crit. Care Med. 199, 342–351. doi: 10.1164/rccm.201806-1018oc
Klip, A., Sun, Y., Chiu, T. T., and Foley, K. P. (2014). Signal transduction meets vesicle traffic: the software and hardware of GLUT4 translocation. Am. J. Physiol. Cell Physiol. 306, C879–C886.
Konrad, K., Scheuing, N., Badenhoop, K., Borkenstein, M. H., Gohlke, B., Schofl, C., et al. (2013). Cystic fibrosis-related diabetes compared with type 1 and type 2 diabetes in adults. Diabetes 29, 568–575.
Lauritzen, H. P., Galbo, H., Toyoda, T., and Goodyear, L. J. (2010). Kinetics of contraction-induced GLUT4 translocation in skeletal muscle fibers from living mice. Diabetes 59, 2134–2144. doi: 10.2337/db10-0233
Leto, D., and Saltiel, A. R. (2012). Regulation of glucose transport by insulin: traffic control of GLUT4. Nat. Rev. Mol. Cell Biol. 13, 383–396. doi: 10.1038/nrm3351
Lim, C. Y., Bi, X., Wu, D., Kim, J. B., Gunning, P. W., Hong, W., et al. (2015). Tropomodulin3 is a novel Akt2 effector regulating insulin-stimulated GLUT4 exocytosis through cortical actin remodeling. Nat. Commun. 6:5951.
Lohr, M., Goertchen, P., Nizze, H., Gould, N. S., Gould, V. E., Oberholzer, M., et al. (1989). Cystic fibrosis associated islet changes may provide a basis for diabetes. An immunocytochemical and morphometrical study. Virchows Arch. A Pathol. Anat. Histopathol. 414, 179–185. doi: 10.1007/bf00718598
Manderson Koivula, F. N., McClenaghan, N. H., Harper, A. G. S., and Kelly, C. (2017). Correction to: islet-intrinsic effects of CFTR mutation. Diabetologia 60, 2544. doi: 10.1007/s00125-017-4474-1
Marshall, B. C., Butler, S. M., Stoddard, M., Moran, A. M., Liou, T. G., and Morgan, W. J. (2005). Epidemiology of cystic fibrosis-related diabetes. J. Pediatr. 146, 681–687.
McCurdy, C. E., and Cartee, G. D. (2005). Akt2 is essential for the full effect of calorie restriction on insulin-stimulated glucose uptake in skeletal muscle. Diabetes 54, 1349–1356. doi: 10.2337/diabetes.54.5.1349
Mohan, K., Miller, H., Dyce, P., Grainger, R., Hughes, R., Vora, J., et al. (2009). Mechanisms of glucose intolerance in cystic fibrosis. Diabet. Med. 26, 582–588. doi: 10.1111/j.1464-5491.2009.02738.x
Moran, A., Becker, D., Casella, S. J., Gottlieb, P. A., Kirkman, M. S., Marshall, B. C., et al. (2010). Committee CFRD consensus conference committee. Epidemiology, pathophysiology, and prognostic implications of cystic fibrosis-related diabetes: a technical review. Diabet. Care 33, 2677–2683. doi: 10.2337/dc10-1279
Moran, A., Dunitz, J., Nathan, B., Saeed, A., Holme, B., and Thomas, W. (2009). Cystic fibrosis-related diabetes: current trends in prevalence, incidence, and mortality. Diabet. Care 32, 1626–1631. doi: 10.2337/dc09-0586
Moran, A., Pyzdrowski, K. L., Weinreb, J., Kahn, B. B., Smith, S. A., Adams, K. S., et al. (1994). Insulin sensitivity in cystic fibrosis. Diabetes 43, 1020–1026.
Noronha, R. M., Calliari, L. E., Damaceno, N., Muramatu, L. H., and Monte, O. (2011). Update on diagnosis and monitoring of cystic fibrosis-related diabetes mellitus (CFRD). Arq. Bras. Endocrinol. Metabol. 55, 613–621. doi: 10.1590/s0004-27302011000800016
Ogilvie, V., Passmore, M., Hyndman, L., Jones, L., Stevenson, B., Wilson, A., et al. (2011). Differential global gene expression in cystic fibrosis nasal and bronchial epithelium. Genomics 98, 327–336. doi: 10.1016/j.ygeno.2011.06.008
Park, K. H., Weisleder, N., Zhou, J., Gumpper, K., Zhou, X., Duann, P., et al. (2014). Assessment of calcium sparks in intact skeletal muscle fibers. J. Vis. Exp. 84:e50898.
Paroni, M., Moalli, F., Nebuloni, M., Pasqualini, F., Bonfield, T., Nonis, A., et al. (2013). Response of CFTR-deficient mice to long-term chronic Pseudomonas aeruginosa infection and PTX3 therapy. J. Infect. Dis. 208, 130–138. doi: 10.1093/infdis/jis636
Randhawa, V. K., Ishikura, S., Talior-Volodarsky, I., Cheng, A. W., Patel, N., Hartwig, J. H., et al. (2008). GLUT4 vesicle recruitment and fusion are differentially regulated by Rac, AS160, and Rab8A in muscle cells. J. Biol. Chem. 283, 27208–27219. doi: 10.1074/jbc.m804282200
Richter, E. A., and Hargreaves, M. (2013). Exercise. GLUT4, and skeletal muscle glucose uptake. Physiol. Rev. 93, 993–1017. doi: 10.1152/physrev.00038.2012
Rong, Y., Liu, M., Ma, L., Du, W., Zhang, H., Tian, Y., et al. (2012). Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nat. Cell Biol. 14, 924–934. doi: 10.1038/ncb2557
Sano, H., Eguez, L., Teruel, M. N., Fukuda, M., Chuang, T. D., Chavez, J. A., et al. (2007). Rab10, a target of the AS160 Rab GAP, is required for insulin-stimulated translocation of GLUT4 to the adipocyte plasma membrane. Cell Metab. 5, 293–303. doi: 10.1016/j.cmet.2007.03.001
Stalvey, M. S., Muller, C., Schatz, D. A., Wasserfall, C. H., Campbell-Thompson, M. L., Theriaque, D. W., et al. (2006). Cystic fibrosis transmembrane conductance regulator deficiency exacerbates islet cell dysfunction after beta-cell injury. Diabetes 55, 1939–1945. doi: 10.2337/db05-1647
Sun, X., Yi, Y., Xie, W., Liang, B., Winter, M. C., He, N., et al. (2017). CFTR influences beta cell function and insulin secretion through non-cell autonomous exocrine-derived factors. Endocrinology 158, 3325–3338. doi: 10.1210/en.2017-00187
Sun, Y., Bilan, P. J., Liu, Z., and Klip, A. (2010). Rab8A and Rab13 are activated by insulin and regulate GLUT4 translocation in muscle cells. Proc. Natl. Acad. Sci. U.S.A. 107, 19909–19914. doi: 10.1073/pnas.1009523107
Tofe, S., Moreno, J. C., Maiz, L., Alonso, M., Escobar, H., and Barrio, R. (2005). Insulin-secretion abnormalities and clinical deterioration related to impaired glucose tolerance in cystic fibrosis. Eur. J. Endocrinol. 152, 241–247. doi: 10.1530/eje.1.01836
Tong, P., Khayat, Z. A., Huang, C., Patel, N., Ueyama, A., and Klip, A. (2001). Insulin-induced cortical actin remodeling promotes GLUT4 insertion at muscle cell membrane ruffles. J. Clin. Invest. 108, 371–381. doi: 10.1172/jci200112348
Wooldridge, J. L., Szczesniak, R. D., Fenchel, M. C., and Elder, D. A. (2015). Insulin secretion abnormalities in exocrine pancreatic sufficient cystic fibrosis patients. J. Cystic Fibr. 14, 792–797. doi: 10.1016/j.jcf.2015.02.009
Keywords: cystic fibrosis, CFTR, CF-related diabetes, glucose homeostasis disorder, GLUT4
Citation: Gu J, Zhang W, Wu L and Gu Y (2021) CFTR Deficiency Affects Glucose Homeostasis via Regulating GLUT4 Plasma Membrane Transportation. Front. Cell Dev. Biol. 9:630654. doi: 10.3389/fcell.2021.630654
Received: 19 November 2020; Accepted: 27 January 2021;
Published: 15 February 2021.
Edited by:
Cesare Indiveri, University of Calabria, ItalyReviewed by:
Valeria Rachela Villella, European Institute for Research in Cystic Fibrosis (IERFC), ItalyCopyright © 2021 Gu, Zhang, Wu and Gu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lida Wu, d2xkcGFwZXJAcGt1LmVkdS5jbg==; d2xkcGFwZXJAZ21haWwuY29t; Yuchun Gu, eWNndUBwa3UuZWR1LmNu
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.